[level-membership-for-critical-care-medicine-category]
124 Agents with Primary Activity Against Gram-Positive Bacteria
The causes of nosocomial infections have changed in recent years. A 25-year study of nosocomial bacteremia demonstrated a change from Staphylococcus aureus and gram-negative bacilli as the predominant pathogens during the 1970s and 1980s to coagulase-negative staphylococci and Enterococcus, along with S. aureus and Pseudomonas aeruginosa, as the most common contemporary pathogens.1 The EPIC II study in 2007 demonstrated gram-positive organisms were associated with 47% of infections in the ICU.2 There can also be differences in the predominance of pathogens in different ICUs and different types of nosocomial infections. Nosocomial bacteremias are caused most often by coagulase-negative staphylococci and S. aureus in the medical ICU.3 S. aureus is the most common pathogen associated with nosocomial pneumonia and the fourth most common cause of skin and soft-tissue infections.3 Along with the increase in prevalence of gram-positive cocci in the ICU, staphylococci are becoming multidrug resistant. This chapter addresses gram-positive organisms and resistance issues associated with each of the antimicrobials with activity against these pathogens.
 Vancomycin
Vancomycin
Vancomycin was discovered in 1956 and marketed in 1958. Early preparations of the drug contained pyrogens and impurities that produced a brownish, muddy appearance that provided vancomycin’s nickname, “Mississippi mud.” In addition, these pyrogens and impurities caused high fevers, hypotension, severe phlebitis, and possibly nephrotoxicity.4
Mechanisms of Action And Resistance
Vancomycin inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Vancomycin is slowly bactericidal against dividing organisms except for Enterococcus and tolerant staphylococci, against which it is bacteriostatic.5 In 2006 the Clinical and Laboratory Standards Institute (CLSI) changed the vancomycin breakpoints against Staphylococcus aureus from ≤4 µg/mL to ≤2 µg/mL for susceptible strains. Intermediate susceptibility is now 4 to 8 µg/mL, and resistance to vancomycin is ≥16 µg/mL.6 The U.S. Food and Drug Administration (FDA) adopted these new breakpoints in 2008. The European Committee on Antimicrobial Susceptibility Testing (EUCAST) changed their vancomycin interpretations against S. aureus to ≤2 µg/mL as susceptible and >2 µg/mL as resistant. These changes in breakpoints will alter how literature is interpreted with respect to the frequency or prevalence of vancomycin-intermediate or vancomycin-resistant S. aureus over the past 30 years.
Five types of resistance for vancomycin have been isolated from enterococci: VanA, VanB, VanC, VanD, and VanE. The VanA phenotype confers high-level resistance to both teicoplanin (minimum inhibitory concentrations [MICs]: 16 to 512 µg/mL) and vancomycin (MICs: 64 to >1000 µg/mL). Vancomycin can induce expression of the VanA gene and has been identified in both Enterococcus faecium and Enterococcus faecalis. The VanB phenotype has also been identified in both E. faecium and E. faecalis and confers low-level resistance primarily to vancomycin. VanA, B, D, and E are all transferable to other organisms. In contrast, the VanC phenotypes are endogenous (constitutively produced) and are components of Enterococcus gallinarum, Enterococcus casseliflavus, and Enterococcus flavescens and confer resistance to vancomycin alone. The VanB gene has been identified in a strain of Streptococcus bovis. This gene showed 96% homology with the prototype VanB gene from E. faecalisV583, indicating the likelihood of the gene transfer from enterococcus to this strain of S. bovis.7
Vancomycin-intermediate S. aureus using the prior breakpoints of MIC 8 to 16 µg/mL was first reported in 1996 from Japan, and by June 2002, eight cases were confirmed in the United States.88 Using the new breakpoints, the incidence of vancomycin-intermediate S. aureus will increase. In June 2002, the first case of vancomycin-resistant S. aureus (MIC > 32 µg/mL) was identified in Michigan, followed in September 2002 by the second case in Pennsylvania.8,9 No mechanism of resistance has yet been identified from the strains of vancomycin-intermediate S. aureus, but the two strains of vancomycin-resistant S. aureus both possessed the VanA gene.
Tolerance is another mechanism by which bactericidal activity is decreased. Tolerance can be measured or assessed by two methods: the ratio of minimum bactericidal concentration to minimum inhibitory concentration (MBC : MIC) and time-kill curves. By definition, a MBC : MIC ratio of 32 or greater or less than 99.9% kill after 24 hours incubation in time-kill studies equates to tolerance. Tolerance to vancomycin has been identified in S. aureus, Streptococcus pneumoniae, and groups C and G streptococci.10–12
Spectrum of Activity
Vancomycin is active primarily against aerobic gram-positive cocci including Corynebacterium and methicillin-resistant S. aureus (MRSA). The MIC90 against methicillin-susceptible S. aureus (MSSA) is 1 µg/mL, and against MRSA it is 1 to 2 µg/mL.13–15 The incidence of vancomycin-intermediate or vancomycin-resistant S. aureus currently is very low and less than 1%. The activity of vancomycin against enterococci varies greatly with the species. E. faecium is the most resistant species of enterococci to vancomycin, with the resistant rates ranging from 30% to 90% depending on the institution. For all enterococci the vancomycin resistance rates are 20% to 25%.16
Most streptococci are susceptible to vancomycin, although it is considered an agent of last resort against these organisms. Vancomycin has been shown to be inferior to nafcillin or oxacillin for the treatment of MSSA infections. Treatment failures, prolonged treatment, and higher mortality rates have been demonstrated when vancomycin was used to treat MSSA infections compared with nafcillin or oxacillin.17,18
Vancomycin is active against anaerobic gram-positive organisms such as Peptostreptococcus spp., Propionibacterium spp., Eubacterium spp., Bifidobacterium spp., and most Clostridium spp., including C. difficile.19
Pharmacokinetics/Pharmacodynamics
Vancomycin is administered orally and intravenously (IV). The drug is poorly absorbed after oral administration, and the majority of the drug is excreted unchanged in feces. Inflammation of the gastrointestinal tract may result in increased absorption of vancomycin, and measurable serum concentrations might be obtained.20 Intramuscular injections are extremely painful and should not be used. Distribution of the drug is complete 1 hour after a 1- to 2-hour IV infusion. Vancomycin is approximately 55% bound to plasma proteins. The volume of distribution corrected for weight ranges from 0.4 to 0.9 L/kg.21–27 Vancomycin does not penetrate well into noninflamed meninges or aqueous humor.28 Distribution into inflamed meninges is variable, with reported ranges of 1% to 37% of serum concentrations29,30 and a mean concentration of 15% of serum or approximately 2.5 µg/mL.31 Penetration into ascitic, pericardial, and synovial fluids is greater than 75% serum concentrations; penetration approximates 50% into pleural fluid, and 30% to 50% into bile.25 Elimination of vancomycin is 80% to 90% unchanged drug in the urine via glomerular filtration and the remaining via nonrenal elimination. The nonrenal elimination rate in healthy individuals is 40 mL/min, and in chronic renal failure patients it is 6 mL/min.32 The half-life of the drug increases with decreased renal function; in patients with creatinine clearances (CrCl) greater than 80 mL/min, the half-life is 4 to 6 hours. The pharmacodynamic effect of vancomycin is time-dependent killing or time above the MIC.33 Therefore, the most important goal of therapy is to maintain a free serum trough concentration above the MIC of the organism. There is no documented correlation between serum peak concentrations and clinical outcomes.
Dosage Regimens
Oral Administration
Oral administration of vancomycin is only for treating C. difficile colitis and is considered second-line therapy for mild/moderate infections and primary therapy for moderate/severe infections. The dose is 125 to 500 mg orally every 6 hours and is not adjusted for renal dysfunction, owing to the poor absorption. Two oral formulations (capsules or liquid) can be used, or the IV solution can be administered orally to treat C. difficile. Table 124-1 lists dosing regimens for the antimicrobials discussed in this chapter.
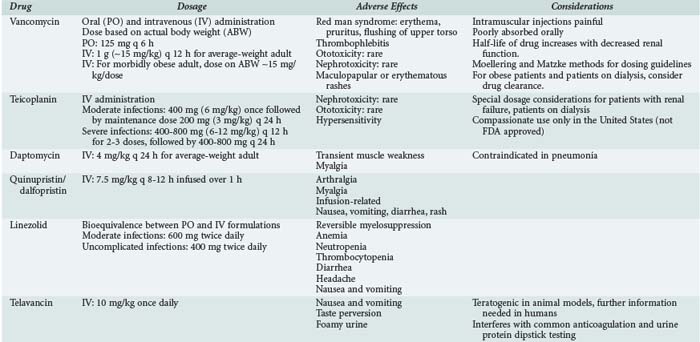
Intravenous Administration in Adults
In nonobese adults with normal renal function, the usual dose of vancomycin is 1 g (∼15 mg/kg) every 12 hours. This dose results in peak serum concentrations of 25 to 40 µg/mL 1 hour after completion of the infusion and trough serum concentration of 5 to 15 µg/mL. Dosing should be based on actual body weight. Several dosing guidelines have been developed to accurately and easily dose vancomycin. The most popular methods include the Moellering23 and Matzke24 nomograms. These methods use body weight and CrCl to calculate vancomycin dose. The weaknesses of these nomograms include the small number of patients used to develop and evaluate the nomogram and the fixed volume of distribution assumed for all patients (0.9 L/kg). Matzke24 found that for patients younger than 65 years of age, a volume of distribution of 0.7 L/kg may be more accurate, and for those older than 65, it is 0.9 L/kg. This variance in volume of distribution does affect the reproducibility of these nomograms when applied to different patient populations. The Cockcroft and Gault and modified Cockcroft and Gault methods of estimating CrCl are relatively reliable and accurate methods in patients of normal body mass.33
Morbidly obese patients are difficult to dose given the lack of pharmacokinetic studies. Doses of approximately 30 mg/kg/d based on actual body weight should provide a peak serum concentration of 25 to 35 µg/mL. Because CrCl is the best correlate to vancomycin clearance, the most accurate method for estimating CrCl should be used and varies with body mass. CrCl estimations in the obese patient are best predicted by the Salazar-Corcoran method.34 Young obese patients with no comorbid conditions affecting renal function often require the dosing interval to be more frequent to achieve a trough serum concentration of 5 to 15 µg/mL. This is due to the faster rate of clearance of the drug (2.3-2.5 times higher) in obese compared with nonobese patients.35,36
Vancomycin Dosing in Critically Ill Patients
Garaud evaluated critically ill patients and found an average volume of distribution of 0.6 L/kg in patients with CrCl greater than 70 mL/min and 0.4 L/kg with CrCl of 10 to 60 mL/min.26 Critically ill patients are often receiving medications to improve hemodynamics, such as dopamine, dobutamine, and furosemide. These medications result in increases in renal function and changes in volume status for the patient. In a study designed to assess the impact of such medications on vancomycin pharmacokinetics, two observations were made.37 First, some of the patients required larger total daily doses of vancomycin to achieve therapeutic concentrations than the Moellering nomogram predicted (26.78 + 3.01 mg/kg/d versus 18.95 + 3.41 mg/kg/d). Second, on discontinuation of these medications, the serum trough concentrations increased despite no change in CrCl or body weight. The theory is that these medications enhanced vancomycin clearance by improving renal blood flow and/or interacting with the renal anion transport system, thus increasing glomerular filtration and renal tubular secretion. Therefore, larger doses of vancomycin may be required while on these medications, and smaller doses may be more appropriate on discontinuation of these medications.
