[level-membership-for-basic-science-category]
5 Adverse drug reactions
Assessing the safety of drugs
When drugs are newly introduced to the market, their safety profile will be provisional. While efficacy and evidence of safety must be demonstrated for regulatory authorities to permit marketing, it is not possible to discover the complete safety profile of a new drug prior to its launch. Pre-marketing clinical trials involve on average 2500 patients, with perhaps a hundred patients using the drug for longer than a year. Therefore, pre-marketing trials do not have the power to detect important reactions that occur at rates of 1 in 10,000, or fewer, drug exposures. Often, only pharmacologically predictable ADRs with short onset times may be identified in clinical trials, nor can pre-marketing trials detect ADRs which are separated in time from drug exposure. Additionally, patients within trials are often carefully selected, without the multiple disease states or complex drug histories of patients in whom the drug will eventually become used. Furthermore, the patient’s perspective is also frequently excluded from clinical trial safety assessments, with ADRs being assessed only by the clinicians who run them (Basch, 2010). For these reasons, rare and potentially serious adverse effects often remain undetected until a wider population is exposed to the drug. The vigilance of health professionals is an essential factor in discovering these new risks, together with regulatory authorities who continuously monitor reports of adverse effects throughout the lifetime of a marketed medicinal product.
Definitions
Having clear definitions of what constitutes an ADR is important. The World Health Organization (WHO) defines an ADR as ‘a response to a drug that is noxious and unintended and occurs at doses normally used in man for the prophylaxis, diagnosis or therapy of disease, or for modification of physiological function’ (WHO, 1972). The use of the phrase ‘at doses normally used in man’ distinguishes the noxious effects of drugs during normal medical use from toxic effects caused by poisoning. Whether an effect is considered noxious depends on both the drug’s beneficial effects and the severity of the disease for which it is being used. There is no need to prove a pharmacological mechanism for any noxious response to be termed an ADR.
The WHO definition has been criticised for excluding the potential for contamination of a product, ADRs that include an element of error, and ADRs associated with pharmacologically inactive excipients in a product. The use of the term ‘drug’ also excluded the use of complementary and alternative treatments, such as herbal products. In an attempt to overcome these points, the following definition of an ADR was proposed, ‘An appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medicinal product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of the dosage regime, or withdrawal of the product’ (Edwards and Aronson, 2000).
Classification of ADRs
Rawlins–Thompson classification
The Rawlins–Thompson system of classification divides ADRs into two main groups: Type A and Type B (Rawlins, 1981). Type A reactions are the normal, but quantitatively exaggerated, pharmacological effects of a drug. They include the primary pharmacological effect of the drug, as well as any secondary pharmacological effects of the drug, for example, ADRs caused by the antimuscarinic activity of tricyclic anti-depressants. Type A reactions are most common, accounting for 80% of reactions.
Type B reactions are qualitatively abnormal effects, which appear unrelated to the drug’s normal pharmacology, such as hepatoxicity from isoniazid. They are more serious in nature, more likely to cause deaths, and are often not discovered until after a drug has been marketed. The Rawlins–Thompson classification has undergone further elaboration over the years (Table 5.1) to take account of ADRs that do not fit within the existing classifications (Edwards and Aronson, 2000).
Table 5.1 Extended Rawlins–Thompson classification of adverse drug reactions
| Type of reaction | Features | Examples |
|---|---|---|
| Type A: Augmented pharmacological effect | Common | Bradycardia associated with a beta-adrenergic receptor antagonist |
| Predictable effect | ||
| Dose-dependent | ||
| Low morbidity | ||
| Low mortality | ||
| Type B: Bizarre effects not related to pharmacological effect | Uncommon | Anaphylaxis associated with a penicillin antibiotic |
| Unpredictable | ||
| Not dose-dependent | ||
| High morbidity | ||
| High mortality | ||
| Type C: Dose-related and time-related | Uncommon | Hypothalamic pituitary–adrenal axis suppression by corticosteroids |
| Related to the cumulative dose | ||
| Type D: Time-related | Uncommon | Carcinogenesis |
| Usually dose-related | ||
| Occurs or becomes apparent some time after use of the drug | ||
| Type E: Withdrawal | Uncommon | Opiate withdrawal syndrome |
| Occurs soon after withdrawal of the drug | ||
| Type F: Unexpected failure of therapy | Common | Failure of oral contraceptive in presence of enzyme inducer |
| Dose-related | ||
| Often cause by drug interactions |
The DoTS system
The DoTS classification is based on Dose relatedness, Timing and patient Susceptibility (Aronson and Ferner, 2003). In contrast to the Rawlins–Thompson classification, which is defined only by the properties of the drug and the reaction, the DoTS classification provides a useful template to examine the various factors that both describe a reaction and influence an individual patient’s susceptibility.
