[level-membership-for-emergency-medicine-category]
section 8 Neurology
8.1 Headache
Introduction
Headache is a common ailment that is often due to a combination of physical and psychological factors. The vast majority are benign and self-limiting and are managed by patients in the community. Only a very small proportion of patients experiencing headache attend emergency departments (ED) for treatment. The challenges are to distinguish potentially life-threatening causes from the more benign, and to effectively manage the pain of headache.
Pathophysiology
The structures in the head capable of producing headache are limited. They include:
The pathological processes that may cause headache are:
The pathophysiological causes of headache are summarized in Table 8.1.1.
| Extracranial | Intracranial | |
|---|---|---|
| Tension/traction | Muscular headache | Intracranial tumour |
| ‘Tension headache’ | Cerebral abscess | |
| Intracranial haematoma | ||
| Vascular | Migraine | Severe hypertension |
| Inflammatory | Temporal arteritis | Meningitis |
| Sinusitis | Subarachnoid haemorrhage | |
| Otitis media | ||
| Mastoiditis | ||
| Tooth abscess | ||
| Neuralgia |
Assessment
Headache patterns
Some headaches have ‘classic’ clinical features: these are listed in Table 8.1.2. It must be remembered that, as with all diseases, there is a spectrum of presenting features and the absence of the classic features does not rule out a particular diagnosis. Every patient must be assessed on their merits and, if symptoms persist without reasonable explanation, further investigation should be undertaken.
Table 8.1.2 Classic clinical complexes and cause of headache
| Preceded by an aura |  |
|
| Throbbing unilateral headache, nausea | Migraine | |
| Family history | ||
| Sudden onset |  |
|
| Severe occipital headache; ‘like a blow’ | Subarachnoid haemorrhage | |
| Worst headache ever | ||
| Throbbing/constant frontal headache |  |
|
| Worse with cough, leaning forward | Sinusitis | |
| Recent URTI | ||
| Pain on percussion of sinuses | ||
| Paroxysmal, fleeting pain |  |
|
| Distribution of a nerve | Neuralgia | |
| Trigger manoeuvres cause pain | ||
| Hyperalgesia of nerve distribution | ||
| Unilateral with superimposed stabbing |  |
|
| Claudication on chewing | Temporal arteritis | |
| Associated malaise, myalgia | ||
| Tender artery with reduced pulsation | ||
| Persistent, deep-seated headache |  |
|
| Increasing duration and intensity | Tumour: primary or secondary | |
| Worse in morning | ||
| Aching in character | ||
| Acute, generalized headache |  |
|
| Fever, nausea and vomiting | Meningitis | |
| Altered level of consciousness | ||
| Neck stiffness +/– rash | ||
| Unilateral, aching, related to eye |  |
|
| Nausea and vomiting | Glaucoma | |
| Raised intraocular pressure | ||
| Aching, facial region |  |
|
| Worse at night | Dental cause | |
| Tooth sensitive to heat, pressure |
Tension headache
The pathological basis of tension headaches remains unclear, but increased tension of the neck or cranial muscles is a prominent feature. A family history of headaches is common, and there is an association with an injury in childhood or adolescence. The most common precipitants are stress and alteration in sleep patterns.
Migraine
Treatment
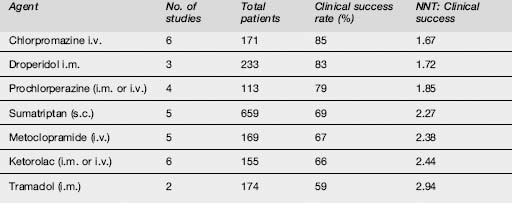
The effectiveness of commonly used agents is summarized in Table 8.1.3. Dosing and administration are summarized in Table 8.1.4. At present the most effective agents seem to be the phenothiazines (chlorpromazine, prochlorperazine, droperidol and possibly haloperidol) and the triptans, each of which has achieved > 70% efficacy in a number of studies. Note that triptans are contraindicated in patients with a history of ischaemic heart disease, uncontrolled hypertension or with the concomitant use of ergot preparations.
