[level-membership-for-gastroenterology-and-hepatology-category]
24 Abnormal liver function test results
Case
A 45-year-old male was found to have abnormal liver function tests on routine investigations performed for an insurance examination. Serum alanine aminotransferase level was elevated at 105 IU/L. International Normalized Ratio (INR), albumin and bilirubin were normal. The patient also had a history of hypertension controlled on antihypertensive therapy and an elevated fasting serum triglyceride concentration. His father had type 2 diabetes, but there was no family history of liver disease. An abdominal ultrasound examination demonstrated changes consistent with fatty liver, but no other abnormality. Further investigations showed no evidence of autoimmune, viral or inherited liver disease. His ferritin was mildly elevated, but with normal transferrin saturation and no mutation of the haemochromatosis (HFE) gene. Physical examination revealed him to be overweight, but otherwise was unremarkable. The patient was advised to reduce his weight and exercise regularly. Under the guidance of a dietician a 5% weight loss was achieved with an improvement in alanine aminotransferase levels. Following that he failed to maintain the intervention, gained weight and discontinued medical review. Ten years later he again was found to have abnormal liver tests but also INR 1.6, albumin 28 gm/L and bilirubin 25 μg/L. He had become diabetic with symptomatic ischaemic heart disease. Abdominal ultrasound on this occasion showed a small irregular liver with splenomegaly, consistent with cirrhosis and an apparent mass in the liver. On a triple phase computed tomography (CT) scan of the liver, the 2-cm mass lesion was typical of a hepatocellular carcinoma and was managed surgically.
An Approach to the Patient with Liver Disease
Liver disease will present as one of a limited number of clinical syndromes. Each syndrome may be caused by a number of specific disease processes. A diagnosis is achieved by paying attention to historical features and physical findings (Box 24.1) and the results of laboratory and other investigations. Only then can specific treatment for the cause of the liver abnormality be planned. Therapy, in many instances, is directed to the management of the clinical problem in addition to treatment directed to the underlying condition.
Box 24.1 Symptoms and signs of liver disease
Signs of poor hepatocellular synthetic function
Bruising, peripheral oedema (reflecting depleted coagulation factors and albumin levels)
In this chapter, liver function tests and their clinical relevance are discussed first because abnormalities are commonly detected in asymptomatic individuals. Next, the common clinical presentations of liver disease are covered. Finally, important liver diseases are described.
Liver Function Test Interpretation
Bilirubin
Elevated serum bilirubin levels may occur in all forms of liver disease. However, it must be remembered that a jaundiced patient does not necessarily have significant hepatic disease. Serum levels of bilirubin reflect production, hepatic uptake, processing (conjugation) and secretion. The physiology of bilirubin and an approach to diagnosis and management of jaundice is described in Chapter 23.
Tests reflecting hepatic function
Serum albumin
In chronic liver disease, low serum albumin concentrations represent a major prognostic indicator (see ‘Child-Pugh score’ later in this chapter). This can reflect the absence of sufficient functioning hepatocytes to maintain albumin production. Furthermore, malnutrition can be a significant contributor to hypoalbuminaemia in patients with chronic liver disease. Alcohol consumption can reduce albumin synthesis. Serum albumin concentrations may also fall without a reduction in hepatic synthesis when the volume of distribution of albumin is expanded by ascites and oedema. As albumin has a half-life of 17–26 days, low levels usually reflect chronic rather than acute hepatitic dysfunction.
Tests of Liver Injury
Transaminases
In the presence of malignant disease, increases in transaminases often indicate hepatic metastases.
Aspartate aminotransferase
Aspartate aminotransferase (AST) is an enzyme found in liver, skeletal muscle, myocardium, kidney, pancreas and erythrocytes. Damage to any of these cell types will result in an elevation of serum AST. ALT levels are usually higher than AST levels in viral liver injury. When liver disease is present, and the AST level is more than twice that of ALT, alcoholic liver disease must be considered (see later). In the presence of cirrhosis, the diagnostic value of the AST:ALT ratio is lost. The mitochondrial isoenzyme of AST is especially elevated in the presence of alcoholic liver injury.
Cholestasis versus hepatocellular disease
Liver function patterns may be used to guide the discrimination between cholestasis (e.g. due to stones or tumour obstructing the large bile ducts) and hepatocellular disease (e.g. cirrhosis or chronic hepatitis). These patterns can be used only as a broad guide and should not be relied on alone. However, if cholestasis is suspected, then looking for evidence of duct obstruction (e.g. by ultrasound) is the next step, while if hepatocellular disease is suspected, then a search for liver disease markers (e.g. virology, immunological tests) is the next step (see Table 23.1 in Ch 23).
Other tests useful in hepatic assessment
A marked elevation (over 1000 U/L) of alpha-fetoprotein is most often due to hepatocellular carcinoma. This test has been used for screening an individual with chronic hepatitis B for the development of hepatocellular carcinoma, having a sensitivity of 70%. It is of limited value, however, because the sensitivity falls to 30% in non-hepatitis B hepatocellular carcinoma. Milder elevations occur in both acute and chronic liver disease of many causes (Ch 25).
Autoantibodies are helpful in diagnosing certain forms of liver disease (Table 24.1). Serum concentration of caeruloplasmin and iron studies are important tools in the diagnosis of Wilson’s disease and haemochromatosis, respectively (Table 24.2), but are also altered by liver injury unrelated to either of these conditions. Tests for viral hepatitis are also important (Table 24.3).
| Test | Condition | Comment |
|---|---|---|
| Antinuclear antibody (ANA) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Smooth muscle antibody (SMA) (anti-actin) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Anti-liver kidney microsomal antibody (anti-LKM1) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Antimitochondrial antibody (AMA) | Primary biliary cirrhosis(95%) | Diagnosis requires liver biopsy |
| Antineutrophil cytoplasmic antibody (pANCA) | Primary sclerosing cholangitis | Diagnosis requires magnetic resonance cholangiopancreatography or endoscopic retrograde cholangiopancreatography |
Table 24.2 Tests for metabolic disorders affecting the liver
| Test | Condition | Comment |
|---|---|---|
| Iron studies | Haemochromatosis | See text |
| Copper and caeruloplasmin | Wilson’s disease | See text |
| Alpha-1 antitrypsin level and phenotype | Alpha-1 antitrypsin deficiency | Heterozygotes may have abnormal liver function tests without significant liver disease |
| Thyroid function tests | Hypothyroidism | Fatty liver |
| Blood glucose level | Diabetes mellitus | Fatty liver, haemochromatosis |
| Insulin/C-peptide | Insulin resistance | Fatty liver, metabolic syndrome |
| Cholesterol and triglycerides | Primary and secondary hyperlipidaemia | Fatty liver, alcoholic liver disease |
Table 24.3 Tests for viral hepatitis
| Test | Meaning of a positive result | Comment |
|---|---|---|
| 1. Tests for hepatitis A virus (HAV) | ||
| Anti-HAV IgM | Recent acquisition of HAV | Acute hepatic illness likely to be HAV |
| Anti-HAV IgG | Past infection/vaccination | Immunity |
| 2. Tests for hepatitis B virus (HBV) | ||
| HBsAg (surface ag) | Present/chronic infection | Structural component of virus |
| Anti-HBsAg (surface ab) | Past infection/vaccination | Immunity |
| Anti-HBc IgM (core ab) | Recent acquisition of HBV | The test for acute HBV infection |
| HBeAg (e ag) | Marker of viral replication | High risk of infectivity |
| Anti-HBeAg (e ab) | Suggests no viral replication | Unlikely to be infectious |
| HBV DNA | Presence of complete virus | High risk of infectivity |
| 3. Tests for hepatitis C virus (HCV) | ||
| Anti-HCV | Exposure to HCV | Interpret in conjunction with other clinical and laboratory data |
| HCV RNA by PCR | Presence of virus | – |
| HCV genotyping | – | Treatment planning and response |
| 4. Tests for hepatitis D virus (HDV) | ||
| Anti-HDV IgG/IgM | Exposure to HDV | Acute or chronic HDV |
| Delta antigen | HDV present | Acute or chronic HDV |
| 5. Tests for hepatitis E virus (HEV) | ||
| Anti-HEV IgM | Recent acquisition of HEV | Acute hepatitic illness likely to be HEV |
| 6. Tests for other organisms | ||
| Cytomegalovirus (CMV) IgM | Recent acquisition of CMV | Acute hepatitic illness likely to be CMV |
| Epstein-Barr virus (EBV) IgM | Recent acquisition of EBV | Acute hepatitic illness likely to be EBV |
| Anti-HIV | HIV-AIDS | Opportunistic hepatobiliary infections |
| Toxoplasmosis serology | – | Consider toxoplasmosis |
| Q fever serology | – | Consider Q fever |
ag = antigen; ab = antibody; PCR = polymerase chain reaction.
Liver biopsy
Histological and, on occasion, chemical analysis and microbiological examination of the liver are essential in the diagnosis of many hepatic disorders. The usual technique for obtaining a liver biopsy involves a percutaneous approach through a lower intercostal space in the right midaxillary line. Ultrasound scanning or CT can be used to optimise the needle entry point or target specific abnormalities. Haemorrhage from the biopsy wound to the liver or injury to adjacent organs are the major risks. These may be significant in approximately 1% of biopsies. The risk of haemorrhage is minimised by ensuring a platelet count greater than 80 × 109/L (80,000/mm3) and an INR less than 1.2. When coagulopathy or gross ascites prevents a percutaneous biopsy, a transjugular approach can be used.
The significance of liver biopsy features is considered in relation to specific disorders later in this chapter. The extent, nature and activity of inflammatory and fibrotic processes are of particular concern. The presence of fibro-inflammatory changes is more likely to be related to a progression to cirrhosis. The absence of such changes carries a better prognosis. Sampling error can be a problem as the tissue sample represents only a tiny proportion of the liver mass and the disease process may not be uniform across the organ. Cirrhosis can be missed in 10–20% of cases, depending on the size and number of samples taken.
