Other laboratory studies that may aid in diagnosis include the red cell count, mean corpuscular volume, and red cell distribution width (RDW), particularly when the hematocrit or hemoglobin levels are less than 60% or 20 g/dL, respectively. Only three situations cause microcytic erythrocytosis: β thalassemia trait, hypoxic erythrocytosis, and PV. With β thalassemia trait, the RDW is normal, whereas with hypoxic erythrocytosis and PV, the RDW may be elevated due to associated iron deficiency. Today, however, the assay for JAK2 V617F has superseded other tests for establishing the diagnosis of PV. Of course, in patients with associated acid-peptic disease, occult gastrointestinal bleeding may lead to a presentation with hypochromic, microcytic anemia, masking the presence of PV.
A bone marrow aspirate and biopsy provide no specific diagnostic information because these may be normal or indistinguishable from ET or PMF. Similarly, no specific cytogenetic abnormality is associated with the disease, and the absence of a cytogenetic marker does not exclude the diagnosis.
COMPLICATIONS
Many of the clinical complications of PV relate directly to the increase in blood viscosity associated with red cell mass elevation and indirectly to the increased turnover of red cells, leukocytes, and platelets with the attendant increase in uric acid and cytokine production. The latter appears to be responsible for constitutional symptoms. Peptic ulcer disease can also be due to Helicobacter pylori infection, the incidence of which is increased in PV, while the pruritus associated with this disorder may be a consequence of mast cell activation by JAK2 V617F. A sudden increase in spleen size can be associated with painful splenic infarction. Myelofibrosis appears to be part of the natural history of the disease but is a reactive, reversible process that does not itself impede hematopoiesis and by itself has no prognostic significance. In approximately 15% of patients, however, myelofibrosis is accompanied by significant extramedullary hematopoiesis, hepatosplenomegaly, and transfusion-dependent anemia, which are manifestations of stem cell failure. The organomegaly can cause significant mechanical discomfort, portal hypertension, and progressive cachexia. Although the incidence of acute nonlymphocytic leukemia is increased in PV, the incidence of acute leukemia in patients not exposed to chemotherapy or radiation therapy is low. Interestingly, chemotherapy, including hydroxyurea, has been associated with acute leukemia in JAK2 V617F–negative stem cells in some PV patients. Erythromelalgia is a curious syndrome of unknown etiology associated with thrombocytosis, primarily involving the lower extremities and usually manifested by erythema, warmth, and pain of the affected appendage and occasionally digital infarction. It occurs with a variable frequency and is usually responsive to salicylates. Some of the central nervous system symptoms observed in patients with PV, such as ocular migraine, appear to represent a variant of erythromelalgia.
Left uncontrolled, erythrocytosis can lead to thrombosis involving vital organs such as the liver, heart, brain, or lungs. Patients with massive splenomegaly are particularly prone to thrombotic events because the associated increase in plasma volume masks the true extent of the red cell mass elevation measured by the hematocrit or hemoglobin level. A “normal” hematocrit or hemoglobin level in a PV patient with massive splenomegaly should be considered indicative of an elevated red cell mass until proven otherwise.
PRIMARY MYELOFIBROSIS
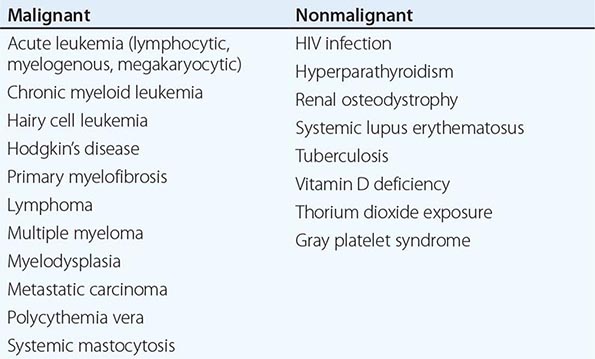
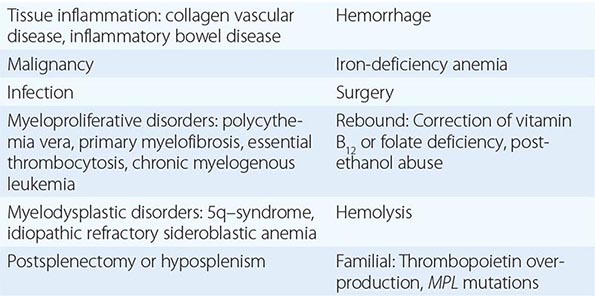
Chronic PMF (other designations include idiopathic myelofibrosis, agnogenic myeloid metaplasia, or myelofibrosis with myeloid metaplasia) is a clonal disorder of a multipotent hematopoietic progenitor cell of unknown etiology characterized by marrow fibrosis, extramedullary hematopoiesis, and splenomegaly. PMF is the least common chronic MPN, and establishing this diagnosis in the absence of a specific clonal marker is difficult because myelofibrosis and splenomegaly are also features of both PV and CML. Furthermore, myelofibrosis and splenomegaly also occur in a variety of benign and malignant disorders (Table 131-3), many of which are amenable to specific therapies not effective in PMF. In contrast to the other chronic MPNs and so-called acute or malignant myelofibrosis, which can occur at any age, PMF primarily afflicts men in their sixth decade or later.
|
DISORDERS CAUSING MYELOFIBROSIS |
ETIOLOGY
![]() The etiology of PMF is unknown. Nonrandom chromosome abnormalities such as 9p, 20q–, 13q–, trisomy 8 or 9, or partial trisomy 1q are common, but no cytogenetic abnormality specific to the disease has been identified. JAK2 V617F is present in approximately 50% of PMF patients, and mutations in the thrombopoietin receptor Mpl occur in about 5%. Most of the rest have mutations in the calreticulin gene (CALR) that alter the carboxy-terminal portion of the gene product. The degree of myelofibrosis and the extent of extramedullary hematopoiesis are also not related. Fibrosis in this disorder is associated with overproduction of transforming growth factor β and tissue inhibitors of metalloproteinases, whereas osteosclerosis is associated with overproduction of osteoprotegerin, an osteoclast inhibitor. Marrow angiogenesis occurs due to increased production of vascular endothelial growth factor. Importantly, fibroblasts in PMF are polyclonal and not part of the neoplastic clone.
The etiology of PMF is unknown. Nonrandom chromosome abnormalities such as 9p, 20q–, 13q–, trisomy 8 or 9, or partial trisomy 1q are common, but no cytogenetic abnormality specific to the disease has been identified. JAK2 V617F is present in approximately 50% of PMF patients, and mutations in the thrombopoietin receptor Mpl occur in about 5%. Most of the rest have mutations in the calreticulin gene (CALR) that alter the carboxy-terminal portion of the gene product. The degree of myelofibrosis and the extent of extramedullary hematopoiesis are also not related. Fibrosis in this disorder is associated with overproduction of transforming growth factor β and tissue inhibitors of metalloproteinases, whereas osteosclerosis is associated with overproduction of osteoprotegerin, an osteoclast inhibitor. Marrow angiogenesis occurs due to increased production of vascular endothelial growth factor. Importantly, fibroblasts in PMF are polyclonal and not part of the neoplastic clone.
CLINICAL FEATURES
No signs or symptoms are specific for PMF. Many patients are asymptomatic at presentation, and the disease is usually detected by the discovery of splenic enlargement and/or abnormal blood counts during a routine examination. However, in contrast to its companion MPN, night sweats, fatigue, and weight loss are common presenting complaints. A blood smear will show the characteristic features of extramedullary hematopoiesis: teardrop-shaped red cells, nucleated red cells, myelocytes, and promyelocytes; myeloblasts may also be present (Fig. 131-1). Anemia, usually mild initially, is the rule, whereas the leukocyte and platelet counts are either normal or increased, but either can be depressed. Mild hepatomegaly may accompany the splenomegaly but is unusual in the absence of splenic enlargement; isolated lymphadenopathy should suggest another diagnosis. Both serum lactate dehydrogenase and alkaline phosphatase levels can be elevated. The LAP score can be low, normal, or high. Marrow is usually inaspirable due to the myelofibrosis (Fig. 131-2), and bone x-rays may reveal osteosclerosis. Exuberant extramedullary hematopoiesis can cause ascites; portal, pulmonary, or intracranial hypertension; intestinal or ureteral obstruction; pericardial tamponade; spinal cord compression; or skin nodules. Splenic enlargement can be sufficiently rapid to cause splenic infarction with fever and pleuritic chest pain. Hyperuricemia and secondary gout may ensue.
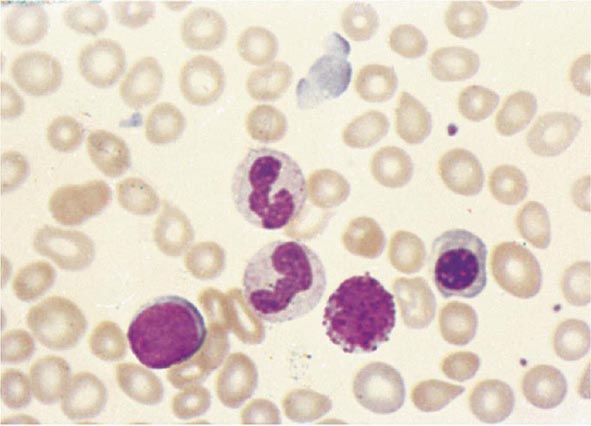
FIGURE 131-1 Teardrop-shaped red blood cells indicative of membrane damage from passage through the spleen, a nucleated red blood cell, and immature myeloid cells indicative of extramedullary hematopoiesis are noted. This peripheral blood smear is related to any cause of extramedullary hematopoiesis.
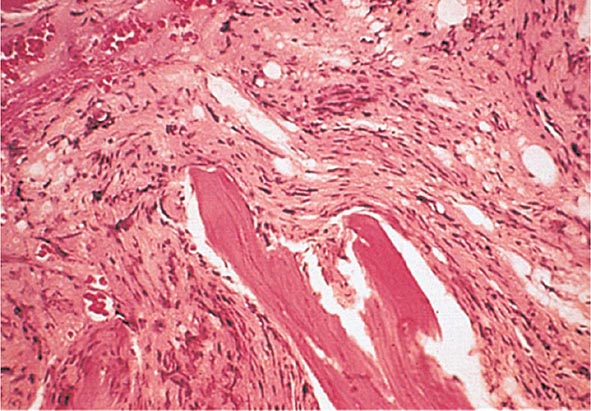
FIGURE 131-2 This marrow section shows the marrow cavity replaced by fibrous tissue composed of reticulin fibers and collagen. When this fibrosis is due to a primary hematologic process, it is called myelofibrosis. When the fibrosis is secondary to a tumor or a granulomatous process, it is called myelophthisis.
DIAGNOSIS
While the clinical picture described above is characteristic of PMF, all of the clinical features described can also be observed in PV or CML. Massive splenomegaly commonly masks erythrocytosis in PV, and reports of intraabdominal thrombosis in PMF most likely represent instances of unrecognized PV. In some patients with PMF, erythrocytosis has developed during the course of the disease. Furthermore, because many other disorders have features that overlap with PMF but respond to distinctly different therapies, the diagnosis of PMF is one of exclusion, which requires that the disorders listed in Table 131-3 be ruled out.
The presence of teardrop-shaped red cells, nucleated red cells, myelocytes, and promyelocytes establishes the presence of extramedullary hematopoiesis, while the presence of leukocytosis, thrombocytosis with large and bizarre platelets, and circulating myelocytes suggests the presence of an MPN as opposed to a secondary form of myelofibrosis (Table 131-3). Marrow is usually inaspirable due to increased marrow reticulin, but marrow biopsy will reveal a hypercellular marrow with trilineage hyperplasia and, in particular, increased numbers of megakaryocytes in clusters and with large, dysplastic nuclei. However, there are no characteristic bone marrow morphologic abnormalities that distinguish PMF from the other chronic MPNs. Splenomegaly due to extramedullary hematopoiesis may be sufficiently massive to cause portal hypertension and variceal formation. In some patients, exuberant extramedullary hematopoiesis can dominate the clinical picture. An intriguing feature of PMF is the occurrence of autoimmune abnormalities such as immune complexes, antinuclear antibodies, rheumatoid factor, or a positive Coombs’ test. Whether these represent a host reaction to the disorder or are involved in its pathogenesis is unknown. Cytogenetic analysis of blood is useful both to exclude CML and for prognostic purposes, because complex karyotype abnormalities portend a poor prognosis in PMF. For unknown reasons, the number of circulating CD34+ cells is markedly increased in PMF (>15,000/μL) compared to the other chronic MPNs, unless they too develop myeloid metaplasia.
Importantly, approximately 50% of PMF patients, like patients with its companion myeloproliferative disorders PV and ET, express the JAK2 V617F mutation, often as homozygotes. Such patients are usually older and have higher hematocrits than the patients who are JAK2 V617F–negative, whereas PMF patients expressing an MPL mutation tend to be more anemic and have lower leukocyte counts. Somatic mutations in exon 9 of the calreticulin gene (CALR) have been found in a majority of patients with PMF and ET who lack mutations in either JAK2 or MPL, and their clinical course appears to be more indolent than patients expressing either a JAK2 or an MPL mutation.
COMPLICATIONS
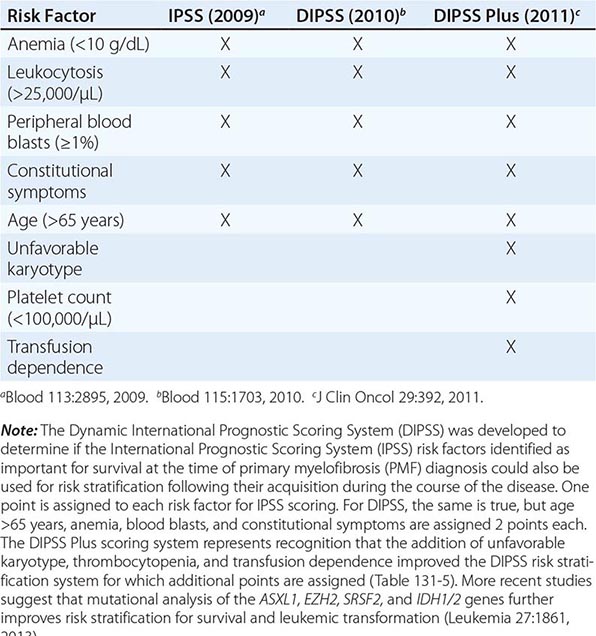
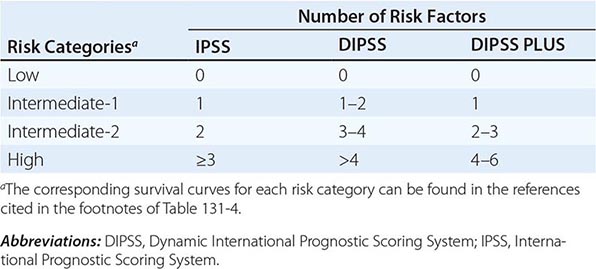
Survival in PMF varies according to specific risk factors at diagnosis (Tables 131-4 and 131-5) but is shorter in most patients than in PV or ET patients. The natural history of PMF is one of increasing marrow failure with transfusion-dependent anemia and increasing organomegaly due to extramedullary hematopoiesis. As with CML, PMF can evolve from a chronic phase to an accelerated phase with constitutional symptoms and increasing marrow failure. About 10% of patients spontaneously transform to an aggressive form of acute leukemia for which therapy is usually ineffective. Additional important prognostic factors for disease acceleration during the course of PMF include the presence of complex cytogenetic abnormalities, thrombocytopenia, and transfusion-dependent anemia. Most recently, mutations in the ASXL1, EZH2, SRSF2, and IDH1/2 genes have been identified as risk factors for early death or transformation to acute leukemia and may prove to be more useful for PMF risk assessment than any clinical scoring system.
|
THREE CURRENT SCORING SYSTEMS FOR ESTIMATING PROGNOSIS IN PMF PATIENTS |
|
IPSS AND DIPSS RISK STRATIFICATION SYSTEMS |
ESSENTIAL THROMBOCYTOSIS
Essential thrombocytosis (other designations include essential thrombocythemia, idiopathic thrombocytosis, primary thrombocytosis, and hemorrhagic thrombocythemia) is a clonal disorder of unknown etiology involving a multipotent hematopoietic progenitor cell manifested clinically by overproduction of platelets without a definable cause. ET is an uncommon disorder, with an incidence of 1–2/100,000 and a distinct female predominance. No clonal marker is available to consistently distinguish ET from the more common nonclonal, reactive forms of thrombocytosis (Table 131-6), making its diagnosis difficult. Once considered a disease of the elderly and responsible for significant morbidity due to hemorrhage or thrombosis, with the widespread use of electronic cell counters, it is now clear that ET can occur at any age in adults and often without symptoms or disturbances of hemostasis. There is an unexplained female predominance in contrast to PMF or the reactive forms of thrombocytosis where no sex difference exists. Because no specific clonal marker is available, clinical criteria have been proposed to distinguish ET from the other chronic MPNs, which may also present with thrombocytosis but have differing prognoses and therapies (Table 131-6). These criteria do not establish clonality; therefore, they are truly useful only in identifying disorders such as CML, PV, or myelodysplasia, which can masquerade as ET, as opposed to actually establishing the presence of ET. Furthermore, as with “idiopathic” erythrocytosis, nonclonal benign forms of thrombocytosis exist (such as hereditary overproduction of thrombopoietin) that are not widely recognized because we currently lack adequate diagnostic tools. Approximately 50% of ET patients carry the JAK2 V617F mutation, but its absence does not exclude the disorder.
|
CAUSES OF THROMBOCYTOSIS |
ETIOLOGY
Megakaryocytopoiesis and platelet production depend on thrombopoietin and its receptor Mpl. As in the case of early erythroid and myeloid progenitor cells, early megakaryocytic progenitors require the presence of interleukin 3 (IL-3) and stem cell factor for optimal proliferation in addition to thrombopoietin. Their subsequent development is also enhanced by the chemokine stromal cell-derived factor 1 (SDF-1). However, megakaryocyte maturation requires thrombopoietin.
Megakaryocytes are unique among hematopoietic progenitor cells because reduplication of their genome is endomitotic rather than mitotic. In the absence of thrombopoietin, endomitotic megakaryocytic reduplication and, by extension, the cytoplasmic development necessary for platelet production are impaired. Like erythropoietin, thrombopoietin is produced in both the liver and the kidneys, and an inverse correlation exists between the platelet count and plasma thrombopoietic activity. Unlike erythropoietin, thrombopoietin is only constitutively produced, and the plasma thrombopoietin level is controlled by the size of its progenitor cell pool. Also, in contrast to erythropoietin, but like its myeloid counterparts, granulocyte and granulocyte-macrophage colony-stimulating factors, thrombopoietin not only enhances the proliferation of its target cells but also enhances the reactivity of their end-stage product, the platelet. In addition to its role in thrombopoiesis, thrombopoietin also enhances the survival of multipotent hematopoietic stem cells and their bone marrow residence.
The clonal nature of ET was established by analysis of glucose-6-phosphate dehydrogenase isoenzyme expression in patients hemizygous for this gene, by analysis of X-linked DNA polymorphisms in informative female patients, and by the expression in patients of nonrandom, though variable, cytogenetic abnormalities. Although thrombocytosis is its principal manifestation, like the other chronic MPNs, a multipotent hematopoietic progenitor cell is involved in ET. Furthermore, a number of families have been described in which ET was inherited, in one instance as an autosomal dominant trait. In addition to ET, PMF and PV have also been observed in some kindreds. Like PMF, most patients who do not have JAK2 mutations have CALR mutations.
CLINICAL FEATURES
Clinically, ET is most often identified incidentally when a platelet count is obtained during the course of a routine medical evaluation. Occasionally, review of previous blood counts will reveal that an elevated platelet count was present but overlooked for many years. No symptoms or signs are specific for ET, but these patients can have hemorrhagic and thrombotic tendencies expressed as easy bruising for the former and microvascular occlusive events for the latter such as erythromelalgia, ocular migraine, or a TIA. Physical examination is generally unremarkable except occasionally for mild splenomegaly. Splenomegaly is indicative of another MPN, in particular PV, PMF, or CML.
Anemia is unusual, but a mild neutrophilic leukocytosis is not. The blood smear is most remarkable for the number of platelets present, some of which may be very large. The large mass of circulating platelets may prevent the accurate measurement of serum potassium due to release of platelet potassium upon blood clotting. This type of hyperkalemia is a laboratory artifact and not associated with electrocardiographic abnormalities. Similarly, arterial oxygen measurements can be inaccurate unless thrombocythemic blood is collected on ice. The prothrombin and partial thromboplastin times are normal, whereas abnormalities of platelet function such as a prolonged bleeding time and impaired platelet aggregation can be present. However, despite much study, no platelet function abnormality is characteristic of ET, and no platelet function test predicts the risk of clinically significant bleeding or thrombosis.
The elevated platelet count may hinder marrow aspiration, but marrow biopsy usually reveals megakaryocyte hypertrophy and hyperplasia, as well as an overall increase in marrow cellularity. If marrow reticulin is increased, another diagnosis should be considered. The absence of stainable iron demands an explanation because iron deficiency alone can cause thrombocytosis, and absent marrow iron in the presence of marrow hypercellularity is a feature of PV.
Nonrandom cytogenetic abnormalities occur in ET but are uncommon, and no specific or consistent abnormality is notable, even those involving chromosomes 3 and 1, where the genes for thrombopoietin and its receptor Mpl, respectively, are located.
DIAGNOSIS
Thrombocytosis is encountered in a broad variety of clinical disorders (Table 131-6), in many of which production of cytokines is increased. The absolute level of the platelet count is not a useful diagnostic aid for distinguishing between benign and clonal causes of thrombocytosis. About 50% of ET patients express the JAK2 V617F mutation. When JAK2 V617F is absent, cytogenetic evaluation is mandatory to determine if the thrombocytosis is due to CML or a myelodysplastic disorder such as the 5q– syndrome. Because the bcr-abl translocation can be present in the absence of the Ph chromosome, and because bcr-abl reverse transcriptase polymerase chain reaction is associated with false-positive results, fluorescence in situ hybridization (FISH) analysis for bcr-abl is the preferred assay in patients with thrombocytosis in whom a cytogenetic study for the Ph chromosome is negative. CALR mutations are present in most patients who do not have JAK2 mutations, but diagnostic tools to detect these mutations are not yet widespread. Anemia and ringed sideroblasts are not features of ET, but they are features of idiopathic refractory sideroblastic anemia, and in some of these patients, the thrombocytosis occurs in association with JAK2 V617F expression. Splenomegaly should suggest the presence of another MPN, and in this setting, a red cell mass determination should be performed because splenomegaly can mask the presence of erythrocytosis. Importantly, what appears to be ET can evolve into PV or PMF after a period of many years, revealing the true nature of the underlying MPN. There is sufficient overlap of the JAK2 V617F neutrophil allele burden between ET and PV that this cannot be used as a distinguishing diagnostic feature; only a red cell mass and plasma volume determination can distinguish PV from ET, and importantly in this regard, 64% of JAK2 V617F–positive ET patients actually were found to have PV when red cell mass and plasma volume determinations were performed.
COMPLICATIONS
Perhaps no other condition in clinical medicine has caused otherwise astute physicians to intervene inappropriately more often than thrombocytosis, particularly if the platelet count is >1 × 106/μL. It is commonly believed that a high platelet count causes intravascular stasis and thrombosis; however, no controlled clinical study has ever established this association, and in patients younger than age 60 years, the incidence of thrombosis was not greater in patients with thrombocytosis than in age-matched controls, and tobacco use appears to be the most important risk factor for thrombosis in ET patients.
To the contrary, very high platelet counts are associated primarily with hemorrhage due to acquired von Willebrand’s disease. This is not meant to imply that an elevated platelet count cannot cause symptoms in an ET patient, but rather that the focus should be on the patient, not the platelet count. For example, some of the most dramatic neurologic problems in ET are migraine-related and respond only to lowering of the platelet count, whereas other symptoms such as erythromelalgia respond simply to platelet cyclooxygenase-1 inhibitors such as aspirin or ibuprofen, without a reduction in platelet number. Still others may represent an interaction between an atherosclerotic vascular system and a high platelet count, and others may have no relationship to the platelet count whatsoever. Recognition that PV can present with thrombocytosis alone as well as the discovery of previously unrecognized causes of hypercoagulability (Chap. 142) make the older literature on the complications of thrombocytosis unreliable.
ET can also evolve into PMF, but whether this is a feature of ET or represents PMF presenting initially with isolated thrombocytosis is unknown.
132 |
Acute Myeloid Leukemia |
INCIDENCE
Acute myeloid leukemia (AML) is a neoplastic disease characterized by infiltration of the blood, bone marrow, and other tissues by proliferative, clonal undifferentiated cells of the hematopoietic system. These leukemias comprise a spectrum of malignancies that, untreated, range from rapidly fatal to slowly growing. In 2013, the estimated number of new AML cases in the United States was 14,590. The incidence of AML is ~3.5 per 100,000 people per year, and the age-adjusted incidence is higher in men than in women (4.5 vs 3.1). AML incidence increases with age; it is 1.7 in individuals age <65 years and 15.9 in those age >65 years. The median age at diagnosis is 67 years.
ETIOLOGY
Heredity, radiation, chemical and other occupational exposures, and drugs have been implicated in the development of AML. No direct evidence suggests a viral etiology.
Heredity Certain syndromes with somatic cell chromosome aneuploidy, such as trisomy 21 noted in Down syndrome, are associated with an increased incidence of AML. Inherited diseases with defective DNA repair, e.g., Fanconi anemia, Bloom syndrome, and ataxia-telangiectasia, are also associated with AML. Congenital neutropenia (Kostmann syndrome) is a disease with mutations in the genes encoding the granulocyte colony-stimulating factor (G-CSF) receptor and, often, neutrophil elastase that may evolve into AML. Germline mutations of CCAAT/enhancer-binding protein α (CEBPA), runt-related transcription factor 1 (RUNX1), and tumor protein p53 (TP53) have also been associated with a higher predisposition to AML in some series.
Radiation High-dose radiation, like that experienced by survivors of the atomic bombs in Japan or nuclear reactor accidents, increases the risk of myeloid leukemias that peaks 5–7 years after exposure. Therapeutic radiation alone seems to add little risk of AML but can increase the risk in people also exposed to alkylating agents.
Chemical and Other Exposures Exposure to benzene, a solvent used in the chemical, plastic, rubber, and pharmaceutical industries, is associated with an increased incidence of AML. Smoking and exposure to petroleum products, paint, embalming fluids, ethylene oxide, herbicides, and pesticides have also been associated with an increased risk of AML.