| Agent | Drug dosing/administration |
|---|---|
| Chlorpromazine i.m. | 12.5 mg intravenously, repeated every 20 minutes as needed to a maximum dose of 37.5 mg, accompanied by 1 L normal saline over 1 hour to avoid hypotension OR 25 mg in 1 L normal saline over 1 hour, repeated if necessary |
| Droperidol (i.m. or i.v.) | 2.5 mg |
| Prochlorperazine (i.m. or i.v.) | 10 mg/12.5 mg (depending on packaging) |
| Sumatriptan (s.c., i.n.) | 6 mg SC, 20 mg i.n. |
| Metoclopramide (i.v.) | 10–20 mg |
| Ketorolac (i.m. or i.v.) | 30 mg i.v.; 60 mg i.m. |
| Tramadol (i.m.) | 100 mg |
Pethidine is not indicated for the treatment of migraine. Its reported effectiveness is only 56%, it has a high rate of rebound headache and it carries a risk of dependence. In two small RCTs haloperidol administered as 5 mg in 500 mL normal saline was reported to give significant pain relief in more than 80% of patients. Lignocaine (lidocaine) has been shown to be no more effective than placebo. The data on dihydroergotamine are difficult to interpret because it is often used in combination with other agents, e.g. metoclopramide; however, it has also been shown to be less effective than chlorpromazine and sumatriptan in acute treatment, and to have a high rate of unpleasant side effects. There are insufficient data to assess the effectiveness of CGRP receptor antagonists. Sodium valproate has also shown moderate effectiveness in small studies, but there are insufficient data to draw a valid conclusion. The efficacy of intravenous magnesium sulphate (1 or 2 mg) remains unclear. It was shown in a small placebo-controlled trial to be effective, but in another study the combination of magnesium with metoclopramide was less effective than metoclopramide and placebo.
Trigeminal neuralgia
Australian and New Zealand College of Anaesthetists and Faculty of Pain Medicine. Acute pain management: scientific evidence, 2nd edn. Canberra: National Health and Medical Council (Australia), 2005.
Friedman BW, Greenwald P, Bania TC, et al. Randomized trial of IV dexamethasone for acute migraine in the emergency department. Neurology. 2007. (Epub ahead of print)
Kelly AM. Specific pain syndromes: Headache. In: Mace S, Ducharme J, Murphy M, editors. Pain management and procedural sedation in the emergency department. New York: McGraw-Hill, 2006.
Kelly AM, Kerr D, Clooney M. Impact of oral dexamethasone versus placebo after ED treatment of migraine with phenothiazines on the rate of recurrent headache: a randomized controlled trial. Emergency Medicine Journal. 2008;25:26-29.
8.2 Stroke and transient ischaemic attacks
Pathophysiology
Ischaemic strokes
These are the results of several pathological processes (Table 8.2.1):
| Ischaemic stroke |
Haemorrhagic stroke
Haemorrhagic stroke is the result of vessel rupture into the surrounding intracerebral tissue or subarachnoid space. Subarachnoid haemorrhage is the subject of a separate chapter in this book (see Chapter 8.3). The neurological defect associated with an intracerebral haemorrhage is the consequence of direct brain injury, secondary occlusion of nearby vessels, reduced cerebral perfusion caused by associated raised intracranial pressure, and cerebral herniation. The causes of intracerebral haemorrhage (ICH) include:
Prevention
This particularly applies to ischaemic strokes. Non-modifiable risk factors for stroke include:
Primary prevention
Hypertension is the most important modifiable risk factor. The benefit of antihypertensive treatment in stroke prevention has been well shown. The other major risk factors for atherosclerosis and its complications – diabetes, smoking and hypercholesterolaemia – often contribute to increased stroke risk. These should be managed according to standard guidelines. The most important cardiac risk factor for TIA and stroke is atrial fibrillation, both chronic and paroxysmal. Warfarin is recommended to prevent cardioembolism, except in unsuitable patients. Those with contraindications to warfarin should initially receive aspirin. Other major cardiac risk factors include endocarditis, mitral stenosis, prosthetic heart valves, recent myocardial infarction and left ventricular aneurysm. Less common risk factors include atrial myxoma, a patent foramen ovale and cardiomyopathies.
A carotid bruit or carotid stenosis found in an otherwise asymptomatic patient is associated with an increased stroke risk. However, the role of carotid endarterectomy in these patients is controversial. In a highly selected patient group, the asymptomatic carotid atherosclerosis study (ACAS)1 showed a small but significant benefit in reduction of stroke or death at 5 years following surgery for angiographically proven stenosis >60% compared to medical therapy. The benefit was much lower than that achieved in symptomatic carotid stenosis shown in the North American Symptomatic Carotid Endarterectomy Study (NASCET 2),2 and can only be achieved with low perioperative mortality and stroke rates.
Ischaemic stroke syndromes
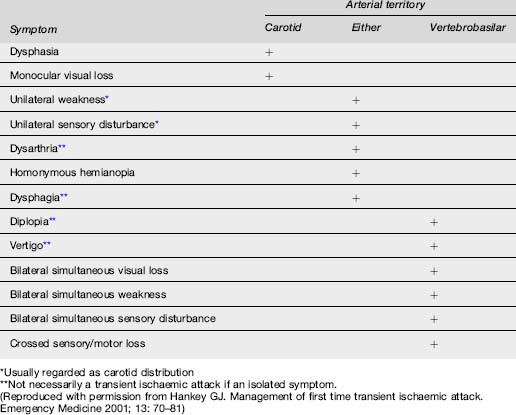
The symptoms and signs of stroke or TIA correspond to the area of the brain affected by ischaemia or haemorrhage (Table 8.2.2).