The final aspect of the DoTS classification system is susceptibility, which includes factors such as genetic predisposition, age, sex, altered physiology, disease and exogenous factors such as drug interactions (Table 5.2)
| Dose relatedness | Time relatedness | Susceptibility |
|---|---|---|
| Toxic effects: ADRs that occur at doses higher than the usual therapeutic dose Collateral effects: ADRs that occur at standard therapeutic doses Hypersusceptability reactions: ADRs that occur at sub-therapeutic doses in susceptible patients |
Time-independent reactions: ADRs that occur at any time during treatment. Time-dependent reactions: Rapid reactions occur when a drug is administered too rapidly. Early reactions occur early in treatment then abate with continuing treatment (tolerance). Intermediate reactions occur after some delay, but if reaction does not occur after a certain time, little or no risk exists. Late reactions risk of ADR increases with continued-to-repeated exposure, including withdrawal reactions. Delayed reactions occur some time after exposure, even if the drug is withdrawn before the ADR occurs. |
Raised susceptibility may be present in some individuals, but not others. Alternatively, susceptibility may follow a continuous distribution – increasing susceptibility with impaired renal function. Factors include: genetic variation, age, sex, altered physiology, exogenous factors (interactions) and disease. |
Factors affecting susceptibility to ADRs
Co-morbidities and concomitant medicines use
Reductions in hepatic and renal function substantially increase the risk of ADRs. A recent study examining factors that predicted repeat admissions to hospital with ADRs in older patients showed that co-morbidities such as congestive cardiac failure, diabetes, and peripheral vascular, chronic pulmonary, rheumatological, hepatic, renal, and malignant diseases were strong predictors of readmissions for ADRs, while advancing age was not. Reasons for this could be pharmacokinetic and pharmacodynamic changes associated with pulmonary, cardiovascular, renal and hepatic insufficiency, or drug interactions because of multiple drug therapy (Zhang et al., 2009).
Ethnicity
Examples of ADRs linked to ethnicity include the increased risk of angioedema with the use of ACE inhibitors in black patients (McDowell et al., 2006), the increased propensity of white and black patients to experience central nervous system ADRs associated with mefloquine compared to patients of Chinese or Japanese origin, and differences in the pharmacokinetics of rosuvastatin in Asian patients which may expose them to an increased risk of myopathy. However, susceptibility based on ethnicity could be associated with genetic or cultural factors and ethnicity can be argued to be a poor marker for a patient’s genotype.
Pharmacogenetics
The narrow therapeutic index of warfarin, its high inter-individual variability in dosing and the serious consequences of toxicity have made it a major target of pharmacogenomic research. Studies of genetic polymorphisms influencing the toxicity of warfarin have focused on CYP2C9, which metabolises warfarin and vitamin K epoxide reductase (VKOR), the target of warfarin anticoagulant activity. Genetic variation in the VKORC1 gene, which encodes VKOR, influences warfarin dosing by a threefold greater extent than CYP2C9 variants. In 2007, the U.S. Food and Drug Administration (FDA) changed the labelling requirement for warfarin, advising that a lower initial dose should be considered in people with certain genetic variations. However, concerns remain because genetic variation only accounts for a proportion of the variability in drug response and clinicians may obtain a false sense of reassurance from genetic testing leading to complacency in monitoring of therapy. In addition, there appears to be little evidence of additional benefit (Laurence, 2009), in terms of preventing major bleeding events, compared to careful monitoring of the INR (see chapter 23)
A success story for pharmacogenetics is the story of the nucleoside analogue reverse transcriptase inhibitor (NRTI) abacavir. Hypersensitivity skin reactions to abacavir are a particular problem in the treatment of human immunodeficiency virus (HIV) infection. Approximately 5–8% of patients taking abacavir develop a severe hypersensitivity reaction, including symptoms such as fever, rash, arthralgia, headache, vomiting and other gastro-intestinal and respiratory disturbances. Early reports that only a subset of patients was affected, a suspected familial predisposition, the short onset time (within 6 weeks of starting therapy), and an apparent lower incidence in African patients led to suspicion of a genetic cause. Subsequent research revealed a strong predictive association with the human leukocyte antigen HLA-B*5701 allele in Caucasian and Hispanic patients. The presence of the allele can be used to stratify the predicted risk of hypersensitivity as high risk (>70%) for carriers of HLA-B*5701 and low risk (<1%) for non-carriers of HLA-B*5701. Evidence from the practical use of HLA-B*5701 screening has shown substantial falls in the incidence of hypersensitivity reactions, as well as a more general improved compliance with the medication (Lucas et al., 2007).
