114 Acute Kidney Injury
Acute kidney injury (AKI) is characterized by an abrupt decrease in the glomerular filtration rate (GFR) that results in accumulation of nitrogenous waste products and an inability to maintain fluid and electrolyte homeostasis.1 AKI can result from decreased renal perfusion not severe enough to cause cellular injury; an ischemic, toxic, or obstructive injury of the renal tubule; a tubulointerstitial process with inflammation and edema; or a primary reduction in the filtering capacity of the glomerulus. If renal tubular and glomerular function is intact, but solute clearance is limited by factors compromising renal perfusion, the injury is termed prerenal azotemia. If renal dysfunction is related to obstruction of the urinary outflow tract, it is termed postrenal azotemia. AKI due to a primary intrarenal cause is called intrinsic renal injury or renal azotemia. Prerenal azotemic and intrinsic renal injury due to ischemia and nephrotoxins are responsible for most episodes of AKI.2,3
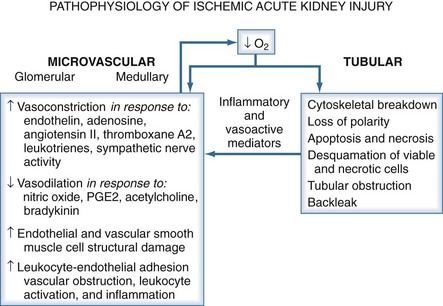
Renal blood flow is approximately 1200 mL/min and constitutes 20% of cardiac output. Given this apparently generous perfusion, it may seem surprising that the kidneys are so susceptible to hemodynamic insults. The majority of this perfusion (80%-90%), however, is to the renal cortex, where glomerular filtration occurs. The medulla is designed to concentrate and dilute urine. During urine concentration, the high osmotic gradient required for reabsorption of water is associated with a low rate of blood flow. In fact, oxygen tension in the outer medulla in the region of the metabolically active thick ascending limb of Henle is only around 10 mm Hg.4 This combination of low blood flow and oxygen tension in a metabolically active environment makes the kidneys very susceptible to ischemic injury.
 Prerenal Causes
Prerenal Causes
Prerenal azotemia is a consequence of reduction in renal perfusion without cellular injury. As such, this is a reversible process if the underlying cause is corrected. It may be secondary to decreased blood volume, as occurs with vomiting, dehydration, and hemorrhage, or it may be due to a reduction in the effective arterial blood volume, as in congestive heart failure and cirrhosis. Further, the administration of medications that interfere with the normal autoregulatory ability of the kidney can contribute to prerenal azotemia. In settings of diminished renal perfusion, administration of nonsteroidal antiinflammatory drugs (NSAIDs) or angiotensin-converting enzyme (ACE) inhibitors can precipitate overt prerenal azotemia.3
During prerenal azotemia, the renin-angiotensin-aldosterone system becomes activated secondary to a decrease in renal blood flow accompanied by increased activity of the adrenergic nervous system. Increased levels of angiotensin II and adrenergic activation serve to increase the proximal reabsorption of sodium, whereas aldosterone increases sodium reabsorption in the distal tubule. Together these actions decrease urine sodium concentration to less than 20 mmol/L and fractional excretion of sodium (FENa) to less than 1%.5
Prerenal azotemia accounts for approximately 70% of community-acquired cases of AKI6 and 40% of hospital-acquired cases.7 Therefore, prerenal causes should be excluded in all cases of AKI. Therapy of prerenal AKI involves reversing the underlying cause, such as volume replacement or discontinuation of offending agents.
 Postrenal Causes
Postrenal Causes
Postrenal AKI occurs when there is bilateral (or unilateral in the case of a single kidney) obstruction of urine flow. Intratubular pressure increases and in turn decreases net glomerular filtration pressure. Obstruction of urine flow is a relatively uncommon cause of AKI and is more common in the community than in the intensive care unit (ICU). Several series have placed the incidence of postrenal AKI at 3% to 25% of all cases of AKI.8,9,10 Postrenal AKI can be divided into renal and extrarenal causes. Extrarenal causes include prostatic disease, pelvic malignancy, and retroperitoneal disorders. Intrarenal causes include crystal deposition, as occurs in ethylene glycol ingestion, or uric acid nephropathy in tumor lysis syndrome. Cast formation and tubular obstruction also occur in light-chain diseases such as multiple myeloma.
Postrenal causes of AKI should be evaluated with renal ultrasonography and measurement of postvoid residual urine in the bladder (>50 mL is abnormal). It is important to rule out these causes rapidly, because the potential for renal recovery is inversely related to the duration of obstruction.11
 Intrarenal Causes
Intrarenal Causes
Intrarenal causes of AKI can be classified according to the anatomic location of the injury: glomerulus, tubule, vasculature, or interstitium. Suspicion of glomerulonephritis or vasculitis should be raised in a patient with renal failure who has an active urine sediment with red cells and red cell casts. In contrast, acute interstitial nephritis classically presents with pyuria and white cell casts in the urine; on occasion, hematuria is also present. Most cases of AKI from interstitial nephritis are drug related, commonly due to antibiotics or NSAIDs. Recovery usually occurs with removal of the offending agent and may be hastened by a short course of steroids, such as 60 to 80 mg of prednisone for 10 days. Tubular injury is most often either ischemic or toxic in nature and presents as acute tubular necrosis (ATN). This is the most common form of AKI encountered in the hospital and ICU10,12,13 and is the focus of this chapter.
In ischemic AKI, there is both tubular and vascular injury. In the tubules, an increase in intracellular calcium after ischemic injury activates the cysteine proteases calpain and caspase. This leads to necrosis and apoptosis as well as relocation of Na+/K+-ATPase from the basolateral membrane to the cytosol. This relocation interferes with normal vectorial transport of sodium and increases distal delivery of sodium chloride (NaCl). An increase in delivery of NaCl to the macula densa in the distal tubule activates tubuloglomerular feedback and further decreases GFR. Further, ischemia increases production of nitric oxide, which also causes cellular damage and detachment of epithelial cells from the basement membrane. Much of the deleterious action of nitric oxide is mediated through the generation of peroxynitrite from the combination of reactive oxygen species and nitric oxide. Cellular detachment is responsible for cast formation and tubular obstruction. These mechanisms all independently contribute to the decrease in renal function seen in ATN.14
In ischemic injury, the vascular endothelium is damaged and displays an exaggerated response to vasoconstrictor stimuli such as angiotensin II and endothelin-1 and a decreased response to vasodilators such as acetylcholine and bradykinin. In addition, there is a loss of autoregulatory capability. This loss of autoregulation in the setting of otherwise minor hemodynamic changes is likely responsible for the fresh ischemic lesions often seen on biopsy when recovery from AKI is delayed.15
The kidney’s susceptibility to toxic injury can be attributed to its functional properties. The kidneys receive 20% to 25% of the cardiac output, and there is extensive reabsorptive capacity as well as concentrating ability. All these factors contribute to the delivery of large amounts of toxin to the tubular epithelial cells. In addition, there is extensive biotransformation, generating toxic metabolites, and the high energy consumption with marginal oxygen delivery renders the tubules susceptible to toxic injury.16
An increasingly common form of AKI in the hospital is secondary to the use of contrast media. Nash and colleagues found contrast nephropathy to be the third most common form of AKI in the hospital.7 The pathogenesis involves both hemodynamic and toxic effects. Contrast media cause renal vasoconstriction and medullary ischemia as well as direct tubular toxicity.17 Patients with preexisting renal disease and diabetes are at high risk, as are patients who are volume depleted.
Differentiation of ATN from prerenal azotemia can be aided by evaluating urinary indices (Table 114-1).18 In established ATN, tubular function is impaired, and tubular sodium reabsorption is hindered. This results in a urine sodium value greater than 40 mmol/L and an FENa greater than 2%. Urine concentrating ability is also abnormal, resulting in isosthenuria with urine osmolality less than 350 mOsm/kg H2O.19 However, a low FENa may be seen in entities causing ATN, such as rhabdomyolysis and myoglobinuria,20 as well as in contrast-mediated AKI21 and sepsis.22 In patients with prerenal azotemia who are treated with diuretics that may obscure the FENa, fractional excretion of urea (FEUrea) or urine-to-plasma ratio of creatinine may be more discriminatory. An FEUrea less than 35% or a urine-to-plasma ratio of creatinine higher than 15 is indicative of prerenal azotemia.23 However, a subsequent study indicates that in patients with AKI administered diuretics, the distinction between transient and persistent AKI cannot be made accurately by means of FEUrea because it lacks specificity.24
TABLE 114-1 Laboratory and Microscopic Findings in Prerenal Azotemia and Acute Tubular Necrosis
| Laboratory Test | Prerenal Azotemia | Acute Tubular Necrosis |
|---|---|---|
| Urine osmolality (mOsm/kg H2O) | >500 | <400 |
| Urine sodium (mEq/L) | <20 | >40 |
| Urine plasma/creatinine ratio | >40 | <20 |
| Fractional excretion of sodium (%) | <1 | >2 |
| Urinary sediment | Normal, occasional hyaline cast | Renal tubular epithelial cells, granular and muddy brown casts |
Data from Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Ann Intern Med 2002;137:744-52.
 Epidemiology
Epidemiology
When the RIFLE criteria (risk, injury, failure, loss, end-stage renal failure) are employed, AKI is a common complication occurring in up to a third of ICU patients and is usually a manifestation of multiorgan failure syndrome.25–27
The most common cause of intrinsic renal failure is ATN.3 Specific causes of ATN can be classified as hemodynamically mediated AKI, such as in prolonged prerenal azotemia, hypotension, and sepsis; toxic AKI, secondary to antibiotics, chemotherapeutic agents, and contrast media; or postsurgical AKI. In a large prospective analysis by Liano and coworkers, sepsis was the most common cause (35%); postsurgical (25%) and toxic (31%) causes were also common.10 Many, if not most, patients have a multifactorial cause of AKI (Figure 114-1). Despite ever-improving supportive interventions in the ICU, the mortality rate for AKI has not changed in the last 3 decades, remaining at 40% to 80% depending on the study.28 It has been hypothesized that this continued poor prognosis is due to the changing patient population cared for in the ICU. Today, patients are older with greater comorbidities, and their renal disease most often develops in the setting of multiorgan failure.10,29 This high incidence of multiorgan failure has made it difficult to discern whether AKI itself causes increased mortality or whether it is a marker of severely ill patients. Several recent studies have found that AKI does in fact contribute to excess mortality in the setting of contrast nephropathy and cardiac surgery.30,31 In those patients who do survive, there is significant morbidity, with about 33% requiring long-term renal replacement therapy (RRT) and 28% requiring long-term institutionalization.32 As explained later, increasing RIFLE severity grades correspond with increasing mortality in patients. Hoste et al. reported that patients with a maximum score of RISK had a mortality rate of 8.8%, compared to 11.4% for INJURY and 26.3% for FAILURE. On the other hand, patients who had no evidence of AKI had a mortality rate of 5.5%.33
The risk of developing AKI in the ICU was evaluated by de Mendonca and associates, who found that seven characteristics, if present on admission, were associated with a high risk of developing AKI (Table 114-2).29 Several other studies addressed risk factors for mortality in the setting of AKI.7,10,12,34 As indicated in Table 114-3, the risk of death in those with AKI is increased by the presence of nonrenal organ failure; more severe renal dysfunction, as indicated by oliguria; sepsis; advanced age; and male gender. Liano and colleagues found that as the number of organ failures increased, mortality increased.10 With two organ failures, mortality was 53%; this increased to 80% with three organ failures and 100% with five organ failures.
Adapted from de Mendonca A et al. Acute renal failure in the ICU: risk factors and outcome evaluated by the SOFA score. Intensive Care Med 2000;26:915-21.