Vancomycin Dosing for Patients on Dialysis/Hemofiltration/Cardiopulmonary Bypass
The percentage of vancomycin removed by low-permeability cellulose hemodialyzers is 4% to 6.9%.38–40 Therefore, no supplemental vancomycin dosing is required after hemodialysis with these older systems. The removal of vancomycin during intradialytic administration has been studied using three types of cellulose membranes: cellulose acetate (CA), cellulose triacetate (CT), and CA high-performance 210 (CAHP-210). With the CA membranes, 0% to 25% (mean of ∼13%) of vancomycin is removed.38,41 The CT membranes remove 16% to 44% (mean of ∼26%) of vancomycin.38,41 Vancomycin removal during intradialytic administration with the CAHP-210 membranes is 0% to 35%, with a mean of 24%.42 High-flux synthetic membranes such as polysulfone or polyacrylonitrile remove significantly more vancomycin than do the cellulose membranes, with 30% to 55% and 25% to 40% of vancomycin removed, respectively.38–40,43–45
Continuous renal replacement therapy (CRRT) is a low-volume (1-2 L/h) therapy. The most frequently used methods of CRRT are continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD), and continuous arteriovenous hemodialysis (CAVHD). Both CVVHD and CAVHD result in a greater total body clearance of vancomycin than does hemofiltration. The clearances achieved with each of these methods vary with blood flow rate, ultrafiltration rate, and the membranes used. The total clearance of vancomycin with CVVHD or CAVHD is 31 to 39 mL/min, and the half-life ranges from 14 to 25 hours.46–49 Clearance of vancomycin in patients with normal renal function (CrCl > 70 mL/min) and with mild renal dysfunction (CrCl 40-70 mL/min) has been reported to be 88 and 48 mL/min, respectively.50 High-volume hemofiltration (HVHF), with an ultrafiltration rate of 6 L/h, increases vancomycin clearance to approximately 60 mL/min.51 Therefore, patients receiving CAVHD or CVVHD should receive vancomycin every 36 to 48 hours, and those undergoing HVHF should receive the drug every 12 to 24 hours.
Cardiopulmonary bypass (CPB) significantly impacts the pharmacokinetic parameters of vancomycin. Immediately after initiating CPB, vancomycin serum concentration decreased by 7 µg/mL (5.7 to 8.4 µg/mL), which represented approximately a 38% decrease in concentration.52 Over the next 30 minutes, serum vancomycin concentration may increase 1 to 2 µg/mL but thereafter gradually and steadily decreases.52 The half-life is not affected by CPB and does not change during the process.
Adverse Effects
Common toxicities that have been associated with vancomycin therapy include red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity. Evidence establishing a clear relationship between these toxicities and vancomycin peak or trough concentrations or the incidence of these events is limited and contradictory.4,53–55
Red man syndrome comprises erythema, pruritus, and flushing of the upper torso and is often associated with too rapid an infusion of the drug. In general, the infusion rate should not exceed 1 g/h. Less frequently, hypotension and angioedema can occur. It is thought that increased histamine release is the cause of this syndrome.4,54–56 A comparative trial of once-daily versus twice-daily vancomycin found the incidence of this syndrome to be 13.7% and 9.6%, respectively.54 The effects of red man syndrome can be relieved by antihistamines.57,58
Thrombophlebitis is reported in 3% to 23% of patients receiving vancomycin and is more common in patients who receive vancomycin for more than 7 days or have peripheral catheter lines for prolonged durations.4,54
Ototoxicity rates range from 0% to 9% in patients receiving vancomycin.4,54 The definition of ototoxicity ranges from tinnitus to hearing loss. The evidence demonstrating any relationship between ototoxicity and high peak serum concentrations of vancomycin is limited. In cancer patients, only 4 of 19 patients with ototoxicity had elevated serum concentrations of vancomycin, and only 1 had a concentration greater than 80 µg/mL.4 Others have reported ototoxicity associated with peak serum concentrations of 37.5 to 152 µg/mL.59,60 A trial comparing once-daily to twice-daily dosing of vancomycin demonstrated more frequent ototoxicity in the twice-daily dosed group (15.6% versus 3.2%), which had a significantly lower peak concentration and similar trough concentration compared to the group receiving daily doses.54 This lack of correlation between serum concentrations of vancomycin and ototoxicity suggests that the observed toxicity was due to either another drug or to the combination of another drug with vancomycin. In the majority of cases, ototoxicity symptoms disappear within a month of discontinuing vancomycin.
The issue of nephrotoxicity associated with vancomycin is complicated by several confounding factors. The original formulation was very impure, and the impurities were associated with toxicities including nephrotoxicity. In addition, many definitions of nephrotoxicity have been used over the years, different patient populations have been studied, and different doses used, making it difficult to compare one study to another. In general, the rate of nephrotoxicity is 5% to 10% when vancomycin is not administered with other nephrotoxic agents and trough concentrations are less than 10 µg/mL.54,61,62 Elting and colleagues identified older age, Acute Physiology and Chronic Health Evaluation (APACHE) score greater than 40, and duration of therapy of greater than 14 days to be the best predictors for a patient to develop nephrotoxicity due solely to vancomycin therapy.4 A number of other studies have found an increased incidence of nephrotoxicity (21%-35%) when vancomycin serum trough concentrations are greater than 10 µg/mL.62–64 In addition, Lodise demonstrated an increased rate of nephrotoxicity (∼35%) when the total daily dose is 4 grams or more compared to total doses less than 4 grams (∼11%).65 Studies have demonstrated higher rates of nephrotoxicity when vancomycin is used in combination with an aminoglycoside compared with either agent alone.62,66,67 Goetz performed a meta-analysis of eight studies and found the incidence of nephrotoxicity associated with combination therapy was 13% greater than with vancomycin alone and 4% greater than with an aminoglycoside alone.67
Other toxicities associated with vancomycin include maculopapular or erythematous rashes (2%-8%)26,68,75 and anecdotal reports of neutropenia and thrombocytopenia.68,69
Therapeutic Drug Monitoring
Routine monitoring of vancomycin serum concentrations has become a highly debated issue over the years. Those who advocate routine monitoring cite the need to ensure therapeutic concentrations as well as minimize toxicities. To date there is only one trial that compared efficacy and toxicity with high-dose once-daily versus twice-daily dosing of vancomycin; with these dosing regimens, peak serum concentrations were vastly different but trough serum concentrations were similar.54 The mean peak serum concentrations in the once-daily and twice-daily dosed groups were 42.8 + 16.1 and 27.0 + 9.2 µg/mL, respectively. There were no differences in clinical efficacy, red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity between the two groups.
Studies over the past 20 years have shown that peak concentrations of vancomycin are not associated with toxicities or clinical efficacy. Therefore, monitoring peak serum concentrations only adds to hospital and healthcare system costs and provides no beneficial clinical information. Some studies have demonstrated a correlation of nephrotoxicity to serum trough concentrations ≥ 10 µg/mL, whereas others have not. Given the lack of consensus, it may be prudent to measure serum trough concentrations until more definitive studies are conducted to address this issue.70
In patients with end-stage renal disease, the fluorescence polarization immunoassay (FPIA) overestimates vancomycin concentrations.71 FPIA is the most common method for determining vancomycin concentrations, and when it was compared with the enzyme multiplied immunoassay technique, it was found to produce higher peak serum concentrations by 7 to 11 µg/mL and higher trough concentrations by 4 to 6 µg/mL.
 Teicoplanin
Teicoplanin
Mechanisms of Action And Resistance
Teicoplanin, like other glycopeptide antibiotics, inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Resistance has been reported in both staphylococci and enterococci. The VanA phenotype confers high-level resistance to both teicoplanin (MIC: 16 to 512 µg/mL) and vancomycin (MIC: 64 to >1000 µg/mL). The VanB phenotype has also been identified in both E. faecium and E. faecalis and usually confers low-level resistance to vancomycin but not to teicoplanin. This may limit the utility of teicoplanin for some vancomycin-resistant enterococcal infections. Several reports of S. aureus resistance developing during therapy with teicoplanin have been reported.72–74 The mechanism of the resistance was determined in one patient to be constitutive and non-plasmid mediated.73
Spectrum of Activity
Teicoplanin is only active against gram-positive organisms. Activity against MSSA and MRSA is comparable to that of vancomycin. Coagulase-negative staphylococci have a varied pattern of susceptibility to teicoplanin. Staphylococcus haemolyticus is the most resistant species to teicoplanin (30%).75 These isolates are 25% more resistant to teicoplanin than to vancomycin. Against methicillin-resistant coagulase-negative staphylococci, 39% of isolates have teicoplanin MICs greater than 8 µg/mL compared with 1% with vancomycin.75,76 Teicoplanin is similar in activity to vancomycin against enterococci, although its reliability in treating infections with VanB resistance to vancomycin may be limited. Teicoplanin is active against other aerobic and anaerobic gram-positive organisms such as Corynebacterium spp., Clostridium spp., including C. difficile and C. perfringens, Peptostreptococcus spp., and Propionibacterium acnes.
Pharmacokinetics/Pharmacodynamics
Teicoplanin is administered orally and intravenously. The drug is poorly absorbed after oral administration, and approximately 40% of the drug is excreted unchanged in feces. The pharmacokinetic model that best describes the elimination of teicoplanin is triexponential. IV administration of 400 mg (6 mg/kg) should provide a peak serum concentration of 20 to 50 µg/mL attained 1 hour after administration.77 The volume of distribution is large at 0.9 to 1.41 L/kg, and teicoplanin is 90% to 95% protein bound.77 Penetration into body fluids and tissues has not been extensively studied. Penetration into noninflamed meninges and fat is poor, but distribution into myocardium and pericardium is good.78,79 Teicoplanin is primarily eliminated via glomerular filtration, and only 3% is metabolized.77 The half-life is approximately 150 hours in patients with normal renal function.77 Because of the long half-life, it takes 14 days to reach steady state. In patients with CrCl of 13 to 25, the half-life was found to be 280 to 667 hours.80,81
Dosage Regimens/Therapeutic Drug Monitoring
Despite the long half-life in patients with normal renal function, teicoplanin should be administered daily, and the dose is dependent on the severity of infection. For less serious infections involving the urinary tract, skin, soft tissue, and lower respiratory tract, a loading dose of 400 mg (6 mg/kg) × 1 is administered, followed by a maintenance dose of 200 mg (3 mg/kg) every 24 hours. For severe infections such as septicemia, endocarditis, and osteomyelitis, 400 mg of teicoplanin is administered every 12 hours for 3 doses, followed by 400 mg every 24 hours.77 Although no therapeutic range has been established for teicoplanin, trough concentrations should be at least 10 µg/mL.77
Renal Failure/Dialysis
Teicoplanin is not removed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD).82,83 The amount removed by CVVHD is dependent on the flow rate but is often minimal.84,85 Several dosing regimens exist for renal dysfunction, and the simplest method is administering a dose of 6-10 mg/kg every 48 to 72 hours.
Adverse Effects
Nephrotoxicity associated with teicoplanin is much lower than with vancomycin. The incidence from published and unpublished studies found the nephrotoxic rate to be 4%.55 Ototoxic rates with teicoplanin are similar to those with vancomycin.55 Hypersensitivity reactions are the most common adverse reaction to teicoplanin (2%-15%).55
 Daptomycin
Daptomycin
Mechanisms of Action And Resistance
Daptomycin has a unique mechanism of action and has been found to inhibit lipoteichoic acid synthesis, owing to binding to the membrane in the presence of calcium.86,87 Minimal information is available on the mechanism(s) of resistance to daptomycin. Limited in vitro studies have been performed attempting to create daptomycin resistance in the laboratory.88 Mechanisms of resistance have not been elucidated, and the clinical relevance of in vitro resistance is unknown.