Immunological reactions
Allergic reactions range from rashes, serum sickness and angioedema to the life-threatening bronchospasm and hypotension associated with anaphylaxis. Patients with a history of atopic or allergic disorders are at higher risk. Immunological (hypersensitivity) reactions are split into four main types (Table 5.3).
Table 5.3 Classification of immunological (hypersensitivity) reactions
| Classification | Mechanism | Symptoms/signs and examples |
|---|---|---|
| Type I (immediate) | Drug/IgE complex to mast cells release of histamine and leukotrienes. | Pruritis, urticaria, bronchoconstriction, angioedema, hypotension, shock, for example, penicillin anaphylaxis. |
| Type II (cytotoxic) | IgG and complement binding to (usually) red blood cell. Cytotoxic T-cells lyse the cell. | Haemolytic anaemia and thrombocytopaenia, for example, associated with cephalosporins, penicillins and rifampicin. |
| Type III (immune complex) | Drug antigen and IgG or IgM form immune complex, attracting macrophages and complement activation. | Cutaneous vasculitis, serum sickness, for example, associated with chlorpromazine and sulphonamides. |
| Type IV (delayed type) | Antigen presentation with major histocompatibility complex protein to T-cells and cytokine and inflammatory mediator release. | Usually occur after 7–20 days. Macular rashes and organ failure, including Stevens–Johnson syndrome and toxic epidermal necrolysis, for example, associated with neomycin and sulphonamides. |
Formulation issues contributing to ADRs
Although excipients are often referred to as inert substances, serious adverse reactions such as anaphylaxis and angioedema have been reported to these substances. Sweeteners, flavourings, colouring agents/dyes and preservatives have all been associated with adverse reactions (Kumar, 2003).
Epidemiology of ADRs
ADRs are widespread, as shown by both systematic reviews and large-scale studies. A review of 69 studies from many countries in 2002 found that ADRs were responsible for an estimated 2.6% of admissions to hospitals and that between 3.5% and 7.3% of in-patients may suffer an ADR. More recent data, however, shows these to be under-estimates. A prospective study (Pirmohamed et al., 2004) found that 6.5% out of 18,820 admissions to medical units were caused by ADRs, with 2.3% of patients dying as a result. A similar prospective study of 3695 in-patient episodes found that 14.7% of those admitted to medical or surgical wards experienced an ADR during their stay. These were more common in women, older patients and in those admitted to surgical wards (Davies et al., 2009).
In primary care, estimates for the incidence of ADRs are more difficult to obtain. Some studies have relied on patients’ reports of ADRs, either to postal questionnaires or telephone surveys. These provide varying estimates in ADR incidence and prevalence, but are hampered by the lack of information about non-responders. Nonetheless, estimates are of the order of 25% in the USA (Ghandi et al., 2003) and 30% in the UK (Jarernsiripornkul et al., 2002). A systematic review in 2007 found an incidence of overall ADEs, including ADRs, of 14.9 per 1000 person-months in primary care settings.
A widely quoted figure is that ADRs are between the fourth and sixth leading cause of death in the USA. This is based on an extrapolation of a meta-analysis of studies carried out in the USA, which showed that the incidence of serious ADRs causing hospital admission or occurring during admissions was 6.7% and resulted in an incidence of fatal ADRs of 0.32% (Lazarou et al., 1998). The study has been criticised for its methodology; however, more recent work from Sweden has identified that ADRs were responsible for 3% of deaths there (Wester et al., 2008), while in England ADRs were shown to occur in 0.4% of all patients admitted to hospital. This latter study showed that mortality was higher in those experiencing an ADR than in those who did not. Furthermore, the median length of stay in patients who experienced an ADR was 20 days compared to 8 days and costs associated with in-patient ADRs were calculated to be £171 million annually for the NHS in England (Davies et al., 2009). Costs to the NHS associated with admissions due to ADRs have been estimated as £466 million annually (Pirmohamed et al., 2004).
