[level-membership-for-pediatrics-category]Chapter 123
Acquired Conditions
Neuroblastoma
Overview: Neuroblastoma is the most common extracranial solid neoplasm in children and accounts for almost 10% of all childhood neoplasms.1 Neuroblastoma arises in the abdomen in two thirds of cases; of these, about two thirds of lesions occur in the adrenal, whereas the remainder may arise anywhere along the sympathetic nerve chains.1
Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma belong to a group of related neoplasms arising from neural crest tissue that are distinguished by their degree of cellular maturation and differentiation. Neuroblastoma accounts for the vast majority of these lesions and has the most primitive and malignant cells.2 Ganglioneuroma represents the more differentiated end of the spectrum and is benign. Ganglioneuroblastomas represent an intermediate group with mixed histology.
Most children with neuroblastoma present between 1 and 5 years of age, with a median age of almost 2 years.2 Neuroblastoma is more common in patients with neurofibromatosis type 1, Beckwith-Wiedemann syndrome, Hirschsprung disease, central hypoventilation syndrome, and DiGeorge syndrome.2
Neuroblastomas typically present as palpable masses or with symptoms and signs related to local tumor invasion, metastatic disease, and the effects of hormone production or autoimmune response (opsoclonus-myoclonus syndrome). Metastatic disease is seen in up to 70% of patients at presentation.3 The most common sites include local and distant lymph nodes, bone, bone marrow, liver, and skin.
The diagnosis of neuroblastoma can be made by tissue biopsy, but a combination of positive bone marrow aspirate and increased urinary catecholamine metabolites (vanillylmandelic acid and homovanillic acid) is sufficient to confirm the diagnosis. The urinary level of catecholamine metabolites is increased in almost 90% of cases of neuroblastoma.4 The International Neuroblastoma Staging System has been the most commonly used worldwide for staging neuroblastoma.1,2,4 However, its application has encountered many difficulties, as it relies on the extent of tumor resected at surgery. For this reason, a new staging system, the International Neuroblastoma Risk Group Staging System, was developed in 2009, and this system relies on preoperative imaging findings (imaging risk factors) and detection of metastatic disease (Box 123-1).5
The prognosis of neuroblastoma depends on stage at presentation, patient’s age (children younger than 12 to 18 months have better prognosis), histologic category, grade of tumor differentiation, status of the MYCN oncogene, chromosome 11q status, and deoxyribonucleic acid ploidy.6
Imaging: Adrenal neuroblastomas and those arising from the adjacent retroperitoneal area are usually easily identified with ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) because the mass is generally quite large by the time of presentation (Fig. 123-1). Very small masses are uncommon, and in those cases seen beyond the neonatal age, CT and MRI play a more important role than ultrasonography (Fig. 123-2). On ultrasonography, the masses have a variety of appearances with the most characteristic being that of a solid, heterogeneous, hyperechoic mass, often with small hyperechoic foci with or without acoustic shadowing caused by calcification (e-Fig. 123-3).1,2,4 Anechoic areas resulting from cystic, hemorrhagic, or necrotic changes may be present (e-Fig. 123-4).
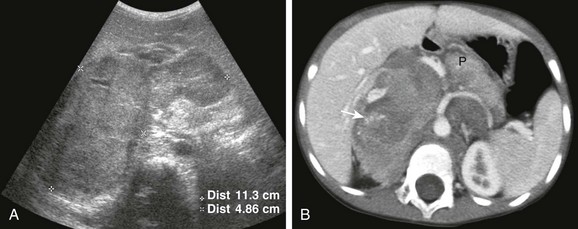
Figure 123-1 Large retroperitoneal neuroblastoma in a 4-year-old boy.
A, A transverse sonogram. B, An axial contrast-enhanced computed tomography image of the abdomen show a large lobulated, solid mass centered in the right upper quadrant but with a significant component crossing the midline and extending into the left hemiabdomen. In A, the mass (between cursors) is relatively hypoechoic compared with the liver. In B, the mass is heterogeneous with hypodense and hyperdense areas as well as foci of calcification (arrow). Note the characteristic encasement of the major vessels by the mass, in this case the aorta, the celiac axis, and its major branches. The pancreas (P) is displaced anteriorly and to the left by the mass.