TABLE 114-3 Risk Factors for Mortality in Acute Kidney Injury
To further stratify the probability of death in critically ill patients, several severity-of-illness scoring systems have been developed. These indices help compare patients enrolled in clinical trials and better utilize finite resources to help those patients with the best chance of recovery. In large populations, these scoring indices have been successful in predicting outcome35; however, they do not discriminate well in patients with AKI.36 The renal parameters used in these scores consist of blood urea nitrogen (BUN), serum creatinine, and total urine output per day. With the latest version of the Acute Physiology and Chronic Health Evaluation (APACHE III), oliguric AKI constitutes just 12.7% of the maximal score, thereby underestimating the effect of AKI on mortality.37 Further, there is no correction for patients with AKI and a low serum creatinine, who also have a poor outcome, probably reflective of poor nutritional status.37 An attempt has therefore been made to develop more disease-specific indices, such as Liano and colleagues’ individual severity index, the Cleveland Clinic Foundation severity score, and the Project to Improve Care in Acute Renal Disease index. The majority of these indices were developed at single centers, and few have been validated outside the original institution. Also, the patient populations to which the indices were applied have differed, such as using all AKI patients or only dialyzed patients. Thus, there is no completely generalizable, validated bedside predictor for mortality in AKI patients.
 Definition
Definition
Acute renal failure (ARF) has traditionally been defined as an abrupt decrease in GFR with resultant retention of urea and other nitrogenous waste products along with dysregulation of body fluids and electrolytes. However, this is only a qualitative definition and not very helpful clinically, where a quantitative definition is required. Until recently, no agreement existed about how to best define, characterize, and study acute renal failure. This lack of a standard definition has been a major hindrance to the progress of clinical and basic research in this field. The term acute kidney injury was proposed by the Acute Kidney Injury Network (AKIN) as an alternative to ARF in order to encompass the entire range of failure based on recent data showing that a small change in serum creatinine influences outcome. The Acute Dialysis Quality Initiative (ADQI) was created to develop consensus and evidence-based guidelines for treatment and prevention of acute renal failure, with the goal of comparing studies and advancing research.38 The ADQI group proposed a consensus categorized definition—the RIFLE criteria39—which were validated and shown to correlate with hospital mortality and patient outcomes in several populations in large international databases. Subsequently, AKIN proposed a revision of the RIFLE criteria40,41,42 to better account for small changes in serum creatinine not captured by RIFLE. The following modifications were made (Table 114-4):
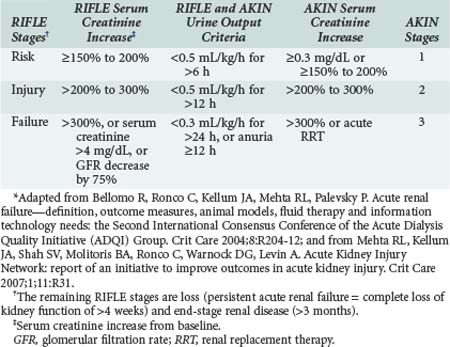
Existing evidence supports the validity of both RIFLE and AKIN criteria to identify groups of hospitalized patients with increased risk of death and/or need for RRT.39,40,46 Staging of AKI is relevant because with increased stage of AKI, the risk of death increases. Moreover, there is now mounting evidence of long-term risk of subsequent development of cardiovascular disease or chronic kidney disease and mortality even after resolution of AKI.47 Lo et al. recently studied the long-term sequelae of AKI in a retrospective analysis of the large Kaiser Permanente database using the years 1996-2003.48 This paper explored AKI and its correlation with long-term kidney disease and mortality in comparison with enrollees of the same healthcare organization who did not develop AKI and served as controls. Compared with controls, patients who suffered dialysis-dependent AKI during their hospitalization had a 28-fold increased risk of developing stage 4 or 5 CKD. There was also a more than twofold long-term risk of death in this group.
Given the difficulties of measuring function as an index of injury, there has been a search for identifying kidney injury markers of critically ill patients. This approach would be optimal because it could identify patients early in the course of AKI who would benefit from intervention. Several biomarkers have been proposed and are currently being investigated.49–51 These biomarkers include:
 Treatment
Treatment
With the increasing use of contrast agents in diagnostic and therapeutic procedures, prevention of contrast-mediated nephropathy has been studied extensively. Intravenous fluids have long been used to prevent contrast nephropathy, but in patients with chronic renal insufficiency, the incidence is still high. Therefore, multiple other agents have been studied. Solomon and coworkers found that both furosemide and mannitol when given with saline produced a worse outcome than saline alone in patients with chronic renal insufficiency.58 Dopamine59 and atrial natriuretic peptide60 have also failed to reduce contrast nephropathy. Two agents, acetylcysteine61 and fenoldopam,62 were found to decrease the incidence of contrast nephropathy in high-risk patients, but these findings were not verified in a study by Allaqaband and associates.63 In that trial, acetylcysteine and fenoldopam offered no additional benefit in patients with chronic renal insufficiency undergoing cardiovascular procedures. Landoni et al. recently performed a meta-analysis of 16 randomized trials of fenoldopam versus placebo or dopamine in 1290 patients who were in a variety of ICU or perioperative settings. They found that fenoldopam reduced the need for renal replacement and mortality in patients with AKI. However, because of the small size and heterogeneity of the studies included, a large multicenter appropriately powered trial will be needed to better define the role of fenoldopam in AKI.64 Currently, our recommendation for preventing contrast nephropathy in high-risk patients (Table 114-5) is adequate hydration, preferably with isotonic sodium bicarbonate,65,66,67 administration of 1200 mg acetylcysteine orally twice daily the day before and day of the procedure (given its tolerability and relative low cost), and the use of low-osmolar68 or iso-osmolar69 contrast media.
Dopamine has long been used to treat AKI. The renal effects of dopamine include an increase in GFR and an increase in sodium and water excretion. Clinically, the first response is an increase in diuresis.70 These responses occur in patients with normal renal function, but it is unknown whether they are also seen in those with AKI. In patients with early renal dysfunction (serum creatinine > 1.8 mg/dL or urine output < 0.5 mL/kg/h), dopamine did not alter peak serum creatinine or the need for RRT.71 This was confirmed in a meta-analysis to determine whether progression of AKI, need for RRT, or mortality were affected by dopamine.72
Aside from its lack of efficacy in AKI, dopamine has deleterious side effects. It hastened the onset of gut ischemia in an experimental model,73 and clinically it worsened contrast nephropathy.74 In cardiac surgery patients, dopamine was independently associated with an increased risk of postoperative atrial fibrillation.75 Higher doses may increase mortality,76 perhaps by worsening myocardial ischemia.77 Therefore, low-dose dopamine currently has no role in the treatment or prevention of AKI.
Diuretics are also frequently used in patients with AKI, especially in an attempt to convert oliguric into nonoliguric AKI, given the improved prognosis of the latter.78–80 Loop diuretics, most commonly furosemide, inhibit Na+/K+-ATPase in the thick ascending loop of Henle and therefore decrease the active reabsorption of sodium. Theoretically, this has some potential benefits, such as decreasing energy expenditure and increasing flow rate to flush out tubular casts. In the experimental setting, loop diuretics can be protective if administered before the insult. However, even when patients are successfully converted to nonoliguria, there is no reduction in the need for RRT or mortality.81,82 Cantarovich and colleagues studied the role of high-dose loop diuretics in a placebo-controlled clinical trial of 388 dialysis-requiring AKI patients. Despite the increase in urine output, there were no differences between the two groups in terms of patient survival, renal recovery rates, number of dialysis sessions required, or time on dialysis. In addition, cardiac surgery patients and patients with contrast nephropathy who were treated with furosemide had a worse outcome.83,84 A study by Mehta and coworkers found an increased mortality in AKI patients treated with diuretics.85 It is unclear why this occurred, but the authors speculated about a possible nephrotoxic effect of diuretics or a delay in the initiation of RRT because of increased urine output. However, the increased mortality occurred in patients who were not diuretic responsive, likely because of more severe AKI. These patients already had a worse prognosis, and whether diuretics may have worsened the outcome is unknown. Ho et al. recently conducted a comprehensive systematic review of the use of furosemide in AKI.86 They have shown that furosemide is not associated with any significant clinical benefits in the prevention or treatment of ARF in adults. High doses may be associated with an increased risk of ototoxicity. Although the use of loop diuretics in early or established AKI facilitates management of fluid balance, hyperkalemia, and hypercalcemia, and is indicated for these clinical purposes, any putative role in prevention or amelioration of AKI course is unproven. Therefore, if diuretics are temporarily employed for such indications, care must be taken to avoid delaying initiation of dialysis if clinically necessary.
Atrial natriuretic peptide is a hormone secreted by the cardiac atria that increases GFR and glomerular filtration pressure by dilating the afferent arteriole and constricting the efferent arteriole.87 It also decreases tubular reabsorption of sodium and chloride,88 redistributes medullary blood flow,89 disrupts tubuloglomerular feedback,90 and reverses endothelin-induced vasoconstriction.91 Mentzer and colleagues studied the perioperative effects of nesiritide (BNP type) in 303 patients with left ventricular dysfunction who were undergoing coronary artery bypass graft.92 They demonstrated short-term benefits of nesiritide on perioperative renal function as assessed by an attenuated increase in levels of serum creatinine, a reduction in calculated GFR loss, and a greater urine output 24 hours after surgery. This trial and other reports that have studied administration of natriuretic peptides during cardiac surgery were recently reviewed by Murray, who emphasized that in addition to such surrogate renal endpoints, future studies must demonstrate beneficial effects on overall survival and/or dialysis-free survival.93 Pending further studies, atrial natriuretic peptide cannot be recommended for prevention or therapy of ATN.
 Hemodynamic Management
Hemodynamic Management
Intravascular volume is critical in maintaining hemodynamic stability, tissue oxygenation, and organ function.94 In critically ill patients, it is increasingly being recognized that accurate assessment of volume status and appropriate use of fluid replacement may lead to better outcomes. In a study by Rivers and associates, it was shown that early goal-directed therapy (EGDT) based on optimizing the mixed control venous oxygen saturation in the first 6 hours resulted in decreased mortality in septic patients.95 Subsequent studies have replicated those results,96,97 and one of them showed a significantly improved prevention of AKI in patients randomized to EGDT compared to the standard care group.98 However, supranormal levels of cardiac index or mixed venous oxygen saturation did not decrease mortality.99 In addition, studies have shown increased mortality in patients with positive fluid balance and acute respiratory distress syndrome (ARDS).100–102
Both pseudo-ARDS and ARDS are frequently associated with sepsis. Sepsis is a vasodilated state in which systemic vascular resistance decreases and cardiac output increases. Studies in renal experimental animals have shown that vasodilatation with an arterial vasodilator such as minoxidil is associated with an increased albumin distribution space and a failure of interstitial hydrostatic pressure to rise during saline administration.103 These changes in interstitial Starling forces favor an increase in interstitial fluid volume during saline infusion. We frequently consult on ventilated ICU patients with AKI who have a 20-L positive fluid balance that has not been recognized in a quantitative sense because the pulmonary capillary wedge pressures are not considered elevated (<18 mm Hg). Excess saline fluid has been administered to resuscitate these vasodilated septic patients, leading to pulmonary edema, hypoxia, and ventilatory support. In the early stages, the majority of these patients do not have decreased pulmonary compliance (i.e., stiff lungs). However, these septic ICU patients with renal failure on prolonged respiratory support ultimately have a mortality as high as 80%. Patient mortality has been reported to begin increasing after 48 hours on a respirator. The potential barotrauma, oxygen toxicity, and pulmonary infections that may occur with prolonged ventilatory support frequently lead to stiff lungs and what virtually all authorities would term bona fide ARDS.
