[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 13 Acquired haemolytic anaemias
Assessing the likelihood of acquired haemolytic anaemia
Haemolytic anaemia may be suspected from either clinical or laboratory abnormalities. Suggestive clinical features include anaemia, jaundice and splenomegaly. Other relevant clinical features that should be sought are a history of autoimmune disease, recent blood transfusion, recent infection, exposure to drugs or toxins, the presence of a cardiac prosthesis and risk of malaria. Previous clinical history and laboratory results will help to establish that the disorder is acquired. The basic laboratory investigations when a haemolytic anaemia is suspected are listed in Chapter 11. In this chapter, tests are described that are more specific for the diagnosis of acquired haemolytic anaemia.
Assessment of the blood film and count in suspected acquired haemolytic anaemia
If haemolytic anaemia is suspected, a full blood count, reticulocyte count and blood film should always be performed. The blood count shows a reduced haemoglobin concentration (Hb) and, usually, an increased mean cell volume (MCV). The increased MCV is attributable to the fact that reticulocytes, which may constitute a significant proportion of total red cells, are larger than mature red cells. The abnormalities that may be detected in the blood film and their possible significance in acquired haemolytic anaemia are shown in Table 13.1. Abnormalities detected in the blood film will direct further investigations. For example, a Heinz body preparation would be relevant if irregularly contracted cells were present, particularly if there appeared to be red cell inclusions. Similarly, a direct antiglobulin test (DAT) would be indicated if the blood film showed spherocytes. Various inherited forms of haemolytic anaemia enter into the differential diagnosis of suspected acquired haemolytic anaemia. Thus, spherocytes could be attributable to hereditary spherocytosis as well as to autoimmune or alloimmune haemolytic anaemia. Haemolysis with irregularly contracted cells could be attributable not only to oxidant exposure but also to an unstable haemoglobin, homozygosity for haemoglobin C or glucose-6-phosphate dehydrogenase (G6PD) deficiency.
Table 13.1 Abnormalities that may be detected on blood film examination and their possible significance
| Morphological abnormality observed on blood film examination | Type of acquired haemolytic anaemia suggested |
|---|---|
| Schistocytes | Fragmentation syndromes including microangiopathic haemolytic anaemia and mechanical haemolytic anaemia |
| Spherocytes | Autoimmune, alloimmune or drug-induced immune haemolytic anaemia, paroxysmal cold haemoglobinuria, burns, Clostridium perfringens sepsis |
| Microspherocytes | Burns, fragmentation syndromes |
| Irregularly contracted cells | Oxidant damage, Zieve’s syndrome |
| Ghost cells, hemi-ghosts and suspicion of Heinz bodies | Acute oxidant damage |
| Marked red cell agglutination | Cold-antibody-induced haemolytic anaemia |
| Minor red cell agglutination | Warm autoimmune haemolytic anaemia, paroxysmal cold haemoglobinuria |
| Red cell agglutination plus erythrophagocytosis | Particularly characteristic of paroxysmal cold haemoglobinuria |
| Hypochromia, microcytosis and basophilic stippling | Lead poisoning |
| Erythrophagocytosis | Paroxysmal cold haemoglobinuria |
| Atypical lymphocytes | Cold-antibody-induced haemolytic anaemia associated with infectious mononucleosis or, less often, other infections |
| Lymphocytosis with mature small lymphocytes and smear cells | Autoimmune haemolytic anaemia associated with chronic lymphocytic leukaemia |
| Thrombocytopenia | Autoimmune haemolytic anaemia (Evans’ syndrome), thrombotic thrombocytopenic purpura, microangiopathic haemolytic anaemia associated with disseminated intravascular coagulation, paroxysmal nocturnal haemoglobinuria |
| Neutropenia | Paroxysmal nocturnal haemoglobinuria |
| No specific red cell features | Paroxysmal nocturnal haemoglobinuria |
Immune haemolytic anaemias
Acquired immune-mediated haemolytic anaemias are the result of autoantibodies to a patient’s own red cell antigens or alloantibodies in a patient’s circulation, either present in the plasma or completely bound to red cells (e.g. transfused or neonatal red cells). Alloantibodies may be present in a patient’s plasma and react with antigens on transfused donor red cells to cause haemolysis. Alloantibodies may also occur in maternal plasma and cause haemolytic disease of the newborn. Autoimmune haemolytic anaemia (AIHA) may be ‘idiopathic’ or secondary, associated mainly with lymphoproliferative disorders and autoimmune diseases, particularly systemic lupus erythematosus. AIHA may also follow atypical (Mycoplasma pneumoniae) pneumonia or infectious mononucleosis and other viral infections. AIHA has also been reported following allogeneic bone marrow transplantation1 and other hematopoietic stem cell transplantation in both adult2 and paediatric patients.3 Paroxysmal cold haemoglobinuria (PCH) also belongs to this group of disorders. Occasionally, drugs may give rise to a haemolytic anaemia of immunological origin that closely mimics idiopathic AIHA both clinically and serologically. This was a relatively common occurrence with α-methyldopa, a drug that is now used very infrequently, but it also occurs occasionally with other drugs. A larger range of drugs give rise to an antibody that is directed primarily against the drug and only secondarily involves the red cells. This is an uncommon occurrence. Such drugs include penicillin, phenacetin, quinidine, quinine, the sodium salt of p-aminosalicylic acid, salicylazosulphapyridine and cephalosporins.4
Types of Autoantibody
The diagnosis of an AIHA requires evidence of anaemia and haemolysis and demonstration of autoantibodies attached to the patient’s red cells (i.e. a positive DAT, see p. 279). A positive DAT may also be caused by the presence of alloantibodies (e.g. owing to a delayed haemolytic transfusion reaction), so details of any transfusion in the past months must be sought.
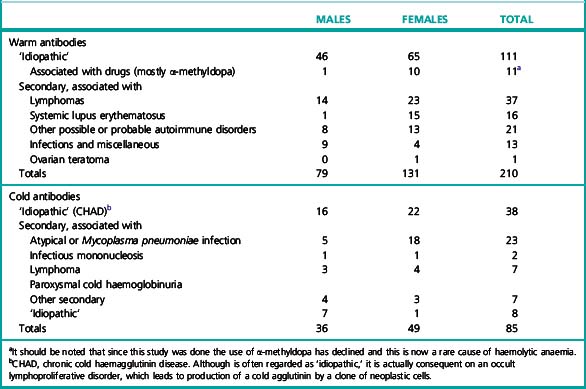
Cases of AIHA can similarly be separated into two broad categories according to the temperature characteristics of the associated autoantibodies: warm-type AIHA and the less frequent cold-type AIHA. The relative frequency of the two categories is illustrated in Table 13.2.5 In unusual instances, both warm autoantibody and cold autoantibody are detected in the patient’s serum and those cases are referred to as mixed-typed AIHA. This can be further classified into idiopathic or secondary, the latter often associated with systemic lupus erythematosus or lymphoma.6,7
Warm Autoantibodies
The most common type of warm autoantibody is an immunoglobulin (Ig) G, which behaves in vitro very similarly to an Rh alloantibody; indeed, many IgG autoantibodies have a mimicking Rh specificity. IgA and IgM warm autoantibodies are much less common and when present they are usually formed in addition to an IgG autoantibody (Table 13.3).8
Table 13.3 Direct antiglobulin test in warm-antibody autoimmune haemolytic anaemia: incidence of different reactions to specific antiglobulin sera8
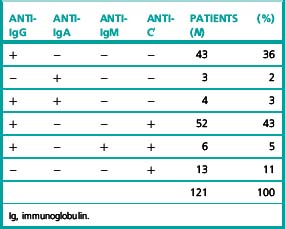
Frequently, patients with warm-type AIHA have complement adsorbed onto their red cells and the red cells are therefore agglutinated by antisera specific for complement or a complement component such as C3d (Table 13.3). In these cases, the complement is probably not being bound by an IgG antibody but is on the cell surface as the result of the action of small and otherwise undetected amounts of IgM autoantibody.
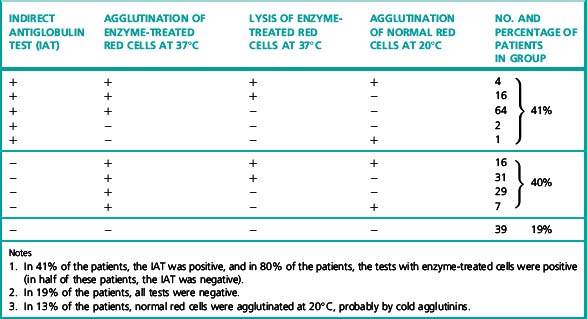
Warm autoantibodies free in the patient’s serum are best detected by means of the indirect antiglobulin test (IAT) or by the use of enzyme-treated (e.g. trypsinized or papainized) red cells. (Antibodies that agglutinate unmodified cells directly in vitro are seldom present.) Not infrequently, antibodies that agglutinate enzyme-treated cells, sometimes at high titres, are present in the sera of patients in whom the IAT using unmodified cells is negative (Table 13.4). Occasionally, too, they are present in the sera of patients in whom the DAT is negative.
Cold Autoantibodies
The red cells of patients suffering from CHAD characteristically give positive antiglobulin reactions only with anticomplement (anti-C′) sera. (The C′ notation is used to distinguish anticomplement antibodies from anti-C antibodies of the Rh system.) This is because of the presence of red cells that have irreversibly adsorbed sublytic amounts of complement; it is an indication of an antigen–antibody reaction that has taken place at a temperature below 37°C. The complement component responsible for the reaction with anti-C′ sera is the C3dg derivative of C3 (see p. 494).
Combined Warm and Cold Autoantibodies
In approximately 7% of cases with AIHA, both warm IgG antibody and cold IgM autoantibody are simultaneously detected in the patient’s serum.6,7 These cases are referred to as ‘combined warm and cold AIHA’ or mixed-type AIHA. The serological characteristics in these patients are the presence of IgM cold autoantibody with a high thermal amplitude (reacting at or above 30°C) in association with a warm IgG autoantibody. In some cases, high- titre cold agglutinins (>1024 at 4°C) were reported9,10 and in others the cold agglutinin titre were reported as >64 at 4°C.11,12
PCH is caused by a biphasic IgG autoantibody, usually with anti-P specificity, and is commonly seen as an acute condition in children. This antibody binds to the red cells in the cold but activates complement and causes haemolysis on rewarming to 37°C. Cases may be idiopathic or can be secondary to acute viral infection in children. Other tests of value in the diagnosis of PCH are discussed on p. 287.