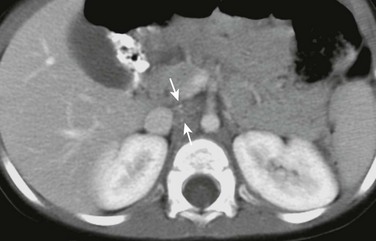
Figure 123-2 Small retroperitoneal neuroblastoma in a 19-month-old boy who presented with ataxia and nystagmus.
Axial contrast-enhanced computed tomography image shows a small hypodense mass with punctate calcifications in the interaortocaval region (arrows). Because of its location and small size, this mass was difficult to appreciate on ultrasonography.
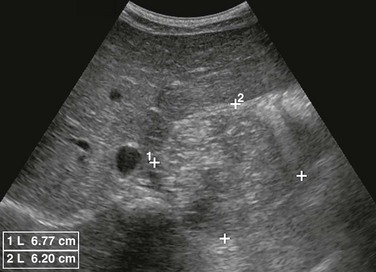
e-Figure 123-3 Left adrenal neuroblastoma in a 7-year-old girl.
A transverse sonogram shows a rounded solid mass (between cursors) posterior to the left lobe of the liver with multiple confluent hyperechoic foci.
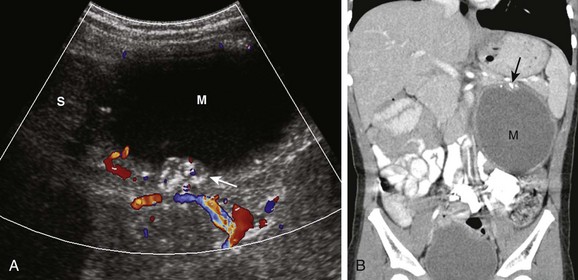
e-Figure 123-4 Left adrenal neuroblastoma in a 3-year-old girl.
A, Transverse sonogram shows a cystic mass (M) anterior and medial to the spleen (S) with foci of mural calcification (arrow). B, Reformatted coronal computed tomography image shows the left upper quadrant cystic mass (M) with mural calcifications (arrow), which on histology proved to be neuroblastoma.
The new International Neuroblastoma Risk Group Staging System requires the use of CT, MRI, or both for staging.5 On CT, neuroblastomas usually show heterogeneous enhancement, depicting solid areas along with areas of necrosis, hemorrhage, and cystic change (see Fig. 123-1). Calcification is present in more than 90% of cases (see Figs. 123-1, 123-2, and e-Figs 123-3 and 123-4).4 On MRI, the lesions are usually heterogeneous, with predominantly low signal on T1-weighted images and high signal on T2-weighted images, and with variable degrees of enhancement (Fig. 123-5).2,4
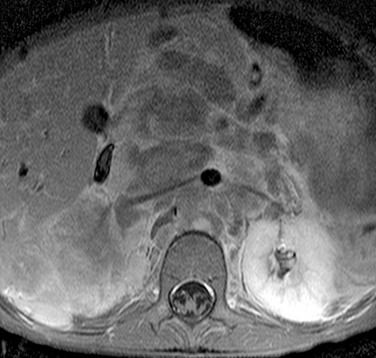
Figure 123-5 Retroperitoneal neuroblastoma in a 4-year-old girl.
An axial gadolinium-enhanced T1 fat-suppressed magnetic resonance image shows a large retroperitoneal mass encasing the aorta and renal arteries and displacing the inferior vena cava to the right. The mass shows heterogeneous enhancement.
A characteristic imaging feature of neuroblastoma is the displacement of adjacent organs and the displacement or encasement of adjacent major vessels (see Figs. 123-1 and 123-5).4 Local spread may be to lymph nodes. Less frequently, direct invasion into the kidneys (Fig. 123-6) or liver may occur, and even less commonly, the tumor may extend into the spinal canal.
