CHAPTER 79 ACUTE DISSEMINATED ENCEPHALOMYELITIS AND PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
Viral infections lead to a variety of demyelinating diseases of the central and peripheral nervous systems in animals and humans. These diseases may be acute or chronic with progressive or relapsing-remitting courses. The pathology and pathogenesis are varied; inflammation may or may not be prominent; the oligodendrocytes or Schwann cells that maintain the myelin may or may not be altered.1
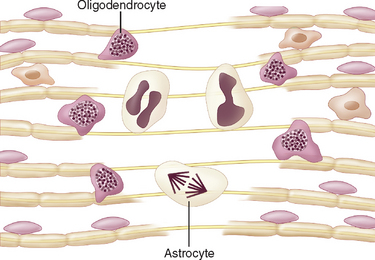
In natural and experimental animal infections, a number of mechanisms have been studied (Table 79-1). Some involve infections of oligodendrocytes, and such infections can expose myelin membranes of the cell to systemic immune responses or can cause direct lysis of the oligodendrocytes; the latter is the apparent mechanism of demyelination in progressive multifocal leukoencephalopathy (PML) in humans. In lentivirus infections of sheep and goats, the microglia and macrophages in the central nervous system are infected, and the astrocytes and oligodendrocytes are not. Cytokines or viral proteins released by the infected cells appear to lead to myelin disruption.2
TABLE 79-1 Mechanisms of Virus-Induced CNS Demyelination
CNS, central nervous system.
From Johnson RT, Major EO: Infectious demyelinating diseases. In Lazzarini R, ed: Myelin Biology and Disorders. San Diego, CA: Academic Press, 2004.
Demyelination can also result from immune responses against myelin in the absence of nervous system infection. When myelin proteins are injected in vaccines made from nerve tissues, an acute, autoimmune, demyelinating encephalomyelitis can be evoked.3 Sequence similarity between infectious agents and encephalitogenic neural proteins may lead to an autoimmune response, a phenomenon called molecular mimicry.4,5 Finally, infection can activate T cells, leading to hindered cell-mediated immune responses and inappropriate cell-mediated immune responses at the same time. This disruption of immune responses in combination with autoimmune responses is seen in human immunodeficiency virus (HIV) infections6 and appears to be the cause of acute postmeasles encephalomyelitis.7
ACUTE DISSEMINATED ENCEPHALOMYELITIS
Definition
Acute hemorrhagic necrotizing leukoencephalitis is regarded as an exaggerated form of ADEM. This rare disease, however, has distinct clinical and pathological features and is associated with a different spectrum of antecedent infections.8
Epidemiology
ADEM occurs worldwide and in all seasons. Immunization policies, however, have dramatically altered the frequency of ADEM and the causal agents. The overall frequency has been reduced by the introduction of vaccines for childhood diseases and by the cessation of vaccination against smallpox. Measles, formerly the commonest cause of ADEM (Table 79-2), has now been eliminated in countries with universal immunization programs. Rubella and mumps virus infections have also been greatly reduced, because many countries use the combined measles-mumps-rubella vaccine. Varicella-zoster virus vaccine has also reduced the cases of ADEM. The second commonest cause of ADEM, vaccination with vaccinia virus, was eliminated after the eradication of smallpox. Before these changes, ADEM was thought to represent one third of all cases of encephalitis; now ADEM represents only about 10% of cases of acute encephalitis. The commonest infections preceding ADEM are nonspecific influenza-like illnesses. ADEM occurs after Epstein-Barr virus, Mycoplasma pneumoniae, and influenza virus infections, but the frequency is uncertain.