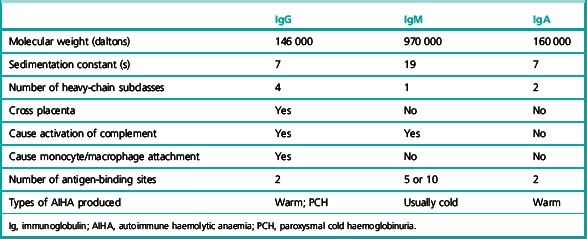
Some of the characteristics of IgG, IgM and IgA antibodies are listed in Table 13.5.
The clinical, haematological and serological aspects of the AIHAs have been summarized by Dacie13 and others.14–18
Methods of Investigation
Many of the methods used in the investigation of a patient suspected of suffering from AIHA are described in Chapter 21. Detailed description is given here of precautions to be taken when collecting blood samples from patients and of methods of particular value in the investigations.
Scheme for Serological Investigation of Haemolytic Anaemia Suspected to be of Immunological Origin
It is important to consider which are the most useful tests to carry out and the order in which they should be done. A suggested scheme has been set out in the form of answers to questions.19 Whereas some information may be helpful in classifying the type of AIHA, the single most important practical consideration is to determine whether, in addition to an autoantibody, there is any underlying alloantibody present. This should be identified before transfusion is undertaken to avoid a delayed haemolytic transfusion reaction that would compound existing haemolysis.
Perform a DAT using a polyspecific ‘broad-spectrum’ reagent, which contains both anti-IgG and anti-C′. (If the DAT is negative, it is unlikely, although not impossible, that the diagnosis is AIHA. See DAT-negative AIHA, p. 281)
Repeat the DAT using monospecific sera (see p. 500) (i.e. anti-IgG and anti-C3d).
Prepare eluates from the patient’s red cells. Test these later (see item 6).
Determine the patient’s ABO and RhD and Kell type. The Rh phenotype is particularly important in warm-type AIHA; other antigens must be determined if alloantibodies are to be differentiated from autoantibodies (see p. 507).
Screen the serum with two or three red cell suspensions suitable for routine pretransfusion antibody screening (see p. 528) looking for agglutination and lysis at 37°C by the IAT (see p. 500). If positive, identify the antibody using an antibody identification panel.
Test the eluate against the antibody identification panel of red cells by IAT and by using enzyme-treated red cells (see p. 498). Titration of autoantibody may be useful in the presence of a strong alloantibody.
The diagnosis of mixed-type AIHA can only be made after appropriate characterization of the serum autoantibodies. The serological characteristic in these cases is the presence of a cold IgM antibody with a high thermal amplitude (reacting at or above 30°C) in association with a warm IgG autoantibody.6,9
Detection of Incomplete Antibodies by Means of the Direct Antiglobulin (Coombs) Test
Significance of Positive Direct Antiglobulin Test
A positive DAT plus anaemia does not necessarily mean that the patient has autoimmune haemolytic anaemia.5,8,20 The causes of a positive test include the following:
Non-specific binding of immunoglobulins to red cells in patients with hypergammaglobulinemia or multiple myeloma and in recipients of antilymphocyte globulin and antithymocyte globulin.24
Szymanski et al.25 used an AutoAnalyser and used Ficoll and polyvinylpyrrolidone (PVP) to enhance agglutination by an anti-IgG serum highly diluted (usually to 1 in 5000) in 0.5% bovine serum albumin. In this sensitive system, the strength of agglutination was positively correlated with the serum γ-globulin concentration, being subnormal in hypogammaglobulinaemia and supranormal in hypergammaglobulinaemia.26
Similar findings were observed in patients with multiple myeloma. Non-specific binding of IgG to red cells was related to the level of monoclonal protein in the patient’s serum.26
Usually, in patients with hypergammaglobulinaemia in whom the DAT is positive, attempts to demonstrate antibodies in eluates fail (i.e. eluates are non-reactive).27,28
There was an association demonstrated between an elevated blood urea nitrogen and a positive DAT.32 Urea may alter the red cell membrane and enhance non-specific IgG adsorption.32
Positive DATs in Normal Subjects
The occurrence of a clearly positive DAT in an apparently healthy subject is a rare but well-known phenomenon. Worlledge20 reported a prevalence in blood donors of approximately 1 in 9000. In a later report, Gorst et al.34 estimated that the prevalence was approximately 1 in 14 000 with an increasing likelihood of a positive test with increasing age. Their report and subsequent reports,25,35 suggest that the finding of a positive DAT, using an anti-IgG serum, in an apparently healthy person is usually of little clinical significance and that, although overt AIHA may subsequently develop, this is infrequent. In some such individuals the DAT eventually becomes negative.
Positive DATs in Hospital Patients
In contrast to the rarity of positive DATs in healthy people, positive tests are much more frequent in hospital patients. Worlledge20 reported that the red cells of 40 out of 489 blood samples (8.9%) submitted for routine tests were agglutinated by anti-C′ sera. Only one sample was agglutinated by an anti-IgG serum and this had been obtained from a patient being treated with α-methyldopa. Freedman36 reported a similar incidence – 7.8% positive tests with anti-C′ sera. Lau et al.37 used anti-IgG sera only. The tests were seldom positive (0.9% positive out of 4664 tests). The probable explanation for the relatively high incidence of positive tests with anti-C′ sera is that the reaction is between anti-C′ antibodies and immune complexes adsorbed to the red cells.
False-Negative Antiglobulin Test Results
There are several causes of false-negative test results:
These phenomena are largely negated by the use of column agglutination technology.
DAT-Negative Autoimmune Haemolytic Anaemia
Most hospital blood banks use polyspecific ‘broad-spectrum’ AHG reagents for screening for diagnosis of AIHA. These reagents contain antibody to human IgG and the C3d component of human complement and have little activity against IgA and IgM proteins. The incidence of IgA-only warm AIHA has been reported as 0.2% to 2.7%,38 and the diagnosis may be missed if such polyspecific AHG is used for the DAT screen. In approximately 2–6% of patients who present with the clinical and haematological features of AIHA, the DAT is negative on repeated testing.20,39,40
Low-affinity IgG autoantibodies dissociate from the red cells during the washing phase if a tube technique is used, resulting in a negative DAT. Alternatively, there may be few IgG molecules coating the red cells and this number may fall below the threshold of detection, which is 300–4000 molecules per red blood cell if a tube technique is used. In such cases, a positive DAT may be demonstrated by a more sensitive technique, such as a column agglutination method, an enzyme-linked immunoabsorbant assay or flow cytometry.41–43
If polyspecific AHG is used and the DAT remains negative with clinical evidence of haemolysis, a more sensitive technique should be used for further investigation.44
Manual Direct Polybrene Test
The following method45 is modified from that of Lalezari and Jiang.46 Polybrene is a polyvalent cationic molecule, hexadimethrine bromide, that can overcome the electrostatic repulsive forces between adjacent red cells, bringing the cells closer together. When low levels of IgG are present on the red cell surface, antibody linkage of adjacent red cells is enhanced. The Polybrene is then neutralized using a negatively charged molecule such as trisodium citrate. Sensitized red cells remain agglutinated after neutralization of the Polybrene. Unsensitized red cells will disaggregate after neutralization.
Reagents
Polybrene stock. 10% Polybrene in 9 g/l NaCl, pH 6.9 (saline).
Working Polybrene solution. Dilute the stock Polybrene solution 1 in 250 in saline.
Resuspending solution. 60 ml of 0.2 mol/l trisodium citrate added to 40 ml of 50 g/l dextrose.
Washing solution. 50 ml of 0.2 mol/l trisodium citrate in 950 ml of saline.
Method
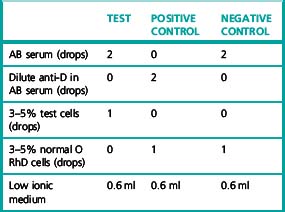
Ensure that all reagents are at room temperature.
Negative control
Normal group AB serum that fails to agglutinate papainized group O, D-positive red cells.
Determination of the Blood Group of a Patient with AIHA
RhD Grouping
When the DAT is positive, monoclonal anti-D reagents should be used; if cold agglutinins are present, perform the test at 37°C. Appropriate controls should be included (see p. 524).
Identification by Adsorption Techniques of Coexisting Alloantibodies in the Presence of Warm Autoantibodies
Use of ZZAP Reagent in Autoadsorption Techniques
‘ZZAP’ reagent is a mixture of dithiothreitol and papain.47 It dissociates an autoantibody already coating the patient’s red cells and enzyme treats the cells, thus increasing the amount of autoantibody that can subsequently be adsorbed onto the patient’s cells in vitro.
Reagents
Dithiothreitol (DTT). 0.2 mol/l.
Phosphate-buffered saline (PBS). pH 6.8–7.2.
Check the pH and adjust to pH 6.0–6.5 using one drop at a time of 0.2 mol/l HCl or 0.2 mol/l NaOH.
Method
Alloadsorption Using Papainized R1R1, R2R2 and rr Cells
Method for Testing Alloadsorbed Sera
Example of alloantibody detection using the alloadsorption technique in a recently transfused patient with AIHA
The patient’s serum when first tested against a panel of group O phenotyped red cells revealed only panreactive antibodies. In contrast, three absorbed sera, A, B and C, obtained by adsorbing the patient’s serum with three selected phenotyped samples of group O cells, were shown to contain anti-E and anti-Jka when tested against a panel of phenotyped group O cells using the IAT. The results of testing the adsorbed sera, A, B and C, are shown in Table 13.7. The patient’s red cell phenotype was R1r Jk (a− b−).