Drugs Anticancer drugs are the leading cause of therapy-associated AML. Alkylating agent–associated leukemias occur on average 4–6 years after exposure, and affected individuals have aberrations in chromosomes 5 and 7. Topoisomerase II inhibitor–associated leukemias occur 1–3 years after exposure, and affected individuals often have aberrations involving chromosome 11q23. Newer agents for treatment of other hematopoietic malignancies and solid tumors are also under scrutiny for increased risk of AML. Chloramphenicol, phenylbutazone, and, less commonly, chloroquine and methoxypsoralen can result in bone marrow failure that may evolve into AML.
CLASSIFICATION
The current categorization of AML uses the World Health Organization (WHO) classification (Table 132-1), which includes different biologically distinct groups based on clinical features and cytogenetic and molecular abnormalities in addition to morphology. In contrast to the previously used French-American-British (FAB) schema, the WHO classification places limited reliance on cytochemistry. A major difference between the WHO and the FAB systems is the blast cutoff for a diagnosis of AML as opposed to myelodysplastic syndrome (MDS); it is 20% in the WHO classification and 30% in the FAB. However, within the WHO classification, specific chromosomal rearrangements, i.e., t(8;21)(q22;q22), inv(16)(p13.1q22), t(16;16)(p13.1;q22), and t(15;17)(q22;q12), define AML even with <20% blasts.
|
WORLD HEALTH ORGANIZATION CLASSIFICATION OF ACUTE MYELOID LEUKEMIA (AML) AND RELATED NEOPLASMSa |
afrom SH Swerdlow et al (eds): World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, IARC Press, 2008. bDiagnosis is AML regardless of blast count.
Immunophenotype and Relevance to the WHO Classification The immunophenotype of human leukemia cells can be studied by multiparameter flow cytometry after the cells are labeled with monoclonal antibodies to cell-surface antigens. This can be important for separating AML from acute lymphoblastic leukemia (ALL) and identifying some subtypes of AML. For example, AML with minimal differentiation that is characterized by immature morphology and no lineage-specific cytochemical reactions may be diagnosed by flow-cytometric demonstration of the myeloid-specific antigens cluster designation (CD) 13 and/or 117. Similarly, acute megakaryoblastic leukemia can often be diagnosed only by expression of the platelet-specific antigens CD41 and/or CD61. Although flow cytometry is useful, widely used, and in some cases essential for the diagnosis of AML, it is supportive only in establishing the different subtypes of AML through the WHO classification.
Clinical Features and Relevance to the WHO Classification The WHO classification also considers clinical features in subdividing AML. For example, it identifies therapy-related AML as a separate entity that develops following prior therapy (e.g., alkylating agents, topoisomerase II inhibitors, ionizing radiation). It also identifies AML with myelodysplasia-related changes based in part on medical history of an antecedent MDS or myelodysplastic/myeloproliferative neoplasm. The clinical features likely contribute to the prognosis of AML and have therefore been included in the classification.
![]() Genetic Findings and Relevance to the WHO Classification The WHO classification uses clinical, morphologic, and cytogenetic and/or molecular criteria to identify subtypes of AML and forces the clinician to take the appropriate steps to correctly identify the entity and thus tailor treatment(s) accordingly. The WHO classification is indeed the first AML classification that incorporates genetic (chromosomal and molecular) information. In this classification, subtypes of AML are recognized based on the presence or absence of specific recurrent genetic abnormalities. For example, the diagnosis of acute promyelocytic leukemia (APL) is based on the presence of either the t(15;17)(q22;q12) cytogenetic rearrangement or the PML-RARA fusion product of the translocation. A similar approach is taken with regard to core binding factor (CBF) AML that is now designated based on the presence of t(8;21)(q22;q22), inv(16)(p13.1q22), or t(16;16)(p13.1;q22) or the respective fusion products RUNX1-RUNX1T1 and CBFB-MYH11.
Genetic Findings and Relevance to the WHO Classification The WHO classification uses clinical, morphologic, and cytogenetic and/or molecular criteria to identify subtypes of AML and forces the clinician to take the appropriate steps to correctly identify the entity and thus tailor treatment(s) accordingly. The WHO classification is indeed the first AML classification that incorporates genetic (chromosomal and molecular) information. In this classification, subtypes of AML are recognized based on the presence or absence of specific recurrent genetic abnormalities. For example, the diagnosis of acute promyelocytic leukemia (APL) is based on the presence of either the t(15;17)(q22;q12) cytogenetic rearrangement or the PML-RARA fusion product of the translocation. A similar approach is taken with regard to core binding factor (CBF) AML that is now designated based on the presence of t(8;21)(q22;q22), inv(16)(p13.1q22), or t(16;16)(p13.1;q22) or the respective fusion products RUNX1-RUNX1T1 and CBFB-MYH11.
The WHO classification incorporates cytogenetics in the AML classification by recognizing a category of AML with recurrent genetic abnormalities and a category of AML with myelodysplasia-related changes (Table 132-1). The latter category is diagnosed not only by morphologic changes, but also in part by selected myelodysplasia-related cytogenetic abnormalities (e.g., complex karyotypes and unbalanced and balanced changes involving, among others, chromosomes 5, 7, and 11). Only one cytogenetic abnormality has been invariably associated with specific morphologic features: t(15;17)(q22;q12) with APL. Other chromosomal abnormalities have been associated primarily with one morphologic/immunophenotypic group, including inv(16)(p13.1q22) with AML with abnormal bone marrow eosinophils; t(8;21)(q22;q22) with slender Auer rods, expression of CD19, and increased normal eosinophils; and t(9;11)(p22;q23), and other translocations involving 11q23, with monocytic features. Recurring chromosomal abnormalities in AML may also be associated with specific clinical characteristics. More commonly associated with younger age are t(8;21) and t(15;17), and with older age, del(5q) and del(7q). Myeloid sarcomas (see below) are associated with t(8;21), and disseminated intravascular coagulation (DIC) is associated with t(15;17).
The WHO classification also incorporates molecular abnormalities by recognizing fusion genes that are products of recurrent cytogenetic aberrations or have been found mutated and may be involved in leukemogenesis. For instance, t(15;17) results in the fusion gene PML-RARA that encodes a chimeric protein, promyelocytic leukemia (Pml)–retinoic acid receptor α (Rarα), which is formed by the fusion of the retinoic acid receptor α (RARA) gene from chromosome 17 and the promyelocytic leukemia (PML) gene from chromosome 15. The RARA gene encodes a member of the nuclear hormone receptor family of transcription factors. After binding retinoic acid, RARA can promote expression of a variety of genes. The 15;17 translocation juxtaposes PML with RARA in a head-to-tail configuration that is under the transcriptional control of PML. Three different breakpoints in the PML gene lead to various fusion protein isoforms. The Pml-Rarα fusion protein tends to suppress gene transcription and blocks differentiation of the cells. Pharmacologic doses of the Rarα ligand, all-trans-retinoic acid (tretinoin), relieve the block and promote hematopoietic cell differentiation (see below). Similar examples of molecular subtypes of the disease included in the category of AML with recurrent genetic abnormalities are those characterized by the leukemogenic fusion genes RUNX1-RUNX1T1, CBFB-MYH11, MLLT3-MLL, and DEK-NUP214, resulting, respectively, from t(8;21), inv(16) or t(16;16), t(9;11), and t(6;9)(p23;q34).
Two new provisional entities defined by the presence of gene mutations, rather than microscopic chromosomal abnormalities, have been added to the category of AML with recurrent genetic abnormalities: AML with mutated nucleophosmin (nucleolar phosphoprotein B23, numatrin) (NPM1) and AML with mutated CEBPA. AML with fms-related tyrosine kinase 3 (FLT3) mutations is not considered a distinct entity, although determining the presence of such mutations is recommended by WHO in patients with cytogenetically normal AML (CN-AML) because the relatively frequent FLT3-internal tandem duplication (ITD) carries a negative prognostic significance and therefore is clinically relevant. FLT3 encodes a tyrosine kinase receptor important in the development of myeloid and lymphoid lineages. Activating mutations of FLT3 are present in ~30% of adult AML patients due to ITDs in the juxtamembrane domain or point mutations of the activating loop of the kinase (called tyrosine kinase domain mutations). Aberrant activation of the FLT3-encoded protein provides increased proliferation and antiapoptotic signals to the myeloid progenitor cell. FLT3-ITD, the more common of the FLT3 mutations, occurs preferentially in patients with CN-AML. The importance of identifying FLT3-ITD at diagnosis relates to the fact that not only is it a useful prognosticator but it also may predict response to specific treatment such as the tyrosine kinase inhibitors that are in clinical investigation.
PROGNOSTIC FACTORS
Several factors have been demonstrated to predict outcome of AML patients treated with chemotherapy, and they can be used for risk stratification and treatment guidance.
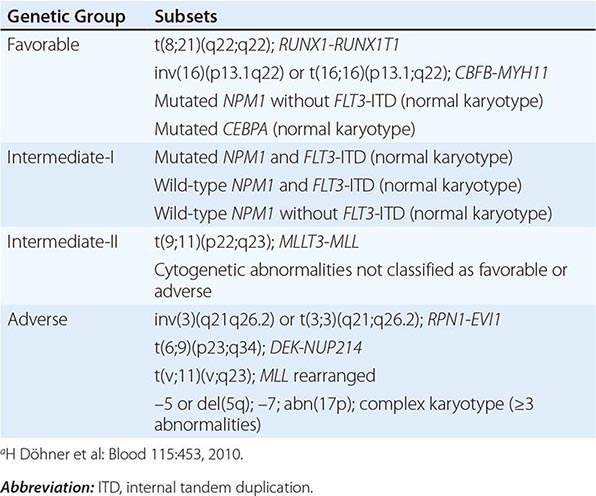
Chromosome findings at diagnosis are currently the most important independent prognostic factors. Several studies have categorized patients as having favorable, intermediate, or poor cytogenetic risk based on the presence of structural and/or numerical aberrations. Patients with t(15;17) have a very good prognosis (~85% cured), and those with t(8;21) and inv(16) have a good prognosis (~55% cured), whereas those with no cytogenetic abnormality have an intermediate outcome risk (~40% cured). Patients with a complex karyotype, t(6;9), inv(3), or –7 have a very poor prognosis. Another cytogenetic subgroup, the monosomal karyotype, has been suggested to adversely impact the outcome of AML patients other than those with t(15;17), t(8;21), or inv(16) or t(16;16). The monosomal karyotype subgroup is defined by the presence of at least two autosomal monosomies (loss of chromosomes other than Y or X) or a single autosomal monosomy with additional structural abnormalities.
For patients lacking prognostic cytogenetic abnormalities, such as those with CN-AML, outcome prediction uses mutated or aberrantly expressed genes. NPM1 mutations without concurrent presence of FLT3-ITD, and CEBPA mutations, especially if concurrently present in two different alleles, have been shown to predict favorable outcome, whereas FLT3-ITD predicts poor outcome. Given the proven prognostic importance of NPM1 and CEBPA mutations and FLT3-ITD, molecular assessment of these genes at diagnosis has been incorporated in AML management guidelines by the National Comprehensive Cancer Network (NCCN) and the European LeukemiaNet (ELN). The same markers have also been incorporated in the definitions of the genetic groups of the ELN standardized reporting system, which are based on both cytogenetic and molecular abnormalities and used for comparing clinical features and treatment response among subsets of patients reported in different studies (Table 132-2). More recently, the prognostic impact of the genetic groups recognized by the ELN reporting system has been demonstrated. Thus, these genetic groups may also be used for risk stratification and treatment guidance.
|
EUROPEAN LEUKEMIANET RECOMMENDED STANDARDIZED REPORTING FOR CORRELATION OF CYTOGENETIC AND MOLECULAR GENETIC DATA IN AML WITH CLINICAL DATAa |
In addition to NPM1 and CEBPA mutations and FLT3-ITD, other molecular aberrations (Table 132-3) may in the future be routinely used for prognostication in AML and incorporated in the WHO classification and the ELN reporting system. Among these prognostic mutated genes are those encoding receptor tyrosine kinases (e.g., v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog [KIT]), transcription factors (i.e., RUNX1 and Wilms tumor 1 [WT1]), and epigenetic modifiers (i.e., additional sex combs like transcriptional regulator 1 [ASXL1], DNA (cytosine-5-)-methyltransferase 3 alpha [DNMT3A], isocitrate dehydrogenase 1 (NADP+), soluble [IDH1] and isocitrate dehydrogenase 2 (NADP+), mitochondrial [IDH2], lysine (K)-specific methyltransferase 2A [KMT2A, also known as MLL], and tet methylcytosine dioxygenase 2 [TET2]). Although KIT mutations are almost exclusively present in CBF AML and impact adversely the outcome, the remaining markers have been reported primarily in CN-AML. These gene mutations have been shown to be associated with outcome in multivariable analyses independently from other prognostic factors. However, for some of them, the prognostic impact (e.g., TET2 mutations) or the type (adverse vs favorable) of prognostic impact (e.g., IDH1, IDH2) has been found in the majority, but not in all, of the reported studies.
|
MOLECULAR PROGNOSTIC MARKERS IN AML |
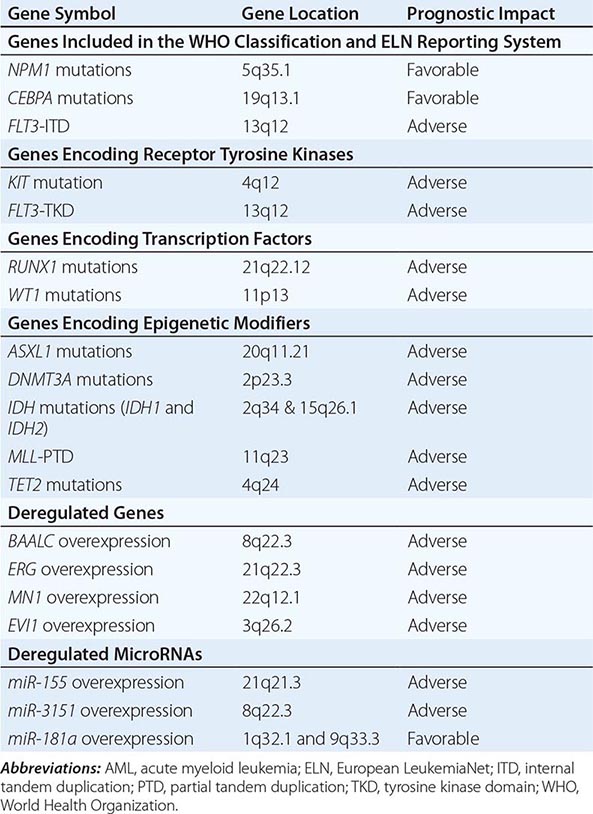
An independent prognostic impact remains to be determined for mutated genes that are either associated primarily with unfavorable cytogenetic aberrations (e.g., TP53) or are found with a relatively lower frequency in AML patients like those encoding epigenetic modifiers (e.g., enhancer of zeste 2 polycomb repressive complex 2 subunit [EZH2]), phosphatases (e.g., protein tyrosine phosphatase, non-receptor type 11 (PTPN11]), putative transcription factors (e.g., PHD finger protein 6 [PHF6]), splicing factors (e.g., U2 small nuclear RNA auxiliary factor 1 [U2AF1]), and proteins involved in chromosome segregation and genome stability (e.g., structural maintenance of chromosomes 1A [SMC1A] or structural maintenance of chromosomes 3 [SMC3]). Finally, other mutated genes are recognized as predictors of treatment response to distinct therapies rather than prognosticators; for example, neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) and Kirsten rat sarcoma viral oncogene homolog (KRAS) predict a better response to high-dose cytarabine in CBF AML.
In addition to gene mutations, deregulation of the expression levels of coding genes and of short noncoding RNAs (microRNAs) have been reported to provide prognostic information (Table 132-3). Overexpression of genes such as brain and acute leukemia, cytoplasmic (BAALC), v-ets avian erythroblastosis virus E26 oncogene homologue (avian) (ERG), meningioma (disrupted in balanced translocation) 1 (MN1), and MDS1 and EVI1 complex locus (MECOM, also known as EVI1) have been found to be predictive for poor outcome, especially in CN-AML. Similarly, deregulated expression levels of microRNAs, naturally occurring noncoding RNAs that have been shown to regulate the expression of proteins involved in hematopoietic differentiation and survival pathways by degradation or translation inhibition of target coding RNAs, have been associated with prognosis in AML. Overexpression of miR-155 and miR-3151 has been found to affect outcome adversely in CN-AML, whereas overexpression of miR-181a predicts a favorable outcome both in CN-AML and cytogenetically abnormal AML.
Because prognostic molecular markers in AML are not mutually exclusive and often occur concurrently (>80% patients have at least two or more prognostic gene mutations), the likelihood that distinct marker combinations may be more informative than single markers is being recognized.
Epigenetic changes (e.g., DNA methylation) and microRNAs are often involved in deregulation of genes involved in hematopoiesis, contribute to leukemogenesis, and are often associated with the previously discussed prognostic gene mutations. These changes not only have been shown to provide biologic insights into leukemogenic mechanisms, but also independent prognostic information. Indeed, it is anticipated that with the enormous progress made in DNA and RNA sequencing technology, additional genetic and epigenetic aberrations will soon be discovered and will contribute to classification and reporting systems and outcome risk determination in AML patients.
In addition to cytogenetics and/or molecular aberrations, several other factors are associated with outcome in AML. Age at diagnosis is one of the most important risk factors. Advancing age is associated with a poorer prognosis not only because of its influence on the ability to survive induction therapy due to coexisting comorbidities, but also because with each successive decade of age, a greater proportion of patients have an intrinsically more resistant disease. A prolonged symptomatic interval with cytopenias preceding diagnosis or a history of antecedent hematologic disorders including myeloproliferative neoplasms is often found in older patients and is a clinical feature associated with a lower complete remission (CR) rate and shorter survival time. The CR rate is lower in patients who have had anemia, leukopenia, and/or thrombocytopenia for >3 months before the diagnosis of AML when compared to those without such a history. Responsiveness to chemotherapy declines as the duration of the antecedent disorder(s) increases. AML developing after treatment with cytotoxic agents for other malignancies is usually difficult to treat successfully. Finally, it is likely that AML in older patients is also associated with poor outcome because of the presence of distinct biologic features that may increase the aggressiveness of the disease and reduce the likelihood of treatment response. The leukemic cells in older patients more commonly express the multidrug resistance 1 (MDR1) efflux pump that conveys resistance to natural product–derived agents such as the anthracyclines that are frequently incorporated into the initial treatment. In addition, older patients less frequently harbor favorable cytogenetic abnormalities [i.e., t(8;21), inv(16), and t(16;16)] and more frequently harbor adverse cytogenetic (e.g., complex and monosomal karyotypes) and/or molecular (e.g., ASXL1, IDH2, RUNX1, TET2) abnormalities.
Other factors independently associated with worse outcome are a low performance status that influences ability to survive induction therapy and thus respond to treatment and a high presenting leukocyte count that in some series is an adverse prognostic factor for attaining a CR. Among patients with hyperleukocytosis (>100,000/μL), early central nervous system bleeding and pulmonary leukostasis contribute to poor outcome with initial therapy.
Achievement of CR is associated with better outcome and longer survival. CR is defined after examination of both blood and bone marrow. The blood neutrophil count must be ≥1000/μL and the platelet count ≥100,000/μL. Hemoglobin concentration is not considered in determining CR. Circulating blasts should be absent. Although rare blasts may be detected in the blood during marrow regeneration, they should disappear on successive studies. The bone marrow should contain <5% blasts, and Auer rods should be absent. Extramedullary leukemia should not be present. Patients who achieve CR after one induction cycle have longer CR durations than those requiring multiple cycles.
CLINICAL PRESENTATION
Symptoms Patients with AML most often present with nonspecific symptoms that begin gradually or abruptly and are the consequence of anemia, leukocytosis, leukopenia or leukocyte dysfunction, or thrombocytopenia. Nearly half have had symptoms for ≤3 months before the leukemia was diagnosed.
Half of patients mention fatigue as the first symptom, but most complain of fatigue or weakness at the time of diagnosis. Anorexia and weight loss are common. Fever with or without an identifiable infection is the initial symptom in approximately 10% of patients. Signs of abnormal hemostasis (bleeding, easy bruising) are noted first in 5% of patients. On occasion, bone pain, lymphadenopathy, nonspecific cough, headache, or diaphoresis is the presenting symptom.
Rarely patients may present with symptoms from a myeloid sarcoma that is a tumor mass consisting of myeloid blasts occurring at anatomic sites other than bone marrow. Sites involved are most commonly the skin, lymph node, gastrointestinal tract, soft tissue, and testis. This rare presentation, often characterized by chromosome aberrations [e.g., monosomy 7, trisomy 8, MLL rearrangement, inv(16), trisomy 4, t(8;21)], may precede or coincide with AML.
Physical Findings Fever, splenomegaly, hepatomegaly, lymphadenopathy, sternal tenderness, and evidence of infection and hemorrhage are often found at diagnosis. Significant gastrointestinal bleeding, intrapulmonary hemorrhage, or intracranial hemorrhage occurs most often in APL. Bleeding associated with coagulopathy may also occur in monocytic AML and with extreme degrees of leukocytosis or thrombocytopenia in other morphologic subtypes. Retinal hemorrhages are detected in 15% of patients. Infiltration of the gingivae, skin, soft tissues, or meninges with leukemic blasts at diagnosis is characteristic of the monocytic subtypes and those with 11q23 chromosomal abnormalities.
Hematologic Findings Anemia is usually present at diagnosis and can be severe. The degree varies considerably, irrespective of other hematologic findings, splenomegaly, or duration of symptoms. The anemia is usually normocytic normochromic. Decreased erythropoiesis often results in a reduced reticulocyte count, and red blood cell (RBC) survival is decreased by accelerated destruction. Active blood loss also contributes to the anemia.
The median presenting leukocyte count is about 15,000/μL. Between 25 and 40% of patients have counts <5000/μL, and 20% have counts >100,000/μL. Fewer than 5% have no detectable leukemic cells in the blood. The morphology of the malignant cell varies in different subsets. In AML, the cytoplasm often contains primary (nonspecific) granules, and the nucleus shows fine, lacy chromatin with one or more nucleoli characteristic of immature cells. Abnormal rod-shaped granules called Auer rods are not uniformly present, but when they are, myeloid lineage is virtually certain (Fig. 132-1). Poor neutrophil function may be noted functionally by impaired phagocytosis and migration and morphologically by abnormal lobulation and deficient granulation.
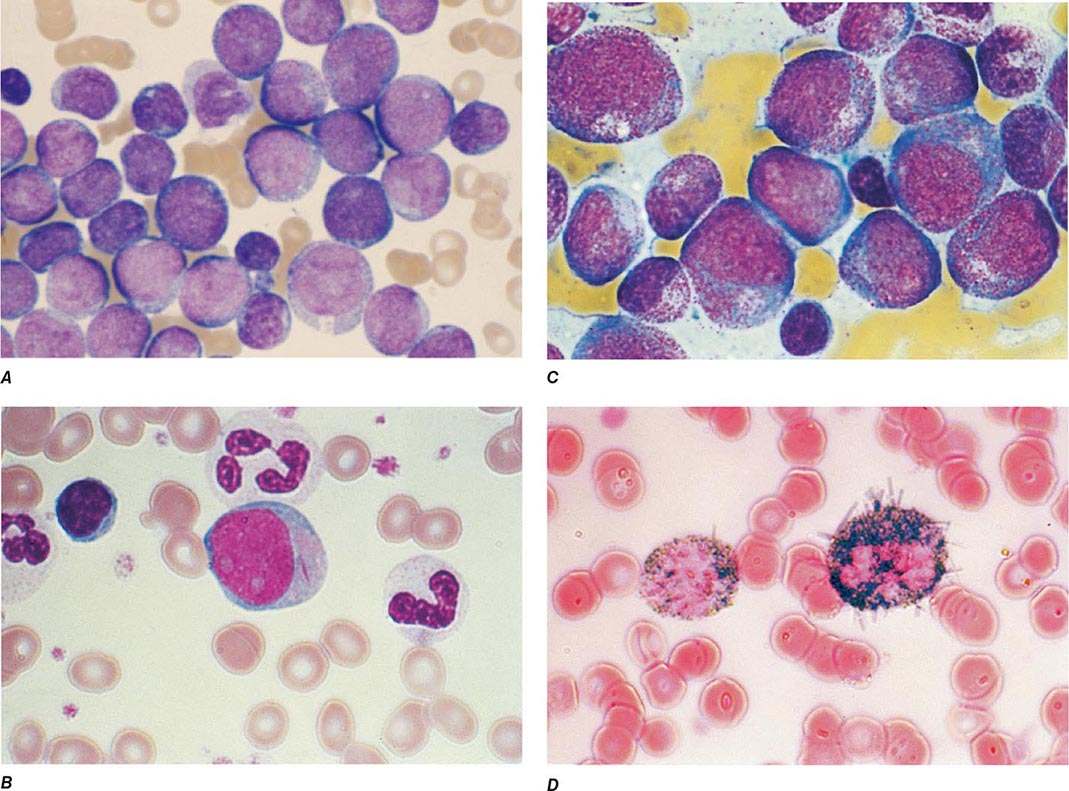
FIGURE 132-1 Morphology of acute myeloid leukemia (AML) cells. A. Uniform population of primitive myeloblasts with immature chromatin, nucleoli in some cells, and primary cytoplasmic granules. B. Leukemic myeloblast containing an Auer rod. C. Promyelocytic leukemia cells with prominent cytoplasmic primary granules. D. Peroxidase stain shows dark blue color characteristic of peroxidase in granules in AML.
Platelet counts <100,000/μL are found at diagnosis in ~75% of patients, and about 25% have counts <25,000/μL. Both morphologic and functional platelet abnormalities can be observed, including large and bizarre shapes with abnormal granulation and inability of platelets to aggregate or adhere normally to one another.
Pretreatment Evaluation Once the diagnosis of AML is suspected, a rapid evaluation and initiation of appropriate therapy should follow. In addition to clarifying the subtype of leukemia, initial studies should evaluate the overall functional integrity of the major organ systems, including the cardiovascular, pulmonary, hepatic, and renal systems (Table 132-4). Factors that have prognostic significance, either for achieving CR or for predicting the duration of CR, should also be assessed before initiating treatment, including cytogenetics and molecular markers (see above). Leukemic cells should be obtained from all patients and cryopreserved for future use as new tests and therapeutics become available. All patients should be evaluated for infection.
|
INITIAL DIAGNOSTIC EVALUATION AND MANAGEMENT OF ADULT PATIENTS WITH AML |
Abbreviations: AML, acute myeloid leukemia; BUN, blood urea nitrogen; CBC, complete blood count; CMV, cytomegalovirus; CNS, central nervous system; DIC, disseminated intravascular coagulation; HLA, human leukocyte antigen; HSCT, hematopoietic stem cell transplantation; HSV, herpes simplex virus; IV, intravenous; LDH, lactate dehydrogenase; MRI, magnetic resonance imaging; MUGA, multigated acquisition; PA, posteroanterior; RBC, red blood (cell) count.