Clinical Features
Symptoms and signs of acute encephalitis (fever, nuchal rigidity, depression of consciousness, focal neurological signs, and/or seizures) usually develop 3 days to 3 weeks after the exanthema or respiratory or gastrointestinal illness. Onset of the encephalitis is typically abrupt and reaches maximal intensity within 24 to 48 hours. Signs of the antecedent illness, such as the fading rash of measles or varicella or the persistent lymphadenopathy of Epstein-Barr virus infection, may still be detectable. The spinal fluid usually exhibits lymphocytic pleocytosis and mild elevation of protein content. Increased immunoglobulin G synthesis and oligoclonal bands are usually not found. Myelin basic protein levels may be elevated early in the disease. Virus cultures usually yield negative results. The spinal fluid is entirely normal in some cases.7,9
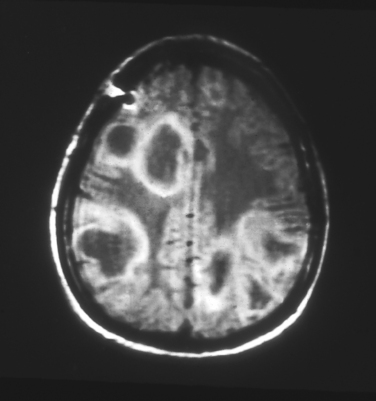
In the absence of a characteristic exanthema preceding the disease, differentiation from acute viral encephalitis is difficult. Magnetic resonance imaging (MRI) can be helpful; in ADEM, multiple white matter lesions of similar age or degree of enhancement are found (Fig. 79-1). Also, in the absence of an exanthem or fever, differentiation from the first attack of multiple sclerosis may be difficult; the presence of nonenhancing periventricular lesions on MRI or the presence of oligoclonal bands are consistent with the diagnosis of multiple sclerosis. Childhood onset, fever, severe depression of consciousness or coma, and seizures all are more consistent with a diagnosis of ADEM.10
Clinical signs vary with some precipitating agents. After chickenpox, one half of the neurological complications are limited to cerebellar ataxia, which may or may not represent a focal form of ADEM. Neurological disease complicates 1% of cases of infectious mononucleosis caused by the Epstein-Barr virus; some are acute demyelinating neuropathies (Guillain-Barré syndrome), some are acute cerebellar ataxia, and others are typical ADEM. Each of these neurological complications characteristically has an onset 1 to 2 weeks after the onset of infectious mononucleosis, which is suggestive of a postinfectious autoimmune process. ADEM and acute demyelinating neuropathy occur as rare complications of HIV infection. These inflammatory, demyelinating complications occur primarily at the time of seroconversion with the initial activation of CD4 cells and are assumed to represent virus-induced autoimmune responses.6
Acute transverse myelitis, in some cases, may also be a localized form of ADEM. It may also occur as a result of vascular disease or tumors or as a first attack of multiple sclerosis.11
Pathophysiology
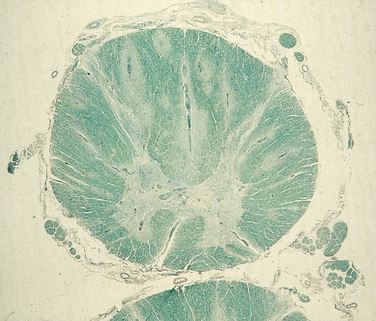
When death occurs during the acute phase, the brain is swollen and congested, and on gross examination, streaks of discoloration are seen along the veins of the white matter. On microscopic examination, mononuclear cells are found along veins and venules, and myelin loss is evident in this area of inflammation; in the spinal cord, this produces a radial pattern (Fig. 79-2). In patients who die later, the flame-shaped demyelinated lesions are more sharply demarcated and contain lipid-laden macrophages.
In the 1930s, the similarity of this pathology to the perivenular demyelinating disease that complicated postexposure rabies prevention was noted. Postexposure rabies vaccine at that time consisted of inactivated virus grown in sheep or rabbit brain and spinal cord tissue, and it was administrated as a series of daily injections over several weeks. Experimentally monkeys were similarly inoculated repeatedly with rabbit brain homogenates without virus, and this led to a disease resembling ADEM and postrabies vaccine encephalomyelitis—an experimental disease called experimental allergic (or autoimmune) encephalomyelitis.12 The experimental disease was shown to result from a cell-mediated immune response against myelin proteins. Subsequent investigations of patients with ADEM complicating measles and varicella and of patients receiving rabies vaccines prepared in central nervous system tissue have shown abnormal lymphocyte proliferation responses to purified human myelin basic protein (Table 79-3).3,7 How a variety of different viral infections leads to an autoimmune response against myelin is unclear. In the case of measles, the virus infects monocytes, which leads to lymphocyte activation and release of several cytokines. The patient cannot respond normally to new antigens and thus is immunocompromised and vulnerable to opportunistic infections. The commonest causes of death secondary to measles are respiratory and gastrointestinal opportunistic infections. Abnormal immune responses also occur, and in about 15% of patients, a lymphocyte proliferative response against myelin can be detected. In a small percentage of these patients, clinical encephalitis develops, even though no virus is found in the brain or spinal cord.7
ACUTE HEMORRHAGIC NECROTIZING LEUKOENCEPHALITIS
This disorder probably represents a hyperacute form of ADEM. However, it rarely follows the exanthems frequently associated in the past with ADEM; it usually follows nonspecific respiratory illnesses. One to 20 days after the prodromal illness, the patient develops fever, severe obtundation, seizures, and neurological signs often suggestive of a mass lesion. Most such patients die within 5 days. The spinal fluid shows polymorphonuclear cells and red blood cells; peripheral leukocytosis and proteinuria are usually present. The pathological specimen demonstrates gross swelling with multiple hemorrhages; microscopically, both veins and arterioles exhibit fibrinoid necrosis with extravasations of fibrin and red blood cells; the inflammatory infiltrates are predominantly polymorphonuclear. Despite the large areas of necrosis, the findings are most intense in the white matter, and myelin loss is more prominent than axonal destruction.13
PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
Epidemiology
PML was originally described in 1958 as “a heretofore unrecognized complication of chronic lymphocytic leukemia and Hodgkin’s disease.”14 It was subsequently reported in patients undergoing aggressive immunosuppression to treat connective tissue diseases and to prevent rejection of transplanted organs. Nevertheless, PML remained a very rare disease. During the first 25 years after its description, only one case was diagnosed at the Johns Hopkins Hospital. With the advent of the acquired immunodeficiency syndrome (AIDS) epidemic in the 1980s, the disease abruptly became common, representing the cause of death in about 4% of patients with AIDS.15 The disease affects patients with AIDS worldwide.