Explanation of the results of testing alloadsorbed sera, A, B and C
Additional notes on adsorption techniques
Notes
Heat Elution
Mix equal volumes of washed packed cells and saline or 6% bovine serum albumin (BSA). Incubate at 56°C for 5 min. Agitate periodically. Centrifuge to pack the red cells. Remove the supernatant (the eluate), which may be haemoglobin stained. Test the eluate48 by appropriate techniques in parallel with the last wash from the red cells.
Freeze and Thaw Elution (Lui)
Mix 0.5 ml of washed packed red cells with 3 drops of PBS or AB serum. Stopper the tube and rotate to coat the glass surface with red cells. Place at –20°C for 10 min. Thaw the red cells rapidly in a 37°C waterbath. Remove the stopper and centrifuge to sediment red cell stroma. Remove the supernatant and test in parallel with the last wash from the red cells by appropriate techniques.49
Determination of the Specificity of Warm Autoantibodies in Eluates and Sera
When tested against a phenotyped panel, about two-thirds of autoantibodies appear to have Rh specificity and in about half of these cases specificity against a particular antigen can be demonstrated.5,15,18 Within the Rh system, anti-e-like is the most common specificity. D − − and Rhnull cells are an advantage.
Titration of Warm Antibodies in Eluates or Sera
The methods used are those described in Chapter 21. The exact technique chosen, and the red cells used, should be those that have given the clearest results in the screening tests. Titration of the eluate can be useful in the presence of a panreacting autoantibody to exclude an underlying alloantibody.
Determination of the Specificity of Cold Autoantibodies
High-titre cold autoantibodies have a well-defined blood-group specificity, which is very often within the I/i system.18,50,51 Because the I antigen is poorly developed in cord-blood red cells, whereas the i antigen is well developed, group O cord blood red cells should be included in the panel used to test for I/i specificity. Adult cells almost always have the I antigen well expressed, but the strength of the antigen varies and it is of considerable advantage to have available adult cells known to possess strong I antigen. (The rare adult i cells, if available, can also be used.)
Titration of Cold Antibodies
If the screening test is positive for cold auto-agglutinins, titrate as follows.
Normal range
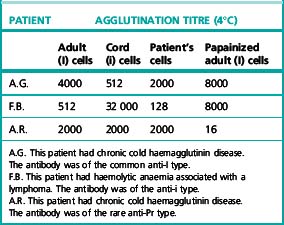
If a cold agglutinin is present at a raised titre, the presence of a cold alloantibody has to be excluded. In this case, the patient’s own red cells will be found to react much less strongly than do normal adult I red cells. It should be noted that in CHAD the patient’s cells commonly react less strongly than do normal adult I cells (Table 13.8).
Cold Agglutinin Titration Patterns
The presence of high-titre cold agglutinins in a patient’s serum will be indicated by the screening procedure described earlier. To demonstrate that the agglutinins are autoantibodies, it is necessary to show that the patient’s own cells are also agglutinated. The titre using the patient’s cells is usually less than that of control normal adult red cells (one-half or one-quarter) (Table 13.8).
In CHAD, whether ‘idiopathic’ or secondary to mycoplasma pneumonia or lymphoma, the autoantibodies usually have anti-I specificity (Patient A.G. in Table 13.8).
In rare cases of haemolytic anaemia associated with infectious mononucleosis, an autoantibody of anti-i specificity has been demonstrated (Patient F.B. in Table 13.8) and this specificity, too, has been found in certain patients with lymphoma. Rarely, in CHAD, the antibody has been shown to have anti-Pr or anti-M specificity: if enzyme-treated red cells are used, then in either type of case the antigen is destroyed by enzyme treatment (Patient A.R. in Table 13.8).
Direct Donath–Landsteiner Test
A false-negative direct Donath–Landsteiner test result is not uncommon for several reasons:
This can be overcome by performing an indirect Donath–Landsteiner test.
Indirect Donath–Landsteiner Test
A false-negative indirect Donath–Landsteiner test can occur. This is a result of the presence of globoside in the serum added as a source of complement. Globoside is the most abundant red cell membrane glycolipid and is present in the serum of all P+ individuals. Addition of ABO-compatible fresh serum as a source of complement could result in cross-reacting with anti-P and this can lead to a false-negative indirect Donath–Landsteiner test. Therefore the indirect Donath–Landsteiner52 test can be modified into two stages.
Two-Stage Indirect Donath–Landsteiner Test
ABO-compatible fresh serum is only added to the red cell–serum suspension after the initial 1-h incubation at 0°C. This prevents antibody inhibition during the cold phase and allows maximum sensitization of the red cells.9
Another possible cause of a false-negative indirect Donath–Landsteiner test result is a low antibody level. Papain treatment of the red cells will expose P antigen and can enhance the sensitization.52
Method
Add a one-tenth volume of EDTA, buffered to pH 7.0 (see p. 620) to the patient’s serum. Prepare doubling dilutions in saline from 1 in 1 to 1 in 28.
After 1 h, wash the red cells four times in a large volume of cold (4°C) saline. Then carry out an antiglobulin test using an anti-IgG reagent, as described on p. 500, but keeping the red cell-antiglobulin serum suspension at 4°C.
Specificity of the Donath–Landsteiner antibody
The D–L antibody appears to have a well-defined specificity within the GLOB blood-group system: namely, anti-P. However, in practice, almost all samples of red cells are acted upon because the cells that will not react (Pk and pp) are extremely rare.53 Cord blood cells are lysed to about the same extent as are adult P1 and P2 cells.
Treatment of Serum with 2-Mercaptoethanol or Dithiothreitol
Weak solutions of 2-mercaptoethanol (2-ME) or dithiothreitol (DTT) destroy the inter-chain sulphydryl bonds of gamma globulins. IgM antibodies treated in this way lose their ability to agglutinate red cells while IgG antibodies do not.18 IgA antibodies may or may not be inhibited depending upon whether or not they are made up of polymers of IgA. Since almost all autoantibodies are either IgM or IgG, treatment of serum or an eluate with 2-ME or DTT gives a reliable indication of the Ig class of autoantibody under investigation.18,54
Drug-Induced Haemolytic Anaemias of Immunological Origin
As already mentioned, acquired haemolytic anaemias may develop as the result of immunological reactions consequent on the administration of certain drugs.15,55–57 Clinically, they often closely mimic AIHA of ‘idiopathic’ origin and for this reason a careful enquiry into the taking of drugs is a necessary part of the interrogation of any patient suspected of having an acquired haemolytic anaemia.
Two immunological mechanisms leading to a drug-induced haemolytic anaemia are recognized. These mechanisms can be referred to as ‘drug-dependent immune’ and ‘drug-induced autoimmune’. Both types of antibody may be present in some patients.58,59 In a unifying concept, the target orientation of these antibodies covers a spectrum in which the primary immune response is initiated by an interaction between the drug or its metabolites and a component of the blood cell membrane to create a neoantigen.60 Drug-dependent antibodies bind to both the drug and the cell membrane but not to either separately. If the drug is withdrawn, the immune reaction subsides. It has been postulated that in the case of the autoantibodies, the greater part of the neoantigen is sufficiently similar to the normal cell membrane to allow binding without the drug being present. Similar mechanisms have been described for drug-induced immune thrombocytopenia and neutropenia of immunological origin (see p. 508).
Drugs that have been shown to cause haemolysis by the previously explained mechanism include quinine, quinidine and rifampicin, chlorpropamide, hydrochlorothiazide, nomifensine, phenacetin, salicylazosulphapyridine, the sodium salt of p-aminosalicylic acid and stibophen. Petz and Branch listed 25 drugs reported to have brought about haemolysis by this mechanism.56
Drug-Induced Autoimmune Haemolytic Anaemias
Other drugs that have been reported to act in a similar fashion to α-methyldopa include L-dopa, chlordiazepoxide, mefenamic acid, flufenamic acid and indometacin.5
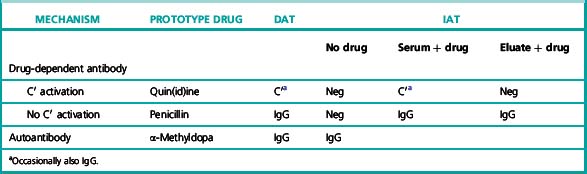
Typical serological features of the different types of drug-induced haemolytic anaemia of immunological origin are summarized in Table 13.9.
Table 13.9 Serological features of the different types of drug-induced haemolytic anaemia of immunological origin
Detection of Antipenicillin Antibodies
The characteristic features of penicillin-induced haemolytic anaemia are as follows:
Reagents
Barbitone buffer. 0.14 mol/l, pH 9.5 (see p. 621).
Penicillin solution. 0.4 g of penicillin G dissolved in 6 ml of barbitone buffer.
Method
Detection of Antibodies against Drugs Other than Penicillin
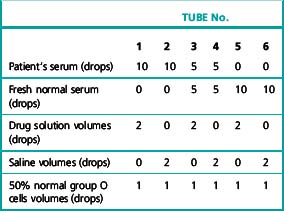
Set up six tubes containing the patient’s serum and the drug solution in the proportions shown in Table 13.10 and add 1 drop of a 50% saline suspension of group O cells to each tube. Incubate at 37°C for 1 h, then examine for agglutination and lysis. Wash the red cells four times in saline, and carry out an IAT using anti-IgG and anti-C′ separately.
Oxidant-induced haemolytic anaemia
Oxidant-induced haemolytic anaemia should be suspected when the blood film of a patient exposed to an oxidant drug or chemical shows irregularly contracted cells. A Heinz-body test (see p. 336) is confirmatory. The oxidant may also cause methaemoglobinaemia or sulphaemoglobinaemia, both of which can be confirmed by spectroscopy (see p. 240) or co-oximetry. The differential diagnosis of haemolysis induced by an exogenous oxidant includes other causes of haemolysis with irregularly contracted cells (e.g. Zieve’s syndrome), G6PD deficiency and the presence of an unstable haemoglobin. In Zieve’s syndrome (haemolysis associated with alcohol excess, fatty liver and hyper-lipidaemia), the plasma may be visibly lipaemic; if this syndrome is suspected, further investigations should include liver function test and serum lipid measurements.