We believe that not distinguishing clinically between pseudo-ARDS and ARDS may be detrimental to ICU patients. Marked improvement in the pulmonary edema of pseudo-ARDS by diuresis or ultrafiltration may allow much earlier extubation and removal of ventilatory support before the development of pulmonary capillary damage and stiff lungs (i.e., ARDS). With ARDS and prolonged ventilatory support, a very high mortality occurs, particularly in the presence of renal failure and thus multiorgan failure. Recently, Bouchard and colleagues have reported results of a prospective multicenter observational study of 618 patients that aimed to determine whether fluid overload (>10% increase in body weight) in critically ill patients with AKI is associated with increased mortality. After adjustment for severity of illness, the study has shown that fluid overload was independently associated with mortality in those AKI patients who did and did not receive dialysis therapy.104 A randomized study by the ARDS clinical trials network demonstrated that pulmonary function in critically ill patients was worse in those treated with a liberal fluid management strategy (to achieve a mean central venous pressure [CVP] of ∼12 mm Hg) than in those who were treated with a conservative strategy (to achieve a mean CVP ∼8 mm Hg).105 Moreover, fewer patients in the conservative strategy group required dialysis than in the liberal strategy group. Several pediatric studies comprising more than 400 children have demonstrated an association between worsening fluid overload (higher than 10% to 20%) and mortality.106–108 Thus, there are reasons to believe that fluid overload is not just a marker but rather a pathologic factor in the high mortality of critically ill patients with AKI. Prospective randomized clinical trials will be needed to confirm this possibility. Until such studies are available, however, we recommend the avoidance of fluid overload in patients with AKI on the basis of knowledge of body weight changes and cumulative fluid balance for these patients.109
To aid in appropriate hemodynamic support, invasive monitoring has been used to guide therapy. Techniques such as the pulmonary artery catheter rely on measurement of filling pressures (e.g., CVP, pulmonary artery occlusion pressure) to estimate preload responsiveness. In critically ill patients, the relation between filling pressures and ventricular end-diastolic volume (preload) is often obscured by changes in ventricular compliance or changes in the pericardium or thorax.110 In addition, the pulmonary artery catheter has been linked to a worse outcome in patients.111,112 A positive response to fluid challenge can be predicted in mechanically ventilated patients by analyzing respiratory variations in pulse pressure. It has been shown that a change in pulse pressure greater than 15% during a single breath is more accurate in predicting an increase in cardiac output in response to volume loading than either right atrial pressure or wedge pressure.113,114
Fluid management in critical illness is aimed at improving organ perfusion. However, in inflammatory states such as sepsis, there may be major fluid shifts resulting in tissue edema despite intravascular depletion. Aside from the inflammatory cascade, vasodilatation itself can result in an increase in interstitial fluid volume, likely secondary to albumin escape from the vasculature.115 There are currently no clinical methods to detect the presence of capillary leak, apart from fluid administration having no effect on intravascular volume.110 Therefore, if only transient improvements in hemodynamics occur with fluid administration, or if there is a continuing need for fluid, it is likely the patient will best be served by a change to vasopressor agents.
When volume replacement is indicated, there is controversy over the optimal type of fluid. Crystalloids are the most common form of volume replacement, but their effect on plasma volume is limited. Each liter of fluid administered increases plasma volume 200 mL, but the intravascular half-life is only 20 to 30 minutes.94
Colloidal substances such as albumin, dextran, and hydroxyethyl starches, because they are macromolecules, are better retained within the intravascular space and have a greater effect on plasma volume. Albumin has been used for decades, but it is expensive and may cause an increase in mortality, according to the Cochrane Injuries Group.116 Nevertheless, a randomized controlled trial was conducted to compare human albumin with crystalloid in ICU patients (Saline versus Albumin Fluid Evaluation [SAFE] study). It indicated that albumin is safe, albeit no more effective than saline for fluid resuscitation. SAFE demonstrated no difference in renal outcomes, at least based on duration of RRT.117 Dextran cannot be recommended for plasma volume expansion because of serious side effects such as coagulation abnormalities118 and AKI.119
Hydroxyethyl starches (HESs) are polymers of amylopectin that vary in molecular weight and number of substitutions of hydroxyethyl groups. As molecular weight and number of substitutions increase, side effects also increase. HES 200/0.5 is a compound with a middle molecular weight and low substitution number. It has been studied in a number of situations such as perioperative volume replacement, cardiac surgery, trauma, and sepsis.120–122 A recent trial compared a “modern” HES preparation with a low-molecular-weight and low-molar substitution and a human albumin solution, given in cardiac surgery patients with preoperative compromised kidney function, showed that this type of HES solution had no negative influence on kidney integrity.123 In another study (Efficacy of Volume Substitution and Insulin Therapy in Severe Sepsis [VISEP]), severely septic patients were randomly assigned to receive either 10% pentastarch, a low-molecular-weight hydroxyethyl starch (HES 200/0.5), or modified Ringer’s lactate for fluid resuscitation. HES appeared to be harmful, leading to higher rate and longer duration of AKI, and its toxicity increased with accumulating doses.124 Aside from coagulation disorders, all hyperoncotic colloids may induce a pathologic entity known as osmotic nephrosis with potential impairment of kidney function.125 A systematic review of randomized controlled trials (RCTs) on the use of HES for fluid management in patients with sepsis (totaling 1062 patients) showed an almost twofold increased risk of AKI with HES compared with crystalloids.126 Lastly, a recent comprehensive Cochrane review concluded there is no evidence from RCTs that resuscitation with colloids instead of crystalloids reduces the risk of death in patients with trauma, burns, or following surgery.127 There is even evidence that colloids may be associated with a higher incidence of AKI. Given the relative efficacy and safety of crystalloids, it is prudent to utilize them in fluid resuscitation and limit colloid use to the framework of clinical trials.
In sepsis and septic shock, there is hypotension despite normal or increased cardiac output.128 The hypotension in sepsis is often unresponsive to fluid and requires administration of vasopressor agents. Because these agents cause vasoconstriction, there has been concern about their use in AKI. Norepinephrine causes a reduction in renal blood flow in healthy animals and humans.129 The ultimate effect of norepinephrine on renal blood flow, however, depends on the resulting increase in blood pressure and vascular resistance. Norepinephrine increases blood pressure via an α1-mediated increase in systemic vascular resistance and a β1-mediated increase in cardiac output. The increase in resistance can potentially decrease cardiac output by increasing afterload. In the kidney, the effect on renal vascular resistance depends on the increase in systemic pressure, with a decreased renal sympathetic tone causing vasodilatation as well as an autoregulatory vasoconstriction secondary to increased perfusion pressure and α1-mediated renal vasoconstriction.130 In a nonrandomized study, it was demonstrated that norepinephrine increased arterial blood pressure, urine output, and GFR.131 A large randomized trial comparing dopamine to norepinephrine as initial vasopressor in patients with septic shock showed no significant differences between groups with regard to renal function or mortality, though norepinephrine was associated with less tachycardia in the first hours and was superior with regard to survival in cardiogenic shock patients (De Backer et al., in press).
Vasopressin is a hormone secreted by the posterior pituitary; it increases systemic vascular resistance by activating V1a receptors on vascular smooth muscle. During septic shock, there is a biphasic response, with early high levels of endogenous vasopressin followed by a decrease.132 The renal effects of vasopressin are complex and involve an interplay between V1 and V2 receptors that regulates the antidiuretic function of vasopressin.132 Vasopressin is gaining attractiveness in the treatment of norepinephrine refractory shock patients.133 It increases blood pressure and enhances diuresis in hypotensive oliguric patients but has not yet been proven to enhance survival nor been shown to prevent or ameliorate AKI in the critically ill.134
It has been proposed that tight glycemic control can reduce the incidence and severity of AKI in critical patients. Recently Schetz et al. combined the renal endpoints of patients in a secondary analysis of two large randomized clinical trials.135 They demonstrated that tight glycemic control significantly reduced the incidence of severe AKI from 7.6% to 4.5%. The need for RRT was not decreased in the overall population, but it was significantly lower in surgical ICU patients than in medical ICU patients. However, further studies have highlighted significant concerns regarding the effectiveness and safety of using intensive insulin therapy with tight glycemic control to prevent or ameliorate morbidity and mortality of AKI and other forms of organ injury. The international Normoglycemia in Intensive Care Evaluation and Survival Using Glucose Algorithm Regulation (NICE-SUGAR) study was recently published.136 This large trial enrolled over 6000 patients and set out to definitively determine the risk/benefit of tight glycemic control in critically ill patients. It showed that in contrast to conventional insulin therapy, intensive glucose control increased mortality among these patients. A blood glucose target of ≤180 mg/dL resulted in lower mortality than a target of 81 to 108 mg/dL. It may therefore be prudent to use conventional insulin therapy in ICU patients at risk of AKI to target plasma glucose of less than 150 mg/dL, using a protocol to avoid hypoglycemia.
 Nutritional Support
Nutritional Support
Nutritional support in patients with AKI does not differ significantly from that of critically ill patients in general. The goals of nutritional support are preservation of lean body mass, stimulation of immune competence, repair, and wound healing. AKI affects water, electrolyte, and acid-base balance, but it also induces a change in protein, carbohydrate, and lipid metabolism.137 In patients with uncomplicated renal failure, oxygen consumption is approximately that of normal subjects. In the presence of sepsis or multiorgan failure, however, oxygen consumption is increased 20% to 30%.138 Therefore, energy expenditure is determined more by the underlying disease. Energy substrate should not exceed this requirement, and it is better to err on the side of slight underfeeding than overfeeding. Patients with AKI should be supplemented with 20 to 30 kcal/kg body weight per day. Even in hypermetabolic states such as sepsis, energy expenditure is rarely greater than 130% of calculated basic energy expenditure. In a randomized trial in AKI patients, comparing 30 and 40 kcal/kg/d energy provision, the higher energy prescription did not induce a more positive nitrogen balance but was associated with a higher incidence of hyperglycemia and hypertriglyceridemia and more positive fluid balance.139 Therefore, supplementation should not exceed 30 kcal/kg body weight per day.
The hallmark of metabolic alterations in AKI is activation of protein catabolism and release of amino acids from skeletal muscle. This process is responsible for the negative nitrogen balance encountered in critically ill patients. An underlying mechanism of protein catabolism is insulin resistance, which may be associated with increased mortality in AKI patients.140 Plasma insulin levels are elevated, but maximal insulin-stimulated glucose uptake is decreased by 50%. This insulin resistance leads to stimulated hepatic gluconeogenesis fueled by protein catabolism.141 The elevated level of gluconeogenesis coupled with insulin resistance also frequently leads to hyperglycemia. Other factors such as inflammatory cytokines (namely, tumor necrosis factor) and catecholamines are also involved in the hypercatabolism.142 To combat malnutrition in this setting, it is often necessary to use nutritional supplementation in the form of enteral or parenteral feeding.
Enteral nutrition has become the standard form of nutritional support in critically ill patients. Enteral feeding helps maintain gastrointestinal function, including acting as a barrier to microorganisms. Two clinical studies have suggested that enteral feeding is associated with improved outcome and survival in ICU patients.143,144 A meta-analysis by Heyland and colleagues reviewed 26 randomized trials comparing total parenteral nutrition with standard care and found no survival benefit and possible harm in medical ICU patients fed parenterally.145 Therefore, enteral support is recommended in critically ill patients with or without AKI.