Pharmacovigilance and epidemiological methods in ADR detection
Spontaneous reporting
Causality assessment
One of the most common methods of causality assessment in use is unstructured clinical assessment, also known as global introspection. Expert review of clinical information is undertaken and a judgement is made about the likelihood of the reaction being due to drug exposure. The assessment of complex situations, often with missing information, is open to variation between different assessors and studies have shown marked disagreement between experts. The WHO international monitoring centre uses global introspection for case assessment, assigning standardised causality categories to suspected ADRs (Table 5.4).
| Category | Description |
|---|---|
| Certain | Pharmacologically definitive, with re-challenge if necessary |
| Probably/likely | Reasonable temporal relationship, unlikely to be attributed to disease processes or other drugs, with reasonable dechallenge response |
| Possible | Reasonable temporal relationship, but could be explained by concurrent disease or drugs. No information on withdrawal |
| Unlikely | Temporal relationship improbable, concurrent disease or drugs provide plausible explanation |
| Conditional/unclassified | An event which requires more data for assessment |
| Unassessable/unclassifiable | An event that cannot be judged because of insufficient/contradictory information which cannot be supplemented or verified |
Yellow Card Scheme
The UK’s Yellow Card Scheme was established in 1964 following the thalidomide tragedy. The Scheme is operated by the Medicines and Health care Products Regulatory Authority (MHRA). Health care professionals and coroners can submit reports of suspected ADRs using a Yellow Card (found in the British National Formulary) or using an on-line form (http://www.yellowcard.gov.uk). An association between the medicine and the event does not have to be confirmed. A suspicion is sufficient for a report to be submitted. The MHRA request that all serious suspected ADRs are reported by health care professionals concerning established medicines (drugs and vaccines). For newer drugs and vaccines, all suspected ADRs should be reported, even if minor events. Newer medicines under intensive surveillance are identified with an inverted black triangle symbol in product information and standard prescribing texts. Black triangle status is generally maintained for at least 2 years, but the period varies, depending on how much information is obtained about a product’s continued safety. All suspected ADRs occurring in children should be reported even if the medicine has been used off-label.
Unfortunately, spontaneous reporting systems, including the Yellow Card Scheme, suffer from severe under-reporting. A systematic review estimated this to be between 82% and 98% (Hazell and Shakir, 2006). There are a variety of reasons for this, including lack of certainty that the medicine caused the symptom, but it is important to emphasise that such certainty is not required. There is also no requirement to provide the patient name or contact details, only those of the actual reporter; hence, confidentiality, also cited as a reason for under-reporting, is no longer an issue. Furthermore, the MHRA have systems in place to check for duplicate reports covering the same incident, thereby eliminating concern about two people submitting reports about the same event in a given patient.
Direct patient reporting
Despite the limited awareness of direct patient reporting, in the main people find it relatively easy to report suspected ADRs (McLernon et al., 2011). The majority of people who reported a suspected ADR identified it as such through issues relating to timing, as outlined in the causality methods used by pharmacovigilance experts, or by accessing information about the medicine from the PIL (Krska et al., 2011). There are a number of countries world-wide which accept patient reports. It has been suggested that these advantages include faster signal generation, avoiding the filtering effect of interpretation of events by health professionals and not least, maintaining the number of reports at a time when reporting by health professionals may be reducing.
Roles of health professionals
Identifying and assessing ADRs in clinical practice
Outside the pharmacovigilance environment of companies and regulatory agencies, the identification of potential ADRs is an essential component of clinical practice. Although assessments in practice may lack the formality of expert or algorithmic assessment, they are likely to take into account similar factors, such as whether the clinical event is commonly drug related, the temporal relationship with drug use, a dose relationship and exclusion of other possible causes. A list of such factors is set out in Box 5.1.
Box 5.1 Factors that may raise or suppress suspicion of a drug-induced event (Shakir, 2004)
The temporal relationship between the exposure to the drug and the subsequent event
The pharmacological plausibility – based on the observer’s knowledge of pharmacology
Concomitant medication – which may be considered the cause of an event
Underlying and concurrent illnesses – may alter the event or be considered the cause of the event
De-challenge – disappearance of symptoms after dose reduction or cessation of therapy
Re-challenge – reappearance of symptoms after dose increases or recommencement of therapy
Every opportunity should be taken to question patients about their experience, to determine whether they perceive any adverse events which could be due to medicines. While routinely asking simple questions is important, it is of equal value to develop a positive attitude towards the patients’ perception of suspected ADRs. There is some evidence that health professionals may dismiss patients who report that they have experienced an ADR, but many patients identify such problems appropriately, using factors such as onset, effect of dose change, effect of de-challenge or even re-challenge, as well as the information sources freely accessible to them (Krska et al., 2011). To ascertain whether a symptom reported by a patient can be reasonably suspected of being an ADR requires careful questioning.