Microangiopathic and mechanical haemolytic anaemias
Microangiopathic or mechanical haemolytic anaemia should be suspected when a blood film shows schistocytes. Examination of the blood film is, in fact, the most important laboratory procedure in making this diagnosis, although some automated blood cell counters will also detect the presence of red cell fragments. Because haemolysis is intravascular, useful confirmatory tests include serum haptoglobin estimation (see p. 233) and, when the condition is chronic, a Perls’ stain of urinary sediment to detect the presence of haemosiderin (see p. 236). Because a microangiopathic haemolytic anaemia is often part of a more generalized syndrome resulting from microvascular damage or fibrin deposition, other tests are also indicated in unexplained cases. They include tests of renal function, a platelet count and a coagulation screen including tests for D-dimer or fibrin degradation products (see p. 440). Tests for verotoxin-secreting E. coli are indicated in cases of microangiopathic haemolytic anaemia with renal failure. If available, quantification of von Willebrand factor-cleaving protease (ADAMTS13) is indicated in suspected thrombotic thrombocytopenic purpura.
Paroxysmal nocturnal haemoglobinuria
Paroxysmal nocturnal haemoglobinuria (PNH) is an acquired clonal disorder of haemopoiesis, in which the patient’s red cells are abnormally sensitive to lysis by normal constituents of plasma. In its classical form, it is characterized by haemoglobinuria during sleep (nocturnal haemoglobinuria), jaundice and haemosiderinuria. Not uncommonly, however, PNH presents as an obscure anaemia without obvious evidence of intravascular haemolysis or it develops in a patient suffering from aplastic anaemia or more rarely from primary myelofibrosis or chronic myeloid leukaemia.61,62
PNH red cells are unusually susceptible to lysis by complement.63,64 This can be demonstrated in vitro by a variety of tests (e.g. the acidified-serum [Ham],65 sucrose,66 thrombin,67 cold-antibody lysis,68 inulin69 and cobra-venom70 tests). In the acidified serum, inulin and cobra-venom tests, complement is activated via the alternative pathway, whereas in the cold-antibody test and probably in the thrombin test, complement is activated by the classical sequence initiated through antigen–antibody interaction. In the sucrose lysis test, a low ionic strength is thought to lead to the binding of IgG molecules non-specifically to the cell membrane and to the subsequent activation of complement via the classical sequence. In addition, the alternative pathway appears to be activated.71 In each test, PNH cells undergo lysis because of their greatly increased sensitivity to lysis by complement.
Minor degrees of lysis may be observed in the cold-antibody lysis and sucrose tests with the red cells from a variety of dyserythropoietic anaemias (e.g. aplastic anaemia, megaloblastic anaemia and primary myelofibrosis).72,73 Weak positive results in these tests thus have to be interpreted with care. PNH red cells, however, almost always undergo considerable lysis in these tests.
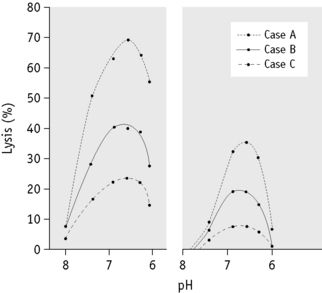
A characteristic feature of a positive test for PNH is that not all the patient’s cells undergo lysis, even if the conditions of the test are made optimal for lysis (Fig. 13.1). This is because only a proportion of any PNH patient’s red cell population is hypersensitive to lysis by complement. This population varies from patient to patient and there is a direct relationship between the proportion of red cells that can be lysed (in any of the diagnostic tests) and the severity of in vivo haemolysis.
The phenomenon of some red cells being sensitive to complement lysis and some being insensitive was studied quantitatively by Rosse and Dacie, who obtained two-component complement sensitivity curves in a series of patients with PNH.64 Later, Rosse reported that in some cases three populations of red cells could be demonstrated.74,75
In vivo the proportion of type III cells parallels the severity of the patient’s haemolysis.
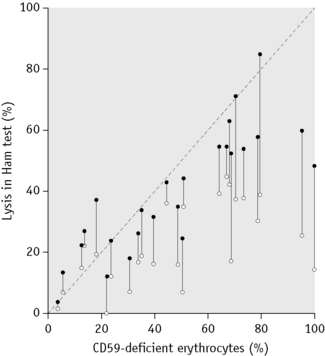
PNH is an acquired clonal disorder76 resulting from a somatic mutation occurring in a haemopoietic stem cell. It has been demonstrated that a proportion of granulocytes, platelets and lymphocytes are also part of the PNH clone.77,78 The characteristic feature of cells belonging to the PNH clone is that they are deficient in several cell-membrane–bound proteins including red cell acetylcholineesterase,79–81 neutrophil alkaline phosphatase,82–84 CD55 (decay accelerating factor or DAF),85,86 homologous restriction factor (HRF),63,87 and CD59 (membrane inhibitor of reactive lysis or MIRL),88–90 among others. CD55, CD59 and HRF all have roles in the protection of the cell against complement-mediated attack. CD59 inhibits the formation of the terminal complex of complement and it has been established that the deficiency of CD59 is largely responsible for the complement sensitivity of PNH red cells. PNH type III red cells have a complete deficiency of CD59, whereas PNH type II red cells have only a partial deficiency and it is this difference that accounts for their variable sensitivities to complement.91,92 The analysis of these deficient proteins on PNH cells by flow cytometry, particularly of the red cells and neutrophils, has become a useful research and diagnostic tool but is only applicable in centres with a significant number of patients requiring investigation for PNH. By comparing the proportion of cells with deficient CD59 to the percentage lysis in the Ham test, it has been possible to assess the sensitivity of the Ham test. The standard Ham test is reasonably good at estimating the proportion of PNH red cells as long as they are PNH type III cells and comprise <20% of the total. In cases in which the PNH cells are type II and >20% are present, the standard Ham test significantly underestimates the proportion of PNH red cells. The standard Ham test can be negative when there are <5% PNH type III cells or <20% PNH type II cells. When the Ham test is supplemented with magnesium, to optimize the activation of complement, the percentage lysis gives a more accurate estimation of the proportion of PNH cells (Fig. 13.2).93
Acidified-Serum Lysis Test (Ham test)
Principle
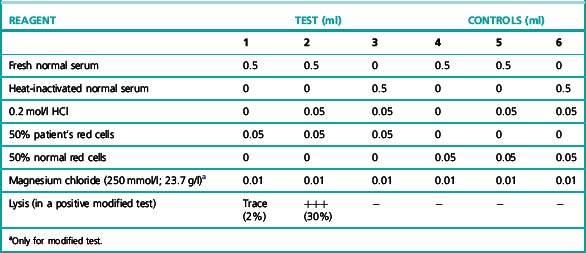
The patient’s red cells are exposed at 37°C to the action of normal or the patient’s own serum suitably acidified to the optimum pH for lysis (pH 6.5–7.0) (Table 13.11).
The patient’s red cells can be obtained from defibrinated, heparinized, oxalated, citrated or EDTA blood and the test can be satisfactorily carried out even on cells that have been stored at 4°C for up to 2–3 weeks in ACD or Alsever’s solution, if kept sterile. The patient’s serum is best obtained by defibrination because in PNH if it is obtained from blood allowed to clot in the ordinary way at 37°C or at room temperature, it will almost certainly be markedly lysed. Normal serum should similarly be obtained by defibrination, although serum derived from blood allowed to clot spontaneously at room temperature or at 37°C can be used. Normal serum known to be strongly lytic to PNH red cells is to be preferred to patient’s serum, the lytic potentiality of which is unknown. However, if the test is positive using normal serum, it is important, particularly if the patient appears not to be suffering from overt intravascular haemolysis, to obtain a positive result using the patient’s serum to exclude hereditary erythroid multinuclearity associated with a positive acidified-serum test (HEMPAS) (see p. 294). The variability between the sera of individuals in their capacity to lyse PNH red cells is shown in Figure 13.1. The activity of a single individual’s serum also varies from time to time,94 and it is always important to include in any test, as a positive control, a sample of known PNH cells or artificially created ‘PNH-like’ cells (see p. 296).
Acidified-Serum Test Lysis with Additional Magnesium (Modified Ham Test)
Method
The method is identical to that for the standard Ham test (see above) with the addition of 10 μl of 250 mmol magnesium chloride (final concentration = 4 mmol) to each tube prior to the incubation (Table 13.11).
Significance of the Acidified-Serum Lysis Test
When the acidified-serum test is positive, a direct antiglobulin test (see p. 500) should also be carried out. If this is positive, it could be the result of a lytic antibody that has given a false-positive acidified-serum test. This can be confirmed by appropriate serological studies. In such complex cases flow cytometry after reaction of the red cells with anti-CD59 is recommended because it is a more definitive test for PNH (see below).
The only disorder other than PNH that may appear to give a clear-cut positive test result is a rare congenital dyserythropoietic anaemia, congenital dyserythropoietic anaemia type II or HEMPAS.95,96 In contrast to PNH, however, HEMPAS red cells undergo lysis in only a proportion (about 30%) of normal sera; moreover, they do not undergo lysis in the patient’s own acidified serum and the sucrose lysis test is negative. In HEMPAS, the expression of glycosylphosphatidylinositol (GPI)-linked proteins, such as CD55 and CD59, is normal. Lysis in HEMPAS appears to be a result of the presence on the red cells of an unusual antigen, which reacts with a complement-fixing IgM antibody (‘anti-HEMPAS’) present in many, but not in all, normal sera.96
PNH red cells are not unduly sensitive to lysis by a lowered pH per se. The addition of the acid adjusts the pH of the serum–cell mixture to the optimum for the activity of the lytic system. As is shown in Figure 13.1, it is possible to construct pH–lysis curves, if different concentrations of acid are used. The optimum pH for lysis is between pH 6.5 and 7.0 (measurements made after the addition of the red cells to the serum).
Sucrose Lysis Test
An iso-osmotic solution of sucrose (92.4 g/l) is required.97 This can be stored at 4°C for up to 3 weeks.