Traditionally, nutrition has been delivered in the form of 50% to 80% carbohydrates. Recently, this has been the subject of study. In addition to providing calories, lipids also provide essential fatty acids. Essential fatty acids such as omega-3 polyunsaturated fatty acids and amino acids such as arginine have been found to stimulate the immune system. A prospective randomized trial of “immune-enhancing” enteral nutrition found that in patients who received adequate nutrition, those who received the immune-enhancing diet had a decrease in mortality and hospital stay.146 This study did not address AKI patients, but it is likely they would also benefit.
In the past, protein restriction was employed in AKI patients to control uremia, but this is likely to be detrimental to the patient and results in a profoundly negative nitrogen balance.147 With the advent of continuous modalities of RRT, it is possible to adequately supplement protein and control uremia. Therefore, some authors recommend aggressive protein replacement at 2.5 g/kg/d, as opposed to the standard 1 to 1.5 g/kg/d.147 However, no compelling data are currently available concerning the efficacy and safety of such high protein intakes. Also, it is important to realize that hypercatabolism cannot be simply overcome by increasing protein or amino acid intake. We suggest administering 0.8 to 1.2 g/kg/d of protein in patients with AKI without the need for dialysis, and 1 to 1.5 g/kg/d in patients with AKI on RRT.
 Indications for Nephrology Consultation
Indications for Nephrology Consultation
Currently there are wide variations in the timing of nephrology consultation in patients with AKI. Some physicians prefer to consult at the first rise in serum creatinine, whereas others wait until RRT is needed. In a study evaluating the effect of nephrology consultation on patient outcome, Mehta and associates found that a delay in nephrology consultation (>48 hours after ICU admission with AKI) led to higher mortality.148 In this study, patients with delayed consultation had a lower serum creatinine concentration and higher urine output but more organ failure and higher total body water. In the multivariate analysis, delayed consultation was no longer significant, but the trend was there. Why would early consultation affect mortality? It could result from delayed recognition of renal failure. Higher total body water likely leads to tissue edema and organ dysfunction (i.e., pulmonary edema), so in ICU patients, early recognition of AKI and its appropriate management may lead to better outcomes.
 Renal Replacement Therapy
Renal Replacement Therapy
Indications
As mentioned previously, up to a third of patients in the ICU develop AKI. Of those, 30% to 70% require RRT.25,26,27 Many practitioners delay initiating RRT as long as possible because of concerns that dialysis may delay the recovery of renal function.149,150 The optimal timing of initiation of dialysis is not defined. There is little disagreement in commencing dialysis in the presence of life-threatening conditions such as diuretic-resistant volume overload, severe hyperkalemia, acidosis, azotemia, or overt symptoms and signs of uremia such as encephalopathy and pericarditis. Medical treatment approaches for hyperkalemia accomplish intracellular shifts. When intermittent hemodialysis is used to correct hyperkalemia after such measures have been utilized, dialytic potassium removal will be reduced, and greater levels of post-dialysis potassium can occur.151 Metabolic acidosis is common in severe AKI but can be corrected with bicarbonate and should rarely require urgent dialysis if not accompanied by volume overload or uremia.152 Some poisons, drug overdose, and toxic compounds can contribute to acid-base disturbances and AKI. In such cases, dialysis can be supportive and facilitate removal of these substances and their metabolites. In acute salicylate poisoning, RRT is indicated when serum concentration is above 100 mg/dL and the patient exhibits altered mental status, pulmonary or cerebral edema, renal impairment, fluid overload that prevents administration of sodium bicarbonate, or clinical deterioration despite aggressive and appropriate supportive care.153 Ethylene glycol and methanol poisoning are important causes of anion-gap metabolic acidosis. Dialysis treatment has been shown to reduce development of subsequent AKI and organ dysfunction.154 Metformin-associated lactic acidosis may be an indication for dialysis, especially in critical patients who are more prone to death. According to a recent study, these are patients who show a low pH (<6.9) and high serum lactate and metformin concentrations.155
The level of azotemia at which RRT should begin is unknown. Several early retrospective studies that used blood urea or BUN suggested that early initiation of RRT resulted in survival improvements.156,157 More recent studies have continued to focus on BUN as the marker for starting dialysis. Single-center observational studies that were restricted to AKI after trauma158 and coronary artery bypass surgery159,160 suggested a benefit to dialysis initiation at lower BUN concentrations. A prospective multicenter observational study analyzed dialysis initiation, as inferred by BUN concentration, in 243 geographically and ethnically diverse patients.161 Survival rates were slightly lower for patients who started dialysis at higher BUN concentrations, despite a lower burden of organ system failure. In a prospective multicenter observational trial study conducted at 54 ICUs in 23 countries, timing of RRT was stratified into “early” or “late” by median urea at the time RRT started and also categorized temporally from ICU admission into early (<2 days), delayed (2-5 days), or late (>5 days).162 Timing by serum urea showed no significant difference in mortality. However, when timing was analyzed in relationship to ICU admission, late RRT (this may also be late AKI) was associated with greater crude mortality and covariate-adjusted mortality. Overall, late RRT was associated with a longer duration of RRT and stay in hospital and greater dialysis dependence.
Serum concentrations of BUN and creatinine are recognized to be inherently subject to a multitude of factors other than kidney function, such as catabolic rate, volume status, age, race, and muscle mass. It would therefore be prudent not to base dialysis initiation decision on a single BUN and creatinine threshold, but rather on the broader clinical context and trends of laboratory tests. Finally, it is important to consider the volume status when deciding the time for initiating RRT, because volume overload, as previously elaborated, emerged as an important factor associated with mortality in AKI. Table 114-6 depicts accepted indications for initiating RRT in the ICU.
TABLE 114-6 Potential Indications for Renal Replacement Therapy in the ICU
* Intermittent hemodialysis removes K+ more efficiently than continuous modalities.
Adapted from Bellomo R, Ronco C. Continuous haemofiltration in the intensive care unit. Crit Care 2000;4:339-45.
Adequate Dosing
In chronic hemodialysis patients, adequacy of dialysis is primarily determined by the level of small-solute (urea) clearance. This is determined by the Kt/V formula, where K is the dialysis membrane clearance of urea, t is the time on dialysis, and V is the volume of distribution of urea, which is equal to total body water. In chronic hemodialysis, a Kt/V of 1.2 per session is considered adequate.163 As can be seen from the formula, to increase urea clearance, one can increase the time on dialysis or increase the dialyzer clearance. Dialyzer clearance depends on blood flow and dialysate flow rates, as well as the inherent properties of the membrane.
In the United States, intermittent hemodialysis and continuous RRT are the most commonly used modalities of RRT, with sustained low-efficiency dialysis and other “hybrid” treatments used in fewer than 10% of patients. Intermittent hemodialysis is most commonly provided on a thrice-weekly or every-other-day schedule.164 Concerning intermittent modalities, there is no standard Kt/V for adequate dialysis in AKI currently, but it has been suggested that a higher target Kt/V confers better patient outcomes. Schiffl and colleagues studied 160 patients with AKI who were divided into two groups: one received daily hemodialysis, and the other alternate-day hemodialysis. It was found that daily hemodialysis resulted in less hypotension, sepsis, gastrointestinal bleeding, and respiratory failure, as well as a significant decrease in mortality.165 This study has been criticized because the Kt/V delivered to the alternate-day group was only 0.94, which is significantly less than the prescribed dose of 1.2. Therefore, the results could be explained by the fact that the alternate-day group received inadequate dialysis. In contrast, the VA/NIH Acute Renal Failure Trial Network Study (ATN study) did not find a benefit for a more intensive dosing strategy for RRT.166 This study compared intermittent hemodialysis (hemodynamically stable patients) or sustained low-efficiency dialysis (hemodynamically unstable patients) performed 3 (less intensive) versus 6 (more intensive) times a week in 563 critically ill patients with AKI and failure of at least one nonrenal organ or sepsis. The prescribed Kt/V per session was 1.2 to 1.4, and the actual delivered mean dose was 1.3 in the less intensive arm. The 60-day mortality rate and percentage of patients recovering renal function were similar in both groups. The Hannover Dialysis Outcomes study was a prospective randomized parallel group study that used intensified extended dialysis (dosed to maintain plasma urea levels <90 mg/dL) versus standard dialysis (dosed to maintain plasma urea levels between 120 and 150 mg/dL) on 14- and 28-day mortality and renal function.167 Mortality and frequency of renal function recovery were similar between the two groups. Based on these two well designed and performed clinical trials, it appears that increasing urea target clearances does not improve mortality or rates of renal recovery. Therefore, at least the smaller dose used in these trials should be pursued, with monitoring of the delivered dose of therapy to ascertain a minimum delivered Kt/V of 1.2 per treatment, or maintenance of plasma urea around 110 mg/dL when using extended or intermittent RRT in AKI patients. The significant difference between prescribed and delivered dialysis dose was studied by Evanson and coworkers, who found that the prescribed dose was a Kt/V of 1.25, whereas the dose delivered was only 1.04.168 These authors found that the most significant factor predicting actual delivered dose was the patient’s predialysis weight. It follows that a higher weight in critically ill patients represents higher total body water and therefore a larger volume of distribution of urea. This would be expected to decrease the Kt/V if it were not accounted for in the prescription of dialysis.
CRRT has been advocated in patients with AKI because of its ability to more efficiently remove solute169 and provide hemodynamic stability,170 but the optimal dosing in CRRT is not known. This form of RRT depends on convection, not diffusion, for solute clearance, which means that there is no dialysate involved, and solute is removed with water during ultrafiltration. Ronco and colleagues randomized 425 patients with AKI to increasing doses of continuous venovenous hemofiltration (CVVH).171 These investigators used three increasing doses of ultrafiltration—20, 35, and 45 mL/kg per hour—and found that mortality was 41%, 57%, and 58%, respectively. Survival was significantly lower in the 20 mL/kg group than in the other two groups. If the patient was septic, using the highest dose was beneficial.170,171 A more recent study by Bouman et al. in severely ill, ventilated patients with high severity scores was unable to detect a difference in mortality between high ultrafiltration volume (48 mL/kg/h) and low ultrafiltration volume (20 mL/kg/h).172 In the mentioned VA/ATN study, an approximate number of 201 patients received predilutional continuous venovenous hemodiafiltration (CVVHDF) in the less intensive arm (mean delivered effluent of 22 mL/kg/min) and 179 in the intensive therapy group (mean delivered effluent of 36 mL/kg/min).166 A higher dose of CRRT did not influence either mortality or renal recovery. The recently reported Randomized Evaluation of Normal versus Augmented Level of RRT (RENAL) study was conducted in 35 centers in Australia and New Zealand.173 It compared the effects of postdilutional CVVHDF doses of 25 and 40 mL/kg/h in 24-day and 90-day mortality rates of 1508 critically ill AKI patients. Treatment with higher-intensity regimen did not reduce mortality at 90 days. In conclusion, the results of these two recent well-designed and executed large clinical trials (ATN and RENAL) did not show any benefit of higher CRRTs doses for critical AKI patients beyond a threshold dose necessary to optimize clinical outcome. Therefore, when using CRRT for treating such patients, a minimum dose to be targeted may be the minimal efficient one proved in those trials: 20 to 25 mL/kg/h. However, it is important to pay careful attention to ensure that interruptions of treatment in the ICU are minimized. Beyond small-solute clearance, other aspects of dialysis adequacy such as volume management should be well attended to.
Modality
When RRT is indicated in the ICU for severe AKI, physicians have to choose between intermittent techniques such as traditional intermittent hemodialysis (IHD; used in end-stage kidney disease), slow low-efficiency dialysis (SLED), or continuous therapies such as CVVH and peritoneal dialysis (PD) (Table 114-7). SLED is performed by utilizing dialysis machines to deliver a slow dialysate flow for periods ranging from 8 to 12 hours per day. Advantages with this technique include high hemodynamic tolerance, excellent solute-removal capability, and capacity to be instituted using regular hemodialysis machines without acquiring new equipment. Availability and expertise with the technique, as well as the hemodynamic status of the patient, are typical determining factors for modality choice for AKI.
