76 Acute Coronary Syndromes
Therapy
 Definition and Clinical Manifestations
Definition and Clinical Manifestations Pathophysiology of Acute Coronary Syndromes
Pathophysiology of Acute Coronary Syndromes
The inciting event underlying the development of an ACS is rupture of an atherosclerotic plaque.1 Possible sequelae of plaque rupture include thrombus formation with total occlusion, with likely development of STEMI; dissolution of thrombus and healing of the fissure, with clinical stabilization; and subtotal occlusion, which can lead to either NSTEMI or UA.
Atherosclerotic plaques are composed of a lipid core that includes cholesterol, oxidized low-density lipoproteins (LDL), macrophages, and smooth muscle cells, covered by a fibrous cap. Plaque rupture occurs when external mechanical forces exceed the tensile strength of the fibrous cap. After plaque rupture, the clinical consequences depend largely on the balance between prothrombotic and antithrombotic forces.2 The lipid core contains tissue factor and other thrombogenic materials that lead to platelet activation and aggregation. Fibrinolytic factors such as tissue plasminogen activator, prostacyclin, and nitric oxide act to counteract the potential for thrombosis. A major factor in the outcome of plaque rupture is blood flow. With subtotal occlusion, high-grade stenosis, or vasospasm, thrombus begins to propagate downstream in the arterial lumen. In contrast to the initial thrombi, which are platelet rich, these thrombi contain large numbers of red cells enmeshed in a web of fibrin. The former would be expected to respond best to antiplatelet therapy, the latter to antithrombotic and fibrinolytic therapy.
 ST-Segment Elevation Myocardial Infarction
ST-Segment Elevation Myocardial Infarction
Patients presenting with suspected myocardial ischemia should undergo a rapid evaluation and should be treated with oxygen, sublingual nitroglycerin (unless systolic pressure is <90 mm Hg), and aspirin, 160 to 325 mg orally.3,4 Opiates relieve pain and also reduce anxiety, the salutary effects of which have been known for decades and should not be underestimated. A 12-lead ECG should be performed and interpreted expeditiously. Figure 76-1 shows a possible treatment algorithm for patients with STEMI.
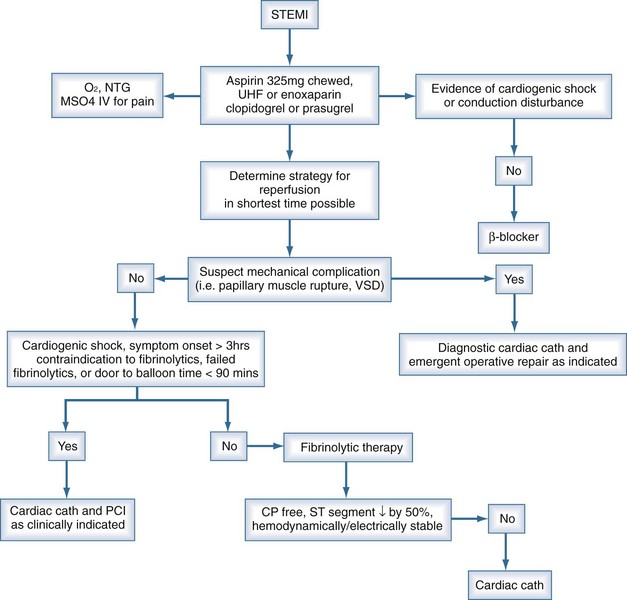
ST-segment elevation of at least 1 mV in two or more contiguous leads provides strong evidence of thrombotic coronary occlusion, and the patient should be considered for immediate reperfusion therapy. The diagnosis of STEMI can be limited in the presence of preexisting left bundle branch block (LBBB) or permanent pacemaker. Nonetheless, new LBBB with a compatible clinical presentation should be treated as acute myocardial infarction (AMI) and treated accordingly. Indeed, recent data suggest that patients with STEMI and new LBBB may stand to gain greater benefit from reperfusion strategies than those with ST elevation.5
Fibrinolytic Therapy
Early reperfusion of an occluded coronary artery is indicated for all eligible candidates. Overwhelming evidence from multiple clinical trials demonstrates the ability of fibrinolytic agents administered early in the course of an acute MI to reduce infarct size, preserve LV function, and reduce short-term and long-term mortality.6–8 Patients treated early derive the most benefit.9 Multiple studies conclude that greatest mortality benefit is seen if fibrinolytics are administered within the first 12 hours of symptom onset,8,10,11 but it is reasonable to administer fibrinolytics to patients whose onset of symptoms exceeds 12 hours but who have continued clinical or ECG evidence of ischemia.
Indications for and contraindications to fibrinolytic therapy are listed in Box 76-1. Because of the small but nonetheless significant risk of a bleeding complication, most notably intracranial hemorrhage, selection of patients with AMI for administration of a fibrinolytic agent should be undertaken with prudence and caution. That is of special importance in ICU patients who may have a predisposition to bleeding complications because of multiple factors. Contraindications can be regarded as absolute or relative. In the surgical patient, thrombolysis may pose a prohibitive risk, and emergent coronary angiography (with percutaneous coronary intervention [PCI] as clinically indicated) may be preferable.
Box 76-1
Indications for and Contraindications to Fibrinolytic Therapy in Acute Myocardial Infarction
Indications
After administration of fibrinolytics for STEMI, the patient should be monitored for signs and symptoms of adequate reperfusion within 90 minutes, as indicated by relief of symptoms and/or hemodynamic/electrical instability coupled with at least a 50% resolution of the highest initial ST elevation.12,13 If signs of adequate reperfusion are not evident within 90 minutes, patients should be taken to the cardiac catheterization lab and considered for PCI. More recent data support the notion that all patients who receive fibrinolytics for STEMI and have at least one high-risk feature should have cardiac catheterization for risk stratification and potential percutaneous revascularization,14,15 even if this involves immediate transfer from the presenting hospital to a PCI-capable facility. Patients not considered high-risk may be observed in the initial facility where fibrinolytics were administered. High-risk features include extensive ST-segment elevation (>2 mm ST elevation in two anterior leads), new-onset LBBB, previous MI, Killip class 2 or 3 or left ventricular ejection fraction (LVEF) ≤ 35%, systolic blood pressure ≤ 100 mm Hg, heart rate ≥ 100 bpm, or right ventricular involvement.
In contrast to the treatment of STEMI, fibrinolytics have shown no benefit and an increased risk of adverse events when used for the treatment of UA/NSTEMI.16 Based on these findings, there is currently no role for fibrinolytic agents in these latter syndromes.
Fibrinolytic Agents
Streptokinase was the original lytic agent used in MI, but it has now been superseded by tissue plasminogen activator (tPA),6 which is more fibrin selective than streptokinase and produces a higher early coronary patency rate (70%-80%).17,18 Administration of tPA usually follows an accelerated regimen consisting of a 15-mg bolus, 0.75 mg/kg (up to 50 mg) IV over the initial 30 minutes, and 0.5 mg/kg (up to 35 mg) over the next 60 minutes. Reteplase (rPA) is a deletion mutant of tPA with an extended half-life, and is given as two 10-U boluses 30 minutes apart. Reteplase was originally evaluated in angiographic trials that demonstrated improved coronary flow at 90 minutes compared to tPA, but subsequent trials showed similar 30-day mortality rates.19 Tenecteplase (TNK-tPA) is a genetically engineered tPA mutant with amino acid substitutions that result in prolonged half-life, resistance to plasminogen activator inhibitor 1, and increased fibrin specificity. TNK-tPA is given as a single bolus adjusted for weight. A single bolus of TNK-tPA has been shown to produced coronary flow rates identical to those seen with accelerated tPA, with equivalent 30-day mortality and bleeding rates.20
Primary Percutaneous Coronary Intervention in Acute Myocardial Infarction
The major advantages of primary PCI over fibrinolytic therapy include a higher rate of normal flow (TIMI grade 3),7 lower risk of intracranial hemorrhage, and the ability to stratify risk based on the severity and distribution of coronary artery disease. Patients ineligible for fibrinolytic therapy should obviously be considered for primary PCI. In addition, data from several randomized trials have suggested that PCI is preferable to fibrinolytic therapy for several subsets of AMI patients at higher risk.21,22 The largest of these trials is the GUSTO-IIb Angioplasty Substudy, which randomized 1138 patients. At 30 days, there was a clinical benefit in the combined primary endpoint of death, nonfatal reinfarction, and nonfatal disabling stroke in the patients treated with percutaneous transluminal coronary angioplasty (PTCA) compared to tPA, but no difference in the “hard” endpoints of death and MI at 30 days.22
Recent meta-analyses comparing direct PTCA with fibrinolytic therapy have suggested lower rates of mortality and reinfarction among those receiving direct PTCA.23,24 Thus direct angioplasty, if performed in a timely manner (ideally within 60 minutes) by highly experienced personnel, may be the preferred method of revascularization, since it offers more complete revascularization with improved restoration of normal coronary blood flow and detailed information about coronary anatomy.3 There are certain subpopulations in which primary PCI is clearly preferred, and other populations in which the data are suggestive of benefit. These subsets are listed in Box 76-2. More important than the method of revascularization is the time to revascularization, and that this should be achieved in the most efficient and expeditious manner possible.25 It is important to keep in mind that early, complete, and sustained reperfusion after MI is known to decrease 30-day mortality. The preferred method for reperfusion in STEMI is PCI only if it can be done within a timely manner. Practical considerations regarding transport to a PCI-capable facility should be carefully reviewed before forgoing thrombolytics for PCI. Early recognition and diagnosis of STEMI are key to achieving the desired door-to-needle (or medical contact–to-needle) time for initiation of fibrinolytic therapy of 30 minutes or door-to-balloon (or medical contact–to-balloon) time for PCI under 90 minutes.3 Achieving reperfusion in timely matter correlates with improvement in ultimate infarct size, LV function, and survival.12,13 The ultimate goal is to restore adequate blood flow through the infarct-related artery to the infarct zone, as well as to limit microvascular damage and reperfusion injury. The latter is accomplished with adjunctive and ancillary treatments that will be discussed in the following sections.
