75 Acute Myocardial Infarction
Angina pectoris was recognized in the 18th century; myocardial infarction (MI), however, was described approximately 200 years later. Simultaneous to the identification of MI was the initial introduction and subsequent application of the electrocardiogram (ECG)—the first objective method of assessing the coronary origin of the presentation. In fact, early clinician investigators described the evolving “electrographic” changes during angina in 1918.1 Over the next 50 years, angina pectoris and MI were further characterized and diagnosed; unfortunately, however, the management of ischemic heart disease did not progress as significantly. From this point in medical history until the 1960s, management consisted primarily of pain relief coupled with strict bed rest for prolonged periods and management of resultant congestive heart failure (CHF); acute complications such as cardiogenic shock and sudden cardiac death were invariably fatal events. Subsequently, the introduction and widespread use of cardiopulmonary resuscitation, external defibrillation, and antidysrhythmic agents gave the clinician powerful new tools in the management of sudden cardiac death and other malignant dysrhythmias. Overall management, however, was still aimed at the complications of ischemic heart disease rather than the syndrome itself.
 Epidemiology
Epidemiology
Globally, cardiovascular disease now ranks as the leading cause of death. It now causes one third of all deaths worldwide. The World Health Organization (WHO) in conjunction with the Centers for Disease Control and Prevention (CDC) published the Atlas of Heart Disease and Stroke; in this report, the WHO/CDC note a combined death toll of 17 million persons per year, with a potential increase to 24 million people per year by 2030.2 In the United States, ischemic heart disease, particularly acute forms of the illness, is the leading cause of death for adults. Unfortunately, half of these deaths result from sudden cardiac death unrelated to ACS, usually within the first 2 hours of symptom onset, either out of hospital or soon after arrival in the ED. Fifteen percent of the fatalities occur prior to age 65 years, with the majority in women. The “burden” placed on medical centers and other acute care facilities is tremendous, with an approximate 8 million people having been admitted to hospital in the past 20 years; 20% of these admissions involve AMI. Furthermore, while death from coronary heart disease has decreased in North America and many western European countries, there is an increased mortality in developing countries.3,4
According to the American Heart Association,5 coronary heart disease caused approximately 1 of every 6 deaths in the United States in 2006. In 2010, an estimated 785,000 Americans will have a new coronary event, and approximately 470,000 will have a recurrent attack. It is estimated that an additional 195,000 “silent” first MIs occur each year. These events usually occur in patients over the age of 40 years, with an increasing occurrence as one ages. Approximately every 25 seconds, someone in the United States will have a coronary event, and approximately every minute someone will die of one such event.5
 Pathophysiology
Pathophysiology
Historically, the two primary intracoronary pathophysiologic events underlying the development of ACS include thrombus formation and vasospasm. In the setting of either a structurally normal artery or preexisting coronary artery disease, initial endothelial damage produces platelet aggregation and resultant thrombus formation. In most cases, disruption of an atherosclerotic plaque provides the endothelial injury. Occlusion of the coronary artery then results, ranging from minimal, transient, asymptomatic obstruction to complete occlusion usually associated with prominent symptomatology, namely AMI. Coronary artery obstruction can lead to myocardial ischemia, hypoxia, acidosis, and ultimately AMI. Vasospasm results when locally active substances are coupled with systemic mediators to produce a cascade of events resulting in worsened myocardial perfusion. Isolated vasospasm followed by thrombus is involved in approximately 10% of AMIs. Refer to Figure 75-1 for a depiction of the acute pathophysiology of AMI.
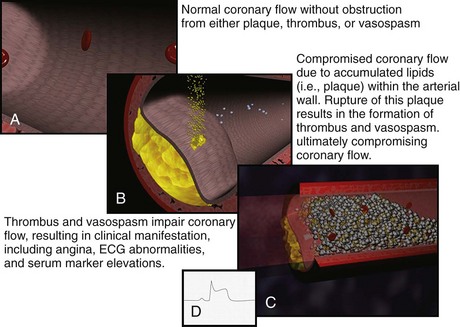
Figure 75-1 Pathophysiology of acute myocardial infarction.
(Figures courtesy Ashok Subramanian, MD.)
In the last decade, the definition of MI has evolved. The European Society of Cardiology and the American College of Cardiology published consensus criteria for “redefinition” of MI in 2000.6 These criteria reflected the improvements in biomarker testing. Then in 2007, working groups from these organizations along with the World Heart Federation and American Heart Association published the “Universal Definition of Myocardial Infarction.”6 This expanded definition classifies infarction based on clinical situations resulting in myocardial necrosis/cell death.6
The term myocardial infarction should be used when there is evidence of myocardial necrosis in a clinical setting consistent with myocardial ischemia. Under these conditions, any one of the following criteria meets the diagnosis for myocardial infarction; the various subcategories of acute myocardial infarction are referred to as types 1 to 56:
 Clinical Features
Clinical Features
There has been disagreement over whether these coronary risk factors should be considered in the clinician’s medical decision making. An early report5 suggested that such factors, which were initially derived because of their ability to predict the development of coronary atherosclerosis and its complications over decades in association with other clinical variables such as ECG interpretation, have minimal predictive value acutely as to whether a patient is currently experiencing an AMI. More contemporary investigation in possible ACS patients suggests that the coronary risk factors do in fact have significant predictive value.7,8,9 This important issue is still debated by the epidemiologists; for the clinician, a consideration of the risk-factor burden is one feature of the overall diagnostic analysis.