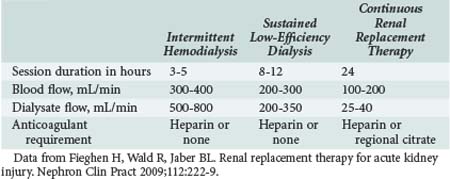
RRT is required in severe AKI to remove uremic toxins and maintain fluid, electrolyte, and acid-base balance. CRRT and IHD are effective therapies that may be utilized and exchanged according to the hemodynamic status or coagulation problems of the patient. The effect of these modalities on patient outcomes has been evaluated. Lins et al. performed a multicenter randomized controlled trial to study the effect of intermittent versus continuous dialysis modalities for the treatment of 316 AKI patients who were admitted to the ICU.174 They demonstrated that ICU stay, hospitalization, mortality, and renal recovery rates were not different between the groups. Moreover, two recent systematic reviews that collectively analyzed 45 studies found that outcomes were similar in critically ill AKI patients (stratified according to severity of illness) with CRRT and IHD for hemodynamically stable patients for relative risk of death, ICU mortality, in-hospital mortality, length of hospitalization, and requirement for chronic dialysis or renal recovery in survivors.175,176
Control of both uremia and volume is the major goal of RRT in AKI. A few studies have suggested that CRRT has advantages over intermittent therapies, including hemodynamic stability, improved survival, greater likelihood of renal recovery,175,177,178 and better fluid balance.179 IHD is complicated by hypotension in 20% to 30% of patients180; in hemodynamically unstable patients, this can significantly limit therapy and delay recovery of renal function. Therefore, some clinicians favor initiating CRRT for hemodynamically unstable patients with AKI, but this has not been supported by a prospective randomized trial181 or the earlier-mentioned systematic reviews.175,176 Moreover, Bagshaw et al. recently performed a systematic review and meta-analysis of nine randomized trials and concluded that it is impossible to make definitive recommendations about the initial RRT modality because of numerous issues related to study design, conduct, and quality of these trials.182 The main disadvantage of CRRT is the need for prolonged anticoagulation. SLED is gaining popularity as an intermittent modality in ICUs because of the aforementioned multiple advantages. Two RCTs that compared SLED with CVVH or CVVHD183,184 found similar outcomes with regard to hemodynamic stability and uremic clearance; furthermore, a decreased anticoagulation requirement was reported for SLED.184 Based on such evidence, all these modalities should be viewed as complementary. CRRT or SLED may be utilized for severe AKI with hemodynamic instability and transitioned to IHD once stability is attained. Peritoneal dialysis is an alternate modality for AKI where vascular access may be difficult, in conditions where anticoagulation may be problematic, in under-resourced regions, or following large disasters with mass casualties.185 A prospective randomized study of daily IHD versus PD in 120 AKI patients showed no difference in survival or recovery of renal function.186 These results contrast to a previous study that showed decreased survival associated with PD in comparison to CVVH187 and suggest that PD remains an acceptable option to CRRT when dosed appropriately.
In certain situations, CRRT is preferable to IHD, including in patients with or at risk for increased intracranial pressure. Studies have shown that CRRT prevents the increase in intracranial pressure associated with intermittent RRT.188,189 The use of CRRT in patients with severe sepsis or septic shock has also received much attention. Sepsis is associated with hemodynamic instability, making CRRT an attractive option. It has been shown that CRRT has beneficial effects on hemodynamics in animal models of sepsis.190 This is thought to be secondary to the removal of inflammatory cytokines by both convective and adsorptive measures. Hemofiltration membranes allow the ultrafiltration of mid-molecular-weight molecules such as cytokines. Further, the continuous blood/membrane contact allows the membrane to adsorb more mediators. There is some evidence that hemofiltration may provide some benefit in those with sepsis and AKI.191,192 Larger and adequately powered studies are needed to better define the role of this modality in AKI and sepsis.
Despite potential hemodynamic advantage over IHD, CRRT has some disadvantages as well. With CRRT, there is generally a need for continuous anticoagulation to prevent clotting of the filter. Although this is usually done with low-dose heparin, there is the risk of bleeding or heparin-induced thrombocytopenia. When a patient is a bleeding risk, a trial of no anticoagulation can be carried out, or regional anticoagulation with citrate is used in some centers. CRRT also requires more nursing support and is considerably more expensive than IHD.193
Dialysis Membrane
Early dialysis membranes were made of cellulose or its derivatives, and it has been shown that the hydroxyl radicals on the cellulose membranes were able to activate the complement system.194 These older membranes were thus not biocompatible. Newer synthetic polymers are less able to activate the complement cascade and also have the ability to bind activated complement, thereby decreasing systemic effects.195 Because of this decrease in immune activation, these membranes are considered biocompatible. In CRRT, there is continuous contact between the blood and membrane, making this interaction quite important. Despite mechanistic advantages, a recent meta-analysis found no advantage to biocompatible versus bio-incompatible membranes in terms of adult patient mortality and kidney function recovery rates.196
Membranes may also play a role in blood purification beyond that of solute clearance. There is interest in the ability of membranes to adsorb and bind cytokines from the blood. This is particularly attractive in sepsis, when there is dysregulation of the immune system with both pro- and antiinflammatory effects. A recent prospective RCT evaluated the use of polymyxin B hemoperfusion in patients with abdominal sepsis to reduce circulating endotoxin levels and improve clinical outcomes. Patients were randomized to conventional therapy or conventional therapy plus two sessions of polymyxin B hemoperfusion. The study showed that polymyxin B hemoperfusion added to conventional therapy significantly improved hemodynamics and organ dysfunction and reduced 28-day mortality.197 Abundant clinical studies show an association between inflammation and mortality in patients with AKI. Some investigators have suggested a potential role for using CRRT198,199 or IHD200 to remove cytokines and/or diminish the inflammatory response in septic patients with AKI. Such an approach is still experimental, and inadvertent removal of other potentially desirable middle molecules may be undertaken.
Buffer
In determining the adequacy of dialysis, factors other than solute clearance must be considered. One goal of RRT is to maintain normal acid-base balance in patients with AKI to prevent the complications of acidemia with regard to cardiovascular performance, hepatic metabolism, and hormonal response. To maintain normal pH, the dialysate must contain a buffer. Traditionally, the buffer choice was between bicarbonate and lactate, which metabolizes in the liver to bicarbonate on an equimolar basis under physiologic conditions. However, in critically ill patients with organ dysfunction and disordered tissue perfusion, it is possible that not all the anion will be converted to bicarbonate, resulting in increased serum lactate levels. Moreover, the increased lactate, without its redox partner pyruvate, can result in increased protein catabolism, myocardial depression,201 and worsening acidosis in patients with preexisting lactic acidosis.202,203
Bicarbonate-based solutions are currently the buffer of choice and are available in separated solutions that are mixed just before use. In a study by Barenbrock and colleagues, bicarbonate-based versus lactate-based fluid replacement was studied in patients with AKI treated with CVVH.204 They found that serum lactate concentration was significantly higher and bicarbonate lower in patients treated with lactate-based solution. In addition, they showed an increase in cardiovascular events and hypotension in patients treated with lactate solution.
Medication Dosing
During AKI, drugs normally eliminated by the kidney exhibit a markedly decreased clearance. The physiochemical characteristics of drugs affect their removal by dialysis and hemofiltration. The amounts of drug removed during these procedures can be sufficient to require supplemental dosing. For patients on hemodialysis, a supplemental dose of drug is most commonly given at the completion of the dialysis session.205 Drug clearance with CVVH is through convective transport, and it approximates the unbound drug concentration in plasma multiplied by the ultrafiltration rate.206 Drugs with molecular weights of less than 500 D are readily removed by either conventional hemodialysis or CVVH, but those with higher weights of 1000 to 5000 D are eliminated more efficiently by CVVH because of the use of high-flux membranes that allow the passage of larger molecules.
The volume of distribution greatly impacts the clearance of a drug, in that those with large distributions are likely to be more bound in the tissues. Therefore, only a small amount has access to the vasculature at any time. For these drugs, clearance with CVVH is greater than with intermittent therapies because of the continuous nature of the clearance.207 The extent of protein binding of a drug is important because the protein-drug complex is generally greater than 50,000 D. At this size, neither intermittent nor continuous therapies will efficiently remove the drug. However, the extent of protein binding is dependent on pH, uremia, concentration of free fatty acids, heparin therapy, and relative concentrations of drug and protein.208 In critically ill patients, serum albumin is often decreased, thereby making more drug available for clearance during RRT. Because of the potential toxicities, as well as the need to maintain therapeutic levels of multiple medications, it is important to consider and adjust medication dose during AKI and its therapy with RRT. Dosages of medications must be adjusted for the type of RRT, as well as for the specific characteristics of the drug.
 Conclusion
Conclusion
Despite extensive clinical experience and improvements in supportive care, the mortality rate of critically ill patients with AKI has not changed over the last 3 decades. However, new information is emerging about the diagnosis and treatment of AKI. Table 114-8 summarizes current recommendations for the care of patients with AKI.18 These recommendations are based on evidence from clinical trials as well as clinical judgment.
TABLE 114-8 Recommendations for Evaluation and Treatment of Acute Kidney Injury
ACE, angiotensin-converting enzyme; ARDS, acute respiratory distress syndrome; AKI, acute renal failure; ATN, acute tubular necrosis; CHF, congestive heart failure; NSAIDs, nonsteroidal antiinflammatory drugs; RBC, red blood cell.
From Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Ann Intern Med 2002;137:744-52.
Key Points
Bellomo R, Ronco C, Kellam JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8:R204-R212.
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Acute Kidney Injury Network. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31.
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365-3370.
Bagshaw SM, Uchino S, Bellomo R, Morimatsu H, Morgera S, Shetz M, et al. Timing of renal replacement therapy and clinical outcomes in critically ill patients with severe acute kidney injury. J Crit Care. 2009;24:129-140.
VA/NIH Acute Renal Failure Trial NetworkPalevsky PM, et al. Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med. 2008;3(359):7-20.
RENAL Replacement Therapy Study InvestigatorsBellomo R, et al. Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med. 2009;22(361):1627-1638.
1 Schrier RW, Edelstein CL. Acute Kidney Injury: Pathogenesis, Diagnosis and Management. In: Schrier RW, editor. Renal and Electrolyte Disorders. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2010:325-388.
2 Liaño F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int. 1996;50(3):811-818.
3 Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365(9457):417-430. 4
4 Brezis M, Rosen S. Hypoxia of the renal medulla—its implications for disease. N Engl J Med. 1995;332:647-655.
5 Blantz RC. Pathophysiology of pre-renal azotemia. Kidney Int. 1998;53:512-523.
6 Kaufman J, et al. Community-acquired acute renal failure. Am J Kidney Dis. 1991;17:191-198.
7 Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39(5):930-936.
8 Behrend T, Miller SB. Acute renal failure in the cardiac care unit: Etiologies, outcomes, and prognostic factors. Kidney Int. 1999;56:238-243.
9 Feest TG, Round A, Hamad S. Incidence of severe acute renal failure in adults: Results of a community based study. BMJ. 1993;306:481-483.
10 Liano F, et al. The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The Madrid Acute Renal Failure Study Group. Kidney Int Suppl. 1998;66:S16-S24.
11 Shapiro SR, Bennett AH. Recovery of renal function after prolonged unilateral ureteral obstruction. J Urol. 1976;115:136-140.