Box 76-2
Situations in Which Primary Angioplasty is Preferred in Acute Myocardial Infarction
Coronary Stenting
Primary angioplasty for AMI results in a significant reduction in mortality but is limited by the possibility of abrupt vessel closure, recurrent in-hospital ischemia, reocclusion of the infarct related artery, and restenosis. The use of coronary stents has been shown to reduce restenosis and adverse cardiac outcomes in both routine and high-risk PCI.26 The PAMI Stent Trial was designed to test the hypothesis that routine implantation of an intracoronary stent in the setting of MI would reduce angiographic restenosis and improve clinical outcomes compared to primary balloon angioplasty alone. This large, randomized, multicenter trial involving 900 patients did not show a difference in mortality at 6 months but did show improvement in ischemia-driven target vessel revascularization and less angina in the stented patients compared to balloon angioplasty alone.27 Despite the lack of definite data demonstrating mortality benefit, virtually all the trials investigating adjunctive therapy for STEMI have employed a strategy of primary stenting, and stenting has becoming the default strategy. Whether to use a bare metal stent or a drug-eluting stent in acute MI is a question that has not yet been addressed definitively by clinical trials; selection is currently based on both patient and angiographic characteristics.
Adjunctive Therapy to Primary PCI
Aspirin
Aspirin is the best known and the most widely used of all the antiplatelet agents because of low cost and relatively low toxicity. Aspirin inhibits the production of thromboxane A2 by irreversibly acetylating the serine residue of the enzyme prostaglandin H2 synthetase. Aspirin has been shown to reduce mortality in acute infarction to the same degree as fibrinolytic therapy, and its effects are additive to fibrinolytics.28 In addition, aspirin reduces the risk of reinfarction.29,30 Unless contraindicated, all patients with a suspected ACS (STEMI, NSTEMI, UA) should be given aspirin as soon as possible.
Thienopyridines
Thienopyridines are a class of oral antiplatelet agents that block the P2Y12 component of the adenosine diphosphate receptor and thus inhibit the activation and aggregation of platelets. Currently used thienopyridines include clopidogrel and prasugrel.32 Clopidogrel is a prodrug that is converted in the liver to the active thiol metabolite via the cytochrome P450 (CYP) 3A, 1A, 2B, and 2C subfamilies. The active metabolite irreversibly binds to the P2Y12 component of the ADP receptor on the platelet surface, which prevents activation of the GPIIb/IIIa receptor complex and reduces platelet aggregation for the remainder of the platelet’s lifespan, approximately 7 to 10 days. Onset of inhibition of platelet aggregation (IPA) is dose dependent, with a 300- to 600-mg loading dose achieving inhibition of platelet within 2 hours, whereas a dose of 50 to 100 mg achieves inhibition of platelets in about 24 to 48 hours. Peak effect (time to maximal IPA) occurs at 6 hours with a loading dose of 300 to 600 mg31 and 5 to 7 days with a dose of 50 to 100 mg.32
The efficacy of clopidogrel in combination with aspirin administered to patients with STEMI prior to PCI was tested in the COMMIT-CCS 2 and CLARITY TIMI-28 studies. CLARITY TIMI-2833 randomized 3491 STEMI patients to clopidogrel (300-mg load followed by 75 mg daily) or placebo. All patients also received a fibrinolytic, aspirin, and when appropriate, heparin. Use of clopidogrel decreased the incidence of the primary composite efficacy endpoint (infarct artery patency or death or recurrent MI before angiography, 15.0 % versus 21.7%, P < 0.001), largely due to a difference in occlusion of the infarct-related artery (12% versus 18%), with no difference in mortality or major bleeding. In the 1863 patients in CLARITY TIMI-28 who underwent PCI (reported as CLARITY-PCI), retreatment with clopidogrel prior to PCI for STEMI resulted in a significant reduction in cardiovascular death, MI, or stroke at 30 days (7.5% versus 12.0%; P = 0.001) without causing excess bleeding.34 It is therefore routine practice to administer a loading dose of clopidogrel, 300 mg or 600 mg, prior to PCI regardless of the physician’s concern that the patient might need coronary artery bypass graft (CABG) in the near future.
Some patients are considered clopidogrel nonresponders, usually defined as a recurrence of cardiovascular events while on the recommended dose. Ex vivo assays measuring the degree of inhibition of platelet aggregation while on clopidogrel have demonstrated that 4% to 30% of patients do not have an adequate platelet response while on clopidogrel.37–39 Despite these findings, testing for clopidogrel resistance has not become routine.
Prasugrel is a recently approved thienopyridine that irreversibly binds to the P2Y12 component of the ADP receptor with a more rapid onset of action.40 Like clopidogrel, prasugrel is a prodrug metabolized to both an active and inactive metabolite, but a higher proportion is metabolized to an active metabolite, resulting in a higher level of inhibition of platelet aggregation than clopidogrel. The onset of inhibition of platelet aggregation is dose dependent and can be achieved in less than 30 minutes at a dose of 60 mg, but peak effect of IPA occurs in approximately 4 hours.35 The randomized double-blind TRITON-TIMI 38 trial compared prasugrel (loading dose of 60 mg followed by maintenance dose of 10 mg) with clopidogrel (300-mg load followed by 75-mg maintenance) in 13,608 patients with UA/NSTEMI (n = 10,074) or STEMI (n = 3534) who underwent PCI.36 All patients also received aspirin, and treatment with prasugrel or clopidogrel was continued for a median of 14.5 months. The primary endpoint, a composite of cardiovascular death, nonfatal MI, and nonfatal stroke, was less frequent among patients who received prasugrel (9.9% versus 12.1 %, P < 0.001). The rate of major bleeding was higher in the prasugrel group (2.4% versus 1.8 %, P = 0.03), as was the rate of life-threatening bleeding. A post hoc analysis of the TRITON TIMI-38 trial identified three ACS subgroups in which prasugrel was found to be harmful or showed no net benefit: patients with a history of transient ischemic attack (TIA) or stroke (net harm), age older than 75 (no net benefit), and body weight less than 60 kg ( no net benefit). The FDA has labeled history of TIA and/or stroke as a contraindication to prasugrel use.36
Dual antiplatelet therapy with aspirin and thienopyridines is given to all patients undergoing PCI, as described above. However, data suggest that even patients not undergoing PCI benefit from the addition of clopidogrel to aspirin. COMMIT-CCS-2 randomized over 45,000 patients with suspected MI to 75 mg of clopidogrel daily (no loading dose).37 The majority of patients had STEMI, but only 54% were treated with fibrinolytics. Clopidogrel was continued after hospital discharge for a mean duration of 14.9 days. The co-primary endpoint of all-cause mortality was reduced from 8.1% in the placebo group to 7.5% in the clopidogrel group (OR, 0.93 [95% CI, 0.87-0.99]; P = 0.03; NNT = 167), without increased bleeding in the clopidogrel group. On the basis of these data, patients presenting with MI should be considered for a thienopyridine regardless of whether or not they underwent reperfusion therapy. The duration of thienopyridine use in this population has yet to be defined.
Glycoprotein IIb/IIIa Receptor Antagonists
Glycoprotein IIb/IIIa receptor antagonists inhibit the final common pathway of platelet aggregation, blocking cross-linking of activated platelets, and are often-used percutaneous interventions.38–42 In the era of dual antiplatelet therapy using a thienopyridine and aspirin, the role of addition of a glycoprotein IIb/IIIa inhibitor in primary angioplasty for STEMI is uncertain. Studies such as the ADMIRAL and CADILLAC trials conducted prior to the use of dual antiplatelet therapy established the efficacy of abciximab in primary PCI (with or without stenting) in patients with STEMI.41 The results of recent clinical trials have raised questions about whether glycoprotein IIb/IIIa antagonists have additional utility when added to dual antiplatelet therapy in patients with STEMI.43–45 The BRAVE-3 trial randomized 800 patients undergoing primary stenting to 600 mg of clopidogrel plus either placebo or abciximab prior to PCI and showed no difference at 30 days in either the primary endpoint of infarct size or the secondary composite endpoint of death, recurrent MI, stroke, or urgent revascularization of the infarct-related artery.43 Similar findings were seen in ON-TIME2, in which 984 patients with STEMI were randomized to either high-dose tirofiban or placebo in addition to dual antiplatelet therapy prior to transport for PCI. Although patients who received high-dose tirofiban had improved resolution of ST-segment elevation before and after PCI, there was no significant difference in TIMI flow or the 30-day composite endpoint of death, recurrent MI, or urgent target-vessel revascularization between the two groups.44 Given the present data, current guidelines suggest that when a STEMI patient is treated with a thienopyridine and aspirin plus an anticoagulant such as UFH or bivalirudin, the use of a glycoprotein IIb/IIIa inhibitor at the time of PCI may be beneficial but cannot be recommended as routine.3
Anticoagulants
Administration of full-dose heparin after fibrinolytic therapy with tPA is essential to diminish reocclusion after successful reperfusion.6,28 Dosing should be adjusted to weight, with a bolus of 60 U/kg up to a maximum of 4000 U and an initial infusion rate of 12 U/kg/h up to a maximum of 1000 U/h, with adjustment to keep the partial thromboplastin time (PTT) between 50 and 70 seconds.4 Heparin should be continued for 24 to 48 hours. For patients undergoing PCI who have already been treated with aspirin and a thienopyridine, both unfractionated heparin or bivalirudin (with or without prior heparin administration) are acceptable anticoagulant regimens.3 Bivalirudin is a direct thrombin inhibitor that inhibits both clot-bound and circulating thrombin. It is administered as an initial bolus of 0.75 mg/kg, followed by a continuous infusion at 1.75 mg/kg/h for the duration of PCI, with adjustments for patients with renal dysfunction. Bivalirudin is an excellent alternative to unfractionated or low-molecular-weight heparin (LMWH) in patients with a history of heparin-induced thrombocytopenia. It is at least equivalent to heparin plus a glycoprotein IIb/IIIa inhibitor in reducing ischemic events associated with UA and/or NSTEMI, with the added benefit of a reduction in bleeding.46 Up until recently, the role of bivalirudin in STEMI was uncertain. The HORIZONS-AMI trial randomized 3602 patients with STEMI undergoing primary PCI to UFH plus a glycoprotein IIb/IIIa inhibitor or to bivalirudin alone (with provisional glycoprotein IIb/IIIa in the cardiac catheterization lab).47 Major adverse cardiac event (MACE) rates were equivalent, but use of bivalirudin alone was associated with a 40% reduction in bleeding (4.9% versus 8.3%, P < 0.001;). However, at 1 year, MACE rates were similar in the two groups (11.9% versus 11.9%, HR 1.00, 0.82-1.21, P = 0.98), but there was a decrease in all-cause mortality with bivalirudin (3.4% versus 4.8%, P = 0.03).48
Enoxaparin is an LMWH with established efficacy as an anticoagulant in patients with STEMI who have received fibrinolytics or are undergoing PCI.49,50 The standard dose of enoxaparin is a 30-mg intravenous (IV) bolus, followed 15 minutes later by subcutaneous injections of 1 mg/kg every 12 hours. Patients with decreased creatinine clearance or older than 75 are at higher risk of bleeding with standard-dose enoxaparin and should not receive a bolus, but can receive a reduced dose of 0.75 mg/kg every 12 hours. Patients undergoing PCI should have an additional bolus if the last dose was given 8 to 12 hours prior. Maintenance dosing of enoxaparin should be given during the hospitalization (up to 8 days).