Because angina is a visceral sensation that is often diffuse, some patients may have an anginal equivalent syndrome. Such anginal equivalent presentations describe patients who are experiencing ACS yet do not complain of typical chest pain; rather, these patients note atypical pain, dyspnea, weakness, diaphoresis, or emesis—these complaints, in fact, are the manifestation of the ACS event. Patients with altered cardiac pain perception (e.g., the elderly or patients with long-standing diabetes mellitus) are potentially at risk to present with anginal equivalent syndromes. A recent large survey of 434,877 confirmed AMI patients reported that a significant minority of these individuals—approximately 30%—lacked chest pain on presentation, noting only the anginal equivalent complaints.10 The most frequently encountered anginal equivalent chief complaint is dyspnea, which is found in 10% to 30% of patients with AMI, often due to pulmonary edema.10,11,12 Isolated emesis and diaphoresis are quite rare.11,12
The geriatric patient may also present atypically with acute weakness (3%–8%) and syncope (3%–5%).13 Unexplained sinus tachycardia, bronchospasm resulting from cardiogenic asthma, and new-onset lower extremity edema have all been reported as anginal equivalent presentations for AMI in this age group. Among the very elderly, anginal equivalent syndromes typically involve neurologic presentations with acute mental status abnormalities and stroke. From the perspective of acute delirium, less than 1% of such patients in an ED population with altered mentation will be found to have AMI. AMI associated with acute stroke is noted in approximately 5% to 9% of patients.13
Physical Examination
The physical examination in the patient with AMI rarely provides diagnostic confirmation of the illness; the examination can certainly suggest MI yet not confirm its presence. The ECG, serum markers, and other investigations interpreted in the context of the clinical event confirm the diagnosis. Specific examination findings resulting directly from ACS include anxiety, pale appearance, and diaphoresis. In fact, the presence of significant diaphoresis as a physical examination finding is strongly suggestive of AMI.14 Significant physical examination findings encountered in the AMI patient most often result indirectly from the coronary event and result directly from complications of the AMI. These findings include hypotension, altered mentation, various other signs of poor perfusion, rales and low oxygen saturations related to pulmonary congestion, and heart sounds related to myocardial and/or valvular dysfunction.15 Both brady- and tachydysrhythmias are seen as well. And, of course, the combination of poor peripheral perfusion—manifested by hypotension unresponsive to hemodynamic support—and pulmonary edema is considered cardiogenic shock.
Caution should be exercised when attributing a chest wall source for pain based on palpation or movement. To safely relate the chest discomfort to a chest wall origin, the pain must be described as sharp or stabbing (i.e., pleuritic in nature) and be completely reproducible by palpation.16 Up to 15% of patients with AMI may have some form of tenderness on chest wall palpation.17
 Diagnostic Strategies
Diagnostic Strategies
Electrocardiogram
The ECG may manifest a range of ECG abnormalities (Figure 75-2) in the patient with potential AMI, including the prominent T wave, T-wave inversion, ST-segment depression, ST-segment elevation, and QA waves, among other findings. The earliest ECG finding resulting from STEMI is the hyperacute T wave, which may appear minutes after the interruption of blood flow; the R wave also increases in amplitude at this stage. The hyperacute T wave, a short-lived structure that evolves rapidly on to ST-segment elevation over a 5- to 30-minute period, is often asymmetric with a broad base; these T waves are also associated not infrequently with reciprocal ST-segment depression in other ECG leads. Such a finding on the ECG is transient in the AMI patient; either apparent or progressive ST-segment elevation is usually encountered at this stage. As the infarction progresses, the hyperacute T wave evolves into the giant R wave, particularly in the anterior wall AMI. The giant R wave is a transition structure from the hyperacute T wave to typical ST-segment elevation; it essentially is a large monophasic R wave with pronounced ST-segment elevation. Prominent T waves may be seen in patients with AMI as well as hyperkalemia, acute myopericarditis, benign early repolarization, left ventricular hypertrophy, and bundle branch block.