12 Brivet FG, et al. Acute renal failure in intensive care units—causes, outcome, and prognostic factors of hospital mortality: A prospective, multicenter study. French Study Group on Acute Renal Failure. Crit Care Med. 1996;24:192-198.
13 Silvester W, Bellomo R, Cole L. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Crit Care Med. 2001;29:1910-1915.
14 Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004 Jul;114(1):5-14.
15 Conger J. Does hemodialysis delay recovery from AKI? Semin Dialysis. 1990;3:146-148.
16 Cummings BS, Schnellmann RG. Pathophysiology of nephrotoxic cell injury. In: Schrier R, editor. Diseases of the Kidney and Urinary Tract. 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2007:962-985. 1034
17 McCullough PA. Radiocontrast-induced acute kidney injury. Nephron Physiol. 2008;109(4):61-72.
18 Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Ann Intern Med. 2002;137:744-752.
19 Miller TR, et al. Urinary diagnostic indices in acute renal failure: A prospective study. Ann Intern Med. 1978;89:47-50.
20 Corwin HL, Schreiber MJ, Fang LS. Low fractional excretion of sodium: Occurrence with hemoglobinuric- and myoglobinuric-induced acute renal failure. Arch Intern Med. 1984;144:981-982.
21 Fang LS, et al. Low fractional excretion of sodium with contrast media-induced acute renal failure. Arch Intern Med. 1980;140:531-533.
22 Vaz AJ. Low fractional excretion of urine sodium in acute renal failure due to sepsis. Arch Intern Med. 1983;143:738-739.
23 Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62:2223-2229.
24 Pépin MN, Bouchard J, Legault L, Éthier J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007;50:566-573.
25 Bagshaw SM, George C, Dinu I, Bellomo R. A multi-centre evaluation of the RIFLE criteria for early acute kidney injury in critically ill patients. Nephrol Dial Transplant. 2008;23(4):1203-1210.
26 Ostermann M, Chang RW. Acute kidney injury in the intensive care unit according to RIFLE. Crit Care Med. 2007;35(8):1837-1843. quiz 1852
27 Cruz DN, Bolgan I, Perazella MA, Bonello M, de Cal M, Corradi V, et alNorth East Italian Prospective Hospital Renal Outcome Survey on Acute Kidney Injury (NEiPHROS-AKI) Investigators. North East Italian Prospective Hospital Renal Outcome Survey on Acute Kidney Injury (NEiPHROS-AKI): targeting the problem with the RIFLE Criteria. Clin J Am Soc Nephrol. 2007;2(3):418-425.
28 Ympa YP, Sakr Y, Reinhart K, Vincent JL. Has mortality from acute renal failure decreased? A systematic review of the literature. Am J Med. 2005;118(8):827-832.
29 de Mendonca A, et al. Acute renal failure in the ICU: Risk factors and outcome evaluated by the SOFA score. Intensive Care Med. 2000;26:915-921.
30 Chertow GM, et al. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med. 1998;104:343-348.
31 Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality: A cohort analysis. JAMA. 1996;275:1489-1494.
32 Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66(4):1613-1621.
33 Hoste EA, Clermont G, Kersten A, Venkataraman R, Angus DC, De Bacquer D, et al. RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care. 2006;10(3):R73.
34 Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et alBeginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;17(294):813-818.
35 Halstenberg WK, Goormastic M, Paganini EP. Utility of risk models for renal failure and critically ill patients. Semin Nephrol. 1994;14:23-32.
36 Fiaccadori E, et al. Predicting patient outcome from acute renal failure comparing three general severity of illness scoring systems. Kidney Int. 2000;58:283-292.
37 Paganini EP, Larive B, Kanagasundaram NS. Severity scores and outcomes with acute renal failure in the ICU setting. Contrib Nephrol. 2001;132:181-195.
38 Ronco C, Kellum JA, Mehta R. Acute dialysis quality initiative (ADQI). Nephrol Dial Transplant. 2001;16:1555.
39 Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P. Acute renal failure—definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204-R212.
40 Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. 1
41 Levin A, Warnock DG, Mehta RL, et al. Improving outcomes from acute kidney injury: report of an initiative. Am J Kidney Dis. 2007;50:1.
42 Molitoris BA, Levin A, Warnock DG, et al. Improving outcomes from acute kidney injury. J Am Soc Nephrol. 2007;18:1992.
43 Schrier RW. Early intervention in acute kidney injury. Nat Rev Nephrol. 2010;6(1):56-59.
44 Murray PT, Palevsky PM. Acute kidney injury and critical care nephrology. Nephrology Self Assess Program. 2009;8:176-177.
45 Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31.
46 Thakar CV, Christianson A, Freyberg R, Almenoff P, Render ML. Incidence and outcomes of acute kidney injury in intensive care units: a Veterans Administration study. Crit Care Med. 2009;37(9):2552-2558.
47 Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;53(6):961-973.
48 Lo LJ, Go AS, Chertow GM, McCulloch CE, Fan D, Ordoñez JD, et al. Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int. 2009;76(8):893-899.
49 Nedelkov D, Nelson RW. Analysis of human urine protein biomarkers via biomolecular interaction analysis mass spectrometry. Am J Kidney Dis. 2001;38(3):481-487.
50 Hewitt SM, Dear J, Star RA. Discovery of protein biomarkers for renal diseases. J Am Soc Nephrol. 2004;15(7):1677-1689.
51 Han WK, Bonventre JV. Biologic markers for the early detection of acute kidney injury. Curr Opin Crit Care. 2004;10(6):476-482.
52 Coll E, Botey A, Alvarez L, Poch E, Quintó L, Saurina A, et al. Serum cystatin C as a new marker for noninvasive estimation of glomerular filtration rate and as a marker for early renal impairment. Am J Kidney Dis. 2000;36(1):29-34.
53 Shimizu-Tokiwa A, Kobata M, Io H, Kobayashi N, Shou I, Funabiki K, et al. Serum cystatin C is a more sensitive marker of glomerular function than serum creatinine. Nephron. 2002;92(1):224-226.
54 Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046-3052.
55 Westhuyzen J, Endre ZH, Reece G, Reith DM, Saltissi D, Morgan TJ. Measurement of tubular enzymuria facilitates early detection of acute renal impairment in the intensive care unit. Nephrol Dial Transplant. 2003;18(3):543-551.
56 Herget-Rosenthal S. One step forward in the early detection of acute renal failure. Lancet. 2005;365(9466):1205-1206. 2-8
57 Han WK, et al. Kidney injury molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62:237-244.
58 Solomon R, et al. Effects of saline, mannitol, and furosemide to prevent acute decreases in renal function induced by radiocontrast agents. N Engl J Med. 1994;331:1416-1420.
59 Hans SS, et al. Effect of dopamine on renal function after arteriography in patients with pre-existing renal insufficiency. Am Surg. 1998;64:432-436.
60 Kurnik BR, Allgren RL, Genter FC, Solomon RJ, Bates ER, Weisberg LS. Prospective study of atrial natriuretic peptide for the prevention of radiocontrast-induced nephropathy. Am J Kidney Dis. 1998;31(4):674-680.
61 Tepel M, et al. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med. 2000;343:180-184.
62 Kini AA, Sharma SK. Managing the high-risk patient: Experience with fenoldopam, a selective dopamine receptor agonist, in prevention of radiocontrast nephropathy during percutaneous coronary intervention. Rev Cardiovasc Med. 2001;2(Suppl 1):S19-S25.
63 Allaqaband S, et al. Prospective randomized study of N-acetylcysteine, fenoldopam, and saline for prevention of radiocontrast-induced nephropathy. Catheter Cardiovasc Interv. 2002;57:279-283.
64 Landoni G, Biondi-Zoccai GG, Tumlin JA, Bove T, De Luca M, Calabrò MG, et al. Beneficial impact of fenoldopam in critically ill patients with or at risk for acute renal failure: a meta-analysis of randomized clinical trials. Am J Kidney Dis. 2007;49(1):56-68.
65 Merten GJ, Burgess WP, Gray LV, Holleman JH, Roush TS, Kowalchuk GJ, et al. Prevention of contrast-induced nephropathy with sodium bicarbonate: a randomized controlled trial. JAMA. 2004 May 19;291(19):2328-2334.
66 Briguori C, Airoldi F, D’Andrea D, Bonizzoni E, Morici N, Focaccio A, et al. Renal Insufficiency Following Contrast Media Administration Trial (REMEDIAL): a randomized comparison of 3 preventive strategies. Circulation. 2007;115(10):1211-1217. 13
67 Wiedermann CJ, Joannidis M. Increasing evidence base for sodium bicarbonate therapy to prevent contrast media-induced acute kidney injury: little role of unpublished studies. Nephrol Dial Transplant. 27, 2009. [Epub ahead of print]
68 Rudnick MR, et al. Nephrotoxicity of ionic and nonionic contrast media in 1196 patients: A randomized trial. The Iohexol Cooperative Study. Kidney Int. 1995;47:254-261.
69 McCullough PA, Bertrand ME, Brinker JA, Stacul F. A meta-analysis of the renal safety of isoosmolar iodixanol compared with low-osmolar contrast media. J Am Coll Cardiol. 2006;48(4):692-699. 15
70 Gambaro G, et al. Diuretics and dopamine for the prevention and treatment of acute renal failure: A critical reappraisal. J Nephrol. 2002;15:213-219.
71 Bellomo R, et al. Low-dose dopamine in patients with early renal dysfunction: A placebo-controlled randomised trial. Australian and New Zealand Intensive Care Society (ANZICS) Clinical Trials Group. Lancet. 2000;356:2139-2143.
72 Friedrich JO, Adhikari N, Herridge MS, Beyene J. Meta-analysis: low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann Intern Med. 2005;142(7):510-524. 5
73 Segal JM, Phang PT, Walley KR. Low-dose dopamine hastens onset of gut ischemia in a porcine model of hemorrhagic shock. J Appl Physiol. 1992;73:1159-1164.
74 Weisberg LS, Kurnik PB, Kurnik BR. Dopamine and renal blood flow in radiocontrast-induced nephropathy in humans. Ren Fail. 1993;15:61-68.
75 Argalious M, Motta P, Khandwala F, Samuel S, Koch CG, Gillinov AM, et al. “Renal dose” dopamine is associated with the risk of new-onset atrial fibrillation after cardiac surgery. Crit Care Med. 2005;33(6):1327-1332.
76 Chertow GM, et al. Is the administration of dopamine associated with adverse or favorable outcomes in acute renal failure? Auriculin Anaritide Acute Renal Failure Study Group. Am J Med. 1996;101:49-53.
77 Rudis MI. Low-dose dopamine in the intensive care unit: DNR or DNRx? Crit Care Med. 2001;29:1638-1639.
78 Anderson RJ, et al. Nonoliguric acute renal failure. N Engl J Med. 1977;296:1134-1138.
79 Bellomo R, Ronco C. Indications and criteria for initiating renal replacement therapy in the intensive care unit. Kidney Int Suppl. 1998;66:S106-S109.
80 Klahr S, Miller SB. Acute oliguria. N Engl J Med. 1998;338:671-675.
81 Kleinknecht D, et al. Furosemide in acute oliguric renal failure: A controlled trial. Nephron. 1976;17:51-58.
82 Brown CB, Ogg CS, Cameron JS. High-dose furosemide in acute renal failure: A controlled trial. Clin Nephrol. 1981;15:90-96.
83 Lassnigg A, Donner E, Grubhofer G, Presterl E, Druml W, Hiesmayr M. Lack of renoprotective effects of dopamine and furosemide during cardiac surgery. J Am Soc Nephrol. 2000;11(1):97-104.
84 Solomon R, et al. Effects of saline, mannitol, and furosemide to prevent acute decreases in renal function induced by radiocontrast agents. N Engl J Med. 1994;331:1416-1420.