The Well Patient with Abnormal Liver Function Profile
History
A history of diabetes mellitus, thyroid disease and high lipid levels should be sought; these together with obesity are associated with hepatic steatosis. The features of the metabolic syndrome with rising body weight and reduced physical activity are common in those with non-alcoholic fatty liver disease, especially when there is a family history of type 2 diabetes.
There is a possibility of genetic predisposition to alcoholic liver disease.
In the otherwise well patient, a history of abdominal pain (e.g. biliary pain: Ch 4), loss of appetite or weight (Ch 17), or a change in the colour of stools or urine (Ch 23) needs to be sought but is almost invariably absent.
Examination
Patients with abnormal liver test results must be regarded as having a liver disorder and thus signs of chronic liver disease should be sought because cirrhosis may be completely asymptomatic (Fig 24.1). The patient’s weight and height should be measured during the examination to allow evaluation of the body mass index (weight in kilograms divided by the square of the height in metres: kg/m2). Truncal obesity is common and waist measurement should be taken.
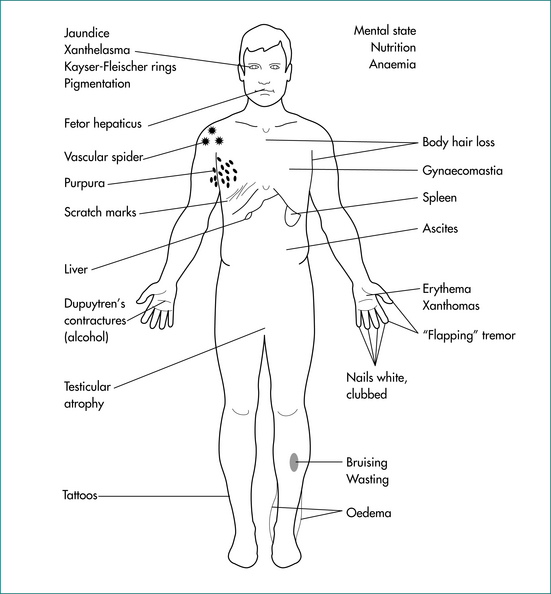
Figure 24.1 Signs of chronic liver disease.
From Sherlock S, Dooley J. Diseases of the liver and biliary system. 11th edn. Oxford: Blackwell; 2002, with permission.
In the usual patient presenting with no history of symptoms and no awareness of liver problems, it is likely that there will be no signs of liver disease. A minor degree of hepatomegaly should be expected in patients with alcohol-related fatty liver or non-alcoholic fatty liver disease (Ch 17). Mild splenomegaly may also be found.
Investigation
Liver imaging is of value (Ch 23). Abdominal ultrasound scans can provide information about the size and shape of the liver and spleen. Often any mass lesions present can also be identified. Ultrasonography will often detect fatty liver. Abdominal CT will provide information on the presence or absence of space-occupying lesions while, in addition, providing insight into liver density, which may be increased in haemochromatosis. Magnetic resonance imaging will usually add little to the information provided by a CT scan but can be of value in certain circumstances, such as in the assessment of mass lesions and of the biliary tract.
Tests for evidence of viral hepatitis (Table 24.3), autoimmune liver disease (Table 24.1) and other causes of chronic liver disease (iron or copper overload, and other inherited liver diseases) (Table 24.2) should be done if hepatocellular disease is possible. If the diagnosis remains unclear and the prognosis of concern, a liver biopsy is required.
Chronic hepatitis is an ongoing inflammatory process. The causes are listed in Table 24.4. If fibrosis follows as part of the healing process consequent to this inflammation, progression to cirrhosis and eventual liver failure is possible. For this reason, potentially controllable chronic hepatitis needs to be identified and treated before irreversible injury has occurred.
Table 24.4 Important causes of chronic hepatitis and cirrhosis
| Chronic hepatitis | Cirrhosis |
|---|---|
Evidence of continuing hepatic inflammation based on a persistent elevation of serum transaminases (6 months or more) is usually required to entertain the diagnosis of a chronic hepatitis. However, it can take many months for serum transaminases to return to normal levels following some acute hepatic illnesses. Once the presence of chronic hepatitis is considered likely, establish the aetiology of the illness, the extent of liver damage and the activity of the inflammatory process. These latter two features can be assessed only by the histological examination of a liver biopsy.
The Sick Patient with Jaundice: Acute Liver Disease
Acute hepatitis
Examination
The patient can appear relatively well or profoundly ill. Jaundice may or may not be present. The liver size is noted. It will often be enlarged and tender. There may be splenomegaly. Look for evidence of an underlying condition (e.g. signs of alcohol excess). Complications (e.g. hepatic encephalopathy) may supervene. Look for signs of chronic liver disease (Fig 24.1).
Investigation
Serum transaminase levels are elevated, usually more than five times the upper limit of normal. In viral hepatitis, transaminase levels of 1000 U/L and over are not unusual. Moderate elevations of alkaline phosphatase also occur. Serum albumin and INR usually remain normal. Specific blood tests are required to diagnose viral illnesses, including viral hepatitis (A, B, C, D and E), cytomegalovirus and Epstein-Barr virus, toxoplasmosis, Q fever and autoimmune disease (Table 24.3). Paracetamol levels should be determined and laboratory evidence of alcohol excess sought. These may be relatively low in patients with chronic, but toxic paracetamol consumption. Abdominal ultrasound scans are useful to document the size of the liver and spleen and to exclude biliary disease.
Management
Alcoholic hepatitis is managed supportively; in certain circumstances specific interventions may be of value. Ethanol is withdrawn, and vitamin (especially thiamine) and nutritional supplementation should be given (enteral feeding may provide a survival benefit). Corticosteroids (prednisone, 40 mg daily for 28 days) may provide a survival benefit when there is encephalopathy (see below) and no gastrointestinal haemorrhage. Propylthiouracil may be of benefit in less severe cases of alcoholic hepatitis, but is rarely used. Abstinence remains the mainstay of therapy.
When gallstone disease is found to be the cause of an acute hepatitic illness, cholecystectomy may be indicated. Choledocholithiasis may require endoscopic clearance of the bile duct (Ch 23). Acute autoimmune hepatitis is managed with corticosteroids (e.g. prednisone, 30–60 mg daily, then taper).
Acute Liver Failure
Assessment and management
Examination is directed towards assessing the possible aetiology and evolving complications of hepatic failure (outlined below). Serial liver function tests can be useful, but a falling ALT level may reflect either recovery or progression to massive hepatic necrosis. A temperature above 38°C or below 36°C, a pulse rate above 90 beats per minute and/or a white cell count above 12,000 or below 4,000 are associated with a greater mortality. The prognosis, and therefore need for liver transplantation, of a patient in acute liver failure can be assessed using the Kings College Criteria (Table 24.5).
Table 24.5 Kings College Criteria for listing of a patient in acute liver failure (ALF) for liver transplantation
| Type | Criteria |
|---|---|
| Paracetamol-induced ALF | Arterial pH < 7.30 after fluid resuscitation or all of: |
INR = International Normalized Ratio.
From O’Grady JG, Alexander GJM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
The patient with fulminant hepatic failure needs to be admitted directly to a unit capable of offering liver transplantation. Admission to intensive care is required with careful management of cerebral oedema, gastrointestinal bleeding, infection and renal failure.
Prognosis
Liver transplantation is a therapeutic option. The decision to transplant and the timing of transplantation are difficult. However, 1-year survival rates of approximately 70% can be achieved. This compares favourably with mortality rates from fulminant hepatic failure without transplantation of up to 90% although with optimal intensive care unit management 30–40% survival can be achieved. N-acetylcysteine may also improve overall survival and transplant-free survival in patients with non-paracetamol-related acute liver failure, especially if encephalopathy is limited to grade I or II.
Patients Presenting with Cirrhosis and its Complications
Cirrhosis represents the non-specific end-stage of hepatic disease that has disrupted the structural organisation of the liver. The presence of cirrhosis is established on a liver biopsy. Fibrosis or scarring surrounding regenerative nodules of hepatocytes is its hallmark. Although cirrhosis is a histological diagnosis, its presence is clinically suspected by finding signs of chronic liver disease and portal hypertension (Box 24.1) or on high quality imaging. Symptomatic liver disease and eventually liver failure occur as the result of impaired hepatocellular function within the distorted architecture of the regenerative nodules and, with progression, eventual loss of an effective liver cell mass. Distortion of the hepatic circulation contributes to portal hypertension and inefficient/ineffective perfusion of parenchymal cells. Portal systemic shunts may develop, allowing blood from the gastrointestinal tract to flow directly to the systemic circulation.
Cirrhosis should be considered as either compensated, with no signs of complications and a relatively good prognosis, or decompensated (Ch 25). Those with compensated cirrhosis are likely to be asymptomatic and the diagnosis may be made incidentally during the investigation of an unrelated condition. In this situation, counselling regarding diet, ethanol consumption and the identification and management of any underlying treatable condition are warranted (see ‘The well patient with abnormal liver function profile’ above). Recent data have indicated that the removal or cure of the underlying disease process can be followed by a reduction in hepatic fibrosis (e.g. after the cure of hepatitis C).
The Child-Pugh score (Table 24.6) provides a useful means of assessing the severity of cirrhosis and the prognosis. A Child-Pugh ‘A’ classification applies to a score of less than 7; ‘B’, a score between 7 and 9; and ‘C’, greater than 9. The 1-year survival of these classification groups is 100%, 80% and 45%, respectively. A higher score also indicates those more likely to succumb to complications during hepatic and no hepatic surgical procedures.