Fondaparinux, also an LMWH, can be dosed daily in patients receiving fibrinolytics for STEMI (initial dose of 2.5 mg IV followed by subcutaneous injections of 2.5 mg once daily). The OASIS-6 trial randomized over 12,000 patients with STEMI to 2.5 mg of fondaparinux or placebo. Death or reinfarction at 30 days was significantly reduced in the fondaparinux group (9.7% versus 11.2%, P = 0.008) and were maintained at 6 months.51 Severe bleeds were reduced with fondaparinux (61 versus 79, P = 0.13), and significant benefit was seen in patients who received fibrinolytics, as well those who were not reperfused. However, in patients undergoing PCI for STEMI, fondaparinux should not be administered alone, owing to an increased rate of catheter-related thrombosis observed in clinical trials.51,52 If fondaparinux has been chosen, unfractionated heparin should be administered with fondaparinux in the catheterization laboratory. Table 76-1 summarizes typical antiplatelet and anticoagulant therapy for ACSs.
TABLE 76-1 Antiplatelet/Anticoagulant Therapy in Acute Coronary Syndromes
| Drug | Initial Medical Treatment |
|---|---|
| Antiplatelet Drugs | |
| Aspirin | 162 to 325 mg nonenteric formulation, orally or chewed |
| Clopidogrel Prasugrel |
LD of 300 to 600 mg orally, MD of 75 mg orally per day LD of 60 mg orally, MD of 10 mg orally per day |
| Ticlopidine | LD of 500 mg orally, MD of 250 mg orally twice daily |
| Anticoagulants | |
| Unfractionated heparin | LD of 60 U per kg (max 4,000 U) as IV bolus MD of IV infusion of 12 U/kg/h (max 1000 U/h) to maintain APTT at 1.5 to 2.0 times control (approximately 50-70 sec) |
| Enoxaparin | LD of 30 mg IV bolus may be given MD of 1 mg/kg subcutaneously every 12 h; extend dosing interval to 1 mg/kg every 24 h if estimated creatinine clearance <30 mL/min |
| Fondaparinux | 2.5 mg subcutaneously once daily. Avoid for creatinine clearance <30 mL/min |
| Eptifibatide | LD of IV bolus of 180 µg/kg MD of IV infusion of 2 µg/kg/min; reduce infusion by 50% in patients with estimated creatinine clearance <50 mL/min |
| Tirofiban | LD of IV infusion of 0.4 µg/g/min for 30 min MD of IV infusion of 0.1 µg/kg/min; reduce rate of infusion by 50% in patients with estimated creatinine clearance <30 mL/min |
| Bivalirudin | 0.1 mg per kg bolus, 0.25 mg/kg/h infusion |
LD, loading dose; MD, maintenance dose.
Adapted from Anderson JL, Adams CD, Antam EM et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non–ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2007;50:e1-157.
Nitrates
Nitrates have a number of beneficial effects in AMI. They reduce myocardial oxygen demand by decreasing preload and afterload, and they may also improve myocardial oxygen supply by increasing subendocardial perfusion and collateral blood flow to the ischemic region.53 Occasional patients with ST elevation due to occlusive coronary artery spasm may have dramatic resolution of ischemia with nitrates. In addition to their hemodynamic effects, nitrates also reduce platelet aggregation. Despite these benefits, the GISSI-3 and ISIS-4 trials failed to show a significant reduction in mortality from routine acute and chronic nitrate therapy.54,55 Nonetheless, nitrates are still first-line agents for the symptomatic relief of angina pectoris and when MI is complicated by congestive heart failure.
Beta-Blockers
Beta-blockers are beneficial both in the early management of MI and as long-term therapy. In the prefibrinolytic era, early IV atenolol was shown to significantly reduce reinfarction, cardiac arrest, cardiac rupture, and death.56 In conjunction with fibrinolytic therapy with tPA, immediate β-blockade with metoprolol resulted in a significant reduction in recurrent ischemia and reinfarction, although mortality was not decreased.57
The COMMIT-CCS 2 trial of 45,852 patients with acute MI had a factorial arm (the clopidogrel arm was discussed earlier) and randomized patients—93% of whom had STEMI and 54% of whom were treated with lytics—to treatment with metoprolol (3 IV injections of 5 mg each followed by oral 200 mg/day for up to 4 weeks) or placebo.58 Surprisingly, there was no difference in the primary endpoint of death, reinfarction, or cardiac arrest by treatment group (9.4% for metoprolol versus 9.9% for placebo, P = NS) or in the co-primary endpoint of all-cause mortality by hospital discharge (7.7% versus 7.8%, P = NS). Although reinfarction was lower in the metoprolol group (2.0% versus 2.5%, P = 0.001). there was an increase in the risk of developing heart failure and cardiogenic shock (5.0% versus 3.9%, P < 0.0001).58 Death due to shock occurred more frequently in the metoprolol group (2.2%, versus 1.7%), while death due to arrhythmia occurred less frequently in the metoprolol group (1.7%, n = 388 versus 2.2%, n = 498). Based on these findings, routine use of intravenous beta-blockers in the absence of systemic hypertension is no longer recommended.59
In contrast to the use of early aggressive beta-blocker therapy, the long-term use of beta-blockers post MI has favorable outcomes on mortality.60,61 The CArvedilol Post-infaRct survIval COntRolled evaluatioN (CAPRICORN) trial was a randomized placebo-controlled trial designed to test the long-term efficacy of carvedilol on morbidity and mortality in patients with LV dysfunction 3 to 21 days after MI who were already treated with angiotensin-converting enzyme (ACE) inhibitors.62 After an average follow-up period of 1.3 years, cardiovascular mortality was lower in the carvedilol arm (11% versus 14% for placebo, P = 0.024), as was all-cause mortality or nonfatal MI (14% versus 20%, P = 0.002).62 This study supports the claim that beta-blocker therapy after acute MI reduces mortality irrespective of reperfusion therapy or ACE inhibitor use. Relative contraindications to oral beta-blockers include heart rate less than 60 bpm, systolic arterial pressure less than 100 mm Hg, moderate or severe LV failure, signs of peripheral hypoperfusion, shock, PR interval greater than 0.24 second, second- or third-degree AV block, active asthma, or reactive airway disease.59
Angiotensin-Converting Enzyme Inhibitors
ACE inhibitors have been shown unequivocally to improve hemodynamics, functional capacity and symptoms, and survival in patients with chronic congestive heart failure.63,64 Moreover, ACE inhibitors prevent the development of congestive heart failure in patients with asymptomatic LV dysfunction.65 This information was the spur for trials evaluating the benefit the prophylactic administration of ACE inhibitors in the post-MI period. The SAVE trial showed that patients with LV dysfunction (LVEF < 40%) after MI had a 21% improvement in survival after treatment with the ACE inhibitor, captopril.66 A smaller but still significant reduction in mortality was seen when all patients were treated with captopril in the ISIS-4 study.55 The HOPE trial randomized 9297 patients with documented vascular disease or those at high risk for atherosclerosis (diabetes plus at least one other risk factor) in the absence of heart failure to treatment with the tissue-selective ACE inhibitor, ramipril (target dose 10 mg/day), or placebo.67 An impressive 22% reduction in the combined endpoint of cardiovascular death, MI, and stroke was observed, and the improved survival was additive to the benefits of aspirin and beta-blockers.67 The mechanisms responsible for the benefits of ACE inhibitors probably include limitation in the progressive LV dysfunction and enlargement (remodeling) that often occur after infarction, but a reduction in ischemic events was seen as well.
Immediate IV ACE inhibition with enalaprilat has not been shown to be beneficial,68 but oral ACE inhibition should be started early in the hospital course. Patients should be started on low doses of oral agents (captopril, 6.25 mg three times daily) and rapidly increased to the range demonstrated beneficial in clinical trials (captopril, 50 mg three times daily; enalapril, 10-20 mg twice daily; lisinopril, 10-20 mg once daily; or ramipril, 10 mg once daily).
