[level-membership-for-hematology-oncology-and-palliative-medicine-category]20
Acute myeloid leukaemia
Classification
The WHO system has now largely superseded the French-American-British (FAB) classification. The newer classification reduces the bone marrow leukaemic blast cell percentage differentiating AML from myelodysplastic syndrome (see p. 50) from 30% to 20%. Other key changes include the creation of specific subtypes with non-random cytogenetic or equivalent molecular abnormalities, and the distinction of patients with multilineage dysplasia and also previous chemotherapy. The major FAB subtypes are included in the ‘other’ category with the exception of acute promyelocytic leukaemia (previously FAB M3) which is now in the ‘recurrent translocations’ group due to the inevitable presence of t(15;17). It can be seen (Table 20.1) that occasional cases of AML show megakaryocytic or erythroid differentiation. Gene mutations are likely to become increasingly important in classification.
Table 20.1
WHO classification of acute myeloid leukaemia
AML with recurrent genetic abnormalities1
AML with inv (16)(p13;q22) or t(16;16) (p13.1;q22) (M4 Eo)
AML with t(15;17)(q22;q12) (M3:M3V)
AML with myelodysplasia-related changes
Therapy related myeloid neoplasms
AML not otherwise specified2
AML with minimal differentiation (M0)
Acute myelomonocytic leukaemia (M4)
Acute monoblastic/ monocytic leukaemia(M4/5)
Acute erythroid leukaemia (M6)
Myeloid sarcoma
Myeloid proliferation related to Down syndrome
Blastic plasmacytoid dendritic cell neoplasm
Clinical features
One subtype of AML deserves special consideration as it must be treated as a medical emergency:
 AML with t(15;17)(q22;q12) (M3, M3V). More traditionally referred to as acute promyelocytic leukaemia, this disease is associated with a high incidence of disseminated intravascular coagulation (DIC) and a high risk of spontaneous bleeding into vital organs.
AML with t(15;17)(q22;q12) (M3, M3V). More traditionally referred to as acute promyelocytic leukaemia, this disease is associated with a high incidence of disseminated intravascular coagulation (DIC) and a high risk of spontaneous bleeding into vital organs.
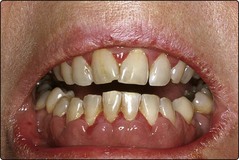
Tissue infiltration is more common in subtypes with monocytic morphology and immunophenotypic features (i.e. FAB M5) – patients often present with gum infiltration (Fig 20.1), lymphadenopathy, skin deposits and hepatosplenomegaly. Central nervous system (CNS) disease is rare in AML but most frequent in monocytic/monoblastic leukaemia.
Diagnosis
Diagnosis depends on a logical sequence of tests.
1. Blood count and film. The white cell count (WCC) is usually elevated (up to 200 × 109/L) but may be normal or low. There is often anaemia and thrombocytopenia. Usually there are leukaemic blast cells although occasionally these are absent. There may be dysplastic changes in other cells.
2. Bone marrow aspirate and trephine. The bone marrow is infiltrated by leukaemic blast cells (Fig 20.2). In more immature forms of AML morphological differentiation from acute lymphoblastic leukaemia (ALL) can be difficult.
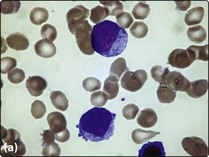
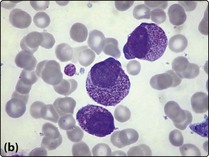
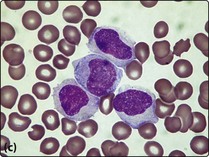
Fig 20.2 Bone marrow appearance in different FAB subtypes of AML.
(a) AML M2: the leukaemic blast cells show some granulocytic differentiation. (b) AML M3 (promyelocytic): the leukaemic cells show marked cytoplasmic granularity. (c) AML M4 (myelomonocytic): some of the leukaemic cells have monocytic features.
3. Cytochemistry. Special stains are used on bone marrow and blood smears to help differentiate myeloid and lymphoid blast cells. In AML there is positivity with Sudan black and myeloperoxidase – these stains are negative in ALL. AML with monocytic features will stain positively with a non-specific esterase stain.
4. Immunophenotyping. Both surface and intracellular antigens are analysed. Characteristic ‘myeloid’ antigens include CD13 and CD33 while CD34 positivity indicates a particularly immature cell of origin. Modern multicolour flow cytometry techniques allow quantitation of blast cells and correlate with both morphology features and the common balanced translocations.
5. Cytogenetics. A bone marrow sample is sent for analysis. Chromosome abnormalities are associated with particular AML subtypes and also give vital prognostic information (see Tables 20.1 and 20.2).