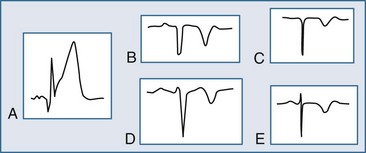
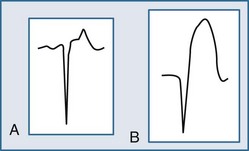
Within moments, the ST segment assumes a more easily recognized morphology. In approximately 85% of STEMI patients, the initial upsloping portion of the ST segment is either convex or flat; if the ST segment is flat, it may be either horizontally or obliquely so. An analysis of the ST-segment waveform can be particularly helpful in distinguishing among the various causes of ST-segment elevation and identifying the AMI case. This technique uses the morphology of the initial portion of the ST segment/T wave—defined as beginning at the J point and ending at the apex of the T wave. Patients with noninfarctional ST-segment elevation (i.e., early repolarization or left ventricular hypertrophy-related change) tend to have a concave morphology of the waveform. Conversely, patients with ST-segment elevation due to AMI have either obliquely flat or convex waveforms. The use of this ST-segment elevation waveform analysis in emergency room chest pain patients increases specificity for the AMI diagnosis.18 This morphologic observation should be used only as a guideline. As with most guidelines, it is not infallible.
Significant ST-segment elevation occurring in at least two anatomically oriented leads is the primary ECG indication for fibrinolysis or urgent PCI. In that ST-segment elevation represents a significant finding, a brief review of the various causes of ST-segment elevation in the chest pain patient is warranted. Unfortunately, ST-segment elevation in the chest pain patient less often results from AMI; in fact, only 20% to 30% of chest pain patients will have STEMI—the remainder of these patients will have noninfarctional causes of the ST-segment elevation.18,19 Patients with chest pain may present electrocardiographically with ST-segment elevation due to AMI, confounding patterns, or masquerading syndromes. In most instances, ST-segment elevation resulting from AMI is easily noted. Confounding patterns such as LBBB, ventricular paced rhythms, and left ventricular hypertrophy may obscure the typical ECG findings of AMI as well as produce noninfarctional ST-segment elevation, which may lead the uninformed clinician astray. Other ST-segment elevation patterns, including benign early repolarization and acute pericarditis, occur in the individual with chest discomfort and may suggest the incorrect diagnosis of AMI, exposing the patient to unnecessary and potentially dangerous therapies.
Reciprocal ST-segment depression, also known as reciprocal change, is defined as ST-segment depression in leads separate and distinct from leads reflecting ST-segment elevation. Importantly, this form of ST-segment depression is not associated with situations in which altered intraventricular conduction produces deviation—such as bundle branch block, left ventricular hypertrophy, and ventricular paced rhythms. Reciprocal change in the setting of a STEMI identifies a patient with an increased chance of poor outcome and, therefore, an individual who may benefit from a more aggressive approach. Furthermore, its presence on the ECG supports the diagnosis of AMI with very high sensitivity and positive predictive values greater than 90%. The use of reciprocal change in both prehospital and emergency room chest pain patients increases the diagnostic accuracy in the ECG recognition of AMI.20,21 Reciprocal change is seen in approximately 75% of cases of inferior wall AMI and much less often in cases of anterior wall MI (30%).20,21
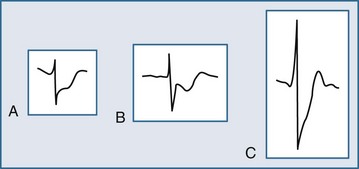
Several ECG patterns confound the diagnosis of AMI, including LBBB, ventricular paced rhythms, and left ventricular hypertrophy. In the patient with LBBB, the anticipated or expected ST-segment/T-wave configurations are discordant, directed on the opposite side of the isoelectric baseline from the terminal portion of the QRS complex. This relationship is called QRS complex–T wave axes discordance (Figure 75-3).22,23 Loss of this discordance in patients with LBBB may imply AMI. The clinician must realize, however, that the ECG is markedly compromised as a diagnostic tool in this setting. As with the LBBB pattern, the right ventricular paced rhythm and left ventricular hypertrophy patterns can both mimic and mask the manifestations of AMI. In ventricular paced rhythms, the principle of appropriate discordance should also be followed. An inspection of the ECG in patients with ventricular paced rhythms must be performed, looking for a loss of this QRS complex–T wave axes discordance. Loss of this normal discordance in patients with ventricular paced rhythms can suggest AMI.24 Left ventricular hypertrophy is not uncommonly encountered on the ECG of chest pain patients. Its presence on the ECG, particularly the repolarization changes that alter the morphology of the ST segment and/or the T wave, can confound the early evaluation. These repolarization changes are seen in approximately 70% of cases and represent the new norm for the patient with electrocardiographic left ventricular hypertrophy.25 Left ventricular hypertrophy is associated with poor R wave progression, producing a QS pattern in the right to mid-precordial leads. In most instances, the ST-segment elevation is seen here along with prominent T waves. ST-segment depression with inverted T wave is also seen in the lateral leads.