Lipid-Lowering Agents
There is extensive epidemiologic, laboratory, and clinical evidence linking cholesterol and coronary artery disease (CAD). Total cholesterol level has been linked to the development of CAD events with a continuous and graded relation.69 Most of this risk is due to LDL cholesterol. A number of large primary and secondary prevention trials have shown that LDL cholesterol lowering is associated with a reduced risk of coronary disease events. Earlier lipid-lowering trials used bile acid sequestrants (cholestyramine), fibric acid derivatives (gemfibrozil and clofibrate), or niacin in addition to diet. The reduction in total cholesterol in these early trials was 6% to 15% and was accompanied by a consistent trend toward a reduction in fatal and nonfatal coronary events.70
More impressive results have been achieved using HMG-CoA reductase inhibitors (statins). Statins have been demonstrated to decrease the rate of adverse ischemic events in patients with documented CAD in the 4S trial,71 as well as in the CARE study72 and the LIPID trial.73
The goal of treatment is an LDL cholesterol level less than 100 mg/dL.74 Maximum benefit may require management of other lipid abnormalities (elevated triglycerides, low HDL cholesterol) and treatment of other atherogenic risk factors.
The use and efficacy of high-dose statin loading prior to PCI for STEMI was addressed in the STATIN STEMI trial; 171 patients were randomized to either 80 mg or 10 mg of atorvastatin in addition to 600 mg of clopidogrel prior to PCI for STEMI.75 The 30-day incidence of death, MI, or target vessel revascularization was 5.8% in the high-dose statin group versus 10.6 % in the low-dose statin group (P = 0.26).75 Although high-dose statin administration prior to PCI is not requisite, all patients with ACS should be started on a statin prior to discharge unless there is a contraindication.
Calcium Channel Blockers
Randomized clinical trials have not demonstrated that routine use of calcium channel blockers improves survival after MI.76 In fact, meta-analyses suggest that high doses of the short-acting dihydropyridine, nifedipine, increase mortality in MI.77 Adverse effects of calcium channel blockers include bradycardia, atrioventricular block, and exacerbation of heart failure. The relative vasodilating, negative inotropic effects, and conduction system effects of the various agents must be considered when they are employed in this setting. Diltiazem is the only calcium channel blocker that has been proven to have tangible benefits, reducing reinfarction and recurrent ischemia in patients with non–Q wave infarctions who do not have evidence of congestive heart failure.78
 Non–ST-Segment Elevation Myocardial Infarction
Non–ST-Segment Elevation Myocardial Infarction
Braunwald has proposed a classification for UA based on severity of symptoms and clinical circumstances for risk stratification.79 The risk of progression to acute MI or death due to ACS increases with age. ST-segment depression on the ECG identifies patients at higher risk for clinical events.79 Conversely, a normal ECG confers an excellent short-term prognosis. Biochemical markers of cardiac injury are also predictive of outcome. Elevated levels of troponin T are associated with an increased risk of cardiac events and a higher 30-day mortality, and in fact, were more strongly correlated with 30-day survival than ECG category or CPK-MB level in an analysis of data from the GUSTO-2 trial.80 Conversely, low levels are associated with low event rates, although the absence of troponin elevation does not guarantee a good prognosis and is not a substitute for good clinical judgment.
Antiplatelet Therapy
As previously noted, aspirin is a mainstay of ACS therapy. Both the VA Cooperative Study Group29 and the Canadian Multicenter Trial81 showed that aspirin reduces the risk of death or MI by approximately 50% in patients with UA or NQMI. Aspirin also reduces events after resolution of an ACS and should be continued indefinitely.
In addition to patients with STEMI, patients with NSTEMI and suspected UA benefit from the use of a thienopyridine in addition to aspirin. This benefit, a decrease in cardiovascular death, MI, or stroke, is seen not only in patients who undergo PCI but also in patients who are managed medically. In the CURE trial, 12,562 patients were randomized to receive clopidogrel or placebo in addition to standard therapy with aspirin within 24 hours of UA symptoms.82 Clopidogrel significantly reduced the risk of MI, stroke, or cardiovascular death from 11.4% to 9.3% (P<0.001).82 It should be noted that this benefit came with a 1% absolute increase in major non-life-threatening bleeds (P = 0.001) as well as a 2.8% absolute increase in major/life-threatening bleeds associated with CABG within 5 days (P=0.07).82 Because percutaneous revascularization was performed on only 23% of patients in the CURE trial during the initial hospitalization, the study provides convincing evidence that clopidogrel is beneficial in patients who are managed medically, in addition to those undergoing PCI. The optimal duration of therapy in this patient population, however, is unknown.
The PCI-CURE report examined the subset of patients (n = 2658) with UA/NSTEMI who underwent PCI.83 Overall, including events before and after PCI, there was a 31% reduction in cardiovascular death or MI (P<0.002). There was no difference between the groups in major bleeding.83 PCI-CURE suggests that patients with UA/NSTEMI who undergo PCI, pretreatment with clopidogrel followed by up to 1 year of clopidogrel therapy is beneficial in reducing major cardiovascular events. However, PCI-CURE did not adequately address the question of dose or timing of clopidogrel in relationship to PCI. The CREDO trial randomized 2116 patients to a 300-mg loading dose of clopidogrel or placebo (3-24 hours before PCI). Both groups received 325 mg of aspirin and were treated with 75 mg of clopidogrel daily for 1 year. Although there was no difference between groups in the 28-day composite endpoint of death, MI, or urgent target vessel revascularization, treatment with clopidogrel was associated with a 26.9% relative risk reduction in the 1-year composite endpoint of death, MI, or stroke.84
Clopidogrel has also been tested for secondary prevention of events. The CAPRIE trial, a multicenter trial of 19,185 patients with known vascular disease (prior stroke, MI, or peripheral vascular disease), randomized patients to either 75 mg/d of clopidogrel or 325 mg aspirin.85 After an average follow-up of 1.6 years, patients treated with clopidogrel had significantly fewer cardiovascular events than patients treated with aspirin (5.8% versus 5.3%, a relative risk reduction of 8.7%).85
The TRITON TIMI-38 trial, as mentioned previously, included both STEMI (n = 3534) and UA/NSTEMI (n = 10,074) patients.36 The primary endpoint, cardiovascular death, nonfatal MI, and nonfatal stroke, was significantly lower in the prasugrel group at the expensive of increased bleeding in the prasugrel-treated patients.36 Although prasugrel is a reasonable choice of thienopyridine in patients with ACS, it should not be used in patients with a history of stroke or TIA, and it should be used with caution in patients older than 75 or weighing less than 60 kg.3 The dosing regimen of prasugrel for patients with UA/NSTEMI is identical to the dose used in STEMI patients (60-mg loading and 10-mg maintenance).
Ticagrelor, which reversibly binds to the P2Y12 platelet receptor, exhibited greater efficacy than clopidogrel in the PLATO trial.86 Major bleeding events did not differ between the groups, although bleeding unrelated to CABG occurred more often with ticagrelor. Both prasugrel and ticagrelor may have a quicker onset of action than clopidogrel and may prove to be very useful in patients who are clopidogrel resistant or have recurrent cardiovascular events while on clopidogrel.
The current guidelines recommend a loading dose of 300 to 600 mg of clopidogrel in patients with UA/NSTEMI, followed by 75 mg daily.3 Prasugrel should be administered as a 60-mg loading dose followed by a 10 mg/d maintenance dose.3 The duration of clopidogrel may depend on whether or not the patient has received a stent. Typically, patients who received bare metal stents (BMS) for at least 4 weeks and those with drug-eluting stents (DES) should remain on clopidogrel for at least 12 months.3,87 For DES, however, adequate long-term data have not been sufficient to formulate a definite recommendation on the duration of therapy.
Anticoagulant Therapy
Heparin is an important component of primary therapy for patients with unstable coronary syndromes without ST elevation. When added to aspirin, heparin has been shown to reduce refractory angina and the development of MI,30 and a meta-analysis of the available data indicates that addition of heparin reduces the composite endpoint of death or MI.88
Several trials have documented beneficial effects of LMWH therapy in unstable coronary syndromes. The ESSENCE trial showed that the LMWH, enoxaparin, reduced the combined endpoint of death, MI, or recurrent ischemia at both 14 and 30 days when compared to heparin.89 Similar results were found in the TIMI 11B trial comparing enoxaparin to heparin.90 A meta-analysis of these two very similar trials demonstrated a 23% 7-day and an 18% 42-day reduction in the harder endpoint of death or MI.90 Dalteparin, another LMWH, is also available, but the evidence for its efficacy is not nearly as compelling as that for enoxaparin.91
Direct Thrombin Inhibitors
Bivalirudin is a 20–amino acid peptide based on the structure of hirudin, a natural anticoagulant isolated from the saliva of the medicinal leech, Hirudo medicinalis. Bivalirudin is the only DTI indicated for use in ACS. The REPLACE 2 trial compared bivalirudin plus provisional glycoprotein IIb/IIIa inhibitor to unfractionated heparin plus planned glycoprotein IIb/IIIa inhibitor in 6010 patients undergoing planned or urgent PCI.46 Although 6-month event rates with bivalirudin were slightly higher (7.6 % versus 7.1%), bleeding with bivalirudin was lower, and the prespecified composite endpoint met statistical criteria for non-inferiority. Similar findings were seen in the ACUITY trial, which compared heparin with glycoprotein IIb/IIIa inhibitor to bivalirudin with glycoprotein IIb/IIIa inhibitor to bivalirudin alone with provisional glycoprotein IIb/IIIa inhibitor.92 Bivalirudin alone, compared with heparin plus glycoprotein IIb/IIIa inhibitors, resulted in non-inferior rates of composite ischemia (7.8% versus 7.3%, P = 0.32). Major bleeding was again significantly reduced with bivalirudin alone. However, patients who got bivalirudin alone without a thienopyridine prior to angiography or PCI had a higher rate of composite ischemic events than patients who received heparin plus a glycoprotein IIb/IIIa inhibitor ( 9.1% versus 7.1%). Therefore, it is not recommended that bivalirudin be administered alone, particularly if there is a going to be a delay to angiography.