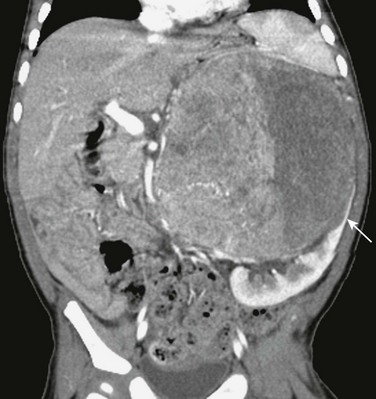
Figure 123-6 Left adrenal neuroblastoma in a 9-month-old girl.
A reformatted coronal computed tomography image shows a large heterogeneous mass invading the left kidney. The left kidney is displaced caudally and laterally with suggestion of a partial “claw sign” (arrow), which may lead to the erroneous interpretation that the mass is arising from the left kidney.
Neuroblastoma may also cause liver metastases. These may be single or multiple nodules or masses or may present with an infiltrative pattern, particularly in neonates (see Chapter 122). Metastases to bone are best identified with metaiodobenzylguanidine scan or with radionuclide bone scan.
Ganglioneuroma
Overview: Ganglioneuroma represents the mature, benign form of neural crest neoplasm. This type of tumor is far less frequent compared with neuroblastoma; it may develop from the maturation of a known malignant neuroblastoma, or it may be found de novo.2
Ganglioneuromas most frequently occur in the posterior mediastinum followed by the extra-adrenal retroperitoneum and the adrenal.2 These tumors usually occur in older children, are frequently asymptomatic, and are often found incidentally on imaging. Occasionally, symptoms may result from tumor growth into the intervertebral foramina, causing cord compression. Urinary catecholamine levels are usually normal.
Imaging: The imaging appearance of ganglioneuroma is most often indistinguishable from that of neuroblastoma (Figs. 123-7 and 123-8).2 Therefore, definitive diagnosis depends on the histologic examination of tumor tissue.
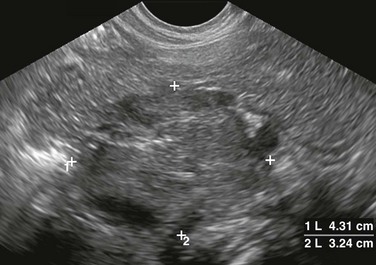
Figure 123-7 Retroperitoneal ganglioneuroma in a 7-year-old girl.
A transverse sonogram shows a large rounded mass (between cursors) of intermediate echogenicity with scattered punctate calcifications in the midline retroperitoneum. The sonographic appearance of ganglioneuroma is indistinguishable from neuroblastoma.
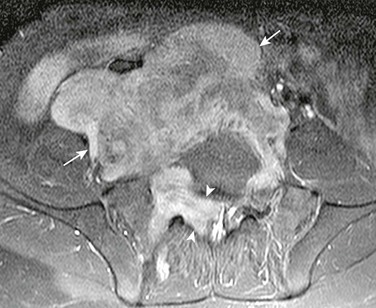
Figure 123-8 Retroperitoneal ganglioneuroma in a 10-year-old girl.
An axial gadolinium-enhanced T1 fat-suppressed magnetic resonance image shows a large, heterogeneous, retroperitoneal mass (arrows), which extends into the sacral intervertebral foramina and into the distal spinal canal (arrowheads). The imaging features are similar to those seen in neuroblastoma.