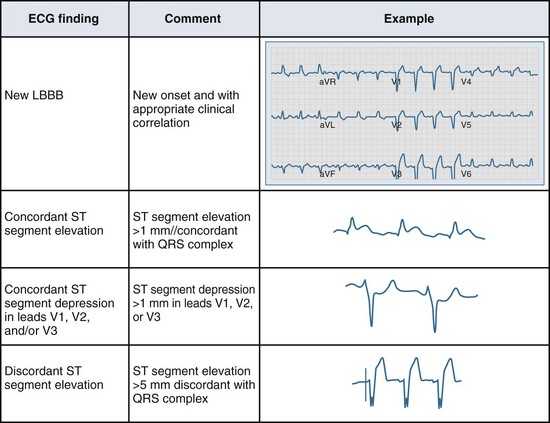
Figure 75-3 Electrocardiographic indications for reperfusion therapy in the left bundle branch block presentation.
Several additional ECG tools can be employed by the clinician to further evaluate the chest pain patient suspected of AMI. These tools include additional ECG leads and ST-segment surveillance. The additional-lead ECG improves the diagnostic power of the standard 12-lead ECG; with the addition of three leads, the 15-lead ECG is produced. In the 15-lead ECG, the posterior leads V8 and V9 image the posterior wall of the left ventricle (posterior AMI) and lead V4R evaluates the right ventricle (right ventricular infarction). The use of the additional leads can not only confirm the presence of AMI but also alter treatment decisions in ACS patients. In a study of all emergency room chest pain patients initially evaluated with a 12-lead ECG, Brady el al.26 reported that the 15-lead ECG provided a more accurate description of myocardial injury in those patients with AMI yet failed to alter rates of diagnoses or the use of reperfusion therapies or change disposition locations. Looking at a more select population of chest pain patients, Zalenski and colleagues27 investigated the use of the 15-lead ECG in chest pain patients with a moderate to high pretest probability of AMI who were already identified as candidates for critical care admission. In this study, the authors reported an approximate 12% increase in sensitivity for the diagnosis of AMI. Potential clinical indications for obtaining the 15-lead ECG in chest pain patients include: (1) ST-segment depression in leads V1 through V3; (2) STEMI of the lateral or inferior wall; (3) isolated ST-segment elevation in lead V1 or ST-segment elevation in leads V1 and V2; and (4) the inferior or lateral AMI complicated by hypotension on presentation or after preload reducing medication administration. Figure 75-4, A is an example of a 15-lead ECG with inferoposterior AMI with right ventricular infarction. Note the ST-segment elevation in leads II, III, and aVf (inferior AMI), RV4 (right ventricular infarction), and leads V8 and V9 (posterior AMI); the ST-segment depression with prominent R wave is also seen in leads V1 to V3.
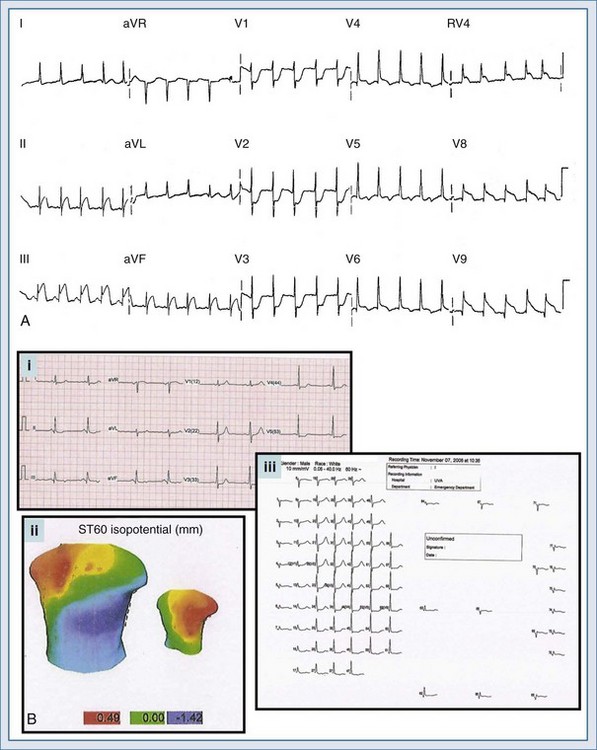
ECG body mapping, an extrapolation of the additional-lead concept, more completely images the heart in an electrical sense. Contemporary body mapping systems rely on a more widely distributed lead distribution, focusing on areas of the myocardium which are not imaged appropriately by the traditional 12-lead ECG, including the electrocardiographically “near-silent” and “silent” areas. The “near-silent” areas include the far inferior and lateral walls as well as the septal region of the left ventricle; the “silent” areas include the posterior wall of the left ventricle and the entire right ventricle. Various systems are available in today’s market, and most rely on a combination of torso mapping with ECG determination. An example of a body map is depicted in Figure 75-4, B; note the torso imaging with colorimetric depictions (green indicating normal ST segments, blue indicating ST-segment depression, and red indicating ST-segment elevation). The various ECG waveforms are also displayed for the entire body map, much more completely describing the heart when compared to the somewhat limited imaging of the 12-lead electrocardiogram. While body mapping has demonstrated increased rates of STEMI diagnosis, at this time, conclusive data noting improved patient outcomes is lacking.28
Serial monitoring of the ST segment can also aid the clinician in the diagnosis of AMI as well as monitor the response to therapy. This can be accomplished using two different approaches: serial 12-lead ECG acquisition or ST-segment trend monitoring. Either technique can demonstrate the evolution of ST-segment/T-wave changes in a number of different clinical scenarios, including the initially nondiagnostic ECG, the continuous chest pain patient with an initially nondiagnostic ECG, and the individual with a confounding or masquerading ECG pattern. This increased level of monitoring may provide earlier evidence of coronary occlusion in patients with non-AMI ACS presentations. Potentially, serial ECGs can furnish an increased level of ECG monitoring in patients presenting with chest pain and a nondiagnostic ECG on presentation.29–33 In the coronary care unit setting, serial ST-segment surveillance initiated at admission offers additional clinical data, with approximately 20% of patients revealing dynamic ECG change in the early stages of the hospital course.34 ST-segment monitoring has proved to be an effective method for noninvasive evaluation of reperfusion after delivery of fibrinolytic therapy in multiple investigations. In one series, Krucoff and colleagues33 noted that angiographically proven reperfusion was detected with a sensitivity of 89% using serial ST-segment trend monitoring, with a corresponding specificity of 82%.