The JC virus is a papovavirus originally isolated in 1971 from the brain of a young man who had PML complicating Hodgkin’s disease.16 The virus is ubiquitous but has not been definitely associated with any other illness. The majority of people develop antibody to JC virus between 1 and 14 years of age; by middle age, more than 70% have antibody. No clinical symptoms or signs have been associated with the primary infection.
Clinical Features
The spinal fluid is generally normal with no pleocytosis, normal protein content, and no abnormalities or increase in immunoglobulins. In patients with HIV infections, however, cell and protein level elevations may result from central nervous system infection coexistent with HIV. The electroencephalogram shows nonspecific findings. Serological tests for JC virus antibody are of no help, inasmuch as antibody is present in most adults, its levels are not unusually elevated in PML patients, and it is not found in spinal fluid. Polymerase chain reaction testing for JC virus in spinal fluid is a useful diagnostic method and has a sensitivity rate of 80% to 90%.17 Computed tomographic imaging or MRI of the brain provides an important clue to the diagnosis of PML. Lucencies on computed tomography or densities on T2-weighted MRI images have a characteristic pattern of multiple nonenhancing lesions of subcortical white matter.
Pathology and Pathogenesis
The neuropathological findings in PML are unique. On gross examination of the brain, discolored lesions in the white matter are evident, ranging from pinpoint size to large confluent areas of leukomalacia. Lesions are most prominent at the cortical white matter junction (Fig. 79-3). Microscopically, foci show a relative sparing of axons and a loss of both oligodendrocytes and myelin. Many oligodendrocytes surrounding the demyelinated foci are greatly enlarged and contain large intranuclear inclusions with a “ground glass” appearance (Fig. 79-4). Within the demyelinated areas, astrocytes are enlarged and have bizarre mitotic forms, multiple nuclei, and multilobular nuclei. In the original description of PML, the authors stated that “astrocytes of this sort are ordinarily met with only in neoplastic processes.”14 Inflammatory cells are usually absent or in minimal numbers. Immunocytochemical staining for JC virus antigens in glial cells confirms the diagnosis on biopsy or autopsy brain tissue.
Speculation that a virus infection might be causative arose from the finding of inclusion bodies and the occurrence of the disease in immunosuppressed patients (Fig. 79-5). In 1965, astonishing electron microscopic findings demonstrating that the inclusion bodies consisted of crystalline arrays of particles corresponding in size to polyomaviruses were published.18 At that time, no human polyomaviruses were known, but through the use of human fetal brain cultures, JC virus, the first known human polyomavirus, was isolated.16
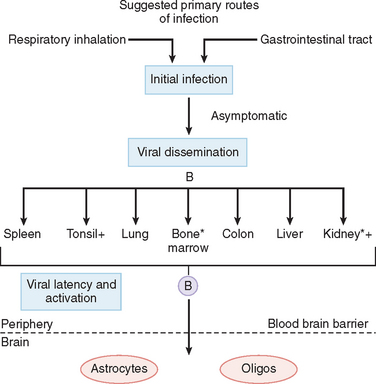
Initial infection with JC virus in childhood is probably through the respiratory route.
Latent virus has been shown in bone marrow and kidneys, and virus has been found in tonsils and urine. B cells have been implicated in the spread of virus, and these cells are thought to carry the virus to the brain after activation in immunosuppressed patients.19
In the brain, JC virus is unique. Highly productive infection occurs only in oligodendrocytes. Astrocytes in lesions express T antigen, have bizarre forms, and appear to proliferate, which are indications of viral transformation; however, contrary to true transformation, astrocytes contain small numbers of virions. In contrast, except for patches of granular cells in the cerebellum, neurons and other cells appear resistant to infection.20
TREATMENT
Both cytarabine (Ara-C), a nucleoside used in cancer therapy, and cidofovir, an antiviral agent effective in the treatment of cytomegalovirus retinitis and genital herpes, have been reported in case reports and small case series to stabilize or to improve PML. Controlled trials of both drugs, however, have failed to show differences in survival between treated and untreated patients.