Glycoprotein IIb/IIIa Antagonists
Given the central role of platelet activation and aggregation in the pathophysiology of unstable coronary syndromes, attention has focused on platelet glycoprotein IIb/IIIa antagonists, which inhibit the final common pathway of platelet aggregation. Three agents are currently available. Abciximab, a monoclonal antibody Fab fragment; tirofiban, a small-molecule, synthetic nonpeptide agent; and eptifibatide, a small-molecule cyclic heptapeptide. The benefits of glycoprotein IIb/IIIa inhibitors as adjunctive treatment in patients with ACS have shown in several trials.93–94 Meta-analyses have found a relative risk reduction of 11% in NSTEMI.38 Additional analysis suggests that glycoprotein IIb/IIIa inhibition is most effective in high-risk patients, those with either ECG changes or elevated troponin.38 The benefits appear to be restricted to patients undergoing PCI, which may not be entirely surprising.
The above mentioned studies were conducted prior to the era of dual antiplatelet therapy. As mentioned previously, it is common practice to administer a thienopyridine and aspirin in conjunction with an anticoagulant in patients with ACS. For patients with UA/NSTEMI undergoing an initial invasive approach, the most recent data suggest that either a glycoprotein IIb/IIIa inhibitor or a thienopyridine can be given in addition to aspirin and an anticoagulant if the patient is considered low risk (troponin negative). However, if the patient is considered high risk (troponin positive, recurrent ischemic features), both a glycoprotein IIb/IIIa inhibitor and clopidogrel can be given in addition to aspirin and an anticoagulant.3,87
Interventional Management
An early invasive approach has now been compared to a conservative approach in several prospective studies. Two earlier trials, the VANQWISH trial95 and the TIMI IIIb16 study, were negative, but the difference in the number of patients who had been revascularized by the end of these trials was small. In addition, they were performed before widespread use of coronary stenting and platelet glycoprotein IIb/IIIa inhibitors, both of which have now been shown to improve outcomes after angioplasty.
The FRISC II,96 TACTICS-TIMI 18,97 and RITA III98 trials each demonstrated that the composite endpoint of death, MI, or refractory angina was less frequent among patients who were randomized to the early invasive strategy, with the greatest benefit observed in high-risk patients, those with elevated cardiac biomarkers, extensive ST-segment depression, and hemodynamic features suggestive of large infarctions.87
The ICTUS trial enrolled 1200 patients with UA/NSTEMI who were initially treated with aspirin and enoxaparin before randomized assignment to one of two strategies: an early invasive strategy within 48 hours that included abciximab for PCI or a selective invasive strategy.99 Patients who were assigned the latter strategy were selected for coronary angiography only if they had refractory angina despite medical treatment, hemodynamic or rhythm instability, or predischarge exercise testing demonstrated clinically significant ischemia. The trial showed no reduction in the composite endpoints of death, nonfatal MI, or rehospitalization for angina at 1 year among patients who were assigned to the early invasive strategy. After 4 years of follow-up, the rates of death and MI among the two groups of patients remained similar.99 It is not clear why the results of ICTUS differ from previous trials. The more recent Timing of Intervention in Acute Coronary Syndromes (TIMACS) study randomized 3031 patients with UA/NSTEMI to undergo cardiac catheterization either within 24 hours of symptom onset or more than 36 hours later.100 The median time to angiography was 14 hours for the early intervention group and 50 hours for the delayed-intervention group. There was no difference between the groups in the composite endpoint of death, MI, or stroke at 6 months.
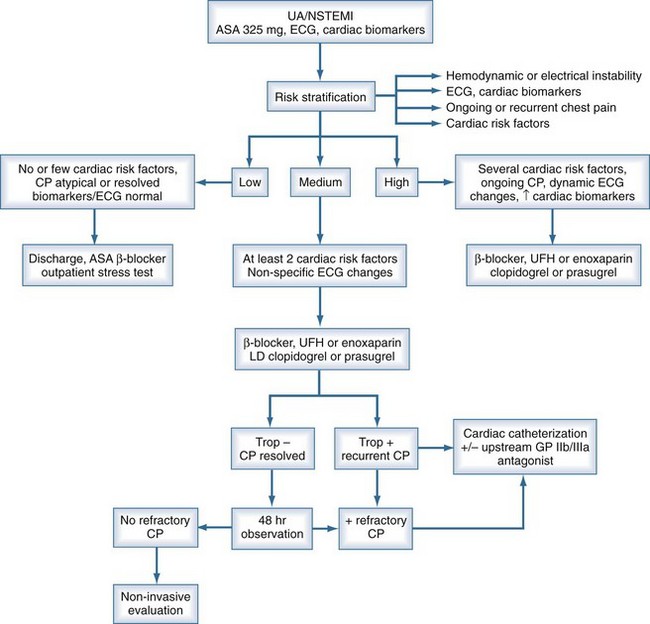
Risk stratification is the key to managing patients with NSTEMI ACS. One possible algorithm for managing patients with NSTEMI is shown in Figure 76-2. An initial strategy of medical management with attempts at stabilization is warranted in patients with lower risk, but patients at higher risk should be considered for cardiac catheterization. Pharmacologic and mechanical strategies are intertwined in the sense that selection of patients for early revascularization will influence the choice of antiplatelet and anticoagulant medication. When good clinical judgment is employed, early coronary angiography in selected ACS patients can lead to better management and lower morbidity and mortality.
 Complications of Acute Myocardial Infarction
Complications of Acute Myocardial Infarction
Ventricular Free Wall Rupture
Ventricular free wall rupture typically occurs during the first week after infarction. The classic patient is elderly, female, and hypertensive. Early use of fibrinolytic therapy reduces the incidence of cardiac rupture, but late use may actually increase the risk. Pseudoaneurysm with leakage may be heralded by chest pain, nausea, and restlessness, but frank free wall rupture presents as a catastrophic event with shock and electromechanical dissociation. Pericardiocentesis may be necessary to relieve acute tamponade, ideally in the operating room, since the pericardial effusion my be tamponading the bleeding. Salvage is possible with expeditious thoracotomy and repair, either with a patch or by direct suturing.101 A pericardial effusion may be seen by echocardiography; contrast ventriculography is not a sensitive way to detect a small rupture.
Ventricular Septal Rupture
Rapid institution of intraaortic balloon pumping and supportive pharmacologic measures are necessary. Operative repair is the only viable option for long-term survival. The timing of surgery has been controversial, but most authorities now suggest that repair should be undertaken early, within 48 hours of the rupture.102
Acute Mitral Regurgitation
Ischemic mitral regurgitation is usually associated with inferior MI and ischemia or infarction of the posterior papillary muscle, although anterior papillary muscle rupture can also occur. Papillary muscle rupture has a bimodal incidence, either within 24 hours or 3 to 7 days after AMI. It presents dramatically with pulmonary edema, hypotension, and cardiogenic shock. When a papillary muscle ruptures, the murmur of acute mitral regurgitation may be limited to early systole because of rapid equalization of pressures in the left atrium and left ventricle. More importantly, the murmur may be soft or inaudible, especially when cardiac output is low.103
Echocardiography is extremely useful in the differential diagnosis, which includes free wall rupture, ventricular septal rupture, and infarct extension with pump failure. Hemodynamic monitoring with pulmonary artery catheterization may also be helpful. Management includes afterload reduction with nitroprusside and intraaortic balloon pumping as temporizing measures. Inotropic or vasopressor therapy may also be needed to support cardiac output and blood pressure. Definitive therapy, however, is surgical valve repair or replacement, which should be undertaken as soon as possible, since clinical deterioration can be sudden.103,104
Right Ventricular Infarction
Right ventricular infarction occurs in up to 30% of patients with inferior infarction and is clinically significant in 10%.105 The combination of a clear chest x-ray with jugular venous distention in a patient with an inferior wall MI should lead to the suspicion of a coexisting right ventricular infarct. The diagnosis is substantiated by demonstration of ST-segment elevation in the right precordial leads (V3R to V5R) or by characteristic hemodynamic findings on right heart catheterization (elevated right atrial and right ventricular end-diastolic pressures, with normal to low pulmonary artery occlusion pressure and low cardiac output). Echocardiography can demonstrate depressed right ventricular contractility.106 Patients with cardiogenic shock on the basis of right ventricular infarction have a better prognosis than those with left-sided pump failure.105 This may be due in part to the fact that right ventricular function tends to return to normal over time with supportive therapy,107 although such therapy may need to be prolonged.
In patients with right ventricular infarction, right ventricular preload should be maintained with fluid administration. In some cases, however, fluid resuscitation may increase pulmonary capillary occlusion pressure but may not increase cardiac output, and overdilation of the right ventricle can compromise LV filling and cardiac output.107 Inotropic therapy with dobutamine may be more effective in increasing cardiac output in some patients, and monitoring with serial echocardiograms may also be useful to detect right ventricular overdistention.107 Maintenance of atrioventricular synchrony is also important in these patients to optimize right ventricular filling.106 For patients with continued hemodynamic instability, intraaortic balloon pumping may be useful, particularly because elevated right ventricular pressures and volumes increase wall stress and oxygen consumption and decrease right coronary perfusion pressure, exacerbating right ventricular ischemia.
Reperfusion of the occluded coronary artery is also crucial. A study using direct angioplasty demonstrated that restoration of normal flow resulted in dramatic recovery of right ventricular function and a mortality rate of only 2%, whereas unsuccessful reperfusion was associated with persistent hemodynamic compromise and a mortality of 58%.108
Cardiogenic Shock
Epidemiology and Pathophysiology
Cardiogenic shock, resulting either from LV pump failure or mechanical complications, represents the leading cause of in-hospital death after MI.109 Despite advances in management of heart failure and AMI, until very recently, clinical outcomes in patients with cardiogenic shock have been poor, with reported mortality rates ranging from 50% to 80%.110 Patients may have cardiogenic shock at initial presentation, but shock often evolves over several hours.111,112
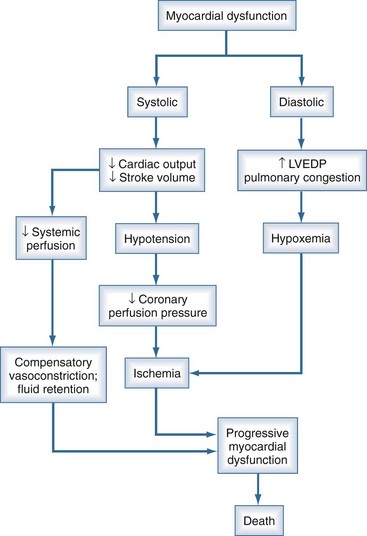
Cardiac dysfunction in patients with cardiogenic shock is usually initiated by MI or ischemia. The myocardial dysfunction resulting from ischemia worsens that ischemia, creating a downward spiral (Figure 76-3). Compensatory mechanisms that retain fluid in an attempt to maintain cardiac output may add to the vicious cycle and further increase diastolic filling pressures. The interruption of this cycle of myocardial dysfunction and ischemia forms the basis for the therapeutic regimens for cardiogenic shock.