Serum Markers
The elevation of serum cardiac markers over several days of hospitalization has traditionally been the standard method for diagnosing AMI. Whereas creatine phosphokinase (CK)-MB fraction once was the typical marker used by most clinical laboratories to indicate myocardial necrosis, now the troponins are the most commonly used serologic tests in the regions with established acute cardiac care. Previously, detection of AMI by enzyme elevations over 48 to 72 hours was sufficient to establish the diagnosis of AMI. Because of the evolution of acute interventional modalities, however, significant time-sensitive pressure now exists to identify patients with AMI earlier after onset of the ailment. Particularly in patients with a nondiagnostic ECG, early serum markers of myocardial necrosis have the potential to alter the diagnostic course and treatment plans. Further, there are now clear data that indicate that elevations in serum markers, even in those not meeting traditional criteria for AMI, independently identify those patients at risk for poor outcome.35–37
Once released into the blood, these markers are then cleared by the kidneys. A baseline elevation of these markers in the absence of MI has been termed a troponin leak and has been noted to occur under multiple clinical conditions (Box 75-1). In fact, previous studies have demonstrated elevated troponin levels in up to half of critical care patients, many of whom do not have evidence of clinically significant coronary artery disease or ACS. However, regardless of etiology, patients with elevated troponin values have a higher incidence of adverse outcome, including mortality. It is the rise or fall of theses values, with one above the 99th percentile of the upper reference limit (URL), coupled with evidence of myocardial ischemia that differentiates MI from other causes of high troponin. Troponin elevations can persist for 1 to 2 weeks following injury. However, they are usually not rising or falling rapidly at his time. A greater than or equal to 20% increase in the value of the sample during this period can indicate re-injury.
As already noted, two myocardial-specific proteins—myocardial troponin T and troponin I—are extremely important in the evaluation of patients suspected of having AMI and have largely replaced CK for biochemical determination of infarction. The cardiac troponins I and T are genetically distinct from those forms found in skeletal muscle, making them highly cardiac-specific markers. The biokinetics of troponin release are related to the location of the protein within the cell. Normally, small quantities of troponins are free in the cytosol, whereas the majority is entwined in the muscle fiber. Following injury, a biphasic rise in serum troponins is seen. This two-component pattern corresponds to the early release of the free cytoplasmic proteins followed by a prolonged rise with disruption of the actual muscle fiber, resulting in a sustained release of the troponins for approximately 7 days. Serum troponin concentrations begin to rise measurably in the serum at about the same time as CK-MB elevations become detectable—as early as 3 hours after onset—and therefore offer no particular benefit over the CK-MB regarding early detection of the event. The troponins, however, remain elevated for prolonged periods of time, ranging from 7 to 10 days. The cardiac-specific troponins are highly sensitive for the early detection of myocardial injury in patients with AMI. A positive test result is associated with significant risk, whereas negative study (i.e., serial troponins) findings predict low risk.38
The sensitivity of the troponins approaches 50% within 3 to 4 hours of the event. The test finding is positive for AMI in about 75% at 6 hours after onset of symptoms; at 12 hours, the test is almost 100% sensitive for AMI.39 Moreover, the presence of a positive troponin, even in the face of a nondiagnostic ECG and negative CK-MB assay, independently confers a prognosis on the patient that is similar to those suffering STEMIs.40,41 Thus, elevated troponin values appear to be excellent indicators of risk of subsequent death, AMI, and acute cardiovascular complications in all ACS patients, even those who do not meet traditional criteria for AMI. A negative test result, however, does not necessarily imply a favorable prognosis. One caveat for the troponins is that a number of systemic diseases can cause elevations in the serum levels of troponins without ACS.