Pheochromocytoma
Overview: Pheochromocytoma is an uncommon neuroendocrine tumor of the adrenal arising from chromaffin cells. If the tumor is extra-adrenal, it is known as a paraganglioma, with most common abdominal locations being in the vicinity of the renal vessels and at the organ of Zuckerkandl, near the origin of the inferior mesenteric artery. In children, 70% of pheochromocytomas occur in the adrenal gland, and 20% to 70% have bilateral adrenal involvement.7 Malignancy is present in 12% of lesions in children.8 The diagnosis of malignancy is often made on the basis of metastases rather than histology.8
Pheochromocytoma usually presents in older children (mean age 11 years). It is often sporadic but may be part of cancer predisposition syndromes, mainly von Hippel-Lindau disease and multiple endocrine neoplasias 2A and 2B, and less commonly neurofibromatosis type 1, multiple endocrine neoplasia 1, and tuberous sclerosis complex.8
The clinical presentation is usually related to the secretion of catecholamines. Patients present with hypertension, headache, tachycardia, diaphoresis, nervousness, and weight loss.7
Imaging: CT and MRI are better imaging modalities than ultrasonography for accurate localization of these tumors. Functional imaging with metaiodobenzylguanidine is more specific and has been reported to be more sensitive in detecting multifocal disease.7,8
On ultrasonography, pheochromocytoma may have a solid homogeneous or heterogeneous appearance (Fig. 123-9 and e-Fig 123-10). Heterogeneity is more commonly seen with larger lesions because of hemorrhage, necrosis, and less commonly calcification.9 On CT, the lesions show avid enhancement except for areas of necrosis or hemorrhage.1 The use of non-ionic low-osmolar intravenous contrast material for CT is safe in patients with pheochromocytoma. On MRI, pheochromocytoma is hypointense to isointense compared with the liver on T1-weighted images and hyperintense on T2-weighted images (see Fig. 123-9 and e-Fig. 123-10), with a pattern of intense enhancement after the administration of gadolinium.1
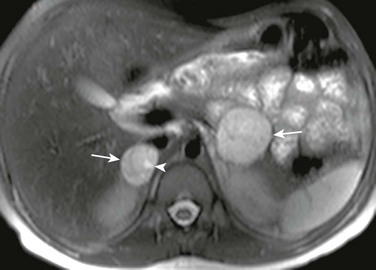
Figure 123-9 Bilateral pheochromocytoma in an 11-year-old boy with von Hippel–Lindau disease and arterial hypertension.
An axial fat-suppressed T2-weighted magnetic resonance image shows bilateral adrenal masses (arrows), larger on the left. The masses are hyperintense with small cystic change on the right medially (arrowhead). See e-Figure 123-10.
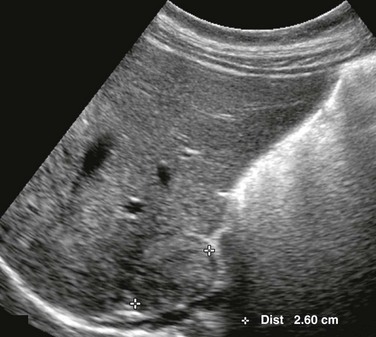
e-Figure 123-10 Bilateral pheochromocytoma in an 11-year-old boy with von Hippel–Lindau disease and arterial hypertension (same patient as shown in Fig. 123-9).
A longitudinal sonogram of the right upper quadrant shows a well-defined rounded solid mass (between cursors) of slightly heterogeneous echogenicity.
Adrenocortical Neoplasms
Overview: Adrenocortical neoplasms comprise two types of tumor: adenomas and carcinomas. Both are uncommon in children. A higher incidence is seen in children under the age of 4 years.10 Girls are affected more commonly compared with boys, and carcinomas are much more common than are adenomas.10 The vast majority of these tumors are functioning tumors, and the most common endocrine abnormality is overproduction of androgens. Girls present with virilization, and boys present with pseudoprecocious puberty. Often, mixed endocrine dysfunction is caused by the overproduction of both glucocorticoids and mineralocorticoids. Two cancer predisposition syndromes, Li-Fraumeni syndrome and Beckwith-Wiedemann syndrome, are strongly associated with adrenocortical neoplasms.10
Imaging: At presentation, adrenocortical neoplasms in children are usually larger than 5 cm.9 Most lesions can be documented with ultrasonography, but CT and MRI are particularly important for assessing large lesions and local invasion and spread. On all three modalities, smaller lesions tend to have a fairly homogeneous appearance. Areas of hemorrhage, necrosis, and calcification are seen more commonly in larger lesions, usually carcinomas (Fig. 123-11 and e-Fig 123-12).9 In larger lesions, the characteristic appearance of a central scar, with radiating linear bands that represent areas of necrosis and calcification (Fig. 123-13), may be seen.1 It is not always possible to predict accurately which masses are malignant, unless local spread and invasion have occurred or metastatic disease is present. Metastatic disease most commonly occurs to the lungs, liver, and bone.