Spectrum of Activity
Daptomycin’s antibacterial activity encompasses most gram-positive bacteria, including vancomycin-resistant isolates and penicillin-resistant pneumococci. The MICs of daptomycin are 8- to 16-fold lower in the presence of calcium. Therefore, all in vitro testing must be supplemented with physiologic concentrations of calcium.89 The breakpoint for susceptible is ≤1 µg/mL for staphylococci and β-hemolytic streptococci. Given the rare number of isolates not susceptible to daptomycin, a resistant breakpoint has yet to be determined. The MIC90 against MSSA, MRSA, Staphylococcus epidermidis, and Staphylococcus saprophyticus are all 0.5 µg/mL or less.89,90 In a recent surveillance study, 7 S. aureus and 6 coagulase-negative staphylococci were non-susceptible to daptomycin.91 Daptomycin also appears active against vancomycin-intermediate and vancomycin-resistant strains of S. aureus.92,93 The breakpoint for susceptible against enterococci is ≤4 µg/mL, and again no resistant breakpoint has been established. Against E. faecalis and E. faecium, including vancomycin-resistant strains, the MIC90 is 2 µg/mL or less.89,90 Daptomycin resistance is higher among E. faecium than E. faecalis.91 The MIC90 is 0.25 µg/mL against S. pneumoniae and β-hemolytic streptococci, and resistance has not been reported with these organisms.89–91
Pharmacokinetics/Pharmacodynamics
Healthy volunteers who received 6 mg/kg of daptomycin given as either a 30- or 2-minute infusion achieved bioequivalent pharmacokinetic results. The maximum plasma concentration (Cmax) was about 94 and 88 µg/mL for the 2- and 30-minute infusions, respectively.94 Daptomycin demonstrates linear kinetics at dosing from 4 to 12 mg/kg, and the half-life is 7 to 9 hours in patients with normal renal function.95 The drug is 90% to 95% protein bound and is primarily eliminated by the renal route. In patients with CrCl less than 30 mL/min, end-stage renal disease/hemodialysis/peritoneal dialysis, a 4 mg/kg dose should provide a peak serum concentrations around 25 to 30 µg/mL and half-life of about 30 hours.95
Daptomycin is rapidly bactericidal and exhibits concentration-dependent killing against gram-positive organisms including enterococci.86,87 Daptomycin also exhibits a post-antibiotic effect which allows for once daily dosing.96
Dosage Regimens and Therapeutic Monitoring
For complicated skin and skin structure infections (cSSSIs), dosing of daptomycin is 4 mg/kg every 24 hours. Dosing for bacteremia or right-sided endocarditis is 6 mg/kg every 24 hours.95
Renal Failure/Dialysis
In patients with CrCl less than 30 mL/min or undergoing hemodialysis or chronic peritoneal dialysis, the dose should be reduced to 4 mg/kg every 48 hours and 6 mg/kg every 48 hours for bacteremia or endocarditis.95 In patients undergoing continuous renal replacement therapy (CRRT), the amount of daptomycin removed is dependent upon the type of filter and the flow rates.97 Dosing recommendations for patients undergoing CRRT are 4 to 6 mg/kg every 48 hours, and there is some speculation that doses may need to be increased to 8 to 10 mg/kg every 48 hours.98,99
Dosing in the Setting of Obesity
Two single-dose studies using 4 mg/kg total body weight have been performed in moderately and morbidly obese patients. The Cmax was increased 25% to 60% compared to normal-weight patients and the area under the curve (AUC) increased 30% to 60% in the obese patients. Half-life was also longer and ranged from 7 to 9 hours; all patients had normal renal function.100,101 Recommendations are to base daptomycin dosage on total body weight, but difficulties in assessing renal function in obese patients have to be considered when selecting the dosing interval.
Burn Patients
One study evaluated single-dose pharmacokinetics (4 mg/kg) in burn patients and found the Cmax was 44% lower, with 47% lower AUC and an increase in volume of distribution and clearance.102 The authors suggest a dose of 10 to 12 mg/kg in burn patients should provide the same drug exposure as 6 mg/kg in healthy volunteers.
 Quinupristin/Dalfopristin
Quinupristin/Dalfopristin
Mechanisms of Action and Resistance
Quinupristin/dalfopristin is a streptogramin antibiotic and is a mix of two different streptogramin components from groups A and B. The individual components are bacteriostatic, but the combination is often bactericidal. Each component binds to different sites on the 50S subunit of the ribosome, inhibiting translation of mRNA at the elongation step.103 The resulting complex of drug and ribosome inhibits protein synthesis.
Streptogramins share similar sites of action with macrolide and lincomycin antibiotics. As a result, mechanisms of resistance are also shared. The most common type of resistance to streptogramins involves the erythromycin resistance methylase (erm) genes, termed MLSB.104 These genes decrease the binding of antibiotics such as streptogramins group B, erythromycin, and clindamycin by dimethylating a residue on the 23S ribosome. Group A streptogramins are not affected, and the combination often retains its synergistic activity.104 Enzymatic modification of both components is another mechanism of resistance to the drug.105,106 The third mechanism involves efflux pumps: one that pumps out both macrolides and streptogramins and one specific for streptogramins.105,107,108
Spectrum of Activity
Quinupristin/dalfopristin is active against a wide variety of gram-positive organisms as well as many anaerobes and oral flora organisms. A MIC of 2 µg/mL or less indicates susceptibility. The MIC90 of most MSSA, MRSA, and coagulase-negative staphylococci is 1 to 2 µg/mL.14,15,90 Against vancomycin-intermediate and vancomycin-resistant S. aureus, the drug is active with MICs of 0.25 to 1 µg/mL.93,109 Both vancomycin-susceptible and vancomycin-resistant E. faecium are susceptible to quinupristin/dalfopristin (MIC90: 1-4 µg/mL); however, E. faecalis is resistant to quinupristin/dalfopristin (MIC90: 4-32 µg/mL).110,111 Against a variety of streptococcal organisms, including penicillin-resistant pneumococci, the MIC90 ranges from 0.5 to 2 µg/mL. Quinupristin/dalfopristin is also active against a variety of other organisms including Chlamydia spp., Mycoplasma pneumoniae, Legionella spp., Peptostreptococcus spp., Fusobacterium spp., Prevotella spp., Actinomyces spp., and Clostridium spp.
Pharmacokinetics/Pharmacodynamics
Quinupristin/dalfopristin infusions should be administered over 1 hour, and the drug is incompatible with saline. In healthy volunteers and in patients undergoing CAPD, the mean peak serum concentration of quinupristin was 2.6 and 2.9 µg/mL, respectively, and for dalfopristin it was 7.1 and 8.5 µg/mL, respectively, following a single 7.5-mg/kg dose.112 Quinupristin/dalfopristin is hepatically metabolized to several active metabolites, and both the parent components and the metabolites are primarily eliminated via bile into feces.113 Urinary excretion of quinupristin/dalfopristin and metabolites is 15% to 19%. The mean half-life ranges from 1.2 to 1.5 hours. The drug is 90% protein bound.114
Adverse Effects
Myalgias (6%-7%) and arthralgias (9%-9.5%) are the most severe adverse effects and are often the reason for discontinuation of the drug.116,117 Elevations in direct and conjugated bilirubin and γ-glutamyl transferase are common. Infusion-related adverse effects occur in 30% to 45% of patients with peripheral lines used for the infusion.116 The reactions include pain, burning, inflammation, and thrombophlebitis. Other toxicities include nausea, diarrhea, vomiting, and rash.
 Linezolid
Linezolid
Mechanisms of Action and Resistance
Linezolid is an oxazolidinone antibiotic, a new class of synthetic agents. Linezolid binds to the 50S ribosome and inhibits the binding of mRNA, thereby preventing protein synthesis.118 Clinical isolates of S. aureus, E. faecium, and E. faecalis resistant to linezolid have been identified but currently are rare. The most common mechanism of resistance is alteration of the 23S rRNA.119 There are three case reports of vancomycin-resistant E. faecium infections in which the organisms were resistant to linezolid without the patient having any prior exposure to linezolid.120,121 A second mechanism of resistance has been identified in S. aureus and involves acquisition of the natural resistance gene, cfr.122,123 The cfr gene confers resistance to chloramphenicol and clindamycin. In animals, this gene is located on a plasmid which could result in propagating the spread of resistance. In humans, the gene has not been identified on a plasmid but rather on the chromosome.122 These resistance issues, although rare, do raise concern and emphasize the importance of appropriate use of linezolid.
Spectrum of Activity
Linezolid’s breakpoint for susceptibility is ≤4 µg/mL for staphylococci and ≤2 µg/mL for enterococci and streptococci. It is active against both methicillin-susceptible and methicillin-resistant staphylococci. The MIC90 against S. aureus and coagulase-negative staphylococci is 2 and 1 µg/mL, respectively.124–126 Against vancomycin-intermediate and vancomycin-resistant S. aureus, the drug is active with MICs of 1 to 2 µg/mL.93,109 Linezolid is equally active against both vancomycin-susceptible and vancomycin-resistant enterococci with an MIC90 of 2 µg/mL.124,126,127 Against both penicillin-susceptible and penicillin-resistant S. pneumoniae, the MIC90 is 1 µg/mL.124,126 Linezolid is also active against a variety of other organisms, including Pasteurella multocida, Peptostreptococcus spp., Fusobacterium spp., and Prevotella spp.
Pharmacokinetics/Pharmacodynamics
Linezolid is available in both oral and IV formulations. Oral absorption is over 90%, making the oral formulation bioequivalent to the IV formulation. The peak serum concentration and half-life at steady state after 600 mg twice daily were 14 to 18 µg/mL and 5 to 6 hours.128–130 Linezolid is approximately 30% protein bound and penetrates quickly into bone, fat, and muscle, achieving 50% to 60% of serum concentrations in bone and 90% to 95% in muscle.131 Elimination of linezolid is 30% renal and 70% metabolized, with essentially no linezolid eliminated in feces as unchanged drug.130 Linezolid is not an inducer of the cytochrome P450 enzyme system.
Linezolid is bacteriostatic against staphylococci and enterococci and is bactericidal against streptococci. It appears that the pharmacodynamic parameter best modeling the killing activity is the area under the concentration time curve to MIC ratio (AUC/MIC).132 The AUC/MIC ratio required to produce a bacteriostatic effect varied from 22 to 97 (mean 48) for pneumococci and 39 to 167 (mean 83) for staphylococci. A dosage regimen of 600 mg twice daily achieves these values for organisms with MICs as high as 4 µg/mL.
Dosage Regimens and Therapeutic Monitoring
Critically Ill Patients
One small study evaluated the pharmacokinetics of linezolid in critically ill patients when administered as standard intermittent bolus therapy or continuous infusion. Standard bolus therapy resulted in free and total trough concentrations below the breakpoint of 4 µg/mL in all patients, and 50% of patients had free trough concentrations less than 1 µg/mL. Standard bolus therapy resulted in only 40% of patients achieving at least 85% of the dosing interval with free concentrations above a MIC of 2 µg/mL (the most common MIC for pathogens identified in the study) compared to 100% in the continuous infusion group. Achieving the target AUC/MIC ratio of at least 80 occurred in only 62.5% of patients given standard bolus therapy compared with 87.5% of patients receiving continuous infusion.133 Wide variability in linezolid pharmacokinetic parameters were observed, and continuous infusion may provide an option for optimizing the pharmacodynamic parameters, but further studies are needed to assess the efficacy and safety of continuous infusion.
Adverse Effects
Reversible myelosuppression is the most significant adverse effect associated with linezolid therapy. Anemia, neutropenia, and thrombocytopenia have all been reported, and the incidence increases with durations of therapy exceeding 14 days.134,135 The decrease in hemoglobin when linezolid therapy is greater than 2 weeks is 18% compared with 13% for comparator agents and linezolid therapy less than 2 weeks’ duration.134 The thrombocytopenia rate is 8% with the longer duration of therapy compared with 5% to 6% in all durations of therapy compared with 3% with comparator agents. Rates of neutropenia also increase to about 10% with extended durations of therapy. Complete blood cell counts should be monitored weekly, especially in patients in whom the duration of therapy is likely to exceed 2 weeks.