Medical decision making regarding serum marker use in the suspected AMI patient is complex. Serum markers are most often used in a serial fashion. Relying solely on the result of a single negative assay can result in a missed diagnosis in up to 74% of patients.42 Single testing strategies, however, may be of value when the clinician is evaluating a nonspecific presentation with illness course lasting greater than 72 to 96 hours. Trending results over time significantly reduces the chance of a missed diagnosis, particularly in acute presentations of short course. A number of studies support the assertion that the troponins approach 100% sensitivity and specificity for cardiac ischemia at 12 hours following an event.39 These studies all caution, however, that such elevations will occur only with cell injury; hence, they are not appropriate markers for non-AMI ACS presentations. In the setting of an appropriate clinical history or diagnostic ECG changes, a strategy of serial cardiac marker testing is relatively straightforward. Depending on the particular investigation employed, the clinician looks for the characteristic rise and fall of serial markers over a time course for the diagnosis of AMI.6 Most literature supports such serial testing in the acute setting for a period of 8 to 12 hours to adequately rule out MI.6,43,44
Chest Radiography
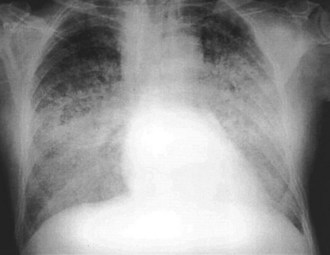
The chest radiograph is obtained in the vast majority of patients who present with AMI. Evidence of pulmonary congestion is noted radiographically in approximately one third of such patients. Radiographic findings often parallel the clinical examination findings. AMI patients who develop CHF based on physical examination have an increased mortality risk, as reported by the Killip classification; the chest radiograph provides prognostic data. The chronicity of the CHF syndrome may also be suggested by the heart size. Patients who present with AMI complicated by pulmonary edema and who have a normal heart size most often have no past history of CHF. In fact, AMI is the most frequent cause of pulmonary edema with a normal cardiac size. In other instances, patients with AMI who manifest an enlarged cardiac silhouette on the chest radiograph frequently have a preexisting history of CHF, anterior wall infarct, and multiple-vessel coronary artery disease (Figure 75-5).45
Invasive Hemodynamic Monitoring
Right heart catheterization, the placement of a pulmonary artery (PA) catheter, allows for precise determination of the patient’s hemodynamic status. Such information allows for determination of the cardiac output pulmonary artery balloon-occluded pressure and mixed venous oxygen saturation (SvO2). Although the array of clinical data provided by right heart catheterization is impressive, the vast majority of AMI patients do not require such extensive and invasive hemodynamic monitoring; in fact, many intensivists have questioned the utility of right heart catheterization.46 More useful monitoring techniques include continuous ECG monitoring (for dysrhythmia), ST-segment trend monitoring (for evolution of ACS), and noninvasive blood pressure determinations. Additionally, serial focused physical examinations provide important clinical data: repeat assessments of the patient’s general appearance, mental status, jugular venous pressure, lung fields, and peripheral perfusion provide (in most instances) appropriate and adequate information regarding the patient’s hemodynamic status.
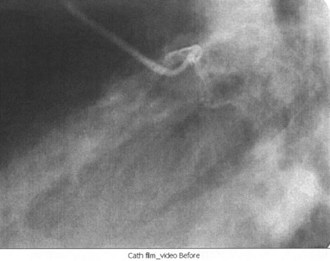
Cardiac Catheterization
Cardiac catheterization, also known as coronary angiography, is used to evaluate the anatomy of the coronary arteries; left ventricular function can also be assessed. Access is usually obtained through the right femoral artery; the left femoral artery and both brachial and radial arteries, however, can be used as well. Once the coronary anatomy has been evaluated, coronary lesions (Figure 75-6) that are appropriate for intervention can be treated with balloon angioplasty or coronary stent placement, or both. Fractional flow reserve is a technique that can be used to evaluate the significance of a lesion by measuring the pressures proximally and distally to the lesion.
Key Points
Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
Hoekstra JW, O’Neill BJ, Pride YB, et al. Acute detection of ST-elevation myocardial infarction missed on standard 12-lead ECG with a novel 80-lead real-time digital body surface map: primary results from the multicenter OCCULT MI trial. Ann Emerg Med. 2009;54:779-788.