Initial Management
When arterial pressure remains inadequate, therapy with vasopressor agents may be required to maintain coronary perfusion pressure. Maintenance of adequate blood pressure is essential to break the vicious cycle of progressive hypotension with further myocardial ischemia. Dopamine increases both blood pressure and cardiac output, but recent data suggest that norepinephrine may be a superior agent in patients with cardiogenic shock.113 Phenylephrine, a selective α1-adrenergic agonist, may be added when tachyarrhythmias limit therapy with other vasopressors. Vasopressor infusions must be titrated carefully in patients with cardiogenic shock to maximize coronary perfusion pressure, with the least possible increase in myocardial oxygen demand. Hemodynamic monitoring with serial measurements of cardiac output, filling pressures, (and other parameters such as mixed venous oxygen saturation) allows for titration of the dosage of vasoactive agents to the minimum dosage required to achieve the chosen therapeutic goals.114
In patients with inadequate tissue perfusion and adequate intravascular volume, cardiovascular support with inotropic agents should be initiated. Dobutamine, a selective β1-adrenergic receptor agonist, can improve myocardial contractility and increase cardiac output; it is the initial agent of choice in patients with systolic pressures above 80 mm Hg. Dobutamine may exacerbate hypotension in some patients and can precipitate tachyarrhythmias. Use of dopamine may be preferable if systolic pressure is less than 80 mm Hg, although tachycardia and increased peripheral resistance may worsen myocardial ischemia. In some situations, a combination of dopamine and dobutamine can be more effective than either agent used alone. Phosphodiesterase inhibitors such as milrinone are less arrhythmogenic than catecholamines, but they have the potential to cause hypotension and should be used with caution in patients with tenuous clinical status. Levosimendan, a calcium sensitizer, has both inotropic and vasodilator properties and does not increase myocardial oxygen consumption. Several relatively small studies have shown hemodynamic benefits with levosimendan in cardiogenic shock after MI,115,116 but survival benefits have not been shown either in cardiogenic shock or acute heart failure.117
Intraaortic balloon pump (IABP) counterpulsation reduces systolic afterload and augments diastolic perfusion pressure, increasing cardiac output and improving coronary blood flow.118 These beneficial effects, in contrast to those of inotropic or vasopressor agents, occur without an increase in oxygen demand. IABP does not, however, produce a significant improvement in blood flow distal to a critical coronary stenosis and has not been shown to improve mortality when used alone without reperfusion therapy or revascularization. In patients with cardiogenic shock and compromised tissue perfusion, IABP can be an essential support mechanism to stabilize patients and allow time for definitive therapeutic measures to be undertaken.118,119 In appropriate settings, more intensive support with mechanical assist devices may also be implemented.
Reperfusion Therapy
Although fibrinolytic therapy reduces the likelihood of subsequent development of shock after initial presentation,112 its role in the management of patients who have already developed shock is less certain. The available randomized trials.6,18,28,120 have not demonstrated that fibrinolytic therapy reduces mortality in patients with established cardiogenic shock. On the other hand, in the SHOCK Registry,121 patients treated with fibrinolytic therapy had a lower in-hospital mortality rate than those who were not (54% versus 64%, P = 0.005), even after adjustment for age and revascularization status (OR 0.70, P = 0.027).
Fibrinolytic therapy is clearly less effective in patients with cardiogenic shock than in those without. The explanation for this lack of efficacy appears to be the low reperfusion rate achieved in this subset of patients. The reasons for decreased fibrinolytic efficacy in patients with cardiogenic shock probably include hemodynamic, mechanical, and metabolic factors that prevent achievement and maintenance of infarct-related artery patency.122 Attempts to increase reperfusion rates by increasing blood pressure with aggressive inotropic and pressor therapy and IABP counterpulsation make theoretic sense, and two small studies support the notion that vasopressor therapy to increase aortic pressure improves fibrinolytic efficacy.122,123 The use of intraaortic balloon pumping to augment aortic diastolic pressure may increase the effectiveness of fibrinolytics as well.
To date, emergency percutaneous revascularization is the only intervention that has been shown to consistently reduce mortality rates in patients with cardiogenic shock. An extensive body of observational and registry studies has shown consistent benefits from revascularization. Notable among these is the GUSTO-1 trial, in which patients treated with an “aggressive” strategy (coronary angiography performed within 24 hours of shock onset with revascularization by PTCA or bypass surgery) had significantly lower mortality (38% compared with 62%).124 The National Registry of Myocardial Infarction–2 (NRMI-2) collected 26,280 shock patients with cardiogenic shock in the setting of MI between 1994 and 1997 and similarly supported the association between revascularization and survival.125 Improved short-term mortality was noted in those who then underwent revascularization during the reference hospitalization, either via PTCA (12.8% mortality versus 43.9%) or CABG (6.5% versus 23.9%).125 These data complement the GUSTO-1 substudy data and are important not only because of the sheer number of patients from whom these values are derived but also because NRMI-2 was a national cross-sectional study which more closely represents general clinical practice than carefully selected trial populations. These studies cannot be regarded as definitive because of their retrospective design, but two randomized controlled trials have now evaluated revascularization for patients with MI.
The SHOCK study was a randomized, multicenter international trial that assigned patients with cardiogenic shock to receive optimal medical management—including IABP and fibrinolytic therapy—or to cardiac catheterization with revascularization using PTCA or CABG The primary endpoint, all-cause mortality at 30 days, was 46.7% in the revascularization group and 56% in the medical therapy group, a difference that did not reach statistical significance (P = 0.11).126 Planned follow-up, however, revealed a significant benefit from early revascularization at 6 months and 1 year (P < 0.03).127 Subgroup analyses also revealed benefit in patients younger than 75, those with prior MI, and those randomized less than 6 hours from onset of infarction.126,127
The SMASH trial was similarly designed but enrolled sicker patients.128 The trial was terminated early owing to difficulties in patient recruitment and enrolled only 55 patients, but it showed a reduction in 30-day absolute mortality similar to that in the SHOCK trial (69% mortality in the invasive group versus 78% in the medically managed group, P = NS), and this benefit was also maintained at 1 year.128
On the basis of these randomized trials, the presence of cardiogenic shock in the setting of acute MI is a class I indication for emergency revascularization, either by percutaneous intervention or CABG.4
Ambrose JA, Martinez EE. A new paradigm for plaque stabilization. Circulation. 2002;105:2000-2004.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2007;106:803-877.
Kushner FG, HM, Smith SC, et al. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (Updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (Updating the 2005 Guideline and 2007 Focused Update): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;120:2271-2306.
Mehta SR, GC, Boden WE, et al. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med. 2009;360:2165-2175.
De Backer D, BP, Devriendt J, et al. Comparison of Dopamine and Norepinephrine in the Treatment of Shock. on behalf of the SOAP II investigators. N Engl J Med. 2010;362:779-789.
Cantor WJ, FD, Borgundvaag B, et al. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med. 2009;360:2705-2718.
Wiviott SD, BE, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13-20.
Stone GW, MB, Cox DA, et al. Bivalirudin for patients with acute coronary syndromes. N Engl J Med. 2006;355:2203-2216.
Wiviott SD, AE, Gibson CM, et al. Evaluation of prasugrel compared with clopidogrel in patients with acute coronary syndromes: design and rationale for the TRial to assess Improvement in Therapeutic Outcomes by optimizing platelet Inhibition with prasugrel Thrombolysis In Myocardial Infarction 38 (TRITON-TIMI 38). Am Heart J. 2006:152.
Chen ZM, JL, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1607-1621.
Chen ZM, PH, Chen YP, et al. COMMIT (CIOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622-1632.
Kim JS, KJ, Choi D, et al. Efficacy of High-Dose Atorvastatin Loading Before Primary Percutaneous Coronary Intervention for ST-Segment Elevation Myocardial Infarction. J Am Coll Caridiol Intv. 2010;3:332-339.
1 Sherman CT, Litvack F, Grundfest W, et al. Coronary angioscopy in patients with unstable angina pectoris. N Engl J Med. 1986;315:913-919.
2 Ambrose JA, Martinez EE. A new paradigm for plaque stabilization. Circulation. 2002;105:2000-2004.
3 Kushner FG, Hand M, Smith SCJr, et al. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (Updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (Updating the 2005 Guideline and 2007 Focused Update): A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2009;120:2271-2306.
4 Ryan TJ, Antman EM, Brooks NH, et al. 1999 update: ACC/AHA guidelines for the management of patients with acute myocardial infarction. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Acute Myocardial Infarction). J AM Coll Cardiol. 1999;34:890-911.
5 Go AS, Barron HV, Rundle AC, Ornato JP, Avins AL. Bundle-branch block and in-hospital mortality in acute myocardial infarction. National Registry of Myocardial Infarction 2 Investigators. Ann Intern Med. 1998;129:690-697.
6 Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet. 1986;1:397-402.
7 TIMI Study Group. The Thrombolysis in Myocardial Infarction (TIMI) trial. Phase I findings. N Engl J Med. 1985;312:932-936.
8 Fibrinolytic Therapy Trialists (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311-322.
9 Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311-322.
10 LATE Study Group. Late Assessment of Thrombolytic Efficacy (LATE) study with alteplase 6-24 hours after onset of acute myocardial infarction. Lancet. 1993;342:759-766.