Linezolid is a reversible nonselective inhibitor of monoamine oxidase; therefore, the potential for interaction with adrenergic and serotonergic agents exists. Several case reports of serotonin syndrome (fever, agitation, tremors, and mental status changes) secondary to an interaction between linezolid and selective serotonin reuptake inhibitors (SSRIs) have been identified.136–138
Other adverse reactions to linezolid include diarrhea (8%), headache (7%), nausea and vomiting (6% and 4%), dizziness, rash, fever, constipation (2%), and abnormal liver function tests (1%). Rare but serious reactions include optic or peripheral neuropathy; optic neuropathy tends to be reversible upon discontinuation of linezolid, but peripheral neuropathy tends to be permanent.139
 Telavancin
Telavancin
Mechanisms of Action And Resistance
Telavancin is a lipoglycopeptide which has a dual mechanism of action. It binds to the D-alanyl-D-alanine terminus of the cell wall precursors as vancomycin does, but additionally it binds to bacterial membranes, resulting in the depolarization and increased permeability of the membrane.140
Spectrum of Activity
Telavancin is active against MSSA, MRSA, coagulase-negative staphylococci, vancomycin-susceptible enterococci, Streptococcus pyogenes, Streptococcus agalactiae, and Streptococcus anginosus. The breakpoint for susceptible against the streptococci is ≤0.12 µg/mL and for staphylococci and enterococci is ≤1 µg/mL. No interpretations for intermediate or resistant exist at this time. Telavancin is active against most anaerobic gram-positive organisms including C. difficile and C. perfringens.141 The MIC90 is ≤1 µg/mL for most clostridia, and against most other anaerobic gram-positive cocci and bacilli it is ≤0.5 µg/mL.141
Pharmacokinetics/Pharmacodynamics
Telavancin demonstrates linear pharmacokinetics over doses of 7.5 to 15 mg/kg. In healthy subjects, doses of 7.5 and 15 mg/kg at steady state resulted in mean Cmax serum concentrations of 88 and 186 µg/mL and trough concentrations of 6 and 16 µg/mL, respectively.142 Approximately 70% of telavancin is renally eliminated, and the half-life was dose dependent and ranged from 6 to 7.5 hours.142 Telavancin is 90% protein bound to albumin and has a volume of distribution of approximately 0.14 L/kg.143 Telavancin penetrates lung epithelial lining fluid and alveolar macrophages well, and concentrations exceeded 0.5 µg/mL during the entire dosing interval.144 Penetration into blister fluid is approximately 40% of serum concentrations.145
Telavancin exhibits rapid concentration-dependent killing. The pharmacodynamic parameter identified in animal models as the best predictor of efficacy is the AUC/MIC ratio.146 The minimum AUC/MIC ratio needed to provide a favorable clinical outcome in humans has not yet been identified.
Dosage Regimens and Therapeutic Monitoring
Renal Failure/Dialysis
Due to the high urinary elimination of telavancin, dosage reductions are required when the patient’s CrCl falls below 50 mL/min. If CrCl is 30 to 50 mL/min, the dose of telavancin is 7.5 mg/kg every 24 hours, and when less than 30 mL/min, the dose is further reduced to 10 mg/kg every 48 hours.143 In vitro studies evaluated the affect of CRRT on telavancin elimination and found high ultrafiltrate or dialysate rates can remove a significant amount of telavancin, which could require supplemental dosing.147
Adverse Effects
Telavancin is a pregnancy category C drug with little information available in pregnant women. In three animal species, telavancin was found to have fetal effects including decreased birth weight and increased digit and limb malformations. A serum pregnancy test should be performed in women of childbearing age prior to starting telavancin. There is a pregnancy exposure registry should there be a need to use telavancin in a pregnant woman.143
The most common adverse effects associated with telavancin are nausea, vomiting, taste disturbance, and foamy urine.148–150 Telavancin interferes with urine protein qualitative dipstick tests, and several anticoagulation tests including PT, APTT, INR, and ACT.143 These tests should be performed when telavancin concentrations are lowest in the bloodstream to minimize the impact on anticoagulation tests.
Key Points
Gerson SL, Kaplan SL, Bruss JB, Le V, Arellano FM, Hafkin B, et al. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother. 2002;46:2723-2726.
Vincent J, Rello J, Marshall J, Silva E, Anzueto A, Martin C, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323-2329.
Rybak M, Lomaestro B, Rotschafer JC, Moellering RJr, Craig W, Billeter M, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009;66:82-98.
Chakraborty A, Roy S, Loeffler J, Chaves RL. Comparison of the pharmacokinetics, safety and tolerability of daptomycin in healthy adult volunteers following intravenous administration by 30 min infusion or 2 min injection. J Antimicrob Chemother. 2009;64:151-158.
Gotfried MH, Shaw JP, Benton BM, Krause KM, Goldberg MR, Kitt MM, et al. Intrapulmonary distribution of intravenous telavancin in healthy subjects and effect of pulmonary surfactant on in vitro activities of telavancin and other antibiotics. Antimicrob Agents Chemother. 2008;52:92-97.
1 Edgeworth J, Treacher D, Eykyn S. A 25-year study of nosocomial bacteremia in an adult intensive care unit. Crit Care Med. 1999;27(8):1421.
2 Vincent J, Rello J, Marshall J, Silva E, Anzueto A, Martin C, et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302(21):2323.
3 Richards M, Edwards J, Culver D, Gaynes R. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect Control Hosp Epidemiol. 2000;21(8):510.
4 Elting L, Rubenstein E, Kurtin D, Rolston K, Fangtang J, Martin C, et al. Mississippi mud in the 1990s: risks and outcomes of vancomycin-associated toxicity in general oncology practice. Cancer. 1998;83(12):2597.
5 Watanakunakorn C. The antibacterial action of vancomycin. Rev Infect Dis. 1981;3(suppl):S210.
6 Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests; approved standards, 9th ed. CLSI document M2-A9. Wayne, PA: Clinical and Laboratory Standards Institute; 2006.
7 Poyart C, Pierre C, Quesne G, Pron B, Berche P, Trieu-Cuot P. Emergence of vancomycin resistance in the genus Streptococcus: characterization of a vanB transferable determinant in Streptococcus bovis. Antimicrob Agents Chemother. 1997;41(1):24.
8 Prevention CfDCa. Staphylococcus aureus resistant to vancomycin–United States, 2002. MMWR: Morb Mortal Wkly Rep. 2002;51(26):565.
9 Prevention CfDCa. Vancomycin-resistant Staphylococcus aureus–Pennsylvania, 2002. MMWR: Morb Mortal Wkly Rep. 2002;51(40):902.
10 Zaoutis T, Schneider B, Steele Moore L, Klein J. Antibiotic susceptibilities of group C and group G streptococci isolated from patients with invasive infections: evidence of vancomycin tolerance among group G serotypes. J Clin Microbiol. 1999;37(10):3380.
11 Henriques Normark B, Normark S. Antibiotic tolerance in pneumococci. Clin Microbiol Infect. 2002;8(10):613.
12 May J, Shannon K, King A, French G. Glycopeptide tolerance in Staphylococcus aureus. J Antimicrob Chemother. 1998;42(2):189.
13 Gales A, Sader HH, Jones R. Respiratory tract pathogens isolated from patients hospitalized with suspected pneumonia in Latin America: frequency of occurrence and antimicrobial susceptibility profile: results from the SENTRY Antimicrobial Surveillance Program (1997-2000). Diag Microbiol Infect Dis. 2002;44(3):301.
14 Rennie R, Jones R, Mutnick A. Occurrence and antimicrobial susceptibility patterns of pathogens isolated from skin and soft tissue infections: report from the SENTRY Antimicrobial Surveillance Program (United States and Canada, 2000). Diag Microbiol Infect Dis. 2003;45(4):287.
15 Hoban D, Biedenbach D, Mutnick A, Jones R. Pathogen of occurrence and susceptibility patterns associated with pneumonia in hospitalized patients in North America: results of the SENTRY Antimicrobial Surveillance Study (2000). Diagn Microbiol Infect Dis. 2003;45(4):279.
16 Murray B. Vancomycin-resistant enterococcal infections. N Engl J Med. 2000;342(10):710.
17 González C, Rubio M, Romero-Vivas J, ález M, Picazo J. Bacteremic pneumonia due to Staphylococcus aureus: A comparison of disease caused by methicillin-resistant and methicillin-susceptible organisms. Clin Infect Dis. 1999;29(5):1171.
18 Gentry C, Rodvold K, Novak R, Hershow R, Naderer O. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy. 1997;17(5):990.
19 Goldstein E, Citron D, Merriam C, Warren Y, Tyrrell K, Fernandez H. In vitro activities of daptomycin, vancomycin, quinupristin- dalfopristin, linezolid, and five other antimicrobials against 307 gram-positive anaerobic and 31 Corynebacterium clinical isolates. Antimicrob Agents Chemother. 2003;47(1):337.
20 ViroPharma Inc. Vancocin. Retrieved February 2, 2010. www.viropharma.com/products/package_insert.aspx.
21 Moellering R. Pharmacokinetics of vancomycin. J Antimicrob Chemother. 1984;14(Suppl D):43.
22 Moellering R, Krogstad D, Greenblatt D. Pharmacokinetics of vancomycin in normal subjects and in patients with reduced renal function. Rev Infect Dis. 1981;3(suppl):S230.
23 Moellering R, Krogstad D, Greenblatt D. Vancomycin therapy in patients with impaired renal function: a nomogram for dosage. Ann Intern Med. 1981;94(3):343.
24 Matzke G, McGory R, Halstenson C, Keane W. Pharmacokinetics of vancomycin in patients with various degrees of renal function. Antimicrob Agents Chemother. 1984;25(4):433.
25 Matzke G, Zhanel G, Guay D. Clinical pharmacokinetics of vancomycin. Clin Pharmacokinet. 1986;11(4):257.
26 Garaud J, Regnier B, Inglebert F, Faurisson F, Bauchet J, Vachon F. Vancomycin pharmacokinetics in critically ill patients. J Antimicrob Chemother. 1984;14(Suppl D):53.
27 James J, Palmer S, Levine D, Rybak M. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother. 1996;40(3):696.
28 MacIlwaine Wt, Sande M, Mandell G. Penetration of antistaphylococcal antibiotics into the human eye. Am J Ophthalmol. 1974;77(4):589.
29 Gump D. Vancomycin for treatment of bacterial meningitis. Rev Infect Dis. 1981;3(suppl):S289.
30 Viladrich P, Gudiol F, Liñares J, Pallarés R, et al. Evaluation of vancomycin for therapy of adult pneumococcal meningitis. Antimicrob Agents Chemother. 1991;35(12):2467.
31 Cunha B. Vancomycin. Med Clin North Am. 1995;79(4):817.
32 Macias W, Mueller B, Scarim S. Vancomycin pharmacokinetics in acute renal failure: preservation of nonrenal clearance. Clin Pharmacol Ther. 1991;50(6):688.
33 Cockcroft D, Gault M. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31.
34 Salazar D, Corcoran G. Predicting creatinine clearance and renal drug clearance in obese patients from estimated fat-free body mass. Am J Med. 1988;84(6):1053.
35 Bauer L, Black D, Lill J. Vancomycin dosing in morbidly obese patients. European J Clin Pharmacol. 1998;54(8):621.
36 Blouin R, Bauer L, Miller D, Record KJr, G W. Vancomycin pharmacokinetics in normal and morbidly obese subjects. Antimicrob Agents Chemother. 1982;21(4):575.
37 Pea F, Porreca L, Baraldo M, Furlanut M. High vancomycin dosage regimens required by intensive care unit patients cotreated with drugs to improve haemodynamics following cardiac surgical procedures. J Antimicrob Chemother. 2000;45(3):329.
38 DeSoi C, Sahm D, Umans J. Vancomycin elimination during high-flux hemodialysis: kinetic model and comparison of four membranes. Am J Kidney Dis. 1992;20(4):354.
39 Lanese D, Alfrey P, Molitoris B. Markedly increased clearance of vancomycin during hemodialysis using polysulfone dialyzers. Kidney Int. 1989;35(6):1409.
40 Torras J, Cao C, Rivas M, Cano M, Fernandez E, Montoliu J. Pharmacokinetics of vancomycin in patients undergoing hemodialysis with polyacrylonitrile. Clin Nephrol. 1991;36(1):35.