The MELD (Model for End Stage Liver Disease) score is a validated means of assessing the prognosis and survival of a patient with chronic liver disease that incorporates bilirubin, INR and creatinine values. This score can be used to prioritise the allocation of organs within a liver transplantation program (see Ch 25).
Maintaining adequate nutrition is vital to the management of patients with cirrhosis and chronic liver disease in general. These patients are often catabolic, requiring greater than usual energy intake. Nausea and dyspepsia are common and may reduce food intake and thus their nutritional status further. Dietary measures should be put in place to ensure a diet of adequate energy and protein intake with attention to micronutrient supplementation as needed (e.g. fat-soluble vitamins). The advice of an experienced dietician should be sought.
The Well Patient with Cirrhosis
Examination
Physical examination includes a search for signs of chronic liver disease (Box 24.1) and evidence of extrahepatic features of possible aetiological factors.
Investigation
Investigations are directed to (1) determining the aetiology of the cirrhosis (Table 24.4); and (2) assessing the severity of hepatic injury. When physical findings and investigations suggest cirrhosis, liver biopsy should be considered to establish its presence, to assess the activity of the fibroinflammatory process and to seek evidence of the aetiology. A past history of excessive ethanol consumption is often identified. In other cases, a specific aetiology can often be established by using special tests for autoantibodies, viral serology and metabolic disorders (Tables 24.1–24.3). Elevations in serum transaminase levels imply ongoing hepatocellular injury. Raised INR (prolonged prothrombin time) and hypoalbuminaemia suggest reduced hepatocellular synthetic function. Abdominal ultrasound scans with Doppler studies will provide evidence of portal hypertension (splenomegaly, portal flow and wave pattern), ascites and liver size (reflecting the remaining liver cell mass). Varices are excluded by upper endoscopy.
Management
Apart from any treatment associated with the underlying cause, the asymptomatic patient with cirrhosis needs to maintain an adequate diet. Sufficient kilojoules, protein and micronutrients should be ingested. A complex carbohydrate snack before retiring in the evenings could help prevent the patient from becoming catabolic overnight as the hepatic glycogen stores become depleted. Regular follow-up is necessary, specifically seeking evidence of evolving complications of cirrhosis (namely, portal hypertension and ascites, portal vein thrombosis, spontaneous bacterial peritonitis, hepatic encephalopathy, hepatorenal syndrome and hepatocellular carcinoma). See Chapter 25 for more details.
Complications of Portal Hypertension in the Patient with Chronic Liver Disease
Portal hypertension
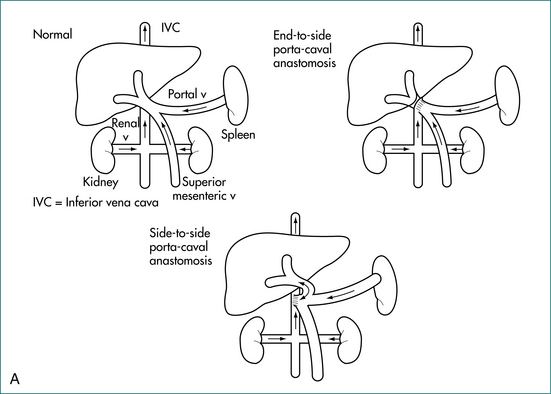
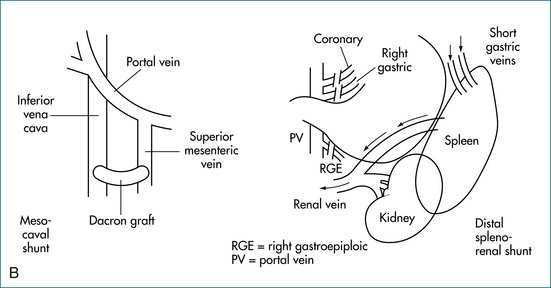
Portal and hepatic vascular disorders can also cause portal hypertension while leaving hepatocyte function relatively intact (Box 24.2). In these situations when liver cell function is maintained, portal systemic shunt surgery, rather than transplantation, may be appropriate for severe disease (Figs 24.2A and 24.2B). When vascular or thrombotic processes are found, an underlying cause for these should be sought and managed accordingly.
Acute upper gastrointestinal haemorrhage in patient with portal hypertension
History
Variceal haemorrhages are usually of large volume, presenting with fresh haematemesis, melaena and shock (Ch 10). Small amounts of overt blood loss without haemodynamic changes are unlikely to be variceal in origin. Bleeding from portal hypertensive gastropathy may be slow, presenting with iron deficiency anaemia, or sufficient to produce overt haematemesis and melaena. Similarly portal hypertensive enteropathy can cause slower gastrointestinal bleeding.
It must be noted that peptic ulceration and other causes of upper gastrointestinal haemorrhage are relatively common in cirrhotic patients and the history should be directed to consider these conditions as well (Ch 5).
Examination
Assessment of the physical signs of blood volume loss is vital (Ch 10). Signs of chronic liver disease (Box 24.1) support the possibility of cirrhosis and variceal bleeding. Splenomegaly suggests portal hypertension. In acute or chronic liver disease, the liver may be palpable and tender. The cirrhotic liver is likely to be small. Blood in the gastrointestinal tract can precipitate encephalopathy in cirrhotic patients. This should be sought and documented. There should be a search for signs of other complications of chronic liver disease including ascites and sepsis.
The chest should be carefully examined for evidence of aspiration, especially in alcoholic patients.
Investigations
Basic haematological investigations are required as with any gastrointestinal haemorrhage (Ch 10). When chronic liver disease is present, particular attention is paid to the factors of the Child-Pugh score (above). Electrolyte disturbances are not uncommon. Renal function must be checked. The source of the upper gastrointestinal haemorrhage is demonstrated by upper gastrointestinal endoscopy as soon as is practical.
Management
Significant upper gastrointestinal haemorrhage requires urgent resuscitation. Once this is achieved, endoscopy can be used to confirm and often control the source of bleeding. Acute variceal bleeding is usually controlled endoscopically by band ligation. The long-acting somatostatin analogue, octreotide, or terlipressin (triglycyl lysine vasopressin) provides an effective pharmacological means of managing acute variceal haemorrhage. These agents are often used to complement endoscopic therapy or to achieve control of blood loss while endoscopy is being arranged. Octreotide is given as a bolus dose of 50 μg intravenously, followed by an infusion of 50 μg/hour for at least 72 hours. When endoscopic therapy is unavailable or not possible, balloon tamponade applied at the cardio-oesophageal junction can be used to control bleeding from oesophageal varices for up to 24 hours. Gastric varices do not respond to endoscopic therapy as well as oesophageal varices, although injection therapy using cyanoacrylate or fibrin glue can be a successful intervention in those with bleeding gastric varices.
Apart from portal systemic shunting, surgical alternatives acutely include transaction of the oesophagus. In cirrhotic patients, an acute surgical shunt has a high mortality. The risk is directly related to the Child-Pugh score.
Liver transplantation should be considered in suitable cirrhotic patients in the long term. When liver cell function is well preserved (as in non-cirrhotic portal hypertension), portal systemic shunting may be viable alternative (Fig 24.2A and B).
Ascites in the Cirrhotic Patient with or without Renal Impairment
Examination
Signs of chronic liver disease (Box 24.1) are expected in patients presenting with ascites and cirrhosis. Peripheral oedema is often present. Other complications of chronic liver disease or of the underlying condition may be present. Their absence should raise the possibility of acute hepatic disease or non-cirrhotic portal hypertension. Alternately, there may be a non-hepatic cause for the ascites. Physical examination may identify other causes of ascites, including cardiac failure and malignant disease. A pleural effusion, particularly on the right, may be present. Umbilical hernias commonly occur in ascites patients and surgical repair in this circumstance carries a high mortality.
Investigation
The diagnosis of the cause of ascites and exclusion of spontaneous bacterial peritonitis by paracentesis (tapping the peritoneal fluid) is mandatory. The gradient of albumin concentration between the serum and the ascitic fluid is particularly useful. A gradient greater than or equal to 11 g/L strongly suggests portal hypertension (as with cirrhosis). On the other hand, a gradient less than 11 g/L suggests malignancy or infection. If the patient is known to have cirrhosis, a low serum–ascites albumin gradient raises the possibility of spontaneous bacterial peritonitis or the development of hepatocellular carcinoma (Ch 20).
Ascites, Fever and Pain: Spontaneous Bacterial Peritonitis
Investigation and mechanisms
Ascitic fluid must be sampled and examined in all patients with ascites and a recent decompensation (Ch 20); this should be done on admission regardless of clinical signs. Of all cirrhotic patients with ascites admitted to hospital, 12–15% will have spontaneous bacterial peritonitis. An ascitic polymorphonuclear leukocyte count greater than or equal to 250 cells/L is indicative of the condition. Ascitic fluid cultures should be collected and the samples inoculated into blood culture bottles, at the bedside. Cefotaxime is adequate antibiotic therapy in most cases. Outcomes may be improved by the addition of intravenous albumin: 1.5 g/kg of body weight at diagnosis, then 1 g/kg of body weight on the third day.
Hepatic Encephalopathy in the Cirrhotic Patient
History and examination
The clinical features and grades of hepatic encephalopathy are shown in Table 24.7 and its classification in Box 24.3.
| Grade | Clinical features |
|---|---|
| I | Personality and mood changes, disturbed sleep pattern, poor hand writing, fetor, asterixis |
| II | Mild disorientation, inappropriate behaviour, slurred speech, ataxia, hyporeflexia |
| III | Disoriented, amnesia, somnolent but rousable, incoherent speech, marked asterixis, hyper-reflexia, clonus, rigidity |
| IV |
HE = hepatic encephalopathy.
∗ Without recognised precipitating factors.
From Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the Working Party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002; 35:716–721.