Most patients are anemic and thrombocytopenic at presentation. Replacement of the appropriate blood components, if necessary, should begin promptly. Because qualitative platelet dysfunction or the presence of an infection may increase the likelihood of bleeding, evidence of hemorrhage justifies the immediate use of platelet transfusion, even if the platelet count is only moderately decreased.
About 50% of patients have a mild to moderate elevation of serum uric acid at presentation. Only 10% have marked elevations, but renal precipitation of uric acid and the nephropathy that may result is a serious but uncommon complication. The initiation of chemotherapy may aggravate hyperuricemia, and patients are usually started immediately on allopurinol and hydration at diagnosis. Rasburicase (recombinant uric oxidase) is also useful for treating uric acid nephropathy and often can normalize the serum uric acid level within hours with a single dose of treatment. The presence of high concentrations of lysozyme, a marker for monocytic differentiation, may be etiologic in renal tubular dysfunction, which could worsen other renal problems that arise during the initial phases of therapy.
TREATMENT ACUTE MYELOID LEUKEMIA
Treatment of the newly diagnosed patient with AML is usually divided into two phases, induction and postremission management (Fig. 132-2). The initial goal is to induce CR. Once CR is obtained, further therapy must be used to prolong survival and achieve cure. The initial induction treatment and subsequent postremission therapy are often chosen based on the patient’s age. Intensifying therapy with traditional chemotherapy agents such as cytarabine and anthracyclines in younger patients (<60 years) appears to increase the cure rate of AML. In older patients, the benefit of intensive therapy is controversial; novel approaches for selecting patients predicted to be responsive to treatment and new therapies are being pursued.
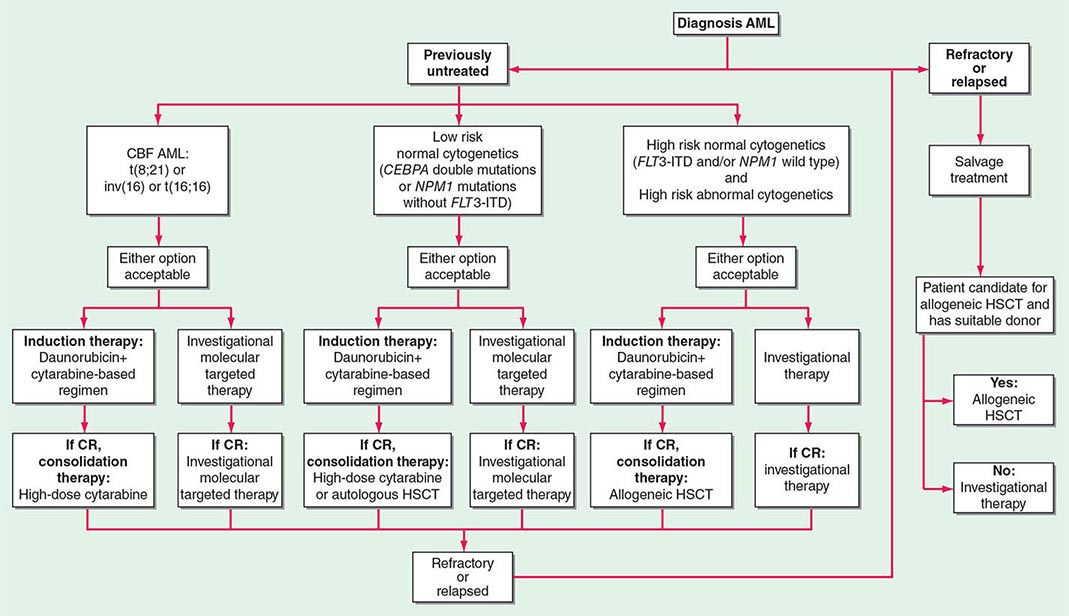
FIGURE 132-2 Flow chart for the therapy of newly diagnosed acute myeloid leukemia (AML). For all forms of AML except acute promyelocytic leukemia (APL), standard therapy includes a regimen based on a 7-day continuous infusion of cytarabine (100–200 mg/m2 per day) and a 3-day course of daunorubicin (60–90 mg/m2 per day) with or without additional drugs. Idarubicin (12–13 mg/m2 per day) could be used in place of daunorubicin (not shown). Patients who achieve complete remission (CR) undergo postremission consolidation therapy, including sequential courses of high-dose cytarabine, autologous hematopoietic stem cell transplantation (HSCT), allogeneic HSCT, or novel therapies, based on their predicted risk of relapse (i.e., risk-stratified therapy). Patients with APL (see text for treatment) usually receive tretinoin and arsenic trioxide–based regimens with or without anthracycline-based chemotherapy and possibly maintenance with tretinoin. CBF, core binding factor; ITD, internal tandem duplication.
INDUCTION CHEMOTHERAPY
The most commonly used CR induction regimens (for patients other than those with APL) consist of combination chemotherapy with cytarabine and an anthracycline (e.g., daunorubicin, idarubicin, mitoxantrone). Cytarabine is a cell cycle S-phase–specific antimetabolite that becomes phosphorylated intracellularly to an active triphosphate form that interferes with DNA synthesis. Anthracyclines are DNA intercalators. Their primary mode of action is thought to be inhibition of topoisomerase II, leading to DNA breaks.
In younger adults (age <60 years), cytarabine is used either at standard dose (100–200 mg/m2) administered as a continuous intravenous infusion for 7 days or higher dose (2 g/m2) administered intravenously every 12 h for 6 days. With standard-dose cytarabine, anthracycline therapy generally consists of daunorubicin (60–90 mg/m2) or idarubicin (12 mg/m2) intravenously on days 1, 2, and 3 (the 7 and 3 regimen). Other agents can be added (i.e., cladribine) when 60 mg/m2 of daunorubicin is used.
High-dose cytarabine-based regimens have also been shown to induce high CR rates. When given in high doses, higher intracellular levels of cytarabine may be achieved, thereby saturating the cytarabine-inactivating enzymes and increasing the intracellular levels of 1-β-D-arabinofuranylcytosine-triphosphate, the active metabolite incorporated into DNA. Thus, higher doses of cytarabine may increase the inhibition of DNA synthesis and thereby overcome resistance to standard-dose cytarabine. With high-dose cytarabine, daunorubicin 60 mg/m2 or idarubicin 12 mg/m2 is generally used.
The hematologic toxicity of high-dose cytarabine-based induction regimens has typically been greater than that associated with 7 and 3 regimens. Toxicity with high-dose cytarabine also includes pulmonary toxicity and significant and occasionally irreversible cerebellar toxicity. All patients treated with high-dose cytarabine must be closely monitored for cerebellar toxicity. Full cerebellar testing should be performed before each dose, and further high-dose cytarabine should be withheld if evidence of cerebellar toxicity develops. This toxicity occurs more commonly in patients with renal impairment and in those older than age 60 years. The increased toxicity observed with high-dose cytarabine has limited the use of this therapy in older AML patients.
Incorporation of novel and molecular targeting agents into these regimens is currently under investigation. For patients with FLT3-ITD AML, trials with tyrosine kinase inhibitors are ongoing. Patients with CBF AML may benefit from the combination of gemtuzumab ozogamicin, a monoclonal CD33 antibody linked to the cytotoxic agent calicheamicin, with induction and consolidation chemotherapies. This agent, initially approved for older patients with relapsed disease, has been withdrawn from the U.S. market at the request of the U.S. Food and Drug Administration due to concerns about the product’s toxicity, including myelosuppression, infusion toxicity, and venoocclusive disease and the clinical benefit of the initially recommended higher doses. However, the aforementioned recent results are encouraging and support the reintroduction of this agent into the therapeutic armamentarium for AML.
In older patients (age ≥60 years), the outcome is generally poor likely due to a higher induction treatment–related mortality rate and frequency of resistant disease, especially in patients with prior hematologic disorders (MDS or myeloproliferative syndromes) or who have received chemotherapy treatment for another malignancy or harbor cytogenetic and genetic abnormalities that adversely impact on clinical outcome. These patients should be considered for clinical trials. Alternatively, older patients can be also treated with the 7 and 3 regimen with standard-dose cytarabine and idarubicin (12 mg/m2), daunorubicin (45–90 mg/m2), or mitoxantrone (12 mg/m2). For patients older than 65 years, higher dose daunorubicin (90 mg/m2) has not shown benefit due to the increased toxicity and is not recommended. The combination of gemtuzumab ozogamicin with chemotherapy reduces the risk of relapse for patients age 50–70 years with previously untreated AML. Finally, older patients may be considered for single-agent therapies with clofarabine or hypomethylating agents (i.e., 5-azacitidine or decitabine). The latter are often used for patients unfit for more intensive therapies.
After one cycle of the 7 and 3 chemotherapy induction regimen, if persistence of leukemia is documented, the patient is usually retreated with the same agents (cytarabine and the anthracycline) for 5 and 2 days, respectively. Our recommendation, however, is to consider changing therapy in this setting.
POSTREMISSION THERAPY
Induction of a durable first CR is critical to long-term disease-free survival in AML. However, without further therapy, virtually all patients experience relapse. Thus, postremission therapy is designed to eradicate residual leukemic cells to prevent relapse and prolong survival. The type of postremission therapy in AML is often based on age and cytogenetic and molecular risk.
For younger patients, most studies include intensive chemotherapy and allogeneic or autologous hematopoietic stem cell transplantation (HSCT). In the postremission setting, high-dose cytarabine for three to four cycles is more effective than standard-dose cytarabine. The Cancer and Leukemia Group B (CALGB), for example, compared the duration of CR in patients randomly assigned after remission to four cycles of high (3 g/m2, every 12 h on days 1, 3, and 5), intermediate (400 mg/m2 for 5 days by continuous infusion), or standard (100 mg/m2 per day for 5 days by continuous infusion) doses of cytarabine. A dose-response effect for cytarabine in patients with AML who were age ≤60 years was demonstrated. High-dose cytarabine significantly prolonged CR and increased the fraction cured in patients with favorable [t(8;21) and inv(16)] and normal cytogenetics, but it had no significant effect on patients with other abnormal karyotypes. As discussed, high-dose cytarabine has increased toxicity in older patients. Therefore, in this age group, for patients without CBF AML, exploration of attenuated chemotherapy regimens has been pursued. However, because the outcome of older patients is poor, allogeneic HSCT, when feasible, should be strongly considered. Postremission therapy is also a setting for introduction of new agents (Table 132-5).
|
SELECTED AGENTS UNDER STUDY FOR THE TREATMENT OF ACUTE MYELOID LEUKEMIA |
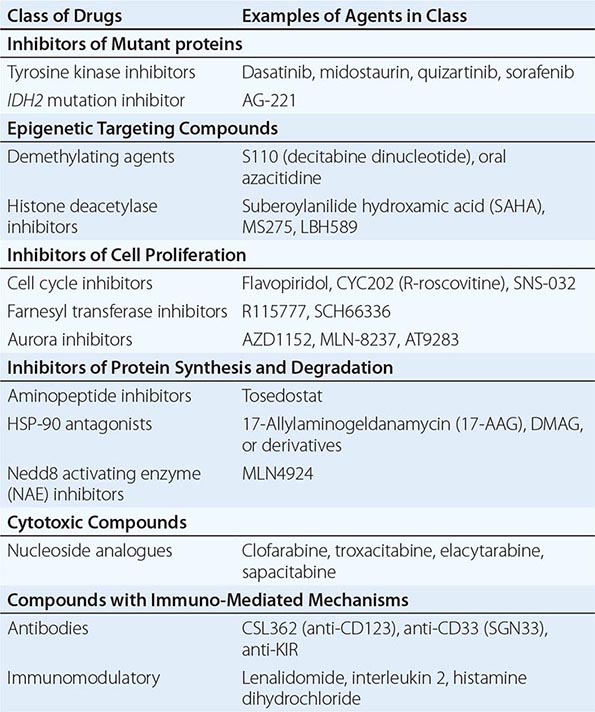
Autologous HSCT preceded by one to two cycles of high-dose cytarabine is also an option for intensive consolidation therapy. Autologous HSCT has been generally applied to AML patients in the context of a clinical trial or when the risk of repetitive intensive chemotherapy represents a higher risk than the autologous HSCT (e.g., in patients with severe platelet alloimmunization) or when other factors including patient age, comorbid conditions, and fertility are considered.
Allogeneic HSCT is used in patients age <70–75 years with a human leukocyte antigen (HLA)-compatible donor who have high-risk cytogenetics. Selected high-risk patients are also considered for alternative donor transplants (e.g., mismatched unrelated, haploidentical related, and unrelated umbilical cord donors). In patients with CN-AML and high-risk molecular features such as FLT3-ITD, allogeneic HSCT is best applied in the context of clinical trials because the impact of aggressive therapy on outcome is unknown. For older patients, exploration of reduced-intensity allogeneic HSCT has been pursued.
Trials comparing intensive chemotherapy and autologous and allogeneic HSCT have shown improved duration of remission with allogeneic HSCT compared to autologous HSCT or chemotherapy alone. However, overall survival is generally not different; the improved disease control with allogeneic HSCT is erased by the increase in fatal toxicity. In fact, relapse following allogeneic HSCT occurs in only a small fraction of patients, but treatment-related toxicity is relatively high; complications include venoocclusive disease, graft-versus-host disease (GVHD), and infections. Autologous HSCT can be administered in young and older patients and uses the same preparative regimens. Patients subsequently receive their own stem cells collected while in remission. The toxicity is relatively low with autologous HSCT (5% mortality rate), but the relapse rate is higher than with allogeneic HSCT, due to the absence of the graft-versus-leukemia (GVL) effect seen with allogeneic HSCT and possible contamination of the autologous stem cells with residual tumor cells.
Prognostic factors may ultimately help to select the appropriate postremission therapy in patients in first CR. Our approach includes allogeneic HSCT in first CR for patients without favorable cytogenetics or genotype (e.g., patients who do not have CEBPA biallelic mutations or NPM1 mutations without FLT3-ITD) and/or with other poor risk factors (e.g., an antecedent hematologic disorder or failure to attain remission with a single induction course). If a suitable HLA donor does not exist, investigational therapeutic approaches are considered. Indeed, postremission therapy is also a setting for introduction of new agents (Table 132-5). Because FLT3-ITD can be targeted with emerging novel inhibitors, patients with this molecular abnormality should be considered for clinical trials with these agents whenever possible.
Patients with the favorable CBF AML [i.e., t(8;21), inv(16), or t(16;16)] are treated with repetitive doses of high-dose cytarabine, which offers a high frequency of cure without the morbidity of transplant. Among AML patients with t(8;21) and inv(16), those with KIT mutations, who have a worse prognosis, may be considered for novel investigational studies, including tyrosine kinase inhibitors. The inclusion of gemtuzumab ozogamicin in induction and consolidation chemotherapy-based treatment has been reported to be beneficial in this subset of patients.
For patients in morphologic CR, immunophenotyping to detect minute populations of blasts or sensitive molecular assays (e.g., reverse transcriptase polymerase chain reaction [RT-PCR]) to detect AML-associated molecular abnormalities (e.g., NPM1 mutation, the CBF AML RUNX1/RUNX1T1 and CBFB/MYH11 transcripts, the APL PML/RARA transcript), and the less sensitive metaphase cytogenetics or interphase cytogenetics by fluorescence in situ hybridization (FISH) to detect AML-associated cytogenetic aberrations, can be performed to assess whether clinically meaningful minimal residual disease (MRD) is present at sequential time points during or after treatment. Detection of MRD may be a reliable discriminator between patients who will continue in CR and those who are destined to experience disease recurrence and therefore require early therapeutic intervention before clinical relapse occurs. Although assessment of MRD in bone marrow and/or blood during CR is routinely used in the clinic to anticipate clinical relapse and initiate timely salvage treatment for APL patients, for other cytogenetic and molecular subtypes of AML, this is an area of current investigation.
SUPPORTIVE CARE
Measures geared to supporting patients through several weeks of neutropenia and thrombocytopenia are critical to the success of AML therapy. Patients with AML should be treated in centers expert in providing supportive measures. Multilumen right atrial catheters should be inserted as soon as patients with newly diagnosed AML have been stabilized. They should be used thereafter for administration of intravenous medications and transfusions, as well as for blood drawing.
Adequate and prompt blood bank support is critical to therapy of AML. Platelet transfusions should be given as needed to maintain a platelet count ≥10,000/μL. The platelet count should be kept at higher levels in febrile patients and during episodes of active bleeding or DIC. Patients with poor posttransfusion platelet count increments may benefit from administration of platelets from HLA-matched donors. RBC transfusions should be administered to keep the hemoglobin level >80 g/L (8 g/dL) in the absence of active bleeding, DIC, or congestive heart failure, which require higher hemoglobin levels. Blood products leukodepleted by filtration should be used to avert or delay alloimmunization as well as febrile reactions. Blood products should also be irradiated to prevent transfusion-associated GVHD. Cytomegalovirus (CMV)-negative blood products should be used for CMV-seronegative patients who are potential candidates for allogeneic HSCT. Leukodepleted products are also effective for these patients if CMV-negative products are not available.
Neutropenia (neutrophils <500/μL or <1000/μL and predicted to decline to <500/μL over the next 48 h) can be part of the initial presentation and/or a side effect of the chemotherapy treatment in AML patients. Thus, infectious complications remain the major cause of morbidity and death during induction and postremission chemotherapy for AML. Antibacterial (i.e., quinolones) and antifungal (i.e., posaconazole) prophylaxis in the absence of fever is likely to be beneficial. For patients who are herpes simplex virus or varicella-zoster seropositive, antiviral prophylaxis should be initiated (e.g., acyclovir, valacyclovir).
Fever develops in most patients with AML, but infections are documented in only half of febrile patients. Early initiation of empirical broad-spectrum antibacterial and antifungal antibiotics has significantly reduced the number of patients dying of infectious complications (Chap. 104). An antibiotic regimen adequate to treat gram-negative organisms should be instituted at the onset of fever in a neutropenic patient after clinical evaluation, including a detailed physical examination with inspection of the indwelling catheter exit site and a perirectal examination, as well as procurement of cultures and radiographs aimed at documenting the source of fever. Specific antibiotic regimens should be based on antibiotic sensitivity data obtained from the institution at which the patient is being treated. Acceptable regimens for empiric antibiotic therapy include monotherapy with imipenem-cilastatin, meropenem, piperacillin/tazobactam, or an extended-spectrum antipseudomonal cephalosporin (cefepime or ceftazidime). The combination of an aminoglycoside with an antipseudomonal penicillin (e.g., piperacillin) or an aminoglycoside in combination with an extended-spectrum antipseudomonal cephalosporin should be considered in complicated or resistant cases. Aminoglycosides should be avoided if possible in patients with renal insufficiency. Empirical vancomycin should be added in neutropenic patients with catheter-related infections, blood cultures positive for gram-positive bacteria before final identification and susceptibility testing, hypotension or shock, or known colonization with penicillin/cephalosporin-resistant pneumococci or methicillin-resistant Staphylococcus aureus. In special situations where decreased susceptibility to vancomycin, vancomycin-resistant organisms, or vancomycin toxicity is documented, other options including linezolid, daptomycin, and quinupristin/dalfopristin need to be considered.
Caspofungin (or a similar echinocandin), voriconazole, or liposomal amphotericin B should be considered for antifungal treatment if fever persists for 4–7 days following initiation of empiric antibiotic therapy. Amphotericin B has long been used for antifungal therapy. Although liposomal formulations have improved the toxicity profile of this agent, its use has been limited to situations with high risk of or documented mold infections. Caspofungin has been approved for empiric antifungal treatment. Voriconazole has also been shown to be equivalent in efficacy and less toxic than amphotericin B. Antibacterial and antifungal antibiotics should be continued until patients are no longer neutropenic, regardless of whether a specific source has been found for the fever.
Recombinant hematopoietic growth factors have been incorporated into clinical trials in AML. These trials have been designed to lower the infection rate after chemotherapy. Both G-CSF and granulocyte-macrophage colony-stimulating factor (GM-CSF) have reduced the median time to neutrophil recovery. This accelerated rate of neutrophil recovery, however, has not generally translated into significant reductions in infection rates or shortened hospitalizations. In most randomized studies, both G-CSF and GM-CSF have failed to improve the CR rate, disease-free survival, or overall survival. Although receptors for both G-CSF and GM-CSF are present on AML blasts, therapeutic efficacy is neither enhanced nor inhibited by these agents. The use of growth factors as supportive care for AML patients is controversial. We favor their use in elderly patients with complicated courses, those receiving intensive postremission regimens, patients with uncontrolled infections, or those participating in clinical trials.
TREATMENT FOR REFRACTORY OR RELAPSED AML
With the 7 and 3 regimen, 65–75% of younger and 50–60% of older patients with primary AML achieve CR. Two-thirds achieve CR after a single course of therapy, and one-third require two courses. Of patients who do not achieve CR, approximately 50% have a drug-resistant leukemia, and 50% do not achieve CR because of fatal complications of bone marrow aplasia or impaired recovery of normal stem cells. Patients with refractory disease after induction should be considered for salvage treatments, preferentially on clinical trials, before receiving allogeneic HSCT usually administered in patients who achieve a disease-free status. Because these patients are usually not cured even if they achieve second CR with salvage chemotherapy, allogeneic HSCT is a necessary therapeutic step.
In patients who relapse after achieving CR, the length of first CR is predictive of response to salvage chemotherapy treatment; patients with longer first CR (>12 months) generally relapse with drug-sensitive disease and have a higher chance of attaining a CR, even with the same chemotherapeutic agents used for first remission induction. Whether initial CR was achieved with one or two courses of chemotherapy and the type of postremission therapy may also predict achievement of second CR. Similar to patients with refractory disease, patients with relapsed disease are rarely cured by the salvage chemotherapy treatments. Therefore, patients who eventually achieve a second CR and are eligible for allogeneic HSCT should be transplanted.
Because achievement of a second CR with routine salvage therapies is relatively uncommon, especially in patients who relapse rapidly after achievement of first CR (<12 months), these patients and those lacking HLA-compatible donors or who are not candidates for allogeneic HSCT should be considered for innovative approaches on clinical trials (Table 132-5). The discovery of novel gene mutations and mechanisms of leukemogenesis that might represent actionable therapeutic targets has prompted the development of new targeting agents. In addition to kinase inhibitors for FLT3– and KIT-mutated AML, other compounds targeting the aberrant activity of mutant proteins (e.g., IDH2 inhibitors) or biologic mechanisms deregulating epigenetics (e.g., histone deacetylase and DNA methyltransferase inhibitors), cell proliferation (e.g., farnesyl transferase inhibitors), protein synthesis (e.g., aminopeptide inhibitors) and folding (e.g., heat shock protein inhibitors), and ubiquitination, or with novel cytotoxic mechanisms (e.g., clofarabine, sapacitabine), are being tested in clinical trials. Furthermore, approaches with antibodies targeting commonly expressed leukemia blasts (e.g., CD33) or leukemia initiating cells (e.g., CD123) and immunomodulatory agents (e.g., lenalidomide) are also under investigation. Once these compounds have demonstrated safety and activity as single agents, investigation of combinations with other molecular targeting compounds and/or chemotherapy should be pursued.
TREATMENT OF ACUTE PROMYELOCYTIC LEUKEMIA
APL is a highly curable subtype of AML, and approximately 85% of these patients achieve long-term survival with current approaches. APL has long been shown to be responsive to cytarabine and daunorubicin, but previously patients treated with these drugs alone frequently died from DIC induced by the release of granule components by the chemotherapy-treated leukemia cells. However, the prognosis of APL patients has changed dramatically from adverse to favorable with the introduction of tretinoin, an oral drug that induces the differentiation of leukemic cells bearing the t(15;17), where disruption of the RARA gene encoding a retinoid acid receptor occurs. Tretinoin decreases the frequency of DIC but produces another complication called the APL differentiation syndrome. Occurring within the first 3 weeks of treatment, it is characterized by fever, fluid retention, dyspnea, chest pain, pulmonary infiltrates, pleural and pericardial effusions, and hypoxemia. The syndrome is related to adhesion of differentiated neoplastic cells to the pulmonary vasculature endothelium. Glucocorticoids, chemotherapy, and/or supportive measures can be effective for management of the APL differentiation syndrome. Temporary discontinuation of tretinoin is necessary in cases of severe APL differentiation syndrome (i.e., patients developing renal failure or requiring admission to the intensive care unit due to respiratory distress). The mortality rate of this syndrome is about 10%.
Tretinoin (45 mg/m2 per day orally until remission is documented) plus concurrent anthracycline-based (i.e., idarubicin or daunorubicin) chemotherapy appears to be among the most effective treatment for APL, leading to CR rates of 90–95%. The role of cytarabine in APL induction and consolidation is controversial. The addition of cytarabine, although not demonstrated to increase the CR rate, seemingly decreases the risk for relapse. Following achievement of CR, patients should receive at least two cycles of anthracycline-based chemotherapy.
Arsenic trioxide has significant antileukemic activity and is being explored as part of initial treatment in clinical trials of APL. In a randomized trial, arsenic trioxide improved outcome if used after achievement of CR and before consolidation therapy with anthracycline-based chemotherapy. Patients receiving arsenic trioxide are at risk of APL differentiation syndrome, especially when it is administered during induction or salvage treatment after disease relapse. In addition, arsenic trioxide may prolong the QT interval, increasing the risk of cardiac arrhythmias.
Given the progress made in APL resulting in high cure rates, in recent years the goal has been to identify patients with low risk of relapse (i.e., those presenting with a leukocyte count ≤10,000/μL) where attempts are being made to decrease the amount of therapy administered and to identify patients at greatest risk of relapse (i.e., those presenting with a leukocyte count ≥10,000/μL) where new approaches can be developed to increase cure. A study compared the gold standard (tretinoin plus chemotherapy) in newly diagnosed non-high-risk APL with a chemotherapy-free combination of tretinoin and arsenic trioxide. An equivalent outcome was demonstrated between the two arms, and the chemotherapy-free regimen will likely become a new standard for non-high-risk APL patients.
Combinations of tretinoin, arsenic trioxide, and/or chemotherapy and/or gemtuzumab ozogamicin have shown favorable responses in high-risk APL patients at diagnosis.
Assessment of residual disease by RT-PCR amplification of the t(15;17) chimeric gene product PML-RARA following the final cycle of chemotherapy is an important step in the management of APL patients. Disappearance of the signal is associated with long-term disease-free survival; its persistence documented by two consecutive tests performed 2 weeks apart invariably predicts relapse. Sequential monitoring of RT-PCR for PML-RARA is now considered standard for postremission monitoring of APL, especially in high-risk patients.
The benefit from maintenance therapy with tretinoin has been documented in some studies and not in others. Thus, the use of tretinoin depends on which regimen has been used for induction and consolidation treatment and the risk category of the patients, with those with high-risk disease seemingly benefiting the most from maintenance therapy.