11 EMERAS (Estudio Multicentrico Estreptoquinasa Republicas de America del Sur) Collaborative Group. Randomized trial of late thrombolytics in patients with suspected acute myocardial infarction. Lancet. 1993;342:767-772.
12 Schroder R, Dissmann R, Bruggemann T, et al. Extent of early ST segment elevation resolution: a simple but strong predictor of outcome in patients with acute myocardial infarction. J Am Coll Cardiol. 1994;24:384-391.
13 Anderson RD, White HD, Ohman EM, et al. Predicting outcome after thrombolysis in acute myocardial infarction according to ST-segment resolution at 90 minutes: a substudy of the GUSTO III trial. Am Heart J. 2002;144:81-88.
14 Di Mario C, Dudek D, Piscione F, et al. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet. 2008;371:559-568.
15 Cantor WJ, Fitchett D, Borgundvaag B, et al. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med. 2009;360:2705-2718.
16 TIMI Investigators. Effects of tissue plasminogen activator and a comparison of early invasive and conservative strategies in unstable angina and non-Q-wave myocardial infarction. Results of the TIMI IIIB Trial. Circulation. 1994;89:1545-1556.
17 GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329:673-682.
18 GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med. 1993;329:1615-1622.
19 Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO III) Investigators. A comparison of reteplase with alteplase for acute myocardial infarction. N Engl J Med. 1997;337:1118-1123.
20 Assessment of the Safety and Efficacy of a New Thrombolytic Investigators. Single-bolus tenecteplase compared with front-loaded alteplase in acute myocardial infarction: the ASSENT-2 double-blind randomised trial. Lancet. 1999;354:716-722.
21 Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med. 1993;328:673-679.
22 GUSTO IIb Investigators. A comparison of recombinant hirudin with heparin for the treatment of acute coronary syndromes. N Engl J Med. 1996;335:775-782.
23 Grines C, Patel A, Zijlstra F, Weaver WD, Granger C, Simes RJ. Primary coronary angioplasty compared with intravenous thrombolytic therapy for acute myocardial infarction: six-month follow up and analysis of individual patient data from randomized trials. Am Heart J. 2003;145:47-57.
24 Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13-20.
25 Cannon CP, Gibson CM, Lambrew CT, et al. Relationship of symptom-onset-to-balloon time and door-to-balloon time with mortality in patients undergoing angioplasty for acute myocardial infarction. JAMA. 2000;283:2941-2947.
26 Rankin JM, Spinelli JJ, Carere RG, et al. Improved clinical outcome after widespread use of coronary-artery stenting in Canada. N Engl J Med. 1999;341:1957-1965.
27 Stone GW, Brodie BR, Griffin JJ, et al. Clinical and angiographic follow-up after primary stenting in acute myocardial infarction: the Primary Angioplasty in Myocardial Infarction (PAMI) stent pilot trial. Circulation. 1999;99:1548-1554.
28 ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet. 1988;2:349-360.
29 Lewis HDJr, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina. Results of a Veterans Administration Cooperative Study. N Engl J Med. 1983;309:396-403.
30 Theroux P, Ouimet H, McCans J, et al. Aspirin, heparin, or both to treat acute unstable angina. N Engl J Med. 1988;319:1105-1111.
31 Montalescot G, Sideris G, Meuleman C, et al. A randomized comparison of high clopidogrel loading doses in patients with non-ST-segment elevation acute coronary syndromes: the ALBION (Assessment of the Best Loading Dose of Clopidogrel to Blunt Platelet Activation, Inflammation and Ongoing Necrosis) trial. J Am Coll Cardiol. 2006;48:931-938.
32 Thebault JJ, Kieffer G, Cariou R. Single-dose pharmacodynamics of clopidogrel. Semin Thromb Hemost. 1999;25(Suppl. 2):3-8.
33 Sabatine MS, Cannon CP, Gibson CM, et al. Addition of clopidogrel to aspirin and fibrinolytic therapy for myocardial infarction with ST-segment elevation. N Engl J Med. 2005;352:1179-1189.
34 Sabatine MS, Cannon CP, Gibson CM, et al. Effect of clopidogrel pretreatment before percutaneous coronary intervention in patients with ST-elevation myocardial infarction treated with fibrinolytics: the PCI-CLARITY study. JAMA. 2005;294:1224-1232.
35 Brandt JT, Payne CD, Wiviott SD, et al. A comparison of prasugrel and clopidogrel loading doses on platelet function: magnitude of platelet inhibition is related to active metabolite formation. Am Heart J. 2007;153:e9-16.
36 Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
37 Chen ZM, Jiang LX, Chen YP, et al. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: random- ised placebo-controlled trial. Lancet. 2005;366:1607-1621.
38 Chew DP, Moliterno DJ. A critical appraisal of platelet glycoprotein IIb/IIIa inhibition. J AM Coll Cardiol. 2000;36:2028-2035.
39 Lincoff AM, Califf RM, Moliterno DJ, et al. Complementary clinical benefits of coronary-artery stenting and blockade of platelet glycoprotein IIb/IIIa receptors. Evaluation of Platelet IIb/IIIa Inhibition in Stenting Investigators. N Engl J Med. 1999;341:319-327.
40 EPISTENT Investigators. Randomised placebo-controlled and balloon-angioplasty-controlled trial to assess safety of coronary stenting with use of platelet glycoprotein- IIb/IIIa blockade. Lancet. 1998;352:87-92.
41 Montalescot G, Barragan P, Wittenberg O, et al. Platelet glycoprotein IIb/IIIa inhibition with coronary stenting for acute myocardial infarction. N Engl J Med. 2001;344:1895-1903.
42 Stone GW, Grines CL, Cox DA, et al. Comparison of angioplasty with stenting, with or without abciximab, in acute myocardial infarction. N Engl J Med. 2002;346:957-966.
43 Mehilli J, Kastrati A, Schulz S, et al. Abciximab in patients with acute ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention after clopidogrel loading: a randomized double-blind trial. Circulation. 2009;119:1933-1940.
44 Van’t Hof AW, Ten Berg J, Heestermans T, et al. Prehospital initiation of tirofiban in patients with ST-elevation myocardial infarction undergoing primary angioplasty (On-TIME 2): a multicentre, double- blind, randomised controlled trial. Lancet. 2008;372:537-546.
45 Stone GW, Witzenbichler B, Guagliumi G, et al. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008;358:2218-2230.
46 Lincoff AM, Bittl JA, Harrington RA, et al. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA. 2003;289:853-863.
47 Mehran R, Brodie B, Cox DA, et al. The Harmonizing Outcomes with RevasculariZatiON and Stents in Acute Myocardial Infarction (HORIZONS-AMI) Trial: study design and rationale. Am Heart J. 2008;156:44-56.
48 Mehran R, Lansky AJ, Witzenbichler B, et al. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet. 2009;374:1149-1159.
49 Antman EM, Morrow DA, McCabe CH, et al. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med. 2006;354:1477-1488.
50 Wallentin L, Goldstein P, Armstrong PW, et al. Efficacy and safety of tenecteplase in combination with the low-molecular-weight heparin enoxaparin or unfractionated heparin in the prehospital setting: the Assessment of the Safety and Efficacy of a New Fibrinolytic Regimen (ASSENT)-3 PLUS randomized trial in acute myocardial infarction. Circulation. 2003;108:135-142.
51 Yusuf S, Mehta SR, Chrolavicius S, et al. Effects of fondaparinux on mortality and reinfarction in patients with acute ST-segment elevation myocardial infarction: the OASIS-6 randomized trial. JAMA. 2006;295:1519-1530.
52 Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cariol. 2007;50:1742-1751.
53 Cohn PF, Gorlin R. Physiologic and clinical actions of nitroglycerin. Med Clin North Am. 1974;58:407-415.
54 Gruppo Italiano per lo Studio della Sopravvinza nell’Infarto Miocardico (GISSI). GISSI-3: effects of lisinopril and transdermal glyceryl trinitrate singly and together on 6-week mortality and ventricular function after acute myocardial infarction. Lancet. 1994;343:1115-1122.
55 ISIS-4 (Fourth International Study of Infarct Survival) Study Group. ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet. 1995;345:669-685.
56 First International Study of Infarct Survival Collaborative Group. Randomised trial of intravenous atenolol among 16 027 cases of suspected acute myocardial infarction: ISIS-1. Lancet. 1986;2:57-66.
57 TIMI Study Group. Comparison of invasive and conservative strategies after treatment with intravenous tissue plasminogen activator in acute myocardial infarction. Results of the thrombolysis in myocardial infarction (TIMI) phase II trial. N Engl J Med. 1989;320:618-627.
58 Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622-1632.
59 Antman EM, Hand M, Armstrong PW, et al. 2007 Focused Update on the ACC/AHA 2004 Guidelines for the Management of Patients with ST-Elevation Myocardial Infarction. J Am Coll Cariol. 2008;51:210-247.
60 MIAMI Trial Research Group. Metoprolol in acute myocardial infarction (MIAMI). A randomised placebo-controlled international trial. Eur Heart J. 1985;6:199-226.
61 The International Collaborative Study Group. Reduction of infarct size with the early use of timolol in acute myocardial infarction. N Engl J Med. 1984;310:9-15.
62 Dargie HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet. 2001;537:1385-1390.
63 CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429-1435.
64 SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293-302.
65 SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685-691.
66 Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the Survival and Ventricular Enlargement Trial. N Engl J Med. 1992;327:669-677.
67 Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342:145-153.
68 Edner M, Bonarjee VV, Nilsen DW, Berning J, Carstensen S, Caidahl K. Effect of enalapril initiated early after acute myocardial infarction on heart failure parameters, with reference to clinical class and echocardiographic determinants. CONSENSUS II Multi-Echo Study Group. Clin Cardiol. 1996;19:543-548.
69 Lipid Research Clinics Program. The lipid research clinics coronary primary prevention trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. JAMA. 1984;251:365-374.
70 Frick MH, Elo O, Haapa K, et al. Helsinki heart study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237-1245.
71 Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344:1383-1389.
72 Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001-1009.
73 Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349-1357.