Hepatic encephalopathy accompanying severe, acute liver disease carries a grave prognosis and is accompanied by the clinical features of acute disease. In chronic liver disease with appropriate clinical features (above), milder, overt long-term encephalopathy may be found. Insomnia can be an early feature. A reversal of normal sleep patterns (sleeping during daylight hours and waking at night) is suggestive of early encephalopathy. More profound encephalopathy accompanies acute episodes of decompensation (e.g. infection and gastrointestinal bleeding) or progression to end-stage liver failure. Drug effects (especially sedatives), electrolyte abnormalities (diuretics), constipation, dietary protein load and renal failure should also be considered as precipitants (Box 24.4). Blood ammonia levels are elevated but vary widely.
Liver Disease in Pregnancy
Hepatic diseases during pregnancy are generally managed as if the patient were not pregnant. Liver disease in pregnancy can conveniently be divided into disease that is incidental to the pregnancy, disease that is exacerbated by the pregnancy, and pregnancy-specific disorders (see Table 24.8). Severe chronic liver disease reduces fertility such that decompensated chronic liver disease is not common in pregnant women. Pregnancy does not preclude hepatic transplantation for acute hepatic failure. Furthermore, past hepatic transplantation does not prevent subsequent successful pregnancy.
| Disease | Cause | Comment |
|---|---|---|
| Incidental to pregnancy |
HELLP = haemolysis, elevated liver function tests and low platelets.
History
Patients presenting with liver disease in pregnancy require prompt assessment and close follow-up because severe life-threatening illness may rapidly follow a seemingly mild prodrome.
Nausea and vomiting with abdominal pain, especially in late pregnancy, indicate a need for complete and careful assessment for preeclampsia and acute fatty liver, especially if there is right upper quadrant pain. However, right upper quadrant pain at any stage in pregnancy could reflect a hepatitic process, biliary disease, a change in a hepatic mass lesion or vascular disease (Table 24.11).
Investigation
When acute fatty liver or severe preeclampsia is considered likely, the need for urgent intervention (delivery) is determined on the basis of the clinical assessment and basic blood test results. In less urgent circumstances, organ imaging with ultrasound may be used to exclude gallstones or hepatic mass lesions or, with Doppler studies, vascular disease (e.g. Budd-Chiari syndrome). Viral and autoimmune serology where the clinical setting is suggestive should also be considered.
Specific Hepatic Diseases
Viral hepatitis
Hepatitis B
Hepatitis B virus (HBV) is a hardy, highly infectious DNA virus. There is an incubation period of 30–180 days (mean: 60–90 days) (Fig 24.3). Acute hepatitis occurs in approximately 25% of those who acquire the virus, with approximately 1% developing fulminant hepatic failure. The majority of cases are asymptomatic and recover completely, unaware of the infection. In hepatitis B infection, liver injury is mediated by T-lymphocytes; the virus itself does not seem to cause hepatocyte death.
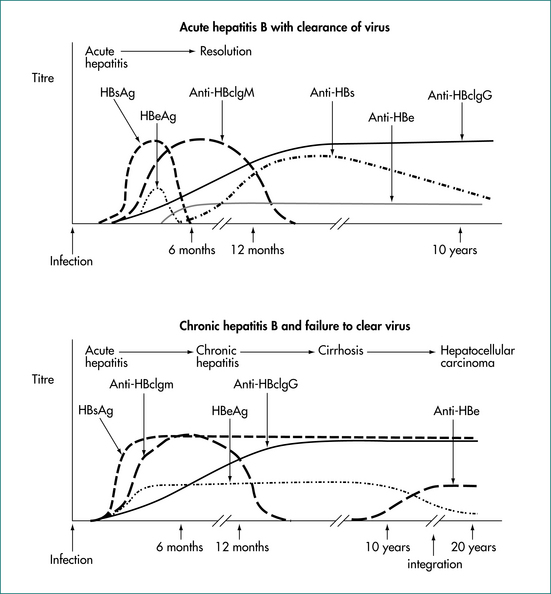
Figure 24.3 Hepatitis B serology in acute infection with viral clearance and chronic infection without viral clearance.
From the Australian Gastroenterology Institute.
The treatment of acute hepatitis B is supportive.
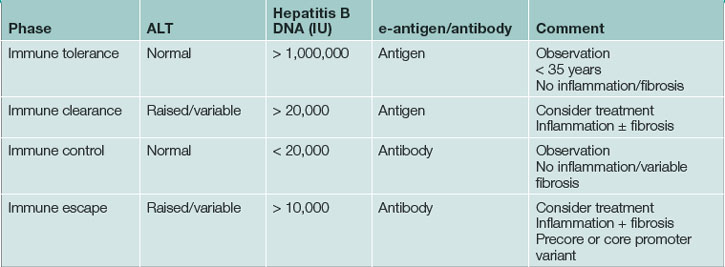
Five to 10% of adults who acquire hepatitis B progress to chronic infection (Figs 24.4 and 24.5). In these, the prognosis is dependent on the degree of liver injury and the duration of infection. Chronic infection is characterised by the persistence of hepatitis B surface antigen, and IgG antibodies to core protein with or without e-antigen. Chronic hepatitis B infection can progress through four phases: immune tolerance, immune clearance, immune control and immune escape. The management of patients depends on which phase they are in (Table 24.9). During active phases of infection, liver biopsy can be of great value in assessing the degree of inflammation and determining whether cirrhosis has developed.
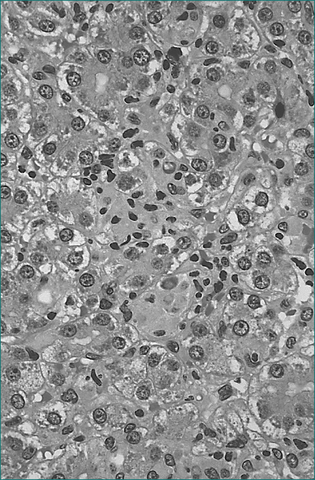
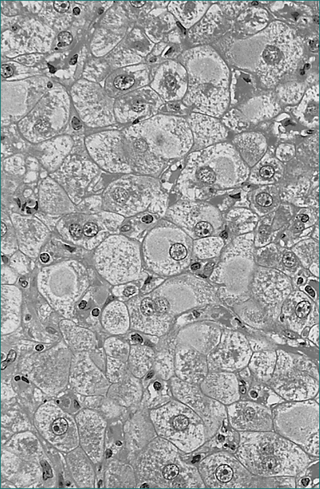
Eventually, chronic hepatitis B results in cirrhosis and/or hepatocellular carcinoma. Well-compensated, chronic hepatitis B liver disease has a 5-year survival of 84%. Five-year survival in decompensated chronic hepatitis B is only 14%. It is possible for a patient with chronic hepatitis B to spontaneously clear the virus as he or she mounts an immune response to the virus. This is usually associated with an episode of clinical hepatitis and is associated with loss of viral antigens and the development of antibodies. The tests used in the assessment of hepatitis B infection are outlined in Table 24.3.
The aim of treatment for chronic hepatitis B infection is to suppress viral replication (reduced hepatitis B DNA levels), thereby reducing liver injury as reflected by normalization of ALT. In hepatitis B e-antigen positive patients seroconversion is associated with improved prognosis.
Hepatitis C virus
Hepatitis C virus (HCV) is an RNA virus, which is thought to be directly cytopathic to the hepatocyte (Figs 24.6 and 24.7). Prior to routine screening for this virus, it was responsible for at least 90% of cases of post-transfusion non-A non-B hepatitis. The carrier rate in the general population is unclear, but is probably between 0.2% and 1%. The incubation period for acute hepatitis C is 15–160 days (mean: 50 days). In the majority of cases, the acquisition of hepatitis C virus is not associated with an acute symptomatic illness. Chronic infection is common, however, occurring in up to 80% of infected cases. Of those who acquire hepatitis C, approximately 10–15% develop severe liver disease, which can take decades to develop. Hepatic failure and hepatocellular carcinoma may then follow. Significant hepatitis or liver injury (on liver biopsy) can be associated with normal serum ALT levels. Conversely, some people with mild histological liver damage have serum ALT levels greater than two or three times the upper limit of normal. From this it is apparent that ALT measurements are of limited value in the assessment of the severity of hepatitis C virus infection.
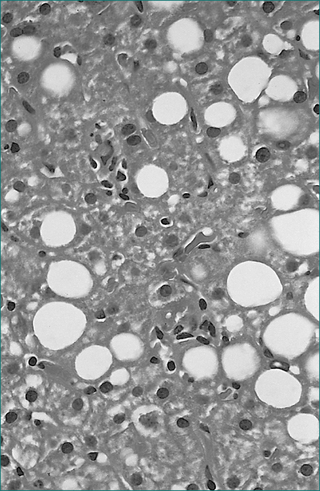
Figure 24.6 Viral hepatitis, chronic, type C. Mild microvesicular fatty change is usually present.
From Kanel GC, Korula K. Liver biopsy evaluation. Philadelphia: WB Saunders; 2000, with permission.
Hepatitis C virus appears to infect and replicate in both hepatocytes and mononuclear cells. Its antigenic structure is variable and appears able to escape immune surveillance. Furthermore, there are multiple genotypes (varieties) of the virus. Prior or current infection with hepatitis C virus does not protect an individual from acquiring further hepatitis C infections. One patient may harbour a number of varieties of hepatitis C virus. The genotype will influence the severity of the infection, the response to therapy and the infection’s prognosis.
HCV therapy failure is associated with excessive alcohol consumption, insulin resistance, obesity, treatment non-compliance, adverse events requiring dose reduction and insufficient duration of therapy. An individual’s genetic make-up may also influence his or her responsiveness to therapy.