Table 20.2
Common genetic abnormalities in AML
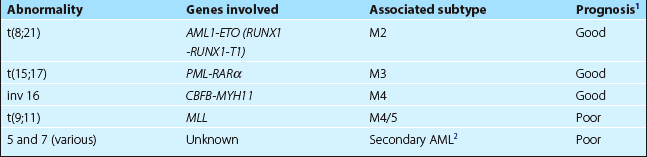
1Compared with AML with no detectable genetic abnormality.
2Antecedent events include chemotherapy, myelodysplastic syndrome and myeloproliferative disorders.
6. Molecular biology. Molecular techniques are increasingly important in classification, determining prognosis, and in monitoring response of disease to treatment (see p. 100). Sequential RT-PCR monitoring (e.g. in patients with the t(15;17) subtype in clinical remission) can predict the likelihood of relapse. Numerous genetic abnormalities are being identified – mutation of the tyrosine kinase receptor gene FLT3 is the commonest finding in patients with normal cytogenetics and carries a poorer prognosis. Other genetic mutations (e.g. nucleophosmin 1 (NPM1), two mutations of CEPPA gene) can favourably influence prognosis.
Prognosis
The major factors determining outcome are age, initial response to treatment and genetic abnormalities. Approximately 80–90% of younger patients will achieve a CR with conventional chemotherapy. Younger patients with ‘standard risk’ disease have 5-year survivals of 40–45% with optimal therapy; this compares with around 70% for ‘good risk’ and 20% for ‘poor risk’ groups. Older patients have a greater incidence of adverse cytogenetics (Fig 20.3) and tolerate chemotherapy less well, and CR and cure rates are much lower (see p. 93). Indeed, it may be kinder not to use chemotherapy in some elderly patients. In children, intensive chemotherapy gives 5-year survival rates of around 50%.
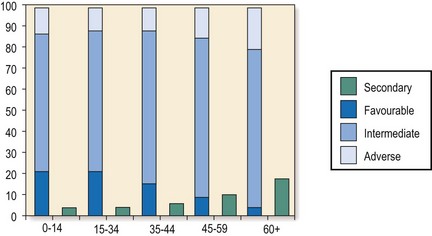
Fig 20.3 Relationship between age of presentation and characteristics of AML.
Older patients have a higher incidence of ‘poor risk’ disease. (Reprinted with permission from Smith ML, Hills RK, Grimwade D 2011 Independent prognostic variables in AML. Blood Reviews 25: 40.)
Acute myeloid leukaemia
 AML arises out of the malignant transformation of a myeloid precursor cell.
AML arises out of the malignant transformation of a myeloid precursor cell.

 Symptoms mainly result from anaemia, neutropenia and thrombocytopenia.
Symptoms mainly result from anaemia, neutropenia and thrombocytopenia.
 Prognosis largely depends on age, initial response to treatment, and genetic abnormalities.
Prognosis largely depends on age, initial response to treatment, and genetic abnormalities.

 Older patients tolerate chemotherapy less well and cure is rarely achievable.
Older patients tolerate chemotherapy less well and cure is rarely achievable.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]20
Acute myeloid leukaemia
Classification
The WHO system has now largely superseded the French-American-British (FAB) classification. The newer classification reduces the bone marrow leukaemic blast cell percentage differentiating AML from myelodysplastic syndrome (see p. 50) from 30% to 20%. Other key changes include the creation of specific subtypes with non-random cytogenetic or equivalent molecular abnormalities, and the distinction of patients with multilineage dysplasia and also previous chemotherapy. The major FAB subtypes are included in the ‘other’ category with the exception of acute promyelocytic leukaemia (previously FAB M3) which is now in the ‘recurrent translocations’ group due to the inevitable presence of t(15;17). It can be seen (Table 20.1) that occasional cases of AML show megakaryocytic or erythroid differentiation. Gene mutations are likely to become increasingly important in classification.
Table 20.1
WHO classification of acute myeloid leukaemia
AML with recurrent genetic abnormalities1
AML with inv (16)(p13;q22) or t(16;16) (p13.1;q22) (M4 Eo)
AML with t(15;17)(q22;q12) (M3:M3V)
AML with myelodysplasia-related changes
Therapy related myeloid neoplasms
AML not otherwise specified2
AML with minimal differentiation (M0)
Acute myelomonocytic leukaemia (M4)
Acute monoblastic/ monocytic leukaemia(M4/5)
Acute erythroid leukaemia (M6)
Myeloid sarcoma
Myeloid proliferation related to Down syndrome
Blastic plasmacytoid dendritic cell neoplasm
Clinical features
One subtype of AML deserves special consideration as it must be treated as a medical emergency:
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