74 Gibbons RJ, Abrams J, Chatterjee K, et al. ACC/AHA 2002 guideline update for the management of patients with chronic stable angina–summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients With Chronic Stable Angina). Circulation. 2003;107:149-158.
75 Kim JS, Kim J, Choi D, et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: the STATIN STEMI trial. J Am Coll Caridiol Intv. 2010;3:332-339.
76 Held PH, Yusuf S, Furberg CD. Calcium channel blockers in acute myocardial infarction and unstable angina: an overview. BMJ. 1989;299:1187-1192.
77 Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
78 Gibson RS, Boden WE, Theroux P, et al. Diltiazem and reinfarction in patients with non-q-wave myocardial infarction. N Engl J Med. 1986;315:423-429.
79 Braunwald E. Unstable angina. A classification. Circulation. 1989;80:410-414.
80 Ohman EM, Armstrong PW, Christenson RH, et al. Cardiac troponin T levels for risk stratification in acute myocardial ischemia. N Engl J Med. 1996;335:133-1341.
81 Cairns JA, Gent M, Singer J, et al. Aspirin, sulfinpyrazone, or both in unstable angina. Results of a Canadian multicenter trial. N Engl J Med. 1985;313:1369-1375.
82 Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494-502.
83 Mehta SR, Yusuf S, Peters RJ, et al. Effects of pretreatment with clopidogrel and aspirin followed by long- term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358:527-533.
84 Steinhubl SR, Berger PB, Mann JT3rd, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;288:2411-2420.
85 CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet. 1996;348:1329-1339.
86 Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045-1057.
87 Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J AM Coll Cardiol. 2007;50:e1-157.
88 Oler A, Whooley MA, Oler J, Grady D. Adding heparin to aspirin reduces the incidence of myocardial infarction and death in patients with unstable angina. A meta-analysis. JAMA. 1996;276:811-815.
89 Cohen M, Demers C, Gurfinkel EP, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for unstable coronary artery disease. Efficacy and Safety of Subcutaneous Enoxaparin in Non-Q-Wave Coronary Events Study Group. N Engl J Med. 1997;337:447-452.
90 Antman EM, Cohen M, Radley D, et al. Assessment of the treatment effect of enoxaparin for unstable angina/non-Q-wave myocardial infarction. TIMI 11B-ESSENCE meta-analysis. Circulation. 1999;100:1602-1608.
91 FRagmin and Fast Revascularisation during InStability in Coronary artery disease Investigators. Long-term low-molecular-mass heparin in unstable coronary-artery disease: FRISC II prospective randomised multicentre study. Lancet. 1999;354:701-707.
92 Stone GW, McLaurin BT, Cox DA, et al. Bivalirudin for patients with acute coronary syndromes. N Engl J Med. 2006;355:2203-2216.
93 Platelet Receptor Inhibition in Ischemic Syndrome Management (PRISM) Study Investigators. A Comparison of Aspirin plus Tirofiban with Aspirin plus Heparin for Unstable Angina. N Engl J Med. 1998;338:1498-1505.
94 CAPTURE Investigators. Randomised placebo-controlled trial of abciximab before and during coronary intervention in refractory unstable angina: the CAPTURE Study. Lancet. 1997;349:1429-1435.
95 Boden WE, O’Rourke RA, Crawford MH, et al. Outcomes in patients with acute non-Q-wave myocardial infarction randomly assigned to an invasive as compared with a conservative management strategy. N Engl J Med. 1998;338:1785-1792.
96 FRagmin and Fast Revascularisation during InStability in Coronary artery disease Investigators. Invasive compared with non-invasive treatment in unstable coronary- artery disease: FRISC II prospective randomised multicentre study. Lancet. 1999;354:708-715.
97 Cannon CP, Weintraub WS, Demopoulos LA, et al. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
98 Fox KA, Poole-Wilson PA, Henderson RA, et al. Interventional versus conservative treatment for patients with unstable angina or non-ST-elevation myocardial infarction: the British Heart Foundation RITA 3 randomised trial. Randomized Intervention Trial of unstable Angina. Lancet. 2002;360:743-751.
99 Hirsch A, Windhausen F, Tijssen JG, Verheugt FW, Cornel JH, de Winter RJ. Long-term outcome after an early invasive versus selective invasive treatment strategy in patients with non-ST-elevation acute coronary syndrome and elevated cardiac troponin T (the ICTUS trial): a follow-up study. Lancet. 2007;369:827-835.
100 Mehta SR, Granger CB, Boden WE, et al. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med. 2009;360:2165-2175.
101 Reardon MJ, Carr CL, Diamond A, et al. Ischemic left ventricular free wall rupture: prediction, diagnosis, and treatment. Ann Thorac Surg. 1997;64:1509-1513.
102 Killen DA, Piehler JM, Borkon AM, Gorton ME, Reed WA. Early repair of postinfarction ventricular septal rupture. Ann Thorac Surg. 1997;63:138-142.
103 Khan SS, Gray RJ. Valvular emergencies. Cardiol Clin. 1991;9:689-709.
104 Bolooki H. Emergency cardiac procedures in patients in cardiogenic shock due to complications of coronary artery disease. Circulation. 1989;79:I137-I148.
105 Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med. 1993;328:981-988.
106 Kinch JW, Ryan TJ. Right ventricular infarction. N Engl J Med. 1994;330:1211-1217.
107 Dell’Italia LJ, Starling MR, Blumhardt R, Lasher JC, O’Rourke RA. Comparative effects of volume loading, dobutamine, and nitroprusside in patients with predominant right ventricular infarction. Circulation. 1985;72:1327-1335.
108 Bowers TR, O’Neill WW, Grines C, Pica MC, Safian RD, Goldstein JA. Effect of reperfusion on biventricular function and survival after right ventricular infarction. N Engl J Med. 1998;338:933-940.
109 Hollenberg SM, Kavinsky CJ, Parrillo JE. Cardiogenic shock. Ann Intern Med. 1999;131:47-59.
110 Goldberg RJ, Samad NA, Yarzebski J, Gurwitz J, Bigelow C, Gore JM. Temporal trends in cardiogenic shock complicating acute myocardial infarction. N Engl J Med. 1999;340:1162-1168.
111 Hochman JS, Boland J, Sleeper LA, et al. Current spectrum of cardiogenic shock and effect of early revascularization on mortality. Results of an International Registry. Circulation. 1995;91:873-881.
112 Holmes DRJr, Bates ER, Kleiman NS, et al. Contemporary reperfusion therapy for cardiogenic shock: the GUSTO-I trial experience. The GUSTO-I Investigators. Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries. J Am Coll Cardiol. 1995;26:668-674.
113 De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779-789.
114 Hollenberg SM, Hoyt JW. Pulmonary artery catheters in cardiovascular disease. New Horizons. 1997;5:207-213.
115 Russ MA, Prondzinsky R, Christoph A, et al. Hemodynamic improvement following levosimendan treatment in patients with acute myocardial infarction and cardiogenic shock. Crit Care Med. 2007;35:2732-2739.
116 Garcia-Gonzalez MJ, Dominguez-Rodriguez A, Ferrer-Hita JJ, Abreu-Gonzalez P, Munoz MB. Cardiogenic shock after primary percutaneous coronary intervention: Effects of levosimendan compared with dobutamine on haemodynamics. Eur J Heart Failure. 2006;8:723-728.
117 Mebazaa A, Nieminen MS, Packer M, et al. Levosimendan vs dobutamine for patients with acute decompensated heart failure: the SURVIVE Randomized Trial. JAMA. 2007;297:1883-1891.
118 Willerson JT, Curry GC, Watson JT, et al. Intraaortic balloon counterpulsation in patients in cardiogenic shock, medically refractory left ventricular failure and/or recurrent ventricular tachycardia. Am J Med. 1975;58:183-191.
119 Bates ER, Stomel RJ, Hochman JS, Ohman EM. The use of intraaortic balloon counterpulsation as an adjunct to reperfusion therapy in cardiogenic shock. Int J Cardiol. 1998;65(Suppl. 1):S37-S42.
120 Gruppo Italiano per lo Studio Della Streptochinasi Nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet. 1986;2:397-402.
121 Sanborn TA, Sleeper LA, Bates ER, et al. Impact of thrombolysis, intra-aortic balloon pump counterpulsation, and their combination in cardiogenic shock complicating acute myocardial infarction: a report from the SHOCK Trial Registry. SHould we emergently revascularize Occluded Coronaries for cardiogenic shocK? J AM Coll Cardiol. 2000;36:1123-1129.
122 Becker RC. Hemodynamic, mechanical, and metabolic determinants of thrombolytic efficacy: a theoretic framework for assessing the limitations of thrombolysis in patients with cardiogenic shock. Am Heart J. 1993;125:919-929.
123 Garber PJ, Mathieson AL, Ducas J, Patton JN, Geddes JS, Prewitt RM. Thrombolytic therapy in cardiogenic shock: effect of increased aortic pressure and rapid tPA administration. Can J Cardiol. 1995;11:30-36.
124 Berger PB, Holmes DRJr, Stebbins AL, Bates ER, Califf RM, Topol EJ. Impact of an aggressive invasive catheterization and revascularization strategy on mortality in patients with cardiogenic shock in the Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Coronary Arteries (GUSTO-I) trial. An observational study. Circulation. 1997;96:122-127.
125 Rogers WJ, Canto JG, Lambrew CT, et al. Temporal trends in the treatment of over 1.5 million patients with myocardial infarction in the US from 1990 through 1999: the National Registry of Myocardial Infarction 1, 2 and 3. J Am Coll Cardiol. 2000;36:2056-2063.
126 Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. N Engl J Med. 1999;341:625-634.
127 Hochman JS, Sleeper LA, White HD, et al. One-year survival following early revascularization for cardiogenic shock. JAMA. 2001;285:190-192.
128 Urban P, Stauffer JC, Bleed D, et al. A randomized evaluation of early revascularization to treat shock complicating acute myocardial infarction. The (Swiss) Multicenter Trial of Angioplasty for Shock-(S)MASH. Eur Heart J. 1999;20:1030-1038.