Non-alcoholic fatty liver disease and steatohepatitis
Usually there is fatty infiltration of the liver on ultrasonography, with no other apparent cause for abnormal liver function test results. CT scan of the abdomen can also identify hepatic fatty infiltration, often with associated marked visceral fat deposition. In the typical case, liver biopsy is not necessary. However, if a biopsy is performed, this condition may have histological findings indistinguishable from alcoholic liver injury. Careful history taking and viral serology are required to clarify the diagnosis in uncertain cases. A carbohydrate-deficient transferrin test will be negative (Table 24.10), but no other biochemical features are diagnostically helpful. High BMI, age greater than 50 years, ALT greater than twice the upper limit of normal and triglycerides greater than 1.7 μmol/L have been associated with a greater risk of hepatic fibrosis in non-alcoholic fatty liver disease.
| Test | Comment |
|---|---|
| Erythrocyte mean cell volume | Round macrocytes, low specificity |
| Gamma-glutamyl transpeptidase | Enzyme-induced by ethanol, level related to long-term prognosis, sensitive but low specificity |
| Urate, high density lipoprotein and triclycerides | Elevated, low specificity |
| Carbohydrate-deficient transferrin | Sensitive and specific, reflects regular excessive ethanol consumption within previous 2 weeks |
Bariatric surgery has been found to improve non-alcoholic fatty liver disease as well as other complications of morbid obesity.
Alcoholic liver disease
Although alcohol is a direct hepatotoxin, a patient’s nutritional state and genetic make-up will influence the degree of hepatic injury caused by excessive intake of this agent. Alcohol-induced liver injury is generally classified into fatty liver, alcoholic hepatitis and cirrhosis (Fig 24.8). The mainstay of therapy is abstinence from alcohol.

Corticosteroids may improve short-term survival in those with severe acute alcoholic hepatitis and jaundice, a high INR or prolonged encephalopathy but no gastrointestinal bleeding. However, the available data do not support the use of corticosteroids in alcoholic hepatitis. In these patients enteral nutritional supplementation may also improve survival. Furthermore, pentoxifylline (400 mg t.d.s.) has been shown to improve survival in severe acute alcoholic hepatitis, by reducing the risk of developing the hepatorenal syndrome. Antioxidants have been shown not to be of benefit.
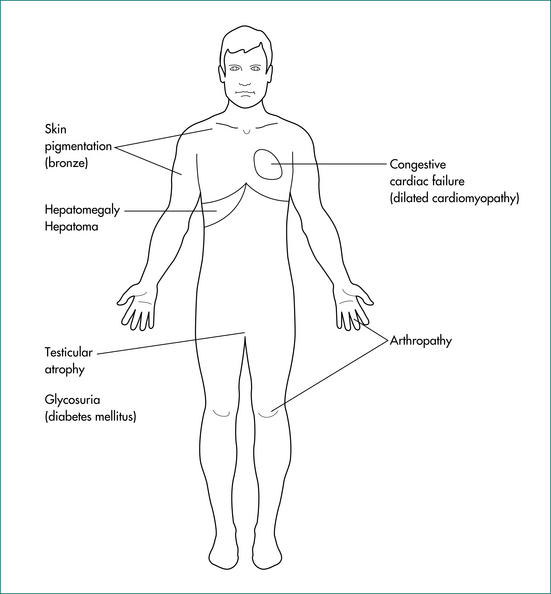
Haemochromatosis
Clinically apparent advanced haemochromatosis presents between 50 and 70 years of age (Fig 24.9). The male to female ratio is between 4:1 and 5:1, depending on the study that measures it. Presenting features may include unexplained hepatomegaly, fatigue, loss of libido and hypogonadism (from pituitary iron deposition), cardiac dysfunction from cardiomyopathy, skin pigmentation (from melatonin and iron) and arthralgias with arthropathy. The arthropathy may involve the second and third metacarpophalangeal joints as well as proximal interphalangeal joints, the knees, wrists and hips. The liver is usually the first organ damaged, and patients may present with cirrhosis or hepatocellular carcinoma. However, a diagnosis of haemochromatosis is not excluded by normal liver function test results.
Iron studies (serum iron, transferrin and ferritin) are used to determine the likelihood of haemochromatosis. In this situation, the transferrin saturation and the serum ferritin concentration are of most value, especially the former. A normal transferring saturation makes haemochromatosis unlikely.
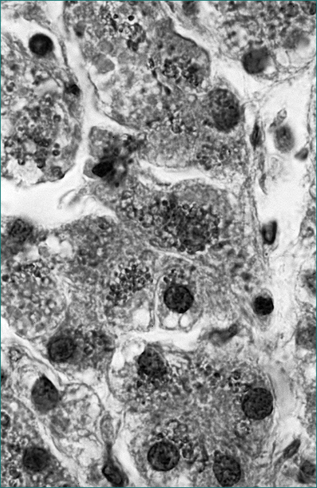
The diagnosis of haemochromatosis when in doubt is established by the measurement of the tissue iron concentration on liver biopsy. The hepatic iron index (HII) is calculated by dividing the liver iron concentration (μmol/g dry weight) by the patient’s age in years. An HII >2 is indicative of haemochromatosis. This is complemented by hepatic histology with staining for iron (Fig 24.10). In haemochromatosis, excess iron occurs predominantly within hepatocytes (as opposed to Kuppfer cells in cases of secondary iron overload). A key role of liver biopsy is also to identify the presence of cirrhosis with its implications for long-term prognosis.
Wilson’s disease
Wilson’s disease occurs in approximately 1 in 40,000 people and is the consequence of mutation of the ATP7B gene on chromosome 13, leading to defective copper trafficking and thereby the progressive accumulation of copper with ongoing liver injury through childhood into adult life. More than 60 polymorphisms have been identified, making genetic testing unhelpful in clinical practice. ATP7B is a trans-Golgi membrane hepatocyte copper-transporting ATPase involved in biliary copper excretion.
Idiopathic, autoimmune chronic hepatitis
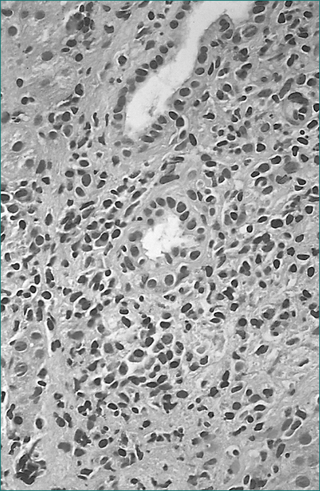
Autoantibodies, commonly anti-actin (anti-smooth muscle) and antinuclear antibodies, are present. There is a prominent polyclonal hypergammaglobulinaemia. In an often aggressive variant that usually affects young females, the smooth muscle antibody and antinuclear antibody can be absent while an anti-liver/kidney microtonal antibody (anti-LKM1) is found.
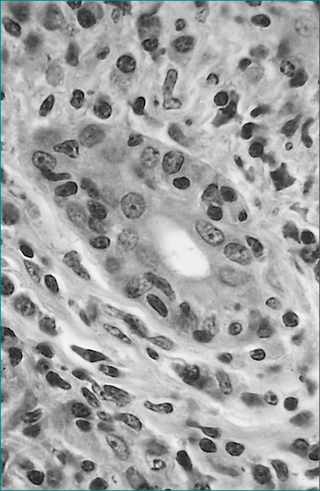
On liver biopsy in aggressive disease, the chronic inflammatory infiltrate, including plasma cells, typically expands the portal areas and extends into the liver lobule, causing erosion of the limiting plate (interface necrosis, Fig 24.11). Varying degrees of fibrosis may be present. Fibrotic linkages between portal tracts or cirrhosis are markers of aggressive disease.
Primary biliary cirrhosis
ALP and, to a lesser extent, serum transaminase levels are usually elevated in patients with primary biliary cirrhosis. AMA is present in the serum in 95% of cases. Its presence is highly suggestive of the diagnosis. IgM levels are often elevated. Differentiation between AMA-negative primary biliary cirrhosis and IACH may be difficult. Smooth muscle antibody and antinuclear antibody may be present in the former, and low titres of AMA can occur in IACH. A true overlap syndrome can occur, and will present features of both conditions. Furthermore, on liver biopsy, primary biliary cirrhosis can share features with IACH, although differentiation is usually possible (Fig 24.12). If uncertainty remains, a therapeutic trial of corticosteroids is likely to induce an improvement in IACH but not in primary biliary cirrhosis.
Adverse prognostic signs in primary biliary cirrhosis include a high serum bilirubin concentration (over 100 μmol/L), a low serum albumin concentration, ascites, gastrointestinal bleeding, advanced age, cirrhosis or central cholestasis on liver biopsy and low serum IgM levels.
Primary sclerosing cholangitis
The diagnosis of primary sclerosing cholangitis relies on cholangiography identifying focal biliary strictures with associated beadlike dilatations. Liver biopsy is of limited diagnostic value (Fig 24.13). Because of the presence of portal inflammation, there may be difficulty in differentiating primary sclerosing cholangitis from other chronic inflammatory conditions on liver biopsy.
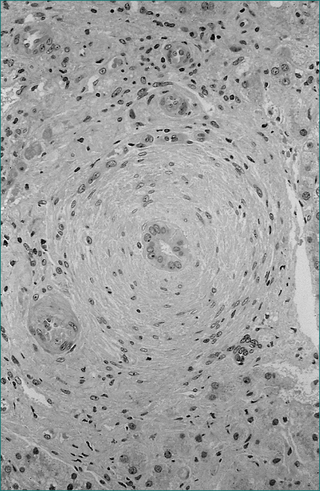
Primary cholangiocarcinoma complicates 10–20% of cases of primary sclerosing cholangitis. Sudden rapid progression of pruritus, jaundice and weight loss suggest malignancy. However, this diagnosis can be difficult and may not be made until postmortem. Brush cytology on ERCP provides a positive result in only 20% of patients with cholangiocarcinoma. If transplant takes place, the outcome is poor when primary cholangiocarcinoma is present.