Lim W, Whitlock R, Khera V, et al. Etiology of troponin elevation in critically ill patients. J Crit Care. 2010;25:322-328.
Body R. Emergent diagnosis of acute coronary syndromes: today’s challenges and tomorrow’s possibilities. Resuscitation. 2008;78:13-20.
Goodacre S, Pett P, Arnold J, et al. Clinical diagnosis of acute coronary syndrome in patients with chest pain and a normal or non-diagnostic electrocardiogram. Emerg Med J. 2009;26:866-870.
1 Bousfield G. Angina pectoris: changes in electrocardiogram during paroxysm. Lancet. 1918;2:475-479.
2 Mackay J, Mensah GA. The Atlas of Heart Disease and Stroke. Geneva: World Health Organization; 2004. www.who.int/cardiovascular_diseases/resources/atlas/en/.
3 Hunink MGM, Goldman L, Tosteson ANA, et al. The recent decline in mortality from coronary heart disease, 1980-1990: the effect of secular trends in risk factors and treatment. JAMA. 1997;277:535-542.
4 Rosamond WD, Chambless LE, Folsom AR, et al. Trends in the incidence of myocardial infarction and in mortality due to coronary heart disease, 1987 to 1994. N Engl J Med. 1998;339:861-867.
5 Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA Guidelines for the management of patients with ST-elevation myocardial infarction—executive summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients With Acute Myocardial Infarction). Circulation. 2004;110:588-636.
6 Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. Circulation. 2007;116:2634-2653.
7 Jayes RL, Beshansky JR, Dagostino RB, et al. Do patients’ coronary risk factor reports predict acute cardiac ischemia in the emergency department? A multicenter study. J Clin Epidemiol. 1992;45:621-626.
8 Ornato JP, Peberdy MA, Jesse RL, et al. Value of coronary artery disease risk factors in judging whether chest pain accompanied by a normal or nondiagnostic ECG in the emergency department is due to acute cardiac ischemia. J Am Coll Cardiol. 1996;27:31A.
9 Han JH, Lindsell CJ, et al. The role of cardiac risk factor burden in diagnosing ACS in the ED setting. Ann Emerg Med. 2007;49:145.
10 Canto JG, Shiplak MG, Rogers WJ, et al. Prevalence, clinical characteristics, and mortality among patients with acute myocardial infarction presenting without chest pain. JAMA. 2000;283:3223-3229.
11 Lusiani L, Perrone A, Pesavento R, Conte G. Prevalence, clinical features, and acute course of atypical myocardial infarction. Angiology. 1994;45:49-55.
12 Uretsky B, Farquhar D, Berenzin A. Symptomatic myocardial infarction without chest pain: prevalence and clinical course. Am J Cardiol. 1977;40:498-503.
13 Bayer AJ, Chadha JS, Farag RR, Pathy MSJ. Changing presentation of myocardial infarction with increasing old age. J Am Geriatr Soc. 1986;34:263-266.
14 Hargarten KM, Aprahamian C, Stueven H, et al. Limitations of prehospital predictors of acute myocardial infarction and unstable angina. Ann Emerg Med. 1987;16:1325-1329.
15 Schillinger M, Domanovits H, et al. Clinical signs of pulmonary congestion predict outcome in patients with acute chest pain. Wien Klin Wochenschr. 2002;114:917-922.
16 Lee T, Cook F, Weisberg M, et al. Acute chest pain in the emergency room: Identification and examination of low risk patients. Arch Intern Med. 1985;145:65-69.
17 Tierney WM, Fitzgerald J, McHenry R. Physicians’ estimates of the probability of myocardial infarction in emergency room patients with chest pain. Med Decis Making. 1986;6:12-17.
18 Brady WJ, Syverud SA, Beagle C, et al. Electrocardiographic ST segment elevation: The diagnosis of AMI by morphologic analysis of the ST segment. Acad Emerg Med. 2001;8:961-967.
19 Brady WJ, Perron AD, Martin ML, et al. Electrocardiographic ST segment elevation in emergency department chest pain center patients: Etiology responsible for the ST segment abnormality. Am J Emerg Med. 2001;19:25-28.
20 Otto LA, Aufderheide TP. Evaluation of ST segment elevation criteria for the prehospital electrocardiographic diagnosis of acute myocardial infarction. Ann Emerg Med. 1994;23:17-24.
21 Brady WJ, Perron AD, Syverud SA, et al. Reciprocal ST segment depression: Impact on the electrocardiographic diagnosis of ST segment elevation acute myocardial infarction. Am J Emerg Med. 2002;20:35-38.
22 Aufderheide TP, Brady WJ. Electrocardiography in the patient with myocardial ischemia or infarction. In: Gibler WB, Aufderheide TP, editors. Emergency Cardiac Care. St. Louis: Mosby; 1994:169-216.