Interpretation
The sucrose lysis test is based on the fact that red cells absorb complement components from serum at low ionic concentrations.94,98 PNH cells, because of their great sensitivity, undergo lysis but normal red cells do not. The red cells from some patients with leukaemia72 or primary myelofibrosis may undergo a small amount of lysis, almost always <10%; in such cases, the acidified-serum test is usually negative and PNH should not be diagnosed. In PNH, lysis is usually between 10% and 80%, but exceptionally may be as little as 5%. Sucrose lysis and acidified-serum lysis of PNH red cells are fairly closely correlated. The sucrose lysis test is usually negative in HEMPAS.
Flow Cytometry Analysis of the GPI-Linked Proteins on Red Cells
Principle
The patient’s red cells can be obtained in any of the anticoagulants described for the Ham test. The cells, if taken into ACD, can be stored for 2–3 weeks prior to analysis. Fluorescein-conjugated anti-CD59 (MEM 43, Cymbus Bioscience Ltd, Southampton, UK) gives excellent results when used for red cell analysis. It is important to use the conjugated antibody because staining with unconjugated anti-CD59 followed by a fluorescein-conjugated second layer antibody may result in artefact, probably owing to red cell agglutination. There is no suitable anti-CD55 antibody commercially available at present. Anti-CD58 (BRIC5, Bioproducts Laboratories, UK, used at 20 μg/ml) gives reproducible results for red cell analysis, but the level of CD58 expression on PNH type-II cells is higher than that of many other GPI-linked proteins and thus studying CD58 expression is useful but not ideal.99
Flow Cytometry Analysis of the GPI-Anchor or GPI-Linked Proteins on Neutrophils
Principle
A proportion of the patient’s neutrophils have been demonstrated to be part of the PNH clone in all patients with PNH. GPI-linked proteins that are suitable for analysis include CD16, CD24, CD55, CD59 and CD67.100,101 There are available numerous fluorescein-conjugated antibodies to CD16 that are suitable for use in this analysis – for example, fluorescein-conjugated anti-Leu-11a (Beckman Coulter). Similarly, fluorescein-conjugated anti-CD59 (Cymbus Biosciences) and anti-CD55 are of value. FLAER (fluorescein-labelled pro-aerolysin) analysis can also be used, pro-aerolysin binding selectively and with high affinity to the GPI anchor.102
Significance of Flow Cytometric Analysis
The presence of a population of cells with a deficiency of more than one GPI-linked protein is diagnostic of PNH (Fig. 13.3). It is important to analyse more than one protein, because there are extremely rare cases in which an inherited deficiency of one protein has been described (i.e. the Inab phenotype,103–105 a deficiency of CD55 owing to a defect of the structural gene encoding this protein, and inherited deficiency of CD59106 due to a defect in the gene encoding CD59). Analysis of the expression of CD59 on erythrocytes allows the identification of PNH type II as well as PNH type III red cells. This is important because, although patients with only PNH type II red cells do not usually suffer from significant haemolysis, they may suffer some of the complications of PNH, such as thrombosis. The analysis of neutrophils for GPI-linked proteins is more difficult than red cell analysis. It is, however, probably more sensitive because the proportion of abnormal neutrophils is usually higher than the proportion of PNH red cells because of the reduced survival of PNH red cells compared to normal cells and because of the effect of transfusions. Thus, flow cytometry applied to neutrophils is a more sensitive method for the diagnosis of PNH than methods relying on the complement sensitivity of PNH red cells.
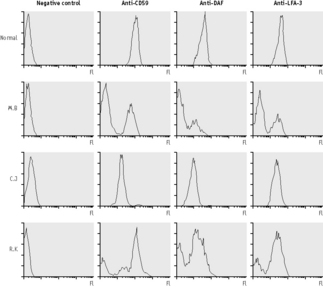
PNH-Like Red Cells
By treating normal red cells with certain chemicals, it is possible to increase their complement sensitivity so that they take on many of the characteristics of PNH cells.107 The chemicals include sulphydryl compounds such as L-cysteine, reduced glutathione, 2-aminoethyl-iso-thiouronium bromide (AET) and 2-mercaptobenzoic acid.108 AET- and 2-mercaptobenzoic acid-treated cells can be used conveniently as a positive control for in vitro lysis tests for PNH.109
Preparation of AET Cells
Prepare an 8 g/l solution of AET and adjust its pH to 8.0 with 5 mol/l NaOH. Collect normal blood into ACD and wash it ×2 in 9 g/l NaCl. Add 1 volume of the packed cells to 4 volumes of the AET solution in a 75 × 12 mm tube, which is then stoppered. Mix the contents gently and place the tube at 37°C for 10–20 min; the optimal time of incubation varies between red cell samples. Then wash the cells repeatedly with large volumes of saline until the supernatant is colourless. The red cells are now ready to use.110
1 Chen F.E., Owen I., Savage D., et al. Late onset haemolysis and red cell autoimmunization after allogeneic bone marrow transplant. Bone Marrow Transplant. 1997;19:491-495.
2 Sanz J., Arriaga F., Montesinos P., et al. Autoimmune hemolytic anemia following allogeneic hematopoietic stem cell transplantation in adult patients. Bone Marrow Transplant. 2007;39:555-561.
3 O’Brien T.A., Eastlund T., Peters C., et al. Autoimmune haemolytic anaemia complicating haematopoietic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated donor transplants for non-malignant diseases. Br J Haematol. 2004;127:67-75.
4 Arndt P.A., Leger R.M., Garratty G. Serology of antibodies to second and third generation cephalosporins associated with immune hemolytic anaemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
5 Dacie J.V., Worlledge S.M. Auto-immune hemolytic anaemias. Prog Hematol. 1969;6:82-120.
6 Sokol R.J., Hewitt S., Stamps B.K. Autoimmune haemolysis: An 18-year study of 865 cases referred to a regional transfusion centre. Br Med J. 1981;282:2023-2027.
7 Sokol R.J., Hewitt S., Stamos B.K. Autoimmune haemolysis: mixed warm and cold antibody type. Acta Haematol. 1983;69:266-274.
8 Dacie J.V. Auto-immune hemolytic anemias. Arch Intern Med. 1975;135:1293-1300.
9 Petz L.D., Garratty G. Acquired Immune Haemolytic Anaemias, 2nd ed. New York: Churchill Livingstone; 2004.
10 Morselli M., Luppi M., Potenza L., et al. Mixed warm and cold autoimmune hemolytic anemia: Complete recovery after 2 courses of rituximab treatment. Blood. 2002;99:3478-3479.
11 Shulman I.A., Branch D.R., Nelson J.M., et al. Autoimmune hemolytic anemia with both cold and warm autoantibodies. J Am Med Assoc. 1985;253:1746-1748.
12 Win N., Tiwari D., Keevil V.L., et al. Mixed-type autoimmune haemolytic anaemia: unusual cases and a case associated with splenic T cell angioimmuno-blastic A non-Hodgkins’ lymphoma. Hematology. 2007;12:159-162.
13 Dacie J. The Haemolytic Anaemias, Vol. 3. The Auto-Immune Haemolytic Anaemias, 3rd ed. Churchill Livingstone, Edinburgh, 1992.
14 Pirofsky B. Autoimmunization and the Autoimmune Hemolytic Anemias. Baltimore: Williams & Wilkins; 1969.
15 Petz L.D., Garratty G. Acquired Immune Hemolytic Anemias. New York: Churchill Livingstone; 1980.
16 Sokol R.J., Hewitt S. Autoimmune hemolysis: a critical review. CRC Critical Reviews in Oncology/Hematology. 1985;4:125-154.
17 Sokol R.J., Hewitt S., Booker D.J., et al. Enzyme linked direct antiglobulin tests in patients with autoimmune haemolysis. J Clin Pathol. 1985;38:912-914.
18 Issitt P.D. Serological diagnosis and characterization of the causative autoantibodies. Methods in Hematology. 1985;12:1-45.
19 Engelfriet C.P., von dem Borne A.E., Beckers D., et al. Auto-immune haemolytic anaemia: serological and immunochemical characteristics of the auto-antibodies: mechanisms of cell destruction. Ser Haematol. 1974;VII:328-347.
20 Worlledge S.M. The interpretation of a positive direct antiglobulin test. Br J Haematol. 1978;39:157-162.
21 Robertson V.M., Dickson L.G., Romond E.H., et al. Positive antiglobulin tests due to intravenous immunoglobulin in patients who received bone marrow transplant. Transfusion. 1987;27:28-31.
22 Ramsey G. Red cell antibodies arising from solid organ transplants. Transfusion. 1991;31:76-86.
23 Petz L.D. Immunohematologic problems associated with bone marrow transplantation. Transfus Med Rev. 1987;1:85-100.
24 Shulman J.A., Petz L.D. Red cell compatibility testing: clinical significance and laboratory methods. In: Petz L.D., Swisher S.N., Kleinman S., et al, editors. Clinical Practice of Transfusion Medicine. 3rd ed. New York: Churchill Livingstone; 1996:199-244.
25 Szymanski I0, Odgren P.R., Fortier N.L., et al. Red blood cell associated IgG in normal and pathologic states. Blood. 1980;55:48-54.
26 Clark J.A., Tranley P.C., Wallas C.H. Evaluation of patients with positive direct antiglobulin tests and non-reactive eluates discovered during pretransfusion testing. Immunohaematol. 1992;8:9-12.
27 Heddle N.M., Kelton J.G., Turchyn K.L., et al. Hypergammaglobulinemia can be associated with a positive direct antiglobulin test, a nonreactive eluate and no evidence of hemolysis. Transfusion. 1988;28:29-33.
28 Huh Y.O., Liu F.J., Rogge K., et al. Positive direct antiglobulin test and high serum immunoglobulin G levels. Am J Clin Pathol. 1988;90:197-200.
29 Vianna J.L., Khamashta M.A., Ordi-Ros J., et al. Comparison of the primary and secondary antiphospholipid syndrome: A European Multicenter Study of 114 patients. Am J Med. 1994;96:3-9.
30 Win N., Islam S.I., Peterkin M.A., et al. Positive direct antiglobulin test due to antiphospholipid antibodies in normal healthy blood donors. Vox Sang. 1997;72:182-184.
31 Petz L.D., Yam P., Wilkinson L., et al. Increased IgG molecules bound to the surface of red blood cells of patients with sickle cell anemia. Blood. 1984;64:301-304.