41 Scott M, Macias W, Kraus M, Clark W, Carfagna M, Mueller B. Effects of dialysis membrane on intradialytic vancomycin administration. Pharmacother. 1997;17(2):256.
42 Lucksiri A, Scott M, Mueller B, Hamburger R, Sowinski K. CAHP-210 dialyzer influence on intra-dialytic vancomycin removal. Nephrol Dialysis Transplant. 2002;17(9):1649.
43 Foote E, Dreitlein W, Steward C, Kapoian T, Walker J, Sherman R. Pharmacokinetics of vancomycin when administered during high flux hemodialysis. Clin Nephrol. 1998;50(1):51.
44 Touchette M, Patel R, Anandan J, Dumler F, Zarowitz B. Vancomycin removal by high-flux polysulfone hemodialysis membranes in critically ill patients with end-stage renal disease. Am J Kidney Dis. 1995;26(3):469.
45 Pollard T, Lampasona V, Akkerman S, Tom K, Hooks M, Mullins R, et al. Vancomycin redistribution: dosing recommendations following high-flux hemodialysis. Kidney Int. 1994;45(1):232.
46 Santré C, Leroy O, Simon M, Georges H, Guery B, Beuscart C, et al. Pharmacokinetics of vancomycin during continuous hemodiafiltration. Intensive Care Med. 1993;19(6):347.
47 Joy M, Matzke G, Frye R, Palevsky P. Determinants of vancomycin clearance by continuous venovenous hemofiltration and continuous venovenous hemodialysis. Am J Kidney Dis. 1998;31(6):1019.
48 Davies S, Azadian B, Kox W, Brown E. Pharmacokinetics of ciprofloxacin and vancomycin in patients with acute renal failure treated by continuous haemodialysis. Nephrol Dialysis Transplant. 1992;7(8):848.
49 Bellomo R, Ernest D, Parkin G, Boyce N. Clearance of vancomycin during continuous arteriovenous hemodiafiltration. Crit Care Med. 1990;18(2):181.
50 Rodvold K, Blum R, Fischer J, Zokufa H, Rotschafer J, Crossley K, et al. Vancomycin pharmacokinetics in patients with various degrees of renal function. Antimicrob Agents Chemother. 1988;32(6):848.
51 Uchino S, Cole L, Morimatsu H, Goldsmith D, Bellomo R. Clearance of vancomycin during high-volume haemofiltration: impact of pre-dilution. Intensive Care Med. 2002;28(11):1664.
52 Ortega GM, Martí-Bonmatí E, Guevara SJ, Gómez IG. Alteration of vancomycin pharmacokinetics during cardiopulmonary bypass in patients undergoing cardiac surgery. Am J Health Syst Pharm. 2003;60(3):260.
53 Pryka R, Rodvold K, Erdman S. An updated comparison of drug dosing methods. Part IV: Vancomycin. Clin Pharmacokinet. 1991;20(6):463.
54 Cohen E, Dadashev A, Drucker M, Samra Z, Rubinstein E, Garty M. Once-daily versus twice-daily intravenous administration of vancomycin for infections in hospitalized patients. J Antimicrob Chemother. 2002;49(1):155.
55 Wilson A. Comparative safety of teicoplanin and vancomycin. Int J Antimicrob Agents. 1998;10(2):143.
56 O’Sullivan T, Ruffing M, Lamp K, Warbasse L, Rybak M. Prospective evaluation of red man syndrome in patients receiving vancomycin. J Infect Dis. 1993;168(3):773.
57 Sahai J, Polk R, Schwartz L, Healy D, Westin E. Severe reaction to vancomycin not mediated by histamine release and documented by rechallenge. J Infect Dis. 1988;158(6):1413.
58 Wallace M, Mascola J3rd, O E. Red man syndrome: incidence, etiology, and prophylaxis. J Infect Dis. 1991;164(6):1180.
59 Sorrell T, Packham D, Shanker S, Foldes M, Munro R. Vancomycin therapy for methicillin-resistant Staphylococcus aureus. Ann Intern Med. 1982;97(3):344.
60 Levine D, Cushing R, Jui J, Brown W. Community-acquired methicillin-resistant Staphylococcus aureus endocarditis in the Detroit Medical Center. Ann Intern Med. 1982;97(3):330.
61 Farber B, Moellering RJ. Retrospective study of the toxicity of preparations of vancomycin from 1974 to 1981. Antimicrob Agents Chemother. 1983;23(1):138.
62 Rybak M, Albrecht L, Boike S, Chandrasekar P. Nephrotoxicity of vancomycin, alone and with an aminoglycoside. J Antimicrob Chemother. 1990;25(4):679.
63 Lodise T, Patel N, Lomaestro B, Rodvold K, Drusano G. Relationship between initial vancomycin concentration-time profile and nephrotoxicity among hospitalized patients. Clin Infect Dis. 2009;49(4):507.
64 Kralovicov áK, Spanik S, Halko J, Netriova J, Studena-Mrazova M, et al. Do vancomycin serum levels predict failures of vancomycin therapy or nephrotoxicity in cancer patients? J Chemother. 1997;9(6):420.
65 Lodise T, Lomaestro B, Graves J, Drusano G. Larger vancomycin doses (at least four grams per day) are associated with an increased incidence of nephrotoxicity. Antimicrob Agents Chemother. 2008;52(4):1330.
66 Cantú T, Yamanaka-Yuen N, Lietman P. Serum vancomycin concentrations: reappraisal of their clinical value. Clin Infect Dis. 1994;18(4):533.
67 Goetz M, Sayers J. Nephrotoxicity of vancomycin and aminoglycoside therapy separately and in combination. J Antimicrob Chemother. 1993;32(2):325.
68 Farwell AJr, K L, Vakil R, Glew R. Delayed appearance of vancomycin-induced neutropenia in a patient with chronic renal failure. South Med J. 1984;77(5):664.
69 Zenon G, Cadle R, Hamill R. Vancomycin-induced thrombocytopenia. Arch Intern Med. 1991;151(5):995.
70 Rybak M, Lomaestro B, Rotschafer JJr, M R, Craig W, Billeter M, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009;66(1):82.
71 Follin S, Mueller B, Scott M, Carfagna M, Kraus M. Falsely elevated serum vancomycin concentrations in hemodialysis patients. Am J Kidney Dis. 1996;27(1):67.
72 Hassan I, Chadwick P, Johnson A. Clinical isolates of methicillin-resistant Staphylococcus aureus (MRSA) with reduced susceptibility to teicoplanin in Northwest England. J Antimicrob Chemother. 2001;48(3):454.
73 Kaatz G, Seo S, Dorman N, Lerner S. Emergence of teicoplanin resistance during therapy of Staphylococcus aureus endocarditis. J Infect Dis. 1990;162(1):103.
74 Elsaghier A, Aucken H, Hamilton-Miller J, Shaw S, Kibbler C. Resistance to teicoplanin developing during treatment of methicillin-resistant Staphylococcus aureus infection. J Antimicrob Chemother. 2002;49(2):423.
75 Del’ Alamo L, Cereda R, Tosin I, Miranda E, Sader H. Antimicrobial susceptibility of coagulase-negative staphylococci and characterization of isolates with reduced susceptibility to glycopeptides. Diagn Microbiol Infect Dis. 1999;34(3):185.
76 Bannerman T, Wadiak D, Kloos W. Susceptibility of Staphylococcus species and subspecies to teicoplanin. Antimicrob Agents Chemother. 1991;35(9):1919.
77 Sanofi-Aventis.co.uk. Targocid. Retrieved March 18, 2010. http://www.sanofi-aventis.co.uk/products/Targocid_SPC.pdf.
78 Bergeron M, Saginur R, Desaulniers D, Trottier S, Goldstein W, Foucault P, et al. Concentrations of teicoplanin in serum and atrial appendages of patients undergoing cardiac surgery. Antimicrob Agents Chemother. 1990;34(9):1699.
79 Wilson A, Shankar S, Felmingham D, Treasure T, Grüneberg R. Serum and tissue levels of teicoplanin during cardiac surgery: the effect of a high dose regimen. J Antimicrob Chemother. 1989;23(4):613.
80 Lam Y, Kapusnik-Uner J, Sachdeva M, Hackbarth C, Gambertoglio J, Sande M. The pharmacokinetics of teicoplanin in varying degrees of renal function. Clin Pharmacol Ther. 1990;47(5):655.
81 Smithers J, Thompson G, Kenny M, Dulworth J, Kulmala H, Lewis E, et al. Applicability of teicoplanin dosage adjustment guidelines for renally impaired patients over the range of 3 to 30 mg kg-1. Biopharm Drug Dispos. 1992;13(8):571.
82 Papaioannou M, Marinaki S, Pappas M, Stamatiadis D, Giamarellos-Bourboulis E, Giamarellou H, et al. Pharmacokinetics of teicoplanin in patients undergoing chronic haemodialysis. Int J Antimicrob Agents. 2002;19(3):233.
83 Stamatiadis D, Papaioannou M, Giamarellos-Bourboulis E, Marinaki S, Giamarellou H, Stathakis C. Pharmacokinetics of teicoplanin in patients undergoing continuous ambulatory peritoneal dialysis. Perit Dial Int. 2003;23(2):127.
84 Wolter K, Claus M, Wagner K, Fritschka E. Teicoplanin pharmacokinetics and dosage recommendations in chronic hemodialysis patients and in patients undergoing continuous veno-venous hemodialysis. Clin Nephrol. 1994;42(6):389.
85 Yagasaki K, Gando S, Matsuda N, Kameue T, Ishitani T, Hirano T, et al. Pharmacokinetics of teicoplanin in critically ill patients undergoing continuous hemodiafiltration. Intensive Care Med. 2003;29(11):2094.
86 Baltz R. Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr Opin Chem Biol. 2009;13(2):144.
87 Carpenter C, Chambers H. Daptomycin: another novel agent for treating infections due to drug-resistant gram-positive pathogens. Clin Infect Dis. 2004;38(7):994.
88 Silverman J, Oliver N, Andrew T, Li T. Resistance studies with daptomycin. Antimicrob Agents Chemother. 2001;45(6):1799.
89 Wise R, Andrews J, Ashby J. Activity of daptomycin against gram-positive pathogens: a comparison with other agents and the determination of a tentative breakpoint. J Antimicrob Chemother. 2001;48(4):563.
90 Barry A, Fuchs P, Brown S. In vitro activities of daptomycin against 2,789 clinical isolates from 11 North American medical centers. Antimicrob Agents Chemother. 2001;45(6):1919.
91 Sader H, Jones R. Antimicrobial susceptibility of gram-positive bacteria isolated from US medical centers: results of the Daptomycin Surveillance Program (2007-2008). Diagn Microbiol Infect Dis. 2009;65(2):158.
92 Petersen P, Bradford P, Weiss W, Murphy T, Sum P, Projan S. In vitro and in vivo activities of tigecycline (GAR-936), daptomycin, and comparative antimicrobial agents against glycopeptide-intermediate Staphylococcus aureus and other resistant gram-positive pathogens. Antimicrob Agents Chemother. 2002;46(8):2595.
93 Rybak M, Hershberger E, Moldovan T, Grucz R. In vitro activities of daptomycin, vancomycin, linezolid, and quinupristin-dalfopristin against staphylococci and enterococci, including vancomycin- intermediate and -resistant strains. Antimicrob Agents Chemother. 2000;44(4):1062.
94 Chakraborty A, Roy S, Loeffler J, Chaves RL. Comparison of the pharmacokinetics, safety and tolerability of daptomycin in healthy adult volunteers following intravenous administration by 30 min infusion or 2 min injection. J Antimicrob Chemother. 2009;64(1):151-158.