Patients in molecular, cytogenetic, or clinical relapse should be salvaged with arsenic trioxide with or without tretinoin; it produces meaningful responses in up to 85% of patients and can be followed by autologous or, less frequently, especially if RT-PCR positive for PML-RARA, allogeneic HSCT.
133 |
Chronic Myeloid Leukemia |
Chronic myeloid leukemia (CML) is a clonal hematopoietic stem cell disorder. The disease is driven by the BCR-ABL1 chimeric gene product, a constitutively active tyrosine kinase, resulting from a reciprocal balanced translocation between the long arms of chromosomes 9 and 22, t(9;22) (q34;q11.2), cytogenetically detected as the Philadelphia chromosome (Ph) (Fig. 133-1). Untreated, the course of CML may be biphasic or triphasic, with an early indolent or chronic phase, followed often by an accelerated phase and a terminal blastic phase. Before the era of selective BCR-ABL1 tyrosine kinase inhibitors (TKIs), the median survival in CML was 3–7 years, and the 10-year survival rate was 30% or less. Introduced into CML therapy in 2000, TKIs have revolutionized the treatment, natural history, and prognosis of CML. Today, the estimated 10-year survival rate with imatinib mesylate, the first BCR-ABL1 TKI approved, is 85%. Allogeneic stem cell transplantation (SCT), a curative but risky treatment approach, is now offered as second- or third-line therapy after failure of TKIs.
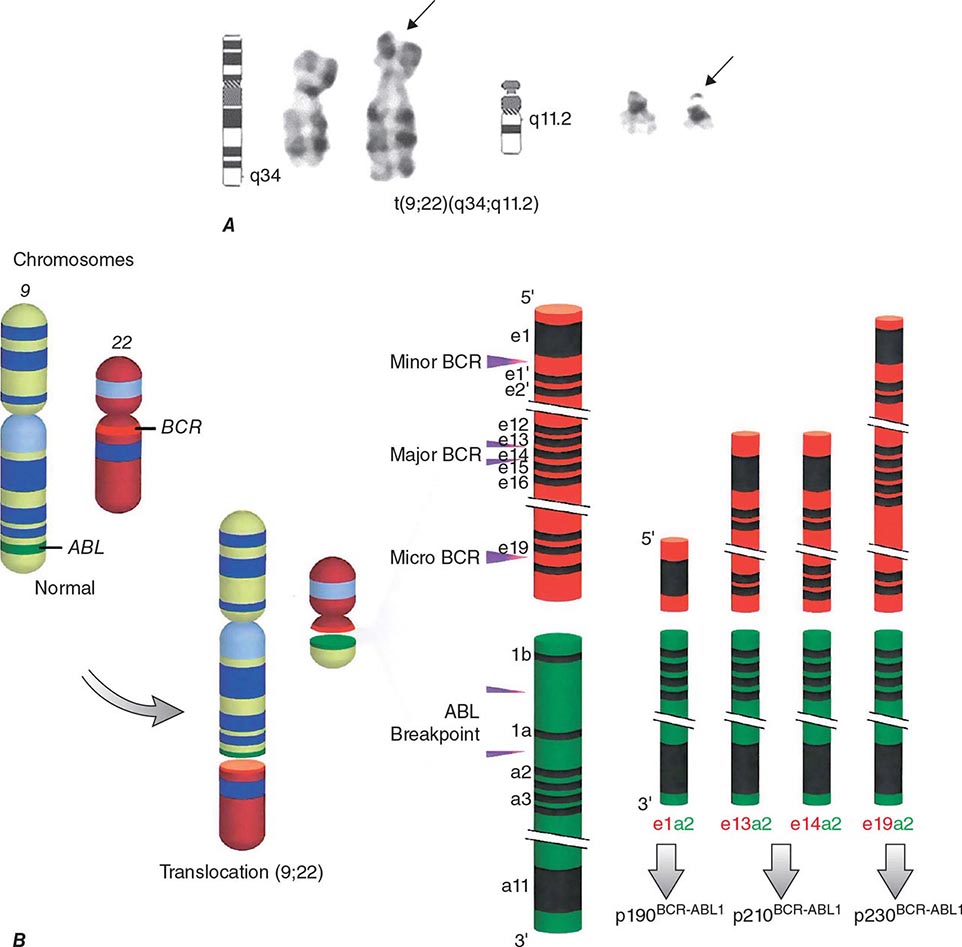
FIGURE 133-1 A. The Philadelphia (Ph) chromosome cytogenetic abnormality. B. Breakpoints in the long arms of chromosome 9 (ABL locus) and chromosome 22 (BCR regions) result in three different BCR-ABL oncoprotein messages, p210BCR-ABL1 (most common message in chronic myeloid leukemia [CML]), p190BCR-ABL1 (present in two-thirds of patients with Ph-positive acute lymphocytic leukemia; rare in CML), and p230BCR-ABL1 (rare in CML and associated with an indolent course). (© 2013 The University of Texas MD Anderson Cancer Center.)
INCIDENCE AND EPIDEMIOLOGY
CML accounts for 15% of all cases of leukemia. There is a slight male preponderance (male:female ratio 1.6:1). The median age at diagnosis is 55–65 years. It is uncommon in children; only 3% of patients with CML are younger than 20 years. CML incidence increases slowly with age, with a steeper increase after the age of 40–50 years. The annual incidence of CML is 1.5 cases per 100,000 individuals. In the United States, this translates into 4500–5000 new cases per year. The incidence of CML has not changed over several decades. By extrapolation, the worldwide annual incidence of CML is about 100,000 cases. With a median survival of 6 years before 2000, the disease prevalence in the United States was 20,000–30,000 cases. With TKI therapy, the annual mortality has been reduced from 10–20% to about 2%. Therefore, the prevalence of CML in the United States is expected to continue to increase (about 80,000 in 2013) and reach a plateau of approximately 180,000 cases around 2030. The worldwide prevalence will depend on the treatment penetration of TKIs and their effect on reduction of worldwide annual mortality. Ideally, with full TKI treatment penetration, the worldwide prevalence should plateau at 35 times the incidence, or around 3 million patients.
ETIOLOGY
There are no familial associations in CML. The risk of developing CML is not increased in monozygotic twins or in relatives of patients. No etiologic agents are incriminated, and no associations exist with exposures to benzene or other toxins, fertilizers, insecticides, or viruses. CML is not a frequent secondary leukemia following therapy of other cancers with alkylating agents and/or radiation. Exposure to ionizing radiation (e.g., nuclear accidents, radiation treatment for ankylosing spondylitis or cervical cancer) has increased the risk of CML, which peaks at 5–10 years after exposure and is dose-related. The median time to development of CML among atomic bomb survivors was 6.3 years. Following the Chernobyl accident, the incidence of CML did not increase, suggesting that only large doses of radiation can cause CML. Because of adequate protection, the risk of CML is not increased in individuals working in the nuclear industry or among radiologists in recent times.
PATHOPHYSIOLOGY
The t(9;22) (q34;q11.2) is present in more than 90% of classical CML cases. It results from a balanced reciprocal translocation between the long arms of chromosomes 9 and 22. It is present in hematopoietic cells (myeloid, erythroid, megakaryocytes, and monocytes; less often mature B lymphocytes; rarely mature T lymphocytes, but not stromal cells), but not in other cells in the human body. As a result of the translocation, DNA sequences from the cellular oncogene ABL1 are translocated next to the major breakpoint cluster region (BCR) gene on chromosome 22, generating a hybrid oncogene, BCR-ABL1. This fusion gene codes for a novel oncoprotein of molecular weight 210 kDa, referred to as p210BCR-ABL1 (Fig. 133-1B). This BCR-ABL1 oncoprotein exhibits constitutive kinase activity that leads to excessive proliferation and reduced apoptosis of CML cells, endowing them with a growth advantage over their normal counterparts. Over time, normal hematopoiesis is suppressed, but normal stem cells can persist and may reemerge following effective therapy, for example with TKIs. In Ph-positive acute lymphocytic leukemia (ALL) and in rare cases of CML, the breakpoint in BCR is more centromeric, in a region called the minor BCR region (mBCR). As a result, a shorter sequence of BCR is fused to ABL1, with a consequent smaller BCR-ABL1 oncoprotein, p190BCR-ABL1. When occurring in Ph-positive CML, this translocation may predict for a worse outcome. A third rare breakpoint in BCR occurs telomeric to the major BCR region and is called micro-BCR (μ-BCR). It juxtaposes a larger fragment of the BCR gene to ABL1 and produces a larger p230BCR-ABL1 oncoprotein, which is associated with a more indolent CML course.
The constitutive activation of BCR-ABL1 results in autophosphorylation and activation of multiple downstream pathways that modify gene transcription, apoptosis, skeletal organization, and degradation of inhibitory proteins. These transduction pathways may involve RAS, mitogen-activated protein (MAP) kinases, signal transducers and activators of transcription (STAT), phosphatidylinositol-3-kinase (PI3k), MYC, and others. These interactions are mostly mediated through tyrosine phosphorylation and require binding of BCR-ABL1 to adapter proteins such as GRB-2, CRK, CRK-like (CRK-L) protein, and Src homology containing proteins (SHC). BCR-ABL1 TKIs bind to the BCR-ABL1 kinase domain (KD), preventing the activation of transformation pathways and inhibiting downstream signaling. As a result, proliferation of CML cells is inhibited and apoptosis induced, leading to the reemergence of normal hematopoiesis. A plethora of signaling pathways have been implicated in BCR-ABL1-mediated cellular transformation. The emerging picture is a complex and redundant transformation network. An additional layer of complexity is related to differences in signal transduction between CML differentiated cells and early progenitors. Beta-catenin, Wnt1, Foxo3a, transforming growth factor β, interleukin-6, PP2A, SIRT1, and others have been implicated in CML stem cell survival.
Experimental models have established the causal relationship between the Ph-related BCR-ABL1 molecular events and the development of CML. In animal models, expression of BCR-ABL1 in normal hematopoietic cells produced CML-like disorders or lymphoid leukemia, demonstrating the leukemogenic potential of BCR-ABL1 as a single oncogenic abnormality.
The cause of the BCR-ABL1 molecular rearrangement is unknown. Molecular techniques that detect BCR-ABL1 at a level of 1 in 108 identify this molecular abnormality in the blood of up to 25% of normal adults and 5% of infants, but 0% of cord blood samples. This suggests that BCR-ABL1 is not sufficient to cause overt CML in the overwhelming majority of individuals in whom it occurs. Because CML develops in only 1.5 of 100,000 individuals annually, it is evident that additional molecular events or poor immune recognition of the rearranged cells are needed to cause overt CML.
CML is defined by the presence of BCR-ABL1 abnormality in a patient with a myeloproliferative neoplasm. In some patients with a typical morphologic picture of CML, the Ph abnormality is not detectable by standard cytogenetic analysis, but fluorescence in situ hybridization (FISH) and molecular studies (polymerase chain reaction [PCR]) detect BCR-ABL1. These patients have a course similar to Ph-positive CML and respond to TKI therapy. Many of the remaining patients have atypical morphologic or clinical features and belong to other diagnostic groups, such as atypical CML or chronic myelomonocytic leukemia. These individuals do not respond to TKI therapy and have a poor prognosis with a median survival of about 2–3 years. Detection of mutations in the granulocyte colony-stimulating factor receptor (CSF3R) in chronic neutrophilic leukemia and in some cases of atypical CML and of mutations in SETBP1 in atypical CML confirmed that they are distinct entities.
The mechanisms associated with the transition of CML from a chronic to accelerated-blastic phase are poorly understood. They are often associated with characteristic chromosomal abnormalities such as a double Ph, trisomy 8, isochromosome 17 or deletion of 17p (loss of TP53), 20q–, and others. Molecular events associated with transformation include mutations in TP53, retinoblastoma 1 (RB1), myeloid transcriptions factors like Runx1, and cell cycle regulators like p16. A plethora of other mutations or functional abnormalities have been implicated in blastic transformation, but no unifying theme has emerged other than that BCR-ABL1 itself induces genetic instability that leads to the acquisition of additional mutations and eventually to blastic transformation. In this frame of thinking, one critical effect of TKIs is their ability to stabilize the CML genome, leading to a much reduced transformation rate. In particular, the previously observed sudden blastic transformations (i.e., abrupt transformation to blastic phase in a patient who had been in cytogenetic response) have become uncommon, occurring rarely in younger patients in the first 1–2 years of TKI therapy (usually sudden lymphoid blastic transformations). Sudden transformations beyond the third year of TKI therapy are rare in patients who continue on TKI therapy. Moreover, initial experience suggests that the course of CML has become significantly more indolent, even without cytogenetic responses, in patients on TKI-based therapy compared to previous experience with hydroxyurea/busulfan.
Among patients developing resistance to TKIs, several resistance mechanisms have been observed. The most clinically relevant one is the development of different ABL1 kinase domain mutations that prevent the binding of TKIs to the catalytic site (ATP binding site) of the kinase. More than 100 BCR-ABL1 mutations have now been described, many of which confer relative or absolute resistance to imatinib. This has resulted in the development of second-generation TKIs (i.e., dasatinib, nilotinib, bosutinib) and of a third-generation TKI (ponatinib) with selective efficacy against T315I, a mutation of the gatekeeper residue of the kinase that causes resistance to all other TKIs.
CLINICAL PRESENTATION
The presenting signs and symptoms in CML depend on the availability of and access to health care procedures, including physical exams and screening tests. In the United States, because of the easy access to health care screening and physical exams, 50–60% of patients are diagnosed on routine blood tests and have minimal symptoms at presentation, such as fatigue. In geographic locations where access to health care is more limited, patients often present with high CML burden including splenomegaly, anemia, and related symptoms (abdominal pain, weight loss, fatigue), as well as a higher frequency of high-risk CML. Presenting findings in patients diagnosed in the United States are shown in Table 133-1.
|
PRESENTING SIGNS AND SYMPTOMS OF NEWLY DIAGNOSED PHILADELPHIA CHROMOSOME–POSITIVE CHRONIC MYELOID LEUKEMIA IN CHRONIC PHASE |
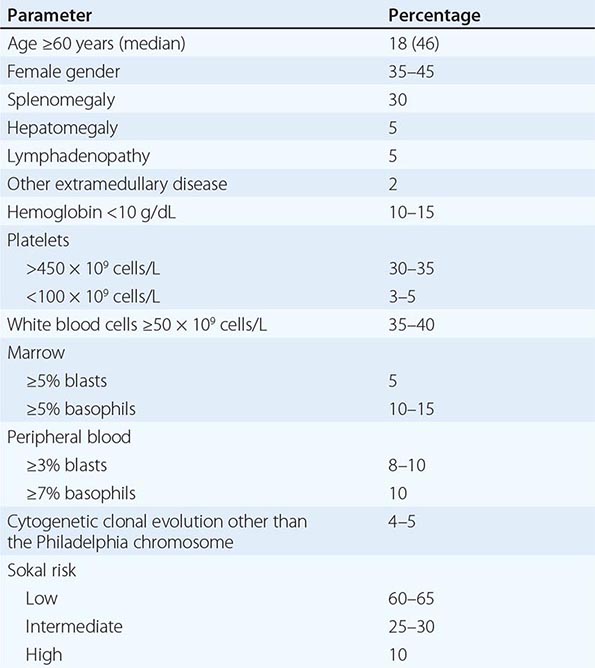
Symptoms Most patients with CML (90%) present in the indolent or chronic phase. Depending on the timing of diagnosis, patients are often asymptomatic (if the diagnosis is discovered during health care screening tests). Common symptoms, when present, are manifestations of anemia and splenomegaly. These may include fatigue, malaise, weight loss (if high leukemia burden), or early satiety and left upper quadrant pain or masses (from splenomegaly). Less common presenting findings include thrombotic or vasoocclusive events (from severe leukocytosis or thrombocytosis). These include priapism, cardiovascular complications, myocardial infarction, venous thrombosis, visual disturbances, dyspnea and pulmonary insufficiency, drowsiness, loss of coordination, confusion, or cerebrovascular accidents. Bleeding diatheses findings include retinal hemorrhages, gastrointestinal bleeding, and others. Patients who present with, or progress to, the accelerated or blastic phases have additional symptoms including unexplained fever, significant weight loss, severe fatigue, bone and joint aches, bleeding and thrombotic events, and infections.
Physical Findings Splenomegaly is the most common physical finding, occurring in 20–70% of patients depending on health care screening frequency. Other less common findings include hepatomegaly (10–20%), lymphadenopathy (5–10%), and extramedullary disease (skin or subcutaneous lesions). The latter indicates CML transformation if a biopsy confirms the presence of sheets of blasts. Other physical findings are manifestations of complications of high tumor burden described earlier (e.g., cardiovascular, cerebrovascular, bleeding). High basophil counts may be associated with histamine overproduction causing pruritus, diarrhea, flushing, and even gastrointestinal ulcers.
Hematologic and Marrow Findings In untreated CML, leukocytosis ranging from 10–500 × 109/L is common. The peripheral blood differential shows left-shifted hematopoiesis with predominance of neutrophils and the presence of bands, myelocytes, metamyelocytes, promyelocytes, and blasts (usually ≤5%). Basophils and/or eosinophils are frequently increased. Thrombocytosis is common, but thrombocytopenia is rare and, when present, suggests a worse prognosis, disease acceleration, or an unrelated etiology. Anemia is present in one-third of patients. Cyclic oscillations of counts are noted in 25% of patients without treatment. Biochemical abnormalities include a low leukocyte alkaline phosphatase score and high levels of vitamin B12, uric acid, lactic dehydrogenase, and lysozyme. The presence of unexplained and sustained leukocytosis, with or without splenomegaly, should lead to a marrow examination and cytogenetic analysis.
The bone marrow is hypercellular with marked myeloid hyperplasia and a high myeloid-to-erythroid ratio of 15–20:1. Marrow blasts are 5% or less; when higher, they carry a worse prognosis or represent acceleration (if they are ≥15%). Increased reticulin fibrosis (by Snook’s silver stain) is common, with 30–40% of patients demonstrating grade 3–4 reticulin fibrosis. This was considered adverse in the pre-TKI era. With TKI therapy, reticulin fibrosis resolves in most patients and is not an indicator of poor prognosis. Collagen fibrosis (Wright-Giemsa stain) is rare at diagnosis. Disease progression with a “spent phase” of myelofibrosis (myelophthisis, or burnt-out marrow) was common with busulfan therapy (20–30%) but is rare with TKI therapy.
Cytogenetic and Molecular Findings The diagnosis of CML is straightforward and depends on documenting t(9;22)(q34;q11.2), which is found in 90% of cases. This is known as the Philadelphia-chromosome abnormality (discovered in Philadelphia) and was initially identified as a shortened chromosome, later identified to be chromosome 22 (22q–) (Fig. 133-1). Some patients may have complex translocations (variant Ph) involving three or more translocations that include chromosomes 9 and 22 and one or more other chromosomes. Others may have a “masked Ph,” involving translocations between chromosome 9 and a chromosome other than 22. The prognosis of these patients and their response to TKI therapy are similar to those in patients with Ph. About 5–10% of patients may have additional chromosomal abnormalities in the Ph-positive cells. These usually involve trisomy 8, a double Ph, isochromosome 17 or 17p deletion, 20q–, or others. This is referred to as clonal evolution and was historically a sign of adverse prognosis, particularly when trisomy 8, double Ph, or chromosome 17 abnormalities were noted.
Techniques such as FISH and PCR are now used to aid in the diagnosis of CML. They are more sensitive approaches to estimate the CML burden in patients on TKI therapy. They can be done on peripheral samples, and thus are less painful and more convenient. Patients with CML at diagnosis should have a FISH analysis to quantify the percentage of Ph-positive cells, if FISH is used to replace marrow cytogenetic analysis in monitoring response to therapy. FISH may not detect additional chromosomal abnormalities (clonal evolution); thus, a cytogenetic analysis is usually recommended at the time of diagnosis. The BCR-ABL1 RNA message is usually one of two variants: e13a2 (formerly b2a2) and e14a2 (formerly b3a2). About 2–5% of patients may have other RNA fusion types (e.g., e1a2, e13a3, or e14a3). In these patients, the routine PCR primers may not amplify the BCR-ABL1 transcripts, thus leading to false-negative results. Therefore, molecular studies at diagnosis are important to document the type and presence of BCR-ABL1 transcripts to avoid erroneously “undetectable” BCR-ABL1 transcripts on follow-up studies, with the misconception of a complete molecular response.
Both FISH and PCR studies can be falsely positive at low levels or falsely negative because of technical issues. Therefore, a diagnosis of CML must always rely on a marrow analysis with routine cytogenetics. The diagnostic bone marrow confirms the presence of the Ph chromosome, detects clonal evolution, i.e., chromosomal abnormalities in the Ph-positive cells (which may be prognostic), and also quantifies the percentage of marrow blasts and basophils. In 10% of patients, the percentage of marrow blasts and basophils can be significantly higher than in the peripheral blood, suggesting poorer prognosis or even disease transformation.
Monitoring patients on TKI therapy by cytogenetics, FISH, and molecular studies has become an important standard practice to assess response to therapy, emphasize compliance, evaluate possible treatment resistance, change TKI therapy, and order mutational analysis studies. It is thus important to recognize the comparability of these measures in monitoring response. A partial cytogenetic response is defined as the presence of 35% less Ph-positive metaphases by routine cytogenetic analysis. This is roughly equivalent to BCR-ABL1 transcripts by the International Scale (IS) of 10% or less. A complete cytogenetic response refers to the absence of Ph-positive metaphases (0% Ph positivity). This is approximately equivalent to BCR-ABL1 transcripts (IS) of 1% or less. A major molecular response refers to BCR-ABL1 transcripts (IS) ≤0.1%, or roughly a 3-log or greater reduction of CML burden from baseline. A complete molecular response usually refers to BCR-ABL1 transcripts (IS) <0.0032% (undetectable by current techniques), roughly equivalent to a more than 4.5-log reduction of CML burden from baseline.
Findings in CML Transformation Progression of CML is usually associated with leukocytosis resistant to therapy, increasing anemia, fever and constitutional symptoms, and increased blasts and basophils in the peripheral blood or marrow. Criteria of accelerated-phase CML, historically associated with median survival of less than 1.5 years, include the presence of 15% or more peripheral blasts, 30% or more peripheral blasts plus promyelocytes, 20% or more peripheral basophils, cytogenetic clonal evolution (presence of chromosomal abnormalities in addition to Ph), and thrombocytopenia <100 × 109/L (unrelated to therapy). About 5–10% of patients present with de novo accelerated phase or blastic phase. The prognosis of de novo accelerated phase with TKI therapy has improved significantly, with an estimated 8-year survival rate of 75%. The median survival of accelerated phase evolving from chronic phase has also improved from a historical median survival of 18 months to an estimated 4-year survival rate of 70% on TKI therapy. Therefore, the criteria for accelerated-phase CML should be revisited because most have lost much of their prognostic significance. Blastic-phase CML is defined by the presence of 30% or more peripheral or marrow blasts or the presence of sheets of blasts in extramedullary disease (usually skin, soft tissues, or lytic bone lesions). Blastic-phase CML is commonly myeloid (60%) but can present uncommonly as erythroid, promyelocytic, monocytic, or megakaryocytic. Lymphoid blastic phase occurs in about 25% of patients. Lymphoblasts are terminal deoxynucleotide transferase positive and peroxidase negative (although occasionally with low positivity up to 3–5%) and express lymphoid markers (CD10, CD19, CD20, CD22). However, they also often express myeloid markers (50–80%), resulting in diagnostic confusion. This is important because, unlike other morphologic blastic phases, lymphoid blastic-phase CML is quite responsive to anti-ALL-type chemotherapy (e.g., hyper-CVAD [cyclophosphamide, vincristine, doxorubicin, and dexamethasone]) in combination with TKIs.
PROGNOSIS AND CML COURSE
Before the imatinib era, the annual mortality in CML was 10% in the first 2 years and 15–20% thereafter. The median survival time in CML was 3–7 years (with hydroxyurea-busulfan and interferon α). Without a curative option of allogeneic SCT, the course of CML was inexorable toward transformation to, and death from, accelerated or blastic phases. The disease stability was unpredictable, with some patients demonstrating sudden transformation to a blastic phase. With imatinib therapy, the annual mortality in CML has decreased to 2% in the first 12 years of observation. Half of the deaths are from factors other than CML, such as old age, accidents, suicides, other cancers, and other medical conditions (e.g., infections, surgical procedures). The estimated 8- to 10-year survival rate is now 85%, or 93% if only CML-related deaths are considered (Fig. 133-2). The course of CML has also become quite predictable. In the first 2 years of TKI therapy, rare sudden transformations are still noted (1–2%), usually lymphoid blastic transformations that respond to combinations of chemotherapy and TKIs followed by allogeneic SCT. These may be explained by the intrinsic mechanisms of sudden transformation already existing in the CML clones before the start of therapy that were not amenable to TKI inhibition, in particular imatinib. Second-generation TKIs (nilotinib, dasatinib) used as frontline therapy have reduced the incidence of transformation in the first 2–3 years from 6–8% with imatinib to 2–4% with nilotinib or dasatinib. Disease transformation to accelerated or blastic phase is rare on continued TKI therapy, estimated at <1% annually in years 4–8 of follow-up on the original imatinib trials. Patients usually develop resistance in the form of cytogenetic relapse, followed by hematologic relapse and subsequent transformation, rather than the previously feared sudden transformations without the warning signals of cytogenetic-hematologic relapse.
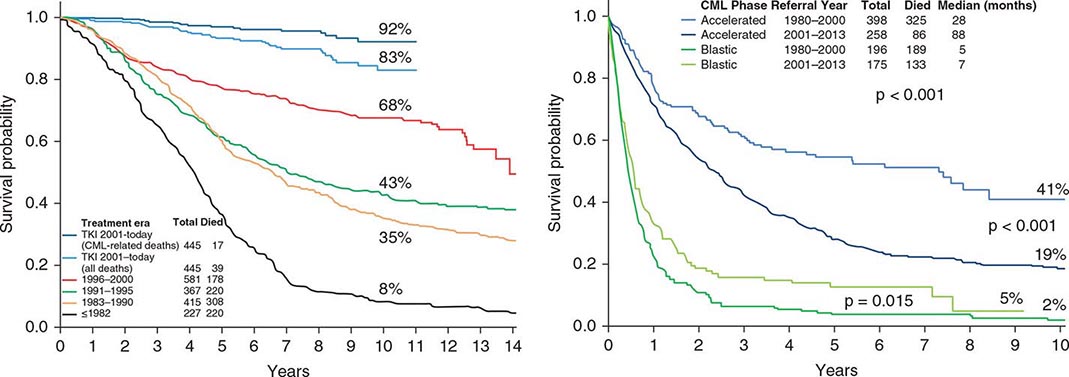
FIGURE 133-2 A. Survival in newly diagnosed chronic-phase chronic myeloid leukemia (CML) by era of therapy (M.D. Anderson Cancer Center experience from 1965 to present). Causes of non-CML deaths in 22 patients were other cancers (n = 7), postsurgical complications (n = 3), car accident (n = 2), suicide (n = 1), neurologic events (n = 3), cardiac (n = 3), pneumonia (n = 1), and unknown (n = 2). B. Survival in patients with accelerated- and blastic-phase CML referred to M.D. Anderson Cancer Center by era of therapy, demonstrating the significant survival benefit in the tyrosine kinase inhibitor (TKI) era in accelerated-phase CML but the modest benefit in blastic-phase CML. Referred cases included de novo and post-chronic-phase transformations.