32 Toy P.T., Chin C.A., Reid M.E., et al. Factors associated with positive direct antiglobulin tests in pretransfusion patients: a case control study. Vox Sang. 1985;49:215-220.
33 Stratton F., Renton P.H. Effect of crystalloid solutions prepared in glass bottles on human red cells. Nature (London). 1955;175:727.
34 Gorst D.W., Rawlinson V.I., Merry A.H., et al. Positive direct antiglobulin test in normal individuals. Vox Sang. 1980;38:99-105.
35 Bareford D., Longster G., Gilks L., et al. Follow-up of normal individuals with a positive antiglobulin test. Scand J Haematol. 1985;35:348-353.
36 Freedman J. False-positive antiglobulin tests in healthy subjects. J Clin Pathol. 1979;32:1014-1018.
37 Lau P., Haesler W.E., Wurzel H.A. Positive direct antiglobulin reaction in a patient population. Am J Clin Pathol. 1976;65:368-375.
38 Sokol R.J., Booker D.J., Stamps R., et al. IgA red cell autoantibodies and auto-immune hemolysis. Transfusion. 1997;37:175-181.
39 Chaplin H.Jr. Clinical usefulness of specific antiglobulin reagents in autoimmune hemolytic anemia. Prog Hematol. 1973;8:25-49.
40 Worlledge S.M., Blajchman M.A. The autoimmune haemolytic anaemias. Br J Haematol. 1972;23(suppl):61-69.
41 Lai M., Rumi C., D’onofrio G., et al. Clinically significant auto-immune hemolytic anemia with a negative direct antiglobulin test by routine tube test and positive by column agglutination method. Immunohematol. 2002;18:112-116.
42 Sokol R.J., Hewitt S., Booker D.J., et al. Small quantities of erythrocyte bound immunoglobulins and auto-immune haemolysis. J Clin Pathol. 1987;40:254-257.
43 Garratty G., Arndt P.A. Application of flow cytofluorometry to red blood cell immunology. Cytometry. 1999;38:259-267.
44 Sokol R.J., Booker D.J., Stamps R., et al. IgA red cell autoantibodies and autoimmune haemolysis. Transfusion. 1997;37:175-181.
45 Owen I., Hows J. Evaluation of the manual Polybrene technique in the investigation of autoimmune hemolytic anemia. Transfusion. 1990;30:814-818.
46 Lalezari P., Jiang A.C. The manual Polybrene test: a simple and rapid procedure for detection of red cell antibodies. Transfusion. 1980;20:206-211.
47 Branch D.R., Petz L.D. Detecting alloantibodies in patients with autoantibodies. Transfusion. 1999;39:6-10.
48 Landsteiner K., Miller C.P.Jr. Serological studies on the blood of primates. II. The blood groups in anthropoid apes. J Exp Med. 1925;42:853-862.
49 Eicher C.A., Wallace M.E., Frank S., et al. The Lui elution: a simple method of antibody elution. Transfusion. 1978;18:647-652.
50 Roelcke D. Cold agglutination. Transfus Med Rev. 1989;3:140-166.
51 Wiener A.S., Unger L.J., Cohen L., et al. Type-specific cold auto-antibodies as a cause of acquired hemolytic anemia and hemolytic transfusion reactions: biologic test with bovine red cells. Ann Intern Med. 1956;44:221-240.
52 Wolach B., Heddle N., Barr R.D., et al. Transient Donath–Landsteiner haemolytic anaemia. Br J Haematol. 1981;48:425-434.
53 Worlledge S.M., Rousso C. Studies on the serology of paroxysmal cold hemoglobinuria (PCH) with special reference to a relationship with the P blood group system. Vox Sang. 1965;10:293-298.
54 Freedman J., Masters C.A., Newlands M., et al. Optimal conditions for the use of sulphydryl compounds in dissociating red cell antibodies. Vox Sang. 1976;30:231-239.
55 Habibi B. Drug-induced immune haemolytic anaemias. Baillières Clinical Immunology and Allergy. 1987;1:343-356.
56 Petz L.D., Branch D.R. Drug-induced immune haemolytic anemias. Methods in Haematology. 1985;12:47-94.
57 Habibi B. Drug induced red blood cell autoantibodies co-developed with drug specific antibodies causing haemolytic anaemias. Br J Haematol. 1985;61:139-143.
58 Salama A., Gottsche B., Mueller-Eckhardt C. Autoantibodies and drug- or metabolite-dependent antibodies in patients with diclofenac-induced immune haemolysis. Br J Haematol. 1991;77:546-549.
59 Salama C., Mueller-Eckhardt C. Cianidanol and its metabolites bind tightly to red cells and are responsible for the production of auto- and/or drug-dependent antibodies against these cells. Br J Haematol. 1987;66:263-266.
60 Salama A., Mueller-Eckhardt C. Immune-mediated blood dyscrasias related to drugs. Semin Hematol. 1992;29:54-63.
61 Dacie J.V., Lewis S.M. Paroxysmal nocturnal haemoglobinuria: clinical manifestations, haematology and nature of the disease. Ser Haematol. 1972;5:3-23.
62 Sirchia G., Lewis S.M. Paroxysmal nocturnal haemoglobinuria. Clin Haematol. 1975;4:199-229.
63 Hansch G.M., Schonermark S., Roeicke D. Paroxysmal nocturnal hemoglobinuria type III: lack of an erythrocyte membrane protein restricting the lysis by C5b–9. J Clin Invest. 1987;80:7-12.
64 Rosse W.F., Dacie J.V. Immune lysis of normal human and paroxysmal nocturnal hemoglobinuria (PNH) red blood cells. 1. The sensitivity of PNH red cells to lysis by complement and specific antibody. J Clin Invest. 1966;45:736-748.
65 Ham T.H., Dingle J.H. Studies on destruction of red blood cells. II. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria: certain immunological aspects of the hemolytic mechanism with special reference to serum complement. J Clin Invest. 1939;18:657-672.
66 Hartmann R.C., Jenkins D.E.Jr, Arnold A.B. Diagnostic specificity of sucrose hemolysis test for paroxysmal nocturnal hemoglobinuria. Blood. 1970;35:462-475.
67 Crosby W.H. Paroxysmal nocturnal hemoglobinuria. A specific test for the disease based on the ability of thrombin to activate the hemolytic factor. Blood. 1950;5:843-846.
68 Dacie J.V., Lewis S.M., Tills D. Comparative sensitivity of the erythrocytes in paroxysmal nocturnal haemoglobinuria to haemolysis by acidified normal serum and by a high-titre cold antibody. Br J Haematol. 1960;6:362-371.
69 Brubaker L.H., Schaberg D.R., Jefferson D.H., et al. A potential rapid screening test for paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1973;288:1059-1060.
70 Kabakci T., Rosse W.F., Logue G.L. The lysis of paroxysmal nocturnal haemoglobinuria red cells by serum and cobra factor. Br J Haematol. 1972;23:693-705.
71 Logue G.L., Rossi W.F., Adams J.P. Mechanisms of immune lysis of red blood cells in vitro. I. Paroxysmal nocturnal hemoglobinuria cells. J Clin Invest. 1973;52:1129-1137.
72 Catovsky D., Lewis S.M., Sherman D. Erythrocyte sensitivity to in-vitro lysis in leukaemia. Br J Haematol. 1971;21:541-550.
73 Lewis S.M., Dacie J.V., Tills D. Comparison of the sensitivity to agglutination and haemolysis by a high-titre cold antibody of the erythrocytes of normal subjects and of patients with a variety of blood diseases including paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1961;7:64-72.
74 Rosse W.F. Variations in the red cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1973;24:327-342.
75 Rosse W.F., Adams J.P., Thorpe A.M. The population of cells in paroxysmal nocturnal haemoglobinuria of intermediate sensitivity to complement lysis: significance and mechanism of increased immune lysis. Br J Haematol. 1974;28:281-290.
76 Oni S.B., Osunkoya B.O., Luzzatto L. Paroxysmal nocturnal hemoglobinuria: evidence for monoclonal origin of abnormal red cells. Blood. 1970;36:145-152.
77 Kinoshita T., Medof M.E., Silber R., et al. Distribution of decay-accelerating factor in the peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1985;162:75-92.
78 Nicholson-Weller A., Spicier D.B., Austen K.F. Deficiency of the complement regulating protein ‘decay accelerating factor’ on membranes of granulocytes, monocytes and platelets in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1985;312:1091-1097.
79 Auditore J.V., Hartmann R.C. Paroxysmal nocturnal hemoglobinuria: II. Erythrocyte acetylcholinesterase defect. Am J Med. 1959;27:401-410.
80 Chow F.-L., Telen M.J., Rosse W.F. The acetylcholinesterase defect in paroxysmal nocturnal hemoglobinuria: evidence that the enzyme is absent from the cell membrane. Blood. 1985;66:940-945.
81 De Sandre G., Ghiotto G. An enzymic disorder in the erythrocytes of paroxysmal nocturnal haemoglobinuria: a deficiency in acetylcholinesterase activity. Br J Haematol. 1960;6:39-42.
82 Beck W.S., Valentine W.N. Biochemical studies on leucocytes. II. Phosphatase activity in chronic lymphatic leucemia, acute leucemia and miscellaneous hematologic conditions. J Lab Clin Med. 1965;38:245-253.
83 Hartmann R.C., Auditore J.V. Paroxysmal nocturnal hemoglobinuria I. Clinical studies. Am J Med. 1959;27:389-400.
84 Lewis S.M., Dacie J.V. Neutrophil (leucocyte) alkaline phosphatase in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1965;11:549-556.
85 Nicholson-Weller A., March J.P., Rosenfield S.I., et al. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc Natl Acad Sci U S A. 1983;80:5066-5070.
86 Pangbum M.K., Schreiber R.D., Müller-Eberhard H.F. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci U S A. 1983;80:5430-5434.
87 Zalman L.S., Wood L.M., Frank M.M., et al. Deficiency of the homologous restriction factor in paroxysmal nocturnal hemoglobinuria. J Exp Med. 1987;165:572-577.
88 Davies A., Simmons D.L., Hale G., et al. CD59 and LY–6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex on homologous cells. J Exp Med. 1989;170:637-654.