95 Cubist Pharmaceuticals Incorporated. Cubicin. Retrieved February 21, 2010. http://cubist.com/products/cubicin.php.
96 Tally F, DeBruin M. Development of daptomycin for gram-positive infections. J Antimicrob Chemother. 2000;46(4):523.
97 Churchwell M, Pasko D, Mueller B. Daptomycin clearance during modeled continuous renal replacement therapy. Blood Purif. 2006;24(5-6):548.
98 Churchwell MD, Mueller BA. Drug Dosing During Continuous Renal Replacement Therapy. Semin Dialysis. 2009;22(2):185-188.
99 Trotman RL, Williamson JC, Shoemaker DM, Salzer WL. Antibiotic dosing in critically ill adult patients receiving continuous renal replacement therapy. Clin Infect Dis. 2005;41(8):1159-1166.
100 Dvorchik B, Damphousse D. The pharmacokinetics of daptomycin in moderately obese, morbidly obese, and matched nonobese subjects. J Clin Pharmacol. 2005;45(1):48.
101 Pai MP, Norenberg JP, Anderson T, Goade DW, Rodvold KA, Telepak RA, et al. Influence of morbid obesity on the single-dose pharmacokinetics of daptomycin. Antimicrob Agents Chemother. 2007;51(8):2741-2747.
102 Mohr JFIII, Ostrosky-Zeichner L, Wainright DJ, Parks DH, Hollenbeck TC, Ericsson CD. Pharmacokinetic evaluation of single-dose intravenous daptomycin in patients with thermal burn injury. Antimicrob Agents Chemother. 2008;52(5):1891-1893.
103 Aumercier M, Bouhallab S, Capmau M, Le Goffic F. RP 59500: a proposed mechanism for its bactericidal activity. J Antimicrob Chemother. 1992;30(Suppl A):9.
104 Leclercq R, Nantas L, Soussy C, Duval J. Activity of RP 59500, a new parenteral semisynthetic streptogramin, against staphylococci with various mechanisms of resistance to macrolide-lincosamide-streptogramin antibiotics. J Antimicrob Chemother. 1992;30(Suppl A):67.
105 Leclercq R, Courvalin P. Intrinsic and unusual resistance to macrolide, lincosamide, and streptogramin antibiotics in bacteria. Antimicrob Agents Chemother. 1991;35(7):1273.
106 Le Goffic F, Capmau M, Abbe J, Cerceau C, Dublanchet A, Duval J. Plasmid mediated pristinamycin resistance: PH 1A, a pristinamycin 1A hydrolase. Ann Microbiol (Paris). 1977;128B(4):471.
107 Allignet J, Loncle V, el Sohl N. Sequence of a staphylococcal plasmid gene, vga, encoding a putative ATP-binding protein involved in resistance to virginiamycin A-like antibiotics. Gene (Amsterdam). 1992;117(1):45.
108 Ross J, Eady E, Cove J, Cunliffe W, Baumberg S, Wootton J. Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol Microbiol. 1990;4(7):1207.
109 Wootton M, Howe R, Walsh T, Bennett P, MacGowan A. In vitro activity of 21 antimicrobials against vancomycin-resistant Staphylococcus aureus (VRSA) and heteroVRSA (hVRSA). J Antimicrob Chemother. 2002;50(5):760.
110 Bonilla H, Perri M, Kauffman C, Zervos M. Comparative in vitro activity of quinupristin/dalfopristin against multidrug resistant Enterococcus faecium. Diagn Microbiol Infect Dis. 1996;25(3):127.
111 Finch R. Antibacterial activity of quinupristin/dalfopristin. Rationale for clinical use. Drugs. 1996;51(Suppl 1):31.
112 Johnson C3rd, T C, Zimmerman S, Bridson W, Chevalier P, Pasquier O, et al. Pharmacokinetics of quinupristin-dalfopristin in continuous ambulatory peritoneal dialysis patients. Antimicrob Agents Chemother. 1999;43(1):152.
113 Bergeron M, Montay G. The pharmacokinetics of quinupristin/dalfopristin in laboratory animals and in humans. J Antimicrob Chemother. 1997;39(Suppl A):129.
114 Griswold M, Lomaestro B, Briceland L. Quinupristin-dalfopristin (RP 59500): an injectable streptogramin combination. Am J Health Syst Pharm. 1996;53(17):2045.
115 Chevalier P, Rey J, Pasquier O, Leclerc V, Baguet J, Meyrier A, et al. Pharmacokinetics of quinupristin/dalfopristin in patients with severe chronic renal insufficiency. Clin Pharmacokinet. 2000;39(1):77.
116 Moellering R, Linden P, Reinhardt J, Blumberg E, Bompart F, Talbot G. The efficacy and safety of quinupristin/dalfopristin for the treatment of infections caused by vancomycin-resistant Enterococcus faecium. Synercid Emergency-Use Study Group. J Antimicrob Chemother. 1999;44(2):251.
117 Rubinstein E, Prokocimer P, Talbot G. Safety and tolerability of quinupristin/dalfopristin: administration guidelines. J Antimicrob Chemother. 1999;44(Suppl A):37.
118 Pfizer. Zyvox. Retrieved March 18, 2010. http://media.pfizer.com/files/products/uspi_zyvox.pdf.
119 Sander P, Belova L, Kidan Y, Pfister P, Mankin A, öttger E. Ribosomal and non-ribosomal resistance to oxazolidinones: species-specific idiosyncrasy of ribosomal alterations. Mol Microbiol. 2002;46(5):1295.
120 Jones R, Della-Latta P, Lee L, Biedenbach D. Linezolid-resistant Enterococcus faecium isolated from a patient without prior exposure to an oxazolidinone: report from the SENTRY Antimicrobial Surveillance Program. Diagn Microbiol Infect Dis. 2002;42(2):137.
121 Rahim S, Pillai S, Gold H, Venkataraman L, Inglima K, Press R. Linezolid-resistant, vancomycin-resistant Enterococcus faecium infection in patients without prior exposure to linezolid. Clin Infect Dis. 2003;36(11):E146.
122 Toh S, Xiong L, Arias C, Villegas M, Lolans K, Quinn J, et al. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol. 2007;64(6):1506.
123 Morales G, Picazo JJ, Baos E, Candel FJ, Arribi A, Pelaez B, et al. Resistance to linezolid is mediated by the cfr gene in the first report of an outbreak of linezolid-resistant Staphylococcus aureus. Clin Infect Dis. 2010;50(6):821-825.
124 Jones R, Ross J, Bell J, Utsuki U, Fumiaki I, Kobayashi I, et al. Zyvox Annual Appraisal of Potency and Spectrum program: linezolid surveillance program results for 2008. Diagn Microbiol Infect Dis. 2009;65(4):404.
125 John M, Pletch C, Hussain Z. In vitro activity of quinupristin/dalfopristin, linezolid, telithromycin and comparator antimicrobial agents against 13 species of coagulase-negative staphylococci. J Antimicrob Chemother. 2002;50(6):933.
126 Farrell D, Mendes R, Ross J, Jones R. Linezolid surveillance program results for 2008 (LEADER Program for 2008). Diagn Microbiol Infect Dis. 2009;65(4):392.
127 Miyazaki S, Fujikawa T, Kobayashi I, Matsumoto T, Tateda K, Yamaguchi K. The in vitro and in vivo antibacterial characterization of vancomycin and linezolid against vancomycin-susceptible and -resistant enterococci. J Antimicrob Chemother. 2002;50(6):971.
128 Burkhardt O, Borner K, von der Höh N, Köppe P, Pletz M, et al. Single- and multiple-dose pharmacokinetics of linezolid and co-amoxiclav in healthy human volunteers. J Antimicrob Chemother. 2002;50(5):707.
129 Gee T, Ellis R, Marshall G, Andrews J, Ashby J, Wise R. Pharmacokinetics and tissue penetration of linezolid following multiple oral doses. Antimicrob Agents Chemother. 2001;45(6):1843.
130 Slatter J, Stalker D, Feenstra K, Welshman I, Bruss J, Sams J, et al. Pharmacokinetics, metabolism, and excretion of linezolid following an oral dose of [(14)C]linezolid to healthy human subjects. Drug Metab Dispos. 2001;29(8):1136.
131 Lovering A, Zhang J, Bannister G, Lankester B, Brown J, Narendra G, et al. Penetration of linezolid into bone, fat, muscle and haematoma of patients undergoing routine hip replacement. J Antimicrob Chemother. 2002;50(1):73.
132 Andes D, van Ogtrop M, Peng J, Craig W. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob Agents Chemother. 2002;46(11):3484.
133 Adembri C, Fallani S, Cassetta M, Arrigucci S, Ottaviano A, Pecile P, et al. Linezolid pharmacokinetic/pharmacodynamic profile in critically ill septic patients: intermittent versus continuous infusion. Int J Antimicrob Agents. 2008;31(2):122.
134 Gerson S, Kaplan S, Bruss J, Le V, Arellano F, Hafkin B, et al. Hematologic effects of linezolid: summary of clinical experience. Antimicrob Agents Chemother. 2002;46(8):2723.
135 Hau T. Efficacy and safety of linezolid in the treatment of skin and soft tissue infections. Eur J Clin Microbiol Infect Dis. 2002;21(7):491.
136 Bernard L, Stern R, Lew D, Hoffmeyer P. Serotonin syndrome after concomitant treatment with linezolid and citalopram. Clin Infect Dis. 2003;36(9):1197.
137 Wigen C, Goetz M. Serotonin syndrome and linezolid. Clin Infect Dis. 2002;34(12):1651.
138 Lavery S, Ravi H, McDaniel W, Pushkin Y. Linezolid and serotonin syndrome. Psychosomatics (Washington, D.C.). 2001;42(5):432.
139 Beekmann S, Gilbert D, Polgreen P. Toxicity of extended courses of linezolid: results of an Infectious Diseases Society of America Emerging Infections Network survey. Diagn Microbiol Infect Dis. 2008;62(4):407.
140 Lunde C, Hartouni S, Janc J, Mammen M, Humphrey P, Benton B. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob Agents Chemother. 2009;53(8):3375.
141 Finegold SM, Bolanos M, Sumannen PH, Molitoris DR. In vitro activities of telavancin and six comparator agents against anaerobic bacterial isolates. Antimicrob Agents Chemother. 2009;53(9):3996-4001.
142 Wong S, Barriere S, Kitt M, Goldberg M. Multiple-dose pharmacokinetics of intravenous telavancin in healthy male and female subjects. J Antimicrob Chemother. 2008;62(4):780.
143 Astellas Pharmaceuticals. Vibativ http://www.astellas.us/docs/us/VIBATIV_PI_Final.pdf
144 Gotfried M, Shaw J, Benton B, Krause K, Goldberg M, Kitt M, et al. Intrapulmonary distribution of intravenous telavancin in healthy subjects and effect of pulmonary surfactant on in vitro activities of telavancin and other antibiotics. Antimicrob Agents Chemother. 2008;52(1):92.
145 Sun H, Duchin K, Nightingale C, Shaw J, Seroogy J, Nicolau D. Tissue penetration of telavancin after intravenous administration in healthy subjects. Antimicrob Agents Chemother. 2006;50(2):788.
146 Hegde S, Reyes N, Wiens T, Vanasse N, Skinner R, McCullough J, et al. Pharmacodynamics of telavancin (TD-6424), a novel bactericidal agent, against gram-positive bacteria. Antimicrob Agents Chemother. 2004;48(8):3043.
147 Patel J, Churchwell M, Seroogy J, Barriere S, Grio M, Mueller B. Telavancin and hydroxy propyl-beta-cyclodextrin clearance during continuous renal replacement therapy: an in vitro study. Int J Artif Organs. 2009;32(10):745.
148 Stryjewski M, Graham D, Wilson S, O’Riordan W, Young D, Lentnek A, et al. Telavancin versus vancomycin for the treatment of complicated skin and skin-structure infections caused by gram-positive organisms. Clin Infect Dis. 2008;46(11):1683.
149 Stryjewski M, O’Riordan W, Lau W, Pien F, Dunbar L, Vallee M, et al. Telavancin versus standard therapy for treatment of complicated skin and soft-tissue infections due to gram-positive bacteria. Clin Infect Dis. 2005;40(11):1601.