Before the imatinib era, several pretreatment prognostic factors predicted for worse outcome in CML and have been incorporated into prognostic models and staging systems. These have included older age, significant splenomegaly, anemia, thrombocytopenia or thrombocytosis, high percentages of blasts and basophils (and/or eosinophils), marrow fibrosis, deletions in the long arm of chromosome 9, clonal evolution, and others. Different risk models and staging systems, derived from multivariate analyses, were proposed to define different risk groups. As with the introduction of cisplatin into testicular cancer therapy, the introduction of TKIs into CML therapy has nullified or lessened the prognostic impact of most of these prognostic factors and the significance of the CML models (e.g., Sokal, Hasford, European Treatment and Outcome Study [EUTOS]). Treatment-related prognostic factors have emerged as the most important prognostic factors in the era of imatinib therapy. Achievement of complete cytogenetic response has become the major therapeutic endpoint and is the only endpoint associated with improvement in survival. Achievement of a major molecular response is associated with decreased risk of events (relapse) and CML progression, may predict for differences in event-free survival (depending on the definition of an event) and for small differences in transformation rates, but has not been associated with survival prolongation. Among patients in complete cytogenetic response, survival is similar independent of whether they achieve a major molecular response or not. This may be due to the efficacy of salvage TKI therapies, which are and should be implemented at the first evidence of cytogenetic relapse. Achievement of complete molecular response (undetectable BCR-ABL1 transcripts), particularly when durable (>2 years), may offer the possibility of durable molecular response (molecular cure rather than functional cure) in the context of investigational trials and may allow temporary therapy interruption in women eager to have babies. The lack of achievement of major or complete molecular responses should not be considered as “failure” of a particular TKI therapy and/or an indication to change the TKI or to consider allogeneic SCT.
Pretreatment prognostic factors and prognostic models have lost much of their clinical relevance to define prognosis and to select different therapies. However TKI-associated therapeutic responses have gained major clinical relevance and dictate appropriate and careful monitoring of patients to optimize their treatment.
TREATMENT CHRONIC MYELOID LEUKEMIA
The introduction of TKI therapy, first in the form of imatinib mesylate in 2001, has revolutionized the treatment and prognosis in CML. Before 2000, allogeneic SCT was frontline therapy, when available, because of its potentially curative capacity. Otherwise, patients were offered interferon α therapy (approved for the treatment of CML in 1986), which had modest benefits (improving survival from a median of 3–4 years with hydroxyurea-busulfan to a median of 6–7 years), but also significant side effects. Other alternatives included hydroxyurea, busulfan, and other nonspecific chemotherapies. With TKI therapy, the estimated 10-year survival in CML is 85%. Since 2001, six agents have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of CML. These include five oral BCR-ABL1-selective TKIs: imatinib (Gleevec), nilotinib (Tasigna), dasatinib (Sprycel), bosutinib (Bosulif), and ponatinib (Iclusig). Imatinib 400 mg orally daily, nilotinib 300 mg orally twice a day (on an empty stomach), and dasatinib 100 mg orally daily are approved for frontline therapy of CML. All three are also approved for salvage therapy (nilotinib 400 mg twice daily), in addition to bosutinib (500 mg daily) and ponatinib (45 mg daily). Imatinib, dasatinib (140 mg daily), bosutinib, and ponatinib are also approved for the treatment of CML transformation (accelerated and blastic phase), whereas nilotinib is only approved for chronic and accelerated phase. Dasatinib, nilotinib, and bosutinib are referred to as second-generation TKIs; ponatinib is referred to as a third-generation TKI. The sixth approved agent is omacetaxine (Synribo), a protein synthesis inhibitor with presumed more selective inhibition of the synthesis of the BCR-ABL1 oncoprotein. It is approved for the treatment of chronic- and accelerated-phase CML after failure of two or more TKIs, at 1.25 mg/m2 subcutaneously twice a day for 14 days for induction and for 7 days for consolidation-maintenance. Nilotinib is similar in structure to imatinib but 30 times more potent. Dasatinib and bosutinib are dual SRC-ABL1 TKIs (dasatinib is reported to be 300 times more potent and bosutinib 30–50 times more potent than imatinib). Ponatinib is effective against wild-type and mutant BCR-ABL1 clones. It is unique in being the only currently available BCR-ABL1 TKI that is active against T315I, a gatekeeper mutant resistant to the other four TKIs (Table 133-2).
|
MEDICAL THERAPEUTIC OPTIONS IN CHRONIC MYELOID LEUKEMIA |
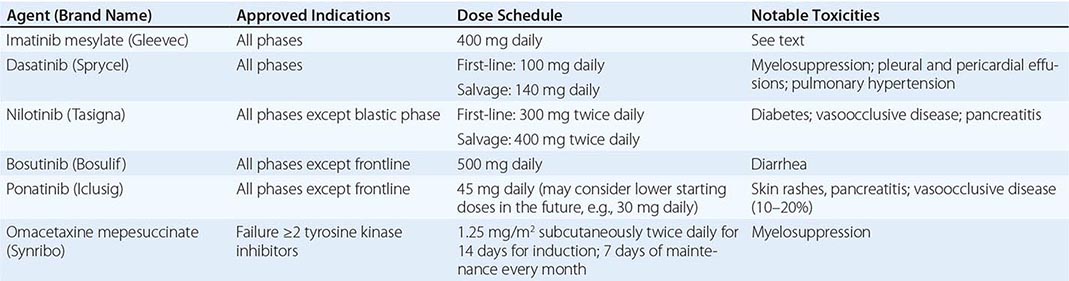
Imatinib, nilotinib, and dasatinib are all acceptable frontline therapies in CML. The long-term results of imatinib are very favorable. The 8-year follow-up results show a cumulative complete cytogenetic response rate (occurring at least once) of 83%, with 60–65% of patients being in complete cytogenetic response at 5-year follow-up. The estimated 8-year event-free survival rate is 81%, and the overall survival rate is 85%. Among patients continuing on imatinib, the annual rate of transformation to accelerated-blastic phase in years 4–8 is <1%. In two randomized studies, one comparing nilotinib 300 mg twice daily or 400 mg twice daily with imatinib (ENEST-nd) and the other comparing dasatinib 100 mg daily with imatinib (DASISION), the second-generation TKIs were associated with better outcomes in early surrogate endpoints, including higher rates of complete cytogenetic responses (85–87% vs 77–82%), major molecular responses (65–76% vs 46–63%), and undetectable BCR-ABL1 transcripts (IS) (32–37% vs 15–30%), and lower rates of transformation to accelerated-blastic phase (2–4% vs 6%). However, neither study showed a survival benefit with second-generation TKIs (median follow-up times of 4–5 years). This may be because salvage therapy with other TKIs (following close observation and treatment change at progression) provides highly effective salvage therapy that rebalances the negative effect of the relapse.
Salvage therapy in chronic phase with dasatinib, nilotinib, bosutinib, or ponatinib is associated with complete cytogenetic response rates of 30–60% of patients, depending on the salvage status (cytogenetic vs hematologic relapse), prior response to other TKIs, and the mutations at the time of relapse. Complete cytogenetic responses are generally durable, particularly in the absence of clonal evolution and mutations. Ponatinib is the only TKI active in the setting of T315I mutation, with complete cytogenetic response rates of 50–70%. The estimated 3- to 5-year survival rates with new TKIs as salvage are 70–80% (compared with <50% before their availability). For example, with dasatinib salvage after imatinib failure in chronic-phase CML, the major molecular response rates were 40–43%, the estimated 6-year survival rates were 74–83%, and progression-free survival rates were 40–51%. Thus, TKIs in the salvage setting have already reduced the annual mortality from the historical rate of 10–15% to ≤5%.
The goal of CML therapy is viewed differently in the context of research versus standard practice. In current practice, functional cure, defined as survival with CML similar to survival among normal individuals, is the current goal of therapy. CML is now considered an indolent disease, which, with appropriate TKI therapy, treatment compliance, careful monitoring, and early change to other TKIs as indicated, can be associated with close to normal survival. Therefore, in standard practice, achievement and maintenance of a complete cytogenetic response are the aims of therapy, because complete cytogenetic response is the only treatment-related factor associated with survival prolongation. Lack of achievement of a major molecular response (protects against events; associated with longer event-free survival) or of negative BCR-ABL1 transcripts (offers the potential of TKI interruption on investigational studies) should not be considered indications to change TKI therapy or to consider allogeneic SCT. A general practice rule is to continue the particular TKI chosen at the most tolerable dose schedule not associated with grade 3–4 side effects or with bothersome chronic side effects, for as long as possible, until either cytogenetic relapse or the persistence of unacceptable side effects. These two factors (i.e., cytogenetic relapse and intolerable side effects as judged by the patient and treating physician) are the indicators of “failure” of a particular TKI therapy. Because of the increasing prevalence of CML (cost of TKI therapy) and the emerging long-term low rates of significant organ toxicities, the ultimate goal of CML therapy in the research setting is to achieve eradication of the disease (molecular cure) that is prolonged and durable, with recovery of nonneoplastic, nonclonal hematopoiesis off TKI therapy. The first step toward this aim is to obtain the highest rates of undetectable BCR-ABL1 transcripts lasting for at least 2 or more years.
Recommendations provided by the National Comprehensive Cancer Network (NCCN) and by the European LeukemiaNet (ELN) discuss optimal/expected, suboptimal/warning, and failure response scenarios at different time points of TKI treatment duration. Unfortunately, they may have been misinterpreted in current practice, because oncologists often report that their aim of treatment is the achievement of major molecular response and disease eradication. Significantly, a substantial proportion of oncologists consider a change of TKI therapy in a patient in complete cytogenetic response if they note “loss of major molecular response” (increase of BCR-ABL1 transcripts ([IS] from <0.1% to >0.1%). This perception may be the result of confusion regarding the NCCN and ELN guidelines, which have been updated often as a result of maturing data and have multiple treatment endpoint considerations. Although such endpoints have been suggested by these recommendations as possible criteria for failure, it is important to emphasize that no randomized study has yet shown that a change of TKI treatment in patients with complete cytogenetic response because of a loss of major molecular response, versus changing at the time of cytogenetic relapse, has been shown to improve survival. This is likely because of the high efficacy of salvage TKI therapy at the time of cytogenetic relapse.
Side effects of TKIs are generally mild to moderate, although with long-term TKI therapy, they could affect the patient’s quality of life. Serious side effects occur in less than 5–10% of patients. With imatinib therapy, common mild to moderate side effects include fluid retention, weight gain, nausea, diarrhea, skin rashes, periorbital edema, bone or muscle aches, fatigue, and others (rates of 10–20%). In general, second-generation TKIs are associated with lower rates of these bothersome adverse events. However, dasatinib is associated with higher rates of myelosuppression (20–30%), particularly thrombocytopenia, and with pleural (10–25%) or pericardial effusions (≤5%). Nilotinib is associated with higher rates of hyperglycemia (10–20%), pruritus and skin rashes, and headaches. Nilotinib is also associated with rare events of pancreatitis (<5%). Bosutinib is associated with higher rates of early and self-limited gastrointestinal complications like diarrhea (50–70%). Ponatinib is associated with higher rates of skin rashes (10–15%), pancreatitis (5%), elevations of amylase/lipase (10%), and vasospastic/vasoocclusive events (10–20%). Nilotinib and dasatinib may cause prolongation of the QTc interval; therefore, they should be evaluated cautiously in patients with prolonged QTc interval on electrocardiogram (>470–480 ms), and drugs given for other medical conditions should have relatively smaller or no effects on QTc. These side effects can often be dose-dependent and are generally reversible with treatment interruptions and dose reductions. Dose reductions can be individualized. However, the lowest estimated effective doses of TKIs (from different studies and treatment practices) are imatinib 300 mg daily; nilotinib 200 mg twice daily; dasatinib 20 mg daily; bosutinib 300 mg daily; and ponatinib 15 mg daily.
With long-term follow-up, rare but clinically relevant serious toxicities are emerging. Renal dysfunction and renal failure (creatinine elevations >2–3 mg/dL) are observed in 2–3% of patients and reverse with TKI discontinuation and empirical use of other TKIs. Pulmonary hypertension has been reported with dasatinib (<1–2%) and should be considered in a patient with shortness of breath and a normal chest x-ray (echocardiogram with emphasis on measurement of pulmonary artery pressure). This may be reversible with dasatinib discontinuation and occasionally the use of sildenafil citrate. Systemic hypertension has been observed more often with ponatinib therapy, as well as other TKIs. Hyperglycemia and diabetes have been noted more frequently with nilotinib. Finally, mid- and small-vessel vasoocclusive and vasospastic events have been reported at low but significant rates with nilotinib and ponatinib and should be considered possibly TKI-related and represent indications to interrupt or reduce the dose of the TKI. These events include angina, coronary artery disease, myocardial infarction, peripheral arterial occlusive disease, transient ischemic attacks, cerebral vascular accidents, Raynaud’s phenomenon, and accelerated atherosclerosis. Although these events are uncommon (<5%), they are clinically significant for the patient’s long-term prognosis and occur at significantly higher rates than in the general population (5–20 times more often).
ALLOGENEIC STEM CELL TRANSPLANT
Allogeneic SCT, a curative modality in CML, is associated with long-term survival rates of 40–60% when implemented in the chronic phase. It is associated with early (1-year) mortality rates of 5–30%. Although the 5- to 10-year survival rates were reported to be around 50–60% (and considered as cure rates), about 10–15% of patients die in the subsequent 1–2 decades from subtle long-term complications of the transplant (rather than from CML relapse). These are related to chronic graft-versus-host disease (GVHD), organ dysfunction, development of second cancers, and hazard ratios for mortality higher than in the normal population. Other significant morbidities include infertility, chronic immune-mediated complications, cataracts, hip necrosis, and other morbidities affecting quality of life. The cure and early mortality rates in chronic-phase CML are also associated with several factors: patient age, duration of chronic phase, whether the donor is related or unrelated, degree of matching, preparative regimen, and others. In accelerated-phase CML, the cure rates with allogeneic SCT are 20–40%, depending on the definition of acceleration. Patients with clonal evolution as the only criterion have cure rates of up to 40–50%. Patients undergoing allogeneic SCT in second chronic phase have cure rates of 40–50%. The cure rates with allogeneic SCT in blastic phase CML are ≤15%. Post–allogeneic SCT strategies are now implemented in the setting of molecular or cytogenetic relapse or in hematologic relapse/transformation. These include the use of TKIs for prevention or treatment of relapse, donor lymphocyte infusions, and second allogeneic SCTs, among others. TKIs appear to be highly successful at reinducing cytogenetic/molecular remissions in the setting of cytogenetic or molecular relapse after allogeneic SCT.
Choice and Timing of Allogeneic SCT Allogeneic SCT was considered first-line CML therapy before 2000. The maturing positive experience with TKIs has now relegated its use to after first-line TKI failures. An important question is the optimal timing and sequence of TKIs and allogeneic SCT (whether allogeneic SCT should be used as second- or third-line therapy). Among patients who present with or evolve to blastic phase, combinations of chemotherapy and TKIs should be used to induce remission, followed by allogeneic SCT as soon as possible. The same applies to patients who evolve from chronic to accelerated phase. Patients with de novo accelerated-phase CML may do well with long-term TKI therapy (estimated 8-year survival rate 75%); the timing of allogeneic SCT depends on their optimal response to TKI (achievement of complete cytogenetic response). Among patients who relapse in chronic phase, the treatment sequence depends on several factors: (1) patient age and availability of appropriate donors; (2) risk of allogeneic SCT; (3) presence or absence of clonal evolution and mutations; (4) patient’s prior history and comorbidities; and (5) patient and physician preferences (Table 133-3). Patients with T315I mutations at relapse should be offered ponatinib and considered for allogeneic SCT (because of the short follow-up with ponatinib). Patients with mutations involving Y253H, E255K/V, and F359V/C/I respond better to dasatinib or bosutinib. Patients with mutations involving V299L, T315A, and F317L/F/I/C respond better to nilotinib. Comorbidities such as diabetes, hypertension, pulmonary hypertension, chronic lung disease, cardiac conditions, and pancreatitis may influence the choice for or against a particular TKI. Patients with clonal evolution, unfavorable mutations, or lack of major/complete cytogenetic response within 1 year of salvage TKI therapy have short remission durations and should consider allogeneic SCT as more urgent in the setting of salvage. Patients without clonal evolution or mutations at relapse and who achieve a complete cytogenetic response with TKI salvage, have long-lasting complete remissions and may delay the option of allogeneic SCT to third-line therapy. Finally, older patients (age 65–70 years or older) and those with high risk of mortality with allogeneic SCT may forgo this curative option for several years of disease control in chronic phase with or without cytogenetic response (Table 133-3). Historically, before the availability of TKIs, patients without cytogenetic response on interferon α or hydroxyurea had expected short median survival times (2–3 years) with expected rapid disease transformation. The maturing experience with TKIs suggests a different course, whereby patients may remain in chronic phase on TKI-based therapies (combinations including hydroxyurea, cytarabine, decitabine, and others), with or without cytogenetic response, for many years. Table 133-3 summarizes a general guidance to the choice of TKIs versus allogeneic SCT.
|
GENERAL SUGGESTIONS REGARDING THE USE OF TYROSINE KINASE INHIBITORS (TKIS) AND ALLOGENEIC STEM CELL TRANSPLANTATION (SCT) IN CHRONIC MYELOID LEUKEMIA (CML) |
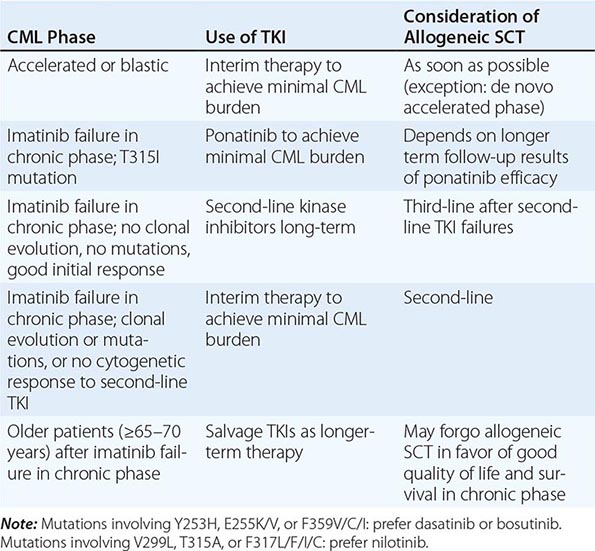
MONITORING THERAPY IN CML
Achievement of complete cytogenetic response by 12 months of imatinib therapy and its persistence later, the only consistent prognostic factor associated with survival, is now the main therapeutic endpoint in CML. Failure to achieve a complete cytogenetic response by 12 months or occurrence of later cytogenetic or hematologic relapse is considered as treatment failure and an indication to change therapy. Because salvage therapy with other TKIs reestablishes good outcome, it is important to ensure patient compliance to continued TKI therapy and change therapy at the first sign of cytogenetic relapse. Patients on frontline imatinib therapy should be closely monitored until documentation of complete cytogenetic response, at which time they can be monitored every 6 months with peripheral blood FISH and PCR studies (to check for concordance of results), or more frequently if there are concerns about changes in BCR-ABL1 transcripts (e.g., every 3 months). Monitoring by molecular studies only is reasonable in patients who are in major molecular response. Cytogenetic relapse on imatinib is an indication of treatment failure and need to change TKI therapy. Mutational analysis in this instance helps in the selection of the next TKI and identifies mutations in 30–50% of patients. Mutational studies in patients in complete cytogenetic response (in whom there may be concerns of increasing BCR-ABL1 transcripts) identify mutations in ≤5% and are therefore not indicated. Earlier response has been identified as a prognostic factor for long-term outcome, including achievement of partial cytogenetic response (BCR-ABL1 transcripts ≤10%) by 3–6 months of therapy. Failure to achieve such a response on imatinib therapy has been associated with significantly worse survival in some studies (particularly when second-generation TKIs were not readily available as salvage therapy), but not in others (when they were).
The use of second-generation TKIs (nilotinib, dasatinib) as frontline therapy changed the monitoring approach slightly. Patients are expected to achieve complete cytogenetic response by 3–6 months of therapy. Failure to do so is associated with worse event-free survival, transformation rates, and survival. However, the 3- to 5-year estimated survival among such patients is still high, around 80–90%, which is better than what would be anticipated if such patients were offered allogeneic SCT at that time. Thus, this adverse response to therapy is considered a warning signal, but it is not known whether changing therapy to other TKIs at that time would improve longer term outcome.
TREATMENT OF ACCELERATED AND BLASTIC PHASES
Patients in accelerated or blastic phase may receive therapy with TKIs, preferably second- or third-generation TKIs (dasatinib, nilotinib, bosutinib, ponatinib), alone or in combination with chemotherapy, to reduce the CML burden, before undergoing allogeneic SCT. Response rates with single-agent TKIs range from 30 to 50% in accelerated phase and from 20 to 30% in blastic phase. Cytogenetic responses, particularly complete cytogenetic responses, are uncommon (10–30%) and transient in blastic phase. Studies of TKIs in combination with chemotherapy are ongoing; the general experience suggests that combined TKI-chemotherapy strategies increase the response rates and their durability and improve survival. In CML lymphoid blastic phase, the combination of anti-ALL chemotherapy with TKIs results in complete response rates of 60–70% and median survival times of 2–3 years (compared with historical response rates of 40–50% and median survival times of 12–18 months). This allows many patients to undergo allogeneic SCT in a state of minimal CML burden or secondary chronic phase, which are associated with higher cure rates. In CML nonlymphoid blastic phase, anti-AML chemotherapy combined with TKIs results in CR rates of 30–50% and median survival times of 9–12 months (compared with historical response rates of 20–30% and median survival times of 3–5 months). In accelerated phase, response to single TKIs is significant in conditions where “softer” accelerated phase criteria are considered (e.g., clonal evolution alone, thrombocytosis alone, significant splenomegaly or resistance to hydroxyurea, but without evidence of high blast and basophil percentages). In accelerated phase, combinations usually include TKIs with low-intensity chemotherapy such as low-dose cytarabine, low-dose idarubicin, decitabine, interferon α, hydroxyurea, or others.
OTHER TREATMENTS AND SPECIAL THERAPEUTIC CONSIDERATIONS
Interferon α Interferon α was a standard of care before 2000. Today, it is considered in combination with TKIs (an investigational approach), sometimes after CML failure on TKIs, occasionally in patients during pregnancy, or as part of investigational strategies with TKIs to eradicate residual molecular disease.
Chemotherapeutic Agents Hydroxyurea and busulfan were commonly used chemotherapeutic agents in the past. Hydroxyurea remains a safe and effective agent (at daily doses of 0.5–10 g) to reduce initial CML burden, as a temporary measure in between definitive therapies, or in combination with TKIs to sustain complete hematologic or cytogenetic responses. Busulfan is often used in allogeneic SCT preparative regimens. Because of its side effects (delayed myelosuppression, Addison-like disease, pulmonary and cardiac fibrosis, myelofibrosis), it is now only rarely used in the chronic management of CML. Low-dose cytarabine, decitabine, anthracyclines, 6-mercaptopurine, 6-thioguanine, thiotepa, anagrelide, and other agents are useful in different CML settings to control the disease burden.
Others Splenectomy is occasionally considered to alleviate symptoms of massive splenomegaly and/or hypersplenism. Splenic irradiation is rarely used, if at all, because of the postirradiation adhesions and complications. Leukapheresis is rarely used in patients presenting with extreme leukocytosis and leukostatic complications. Single doses of high-dose cytarabine or high doses of hydroxyurea, with tumor lysis management, may be as effective and less cumbersome.
Special Considerations Women with CML who become pregnant should discontinue TKI therapy immediately. Among 125 babies delivered to women with CML who discontinued TKI therapy as soon as the pregnancy was known, three babies were born with ocular, skeletal, and renal malformations, suggesting the uncommon teratogenicity of imatinib. There are no or little data with other TKIs. Control of CML during pregnancy can be managed with leukapheresis for severe symptomatic leukocytosis in the first trimester and with hydroxyurea subsequently until delivery. There are case reports of successful pregnancies and deliveries of normal babies with interferon α therapy and registry studies in essential thrombocytosis of its safety, but interferon α can be antiangiogenic and may increase the risk of spontaneous abortions.
Patients on TKI therapy may develop chromosomal abnormalities in the Ph-negative cells. These may involve loss of chromosome Y, trisomy 8, 20q–, chromosome 5 or 7 abnormalities, and others. Most chromosomal abnormalities disappear spontaneously on follow-up and may be indicative of the genetic instability of the hematopoietic stem cells that predispose the patient to develop CML in the first place. Rarely, abnormalities involving chromosomes 5 or 7 may be truly clonal and evolve into myelodysplastic syndrome or acute myeloid leukemia. This is thought to be part of the natural course of patients in whom CML was suppressed and who live long enough to develop other hematologic malignancies.
GLOBAL ASPECTS OF CHRONIC MYELOID LEUKEMIA
![]() Routine physical exams and blood tests in the United States and advanced countries result in early detection of CML in most patients. About 50–70% of patients with CML are diagnosed accidentally, and high-risk CML as defined by prognostic models (e.g., Sokal risk groups) is found in only 10–20% of patients. This is not the same situation in emerging nations (e.g., India, China, African countries, the Middle East), where most patients are diagnosed following evaluation for symptoms and many present with high tumor burdens, such as massive splenomegaly, and advanced phases of CML (high-risk CML documented in 30–50%). Therefore, the prognosis of such patients on TKI therapy may be worse than the published experience.
Routine physical exams and blood tests in the United States and advanced countries result in early detection of CML in most patients. About 50–70% of patients with CML are diagnosed accidentally, and high-risk CML as defined by prognostic models (e.g., Sokal risk groups) is found in only 10–20% of patients. This is not the same situation in emerging nations (e.g., India, China, African countries, the Middle East), where most patients are diagnosed following evaluation for symptoms and many present with high tumor burdens, such as massive splenomegaly, and advanced phases of CML (high-risk CML documented in 30–50%). Therefore, the prognosis of such patients on TKI therapy may be worse than the published experience.