Preventing ADRs
The majority of ADRs are thought to be preventable; hence, there is potential to dramatically reduce the costs associated with ADRs and possibly also deaths. Assessing preventability is a difficult area, since it involves judgements and many different methods have been developed for making these judgements. The approach of Hallas et al. (1990) is widely used, providing definitions of avoidability which range from definite (due to a procedure inconsistent with present-day knowledge or good medical practice) to unevaluable (poor data or conflicting evidence). Recent estimates suggest that between 53% and 72% of hospital admissions due to ADEs are preventable, while a meta-analysis (Beijer and de Blaey, 2002) showed that 88% of ADRs causing hospital admission in the elderly were preventable. However, not all ADRs are absolutely preventable and assessments using hindsight are unlikely to replicate clinical decision making at the point of prescribing. Preventability also varies from those with clear solutions, such as the prescribing of a teratogenic drug to a female of child bearing age, to those where the drug increases the risk of an event that occurs within the population.
Monitoring therapy
Monitoring the effects of drugs either by direct measurement of serum concentration or by measurement of physiological markers is another potential mechanism to reduce the risk of ADRs. For example, it has been estimated that one in four of preventable drug-related hospital admissions are caused by failure to monitor renal function and electrolytes (Howard et al., 2003).
Ideally, advice on monitoring should be clear, provide an evidence-based frequency of monitoring and acceptable values. However, robust evidence for the optimal monitoring frequency is limited, hampering specific guidance on monitoring. Guidelines vary between various expert bodies and drug information sources. An examination of the adequacy of manufacturers’ advice on monitoring for haematological ADRs found that advice was too vague to be useful to prescribers (Ferner et al., 2005).
Currently, monitoring is often neglected, although practitioners may take greater care when treating the elderly and those with more co-morbidities (McDowell., 2010). Warfarin remains one of the top 10 drugs involved in drug-induced admissions, despite a clearly defined monitoring requirement.
Explaining risks to patients
Another approach is the use of pictures, such as faces, graphs or charts. One example is the ‘Paling palette’, which is a grid of 1000 stick figures to convey information on the chances of experiencing a particular outcome. A similar method is a ‘Cates plot’ which is a grid of 100 faces or 1000 faces for rarer events, coloured differently and either smiling or downcast depending on the outcome. An example of a Cates plot is provided in Fig. 5.1. These types of icon grids are mainly used to convey the potential benefits and risks of a particular action, but can also be used to explain the risks of getting a side effect. Cates plots have been used to good effect by the UK’s National Prescribing Centre. However, there are people who do not find these easy to understand (Ancker et al., 2006).
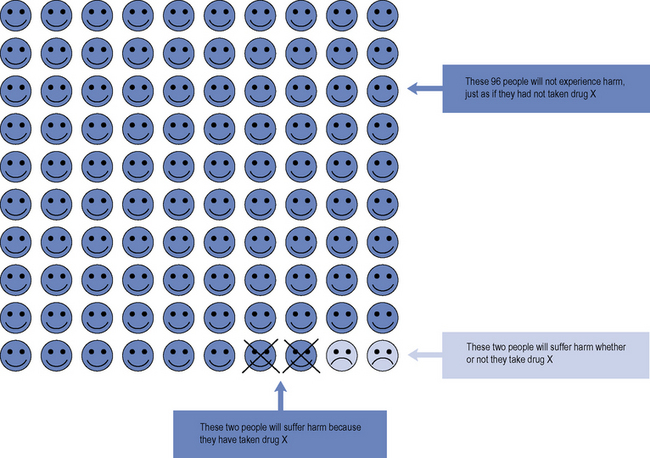
Fig. 5.1 Acute coronary syndrome (ACS) patient decision aid: aspirin plus clopidogrel versus aspirin alone.
Copyright National Prescribing Centre, reproduced by permission.
Answers
Answers
Answers
Answers
Ancker J., Senathirajah Y., Kukafka R., et al. Design features of graphs in health risk communication: a systematic review. J. Am. Med. Inform. Assoc.. 2006;13:608-618.
Aronson J.K., Ferner R.E. Joining the DoTS: new approach to classifying adverse drug reactions. Br. Med. J.. 2003;327:1222-1225.
Basch E. The missing voice of patients in drug-safety reporting. N. Engl. J. Med.. 2010;362:865-869.
Beijer H.J., de Blaey C.J. Hospitalisations caused by adverse drug reactions (ADR): a meta-analysis of observational studies. Pharm. World Sci.. 2002;24:46-54.
Davies E.C., Green C.F., Taylor S., et al. Adverse drug reactions in hospital in-patients: a prospective analysis of 3965 patient-episodes. PLoS ONE. 2009;4:e4439. doi:10.1371/journal.pone.0004439
Edwards I.R., Aronson J.K. Adverse drug reactions: definitions, diagnosis, and management. Lancet. 2000;356:1255-1259.