89 Holguin M.H., Wilcox L.A., Bernshaw N.J., et al. Relationship between the membrane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84:1387-1394.
90 Holguin M.H., Fredrick N.J., Bernshaw L.A., et al. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest. 1989;84:7-17.
91 Rosse W.F., Hoffman S., Campbell M., et al. The erythrocytes in paroxysmal nocturnal haemoglobinuria of intermediate sensitivity to complement lysis. Br J Haematol. 1991;79:99-107.
92 Shichishima T., Terasawa T., Hashimoto C., et al. Heterogenous expression of decay accelerating factor and CD59/membrane attack complex inhibition factor on paroxysmal nocturnal haemoglobinuria (PNH) erythrocytes. Br J Haematol. 1991;78:545-550.
93 May J.E., Rosse W.F., Frank M.M. Paroxysmal nocturnal hemoglobinuria: alternative-complement-pathway-mediated lysis induced by magnesium. N Engl J Med. 1973;289:705-709.
94 Packman C.H., Rosenfeld S.I., Jenkins D.E.Jr, et al. Complement lysis of human erythrocytes. Differing susceptibility of two types of paroxysmal nocturnal hemoglobinuria cells to C5b-9. J Clin Invest. 1979;64:428-433.
95 Crookston J.H., Crookston M.C., Burnie K.L., et al. Hereditary erythroblastic multinuclearity associated with a positive acidified-serum test: a type of congenital dyserythropoietic anaemia. Br J Haematol. 1969;17:11-26.
96 Verwilghen R.L., Lewis S.M., Dacie J.V. HEMPAS: congenital dyserythropoietic anaemia (type II). Q J Med. 1973;42:257-278.
97 Hartmann R.C., Jenkins D.E.Jr. The ‘sugar water’ test for paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1966;275:155-157.
98 Mollison P.L., Polley M.J. Uptake of γ-globulin and complement by red cells exposed to serum at low ionic strength. Nature (London). 1964;203:535.
99 Hillmen P., Hows J.M., Luzzatto L. Two distinct patterns of glycosylphosphatidylinositol (GPI) linked protein deficiency in the red cells of patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1992;80:399-405.
100 Plesner T., Hansen N.E., Carlsen K. Estimation of PI-bound proteins on blood cells from PNH patients by quantitative flow cytometry. Br J Haematol. 1990;75:585-590.
101 van der Schoot C.E., Huizinga T.W., van’t Veer-Korthof E.T., et al. Deficiency of glycosyl-phosphatidylinositol-linked membrane glycoproteins of leukocytes in paroxysmal nocturnal hemoglobinuria, description of a new diagnostic cytofluorometric assay. Blood. 1990;76:1853-1859.
102 Brodsky R.A. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22:65-74.
103 Merry A.H., Rawlinson V.I., Uchikawa M., et al. Studies on the sensitivity to complement-mediated lysis of erythrocytes (Inab phenotype) with a deficiency of DAF (decay accelerating factor). Br J Haematol. 1989;73:248-253.
104 Merry A.H., Rawlinson V.I., Uchikawa M., et al. Lack of abnormal sensitivity to complement-mediated lysis in erythrocytes deficient only in decay accelerating factor. Biochem Soc Trans. 1989;17:514.
105 Telen M.J., Green A.M. The Inab phenotype: characterization of the membrane protein and complement regulatory defect. Blood. 1989;74:437-441.
106 Yamashina M., Ueda E., Kinoshita T., et al. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1990;323:1184-1189.
107 Sirchia G., Ferrone S. The laboratory substitutes of the red cell of paroxysmal nocturnal haemoglobinuria (PNH): PNH-like red cells. Ser Haematol. 1972;5:137-175.
108 Francis D.A. Production of PNH-like red cells using 2-mercaptobenzoic acid. Med Lab Sci. 1983;40:33-38.
109 Sirchia G., Marubini E., Mercuriali F., et al. Study of two in vitro diagnostic tests for paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1973;24:751-759.
110 Sirchia G., Ferrone S., Mercuriali F. The action of two sulfhydryl compounds on normal human red cells. Relationship to red cells of paroxysmal nocturnal haemoglobinuria. Blood. 1965;25:502-510.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 13 Acquired haemolytic anaemias
Assessing the likelihood of acquired haemolytic anaemia
Haemolytic anaemia may be suspected from either clinical or laboratory abnormalities. Suggestive clinical features include anaemia, jaundice and splenomegaly. Other relevant clinical features that should be sought are a history of autoimmune disease, recent blood transfusion, recent infection, exposure to drugs or toxins, the presence of a cardiac prosthesis and risk of malaria. Previous clinical history and laboratory results will help to establish that the disorder is acquired. The basic laboratory investigations when a haemolytic anaemia is suspected are listed in Chapter 11. In this chapter, tests are described that are more specific for the diagnosis of acquired haemolytic anaemia.
Assessment of the blood film and count in suspected acquired haemolytic anaemia
If haemolytic anaemia is suspected, a full blood count, reticulocyte count and blood film should always be performed. The blood count shows a reduced haemoglobin concentration (Hb) and, usually, an increased mean cell volume (MCV). The increased MCV is attributable to the fact that reticulocytes, which may constitute a significant proportion of total red cells, are larger than mature red cells. The abnormalities that may be detected in the blood film and their possible significance in acquired haemolytic anaemia are shown in Table 13.1. Abnormalities detected in the blood film will direct further investigations. For example, a Heinz body preparation would be relevant if irregularly contracted cells were present, particularly if there appeared to be red cell inclusions. Similarly, a direct antiglobulin test (DAT) would be indicated if the blood film showed spherocytes. Various inherited forms of haemolytic anaemia enter into the differential diagnosis of suspected acquired haemolytic anaemia. Thus, spherocytes could be attributable to hereditary spherocytosis as well as to autoimmune or alloimmune haemolytic anaemia. Haemolysis with irregularly contracted cells could be attributable not only to oxidant exposure but also to an unstable haemoglobin, homozygosity for haemoglobin C or glucose-6-phosphate dehydrogenase (G6PD) deficiency.
Table 13.1 Abnormalities that may be detected on blood film examination and their possible significance
| Morphological abnormality observed on blood film examination | Type of acquired haemolytic anaemia suggested |
|---|---|
| Schistocytes | Fragmentation syndromes including microangiopathic haemolytic anaemia and mechanical haemolytic anaemia |
| Spherocytes | Autoimmune, alloimmune or drug-induced immune haemolytic anaemia, paroxysmal cold haemoglobinuria, burns, Clostridium perfringens sepsis |
| Microspherocytes | Burns, fragmentation syndromes |
| Irregularly contracted cells | Oxidant damage, Zieve’s syndrome |
| Ghost cells, hemi-ghosts and suspicion of Heinz bodies | Acute oxidant damage |
| Marked red cell agglutination | Cold-antibody-induced haemolytic anaemia |
| Minor red cell agglutination | Warm autoimmune haemolytic anaemia, paroxysmal cold haemoglobinuria |
| Red cell agglutination plus erythrophagocytosis | Particularly characteristic of paroxysmal cold haemoglobinuria |
| Hypochromia, microcytosis and basophilic stippling | Lead poisoning |
| Erythrophagocytosis | Paroxysmal cold haemoglobinuria |
| Atypical lymphocytes | Cold-antibody-induced haemolytic anaemia associated with infectious mononucleosis or, less often, other infections |
| Lymphocytosis with mature small lymphocytes and smear cells | Autoimmune haemolytic anaemia associated with chronic lymphocytic leukaemia |
| Thrombocytopenia | Autoimmune haemolytic anaemia (Evans’ syndrome), thrombotic thrombocytopenic purpura, microangiopathic haemolytic anaemia associated with disseminated intravascular coagulation, paroxysmal nocturnal haemoglobinuria |
| Neutropenia | Paroxysmal nocturnal haemoglobinuria |
| No specific red cell features | Paroxysmal nocturnal haemoglobinuria |
Immune haemolytic anaemias
Acquired immune-mediated haemolytic anaemias are the result of autoantibodies to a patient’s own red cell antigens or alloantibodies in a patient’s circulation, either present in the plasma or completely bound to red cells (e.g. transfused or neonatal red cells). Alloantibodies may be present in a patient’s plasma and react with antigens on transfused donor red cells to cause haemolysis. Alloantibodies may also occur in maternal plasma and cause haemolytic disease of the newborn. Autoimmune haemolytic anaemia (AIHA) may be ‘idiopathic’ or secondary, associated mainly with lymphoproliferative disorders and autoimmune diseases, particularly systemic lupus erythematosus. AIHA may also follow atypical (Mycoplasma pneumoniae) pneumonia or infectious mononucleosis and other viral infections. AIHA has also been reported following allogeneic bone marrow transplantation1 and other hematopoietic stem cell transplantation in both adult2 and paediatric patients.3 Paroxysmal cold haemoglobinuria (PCH) also belongs to this group of disorders. Occasionally, drugs may give rise to a haemolytic anaemia of immunological origin that closely mimics idiopathic AIHA both clinically and serologically. This was a relatively common occurrence with α-methyldopa, a drug that is now used very infrequently, but it also occurs occasionally with other drugs. A larger range of drugs give rise to an antibody that is directed primarily against the drug and only secondarily involves the red cells. This is an uncommon occurrence. Such drugs include penicillin, phenacetin, quinidine, quinine, the sodium salt of p-aminosalicylic acid, salicylazosulphapyridine and cephalosporins.4
Types of Autoantibody
The diagnosis of an AIHA requires evidence of anaemia and haemolysis and demonstration of autoantibodies attached to the patient’s red cells (i.e. a positive DAT, see p. 279). A positive DAT may also be caused by the presence of alloantibodies (e.g. owing to a delayed haemolytic transfusion reaction), so details of any transfusion in the past months must be sought.