In patients with PML complicating immunosuppressive therapy, the reduction of the immunosuppressant medication is thought to stabilize or retard the disease. The institution of highly active antiretroviral therapy (HAART) for HIV infections has shown the validity of this strategy. The immune status often improves with HAART, and clinical symptoms, radiological findings, and survival times of PML can improve with treatment.21 This is not a panacea, however, inasmuch as PML can develop in patients while they are receiving HAART, and the decrease in HIV load and increase in CD4 cell counts provided by HAART are not always correlated with improvement in PML.22
CONCLUSIONS
Garg RK. Acute disseminated encephalomyelitis. Postgrad Med J. 2003;79:11-17.
Johnson RT. Viral Infections of the Nervous System, 2nd ed. Philadelphia: Lippincott-Raven, 1998.
Johnson RT, Major EO. Infectious demyelinating diseases. In: Lazzarini RA, editor. Myelin Biology and Disorders. San Diego, CA: Elsevier; 2004:953-983.
Koralnik IJ. New insights into progressive multifocal leukoencephalopathy. Curr Opin Neurol. 2004;17:365-370.
1 Johnson RT, Major EO. Infectious demyelinating diseases. In: Lazzarini RA, editor. Myelin Biology and Disorders. San Diego, CA: Elsevier; 2004:953-983.
2 Kennedy PG, Narayan O, Ghotbi Z, et al. Persistent expression of Ia antigen and viral genome in visna-maedi virus–induced inflammatory cells. Possible role of lentivirus-induced inter-feron. J Exp Med. 1985;162:1970-1982.
3 Hemachudha T, Phanuphak P, Johnson RT, et al. Neurologic complications of Semple-type rabies vaccine. Neurology. 1987;37:550-556.
4 Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043-1045.
5 Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108:1097-1104.
6 Jones HR, Ho DD, Forgacs P, et al. Acute fulminating fatal leukoencephalopathy as the only manifestation of human immunodeficiency virus infection. Ann Neurol. 1988;23:519-522.
7 Johnson RT, Griffin DE, Hirsch RL, et al. Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med. 1984;310:137-141.
8 Johnson RT. Viral Infections of the Nervous System, 2nd ed. Philadelphia: Lippincott-Raven, 1998.
9 Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis. A long-term follow-up study of 84 pediatric patients. Neurology. 2002;59:1224-1231.
10 Dale RC, de Sousa C, Chong WK, et al. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. 2000;123:2407-2422.
11 Al Deeb SM, Yaqub BA, Bruyn GW, et al. Acute transverse myelitis. A localized form of postinfectious encephalomyelitis. Brain. 1997;120:1115-1122.
12 Rivers TM, Schwentker FF. Encephalomyelitis accompanied by myelin destruction experimentally produced in monkeys. J Exp Med. 1935;61:689-702.
13 Sharmeela K, Ostrow P, Landi MK, et al. Acute hemorrhagic leukoencephalitis vs ADEM: FLAIR MRI and neuropathology findings. Neurology. 2003;60:721-722.
14 Astrom KE, Mancall EL, Richardson EP. Progressive multifocal leukoencephalopathy. Brain. 1958;81:93-127.
15 Berger JR, Kaszovitz B, Post MJD, et al. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infections. A review of the literature with a report of sixteen cases. Ann Intern Med. 1987;107:78-87.
16 Padgett BL, Walker DL, ZuRhein GM, et al. Cultivation of papova-like virus from human brain with progressive multifocal leukoencephalopathy. Lancet. 1971;1:1257-1260.
17 McGuire NM, Barhite S, Hollander H, et al. JC virus DNA in cerebrospinal fluid of human immunodeficiency virus–infected patients: predictive value for progressive multifocal leukoencephalopathy. Ann Neurol. 1995;37:395-399.
18 ZuRhein GM, Chou S-M. Particles resembling papovaviruses in human cerebral demyelinating disease. Science. 1965;148:1477-1479.
19 Tornatore C, Berger EO, Houff SA, et al. Detection of JC virus DNA in peripheral lymphocytes for patients with and without progressive multifocal leukoencephalopathy. Ann Neurol. 1992;31:454-462.
20 Koralnik IJ, Wuthrich C, Dang X, et al. JC virus granular cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol. 2005;57:576-580.
21 Clifford DB, Yiannoutsos C, Glicksman M, et al. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623-625.
22 Cinque P, Bossolasco S, Brambilla AM, et al. The effect of highly active antiretroviral therapy-induced immune reconstitution on development and outcome of progressive multifocal leukoencephalopathy: study of 43 cases with review of the literature. J Neurovirol. 2003;9(Suppl 1):73-80.