Ferner R.E., Coleman J., Pirmohammed M., et al. The quality of information on monitoring for haemotological adverse drug reactions. Br. J. Pharmacol.. 2005;60:448-451.
Ghandi T.K., Weingart S.N., Borus J., et al. Adverse drug events in ambulatory care. N. Engl. J. Med.. 2003;348:1556-1564.
Hallas J., Harvald B., Gran L.F., et al. Drug-related hospital admissions: the role of definitions and intensity of data collection and the possibility of prevention. J. Intern. Med.. 1990;228:83-90.
Hazell L., Shakir S.A. Under-reporting of adverse drug reactions: a systematic review. Drug Saf.. 2006;29:385-396.
Howard R., Avery A.J., Howard P.D., et al. Investigations into the reasons for preventable admissions to a medical admissions unit. Qual. Saf. Health Care. 2003;12:280-285.
Jarernsiripornkul N., Krska J., Capps P.A.G., et al. Patient reporting of potential adverse drug reactions: A methodological study. Br. J. Clin. Pharmacol.. 2002;53:318-325.
Kumar A. Adverse effects of pharmaceutical excipients. Adverse Drug React. Bull.. 2003;222:851-854.
Krska J., Anderson C.A., Murphy E., Avery A.J., on behalf of the Yellow Card Study Collaboration. How do patient reporters identify adverse drug reactions? A qualitative study of reporting via the UK Yellow Card Scheme. Drug Saf.. 2011. In press
Laurence J. Getting personal: the promises and pitfalls of personalized medicine. Transl. Med.. 2009;154:269-271.
Lazarou J., Pomeranz B.H., Corey P.N. Incidence of adverse drug reactions in hospitalized patients. J. Am. Med. Assoc.. 1998;279:1200-1205.
Lucas A., Nolan D., Mallal S. HLA-B*5701 screening for susceptibility to a bacavir hypersensitivity. J. Antimicrob. Chemother.. 2007;59:591-595.
McDowell S.E. Monitoring of patients treated with antihypertensive therapy for adverse drug reactions. Adverse Drug React. Bull.. 2010;261:1003-1006.
McDowell S.E., Coleman J.J., Ferner R.E. Systematic review and meta-analysis of ethnic differences in risks of adverse reactions to drugs used in cardiovascular medicine. Br. Med. J.. 2006;332:1177-1181.
McLernon D.J., Bond C.M., Fortnum H., Hannaford P.C., Krska J., Lee A.J., Watson M.C., Avery A.J., on behalf of the Yellow Card Study Collaboration. Patient experience of reporting adverse drug reactions via the Yellow Card Scheme in the UK. Pharmacoepidemiol. Drug Saf.. 2011. Published early on-line: doi: 10.1002/pds.2117
Pirmohamed M., James S., Meakin S., et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. Br. Med. J.. 2004;329:15-19.
Rawlins M.D. Clinical pharmacology: adverse reactions to drugs. Br. Med. J.. 1981;282:974-976.
Shakir S.A.W. Causality and correlation in pharmacovigilance. In: Talbot T., Waller P., editors. Stephens’ Detection of New Adverse Drug Reactions. fifth ed. Chichester: John Wiley and Sons Ltd; 2004:329-343.
Wester K., Jonnson A.K., Sigset O., et al. Incidence of fatal adverse drug reactions: a population based study. Br. J. Clin. Pharmacol.. 2008;65:573-579.
WHO. International drug monitoring: the role of national centres. Tech. Rep. Ser.. 1972;498:1-25.
Zhang M., Holman C.D.J., Price S.D., et al. Co-morbidity and repeat admission to hospital for adverse drug reactions in older adults: retrospective cohort study. Br. Med. J.. 2009;338:a2752.
Aronson J.K., Ferner R.E. Clarification of terminology in drug safety. Drug Saf.. 2005;28:851-870.
Cates Plot. Dr. Chris Cates’ EBM Website. http://www.nntonline.net/visualrx/cates_plot/. (Accessed 16th March 2011)
Drug Safety Update. http://www.mhra.gov.uk/Publications/Safetyguidance/DrugSafetyUpdate. MHRA, London. Available at:
Lee A. Adverse Drug Reactions, second ed., London: Pharmaceutical Press, 2006.
Talbot J., Waller P. Stephens’ Detection of New Adverse Drug Reactions, fifth ed, Chichester: John Wiley & Sons Ltd, 2004.