The high cost of TKI therapies (annual costs of $90,000–$140,000 in the United States; lower but variable in the rest of the world) makes the general affordability of such treatments difficult. Although TKI treatment penetration is high in nations where cost of therapy is not an issue (e.g., Sweden, European Union), it may be less so in other nations, even in advanced ones like the United States, where out-of-pocket expenses may be prohibitive to a subset of patients (perhaps 10–20%). Based on the sales of imatinib worldwide and charity free drug supplies, it is estimated that less than 30% of patients are treated with imatinib (or other TKIs) consistently. Although the estimated 10-year survival in CML is 85% in single-institution studies (e.g., M.D. Anderson Cancer Center), in national studies in countries with TKI affordability (Sweden) (Figs. 133-2 and 133-3) or in company-sponsored studies (where all patients have access to TKIs throughout their care), the estimated 10-year survival worldwide, even 12 years after the introduction of TKI therapies, is likely to be less than 50%. The Surveillance, Epidemiology, and End Results (SEER) data from the United States report an estimated 5-year survival rate of 60% in the era of TKIs.
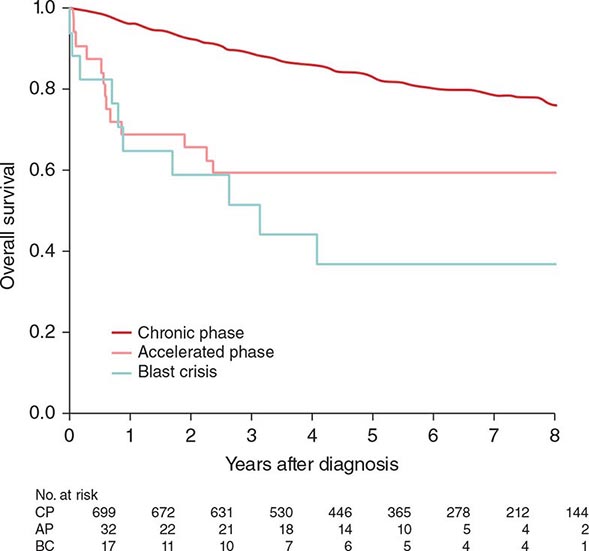
FIGURE 133-3 Survival in chronic (CP), accelerated (AP), and blastic crisis (BC) phases of chronic myeloid leukemia (CML) in the population-based Swedish national registry study. The accelerated- and blastic-phase cases are de novo presentations. The favorable outcome with de novo blastic phase may be due to use of 20% blasts or more to define blastic phase. (With permission from Dr. Martin Hoglund, Swedish CML Registry, 2013.)
The current high cost of TKI therapies poses two additional considerations. The first are the treatment pathways and guidelines in nations where TKIs may not be affordable by patients or the health care system. In these conditions, there are trends of pathways advocating frontline allogeneic SCT (a one-time cost of $30,000–$50,000) despite the associated mortality and morbidities. The second is the choice of frontline TKI therapy once imatinib becomes available in generic forms (hopefully at much lower annual prices, e.g., $2,000–$10,000). This will depend on the maturing data in randomized studies of second-generation TKIs versus imatinib in relation to important long-term outcome endpoints, particularly survival, but also event-free survival and transformation-free survival.
134 |
Malignancies of Lymphoid Cells |
Malignancies of lymphoid cells range from the most indolent to the most aggressive human malignancies. These cancers arise from cells of the immune system at different stages of differentiation, resulting in a wide range of morphologic, immunologic, and clinical findings. Insights on the normal immune system have allowed a better understanding of these sometimes confusing disorders.
Some malignancies of lymphoid cells almost always present as leukemia (i.e., primary involvement of bone marrow and blood), while others almost always present as lymphomas (i.e., solid tumors of the immune system). However, other malignancies of lymphoid cells can present as either leukemia or lymphoma. In addition, the clinical pattern can change over the course of the illness. This change is more often seen in a patient who seems to have a lymphoma and then develops the manifestations of leukemia over the course of the illness.
BIOLOGY OF LYMPHOID MALIGNANCIES: CONCEPTS OF THE WORLD HEALTH ORGANIZATION CLASSIFICATION OF LYMPHOID MALIGNANCIES
The classification of lymphoid cancers evolved steadily throughout the twentieth century. The distinction between leukemia and lymphoma was made early, and separate classification systems were developed for each. Leukemias were first divided into acute and chronic subtypes based on average survival. Chronic leukemias were easily subdivided into those of lymphoid or myeloid origin based on morphologic characteristics. However, a spectrum of diseases that were formerly all called chronic lymphoid leukemia has become apparent (Table 134-1). The acute leukemias were usually malignancies of blast cells with few identifying characteristics. When cytochemical stains became available, it was possible to divide these objectively into myeloid malignancies and acute leukemias of lymphoid cells. Acute leukemias of lymphoid cells have been subdivided based on morphologic characteristics by the French-American-British (FAB) group (Table 134-2). Using this system, lymphoid malignancies of small uniform blasts (e.g., typical childhood acute lymphoblastic leukemia) were called L1, lymphoid malignancies with larger and more variable size cells were called L2, and lymphoid malignancies of uniform cells with basophilic and sometimes vacuolated cytoplasm were called L3 (e.g., typical Burkitt’s lymphoma cells). Acute leukemias of lymphoid cells have also been subdivided based on immunologic (i.e., T cell vs B cell) and cytogenetic abnormalities (Table 134-2). Major cytogenetic subgroups include the t(9;22) (e.g., Philadelphia chromosome–positive acute lymphoblastic leukemia) and the t(8;14) found in the L3 or Burkitt’s leukemia.
|
LYMPHOID DISORDERS THAT CAN PRESENT AS “CHRONIC LEUKEMIA” AND BE CONFUSED WITH TYPICAL B-CELL CHRONIC LYMPHOID LEUKEMIA |
|
CLASSIFICATION OF ACUTE LYMPHOID LEUKEMIA (ALL) |
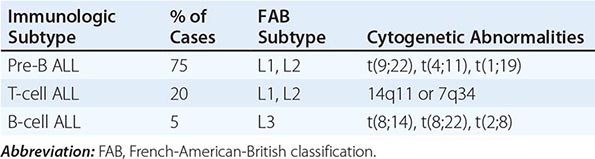
Non-Hodgkin’s lymphomas were separated from Hodgkin’s lymphoma by recognition of the Sternberg-Reed cells early in the twentieth century. The histologic classification for non-Hodgkin’s lymphomas has been one of the most contentious issues in oncology. Imperfect morphologic systems were supplanted by imperfect immunologic systems, and poor reproducibility of diagnosis has hampered progress. In 1999, the World Health Organization (WHO) classification of lymphoid malignancies was devised through a process of consensus development among international leaders in hematopathology and clinical oncology. The WHO classification takes into account morphologic, clinical, immunologic, and genetic information and attempts to divide non-Hodgkin’s lymphomas and other lymphoid malignancies into clinical/pathologic entities that have clinical and therapeutic relevance. This system is presented in Table 134-3. This system is clinically relevant and has a higher degree of diagnostic accuracy than those used previously. The possibilities for subdividing lymphoid malignancies are extensive. However, Table 134-3 presents in bold those malignancies that occur in at least 1% of patients. Specific lymphoma subtypes will be dealt with in more detail below.
|
WHO CLASSIFICATION OF LYMPHOID MALIGNANCIES |
Lymphomas occurring in fewer than 1% of patients with lymphoproliferative diseases are discussed in Chap. 135e, and lymphomas associated with HIV infection are discussed in Chap. 226.
GENERAL ASPECTS OF LYMPHOID MALIGNANCIES
ETIOLOGY AND EPIDEMIOLOGY
The relative frequency of the various lymphoid malignancies is shown in Fig. 134-1. Chronic lymphoid leukemia (CLL) is the most prevalent form of leukemia in Western countries. It occurs most frequently in older adults and is exceedingly rare in children. In 2014, 15,720 new cases were diagnosed in the United States, but because of the prolonged survival associated with this disorder, the total prevalence is many times higher. CLL is more common in men than in women and more common in whites than in blacks. This is an uncommon malignancy in Asia. The etiologic factors for typical CLL are unknown.
FIGURE 134-1 Relative frequency of lymphoid malignancies. ALL, acute lymphoid leukemia; CLL, chronic lymphoid leukemia; MALT, mucosa-associated lymphoid tissue.
In contrast to CLL, acute lymphoid leukemias (ALLs) are predominantly cancers of children and young adults. The L3 or Burkitt’s leukemia occurring in children in developing countries seems to be associated with infection by the Epstein-Barr virus (EBV) in infancy. However, the explanation for the etiology of more common subtypes of ALL is much less certain. Childhood ALL occurs more often in higher socioeconomic subgroups. Children with trisomy 21 (Down’s syndrome) have an increased risk for childhood ALL as well as acute myeloid leukemia (AML). Exposure to high-energy radiation in early childhood increases the risk of developing T-cell ALL.
The etiology of ALL in adults is also uncertain. ALL is unusual in middle-aged adults but increases in incidence in the elderly. However, AML is still much more common in older patients. Environmental exposures, including certain industrial exposures, exposure to agricultural chemicals, and smoking, might increase the risk of developing ALL as an adult. ALL was diagnosed in 6020 persons and AML in 18,860 persons in the United States in 2014.
The preponderance of evidence suggests that Hodgkin’s lymphoma is of B-cell origin. The incidence of Hodgkin’s lymphoma appears fairly stable, with 9190 new cases diagnosed in 2014 in the United States. Hodgkin’s lymphoma is more common in whites than in blacks and more common in males than in females. A bimodal distribution of age at diagnosis has been observed, with one peak incidence occurring in patients in their twenties and the other in those in their eighties. Some of the late age peak may be attributed to confusion among entities with similar appearance such as anaplastic large cell lymphoma and T-cell–rich B-cell lymphoma. Patients in the younger age groups diagnosed in the United States largely have the nodular sclerosing subtype of Hodgkin’s lymphoma. Elderly patients, patients infected with HIV, and patients in Third World countries more commonly have mixed-cellularity Hodgkin’s lymphoma or lymphocyte-depleted Hodgkin’s lymphoma. Infection by HIV is a risk factor for developing Hodgkin’s lymphoma. In addition, an association between infection by EBV and Hodgkin’s lymphoma has been suggested. A monoclonal or oligoclonal proliferation of EBV-infected cells in 20–40% of the patients with Hodgkin’s lymphoma has led to proposals for this virus having an etiologic role in Hodgkin’s lymphoma. However, the matter is not settled definitively.
For unknown reasons, non-Hodgkin’s lymphomas increased in frequency in the United States at the rate of 4% per year and increased 2–8% per year globally between 1950 and the late 1990s. The rate of increase in the past few years seems to be decreasing. About 70,800 new cases of non-Hodgkin’s lymphoma were diagnosed in the United States in 2014 and nearly 360,000 cases worldwide. Non-Hodgkin’s lymphomas are more frequent in the elderly and more frequent in men. Patients with both primary and secondary immunodeficiency states are predisposed to developing non-Hodgkin’s lymphomas. These include patients with HIV infection; patients who have undergone organ transplantation; and patients with inherited immune deficiencies, the sicca syndrome, and rheumatoid arthritis.
The incidence of non-Hodgkin’s lymphomas and the patterns of expression of the various subtypes differ geographically. T-cell lymphomas are more common in Asia than in Western countries, while certain subtypes of B-cell lymphomas such as follicular lymphoma are more common in Western countries. A specific subtype of non-Hodgkin’s lymphoma known as the angiocentric nasal T/natural killer (NK) cell lymphoma has a striking geographic occurrence, being most frequent in Southern Asia and parts of Latin America. Another subtype of non-Hodgkin’s lymphoma associated with infection by human T-cell lymphotropic virus (HTLV) 1 is seen particularly in southern Japan and the Caribbean (Chap. 225e).
A number of environmental factors have been implicated in the occurrence of non-Hodgkin’s lymphoma, including infectious agents, chemical exposures, and medical treatments. Several studies have demonstrated an association between exposure to agricultural chemicals and an increased incidence of non-Hodgkin’s lymphoma. Patients treated for Hodgkin’s lymphoma can develop non-Hodgkin’s lymphoma; it is unclear whether this is a consequence of the Hodgkin’s lymphoma or its treatment. However, a number of non-Hodgkin’s lymphomas are associated with infectious agents (Table 134-4). HTLV-1 infects T cells and leads directly to the development of adult T-cell lymphoma in a small percentage of infected patients. The cumulative lifetime risk of developing lymphoma in an infected patient is 2.5%. The virus is transmitted by infected lymphocytes ingested by nursing babies of infected mothers, bloodborne transmission, or sexually. The median age of patients with adult T-cell lymphoma is ~56 years, emphasizing the long latency. HTLV-1 is also the cause of tropical spastic paraparesis—a neurologic disorder that occurs somewhat more frequently than lymphoma and with shorter latency and usually from transfusion-transmitted virus (Chap. 225e).
|
INFECTIOUS AGENTS ASSOCIATED WITH THE DEVELOPMENT OF LYMPHOID MALIGNANCIES |
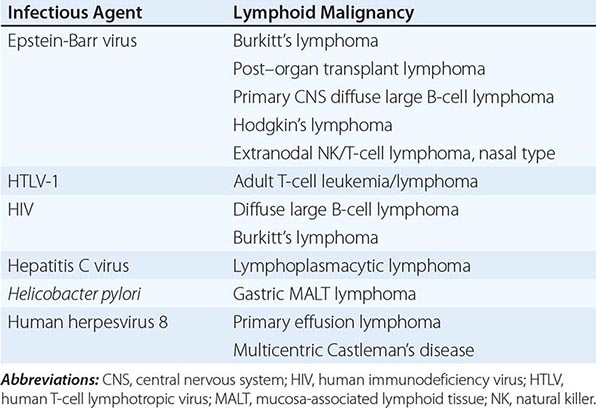
EBV is associated with the development of Burkitt’s lymphoma in Central Africa and the occurrence of aggressive non-Hodgkin’s lymphomas in immunosuppressed patients in Western countries. The majority of primary central nervous system (CNS) lymphomas are associated with EBV. EBV infection is strongly associated with the occurrence of extranodal nasal T/NK cell lymphomas in Asia and South America. Infection with HIV predisposes to the development of aggressive, B-cell non-Hodgkin’s lymphoma. This may be through overexpression of interleukin 6 by infected macrophages. Infection of the stomach by the bacterium Helicobacter pylori induces the development of gastric MALT (mucosa-associated lymphoid tissue) lymphomas. This association is supported by evidence that patients treated with antibiotics to eradicate H. pylori have regression of their MALT lymphoma. The bacterium does not transform lymphocytes to produce the lymphoma; instead, a vigorous immune response is made to the bacterium, and the chronic antigenic stimulation leads to the neoplasia. MALT lymphomas of the skin may be related to Borrelia sp. infections, those of the eyes to Chlamydophila psittaci, and those of the small intestine to Campylobacter jejuni.
Chronic hepatitis C virus infection has been associated with the development of lymphoplasmacytic lymphoma. Human herpesvirus 8 is associated with primary effusion lymphoma in HIV-infected persons and multicentric Castleman’s disease, a diffuse lymphadenopathy associated with systemic symptoms of fever, malaise, and weight loss.
In addition to infectious agents, a number of other diseases or exposures may predispose to developing lymphoma (Table 134-5).
|
DISEASES OR EXPOSURES ASSOCIATED WITH INCREASED RISK OF DEVELOPMENT OF MALIGNANT LYMPHOMA |
IMMUNOLOGY
All lymphoid cells are derived from a common hematopoietic progenitor that gives rise to lymphoid, myeloid, erythroid, monocyte, and megakaryocyte lineages. Through the ordered and sequential activation of a series of transcription factors, the cell first becomes committed to the lymphoid lineage and then gives rise to B and T cells. About 75% of all lymphoid leukemias and 90% of all lymphomas are of B-cell origin. A cell becomes committed to B-cell development when it begins to rearrange its immunoglobulin genes. The sequence of cellular changes, including changes in cell-surface phenotype, that characterizes normal B-cell development is shown in Fig. 134-2. A cell becomes committed to T-cell differentiation upon migration to the thymus and rearrangement of T-cell antigen receptor genes. The sequence of the events that characterize T-cell development is depicted in Fig. 134-3.
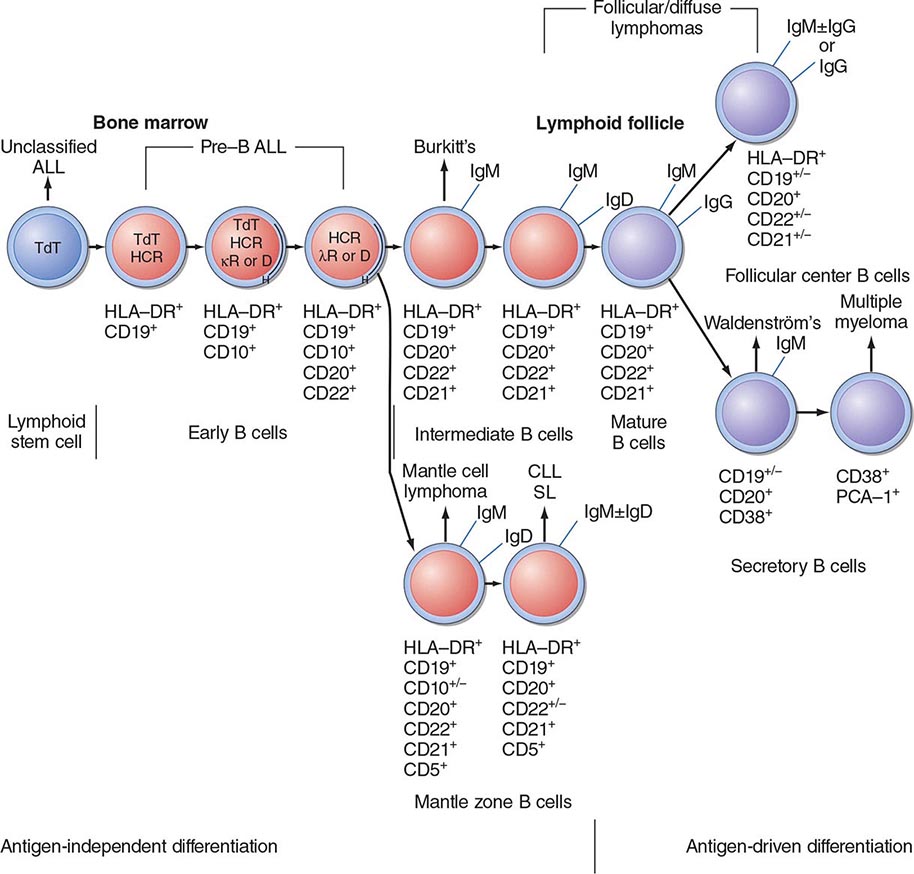
FIGURE 134-2 Pathway of normal B-cell differentiation and relationship to B-cell lymphomas. HLA-DR, CD10, CD19, CD20, CD21, CD22, CD5, and CD38 are cell markers used to distinguish stages of development. Terminal transferase (TdT) is a cellular enzyme. Immunoglobulin heavy chain gene rearrangement (HCR) and light chain gene rearrangement or deletion (κR or D, λR or D) occur early in B-cell development. The approximate normal stage of differentiation associated with particular lymphomas is shown. ALL, acute lymphoid leukemia; CLL, chronic lymphoid leukemia; SL, small lymphocytic lymphoma.
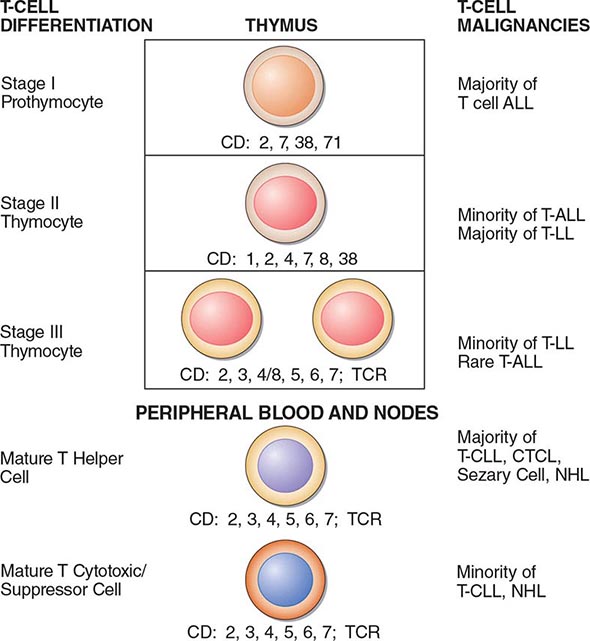
FIGURE 134-3 Pathway of normal T-cell differentiation and relationship to T-cell lymphomas. CD1, CD2, CD3, CD4, CD5, CD6, CD7, CD8, CD38, and CD71 are cell markers used to distinguish stages of development. T-cell antigen receptors (TCR) rearrange in the thymus, and mature T cells emigrate to nodes and peripheral blood. ALL, acute lymphoid leukemia; T-ALL, T-cell ALL; T-LL, T-cell lymphoblastic lymphoma; T-CLL, T-cell chronic lymphoid leukemia; CTCL, cutaneous T-cell lymphoma; NHL, non-Hodgkin’s lymphoma.
Although lymphoid malignancies often retain the cell-surface phenotype of lymphoid cells at particular stages of differentiation, this information is of little consequence. The so-called stage of differentiation of a malignant lymphoma does not predict its natural history. For example, the clinically most aggressive lymphoid leukemia is Burkitt’s leukemia, which has the phenotype of a mature follicle center IgM-bearing B cell. Leukemias bearing the immunologic cell-surface phenotype of more primitive cells (e.g., pre-B ALL, CD10+) are less aggressive and more amenable to curative therapy than the “more mature” appearing Burkitt’s leukemia cells. Furthermore, the apparent stage of differentiation of the malignant cell does not reflect the stage at which the genetic lesions that gave rise to the malignancy developed. For example, follicular lymphoma has the cell-surface phenotype of a follicle center cell, but its characteristic chromosomal translocation, the t(14;18), which involves juxtaposition of the antiapoptotic bcl-2 gene next to the immunoglobulin heavy chain gene (see below), had to develop early in ontogeny as an error in the process of immunoglobulin gene rearrangement. Why the subsequent steps that led to transformation became manifest in a cell of follicle center differentiation is not clear.
The major value of cell-surface phenotyping is to aid in the differential diagnosis of lymphoid tumors that appear similar by light microscopy. For example, benign follicular hyperplasia may resemble follicular lymphoma; however, the demonstration that all the cells bear the same immunoglobulin light chain isotype strongly suggests the mass is a clonal proliferation rather than a polyclonal response to an exogenous stimulus.
Malignancies of lymphoid cells are associated with recurring genetic abnormalities. While specific genetic abnormalities have not been identified for all subtypes of lymphoid malignancies, it is presumed that they exist. Genetic abnormalities can be identified at a variety of levels including gross chromosomal changes (i.e., translocations, additions, or deletions); rearrangement of specific genes that may or may not be apparent from cytogenetic studies; and overexpression, underexpression, or mutation of specific oncogenes. Altered expression or mutation of specific proteins is particularly important. Many lymphomas contain balanced chromosomal translocations involving the antigen receptor genes; immunoglobulin genes on chromosomes 2, 14, and 22 in B cells; and T-cell antigen receptor genes on chromosomes 7 and 14 in T cells. The rearrangement of chromosome segments to generate mature antigen receptors must create a site of vulnerability to aberrant recombination. B cells are even more susceptible to acquiring mutations during their maturation in germinal centers; the generation of antibody of higher affinity requires the introduction of mutations into the variable region genes in the germinal centers. Other nonimmunoglobulin genes, e.g., bcl-6, may acquire mutations as well.
In the case of diffuse large B-cell lymphoma, the translocation t(14;18) occurs in ~30% of patients and leads to overexpression of the bcl-2 gene found on chromosome 18. Some other patients without the translocation also overexpress the BCL-2 protein. This protein is involved in suppressing apoptosis—i.e., the mechanism of cell death most often induced by cytotoxic chemotherapeutic agents. A higher relapse rate has been observed in patients whose tumors overexpress the BCL-2 protein, but not in those patients whose lymphoma cells show only the translocation. Thus, particular genetic mechanisms have clinical ramifications.
Table 134-6 presents the most common translocations and associated oncogenes for various subtypes of lymphoid malignancies. In some cases, such as the association of the t(14;18) in follicular lymphoma, the t(2;5) in anaplastic large T/null cell lymphoma, the t(8;14) in Burkitt’s lymphoma, and the t(11;14) in mantle cell lymphoma, the great majority of tumors in patients with these diagnoses display these abnormalities. In other types of lymphoma where a minority of the patients have tumors expressing specific genetic abnormalities, the defects may have prognostic significance. No specific genetic abnormalities have been identified in Hodgkin’s lymphoma other than aneuploidy.
|
CYTOGENETIC TRANSLOCATION AND ASSOCIATED ONCOGENES OFTEN SEEN IN LYMPHOID MALIGNANCIES |
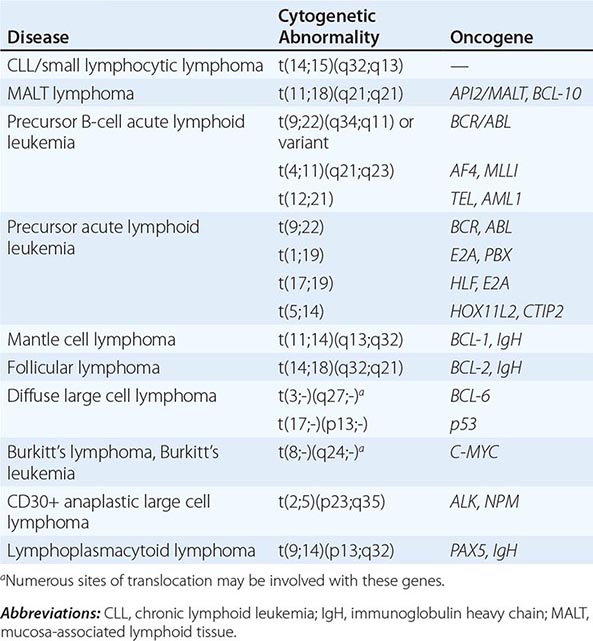
In typical B-cell CLL, trisomy 12 conveys a poorer prognosis. In ALL in both adults and children, genetic abnormalities have important prognostic significance. Patients whose tumor cells display the t(9;22) and translocations involving the MLL gene on chromosome 11q23 have a much poorer outlook than patients who do not have these translocations. Other genetic abnormalities that occur frequently in adults with ALL include the t(4;11) and the t(8;14). The t(4;11) is associated with younger age, female predominance, high white cell counts, and L1 morphology. The t(8;14) is associated with older age, male predominance, frequent CNS involvement, and L3 morphology. Both are associated with a poor prognosis. In childhood ALL, hyperdiploidy has been shown to have a favorable prognosis.