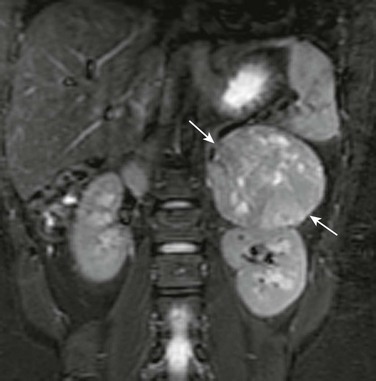
Figure 123-11 Adrenocortical carcinoma in a 14-year-old girl who presented with virilization signs and elevated testosterone levels.
A coronal short inversion time inversion recovery magnetic resonance image shows a large heterogeneous left suprarenal mass (arrows) causing inferior displacement and flattening of the upper pole of the adjacent left kidney. The mass contains multiple hyperintense foci due to areas of necrosis. See e-Figure 123-12.
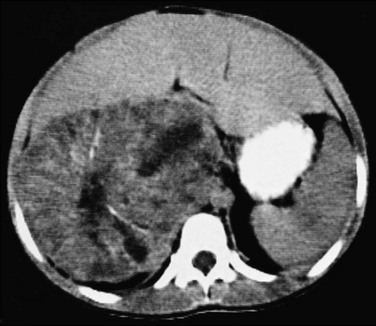
Figure 123-13 Adrenocortical carcinoma in an 8-year-old girl with precocious puberty.
A contrast-enhanced axial computed tomography image shows a large, relatively well-defined right adrenal mass with heterogeneous attenuation. A radial pattern of calcification in the mass is seen, with adjacent areas of decreased attenuation caused by necrosis, which is characteristic of adrenocortical carcinoma.
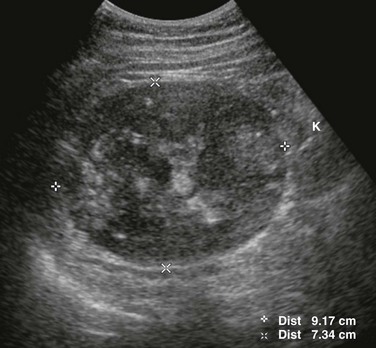
e-Figure 123-12 Adrenocortical carcinoma in a 14-year-old girl who presented with virilization signs and elevated testosterone levels (same patient as shown in Fig. 123-11).
A longitudinal sonogram shows a large, heterogeneous mass (between cursors) in the left upper quadrant superior to the left kidney (K). The mass contains anechoic areas due to necrosis as well as hyperechoic foci.
Treatment: Complete resection of the primary tumor is essential for survival.10 Chemotherapy has had a major effect on improved outcome. Adenomas clearly have a good prognosis if they are removed completely, but clinical follow-up and imaging follow-up are essential because the histologic differences between adenoma and carcinoma are not clear in all cases.
Other Retroperitoneal Conditions
Overview: Apart from pathology arising from the urinary tract and adrenal and sympathetic chains, other less common retroperitoneal conditions include lymphadenopathy (mainly lymphoma or metastatic lymphadenopathy), vascular malformations, hemangiomas, neurogenic tumors (schwannoma, neurofibroma, malignant peripheral nerve sheath tumor), adipocytic tumors, germ-cell tumors (Fig. 123-14), rhabdomyosarcoma, fibromatosis, idiopathic fibrosis, hematomas, and abscesses.
 What the Clinician Needs to Know
What the Clinician Needs to Know
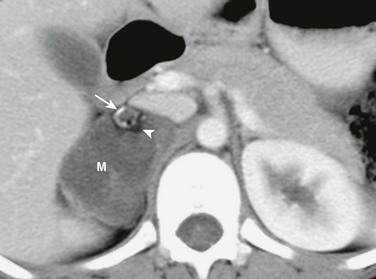
Figure 123-14 Retroperitoneal teratoma in an 11-year-old girl.