Drugs and the liver
A careful history of medication use is vital in the assessment of the patient with abnormal liver function test results. Toxic or idiosyncratic adverse reactions may occur with many therapeutic agents (Table 24.11). Immune mechanisms are responsible for many of these reactions. The contraceptive pill may be associated with a number of liver diseases including hepatic adenoma (rare—usually single), hepatocellular carcinoma (very rare), peliosis hepatis (large blood-filled cavities), Budd-Chiari syndrome (see below), cholestasis and cholesterol gallstones (because of more lithogenic bile).
| Liver disease | Clinicopathological features | Drug examples |
|---|---|---|
| 1. Altered LFTs without liver disease | ||
| Due to microsomal enzyme induction | No clinical features; raised GGT and ALP; ground glass cells | Phenytoin, warfarin |
| Hyperbilirubinaemia | Jaundice rare | Rifampicin |
| 2. Hepatocellular necrosis: hepatocellular necrosis with varying inflammatory change, ALT > 5 × normal | ||
| Focal necrosis | Lobular hepatitis, resembles viral hepatitis | Isoniazid, cloxacillin, halothane (mild) |
| Bridging necrosis | Traces of necrosis connect adjacent portal and/or central veins | Isoniazid, alpha-methyldopa |
| Zonal necrosis | Well-demarcated zone of necrotic hepatocytes; less conspicuous inflammation | Paracetamol, halothane (severe) |
| Massive necrosis | Entire hepatic acini necrotic; fulminant hepatic failure | Halothane (fatal), valproic acid NSAIDs |
| 3. Fatty liver | ||
| Acute fatty change | Usually microvesicular; clinical features of hepatitis, liver failure | Tetracycline, valproic acid, corticosteroids, NSAIDs, L-asparaginase |
| Steatohepatitis | Resembles alcoholic hepatitis histologically; clinical features of chronic liver disease | Perhexiline maleate, amiodarone |
| 4. Granulomatous reactions | ||
| Non-caseating granulomas | Varying lobular hepatitis, cholestasis or pericholangitis; usually mixed LFT results, may be hepatocellular | Allopurinol, chlorpromazine, chlorothiazide, isoniazid, phenytoin, sulfonamides |
| 5. Acute cholestasis | ||
| Cholestasis without hepatitis | Cholestasis, no inflammation; pruritus with minimal systemic symptoms ALP > 2 × normal | OCS, anabolic androgens |
| Cholestasis with hepatitis | Cholestasis with portal and lobular inflammation; systemic symptoms; ALT elevated as well as ALP | Chlorpromazine, erythromycin estolate, flucloxacillin |
| Cholestasis with bile duct injury | Destructive lesions of bile duct epithelium; clinically similar to acute cholangitis | Flucloxacillin, chlorpromazine |
| 6. Chronic cholestasis: cholestasis > 3 months | ||
| Vanishing bile duct syndrome | Paucity of small bile ducts, varying fibrosis; clinically resembles primary biliary cirrhosis; AMA negative | Chlorpromazine, amitriptyline, flucloxacillin |
| 7. Chronic parenchymal liver disease: abnormalities present > 3 months | ||
| Chronic hepatitis | Periportal and/or bridging necrosis, fibrosis or cirrhosis; clinical and biochemical features of chronic liver disease; liver failure may occur | Alpha-methyldopa, nitrofurantoin, dantrolene |
| Fibrosis and cirrhosis | Portal hypertension; LFTs often normal | Methotrexate, hypervitaminosis A |
| 8. Vascular disorders | ||
| Sinusoidal dilatation | Isolated finding or adjacent to tumours; hepatomegaly is the only clinical feature | OCS |
| Peliosis hepatis | Destructive lesions of sinusoids resulting in blood-filled lakes | Anabolic androgens |
| Non-cirrhotic portal hypertension | Portal and perisinusoidal fibrosis; splenomegaly, oesophageal varices | Vinyl chloride, azathioprine, hypervitaminosis A |
| Hepatic venous outflow obstruction | Budd-Chiari syndrome, veno-occlusive disease and overlap syndromes | 6-thioguanine, OCS, pyrrolizidine alkaloids |
| Nodular regenerative hyperplasia | Regeneration nodules with minimal fibrosis; portal hypertension | Azathioprine, dactinomycin |
| 9. Hepatic tumours | ||
| Focal nodular hyperplasia | Hamartoma, associated vascularity attributable to oestrogens | OCS |
| Hepatocellular adenoma | Benign neoplasm of hepatocytes | OCS, androgens |
| Hepatocellular carcinoma | Primary liver cancer (hepatoma) | OCS, androgens |
| Rarer carcinomas | Fibrolamellar variant, hepatoblastoma, cholangiocarcinoma, cholangiohepatocellular tumours, carcinosarcoma | OCS |
| Angiosarcoma | Malignant tumour, possibly arises from sinusoidal lining cells | Arsenic, vinyl chloride, thorium dioxide |
ALP = alkaline phosphatase; ALT = alanine aminotransferase; AMA = anti-mitochondrial antibody; LFTs = liver function tests; NSAIDs = non-steroidal anti-inflammatory drugs; OCS = oral contraceptive steroids.
From Farrell G. Drug induced liver disease. Edinburgh: Churchill Livingstone; 1994, with permission.
Vascular and perfusion disorders of the liver
Veno-occlusive disease of the liver is associated with obstruction of the terminal hepatic venules. Precipitating events include bone marrow transplantation and exposure to chemotherapeutic agents and certain plant alkaloids. Clinically, there is right upper quadrant tenderness, hepatomegaly, ascites and jaundice. Progression to liver failure may occur.
Budd-Chiari syndrome involves thrombosis of the hepatic veins. It presents with pain, hepatomegaly and ascites (Ch 4). There is portal hypertension and loss of hepatocyte function. There are acute and chronic forms of this syndrome. Progression to liver failure is not unusual.
Hepatocellular carcinoma
Both the incidence of and mortality from hepatocellular carcinoma are increasing in Australia and other Western countries, primarily as a consequence of chronic viral hepatitis. Treatment options depend on the size and location of the tumour and as such early detection is important. The diagnosis of hepatocellular carcinoma can be made on good quality imaging, such as triple phase CT scan or MRI, without the need for biopsy.
When there is a single hepatocellular carcinoma (less than 5 cm) or up to three lesions less than 3 cm in diameter, liver transplantation can be potentially curative and should be considered. If transplantation is contraindicated local ablative therapy (radiofrequency ablation or ethanol injection) can be of value. With advanced hepatocellular carcinoma transarterial chemoembolisation and for those with more severe underlying liver disease, oral sorefenib can slow the progression of the disease. Patients with terminal disease should be managed symptomatically (see Ch 28).
Benign tumours
Benign hepatic mass lesions are often identified incidentally during investigation (imaging) for unrelated symptoms. Adenomas, focal nodular hyperplasia and focal fatty change produce ‘masses’. Normal liver function test results are expected. Intervention is not required for focal nodular hyperplasia or fatty change. Hepatic adenoma may bleed (50% risk), may be difficult to differentiate from low-grade hepatocellular carcinoma and can progress to carcinoma. Imaging is often non-specific in the differentiation between focal nodular hyperplasia and adenoma. If an adenoma is suspected, segmental resection is usually indicated (Ch 26).
Haemangiomas and hepatic cysts are common benign conditions often found incidentally during hepatic imaging. Simple fluid-filled cysts need no further investigation. If a lesion is typical of haemangioma on ultrasound or computed tomography, again no further tests should be necessary. If, however, there remains uncertainty as to the nature of the lesion, a nuclear-labelled red cell scan or MRI can be helpful. Angiography is not usually necessary. Small lesions require no treatment but large symptomatic lesions may need segmental resection (Ch 26).
Acute fatty liver of pregnancy
Abdominal ultrasound scans have poor sensitivity in identifying acute fatty liver but are of use in excluding Budd-Chiari syndrome and gallstones. If necessary, a fine-needle aspiration biopsy will confirm the diagnosis. However, once the clinical diagnosis is made, performing a biopsy could waste valuable time and in so doing adversely affect the outcome.
Key Points
Amin J., Law M.G., Bartlett M., et al. Causes of death after diagnosis of hepatitis b or hepatitis C infection: a large community-based linkage study. Lancet. 2006;48:938-945.
Bambha K., Kim W.R., Talwalker J., et al. Incidence, clinical spectrum and outcome of primary sclerosing cholangitis in a United States community. Gastroenterol. 2003;125:1364-1369.
Black M. Drug hepatoltoxicity. Clin Liver Dis. 2003;7:295-512.
Buster E., Hansen B., Lau G., et al. Factors that predict response of patients with hepatitis B e antigen-positive chronic hepatitis B to peginterferon-alfa. Gastroenterology. 2009;137:2002-2009.
Cabre E., Rodriguez Iglesias P., Caballeria J., et al. Short and long term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology. 2000;32:36-42.
Chitturi S., et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373-379.
Deugnier Y., Brissot P., Loreal O. Iron and the liver: update 2008. J Hepatol. 2008;48:S113-S123.
Farrell G.C., Larter C.Z. Non-alcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(suppl 1):S99-S112.
Ferenci P., Lockwood A., Mullen K., et al. Hepatic Encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the Working Party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
Green R.M., Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123:1367-2384.
Hay J.E. Liver disease in pregnancy. Hepatology. 2008;47:1067-1076.
Hézode C., Forestier N., Dusheiko G., et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. NEJM. 2009;360(18):1839-1850.
Lok A.S.F., MaMahon B.J. Chronic hepatitis B. Hepatology. 2007;45:507-539.
Munoz S.J. The hepatorenal syndrome. Med Clin N Am.. 2008;92:813-837.