Gene profiling using array technology allows the simultaneous assessment of the expression of thousands of genes. This technology provides the possibility to identify new genes with pathologic importance in lymphomas, the identification of patterns of gene expression with diagnostic and/or prognostic significance, and the identification of new therapeutic targets. Recognition of patterns of gene expression is complicated and requires sophisticated mathematical techniques. Early successes using this technology in lymphoma include the identification of previously unrecognized subtypes of diffuse large B-cell lymphoma whose gene expression patterns resemble either those of follicular center B cells or activated peripheral blood B cells. Patients whose lymphomas have a germinal center B-cell pattern of gene expression have a considerably better prognosis than those whose lymphomas have a pattern resembling activated peripheral blood B cells. This improved prognosis is independent of other known prognostic factors. Similar information is being generated in follicular lymphoma and mantle cell lymphoma. The challenge remains to provide information from such techniques in a clinically useful time frame.
APPROACH TO THE PATIENT:
Lymphoid Cell Malignancies
Regardless of the type of lymphoid malignancy, the initial evaluation of the patient should include performance of a careful history and physical examination. These will help confirm the diagnosis, identify those manifestations of the disease that might require prompt attention, and aid in the selection of further studies to optimally characterize the patient’s status to allow the best choice of therapy. It is difficult to overemphasize the importance of a carefully done history and physical examination. They might provide observations that lead to reconsidering the diagnosis, provide hints at etiology, clarify the stage, and allow the physician to establish rapport with the patient that will make it possible to develop and carry out a therapeutic plan.
For patients with ALL, evaluation is usually completed after a complete blood count, chemistry studies reflecting major organ function, a bone marrow biopsy with genetic and immunologic studies, and a lumbar puncture. The latter is necessary to rule out occult CNS involvement. At this point, most patients would be ready to begin therapy. In ALL, prognosis is dependent on the genetic characteristics of the tumor, the patient’s age, the white cell count, and the patient’s overall clinical status and major organ function.
In CLL, the patient evaluation should include a complete blood count, chemistry tests to measure major organ function, serum protein electrophoresis, and a bone marrow biopsy. However, some physicians believe that the diagnosis would not always require a bone marrow biopsy. Patients often have imaging studies of the chest and abdomen looking for pathologic lymphadenopathy. Patients with typical B-cell CLL can be subdivided into three major prognostic groups. Those patients with only blood and bone marrow involvement by leukemia but no lymphadenopathy, organomegaly, or signs of bone marrow failure have the best prognosis. Those with lymphadenopathy and organomegaly have an intermediate prognosis, and patients with bone marrow failure, defined as hemoglobin <100 g/L (10 g/dL) or platelet count <100,000/μL, have the worst prognosis. The pathogenesis of the anemia or thrombocytopenia is important to discern. The prognosis is adversely affected when either or both of these abnormalities are due to progressive marrow infiltration and loss of productive marrow. However, either or both may be due to autoimmune phenomena or to hypersplenism that can develop during the course of the disease. These destructive mechanisms are usually completely reversible (glucocorticoids for autoimmune disease; splenectomy for hypersplenism) and do not influence disease prognosis.
Two popular staging systems have been developed to reflect these prognostic groupings (Table 134-7). Patients with typical B-cell CLL can have their course complicated by immunologic abnormalities, including autoimmune hemolytic anemia, autoimmune thrombocytopenia, and hypogammaglobulinemia. Patients with hypogammaglobulinemia benefit from regular (monthly) γ globulin administration. Because of expense, γ globulin is often withheld until the patient experiences a significant infection. These abnormalities do not have a clear prognostic significance and should not be used to assign a higher stage.
|
STAGING OF TYPICAL B-CELL LYMPHOID LEUKEMIA |
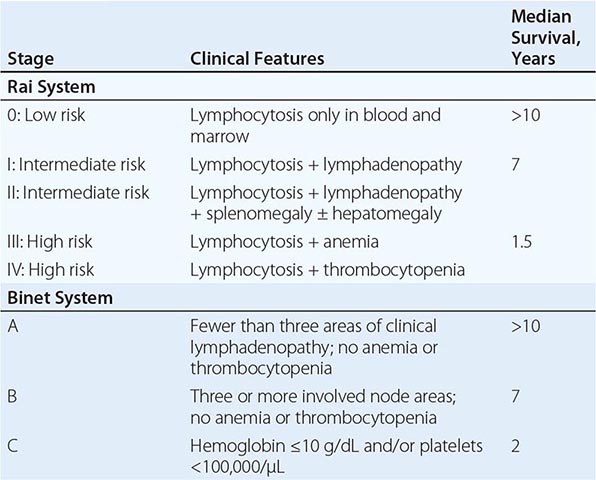
Two other features may be used to assess prognosis in B-cell CLL, but neither has yet been incorporated into a staging classification. At least two subsets of CLL have been identified based on the cytoplasmic expression of ZAP-70; expression of this protein, which is usually expressed in T cells, identifies a subgroup with poorer prognosis. A less powerful subsetting tool is CD38 expression. CD38+ tumors tend to have a poorer prognosis than CD38– tumors. A less easily measured feature, the presence of immunoglobulin variable region gene mutations, is also able to separate prognostic groups; patients with mutated immunoglobulin variable region genes respond better to treatment and have better survival than those with unmutated immunoglobulin variable region genes.
The initial evaluation of a patient with Hodgkin’s lymphoma or non-Hodgkin’s lymphoma is similar. In both situations, the determination of an accurate anatomic stage is an important part of the evaluation. Staging is done using the Ann Arbor staging system originally developed for Hodgkin’s lymphoma (Table 134-8).
|
THE ANN ARBOR STAGING SYSTEM FOR HODGKIN’S LYMPHOMA |
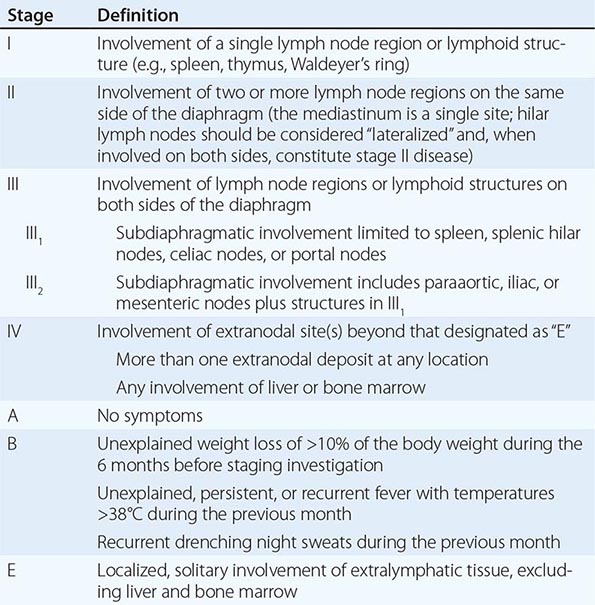
Evaluation of patients with Hodgkin’s lymphoma will typically include a complete blood count; erythrocyte sedimentation rate; chemistry studies reflecting major organ function; computed tomography (CT) scans of the chest, abdomen, and pelvis; and a bone marrow biopsy. Neither a positron emission tomography (PET) scan nor a gallium scan is absolutely necessary for primary staging, but one performed at the completion of therapy allows evaluation of persisting radiographic abnormalities, particularly the mediastinum. Knowing that the PET scan or gallium scan is abnormal before treatment can help in this assessment. In most cases, these studies will allow assignment of anatomic stage and the development of a therapeutic plan.
In patients with non-Hodgkin’s lymphoma, the same evaluation described for patients with Hodgkin’s lymphoma is usually carried out. In addition, serum levels of lactate dehydrogenase (LDH) and β2-microglobulin and serum protein electrophoresis are often included in the evaluation. Anatomic stage is assigned in the same manner as used for Hodgkin’s lymphoma. However, the prognosis of patients with non-Hodgkin’s lymphoma is best assigned using the International Prognostic Index (IPI) (Table 134-9). This is a powerful predictor of outcome in all subtypes of non-Hodgkin’s lymphoma. Patients are assigned an IPI score based on the presence or absence of five adverse prognostic factors and may have none or all five of these adverse prognostic factors. Figure 134-4 shows the prognostic significance of this score in 1300 patients with all types of non-Hodgkin’s lymphoma. With the addition of rituximab to CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), treatment outcomes have improved and the original IPI has lost some of its discrimination power. A revised IPI has been proposed that better predicts outcome of rituximab plus chemotherapy-based programs (Table 134-9). CT scans are routinely used in the evaluation of patients with all subtypes of non-Hodgkin’s lymphoma, but PET and gallium scans are much more useful in aggressive subtypes such as diffuse large B-cell lymphoma than in more indolent subtypes such as follicular lymphoma or small lymphocytic lymphoma. Although the IPI does divide patients with follicular lymphoma into subsets with distinct prognoses, the distribution of such patients is skewed toward lower-risk categories. A follicular lymphoma–specific IPI (FLIPI) has been proposed that replaces performance status with hemoglobin level (<120 g/L [<12 g/dL]) and number of extranodal sites with number of nodal sites (more than four). Low risk (zero or one factor) was assigned to 36% of patients, intermediate risk (two factors) to 37%, and poor risk (more than two factors) to 27% of patients.
|
INTERNATIONAL PROGNOSTIC INDEX FOR NON-HODGKIN’S LYMPHOMA |
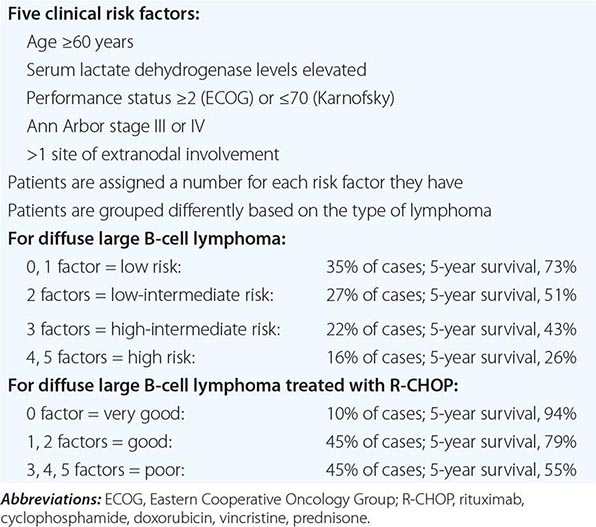
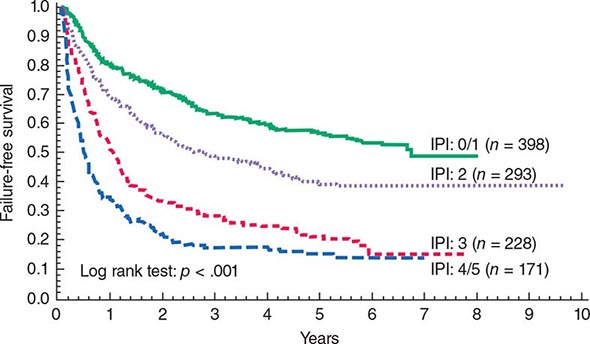
FIGURE 134-4 Relationship of International Prognostic Index (IPI) to survival. Kaplan-Meier survival curves for 1300 patients with various kinds of lymphoma stratified according to the IPI.
CLINICAL FEATURES, TREATMENT, AND PROGNOSIS OF SPECIFIC LYMPHOID MALIGNANCIES
PRECURSOR CELL B-CELL NEOPLASMS
Precursor B-Cell Lymphoblastic Leukemia/Lymphoma The most common cancer in childhood is B-cell ALL. Although this disorder can also present as a lymphoma in either adults or children, presentation as lymphoma is rare.
The malignant cells in patients with precursor B-cell lymphoblastic leukemia are most commonly of pre–B cell origin. Patients typically present with signs of bone marrow failure such as pallor, fatigue, bleeding, fever, and infection related to peripheral blood cytopenias. Peripheral blood counts regularly show anemia and thrombocytopenia but might show leukopenia, a normal leukocyte count, or leukocytosis based largely on the number of circulating malignant cells (Fig. 134-5). Extramedullary sites of disease are frequently involved in patients who present with leukemia, including lymphadenopathy, hepato- or splenomegaly, CNS disease, testicular enlargement, and/or cutaneous infiltration.
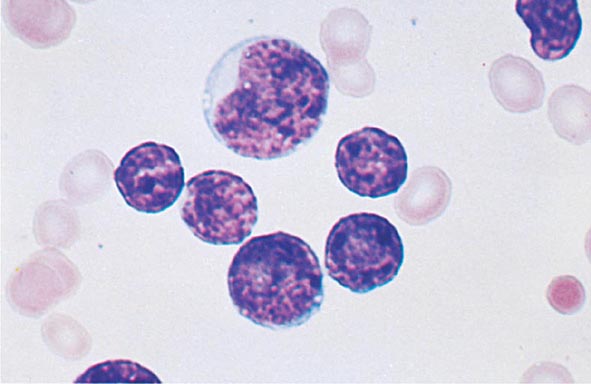
FIGURE 134-5 Acute lymphoblastic leukemia. The cells are heterogeneous in size and have round or convoluted nuclei, high nuclear/cytoplasmic ratio, and absence of cytoplasmic granules.
The diagnosis is usually made by bone marrow biopsy, which shows infiltration by malignant lymphoblasts. Demonstration of a pre–B cell immunophenotype (Fig. 134-2) and, often, characteristic cytogenetic abnormalities (Table 134-6) confirm the diagnosis. An adverse prognosis in patients with precursor B-cell ALL is predicted by a very high white cell count, the presence of symptomatic CNS disease, and unfavorable cytogenetic abnormalities. For example, t(9;22), frequently found in adults with B-cell ALL, has been associated with a very poor outlook. The bcr/abl kinase inhibitors have improved the prognosis.
MATURE (PERIPHERAL) B-CELL NEOPLASMS
B-Cell Chronic Lymphoid Leukemia/Small Lymphocytic Lymphoma B-cell CLL/small lymphocytic lymphoma represents the most common lymphoid leukemia, and when presenting as a lymphoma, it accounts for ~7% of non-Hodgkin’s lymphomas. Presentation can be as either leukemia or lymphoma. The major clinical characteristics of B-cell CLL/small lymphocytic lymphoma are presented in Table 134-10.
|
CLINICAL CHARACTERISTICS OF PATIENTS WITH COMMON TYPES OF NON-HODGKIN’S LYMPHOMA (NHL) |
The diagnosis of typical B-cell CLL is made when an increased number of circulating lymphocytes (i.e., >4 × 109/L and usually >10 × 109/L) is found (Fig. 134-6) that are monoclonal B cells expressing the CD5 antigen. Finding bone marrow infiltration by the same cells confirms the diagnosis. The peripheral blood smear in such patients typically shows many “smudge” or “basket” cells, nuclear remnants of cells damaged by the physical shear stress of making the blood smear. If cytogenetic studies are performed, trisomy 12 is found in 25–30% of patients. Abnormalities in chromosome 13 are also seen.
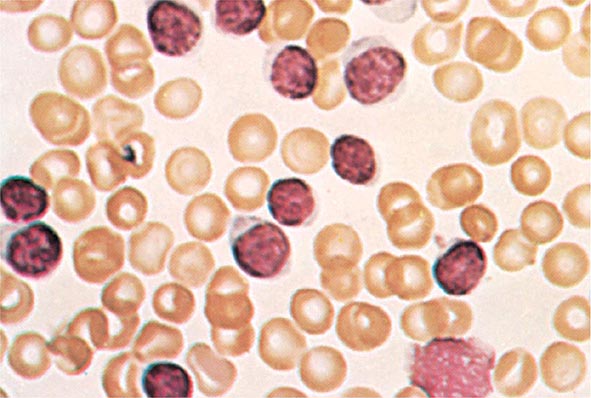
FIGURE 134-6 Chronic lymphocytic leukemia. The peripheral white blood cell count is high due to increased numbers of small, well-differentiated, normal-appearing lymphocytes. The leukemia lymphocytes are fragile, and substantial numbers of broken, smudged cells are usually also present on the blood smear.
If the primary presentation is lymphadenopathy and a lymph node biopsy is performed, pathologists usually have little difficulty in making the diagnosis of small lymphocytic lymphoma based on morphologic findings and immunophenotype. However, even in these patients, 70–75% will be found to have bone marrow involvement and circulating monoclonal B lymphocytes are often present.
The differential diagnosis of typical B-cell CLL is extensive (Table 134-1). Immunophenotyping will eliminate the T-cell disorders and can often help sort out other B-cell malignancies. For example, only mantle cell lymphoma and typical B-cell CLL are usually CD5 positive. Typical B-cell small lymphocytic lymphoma can be confused with other B-cell disorders, including lymphoplasmacytic lymphoma (i.e., the tissue manifestation of Waldenström’s macroglobulinemia), nodal marginal zone B-cell lymphoma, and mantle cell lymphoma. In addition, some small lymphocytic lymphomas have areas of large cells that can lead to confusion with diffuse large B-cell lymphoma. An expert hematopathologist is vital for making this distinction.
Typical B-cell CLL is often found incidentally when a complete blood count is done for another reason. However, complaints that might lead to the diagnosis include fatigue, frequent infections, and new lymphadenopathy. The diagnosis of typical B-cell CLL should be considered in a patient presenting with an autoimmune hemolytic anemia or autoimmune thrombocytopenia. B-cell CLL has also been associated with red cell aplasia. When this disorder presents as lymphoma, the most common abnormality is asymptomatic lymphadenopathy, with or without splenomegaly. The staging systems predict prognosis in patients with typical B-cell CLL (Table 134-7). The evaluation of a new patient with typical B-cell CLL/small lymphocytic lymphoma will include many of the studies (Table 134-11) that are used in patients with other non-Hodgkin’s lymphomas. In addition, particular attention needs to be given to detecting immune abnormalities such as autoimmune hemolytic anemia, autoimmune thrombocytopenia, hypogammaglobulinemia, and red cell aplasia. Molecular analysis of immunoglobulin gene sequences in CLL has demonstrated that about half the patients have tumors expressing mutated immunoglobulin genes and half have tumors expressing unmutated or germline immunoglobulin sequences. Patients with unmutated immunoglobulins tend to have a more aggressive clinical course and are less responsive to therapy. Unfortunately, immunoglobulin gene sequencing is not routinely available. CD38 expression is said to be low in the better-prognosis patients expressing mutated immunoglobulin and high in poorer-prognosis patients expressing unmutated immunoglobulin, but this test has not been confirmed as a reliable means of distinguishing the two groups. ZAP-70 expression correlates with the presence of unmutated immunoglobulin genes, but the assay is not yet standardized and widely available.
|
STAGING EVALUATION FOR NON-HODGKIN’S LYMPHOMA |
Abbreviations: CT, computed tomography; PET, positron emission tomography; SPECT, single-photon emission computed tomography.
Extranodal Marginal Zone B-Cell Lymphoma of MALT Type Extranodal marginal zone B-cell lymphoma of MALT type (MALT lymphoma) makes up ~8% of non-Hodgkin’s lymphomas. This small cell lymphoma presents in extranodal sites. It was previously considered a small lymphocytic lymphoma or sometimes a pseudolymphoma. The recognition that the gastric presentation of this lymphoma was associated with H. pylori infection was an important step in recognizing it as a separate entity. The clinical characteristics of MALT lymphoma are presented in Table 134-10.
The diagnosis of MALT lymphoma can be made accurately by an expert hematopathologist based on a characteristic pattern of infiltration of small lymphocytes that are monoclonal B cells and CD5 negative. In some cases, transformation to diffuse large B-cell lymphoma occurs, and both diagnoses may be made in the same biopsy. The differential diagnosis includes benign lymphocytic infiltration of extranodal organs and other small cell B-cell lymphomas.
MALT lymphoma may occur in the stomach, orbit, intestine, lung, thyroid, salivary gland, skin, soft tissues, bladder, kidney, and CNS. It may present as a new mass, be found on routine imaging studies, or be associated with local symptoms such as upper abdominal discomfort in gastric lymphoma. Most MALT lymphomas are gastric in origin. At least two genetic forms of gastric MALT exist: one (accounting for ~50% of cases) characterized by t(11;18)(q21;q21) that juxtaposes the amino terminal of the API2 gene with the carboxy terminal of the MALT1 gene creating an API2/MALT1 fusion product, and the other characterized by multiple sites of genetic instability including trisomies of chromosomes 3, 7, 12, and 18. About 95% of gastric MALT lymphomas are associated with H. pylori infection, and those that are do not usually express t(11;18). The t(11;18) usually results in activation of nuclear factor-κB (NF-κB), which acts as a survival factor for the cells. Lymphomas with t(11;18) translocations are genetically stable and do not evolve to diffuse large B-cell lymphoma. By contrast, t(11;18)-negative MALT lymphomas often acquire BCL6 mutations and progress to aggressive histology lymphoma. MALT lymphomas are localized to the organ of origin in ~40% of cases and to the organ and regional lymph nodes in ~30% of patients. However, distant metastasis can occur—particularly with transformation to diffuse large B-cell lymphoma. Many patients who develop this lymphoma will have an autoimmune or inflammatory process such as Sjögren’s syndrome (salivary gland MALT), Hashimoto’s thyroiditis (thyroid MALT), Helicobacter gastritis (gastric MALT), C. psittaci conjunctivitis (ocular MALT), or Borrelia skin infections (cutaneous MALT).
Evaluation of patients with MALT lymphoma follows the pattern (Table 134-11) for staging a patient with non-Hodgkin’s lymphoma. In particular, patients with gastric lymphoma need to have studies performed to document the presence or absence of H. pylori infection. Endoscopic studies including ultrasound can help define the extent of gastric involvement. Most patients with MALT lymphoma have a good prognosis, with a 5-year survival of ~75%. In patients with a low IPI score, the 5-year survival is ~90%, whereas it drops to ~40% in patients with a high IPI score.
Mantle Cell Lymphoma Mantle cell lymphoma makes up ~6% of all non-Hodgkin’s lymphomas. This lymphoma was previously placed in a number of other subtypes. Its existence was confirmed by the recognition that these lymphomas have a characteristic chromosomal translocation, t(11;14), between the immunoglobulin heavy chain gene on chromosome 14 and the bcl-1 gene on chromosome 11, and regularly overexpress the BCL-1 protein, also known as cyclin D1. Table 134-10 shows the clinical characteristics of mantle cell lymphoma.
The diagnosis of mantle cell lymphoma can be made accurately by an expert hematopathologist. As with all subtypes of lymphoma, an adequate biopsy is important. The differential diagnosis of mantle cell lymphoma includes other small cell B-cell lymphomas. In particular, mantle cell lymphoma and small lymphocytic lymphoma share a characteristic expression of CD5. Mantle cell lymphoma usually has a slightly indented nucleus.
The most common presentation of mantle cell lymphoma is with palpable lymphadenopathy, frequently accompanied by systemic symptoms. The median age is 63 years, and men are affected four times as commonly as women. Approximately 70% of patients will be stage IV at the time of diagnosis, with frequent bone marrow and peripheral blood involvement. Of the extranodal organs that can be involved, gastrointestinal involvement is particularly important to recognize. Patients who present with lymphomatosis polyposis in the large intestine usually have mantle cell lymphoma. Table 134-11 outlines the evaluation of patients with mantle cell lymphoma. Patients who present with gastrointestinal tract involvement often have Waldeyer’s ring involvement, and vice versa. The 5-year survival for all patients with mantle cell lymphoma is ~25%, with only occasional patients who present with a high IPI score surviving 5 years and ~50% of patients with a low IPI score surviving 5 years.
Follicular Lymphoma Follicular lymphomas make up 22% of non-Hodgkin’s lymphomas worldwide and at least 30% of non-Hodgkin’s lymphomas diagnosed in the United States. This type of lymphoma can be diagnosed accurately on morphologic findings alone and has been the diagnosis in the majority of patients in therapeutic trials for “low-grade” lymphoma in the past. The clinical characteristics of follicular lymphoma are presented in Table 134-10.
Evaluation of an adequate biopsy by an expert hematopathologist is sufficient to make a diagnosis of follicular lymphoma. The tumor is composed of small cleaved and large cells in varying proportions organized in a follicular pattern of growth (Fig. 134-7). Confirmation of B-cell immunophenotype and the existence of the t(14;18) and abnormal expression of BCL-2 protein are confirmatory. The major differential diagnosis is between lymphoma and reactive follicular hyperplasia. The coexistence of diffuse large B-cell lymphoma must be considered. Patients with follicular lymphoma are often subclassified into those with predominantly small cells, those with a mixture of small and large cells, and those with predominantly large cells. Although this distinction cannot be made simply or very accurately, these subdivisions do have prognostic significance. Patients with follicular lymphoma with predominantly large cells have a higher proliferative fraction, progress more rapidly, and have a shorter overall survival with simple chemotherapy regimens.
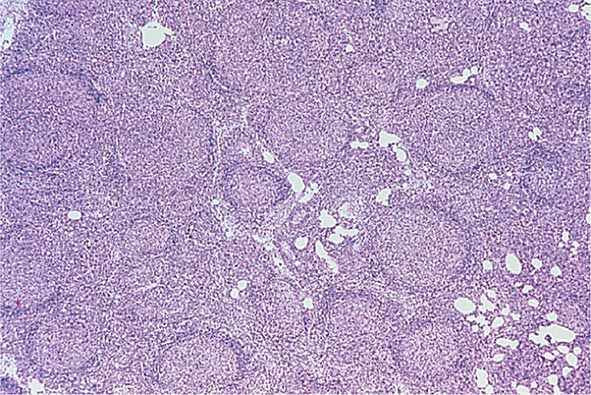
FIGURE 134-7 Follicular lymphoma. The normal nodal architecture is effaced by nodular expansions of tumor cells. Nodules vary in size and contain predominantly small lymphocytes with cleaved nuclei along with variable numbers of larger cells with vesicular chromatin and prominent nucleoli.
The most common presentation for follicular lymphoma is with new, painless lymphadenopathy. Multiple sites of lymphoid involvement are typical, and unusual sites such as epitrochlear nodes are sometimes seen. However, essentially any organ can be involved, and extranodal presentations do occur. Most patients do not have fevers, sweats, or weight loss, and an IPI score of 0 or 1 is found in ~50% of patients. Fewer than 10% of patients have a high (i.e., 4 or 5) IPI score. The staging evaluation for patients with follicular lymphoma should include the studies shown in Table 134-11.
Diffuse Large B-Cell Lymphoma Diffuse large B-cell lymphoma is the most common type of non-Hodgkin’s lymphoma, representing approximately one-third of all cases. This lymphoma makes up the majority of cases in previous clinical trials of “aggressive” or “intermediate-grade” lymphoma. Table 134-10 shows the clinical characteristics of diffuse large B-cell lymphoma.