A contrast-enhanced axial computed tomography image shows a well-defined right suprarenal mass (M), with mainly fluid attenuation and some soft tissue septa. Calcification (arrow) and fatty attenuation (arrowhead) are seen anteriorly within the mass.
Balassy, C, Navarro, OM, Daneman, A. Adrenal masses in children. Radiol Clin North Am. 2011;49(4):711–727. 49
Hiorns, MP, Owens, CM. Radiology of neuroblastoma in children. Eur Radiol. 11, 2001. 2071-2081
Lonergan, GJ, Schwab, CM, Suarez, ES, et al. Neuroblastoma, ganglioneuroblastoma and ganglioneuroma: radiologic-pathologic correlation. RadioGraphics. 2002;22:911–934.
Paterson, A. Adrenal pathology in childhood spectrum of disease. Eur Radiol. 2002;12:2491–2508.
Rescorla, FJ. Malignant adrenal tumors. Semin Pediatr Surg. 2006;15:48–56.
References
1. Paterson, A. Adrenal pathology in childhood, a spectrum of disease. Eur Radiol. 2002;12:2491–2508.
2. Lonergan, GJ, Schwab, CM, Suarez, ES, et al. Neuroblastoma, ganglioneuroblastoma and ganglioneuroma: radiologic-pathologic correlation. RadioGraphics. 2002;22:911–934.
3. McHugh, K, Pritchard, J. Problems in the imaging of three common paediatric solid tumours. Eur J Radiol. 2001;37:72–78.
4. Hiorns, MP, Owens, CM. Radiology of neuroblastoma in children. Eur Radiol. 11, 2001. [2071-2081].
5. Monclair, T, Brodeur, GM, Ambros, PF, et al. The international neuroblastoma risk group (INRG) staging system: an INRG task force report. J Clin Oncol. 2009;27:298–303.
6. Cohn, SL, Pearson, ADJ, London, WB, et al. The international neuroblastoma risk group (INRG) staging system: an INRG task force report. J Clin Oncol. 2009;27:289–297.
7. Rescorla, FJ. Malignant adrenal tumors. Semin Pediatr Surg. 2006;15:48–56.
8. Waguespack, SG, Rich, T, Grubbs, E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010;95:2023–2037.
9. Siegel, MJ, Chung, EM. Adrenal glands, pancreas, and other retroperitoneal structures. In: Siegel MJ, ed. Pediatric sonography. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2011:461–508.
10. Ribeiro, RC, Figueiredo, B. Childhood adrenocortical tumors. Eur J Cancer. 2004;40:1117–1126.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 123
Acquired Conditions
Neuroblastoma
Overview: Neuroblastoma is the most common extracranial solid neoplasm in children and accounts for almost 10% of all childhood neoplasms.1 Neuroblastoma arises in the abdomen in two thirds of cases; of these, about two thirds of lesions occur in the adrenal, whereas the remainder may arise anywhere along the sympathetic nerve chains.1
Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma belong to a group of related neoplasms arising from neural crest tissue that are distinguished by their degree of cellular maturation and differentiation. Neuroblastoma accounts for the vast majority of these lesions and has the most primitive and malignant cells.2 Ganglioneuroma represents the more differentiated end of the spectrum and is benign. Ganglioneuroblastomas represent an intermediate group with mixed histology.
Most children with neuroblastoma present between 1 and 5 years of age, with a median age of almost 2 years.2 Neuroblastoma is more common in patients with neurofibromatosis type 1, Beckwith-Wiedemann syndrome, Hirschsprung disease, central hypoventilation syndrome, and DiGeorge syndrome.2
Neuroblastomas typically present as palpable masses or with symptoms and signs related to local tumor invasion, metastatic disease, and the effects of hormone production or autoimmune response (opsoclonus-myoclonus syndrome). Metastatic disease is seen in up to 70% of patients at presentation.3 The most common sites include local and distant lymph nodes, bone, bone marrow, liver, and skin.