Ratziu V., Giral P., Charlotte F., et al. Liver fibrosis in overweight patients. Gastroenterol. 2000;118:1117-1123.
Rauch A., Kutalik Z., Descombes P., et al. Genetic variation in IL28B is associated with hepatitis C and treatment failure. Gastroenterology. 2010;138:1338-1345.
Roberts E.A., Schilsky M.L. Practice guideline on Wilson’s disease. Hepatology. 2003;37:1475-1492.
Runyon B.A. Care of patients with ascites. N Eng J Med. 1994;330:337-342.
Sherlock S. Alcoholic liver disease. Lancet. 1995;345:227-229.
Sorrell M.F., Belongia Edward A., et al. National Institutes of Health consensus development conference statement: management of Hepatitis B. Ann Intern Med. 2009;150:104-110.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
24 Abnormal liver function test results
Case
A 45-year-old male was found to have abnormal liver function tests on routine investigations performed for an insurance examination. Serum alanine aminotransferase level was elevated at 105 IU/L. International Normalized Ratio (INR), albumin and bilirubin were normal. The patient also had a history of hypertension controlled on antihypertensive therapy and an elevated fasting serum triglyceride concentration. His father had type 2 diabetes, but there was no family history of liver disease. An abdominal ultrasound examination demonstrated changes consistent with fatty liver, but no other abnormality. Further investigations showed no evidence of autoimmune, viral or inherited liver disease. His ferritin was mildly elevated, but with normal transferrin saturation and no mutation of the haemochromatosis (HFE) gene. Physical examination revealed him to be overweight, but otherwise was unremarkable. The patient was advised to reduce his weight and exercise regularly. Under the guidance of a dietician a 5% weight loss was achieved with an improvement in alanine aminotransferase levels. Following that he failed to maintain the intervention, gained weight and discontinued medical review. Ten years later he again was found to have abnormal liver tests but also INR 1.6, albumin 28 gm/L and bilirubin 25 μg/L. He had become diabetic with symptomatic ischaemic heart disease. Abdominal ultrasound on this occasion showed a small irregular liver with splenomegaly, consistent with cirrhosis and an apparent mass in the liver. On a triple phase computed tomography (CT) scan of the liver, the 2-cm mass lesion was typical of a hepatocellular carcinoma and was managed surgically.
An Approach to the Patient with Liver Disease
Liver disease will present as one of a limited number of clinical syndromes. Each syndrome may be caused by a number of specific disease processes. A diagnosis is achieved by paying attention to historical features and physical findings (Box 24.1) and the results of laboratory and other investigations. Only then can specific treatment for the cause of the liver abnormality be planned. Therapy, in many instances, is directed to the management of the clinical problem in addition to treatment directed to the underlying condition.
Box 24.1 Symptoms and signs of liver disease
Signs of poor hepatocellular synthetic function
Bruising, peripheral oedema (reflecting depleted coagulation factors and albumin levels)
In this chapter, liver function tests and their clinical relevance are discussed first because abnormalities are commonly detected in asymptomatic individuals. Next, the common clinical presentations of liver disease are covered. Finally, important liver diseases are described.
Liver Function Test Interpretation
Bilirubin
Elevated serum bilirubin levels may occur in all forms of liver disease. However, it must be remembered that a jaundiced patient does not necessarily have significant hepatic disease. Serum levels of bilirubin reflect production, hepatic uptake, processing (conjugation) and secretion. The physiology of bilirubin and an approach to diagnosis and management of jaundice is described in Chapter 23.
Tests reflecting hepatic function
Serum albumin
In chronic liver disease, low serum albumin concentrations represent a major prognostic indicator (see ‘Child-Pugh score’ later in this chapter). This can reflect the absence of sufficient functioning hepatocytes to maintain albumin production. Furthermore, malnutrition can be a significant contributor to hypoalbuminaemia in patients with chronic liver disease. Alcohol consumption can reduce albumin synthesis. Serum albumin concentrations may also fall without a reduction in hepatic synthesis when the volume of distribution of albumin is expanded by ascites and oedema. As albumin has a half-life of 17–26 days, low levels usually reflect chronic rather than acute hepatitic dysfunction.
Tests of Liver Injury
Transaminases
In the presence of malignant disease, increases in transaminases often indicate hepatic metastases.
Aspartate aminotransferase
Aspartate aminotransferase (AST) is an enzyme found in liver, skeletal muscle, myocardium, kidney, pancreas and erythrocytes. Damage to any of these cell types will result in an elevation of serum AST. ALT levels are usually higher than AST levels in viral liver injury. When liver disease is present, and the AST level is more than twice that of ALT, alcoholic liver disease must be considered (see later). In the presence of cirrhosis, the diagnostic value of the AST:ALT ratio is lost. The mitochondrial isoenzyme of AST is especially elevated in the presence of alcoholic liver injury.
Cholestasis versus hepatocellular disease
Liver function patterns may be used to guide the discrimination between cholestasis (e.g. due to stones or tumour obstructing the large bile ducts) and hepatocellular disease (e.g. cirrhosis or chronic hepatitis). These patterns can be used only as a broad guide and should not be relied on alone. However, if cholestasis is suspected, then looking for evidence of duct obstruction (e.g. by ultrasound) is the next step, while if hepatocellular disease is suspected, then a search for liver disease markers (e.g. virology, immunological tests) is the next step (see Table 23.1 in Ch 23).
Other tests useful in hepatic assessment
A marked elevation (over 1000 U/L) of alpha-fetoprotein is most often due to hepatocellular carcinoma. This test has been used for screening an individual with chronic hepatitis B for the development of hepatocellular carcinoma, having a sensitivity of 70%. It is of limited value, however, because the sensitivity falls to 30% in non-hepatitis B hepatocellular carcinoma. Milder elevations occur in both acute and chronic liver disease of many causes (Ch 25).
Autoantibodies are helpful in diagnosing certain forms of liver disease (Table 24.1). Serum concentration of caeruloplasmin and iron studies are important tools in the diagnosis of Wilson’s disease and haemochromatosis, respectively (Table 24.2), but are also altered by liver injury unrelated to either of these conditions. Tests for viral hepatitis are also important (Table 24.3).
| Test | Condition | Comment |
|---|---|---|
| Antinuclear antibody (ANA) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Smooth muscle antibody (SMA) (anti-actin) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Anti-liver kidney microsomal antibody (anti-LKM1) | Autoimmune chronic hepatitis | Diagnosis requires liver biopsy |
| Antimitochondrial antibody (AMA) | Primary biliary cirrhosis(95%) | Diagnosis requires liver biopsy |
| Antineutrophil cytoplasmic antibody (pANCA) | Primary sclerosing cholangitis | Diagnosis requires magnetic resonance cholangiopancreatography or endoscopic retrograde cholangiopancreatography |
Table 24.2 Tests for metabolic disorders affecting the liver
| Test | Condition | Comment |
|---|---|---|
| Iron studies | Haemochromatosis | See text |
| Copper and caeruloplasmin | Wilson’s disease | See text |
| Alpha-1 antitrypsin level and phenotype | Alpha-1 antitrypsin deficiency | Heterozygotes may have abnormal liver function tests without significant liver disease |
| Thyroid function tests | Hypothyroidism | Fatty liver |
| Blood glucose level | Diabetes mellitus | Fatty liver, haemochromatosis |
| Insulin/C-peptide | Insulin resistance | Fatty liver, metabolic syndrome |
| Cholesterol and triglycerides | Primary and secondary hyperlipidaemia | Fatty liver, alcoholic liver disease |
Table 24.3 Tests for viral hepatitis
| Test | Meaning of a positive result | Comment |
|---|---|---|
| 1. Tests for hepatitis A virus (HAV) | ||
| Anti-HAV IgM | Recent acquisition of HAV | Acute hepatic illness likely to be HAV |
| Anti-HAV IgG | Past infection/vaccination | Immunity |
| 2. Tests for hepatitis B virus (HBV) | ||
| HBsAg (surface ag) | Present/chronic infection | Structural component of virus |
| Anti-HBsAg (surface ab) | Past infection/vaccination | Immunity |
| Anti-HBc IgM (core ab) | Recent acquisition of HBV | The test for acute HBV infection |
| HBeAg (e ag) | Marker of viral replication | High risk of infectivity |
| Anti-HBeAg (e ab) | Suggests no viral replication | Unlikely to be infectious |
| HBV DNA | Presence of complete virus | High risk of infectivity |
| 3. Tests for hepatitis C virus (HCV) | ||
| Anti-HCV | Exposure to HCV | Interpret in conjunction with other clinical and laboratory data |
| HCV RNA by PCR | Presence of virus | – |
| HCV genotyping | – | Treatment planning and response |
| 4. Tests for hepatitis D virus (HDV) | ||
| Anti-HDV IgG/IgM | Exposure to HDV | Acute or chronic HDV |
| Delta antigen | HDV present | Acute or chronic HDV |
| 5. Tests for hepatitis E virus (HEV) | ||
| Anti-HEV IgM | Recent acquisition of HEV | Acute hepatitic illness likely to be HEV |
| 6. Tests for other organisms | ||
| Cytomegalovirus (CMV) IgM | Recent acquisition of CMV | Acute hepatitic illness likely to be CMV |
| Epstein-Barr virus (EBV) IgM | Recent acquisition of EBV | Acute hepatitic illness likely to be EBV |
| Anti-HIV | HIV-AIDS | Opportunistic hepatobiliary infections |
| Toxoplasmosis serology | – | Consider toxoplasmosis |
| Q fever serology | – | Consider Q fever |
ag = antigen; ab = antibody; PCR = polymerase chain reaction.