The diagnosis of diffuse large B-cell lymphoma can be made accurately by an expert hematopathologist (Fig. 134-8). Cytogenetic and molecular genetic studies are not necessary for diagnosis, but some evidence has accumulated that patients whose tumors overexpress the BCL-2 protein might be more likely to relapse than others. A subset of patients have tumors with mutations in BCL6 and translocations involving MYC; these are called “double-hit” lymphomas and typically have more aggressive growth and are more poorly responsive to treatment than other diffuse large B-cell lymphomas. Patients with prominent mediastinal involvement are sometimes diagnosed as a separate subgroup having primary mediastinal diffuse large B-cell lymphoma. This latter group of patients has a younger median age (i.e., 37 years) and a female predominance (66%). Subtypes of diffuse large B-cell lymphoma, including those with an immunoblastic subtype and tumors with extensive fibrosis, are recognized by pathologists but do not appear to have important independent prognostic significance.
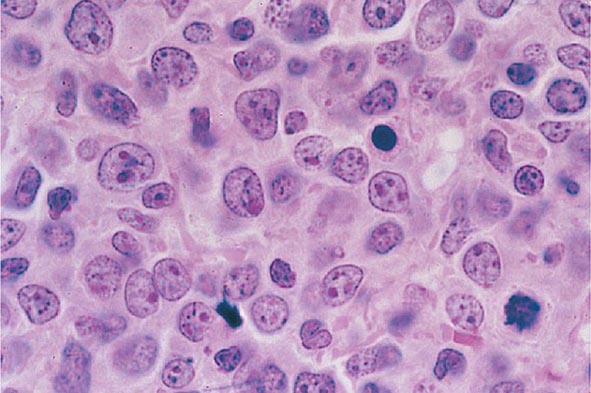
FIGURE 134-8 Diffuse large B-cell lymphoma. The neoplastic cells are heterogeneous but predominantly large cells with vesicular chromatin and prominent nucleoli.
Diffuse large B-cell lymphoma can present as either primary lymph node disease or at extranodal sites. More than 50% of patients will have some site of extranodal involvement at diagnosis, with the most common sites being the gastrointestinal tract and bone marrow, each being involved in 15–20% of patients. Essentially any organ can be involved, making a diagnostic biopsy imperative. For example, diffuse large B-cell lymphoma of the pancreas has a much better prognosis than pancreatic carcinoma but would be missed without biopsy. Primary diffuse large B-cell lymphoma of the brain is being diagnosed with increasing frequency. Other unusual subtypes of diffuse large B-cell lymphoma such as pleural effusion lymphoma and intravascular lymphoma have been difficult to diagnose and associated with a very poor prognosis.
Table 134-11 shows the initial evaluation of patients with diffuse large B-cell lymphoma. After a careful staging evaluation, ~50% of patients will be found to have stage I or II disease, and ~50% will have widely disseminated lymphoma. Bone marrow biopsy shows involvement by lymphoma in ~15% of cases, with marrow involvement by small cells more frequent than by large cells.
Burkitt’s Lymphoma/Leukemia Burkitt’s lymphoma/leukemia is a rare disease in adults in the United States, making up <1% of non-Hodgkin’s lymphomas, but it makes up ~30% of childhood non-Hodgkin’s lymphoma. Burkitt’s leukemia, or L3 ALL, makes up a small proportion of childhood and adult acute leukemias. Table 134-10 shows the clinical features of Burkitt’s lymphoma.
Burkitt’s lymphoma can be diagnosed morphologically by an expert hematopathologist with a high degree of accuracy. The cells are homogeneous in size and shape (Fig. 134-9). Demonstration of a very high proliferative fraction and the presence of the t(8;14) or one of its variants, t(2;8) (c-myc and the λ light chain gene) or t(8;22) (c-myc and the κ light chain gene), can be confirmatory. Burkitt’s cell leukemia is recognized by the typical monotonous mass of medium-sized cells with round nuclei, multiple nucleoli, and basophilic cytoplasm with cytoplasmic vacuoles. Demonstration of surface expression of immunoglobulin and one of the above-noted cytogenetic abnormalities is confirmatory.
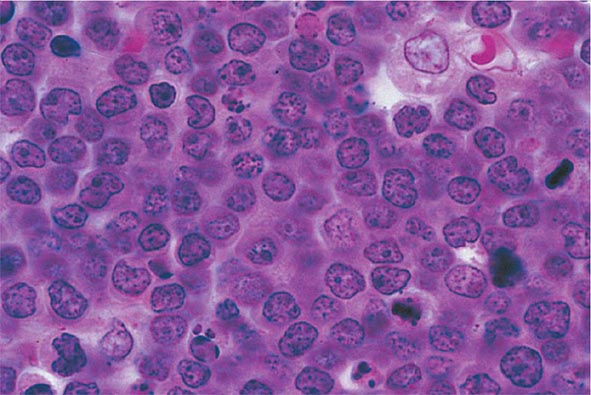
FIGURE 134-9 Burkitt’s lymphoma. The neoplastic cells are homogeneous, medium-sized B cells with frequent mitotic figures, a morphologic correlate of high growth fraction. Reactive macrophages are scattered through the tumor, and their pale cytoplasm in a background of blue-staining tumor cells gives the tumor a so-called starry sky appearance.
Three distinct clinical forms of Burkitt’s lymphoma are recognized: endemic, sporadic, and immunodeficiency-associated. Endemic and sporadic Burkitt’s lymphomas occur frequently in children in Africa, and the sporadic form occurs in Western countries. Immunodeficiency-associated Burkitt’s lymphoma is seen in patients with HIV infection.
Pathologists sometimes have difficulty distinguishing between Burkitt’s lymphoma and diffuse large B-cell lymphoma. In the past, a separate subgroup of non-Hodgkin’s lymphoma intermediate between the two was recognized. When tested, this subgroup could not be diagnosed accurately. Distinction between the two major types of B-cell aggressive non-Hodgkin’s lymphoma can sometimes be made based on the extremely high proliferative fraction seen in patients with Burkitt’s lymphoma (i.e., essentially 100% of tumor cells are in cycle) caused by c-myc deregulation.
Most patients in the United States with Burkitt’s lymphoma present with peripheral lymphadenopathy or an intraabdominal mass. The disease is rapidly progressive and has a propensity to metastasize to the CNS. Initial evaluation should always include an examination of cerebrospinal fluid to rule out metastasis in addition to the other staging evaluations noted in Table 134-11. Once the diagnosis of Burkitt’s lymphoma is suspected, a diagnosis must be made promptly, and staging evaluation must be accomplished expeditiously. This is the most rapidly progressive human tumor, and any delay in initiating therapy can adversely affect the patient’s prognosis.
Other B-Cell Lymphoid Malignancies B-cell prolymphocytic leukemia involves blood and marrow infiltration by large lymphocytes with prominent nucleoli. Patients typically have a high white cell count, splenomegaly, and minimal lymphadenopathy. The chances for a complete response to therapy are poor.
Hairy cell leukemia is a rare disease that presents predominantly in older males. Typical presentation involves pancytopenia, although occasional patients will have a leukemic presentation. Splenomegaly is usual. The malignant cells appear to have “hairy” projections on light and electron microscopy and show a characteristic staining pattern with tartrate-resistant acid phosphatase. Bone marrow is typically not able to be aspirated, and biopsy shows a pattern of fibrosis with diffuse infiltration by the malignant cells. Patients with this disorder have monocytopenia and are prone to unusual infections, including infection by Mycobacterium avium intracellulare, and to vasculitic syndromes. Hairy cell leukemia is responsive to chemotherapy with interferon α, pentostatin, or cladribine, with the latter being the usually preferred treatment. Clinical complete remissions with cladribine occur in the majority of patients, and long-term disease-free survival is frequent. Many of these tumors have the V600E BRAF mutation and accordingly are responsive to BRAF inhibitors like vemurafenib.
Splenic marginal zone lymphoma involves infiltration of the splenic white pulp by small, monoclonal B cells. This is a rare disorder that can present as leukemia as well as lymphoma. Definitive diagnosis is often made at splenectomy, which is also an effective therapy. This is an extremely indolent disorder, but when chemotherapy is required, the most usual treatment has been chlorambucil.
Lymphoplasmacytic lymphoma is the tissue manifestation of Waldenström’s macroglobulinemia (Chap. 136). Many of these tumors harbor a specific mutation, L265P, in MYD88, a change that leads to NF-κB activation. This type of lymphoma has been associated with chronic hepatitis C virus infection, and an etiologic association has been proposed. Patients typically present with lymphadenopathy, splenomegaly, bone marrow involvement, and occasionally peripheral blood involvement. The tumor cells do not express CD5. Patients often have a monoclonal IgM protein, high levels of which can dominate the clinical picture with the symptoms of hyperviscosity. Treatment of lymphoplasmacytic lymphoma can be aimed primarily at reducing the abnormal protein, if present, but will usually also involve chemotherapy. Chlorambucil, fludarabine, and cladribine have been used. The median 5-year survival for patients with this disorder is ~60%.
Nodal marginal zone lymphoma, also known as monocytoid cell lymphoma, represents ~1% of non-Hodgkin’s lymphomas. This lymphoma has a slight female predominance and presents with disseminated disease (i.e., stage III or IV) in 75% of patients. Approximately one-third of patients have bone marrow involvement, and a leukemic presentation occasionally occurs. The staging evaluation and therapy should use the same approach as used for patients with follicular lymphoma. Approximately 60% of the patients with nodal marginal zone lymphoma will survive 5 years after diagnosis.
Other more uncommon B-cell malignancies are discussed in Chap. 135e.
PRECURSOR T-CELL MALIGNANCIES
Precursor T-Cell Lymphoblastic Leukemia/Lymphoma Precursor T-cell malignancies can present either as ALL or as an aggressive lymphoma. These malignancies are more common in children and young adults, with males more frequently affected than females.
Precursor T-cell ALL can present with bone marrow failure, although the severity of anemia, neutropenia, and thrombocytopenia is often less than in precursor B-cell ALL. These patients sometimes have very high white cell counts, a mediastinal mass, lymphadenopathy, and hepatosplenomegaly. Precursor T-cell lymphoblastic lymphoma is most often found in young men presenting with a large mediastinal mass and pleural effusions. Both presentations have a propensity to metastasize to the CNS, and CNS involvement is often present at diagnosis.
MATURE (PERIPHERAL) T-CELL DISORDERS
Mycosis Fungoides Mycosis fungoides is also known as cutaneous T-cell lymphoma. This lymphoma is more often seen by dermatologists than internists. The median age of onset is in the mid-fifties, and the disease is more common in males and in blacks.
Mycosis fungoides is an indolent lymphoma with patients often having several years of eczematous or dermatitic skin lesions before the diagnosis is finally established. The skin lesions progress from patch stage to plaque stage to cutaneous tumors. Early in the disease, biopsies are often difficult to interpret, and the diagnosis may only become apparent by observing the patient over time. In advanced stages, the lymphoma can spread to lymph nodes and visceral organs. Patients with this lymphoma may develop generalized erythroderma and circulating tumor cells, called Sézary’s syndrome.
Rare patients with localized early-stage mycosis fungoides can be cured with radiotherapy, often total-skin electron beam irradiation. More advanced disease has been treated with topical glucocorticoids, topical nitrogen mustard, phototherapy, psoralen with ultraviolet A (PUVA), extracorporeal photopheresis, retinoids (bexarotene), electron beam radiation, interferon, antibodies, fusion toxins, histone deacetylase inhibitors, and systemic cytotoxic therapy. Unfortunately, these treatments are palliative.
Adult T-Cell Lymphoma/Leukemia Adult T-cell lymphoma/leukemia is one manifestation of infection by the HTLV-1 retrovirus. Patients can be infected through transplacental transmission, mother’s milk, blood transfusion, and by sexual transmission of the virus. Patients who acquire the virus from their mother through breast milk are most likely to develop lymphoma, but the risk is still only 2.5% and the latency averages 55 years. Nationwide testing for HTLV-1 antibodies and the aggressive implementation of public health measures could theoretically lead to the disappearance of adult T-cell lymphoma/leukemia. Tropical spastic paraparesis, another manifestation of HTLV-1 infection (Chap. 225e), occurs after a shorter latency (1–3 years) and is most common in individuals who acquire the virus during adulthood from transfusion or sex.
The diagnosis of adult T-cell lymphoma/leukemia is made when an expert hematopathologist recognizes the typical morphologic picture, a T-cell immunophenotype (i.e., CD4 positive), and the presence in serum of antibodies to HTLV-1. Examination of the peripheral blood will usually reveal characteristic, pleomorphic abnormal CD4-positive cells with indented nuclei, which have been called “flower” cells (Fig. 134-10).
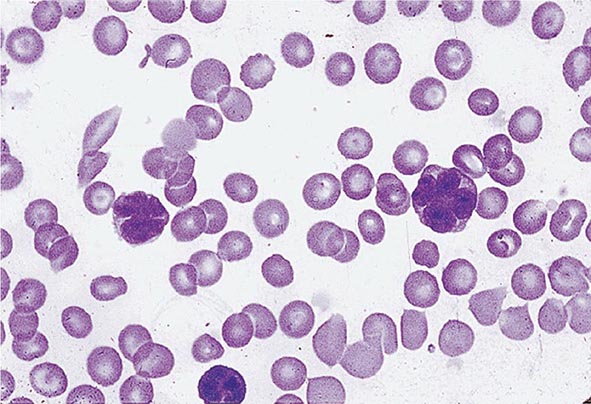
FIGURE 134-10 Adult T-cell leukemia/lymphoma. Peripheral blood smear showing leukemia cells with typical “flower-shaped” nucleus.
A subset of patients have a smoldering clinical course and long survival, but most patients present with an aggressive disease manifested by lymphadenopathy, hepatosplenomegaly, skin infiltration, pulmonary infiltrates, hypercalcemia, lytic bone lesions, and elevated LDH levels. The skin lesions can be papules, plaques, tumors, and ulcerations. Lung lesions can be either tumor or opportunistic infection in light of the underlying immunodeficiency in the disease. Bone marrow involvement is not usually extensive, and anemia and thrombocytopenia are not usually prominent. Although treatment with combination chemotherapy regimens can result in objective responses, true complete remissions are unusual, and the median survival of patients is ~7 months. A small phase II study reported a high response rate with interferon plus zidovudine and arsenic trioxide.
Anaplastic Large T/Null-Cell Lymphoma Anaplastic large T/null-cell lymphoma was previously usually diagnosed as undifferentiated carcinoma or malignant histiocytosis. Discovery of the CD30 (Ki-1) antigen and the recognition that some patients with previously unclassified malignancies displayed this antigen led to the identification of a new type of lymphoma. Subsequently, discovery of the t(2;5) and the resultant frequent overexpression of the anaplastic lymphoma kinase (ALK) protein confirmed the existence of this entity. This lymphoma accounts for ~2% of all non-Hodgkin’s lymphomas. Table 134-10 shows the clinical characteristics of patients with anaplastic large T/null cell lymphoma.
The diagnosis of anaplastic large T/null-cell lymphoma is made when an expert hematopathologist recognizes the typical morphologic picture and a T-cell or null-cell immunophenotype with CD30 positivity. Documentation of the t(2;5) and/or overexpression of ALK protein confirm the diagnosis. Some diffuse large B-cell lymphomas can also have an anaplastic appearance but have the same clinical course or response to therapy as other diffuse large B-cell lymphomas. A small percentage of anaplastic lymphomas are ALK negative.
Patients with anaplastic large T/null-cell lymphoma are typically young (median age, 33 years) and male (~70%). Some 50% of patients present in stage I/II, and the remainder present with more extensive disease. Systemic symptoms and elevated LDH levels are seen in about one-half of patients. Bone marrow and the gastrointestinal tract are rarely involved, but skin involvement is frequent. Some patients with disease confined to the skin have a different and more indolent disorder that has been termed cutaneous anaplastic large T/null-cell lymphoma and might be related to lymphomatoid papulosis.
Peripheral T-Cell Lymphoma The peripheral T-cell lymphomas make up a heterogeneous morphologic group of aggressive neoplasms that share a mature T-cell immunophenotype. They represent ~7% of all cases of non-Hodgkin’s lymphoma. A number of distinct clinical syndromes are included in this group of disorders. Table 134-10 shows the clinical characteristics of patients with peripheral T-cell lymphoma.
The diagnosis of peripheral T-cell lymphoma, or any of its specific subtypes, requires an expert hematopathologist, an adequate biopsy, and immunophenotyping. Most peripheral T-cell lymphomas are CD4+, but a few will be CD8+, both CD4+ and CD8+, or have an NK cell immunophenotype. No characteristic genetic abnormalities have yet been identified, but translocations involving the T-cell antigen receptor genes on chromosomes 7 or 14 may be detected. The differential diagnosis of patients suspected of having peripheral T-cell lymphoma includes reactive T-cell infiltrative processes. In some cases, demonstration of a monoclonal T-cell population using T-cell receptor gene rearrangement studies will be required to make a diagnosis.
The initial evaluation of a patient with a peripheral T-cell lymphoma should include the studies in Table 134-11 for staging patients with non-Hodgkin’s lymphoma. Unfortunately, patients with peripheral T-cell lymphoma usually present with adverse prognostic factors, with >80% of patients having an IPI score ≥2 and >30% having an IPI score ≥4. As this would predict, peripheral T-cell lymphomas are associated with a poor outcome, and only 25% of the patients survive 5 years after diagnosis. Treatment regimens are the same as those used for diffuse large B-cell lymphoma (omitting rituximab), but patients with peripheral T-cell lymphoma have a poorer response to treatment. Because of this poor treatment outcome, hematopoietic stem cell transplantation is often considered early in the care of young patients.
A number of specific clinical syndromes are seen in the peripheral T-cell lymphomas. Angioimmunoblastic T-cell lymphoma is one of the more common subtypes, making up ~20% of T-cell lymphomas. These patients typically present with generalized lymphadenopathy, fever, weight loss, skin rash, and polyclonal hypergammaglobulinemia. In some cases, it is difficult to separate patients with a reactive disorder from those with true lymphoma.
Extranodal T/NK-cell lymphoma of nasal type has also been called angiocentric lymphoma and was previously termed lethal midline granuloma. This disorder is more frequent in Asia and South America than in the United States and Europe. EBV is thought to play an etiologic role. Although most frequent in the upper airway, it can involve other organs. The course is aggressive, and patients frequently have the hemophagocytic syndrome. When marrow and blood involvement occur, distinction between this disease and leukemia might be difficult. Some patients will respond to aggressive combination chemotherapy regimens, but the overall outlook is poor.
Enteropathy-type intestinal T-cell lymphoma is a rare disorder that occurs in patients with untreated gluten-sensitive enteropathy. Patients are frequently wasted and sometimes present with intestinal perforation. The prognosis is poor. Hepatosplenic γδ T-cell lymphoma is a systemic illness that presents with sinusoidal infiltration of the liver, spleen, and bone marrow by malignant T cells. Tumor masses generally do not occur. The disease is associated with systemic symptoms and is often difficult to diagnose. Treatment outcome is poor. Subcutaneous panniculitis-like T-cell lymphoma is a rare disorder that is often confused with panniculitis. Patients present with multiple subcutaneous nodules, which progress and can ulcerate. Hemophagocytic syndrome is common. Response to therapy is poor. The development of the hemophagocytic syndrome (profound anemia, ingestion of erythrocytes by monocytes and macrophages, elevated ferritin levels) in the course of any peripheral T-cell lymphoma is generally associated with a fatal outcome.
HODGKIN’S LYMPHOMA
Classical Hodgkin’s Lymphoma Hodgkin’s lymphoma occurs in 9000 patients in the United States each year, and the disease does not appear to be increasing in frequency. Most patients present with palpable lymphadenopathy that is nontender; in most patients, these lymph nodes are in the neck, supraclavicular area, and axilla. More than half the patients will have mediastinal adenopathy at diagnosis, and this is sometimes the initial manifestation. Subdiaphragmatic presentation of Hodgkin’s lymphoma is unusual and more common in older males. One-third of patients present with fevers, night sweats, and/or weight loss—B symptoms in the Ann Arbor staging classification (Table 134-8). Occasionally, Hodgkin’s lymphoma can present as a fever of unknown origin. This is more common in older patients who are found to have mixed-cellularity Hodgkin’s lymphoma in an abdominal site. Rarely, the fevers persist for days to weeks, followed by afebrile intervals and then recurrence of the fever. This pattern is known as Pel-Ebstein fever. Hodgkin’s lymphoma can occasionally present with unusual manifestations. These include severe and unexplained itching, cutaneous disorders such as erythema nodosum and ichthyosiform atrophy, paraneoplastic cerebellar degeneration and other distant effects on the CNS, nephrotic syndrome, immune hemolytic anemia and thrombocytopenia, hypercalcemia, and pain in lymph nodes on alcohol ingestion.
The diagnosis of Hodgkin’s lymphoma is established by review of an adequate biopsy specimen by an expert hematopathologist. In the United States, most patients have nodular sclerosing Hodgkin’s lymphoma, with a minority of patients having mixed-cellularity Hodgkin’s lymphoma. Lymphocyte-predominant and lymphocyte-depleted Hodgkin’s lymphoma are rare. Mixed-cellularity Hodgkin’s lymphoma or lymphocyte-depletion Hodgkin’s lymphoma are seen more frequently in patients infected by HIV (Fig. 134-11). Hodgkin’s lymphoma is a tumor characterized by rare neoplastic cells of B-cell origin (immunoglobulin genes are rearranged but not expressed) in a tumor mass that is largely polyclonal inflammatory infiltrate, probably a reaction to cytokines produced by the tumor cells. The differential diagnosis of a lymph node biopsy suspicious for Hodgkin’s lymphoma includes inflammatory processes, mononucleosis, non-Hodgkin’s lymphoma, phenytoin-induced adenopathy, and nonlymphomatous malignancies.
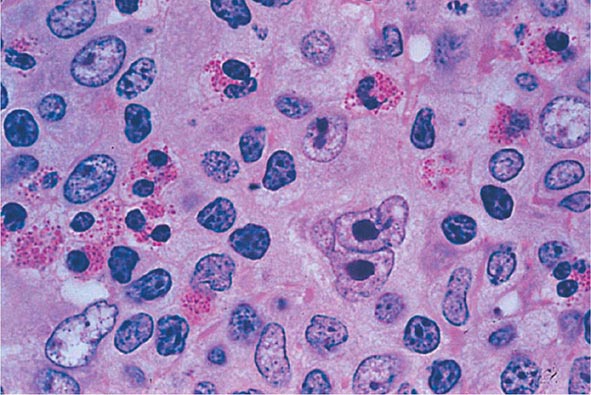
FIGURE 134-11 Mixed-cellularity Hodgkin’s lymphoma. A Reed-Sternberg cell is present near the center of the field; a large cell with a bilobed nucleus and prominent nucleoli giving an “owl’s eyes” appearance. The majority of the cells are normal lymphocytes, neutrophils, and eosinophils that form a pleomorphic cellular infiltrate.
The staging evaluation for a patient with Hodgkin’s lymphoma would typically include a careful history and physical examination; complete blood count; erythrocyte sedimentation rate; serum chemistry studies including LDH; chest radiograph; CT scan of the chest, abdomen, and pelvis; and bone marrow biopsy. Many patients would also have a PET scan or a gallium scan. Although rarely used, a bipedal lymphangiogram can be helpful. PET and gallium scans are most useful to document remission. Staging laparotomies were once popular for most patients with Hodgkin’s lymphoma but are now done rarely because of an increased reliance on systemic rather than local therapy.
Nodular Lymphocyte-Predominant Hodgkin’s Lymphoma Nodular lymphocyte-predominant Hodgkin’s lymphoma is now recognized as an entity distinct from classical Hodgkin’s lymphoma. Previous classification systems recognized that biopsies from a subset of patients diagnosed as having Hodgkin’s lymphoma contained a predominance of small lymphocytes and rare Reed-Sternberg cells (Fig. 134-11). A subset of these patients have tumors with nodular growth pattern and a clinical course that varied from that of patients with classical Hodgkin’s lymphoma. This is an unusual clinical entity and represents <5% of cases of Hodgkin’s lymphoma.
Nodular lymphocyte-predominant Hodgkin’s lymphoma has a number of characteristics that suggest its relationship to non-Hodgkin’s lymphoma. These include a clonal proliferation of B cells and a distinctive immunophenotype; tumor cells express J chain and display CD45 and epithelial membrane antigen (EMA) and do not express two markers normally found on Reed-Sternberg cells, CD30 and CD15. This lymphoma tends to have a chronic, relapsing course and sometimes transforms to diffuse large B-cell lymphoma.
The treatment of patients with nodular lymphocyte-predominant Hodgkin’s lymphoma is controversial. Some clinicians favor no treatment and merely close follow-up. In the United States, most physicians will treat localized disease with radiotherapy and disseminated disease with regimens used for patients with classical Hodgkin’s lymphoma. Regardless of the therapy used, most series report a long-term survival of >80%.
LYMPHOMA-LIKE DISORDERS
The most common condition that pathologists and clinicians might confuse with lymphoma is reactive, atypical lymphoid hyperplasia. Patients might have localized or disseminated lymphadenopathy and might have the systemic symptoms characteristic of lymphoma. Underlying causes include a drug reaction to phenytoin or carbamazepine. Immune disorders such as rheumatoid arthritis and lupus erythematosus, viral infections such as cytomegalovirus and EBV, and bacterial infections such as cat-scratch disease may cause adenopathy (Chap. 79). In the absence of a definitive diagnosis after initial biopsy, continued follow-up, further testing, and repeated biopsies, if necessary, constitute the appropriate approach, rather than instituting therapy.
Specific conditions that can be confused with lymphoma include Castleman’s disease, which can present with localized or disseminated lymphadenopathy; some patients have systemic symptoms. The disseminated form is often accompanied by anemia and polyclonal hypergammaglobulinemia, and the condition has been associated with overproduction of interleukin 6 (IL-6), in some cases produced by human herpesvirus 8 infection. Patients with localized disease can be treated effectively with local therapy, whereas the initial treatment for patients with disseminated disease is usually with systemic glucocorticoids. IL-6-directed therapy (tocilizumab) has produced short-term responses. Rituximab appears to produce longer remissions than tocilizumab.
Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) usually presents with bulky lymphadenopathy in children or young adults. The disease is usually nonprogressive and self-limited, but patients can manifest autoimmune hemolytic anemia.
Lymphomatoid papulosis is a cutaneous lymphoproliferative disorder that is often confused with anaplastic large cell lymphoma involving the skin. The cells of lymphomatoid papulosis are similar to those seen in lymphoma and stain for CD30, and T-cell receptor gene rearrangements are sometimes seen. However, the condition is characterized by waxing and waning skin lesions that usually heal, leaving small scars. In the absence of effective communication between the clinician and the pathologist regarding the clinical course in the patient, this disease will be misdiagnosed. Since the clinical picture is usually benign, misdiagnosis is a serious mistake.
ACKNOWLEDGMENT
James Armitage was a coauthor of this chapter in prior editions, and substantial material from those editions has been included here.