Waller P. An Introduction to Pharmacovigilance. Oxford: Wiley-Blackwell, 2010.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
5 Adverse drug reactions
Assessing the safety of drugs
When drugs are newly introduced to the market, their safety profile will be provisional. While efficacy and evidence of safety must be demonstrated for regulatory authorities to permit marketing, it is not possible to discover the complete safety profile of a new drug prior to its launch. Pre-marketing clinical trials involve on average 2500 patients, with perhaps a hundred patients using the drug for longer than a year. Therefore, pre-marketing trials do not have the power to detect important reactions that occur at rates of 1 in 10,000, or fewer, drug exposures. Often, only pharmacologically predictable ADRs with short onset times may be identified in clinical trials, nor can pre-marketing trials detect ADRs which are separated in time from drug exposure. Additionally, patients within trials are often carefully selected, without the multiple disease states or complex drug histories of patients in whom the drug will eventually become used. Furthermore, the patient’s perspective is also frequently excluded from clinical trial safety assessments, with ADRs being assessed only by the clinicians who run them (Basch, 2010). For these reasons, rare and potentially serious adverse effects often remain undetected until a wider population is exposed to the drug. The vigilance of health professionals is an essential factor in discovering these new risks, together with regulatory authorities who continuously monitor reports of adverse effects throughout the lifetime of a marketed medicinal product.
Definitions
Having clear definitions of what constitutes an ADR is important. The World Health Organization (WHO) defines an ADR as ‘a response to a drug that is noxious and unintended and occurs at doses normally used in man for the prophylaxis, diagnosis or therapy of disease, or for modification of physiological function’ (WHO, 1972). The use of the phrase ‘at doses normally used in man’ distinguishes the noxious effects of drugs during normal medical use from toxic effects caused by poisoning. Whether an effect is considered noxious depends on both the drug’s beneficial effects and the severity of the disease for which it is being used. There is no need to prove a pharmacological mechanism for any noxious response to be termed an ADR.
The WHO definition has been criticised for excluding the potential for contamination of a product, ADRs that include an element of error, and ADRs associated with pharmacologically inactive excipients in a product. The use of the term ‘drug’ also excluded the use of complementary and alternative treatments, such as herbal products. In an attempt to overcome these points, the following definition of an ADR was proposed, ‘An appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medicinal product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of the dosage regime, or withdrawal of the product’ (Edwards and Aronson, 2000).
Classification of ADRs
Rawlins–Thompson classification
The Rawlins–Thompson system of classification divides ADRs into two main groups: Type A and Type B (Rawlins, 1981). Type A reactions are the normal, but quantitatively exaggerated, pharmacological effects of a drug. They include the primary pharmacological effect of the drug, as well as any secondary pharmacological effects of the drug, for example, ADRs caused by the antimuscarinic activity of tricyclic anti-depressants. Type A reactions are most common, accounting for 80% of reactions.
Type B reactions are qualitatively abnormal effects, which appear unrelated to the drug’s normal pharmacology, such as hepatoxicity from isoniazid. They are more serious in nature, more likely to cause deaths, and are often not discovered until after a drug has been marketed. The Rawlins–Thompson classification has undergone further elaboration over the years (Table 5.1) to take account of ADRs that do not fit within the existing classifications (Edwards and Aronson, 2000).
Table 5.1 Extended Rawlins–Thompson classification of adverse drug reactions
| Type of reaction | Features | Examples |
|---|---|---|
| Type A: Augmented pharmacological effect | Common | Bradycardia associated with a beta-adrenergic receptor antagonist |
| Predictable effect | ||
| Dose-dependent | ||
| Low morbidity | ||
| Low mortality | ||
| Type B: Bizarre effects not related to pharmacological effect | Uncommon | Anaphylaxis associated with a penicillin antibiotic |
| Unpredictable | ||
| Not dose-dependent | ||
| High morbidity | ||
| High mortality | ||
| Type C: Dose-related and time-related | Uncommon | Hypothalamic pituitary–adrenal axis suppression by corticosteroids |
| Related to the cumulative dose | ||
| Type D: Time-related | Uncommon | Carcinogenesis |
| Usually dose-related | ||
| Occurs or becomes apparent some time after use of the drug | ||
| Type E: Withdrawal | Uncommon | Opiate withdrawal syndrome |
| Occurs soon after withdrawal of the drug | ||
| Type F: Unexpected failure of therapy | Common | Failure of oral contraceptive in presence of enzyme inducer |
| Dose-related | ||
| Often cause by drug interactions |
The DoTS system
The DoTS classification is based on Dose relatedness, Timing and patient Susceptibility (Aronson and Ferner, 2003). In contrast to the Rawlins–Thompson classification, which is defined only by the properties of the drug and the reaction, the DoTS classification provides a useful template to examine the various factors that both describe a reaction and influence an individual patient’s susceptibility.
