Acute lymphoblastic leukaemia
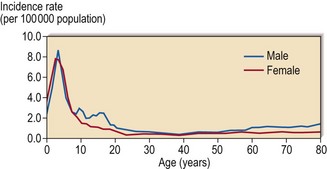
ALL has a peak incidence in childhood with a gradual rise in incidence in later years (Fig 21.1). The disease has distinct characteristics in children and adults. Childhood ALL is often curable by chemotherapy whereas cure is elusive in adult ALL. Poorer outcome in adult ALL is due to a combination of a greater frequency of high-risk leukaemia with more drug resistance, and less effective treatment regimens.
Classification
The French-American-British (FAB) morphological classification is based on characteristics of the blast cells, including cell size, nuclear–cytoplasmic ratio, number and size of nucleoli and the degree of cytoplasmic basophilia (Fig 21.2). Morphological classification is now less important than that based on immunophenotyping, cytogenetics and molecular analysis (Table 21.1).
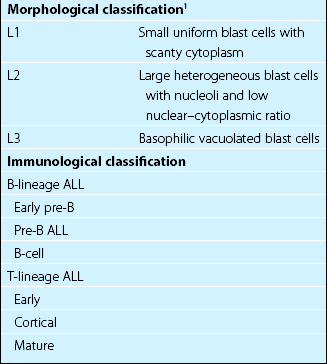
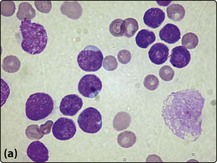
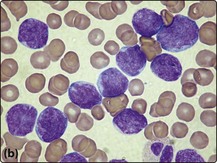
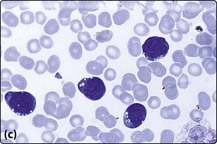
Fig 21.2 Morphology of ALL blast cells.
(a) L1 type; (b) L2 type; (c) L3 type. Note that the L2 cells have more cytoplasm and more prominent nucleoli than L1 cells. L3 type cells have cytoplasmic vacuolation.
In the selection of treatment it is important to differentiate between three broad groups; T-cell ALL, mature B-ALL and all other types of B-lineage ALL. Genetic abnormalities are becoming increasingly important in classification of ALL as they give vital prognostic information (Table 21.2).
Table 21.2
Chromosomal abnormalities in ALL
| Abnormality | Prognostic significance | |
| Numerical change | ||
| High hyperdiploidy (over 50 chromosomes) | Favourable | |
| Hyperdiploidy (47–50) | Intermediate | |
| Pseudodiploidy (46 with structural/numerical change) | Intermediate | |
| Hypodiploidy (less than 46) | Poor | |
| Structural abnormality | Genes involved | |
| Philadelphia chromosome, t(9;22)1 | BCR-ABL | Poor |
| t(12;21)2 | TEL-AML1 (ETV6-RUNX1) | Good |
| t(1;19) | E2A-PBX1 | Good |
| t(v;11q23) | MLL-AF4, ENL-MLL | Poor |
| t(8;14)3 | MYC | Good |
1Must be distinguished from the lymphoid blast crisis of chronic myeloid leukaemia.
2Occurs in 20% cases of childhood ALL. Not detectable by standard cytogenetics.
Diagnosis
2 Bone marrow aspirate and trephine
This is essential to confirm the diagnosis and for classification.
5 Cytogenetics
Cytogenetic analysis is doubly useful as structural abnormalities correlate with particular subtypes of ALL and both structural and numerical abnormalities give prognostic information (see Table 21.2). Varying patterns of cytogenetic abnormality may partly explain the different prognosis in children and adults. The Philadelphia chromosome, regarded as a marker of ‘incurability’ by chemotherapy, is found in 20–30% of adult cases but in only 2% of children.
6 Molecular techniques
Molecular analysis yields complementary and additional information to conventional cytogenetics (see Table 21.2). The cryptic t(12;21) creates a TEL-AML1 (ETV6-RUNX1) fusion gene – this is the commonest genetic rearrangement in childhood ALL and it can only be detected by molecular techniques. Although not yet routinely available in most laboratories, global gene expression profiling reveals distinct patterns in specific subtypes of ALL (see p. 100).
Management and outcome
The ultimate choice of management is influenced by a number of prognostic factors which have changed with improving treatment (Table 21.3). Where clinical and laboratory features predict a poor response to chemotherapy alone, more intensive treatments such as allogeneic stem cell transplantation (SCT) are considered. Of all the prognostic indices the most influential is age.
Table 21.3
Factors predicting poor prognosis in ALL




1With the exception of children under 1 year who have a worse prognosis than older children.
2Assessed from the bone marrow appearance after 14 days of chemotherapy.