150 Stryjewski ME, Chu VH, O’Riordan WD, Warren BL, Dunbar LM, Young DM, et al. Telavancin versus standard therapy for treatment of complicated skin and skin structure infections caused by gram-positive bacteria: FAST 2 study. Antimicrob Agents Chemother. 2006;50(3):862-867.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
124 Agents with Primary Activity Against Gram-Positive Bacteria
The causes of nosocomial infections have changed in recent years. A 25-year study of nosocomial bacteremia demonstrated a change from Staphylococcus aureus and gram-negative bacilli as the predominant pathogens during the 1970s and 1980s to coagulase-negative staphylococci and Enterococcus, along with S. aureus and Pseudomonas aeruginosa, as the most common contemporary pathogens.1 The EPIC II study in 2007 demonstrated gram-positive organisms were associated with 47% of infections in the ICU.2 There can also be differences in the predominance of pathogens in different ICUs and different types of nosocomial infections. Nosocomial bacteremias are caused most often by coagulase-negative staphylococci and S. aureus in the medical ICU.3 S. aureus is the most common pathogen associated with nosocomial pneumonia and the fourth most common cause of skin and soft-tissue infections.3 Along with the increase in prevalence of gram-positive cocci in the ICU, staphylococci are becoming multidrug resistant. This chapter addresses gram-positive organisms and resistance issues associated with each of the antimicrobials with activity against these pathogens.
Vancomycin
Vancomycin was discovered in 1956 and marketed in 1958. Early preparations of the drug contained pyrogens and impurities that produced a brownish, muddy appearance that provided vancomycin’s nickname, “Mississippi mud.” In addition, these pyrogens and impurities caused high fevers, hypotension, severe phlebitis, and possibly nephrotoxicity.4
Mechanisms of Action And Resistance
Vancomycin inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Vancomycin is slowly bactericidal against dividing organisms except for Enterococcus and tolerant staphylococci, against which it is bacteriostatic.5 In 2006 the Clinical and Laboratory Standards Institute (CLSI) changed the vancomycin breakpoints against Staphylococcus aureus from ≤4 µg/mL to ≤2 µg/mL for susceptible strains. Intermediate susceptibility is now 4 to 8 µg/mL, and resistance to vancomycin is ≥16 µg/mL.6 The U.S. Food and Drug Administration (FDA) adopted these new breakpoints in 2008. The European Committee on Antimicrobial Susceptibility Testing (EUCAST) changed their vancomycin interpretations against S. aureus to ≤2 µg/mL as susceptible and >2 µg/mL as resistant. These changes in breakpoints will alter how literature is interpreted with respect to the frequency or prevalence of vancomycin-intermediate or vancomycin-resistant S. aureus over the past 30 years.
Five types of resistance for vancomycin have been isolated from enterococci: VanA, VanB, VanC, VanD, and VanE. The VanA phenotype confers high-level resistance to both teicoplanin (minimum inhibitory concentrations [MICs]: 16 to 512 µg/mL) and vancomycin (MICs: 64 to >1000 µg/mL). Vancomycin can induce expression of the VanA gene and has been identified in both Enterococcus faecium and Enterococcus faecalis. The VanB phenotype has also been identified in both E. faecium and E. faecalis and confers low-level resistance primarily to vancomycin. VanA, B, D, and E are all transferable to other organisms. In contrast, the VanC phenotypes are endogenous (constitutively produced) and are components of Enterococcus gallinarum, Enterococcus casseliflavus, and Enterococcus flavescens and confer resistance to vancomycin alone. The VanB gene has been identified in a strain of Streptococcus bovis. This gene showed 96% homology with the prototype VanB gene from E. faecalisV583, indicating the likelihood of the gene transfer from enterococcus to this strain of S. bovis.7
Vancomycin-intermediate S. aureus using the prior breakpoints of MIC 8 to 16 µg/mL was first reported in 1996 from Japan, and by June 2002, eight cases were confirmed in the United States.88 Using the new breakpoints, the incidence of vancomycin-intermediate S. aureus will increase. In June 2002, the first case of vancomycin-resistant S. aureus (MIC > 32 µg/mL) was identified in Michigan, followed in September 2002 by the second case in Pennsylvania.8,9 No mechanism of resistance has yet been identified from the strains of vancomycin-intermediate S. aureus, but the two strains of vancomycin-resistant S. aureus both possessed the VanA gene.
Tolerance is another mechanism by which bactericidal activity is decreased. Tolerance can be measured or assessed by two methods: the ratio of minimum bactericidal concentration to minimum inhibitory concentration (MBC : MIC) and time-kill curves. By definition, a MBC : MIC ratio of 32 or greater or less than 99.9% kill after 24 hours incubation in time-kill studies equates to tolerance. Tolerance to vancomycin has been identified in S. aureus, Streptococcus pneumoniae, and groups C and G streptococci.10–12
Spectrum of Activity
Vancomycin is active primarily against aerobic gram-positive cocci including Corynebacterium and methicillin-resistant S. aureus (MRSA). The MIC90 against methicillin-susceptible S. aureus (MSSA) is 1 µg/mL, and against MRSA it is 1 to 2 µg/mL.13–15 The incidence of vancomycin-intermediate or vancomycin-resistant S. aureus currently is very low and less than 1%. The activity of vancomycin against enterococci varies greatly with the species. E. faecium is the most resistant species of enterococci to vancomycin, with the resistant rates ranging from 30% to 90% depending on the institution. For all enterococci the vancomycin resistance rates are 20% to 25%.16
Most streptococci are susceptible to vancomycin, although it is considered an agent of last resort against these organisms. Vancomycin has been shown to be inferior to nafcillin or oxacillin for the treatment of MSSA infections. Treatment failures, prolonged treatment, and higher mortality rates have been demonstrated when vancomycin was used to treat MSSA infections compared with nafcillin or oxacillin.17,18
Vancomycin is active against anaerobic gram-positive organisms such as Peptostreptococcus spp., Propionibacterium spp., Eubacterium spp., Bifidobacterium spp., and most Clostridium spp., including C. difficile.19
Pharmacokinetics/Pharmacodynamics
Vancomycin is administered orally and intravenously (IV). The drug is poorly absorbed after oral administration, and the majority of the drug is excreted unchanged in feces. Inflammation of the gastrointestinal tract may result in increased absorption of vancomycin, and measurable serum concentrations might be obtained.20 Intramuscular injections are extremely painful and should not be used. Distribution of the drug is complete 1 hour after a 1- to 2-hour IV infusion. Vancomycin is approximately 55% bound to plasma proteins. The volume of distribution corrected for weight ranges from 0.4 to 0.9 L/kg.21–27 Vancomycin does not penetrate well into noninflamed meninges or aqueous humor.28 Distribution into inflamed meninges is variable, with reported ranges of 1% to 37% of serum concentrations29,30 and a mean concentration of 15% of serum or approximately 2.5 µg/mL.31 Penetration into ascitic, pericardial, and synovial fluids is greater than 75% serum concentrations; penetration approximates 50% into pleural fluid, and 30% to 50% into bile.25 Elimination of vancomycin is 80% to 90% unchanged drug in the urine via glomerular filtration and the remaining via nonrenal elimination. The nonrenal elimination rate in healthy individuals is 40 mL/min, and in chronic renal failure patients it is 6 mL/min.32 The half-life of the drug increases with decreased renal function; in patients with creatinine clearances (CrCl) greater than 80 mL/min, the half-life is 4 to 6 hours. The pharmacodynamic effect of vancomycin is time-dependent killing or time above the MIC.33 Therefore, the most important goal of therapy is to maintain a free serum trough concentration above the MIC of the organism. There is no documented correlation between serum peak concentrations and clinical outcomes.
Dosage Regimens
Oral Administration
Oral administration of vancomycin is only for treating C. difficile colitis and is considered second-line therapy for mild/moderate infections and primary therapy for moderate/severe infections. The dose is 125 to 500 mg orally every 6 hours and is not adjusted for renal dysfunction, owing to the poor absorption. Two oral formulations (capsules or liquid) can be used, or the IV solution can be administered orally to treat C. difficile. Table 124-1 lists dosing regimens for the antimicrobials discussed in this chapter.
Intravenous Administration in Adults
In nonobese adults with normal renal function, the usual dose of vancomycin is 1 g (∼15 mg/kg) every 12 hours. This dose results in peak serum concentrations of 25 to 40 µg/mL 1 hour after completion of the infusion and trough serum concentration of 5 to 15 µg/mL. Dosing should be based on actual body weight. Several dosing guidelines have been developed to accurately and easily dose vancomycin. The most popular methods include the Moellering23 and Matzke24 nomograms. These methods use body weight and CrCl to calculate vancomycin dose. The weaknesses of these nomograms include the small number of patients used to develop and evaluate the nomogram and the fixed volume of distribution assumed for all patients (0.9 L/kg). Matzke24 found that for patients younger than 65 years of age, a volume of distribution of 0.7 L/kg may be more accurate, and for those older than 65, it is 0.9 L/kg. This variance in volume of distribution does affect the reproducibility of these nomograms when applied to different patient populations. The Cockcroft and Gault and modified Cockcroft and Gault methods of estimating CrCl are relatively reliable and accurate methods in patients of normal body mass.33
Morbidly obese patients are difficult to dose given the lack of pharmacokinetic studies. Doses of approximately 30 mg/kg/d based on actual body weight should provide a peak serum concentration of 25 to 35 µg/mL. Because CrCl is the best correlate to vancomycin clearance, the most accurate method for estimating CrCl should be used and varies with body mass. CrCl estimations in the obese patient are best predicted by the Salazar-Corcoran method.34 Young obese patients with no comorbid conditions affecting renal function often require the dosing interval to be more frequent to achieve a trough serum concentration of 5 to 15 µg/mL. This is due to the faster rate of clearance of the drug (2.3-2.5 times higher) in obese compared with nonobese patients.35,36
Vancomycin Dosing in Critically Ill Patients
Garaud evaluated critically ill patients and found an average volume of distribution of 0.6 L/kg in patients with CrCl greater than 70 mL/min and 0.4 L/kg with CrCl of 10 to 60 mL/min.26 Critically ill patients are often receiving medications to improve hemodynamics, such as dopamine, dobutamine, and furosemide. These medications result in increases in renal function and changes in volume status for the patient. In a study designed to assess the impact of such medications on vancomycin pharmacokinetics, two observations were made.37 First, some of the patients required larger total daily doses of vancomycin to achieve therapeutic concentrations than the Moellering nomogram predicted (26.78 + 3.01 mg/kg/d versus 18.95 + 3.41 mg/kg/d). Second, on discontinuation of these medications, the serum trough concentrations increased despite no change in CrCl or body weight. The theory is that these medications enhanced vancomycin clearance by improving renal blood flow and/or interacting with the renal anion transport system, thus increasing glomerular filtration and renal tubular secretion. Therefore, larger doses of vancomycin may be required while on these medications, and smaller doses may be more appropriate on discontinuation of these medications.