85 Mehta RL, et al. Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA. 2002;288:2547-2553.
86 Ho KM, Sheridan DJ. Meta-analysis of frusemide to prevent or treat acute renal failure. BMJ. 2006;333(7565):420. 26
87 Lanese DM, et al. Effects of atriopeptin III on isolated rat afferent and efferent arterioles. Am J Physiol. 1991;261(6 Pt 2):F1102-F1109.
88 Roy DR. Effect of synthetic ANP on renal and loop of Henle functions in the young rat. Am J Physiol. 1986;251(2 Pt 2):F220-F225.
89 Davis CL, Briggs JP. Effect of atrial natriuretic peptides on renal medullary solute gradients. Am J Physiol. 1987;253(4 Pt 2):F679-F684.
90 Margulies KB, Burnett JCJr. Atrial natriuretic factor modulates whole kidney tubuloglomerular feedback. Am J Physiol. 1990;259(1 Pt 2):R97-R101.
91 Opgenorth TJ, Novosadb EI. Atrial natriuretic factor and endothelin interactions in control of vascular tone. Eur J Pharmacol. 1990;191:351-357.
92 Mentzer RMJr, Oz MC, Sladen RN, Graeve AH, Hebeler RFJr, Luber JMJr, et alNAPA Investigators. Effects of perioperative nesiritide in patients with left ventricular dysfunction undergoing cardiac surgery: the NAPA Trial. J Am Coll Cardiol. 2007;49(6):716-726. 13
93 Murray P. Brain natriuretic peptide therapy to prevent acute kidney injury after cardiac surgery. Am J Kidney Dis. 2008;51(1):5-9.
94 Ragaller MJ, Theilen H, Koch T. Volume replacement in critically ill patients with acute renal failure. J Am Soc Nephrol. 2001;12(Suppl 17):S33-S39.
95 Rivers E, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368-1377.
96 Nguyen HB, Corbett SW, Menes K, Cho T, Daugharthy J, Klein W, et al. Early goal-directed therapy, corticosteroid, and recombinant human activated protein C for the treatment of severe sepsis and septic shock in the emergency department. Acad Emerg Med. 2006;13(1):109-113.
97 Jones AE, Focht A, Horton JM, Kline JA. Prospective external validation of the clinical effectiveness of an emergency department-based early goal-directed therapy protocol for severe sepsis and septic shock. Chest. 2007;132(2):425-432.
98 Lin SM, Huang CD, Lin HC, Liu CY, Wang CH, Kuo HP. A modified goal-directed protocol improves clinical outcomes in intensive care unit patients with septic shock: a randomized controlled trial. Shock. 2006;26(6):551-557.
99 Gattinoni L, et al. A trial of goal-oriented hemodynamic therapy in critically ill patients. SvO2 Collaborative Group. N Engl J Med. 1995;333:1025-1032.
100 Martin GS. Fluid balance and colloid osmotic pressure in acute respiratory failure: Emerging clinical evidence. Crit Care. 2000;4(Suppl 2):S21-S25.
101 Simmons RS, et al. Fluid balance and the adult respiratory distress syndrome. Am Rev Respir Dis. 1987;135:924-929.
102 Schuller D, et al. Fluid balance during pulmonary edema: Is fluid gain a marker or a cause of poor outcome? Chest. 1991;100:1068-1075.
103 Ragaller MJ, Theilen H, Koch T. Volume replacement in critically ill patients with acute renal failure. J Am Soc Nephrol. 2001;12(Suppl 17):S33-S39.
104 Bouchard J, Soroko SB, Chertow GM, Himmelfarb J, Ikizler TA, Paganini EP, et alProgram to Improve Care in Acute Renal Disease (PICARD) Study Group. Fluid accumulation, survival and recovery of kidney function in critically ill patients with acute kidney injury. Kidney Int. 2009;76(4):422-427.
105 National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials NetworkWiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354(24):2564-2575. 15
106 Foland JA, Fortenberry JD, Warshaw BL, Pettignano R, Merritt RK, Heard ML, et al. Fluid overload before continuous hemofiltration and survival in critically ill children: a retrospective analysis. Crit Care Med. 2004;32(8):1771-1776.
107 Hayes LW, Oster RA, Tofil NM, Tolwani AJ. Outcomes of critically ill children requiring continuous renal replacement therapy. J Crit Care. 2009;24(3):394-400.
108 Gillespie RS, Seidel K, Symons JM. Effect of fluid overload and dose of replacement fluid on survival in hemofiltration. Pediatr Nephrol. 2004;19(12):1394-1399.
109 Schrier RW. Fluid Administration in Critically Ill Patients with Acute Kidney Injury. Clin J Am Soc Nephrol. 2010 Feb 18. [Epub ahead of print]
110 Mehta RL, Clark WC, Schetz M. Techniques for assessing and achieving fluid balance in acute renal failure. Curr Opin Crit Care. 2002;8:535-543.
111 Connors AFJr, et al. The effectiveness of right heart catheterization in the initial care of critically ill patients. SUPPORT Investigators. JAMA. 1996;276:889-897.
112 Porter JM, Ivatury RR. In search of the optimal end points of resuscitation in trauma patients: A review. J Trauma. 1998;44:908-914.
113 Michard F, et al. Clinical use of respiratory changes in arterial pulse pressure to monitor the hemodynamic effects of PEEP. Am J Respir Crit Care Med. 1999;159:935-939.
114 Michard F, et al. Relation between respiratory changes in arterial pulse pressure and fluid responsiveness in septic patients with acute circulatory failure. Am J Respir Crit Care Med. 2000;162:134-138.
115 Sanz E, et al. Intravascular and interstitial fluid dynamics in rats treated with minoxidil. J Cardiovasc Pharmacol. 1990;15:485-492.
116 Human albumin administration in critically ill patients: Systematic review of randomised controlled trials. Cochrane Injuries Group Albumin Reviewers. BMJ. 1998;317:235-240.
117 SAFE Study InvestigatorsFinfer S, Bellomo R, McEvoy S, Lo SK, Myburgh J, Neal B, et al. Effect of baseline serum albumin concentration on outcome of resuscitation with albumin or saline in patients in intensive care units: analysis of data from the saline versus albumin fluid evaluation (SAFE) study. BMJ. 2006;333(7577):1044. 18
118 Messmer KF. The use of plasma substitutes with special attention to their side effects. World J Surg. 1987;11:69-74.
119 Mailloux L, et al. Acute renal failure after administration of low-molecular-weight dextran. N Engl J Med. 1967;277:1113-1118.
120 Boldt J, et al. The effects of albumin versus hydroxyethyl starch solution on cardiorespiratory and circulatory variables in critically ill patients. Anesth Analg. 1996;83:254-261.
121 Boldt J, et al. Influence of volume replacement with different HES-solutions on microcirculatory blood flow in cardiac surgery. Acta Anaesthesiol Scand. 1994;38:432-438.
122 Boldt J, et al. The influence of volume therapy and pentoxifylline infusion on circulating adhesion molecules in trauma patients. Anaesthesia. 1996;51:529-535.
123 Boldt J, Brosch C, Ducke M, Papsdorf M, Lehmann A. Influence of volume therapy with a modern hydroxyethyl starch preparation on kidney function in cardiac surgery patients with compromised renal function: a comparison with human albumin. Crit Care Med. 2007;35(12):2740-2746.
124 Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, et al. German Competence Network Sepsis (SepNet). Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358(2):125-139. 10
125 Dickenmann M, Oettl T, Mihatsch MJ. Osmotic nephrosis: acute kidney injury with accumulation of proximal tubular lysosomes due to administration of exogenous solutes. Am J Kidney Dis. 2008;51(3):491-503.
126 Wiedermann CJ. Systematic review of randomized clinical trials on the use of hydroxyethyl starch for fluid management in sepsis. BMC Emerg Med. 2008;8:1. 24
127 Perel P, Roberts I. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev 2007 17;(4):CD000567.
128 American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864-874.
129 Schetz M. Vasopressors and the kidney. Blood Purif. 2002;20:243-251.
130 De Vriese AS. Prevention and treatment of acute renal failure in sepsis. J Am Soc Nephrol. 2003;14:792-805.
131 Albanèse J, Leone M, Garnier F, Bourgoin A, Antonini F, Martin C. Renal effects of norepinephrine in septic and nonseptic patients. Chest. 2004;126(2):534-539.
132 Holmes CL, et al. Physiology of vasopressin relevant to management of septic shock. Chest. 2001;120:989-1002.
133 Albanèse J, Leone M, Delmas A, Martin C. Terlipressin or norepinephrine in hyperdynamic septic shock: a prospective, randomized study. Crit Care Med. 2005;33(9):1897-1902.
134 Russell JA, Walley KR, Singer J, Gordon AC, Hébert PC, Cooper DJ, et alVASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877-887. 28
135 Schetz M, Vanhorebeek I, Wouters PJ, Wilmer A, Van den Berghe G. Tight blood glucose control is renoprotective in critically ill patients. J Am Soc Nephrol. 2008;19(3):571-578.
136 NICE-SUGAR Study InvestigatorsFinfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360(13):1283-1297. 26
137 Druml W. Nutritional management of acute renal failure. Am J Kidney Dis. 2001;37(Suppl 2):S89-S94.
138 Schneeweiss B, et al. Energy metabolism in acute and chronic renal failure. Am J Clin Nutr. 1990;52:596-601.
139 Fiaccadori E, Maggiore U, Rotelli C, Giacosa R, Picetti E, Parenti E, et al. Effects of different energy intakes on nitrogen balance in patients with acute renal failure: a pilot study. Nephrol Dial Transplant. 2005;20(9):1976-1980.
140 Basi S, Pupim LB, Simmons EM, Sezer MT, Shyr Y, Freedman S, et al. Insulin resistance in critically ill patients with acute renal failure. Am J Physiol Renal Physiol. 2005;289(2):F259-F264.
141 Cianciaruso B, et al. Hepatic uptake and release of glucose, lactate, and amino acids in acutely uremic dogs. Metabolism. 1991;40:261-269.
142 Druml W. Protein metabolism in acute renal failure. Miner Electrolyte Metab. 1998;24:47-54.
143 Scheinkestel CD, Kar L, Marshall K, Bailey M, Davies A, Nyulasi I, et al. Prospective randomized trial to assess caloric and protein needs of critically Ill, anuric, ventilated patients requiring continuous renal replacement therapy. Nutrition. 2003;19(11-12):909-916.
144 Metnitz PG, Krenn CG, Steltzer H, Lang T, Ploder J, Lenz K, et al. Effect of acute renal failure requiring renal replacement therapy on outcome in critically ill patients. Crit Care Med. 2002;30(9):2051-2058.
145 Heyland DK, et al. Total parenteral nutrition in the critically ill patient: A meta-analysis. JAMA. 1998;280:2013-2019.
146 Atkinson S, Sieffert E, Bihari D. A prospective, randomized, double-blind, controlled clinical trial of enteral immunonutrition in the critically ill. Guy’s Hospital Intensive Care Group. Crit Care Med. 1998;26:1164-1172.
147 Bellomo R. How to feed patients with renal dysfunction. Blood Purif. 2002;20:296-303.
148 Mehta RL, et al. Nephrology consultation in acute renal failure: Does timing matter? Am J Med. 2002;113:456-461.
149 Solez K, Morel-Maroger L, Sraer JD. The morphology of “acute tubular necrosis” in man: Analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine (Baltimore). 1979;58:362-376.
150 Conger JD, Hammond WS. Renal vasculature and ischemic injury. Ren Fail. 1992;14:307-310.
151 Allon M, Shanklin N. Effect of albuterol treatment on subsequent dialytic potassium removal. Am J Kidney Dis. 1995;26(4):607-613.