23 Brady WJ, Aufderheide TP. Left bundle branch block pattern complicating the evaluation of acute myocardial infarction in the emergency department. Acad Emerg Med. 1997;4:56-62.
24 Kozlowski FH, Brady WJ, Aufderheide TP, Buckley RS. The electrocardiographic diagnosis of acute myocardial infarction in patients with ventricular paced rhythms. Acad Emerg Med. 1998;5:52-57.
25 Huwez FU, Pringle SD, Macfarlane FW. Variable patterns of ST-T abnormalities in patients with left ventricular hypertrophy and normal coronary arteries. Br Heart J. 1992;67:304-307.
26 Brady WJ, Hwang V, Sullivan R, et al. A comparison of the 12-lead ECG to the 15-lead ECG in emergency department chest pain patients: Impact on diagnosis, therapy, and disposition. Am J Emerg Med. 2000;18:239-243.
27 Zalenski RJ, Cook D, Rydman R. Assessing the diagnostic value of an ECG containing leads V4R, V8, and V9: the 15-lead ECG. Ann Emerg Med. 1993;22:786-793.
28 Self WH, Mattu A, Martin M, et al. Body surface mapping in the ED evaluation of the patient with chest pain: use of the 80-lead electrocardiogram system. Am J Emerg Med. 2006;24:87-112.
29 Fesmire FM, Smith EE. Continuous 12-lead electrocardiograph monitoring in the emergency department. Am J Emerg Med. 1993;11:54-60.
30 Gibler WB, Sayre MR, Levy RC, et al. Serial 12-lead electrocardiographic monitoring in patients presenting to the emergency department with chest pain. J Electrocardiol. 1993;26(Suppl):238-243.
31 Fesmire FM, Percy RF, Bardoner JB, et al. Usefulness of automated serial 12-lead ECG monitoring during the initial emergency department evaluation of patients with chest pain. Ann Emerg Med. 1998;31:3-11.
32 Fesmire FM. ECG diagnosis of acute myocardial infarction in the presence of left bundle branch block in patients undergoing continuous ECG monitoring. Ann Emerg Med. 1995;26:69-82.
33 Krucoff MW, Green CE, Satler LF, et al. Noninvasive detection of coronary artery patency using continuous ST-segment monitoring. Am J Cardiol. 1986;57:916-922.
34 Jernberg T, Lindahl B, Wallentin L. ST-segment monitoring with continuous 12-lead ECG improves early risk stratification in patients with chest pain and ECG nondiagnostic of acute myocardial infarction. J Am Coll Cardiol. 1999;34:1413-1419.
35 deFilippi CR, Tocchi M, Parmar RJ, et al. Cardiac troponin T in chest pain unit patients without ischemic electrocardiographic changes: angiographic correlates and long-term clinical outcomes. J Am Coll Cardiol. 2000;35:1827-1834.
36 Lindahl B, Toss H, Siegbahn A, et al. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. N Engl J Med. 2000;343:1139-1147.
37 Apple FS, Murakami MM, Pearce LA, et al. Predictive value of cardiac troponin I and T for subsequent death in end-stage renal disease. Circulation. 2002;106:2941-2945.
38 Hamm CW, Goldmann BU, Heeschen C, et al. Emergency room triage of patients with acute chest pain by means of rapid testing for cardiac troponin T or troponin I. N Engl J Med. 1997;337:1648-1653.
39 Balk EM, Ioannidis JP, Salem D, et al. Accuracy of biomarkers to diagnose acute cardiac ischemia in the emergency department: a meta-analysis. Ann Emerg Med. 2001;37:478-494.
40 Antman EM, Tanasijevic MJ, Thompson B, et al. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med. 1996;335:1342-1349.
41 Ohman EM, Armstrong PW, Christenson RH, et al. Cardiac troponin T levels for risk stratification in acute myocardial ischemia. N Engl J Med. 1996;335:1333-1341.
42 Zimmerman J, Fromm R, Meyer D, et al. Diagnostic marker cooperative study for the diagnosis of myocardial infarction. Circulation. 1999;99:1671-1677.
43 Panteghini M, Apple FS, Christenson RH, et al. Use of biochemical markers in acute coronary syndromes. IFCC Scientific Division, Committee on Standardization of Markers of Cardiac Damage. Clin Chem Lab Med. 1999;37:687-693.
44 Wu AHB, Apple FS, Gibler WG, et al. National Academy of Clinical Biochemistry Standards of Laboratory Practice: Recommendations for the use of cardiac markers in coronary artery diseases. Clin Chem. 1999;45:1104-1121.
45 Brady WJ, Aufderheide TP, Kaplan P. Cardiovascular chest radiography. In: Reisdorff E, Schwartz D, Williamson B, editors. Emergency Radiology. New York, NY: McGraw-Hill, Inc., 1999.
46 Dalen JE, Bone RC. Is it time to pull the pulmonary artery catheter? JAMA. 1996;276:916-918.