Liver biopsy
Histological and, on occasion, chemical analysis and microbiological examination of the liver are essential in the diagnosis of many hepatic disorders. The usual technique for obtaining a liver biopsy involves a percutaneous approach through a lower intercostal space in the right midaxillary line. Ultrasound scanning or CT can be used to optimise the needle entry point or target specific abnormalities. Haemorrhage from the biopsy wound to the liver or injury to adjacent organs are the major risks. These may be significant in approximately 1% of biopsies. The risk of haemorrhage is minimised by ensuring a platelet count greater than 80 × 109/L (80,000/mm3) and an INR less than 1.2. When coagulopathy or gross ascites prevents a percutaneous biopsy, a transjugular approach can be used.
The significance of liver biopsy features is considered in relation to specific disorders later in this chapter. The extent, nature and activity of inflammatory and fibrotic processes are of particular concern. The presence of fibro-inflammatory changes is more likely to be related to a progression to cirrhosis. The absence of such changes carries a better prognosis. Sampling error can be a problem as the tissue sample represents only a tiny proportion of the liver mass and the disease process may not be uniform across the organ. Cirrhosis can be missed in 10–20% of cases, depending on the size and number of samples taken.
The Well Patient with Abnormal Liver Function Profile
History
A history of diabetes mellitus, thyroid disease and high lipid levels should be sought; these together with obesity are associated with hepatic steatosis. The features of the metabolic syndrome with rising body weight and reduced physical activity are common in those with non-alcoholic fatty liver disease, especially when there is a family history of type 2 diabetes.
There is a possibility of genetic predisposition to alcoholic liver disease.
In the otherwise well patient, a history of abdominal pain (e.g. biliary pain: Ch 4), loss of appetite or weight (Ch 17), or a change in the colour of stools or urine (Ch 23) needs to be sought but is almost invariably absent.
Examination
Patients with abnormal liver test results must be regarded as having a liver disorder and thus signs of chronic liver disease should be sought because cirrhosis may be completely asymptomatic (Fig 24.1). The patient’s weight and height should be measured during the examination to allow evaluation of the body mass index (weight in kilograms divided by the square of the height in metres: kg/m2). Truncal obesity is common and waist measurement should be taken.
Figure 24.1 Signs of chronic liver disease.
From Sherlock S, Dooley J. Diseases of the liver and biliary system. 11th edn. Oxford: Blackwell; 2002, with permission.
In the usual patient presenting with no history of symptoms and no awareness of liver problems, it is likely that there will be no signs of liver disease. A minor degree of hepatomegaly should be expected in patients with alcohol-related fatty liver or non-alcoholic fatty liver disease (Ch 17). Mild splenomegaly may also be found.
Investigation
Liver imaging is of value (Ch 23). Abdominal ultrasound scans can provide information about the size and shape of the liver and spleen. Often any mass lesions present can also be identified. Ultrasonography will often detect fatty liver. Abdominal CT will provide information on the presence or absence of space-occupying lesions while, in addition, providing insight into liver density, which may be increased in haemochromatosis. Magnetic resonance imaging will usually add little to the information provided by a CT scan but can be of value in certain circumstances, such as in the assessment of mass lesions and of the biliary tract.
Tests for evidence of viral hepatitis (Table 24.3), autoimmune liver disease (Table 24.1) and other causes of chronic liver disease (iron or copper overload, and other inherited liver diseases) (Table 24.2) should be done if hepatocellular disease is possible. If the diagnosis remains unclear and the prognosis of concern, a liver biopsy is required.
Chronic hepatitis is an ongoing inflammatory process. The causes are listed in Table 24.4. If fibrosis follows as part of the healing process consequent to this inflammation, progression to cirrhosis and eventual liver failure is possible. For this reason, potentially controllable chronic hepatitis needs to be identified and treated before irreversible injury has occurred.
Table 24.4 Important causes of chronic hepatitis and cirrhosis
| Chronic hepatitis | Cirrhosis |
|---|---|
Evidence of continuing hepatic inflammation based on a persistent elevation of serum transaminases (6 months or more) is usually required to entertain the diagnosis of a chronic hepatitis. However, it can take many months for serum transaminases to return to normal levels following some acute hepatic illnesses. Once the presence of chronic hepatitis is considered likely, establish the aetiology of the illness, the extent of liver damage and the activity of the inflammatory process. These latter two features can be assessed only by the histological examination of a liver biopsy.
The Sick Patient with Jaundice: Acute Liver Disease
Acute hepatitis
Examination
The patient can appear relatively well or profoundly ill. Jaundice may or may not be present. The liver size is noted. It will often be enlarged and tender. There may be splenomegaly. Look for evidence of an underlying condition (e.g. signs of alcohol excess). Complications (e.g. hepatic encephalopathy) may supervene. Look for signs of chronic liver disease (Fig 24.1).
Investigation
Serum transaminase levels are elevated, usually more than five times the upper limit of normal. In viral hepatitis, transaminase levels of 1000 U/L and over are not unusual. Moderate elevations of alkaline phosphatase also occur. Serum albumin and INR usually remain normal. Specific blood tests are required to diagnose viral illnesses, including viral hepatitis (A, B, C, D and E), cytomegalovirus and Epstein-Barr virus, toxoplasmosis, Q fever and autoimmune disease (Table 24.3). Paracetamol levels should be determined and laboratory evidence of alcohol excess sought. These may be relatively low in patients with chronic, but toxic paracetamol consumption. Abdominal ultrasound scans are useful to document the size of the liver and spleen and to exclude biliary disease.
Management
Alcoholic hepatitis is managed supportively; in certain circumstances specific interventions may be of value. Ethanol is withdrawn, and vitamin (especially thiamine) and nutritional supplementation should be given (enteral feeding may provide a survival benefit). Corticosteroids (prednisone, 40 mg daily for 28 days) may provide a survival benefit when there is encephalopathy (see below) and no gastrointestinal haemorrhage. Propylthiouracil may be of benefit in less severe cases of alcoholic hepatitis, but is rarely used. Abstinence remains the mainstay of therapy.
When gallstone disease is found to be the cause of an acute hepatitic illness, cholecystectomy may be indicated. Choledocholithiasis may require endoscopic clearance of the bile duct (Ch 23). Acute autoimmune hepatitis is managed with corticosteroids (e.g. prednisone, 30–60 mg daily, then taper).
Acute Liver Failure
Assessment and management
Examination is directed towards assessing the possible aetiology and evolving complications of hepatic failure (outlined below). Serial liver function tests can be useful, but a falling ALT level may reflect either recovery or progression to massive hepatic necrosis. A temperature above 38°C or below 36°C, a pulse rate above 90 beats per minute and/or a white cell count above 12,000 or below 4,000 are associated with a greater mortality. The prognosis, and therefore need for liver transplantation, of a patient in acute liver failure can be assessed using the Kings College Criteria (Table 24.5).
Table 24.5 Kings College Criteria for listing of a patient in acute liver failure (ALF) for liver transplantation
| Type | Criteria |
|---|---|
| Paracetamol-induced ALF | Arterial pH < 7.30 after fluid resuscitation or all of: |
INR = International Normalized Ratio.
From O’Grady JG, Alexander GJM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
The patient with fulminant hepatic failure needs to be admitted directly to a unit capable of offering liver transplantation. Admission to intensive care is required with careful management of cerebral oedema, gastrointestinal bleeding, infection and renal failure.
Prognosis
Liver transplantation is a therapeutic option. The decision to transplant and the timing of transplantation are difficult. However, 1-year survival rates of approximately 70% can be achieved. This compares favourably with mortality rates from fulminant hepatic failure without transplantation of up to 90% although with optimal intensive care unit management 30–40% survival can be achieved. N-acetylcysteine may also improve overall survival and transplant-free survival in patients with non-paracetamol-related acute liver failure, especially if encephalopathy is limited to grade I or II.
Patients Presenting with Cirrhosis and its Complications
Cirrhosis represents the non-specific end-stage of hepatic disease that has disrupted the structural organisation of the liver. The presence of cirrhosis is established on a liver biopsy. Fibrosis or scarring surrounding regenerative nodules of hepatocytes is its hallmark. Although cirrhosis is a histological diagnosis, its presence is clinically suspected by finding signs of chronic liver disease and portal hypertension (Box 24.1) or on high quality imaging. Symptomatic liver disease and eventually liver failure occur as the result of impaired hepatocellular function within the distorted architecture of the regenerative nodules and, with progression, eventual loss of an effective liver cell mass. Distortion of the hepatic circulation contributes to portal hypertension and inefficient/ineffective perfusion of parenchymal cells. Portal systemic shunts may develop, allowing blood from the gastrointestinal tract to flow directly to the systemic circulation.
Cirrhosis should be considered as either compensated, with no signs of complications and a relatively good prognosis, or decompensated (Ch 25). Those with compensated cirrhosis are likely to be asymptomatic and the diagnosis may be made incidentally during the investigation of an unrelated condition. In this situation, counselling regarding diet, ethanol consumption and the identification and management of any underlying treatable condition are warranted (see ‘The well patient with abnormal liver function profile’ above). Recent data have indicated that the removal or cure of the underlying disease process can be followed by a reduction in hepatic fibrosis (e.g. after the cure of hepatitis C).
The Child-Pugh score (Table 24.6) provides a useful means of assessing the severity of cirrhosis and the prognosis. A Child-Pugh ‘A’ classification applies to a score of less than 7; ‘B’, a score between 7 and 9; and ‘C’, greater than 9. The 1-year survival of these classification groups is 100%, 80% and 45%, respectively. A higher score also indicates those more likely to succumb to complications during hepatic and no hepatic surgical procedures.
The MELD (Model for End Stage Liver Disease) score is a validated means of assessing the prognosis and survival of a patient with chronic liver disease that incorporates bilirubin, INR and creatinine values. This score can be used to prioritise the allocation of organs within a liver transplantation program (see Ch 25).
[/not-level-membership-for-gastroenterology-and-hepatology-category]