The final aspect of the DoTS classification system is susceptibility, which includes factors such as genetic predisposition, age, sex, altered physiology, disease and exogenous factors such as drug interactions (Table 5.2)
| Dose relatedness | Time relatedness | Susceptibility |
|---|---|---|
| Toxic effects: ADRs that occur at doses higher than the usual therapeutic dose Collateral effects: ADRs that occur at standard therapeutic doses Hypersusceptability reactions: ADRs that occur at sub-therapeutic doses in susceptible patients |
Time-independent reactions: ADRs that occur at any time during treatment. Time-dependent reactions: Rapid reactions occur when a drug is administered too rapidly. Early reactions occur early in treatment then abate with continuing treatment (tolerance). Intermediate reactions occur after some delay, but if reaction does not occur after a certain time, little or no risk exists. Late reactions risk of ADR increases with continued-to-repeated exposure, including withdrawal reactions. Delayed reactions occur some time after exposure, even if the drug is withdrawn before the ADR occurs. |
Raised susceptibility may be present in some individuals, but not others. Alternatively, susceptibility may follow a continuous distribution – increasing susceptibility with impaired renal function. Factors include: genetic variation, age, sex, altered physiology, exogenous factors (interactions) and disease. |
Factors affecting susceptibility to ADRs
Co-morbidities and concomitant medicines use
Reductions in hepatic and renal function substantially increase the risk of ADRs. A recent study examining factors that predicted repeat admissions to hospital with ADRs in older patients showed that co-morbidities such as congestive cardiac failure, diabetes, and peripheral vascular, chronic pulmonary, rheumatological, hepatic, renal, and malignant diseases were strong predictors of readmissions for ADRs, while advancing age was not. Reasons for this could be pharmacokinetic and pharmacodynamic changes associated with pulmonary, cardiovascular, renal and hepatic insufficiency, or drug interactions because of multiple drug therapy (Zhang et al., 2009).
Ethnicity
Examples of ADRs linked to ethnicity include the increased risk of angioedema with the use of ACE inhibitors in black patients (McDowell et al., 2006), the increased propensity of white and black patients to experience central nervous system ADRs associated with mefloquine compared to patients of Chinese or Japanese origin, and differences in the pharmacokinetics of rosuvastatin in Asian patients which may expose them to an increased risk of myopathy. However, susceptibility based on ethnicity could be associated with genetic or cultural factors and ethnicity can be argued to be a poor marker for a patient’s genotype.
Pharmacogenetics
The narrow therapeutic index of warfarin, its high inter-individual variability in dosing and the serious consequences of toxicity have made it a major target of pharmacogenomic research. Studies of genetic polymorphisms influencing the toxicity of warfarin have focused on CYP2C9, which metabolises warfarin and vitamin K epoxide reductase (VKOR), the target of warfarin anticoagulant activity. Genetic variation in the VKORC1 gene, which encodes VKOR, influences warfarin dosing by a threefold greater extent than CYP2C9 variants. In 2007, the U.S. Food and Drug Administration (FDA) changed the labelling requirement for warfarin, advising that a lower initial dose should be considered in people with certain genetic variations. However, concerns remain because genetic variation only accounts for a proportion of the variability in drug response and clinicians may obtain a false sense of reassurance from genetic testing leading to complacency in monitoring of therapy. In addition, there appears to be little evidence of additional benefit (Laurence, 2009), in terms of preventing major bleeding events, compared to careful monitoring of the INR (see chapter 23)
A success story for pharmacogenetics is the story of the nucleoside analogue reverse transcriptase inhibitor (NRTI) abacavir. Hypersensitivity skin reactions to abacavir are a particular problem in the treatment of human immunodeficiency virus (HIV) infection. Approximately 5–8% of patients taking abacavir develop a severe hypersensitivity reaction, including symptoms such as fever, rash, arthralgia, headache, vomiting and other gastro-intestinal and respiratory disturbances. Early reports that only a subset of patients was affected, a suspected familial predisposition, the short onset time (within 6 weeks of starting therapy), and an apparent lower incidence in African patients led to suspicion of a genetic cause. Subsequent research revealed a strong predictive association with the human leukocyte antigen HLA-B*5701 allele in Caucasian and Hispanic patients. The presence of the allele can be used to stratify the predicted risk of hypersensitivity as high risk (>70%) for carriers of HLA-B*5701 and low risk (<1%) for non-carriers of HLA-B*5701. Evidence from the practical use of HLA-B*5701 screening has shown substantial falls in the incidence of hypersensitivity reactions, as well as a more general improved compliance with the medication (Lucas et al., 2007).
[/not-level-membership-for-basic-science-category]