Cases of AIHA can similarly be separated into two broad categories according to the temperature characteristics of the associated autoantibodies: warm-type AIHA and the less frequent cold-type AIHA. The relative frequency of the two categories is illustrated in Table 13.2.5 In unusual instances, both warm autoantibody and cold autoantibody are detected in the patient’s serum and those cases are referred to as mixed-typed AIHA. This can be further classified into idiopathic or secondary, the latter often associated with systemic lupus erythematosus or lymphoma.6,7
Warm Autoantibodies
The most common type of warm autoantibody is an immunoglobulin (Ig) G, which behaves in vitro very similarly to an Rh alloantibody; indeed, many IgG autoantibodies have a mimicking Rh specificity. IgA and IgM warm autoantibodies are much less common and when present they are usually formed in addition to an IgG autoantibody (Table 13.3).8
Table 13.3 Direct antiglobulin test in warm-antibody autoimmune haemolytic anaemia: incidence of different reactions to specific antiglobulin sera8
Frequently, patients with warm-type AIHA have complement adsorbed onto their red cells and the red cells are therefore agglutinated by antisera specific for complement or a complement component such as C3d (Table 13.3). In these cases, the complement is probably not being bound by an IgG antibody but is on the cell surface as the result of the action of small and otherwise undetected amounts of IgM autoantibody.
Warm autoantibodies free in the patient’s serum are best detected by means of the indirect antiglobulin test (IAT) or by the use of enzyme-treated (e.g. trypsinized or papainized) red cells. (Antibodies that agglutinate unmodified cells directly in vitro are seldom present.) Not infrequently, antibodies that agglutinate enzyme-treated cells, sometimes at high titres, are present in the sera of patients in whom the IAT using unmodified cells is negative (Table 13.4). Occasionally, too, they are present in the sera of patients in whom the DAT is negative.
Cold Autoantibodies
The red cells of patients suffering from CHAD characteristically give positive antiglobulin reactions only with anticomplement (anti-C′) sera. (The C′ notation is used to distinguish anticomplement antibodies from anti-C antibodies of the Rh system.) This is because of the presence of red cells that have irreversibly adsorbed sublytic amounts of complement; it is an indication of an antigen–antibody reaction that has taken place at a temperature below 37°C. The complement component responsible for the reaction with anti-C′ sera is the C3dg derivative of C3 (see p. 494).
Combined Warm and Cold Autoantibodies
In approximately 7% of cases with AIHA, both warm IgG antibody and cold IgM autoantibody are simultaneously detected in the patient’s serum.6,7 These cases are referred to as ‘combined warm and cold AIHA’ or mixed-type AIHA. The serological characteristics in these patients are the presence of IgM cold autoantibody with a high thermal amplitude (reacting at or above 30°C) in association with a warm IgG autoantibody. In some cases, high- titre cold agglutinins (>1024 at 4°C) were reported9,10 and in others the cold agglutinin titre were reported as >64 at 4°C.11,12
PCH is caused by a biphasic IgG autoantibody, usually with anti-P specificity, and is commonly seen as an acute condition in children. This antibody binds to the red cells in the cold but activates complement and causes haemolysis on rewarming to 37°C. Cases may be idiopathic or can be secondary to acute viral infection in children. Other tests of value in the diagnosis of PCH are discussed on p. 287.
Some of the characteristics of IgG, IgM and IgA antibodies are listed in Table 13.5.
The clinical, haematological and serological aspects of the AIHAs have been summarized by Dacie13 and others.14–18
Methods of Investigation
Many of the methods used in the investigation of a patient suspected of suffering from AIHA are described in Chapter 21. Detailed description is given here of precautions to be taken when collecting blood samples from patients and of methods of particular value in the investigations.
Scheme for Serological Investigation of Haemolytic Anaemia Suspected to be of Immunological Origin
It is important to consider which are the most useful tests to carry out and the order in which they should be done. A suggested scheme has been set out in the form of answers to questions.19 Whereas some information may be helpful in classifying the type of AIHA, the single most important practical consideration is to determine whether, in addition to an autoantibody, there is any underlying alloantibody present. This should be identified before transfusion is undertaken to avoid a delayed haemolytic transfusion reaction that would compound existing haemolysis.
Perform a DAT using a polyspecific ‘broad-spectrum’ reagent, which contains both anti-IgG and anti-C′. (If the DAT is negative, it is unlikely, although not impossible, that the diagnosis is AIHA. See DAT-negative AIHA, p. 281)
Repeat the DAT using monospecific sera (see p. 500) (i.e. anti-IgG and anti-C3d).
Prepare eluates from the patient’s red cells. Test these later (see item 6).
Determine the patient’s ABO and RhD and Kell type. The Rh phenotype is particularly important in warm-type AIHA; other antigens must be determined if alloantibodies are to be differentiated from autoantibodies (see p. 507).
Screen the serum with two or three red cell suspensions suitable for routine pretransfusion antibody screening (see p. 528) looking for agglutination and lysis at 37°C by the IAT (see p. 500). If positive, identify the antibody using an antibody identification panel.
Test the eluate against the antibody identification panel of red cells by IAT and by using enzyme-treated red cells (see p. 498). Titration of autoantibody may be useful in the presence of a strong alloantibody.
The diagnosis of mixed-type AIHA can only be made after appropriate characterization of the serum autoantibodies. The serological characteristic in these cases is the presence of a cold IgM antibody with a high thermal amplitude (reacting at or above 30°C) in association with a warm IgG autoantibody.6,9
Detection of Incomplete Antibodies by Means of the Direct Antiglobulin (Coombs) Test
Significance of Positive Direct Antiglobulin Test
A positive DAT plus anaemia does not necessarily mean that the patient has autoimmune haemolytic anaemia.5,8,20 The causes of a positive test include the following:
Non-specific binding of immunoglobulins to red cells in patients with hypergammaglobulinemia or multiple myeloma and in recipients of antilymphocyte globulin and antithymocyte globulin.24
Szymanski et al.25 used an AutoAnalyser and used Ficoll and polyvinylpyrrolidone (PVP) to enhance agglutination by an anti-IgG serum highly diluted (usually to 1 in 5000) in 0.5% bovine serum albumin. In this sensitive system, the strength of agglutination was positively correlated with the serum γ-globulin concentration, being subnormal in hypogammaglobulinaemia and supranormal in hypergammaglobulinaemia.26
Similar findings were observed in patients with multiple myeloma. Non-specific binding of IgG to red cells was related to the level of monoclonal protein in the patient’s serum.26
Usually, in patients with hypergammaglobulinaemia in whom the DAT is positive, attempts to demonstrate antibodies in eluates fail (i.e. eluates are non-reactive).27,28
There was an association demonstrated between an elevated blood urea nitrogen and a positive DAT.32 Urea may alter the red cell membrane and enhance non-specific IgG adsorption.32
Positive DATs in Normal Subjects
The occurrence of a clearly positive DAT in an apparently healthy subject is a rare but well-known phenomenon. Worlledge20 reported a prevalence in blood donors of approximately 1 in 9000. In a later report, Gorst et al.34 estimated that the prevalence was approximately 1 in 14 000 with an increasing likelihood of a positive test with increasing age. Their report and subsequent reports,25,35 suggest that the finding of a positive DAT, using an anti-IgG serum, in an apparently healthy person is usually of little clinical significance and that, although overt AIHA may subsequently develop, this is infrequent. In some such individuals the DAT eventually becomes negative.
Positive DATs in Hospital Patients
In contrast to the rarity of positive DATs in healthy people, positive tests are much more frequent in hospital patients. Worlledge20 reported that the red cells of 40 out of 489 blood samples (8.9%) submitted for routine tests were agglutinated by anti-C′ sera. Only one sample was agglutinated by an anti-IgG serum and this had been obtained from a patient being treated with α-methyldopa. Freedman36 reported a similar incidence – 7.8% positive tests with anti-C′ sera. Lau et al.37 used anti-IgG sera only. The tests were seldom positive (0.9% positive out of 4664 tests). The probable explanation for the relatively high incidence of positive tests with anti-C′ sera is that the reaction is between anti-C′ antibodies and immune complexes adsorbed to the red cells.
False-Negative Antiglobulin Test Results
There are several causes of false-negative test results:
These phenomena are largely negated by the use of column agglutination technology.
DAT-Negative Autoimmune Haemolytic Anaemia
Most hospital blood banks use polyspecific ‘broad-spectrum’ AHG reagents for screening for diagnosis of AIHA. These reagents contain antibody to human IgG and the C3d component of human complement and have little activity against IgA and IgM proteins. The incidence of IgA-only warm AIHA has been reported as 0.2% to 2.7%,38 and the diagnosis may be missed if such polyspecific AHG is used for the DAT screen. In approximately 2–6% of patients who present with the clinical and haematological features of AIHA, the DAT is negative on repeated testing.20,39,40
Low-affinity IgG autoantibodies dissociate from the red cells during the washing phase if a tube technique is used, resulting in a negative DAT. Alternatively, there may be few IgG molecules coating the red cells and this number may fall below the threshold of detection, which is 300–4000 molecules per red blood cell if a tube technique is used. In such cases, a positive DAT may be demonstrated by a more sensitive technique, such as a column agglutination method, an enzyme-linked immunoabsorbant assay or flow cytometry.41–43
If polyspecific AHG is used and the DAT remains negative with clinical evidence of haemolysis, a more sensitive technique should be used for further investigation.44
Manual Direct Polybrene Test
The following method45 is modified from that of Lalezari and Jiang.46 Polybrene is a polyvalent cationic molecule, hexadimethrine bromide, that can overcome the electrostatic repulsive forces between adjacent red cells, bringing the cells closer together. When low levels of IgG are present on the red cell surface, antibody linkage of adjacent red cells is enhanced. The Polybrene is then neutralized using a negatively charged molecule such as trisodium citrate. Sensitized red cells remain agglutinated after neutralization of the Polybrene. Unsensitized red cells will disaggregate after neutralization.
Reagents
Polybrene stock. 10% Polybrene in 9 g/l NaCl, pH 6.9 (saline).
Working Polybrene solution. Dilute the stock Polybrene solution 1 in 250 in saline.
Resuspending solution. 60 ml of 0.2 mol/l trisodium citrate added to 40 ml of 50 g/l dextrose.
Washing solution. 50 ml of 0.2 mol/l trisodium citrate in 950 ml of saline.
Method
Ensure that all reagents are at room temperature.
Negative control
Normal group AB serum that fails to agglutinate papainized group O, D-positive red cells.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