The diagnosis of neuroblastoma can be made by tissue biopsy, but a combination of positive bone marrow aspirate and increased urinary catecholamine metabolites (vanillylmandelic acid and homovanillic acid) is sufficient to confirm the diagnosis. The urinary level of catecholamine metabolites is increased in almost 90% of cases of neuroblastoma.4 The International Neuroblastoma Staging System has been the most commonly used worldwide for staging neuroblastoma.1,2,4 However, its application has encountered many difficulties, as it relies on the extent of tumor resected at surgery. For this reason, a new staging system, the International Neuroblastoma Risk Group Staging System, was developed in 2009, and this system relies on preoperative imaging findings (imaging risk factors) and detection of metastatic disease (Box 123-1).5
The prognosis of neuroblastoma depends on stage at presentation, patient’s age (children younger than 12 to 18 months have better prognosis), histologic category, grade of tumor differentiation, status of the MYCN oncogene, chromosome 11q status, and deoxyribonucleic acid ploidy.6
Imaging: Adrenal neuroblastomas and those arising from the adjacent retroperitoneal area are usually easily identified with ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) because the mass is generally quite large by the time of presentation (Fig. 123-1). Very small masses are uncommon, and in those cases seen beyond the neonatal age, CT and MRI play a more important role than ultrasonography (Fig. 123-2). On ultrasonography, the masses have a variety of appearances with the most characteristic being that of a solid, heterogeneous, hyperechoic mass, often with small hyperechoic foci with or without acoustic shadowing caused by calcification (e-Fig. 123-3).1,2,4 Anechoic areas resulting from cystic, hemorrhagic, or necrotic changes may be present (e-Fig. 123-4).
Figure 123-1 Large retroperitoneal neuroblastoma in a 4-year-old boy.
A, A transverse sonogram. B, An axial contrast-enhanced computed tomography image of the abdomen show a large lobulated, solid mass centered in the right upper quadrant but with a significant component crossing the midline and extending into the left hemiabdomen. In A, the mass (between cursors) is relatively hypoechoic compared with the liver. In B, the mass is heterogeneous with hypodense and hyperdense areas as well as foci of calcification (arrow). Note the characteristic encasement of the major vessels by the mass, in this case the aorta, the celiac axis, and its major branches. The pancreas (P) is displaced anteriorly and to the left by the mass.
Figure 123-2 Small retroperitoneal neuroblastoma in a 19-month-old boy who presented with ataxia and nystagmus.
Axial contrast-enhanced computed tomography image shows a small hypodense mass with punctate calcifications in the interaortocaval region (arrows). Because of its location and small size, this mass was difficult to appreciate on ultrasonography.
e-Figure 123-3 Left adrenal neuroblastoma in a 7-year-old girl.
A transverse sonogram shows a rounded solid mass (between cursors) posterior to the left lobe of the liver with multiple confluent hyperechoic foci.
e-Figure 123-4 Left adrenal neuroblastoma in a 3-year-old girl.
A, Transverse sonogram shows a cystic mass (M) anterior and medial to the spleen (S) with foci of mural calcification (arrow). B, Reformatted coronal computed tomography image shows the left upper quadrant cystic mass (M) with mural calcifications (arrow), which on histology proved to be neuroblastoma.
The new International Neuroblastoma Risk Group Staging System requires the use of CT, MRI, or both for staging.5 On CT, neuroblastomas usually show heterogeneous enhancement, depicting solid areas along with areas of necrosis, hemorrhage, and cystic change (see Fig. 123-1). Calcification is present in more than 90% of cases (see Figs. 123-1, 123-2, and e-Figs 123-3 and 123-4).4 On MRI, the lesions are usually heterogeneous, with predominantly low signal on T1-weighted images and high signal on T2-weighted images, and with variable degrees of enhancement (Fig. 123-5).2,4
Figure 123-5 Retroperitoneal neuroblastoma in a 4-year-old girl.
An axial gadolinium-enhanced T1 fat-suppressed magnetic resonance image shows a large retroperitoneal mass encasing the aorta and renal arteries and displacing the inferior vena cava to the right. The mass shows heterogeneous enhancement.
[/not-level-membership-for-pediatrics-category]