Vancomycin Dosing for Patients on Dialysis/Hemofiltration/Cardiopulmonary Bypass
The percentage of vancomycin removed by low-permeability cellulose hemodialyzers is 4% to 6.9%.38–40 Therefore, no supplemental vancomycin dosing is required after hemodialysis with these older systems. The removal of vancomycin during intradialytic administration has been studied using three types of cellulose membranes: cellulose acetate (CA), cellulose triacetate (CT), and CA high-performance 210 (CAHP-210). With the CA membranes, 0% to 25% (mean of ∼13%) of vancomycin is removed.38,41 The CT membranes remove 16% to 44% (mean of ∼26%) of vancomycin.38,41 Vancomycin removal during intradialytic administration with the CAHP-210 membranes is 0% to 35%, with a mean of 24%.42 High-flux synthetic membranes such as polysulfone or polyacrylonitrile remove significantly more vancomycin than do the cellulose membranes, with 30% to 55% and 25% to 40% of vancomycin removed, respectively.38–40,43–45
Continuous renal replacement therapy (CRRT) is a low-volume (1-2 L/h) therapy. The most frequently used methods of CRRT are continuous venovenous hemofiltration (CVVH), continuous venovenous hemodialysis (CVVHD), and continuous arteriovenous hemodialysis (CAVHD). Both CVVHD and CAVHD result in a greater total body clearance of vancomycin than does hemofiltration. The clearances achieved with each of these methods vary with blood flow rate, ultrafiltration rate, and the membranes used. The total clearance of vancomycin with CVVHD or CAVHD is 31 to 39 mL/min, and the half-life ranges from 14 to 25 hours.46–49 Clearance of vancomycin in patients with normal renal function (CrCl > 70 mL/min) and with mild renal dysfunction (CrCl 40-70 mL/min) has been reported to be 88 and 48 mL/min, respectively.50 High-volume hemofiltration (HVHF), with an ultrafiltration rate of 6 L/h, increases vancomycin clearance to approximately 60 mL/min.51 Therefore, patients receiving CAVHD or CVVHD should receive vancomycin every 36 to 48 hours, and those undergoing HVHF should receive the drug every 12 to 24 hours.
Cardiopulmonary bypass (CPB) significantly impacts the pharmacokinetic parameters of vancomycin. Immediately after initiating CPB, vancomycin serum concentration decreased by 7 µg/mL (5.7 to 8.4 µg/mL), which represented approximately a 38% decrease in concentration.52 Over the next 30 minutes, serum vancomycin concentration may increase 1 to 2 µg/mL but thereafter gradually and steadily decreases.52 The half-life is not affected by CPB and does not change during the process.
Adverse Effects
Common toxicities that have been associated with vancomycin therapy include red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity. Evidence establishing a clear relationship between these toxicities and vancomycin peak or trough concentrations or the incidence of these events is limited and contradictory.4,53–55
Red man syndrome comprises erythema, pruritus, and flushing of the upper torso and is often associated with too rapid an infusion of the drug. In general, the infusion rate should not exceed 1 g/h. Less frequently, hypotension and angioedema can occur. It is thought that increased histamine release is the cause of this syndrome.4,54–56 A comparative trial of once-daily versus twice-daily vancomycin found the incidence of this syndrome to be 13.7% and 9.6%, respectively.54 The effects of red man syndrome can be relieved by antihistamines.57,58
Thrombophlebitis is reported in 3% to 23% of patients receiving vancomycin and is more common in patients who receive vancomycin for more than 7 days or have peripheral catheter lines for prolonged durations.4,54
Ototoxicity rates range from 0% to 9% in patients receiving vancomycin.4,54 The definition of ototoxicity ranges from tinnitus to hearing loss. The evidence demonstrating any relationship between ototoxicity and high peak serum concentrations of vancomycin is limited. In cancer patients, only 4 of 19 patients with ototoxicity had elevated serum concentrations of vancomycin, and only 1 had a concentration greater than 80 µg/mL.4 Others have reported ototoxicity associated with peak serum concentrations of 37.5 to 152 µg/mL.59,60 A trial comparing once-daily to twice-daily dosing of vancomycin demonstrated more frequent ototoxicity in the twice-daily dosed group (15.6% versus 3.2%), which had a significantly lower peak concentration and similar trough concentration compared to the group receiving daily doses.54 This lack of correlation between serum concentrations of vancomycin and ototoxicity suggests that the observed toxicity was due to either another drug or to the combination of another drug with vancomycin. In the majority of cases, ototoxicity symptoms disappear within a month of discontinuing vancomycin.
The issue of nephrotoxicity associated with vancomycin is complicated by several confounding factors. The original formulation was very impure, and the impurities were associated with toxicities including nephrotoxicity. In addition, many definitions of nephrotoxicity have been used over the years, different patient populations have been studied, and different doses used, making it difficult to compare one study to another. In general, the rate of nephrotoxicity is 5% to 10% when vancomycin is not administered with other nephrotoxic agents and trough concentrations are less than 10 µg/mL.54,61,62 Elting and colleagues identified older age, Acute Physiology and Chronic Health Evaluation (APACHE) score greater than 40, and duration of therapy of greater than 14 days to be the best predictors for a patient to develop nephrotoxicity due solely to vancomycin therapy.4 A number of other studies have found an increased incidence of nephrotoxicity (21%-35%) when vancomycin serum trough concentrations are greater than 10 µg/mL.62–64 In addition, Lodise demonstrated an increased rate of nephrotoxicity (∼35%) when the total daily dose is 4 grams or more compared to total doses less than 4 grams (∼11%).65 Studies have demonstrated higher rates of nephrotoxicity when vancomycin is used in combination with an aminoglycoside compared with either agent alone.62,66,67 Goetz performed a meta-analysis of eight studies and found the incidence of nephrotoxicity associated with combination therapy was 13% greater than with vancomycin alone and 4% greater than with an aminoglycoside alone.67
Other toxicities associated with vancomycin include maculopapular or erythematous rashes (2%-8%)26,68,75 and anecdotal reports of neutropenia and thrombocytopenia.68,69
Therapeutic Drug Monitoring
Routine monitoring of vancomycin serum concentrations has become a highly debated issue over the years. Those who advocate routine monitoring cite the need to ensure therapeutic concentrations as well as minimize toxicities. To date there is only one trial that compared efficacy and toxicity with high-dose once-daily versus twice-daily dosing of vancomycin; with these dosing regimens, peak serum concentrations were vastly different but trough serum concentrations were similar.54 The mean peak serum concentrations in the once-daily and twice-daily dosed groups were 42.8 + 16.1 and 27.0 + 9.2 µg/mL, respectively. There were no differences in clinical efficacy, red man syndrome, thrombophlebitis, ototoxicity, and nephrotoxicity between the two groups.
Studies over the past 20 years have shown that peak concentrations of vancomycin are not associated with toxicities or clinical efficacy. Therefore, monitoring peak serum concentrations only adds to hospital and healthcare system costs and provides no beneficial clinical information. Some studies have demonstrated a correlation of nephrotoxicity to serum trough concentrations ≥ 10 µg/mL, whereas others have not. Given the lack of consensus, it may be prudent to measure serum trough concentrations until more definitive studies are conducted to address this issue.70
In patients with end-stage renal disease, the fluorescence polarization immunoassay (FPIA) overestimates vancomycin concentrations.71 FPIA is the most common method for determining vancomycin concentrations, and when it was compared with the enzyme multiplied immunoassay technique, it was found to produce higher peak serum concentrations by 7 to 11 µg/mL and higher trough concentrations by 4 to 6 µg/mL.
Teicoplanin
Mechanisms of Action And Resistance
Teicoplanin, like other glycopeptide antibiotics, inhibits synthesis of the cell wall by binding to the D-alanyl-D-alanine terminus of cell wall precursor units. Resistance has been reported in both staphylococci and enterococci. The VanA phenotype confers high-level resistance to both teicoplanin (MIC: 16 to 512 µg/mL) and vancomycin (MIC: 64 to >1000 µg/mL). The VanB phenotype has also been identified in both E. faecium and E. faecalis and usually confers low-level resistance to vancomycin but not to teicoplanin. This may limit the utility of teicoplanin for some vancomycin-resistant enterococcal infections. Several reports of S. aureus resistance developing during therapy with teicoplanin have been reported.72–74 The mechanism of the resistance was determined in one patient to be constitutive and non-plasmid mediated.73
Spectrum of Activity
Teicoplanin is only active against gram-positive organisms. Activity against MSSA and MRSA is comparable to that of vancomycin. Coagulase-negative staphylococci have a varied pattern of susceptibility to teicoplanin. Staphylococcus haemolyticus is the most resistant species to teicoplanin (30%).75 These isolates are 25% more resistant to teicoplanin than to vancomycin. Against methicillin-resistant coagulase-negative staphylococci, 39% of isolates have teicoplanin MICs greater than 8 µg/mL compared with 1% with vancomycin.75,76 Teicoplanin is similar in activity to vancomycin against enterococci, although its reliability in treating infections with VanB resistance to vancomycin may be limited. Teicoplanin is active against other aerobic and anaerobic gram-positive organisms such as Corynebacterium spp., Clostridium spp., including C. difficile and C. perfringens, Peptostreptococcus spp., and Propionibacterium acnes.
Pharmacokinetics/Pharmacodynamics
Teicoplanin is administered orally and intravenously. The drug is poorly absorbed after oral administration, and approximately 40% of the drug is excreted unchanged in feces. The pharmacokinetic model that best describes the elimination of teicoplanin is triexponential. IV administration of 400 mg (6 mg/kg) should provide a peak serum concentration of 20 to 50 µg/mL attained 1 hour after administration.77 The volume of distribution is large at 0.9 to 1.41 L/kg, and teicoplanin is 90% to 95% protein bound.77 Penetration into body fluids and tissues has not been extensively studied. Penetration into noninflamed meninges and fat is poor, but distribution into myocardium and pericardium is good.78,79 Teicoplanin is primarily eliminated via glomerular filtration, and only 3% is metabolized.77 The half-life is approximately 150 hours in patients with normal renal function.77 Because of the long half-life, it takes 14 days to reach steady state. In patients with CrCl of 13 to 25, the half-life was found to be 280 to 667 hours.80,81
Dosage Regimens/Therapeutic Drug Monitoring
Despite the long half-life in patients with normal renal function, teicoplanin should be administered daily, and the dose is dependent on the severity of infection. For less serious infections involving the urinary tract, skin, soft tissue, and lower respiratory tract, a loading dose of 400 mg (6 mg/kg) × 1 is administered, followed by a maintenance dose of 200 mg (3 mg/kg) every 24 hours. For severe infections such as septicemia, endocarditis, and osteomyelitis, 400 mg of teicoplanin is administered every 12 hours for 3 doses, followed by 400 mg every 24 hours.77 Although no therapeutic range has been established for teicoplanin, trough concentrations should be at least 10 µg/mL.77
Renal Failure/Dialysis
Teicoplanin is not removed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD).82,83 The amount removed by CVVHD is dependent on the flow rate but is often minimal.84,85 Several dosing regimens exist for renal dysfunction, and the simplest method is administering a dose of 6-10 mg/kg every 48 to 72 hours.
Adverse Effects
Nephrotoxicity associated with teicoplanin is much lower than with vancomycin. The incidence from published and unpublished studies found the nephrotoxic rate to be 4%.55 Ototoxic rates with teicoplanin are similar to those with vancomycin.55 Hypersensitivity reactions are the most common adverse reaction to teicoplanin (2%-15%).55
Daptomycin
Mechanisms of Action And Resistance
Daptomycin has a unique mechanism of action and has been found to inhibit lipoteichoic acid synthesis, owing to binding to the membrane in the presence of calcium.86,87 Minimal information is available on the mechanism(s) of resistance to daptomycin. Limited in vitro studies have been performed attempting to create daptomycin resistance in the laboratory.88 Mechanisms of resistance have not been elucidated, and the clinical relevance of in vitro resistance is unknown.
Spectrum of Activity
Daptomycin’s antibacterial activity encompasses most gram-positive bacteria, including vancomycin-resistant isolates and penicillin-resistant pneumococci. The MICs of daptomycin are 8- to 16-fold lower in the presence of calcium. Therefore, all in vitro testing must be supplemented with physiologic concentrations of calcium.89 The breakpoint for susceptible is ≤1 µg/mL for staphylococci and β-hemolytic streptococci. Given the rare number of isolates not susceptible to daptomycin, a resistant breakpoint has yet to be determined. The MIC90 against MSSA, MRSA, Staphylococcus epidermidis, and Staphylococcus saprophyticus are all 0.5 µg/mL or less.89,90 In a recent surveillance study, 7 S. aureus and 6 coagulase-negative staphylococci were non-susceptible to daptomycin.91
[/not-level-membership-for-critical-care-medicine-category]