152 Gauthier PM, Szerlip HM. Metabolic acidosis in the intensive care unit. Crit Care Clin. 2002;18(2):289-308. vi
153 Traub SJ. Aspirin poisoning in adults. In: UpToDate. Waltham, MA: UpToDate; 2009.
154 Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol. 2008;3(1):208-225.
155 Dell’Aglio DM, Perino LJ, Kazzi Z, Abramson J, Schwartz MD, Morgan BW. Acute metformin overdose: examining serum pH, lactate level, and metformin concentrations in survivors versus nonsurvivors: a systematic review of the literature. Ann Emerg Med. 2009;54(6):818-823.
156 Fischer RP, et al. Early dialysis in the treatment of acute renal failure. Surg Gynecol Obstet. 1966;123:1019-1023.
157 Kleinknecht D, et al. Uremic and non-uremic complications in acute renal failure: Evaluation of early and frequent dialysis on prognosis. Kidney Int. 1972;1:190-196.
158 Gettings LG, Reynolds HN, Scalea T. Outcome in post-traumatic acute renal failure when continuous renal replacement therapy is applied early vs late. Intensive Care Med. 1999;25:805-813.
159 Demirkiliç U, Kuralay E, Yenicesu M, Cağlar K, Oz BS, Cingöz F, et al. Timing of replacement therapy for acute renal failure after cardiac surgery. J Card Surg. 2004;19(1):17-20.
160 Elahi MM, Lim MY, Joseph RN, Dhannapuneni RR, Spyt TJ. Early hemofiltration improves survival in post-cardiotomy patients with acute renal failure. Eur J Cardiothorac Surg. 2004;26(5):1027-1031.
161 Liu KD, Himmelfarb J, Paganini E, Ikizler TA, Soroko SH, Mehta RL, et al. Timing of initiation of dialysis in critically ill patients with acute kidney injury. Clin J Am Soc Nephrol. 2006;1(5):915-919.
162 Bagshaw SM, Uchino S, Bellomo R, Morimatsu H, Morgera S, Schetz M, et al. Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Timing of renal replacement therapy and clinical outcomes in critically ill patients with severe acute kidney injury. J Crit Care. 2009;24(1):129-140.
163 I. NKF-K/DOQI Clinical Practice Guidelines for Hemodialysis Adequacy: Update 2000. Am J Kidney Dis. 2001;37(1 Suppl 1):S7-S64.
164 Overberger P, Pesacreta M, Palevsky PM, VA/NIH Acute Renal Failure Trial Network. Management of renal replacement therapy in acute kidney injury: a survey of practitioner prescribing practices. Clin J Am Soc Nephrol. 2007;2(4):623-630.
165 Schiffl H, Lang SM, Fischer R. Daily hemodialysis and the outcome of acute renal failure. N Engl J Med. 2002;346:305.
166 VA/NIH Acute Renal Failure Trial NetworkPalevsky PM, Zhang JH, O’Connor TZ, Chertow GM, Crowley ST, Choudhury D, et al. Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med. 2008;359(1):7-20. 3
167 Faulhaber-Walter R, Hafer C, Jahr N, Vahlbruch J, Hoy L, Haller H, et al. The Hannover Dialysis Outcome study: comparison of standard versus intensified extended dialysis for treatment of patients with acute kidney injury in the intensive care unit. Nephrol Dial Transplant. 2009;24(7):2179-2186.
168 Evanson JA, et al. Prescribed versus delivered dialysis in acute renal failure patients. Am J Kidney Dis. 1998;32:731-738.
169 Clark WR, et al. A comparison of metabolic control by continuous and intermittent therapies in acute renal failure. J Am Soc Nephrol. 1994;4:1413-1420.
170 Bellomo R, Ronco C. Continuous haemofiltration in the intensive care unit. Crit Care. 2000;4:339-345.
171 Ronco C, et al. Effects of different doses in continuous veno-venous haemofiltration on outcomes of acute renal failure: A prospective randomised trial. Lancet. 2000;356:26-30.
172 Bouman CS, et al. Effects of early high-volume continuous venovenous hemofiltration on survival and recovery of renal function in intensive care patients with acute renal failure: A prospective, randomized trial. Crit Care Med. 2002;30:2205-2211.
173 RENAL Replacement Therapy Study InvestigatorsBellomo R, Cass A, Cole L, Finfer S, Gallagher M, Lo S, et al. Randomized Evaluation of Normal versus Augmented Level of Intensity of continuous renal-replacement therapy in critically ill patients. N Engl J Med. 2009;361(17):1627-1638. 22
174 Lins RL, Elseviers MM, Van der Niepen P, Hoste E, Malbrain ML, Damas P, et alSHAKI investigators. Intermittent versus continuous renal replacement therapy for acute kidney injury patients admitted to the intensive care unit: results of a randomized clinical trial. Nephrol Dial Transplant. 2009;24(2):512-518.
175 Pannu N, Klarenbach S, Wiebe N, Manns B, Tonelli M, Alberta Kidney Disease Network. Renal replacement therapy in patients with acute renal failure: a systematic review. JAMA. 2008;299(7):793-805.
176 Rabindranath K, Adams J, Macleod AM, Muirhead N. Intermittent versus continuous renal replacement therapy for acute renal failure in adults. Cochrane Database Syst Rev 2007;(3):CD003773.
177 Kruczynski K, Irvine-Bird K, Toffelmire EB, Morton AR. A comparison of continuous arteriovenous hemofiltration and intermittent hemodialysis in acute renal failure patients in the intensive care unit. ASAIO J. 1993;39(3):M778-M781.
178 Bellomo R, Farmer M, Parkin G, Wright C, Boyce N. Severe acute renal failure: a comparison of acute continuous hemodiafiltration and conventional dialytic therapy. Nephron. 1995;71(1):59-64.
179 Bagshaw SM, Berthiaume LR, Delaney A, Bellomo R. Continuous versus intermittent renal replacement therapy for critically ill patients with acute kidney injury: a meta-analysis. Crit Care Med. 2008;36(2):610-617.
180 Hakim RM, Wingard RL, Parker RA. Effect of the dialysis membrane in the treatment of patients with acute renal failure. N Engl J Med. 1994;331:1338-1342.
181 Uehlinger DE, Jakob SM, Ferrari P, Eichelberger M, Huynh-Do U, Marti HP, et al. Comparison of continuous and intermittent renal replacement therapy for acute renal failure. Nephrol Dial Transplant. 2005;20(8):1630-1637.
182 Bagshaw SM, Berthiaume LR, Delaney A, Bellomo R. Continuous versus intermittent renal replacement therapy for critically ill patients with acute kidney injury: a meta-analysis. Crit Care Med. 2008;36(2):610-617.
183 Kielstein JT, Kretschmer U, Ernst T, Hafer C, Bahr MJ, Haller H, et al. Efficacy and cardiovascular tolerability of extended dialysis in critically ill patients. Am J Kidney Dis. 2004;43(2):342-349.
184 Kumar VA, Yeun JY, Depner TA, Don BR. Extended daily dialysis vs. continuous hemodialysis for ICU patients with acute renal failure: a two-year single center report. Int J Artif Organs. 2004;27(5):371-379.
185 Gabriel DP, Fernández-Cean J, Balbi AL. Utilization of peritoneal dialysis in the acute setting. Perit Dial Int. 2007;27(3):328-331.
186 Gabriel DP, Caramori JT, Martin LC, Barretti P, Balbi AL. Continuous peritoneal dialysis compared with daily hemodialysis in patients with acute kidney injury. Perit Dial Int. 2009;29(Suppl 2):S62-S71.
187 Phu NH, Hien TT, Mai NT, Chau TT, Chuong LV, Loc PP, et al. Hemofiltration and peritoneal dialysis in infection-associated acute renal failure in Vietnam. N Engl J Med. 2002;347(12):895-902. 19
188 Davenport A, Will EJ, Davison AM. Effect of renal replacement therapy on patients with combined acute renal and fulminant hepatic failure. Kidney Int Suppl. 1993;41:S245-S251.
189 Davenport A, Finn R, Goldsmith HJ. Management of patients with renal failure complicated by cerebral oedema. Blood Purif. 1989;7:203-209.
190 Bellomo R. Continuous hemofiltration as blood purification in sepsis. New Horiz. 1995;3:732-737.
191 Ronco C, Ricci Z, Bellomo R. Importance of increased ultrafiltration volume and impact on mortality: Sepsis and cytokine story and the role of continuous veno-venous haemofiltration. Curr Opin Nephrol Hypertens. 2001;10:755-761.
192 Boussekey N, Chiche A, Faure K, Devos P, Guery B, d’Escrivan T, et al. Pilot randomized study comparing high and low volume hemofiltration on vasopressor use in septic shock. Intensive Care Med. 2008;34(9):1646-1653.
193 Rauf AA, Long KH, Gajic O, Anderson SS, Swaminathan L, Albright RC. Intermittent hemodialysis versus continuous renal replacement therapy for acute renal failure in the intensive care unit: an observational outcomes analysis. J Intensive Care Med. 2008;23(3):195-203.
194 Cheung AK. Interactions between plasma proteins and hemodialysis membranes. Adv Nephrol Necker Hosp. 1993;22:417-437.
195 Cheung AK, et al. Compartmental distribution of complement activation products in artificial kidneys. Kidney Int. 1986;30:74-80.
196 Alonso A, Lau J, Jaber BL. Biocompatible hemodialysis membranes for acute renal failure. Cochrane Database Syst Rev 2008;23;(1):CD005283.
197 Cruz DN, Antonelli M, Fumagalli R, Foltran F, Brienza N, Donati A, et al. Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial. JAMA. 2009;301(23):2445-2452. 17
198 Honoré PM, Matson JR. Hemofiltration, adsorption, sieving and the challenge of sepsis therapy design. Crit Care. 2002;6(5):394-396.
199 Bellomo R, Honoré PM, Matson J, Ronco C, Winchester J. Extracorporeal blood treatment (EBT) methods in SIRS/Sepsis. Int J Artif Organs. 2005;28(5):450-458.
200 Haase M, Bellomo R, Baldwin I, Haase-Fielitz A, Fealy N, Davenport P, et al. Hemodialysis membrane with a high-molecular-weight cutoff and cytokine levels in sepsis complicated by acute renal failure: a phase 1 randomized trial. Am J Kidney Dis. 2007;50(2):296-304.
201 Veech RL. The toxic impact of parenteral solutions on the metabolism of cells: A hypothesis for physiological parenteral therapy. Am J Clin Nutr. 1986;44:519-551.
202 Davenport A, Will EJ, Davison AM. The effect of lactate-buffered solutions on the acid-base status of patients with renal failure. Nephrol Dial Transplant. 1989;4:800-804.
203 Davenport A, Will EJ, Davison AM. Hyperlactataemia and metabolic acidosis during haemofiltration using lactate-buffered fluids. Nephron. 1991;59:461-465.
204 Barenbrock M, et al. Effects of bicarbonate- and lactate-buffered replacement fluids on cardiovascular outcome in CVVH patients. Kidney Int. 2000;58:1751-1757.
205 Brater DC. Drug dosing in patients with impaired renal function. Clin Pharmacol Ther. 2009;86(5):483-489.
206 Golper TA. Drug removal during continuous hemofiltration or hemodialysis. Contrib Nephrol. 1991;93:110-116.
207 Joy MS, et al. A primer on continuous renal replacement therapy for critically ill patients. Ann Pharmacother. 1998;32:362-375.
208 Bressolle F, Kinowski JM, de la Coussaye JE, Wynn N, Eledjam JJ, Galtier M. Clinical pharmacokinetics during continuous haemofiltration. Clin Pharmacokinet. 1994;26:457-471.