Anterior circulation ischaemia
The anterior circulation supplies blood to 80% of the brain and consists of the ICA and its branches, principally the ophthalmic, middle cerebral and anterior cerebral arteries. Hence this system supplies the optic nerve, retina, frontoparietal and most of the temporal lobes. Ischaemic injury involving the anterior cerebral circulation commonly has its origins in atherothrombotic disease of the ICA. Atherosclerosis of this artery usually affects the proximal 2 cm, just distal to the division of the common carotid artery. Advanced lesions may be the source of embolism to other parts of the anterior circulation, or cause severe stenosis with resultant hypoperfusion distally if there is inadequate collateral supply via the Circle of Willis. This is usually manifest by signs and symptoms in the middle cerebral artery (MCA) territory (Table 8.2.3). Less commonly, lesions of the intracranial ICA and MCA may cause similar clinical features.
| Homonymous hemianopia |
| Contralateral hemiplegia affecting face and arm more than leg |
| Contralateral hemisensory loss |
| Dysphasias with dominant hemispheric involvement (usually left) |
| Spatial neglect and dressing apraxia with non-dominant hemispheric involvement. |
Embolism to the ophthalmic artery or its branches causes monocular visual symptoms of blurring, loss of vision and field defects. When transient, this is referred to as amaurosis fugax, or transient monocular blindness.
Posterior circulation ischaemia
Specific brainstem syndromes include:
Pre-hospital care
A pre-hospital evaluation tool that has been developed and validated is the Cincinatti Prehospital Stroke Scale or FAST: F –facial movements, A –arm movements, S – speech, and T – test.3 Pre-hospital personnel who identify patients with acute onset of neurological deficits identified using this simple scale can then notify the ED in order to mobilize appropriate staff and forewarn Radiology, so as to expedite assessment and imaging, particularly if thrombolysis is being considered.
Clinical evaluation in the ED
Examination
Central nervous system
This includes assessing the level of consciousness, pupil size and reactivity, extent of neurological deficit, presence of neck stiffness and funduscopy for signs of papilloedema and retinal haemorrhage. Quantifying the neurological deficit using a stroke scale such as the 42-point National Institute of Health Stroke Scale (NIHSS)4 is useful in the initial assessment, and also for monitoring progress in a more objective way than clinical description alone. Strokes with a NIHSS score >22 are classified as severe.
In the case of TIA all clinical signs may have resolved. The average TIA lasts less than 15 minutes.
Differential diagnosis (Table 8.2.4)
| Intracranial space-occupying lesion |
| Subdural haematoma |
| Brain tumour |
| Brain abscess |
| Postictal neurological deficit – Todd’s paresis |
| Head injury |
| Encephalitis |
| Metabolic or drug-induced encephalopathy |
| Hypoglycaemia, hyponatraemia etc. |
| Wernicke–Korsakoff syndrome |
| Drug toxicity |
| Hypertensive encephalopathy |
| Multiple sclerosis |
| Migraine |
| Peripheral nerve lesions |
| Functional |
Complications of stroke
Investigations
General
TIAs and non-disabling strokes should be evaluated similarly in order to promptly diagnose and manage a potentially treatable process that might lead to a subsequent major stroke. The risk of a stroke following a TIA is now appreciated to be much higher than previously thought, and may be as high as 30% in the first week. The ABCD stroke risk score from TIA has been developed and validated to evaluate the risk of a stroke in the first 7 days following a TIA.5 This has the potential to guide the urgency of investigations, such as carotid ultrasound, required to determine the underlying causes of the TIA. The scoring system is outlined in Table 8.2.5. In patients with an ABCD score <4 there is minimal short-term risk of stroke. With scores of 4, 5 and 6 the risk is 2.2%, 16.5% and 35%, respectively. Other patient groups are at increased risk of stroke independent of the ABCD scoring system. These include patients with diabetes, multiple TIAs within a short period, and patients with a probable or proven cardioembolic source. Diabetes has been incorporated in the recently published ABCD2 scoring system (see Further Reading).
| ABCD | Risk factor | Score |
|---|---|---|
| Age | Below 60 | 0 |
| Above 60 | 1 | |
| Blood pressure | BP > systolic 140 mmHg, and/or diastolic 90 mmHg | 1 |
| Clinical | Unilateral weakness of face, arm, hand or leg | 2 |
| Speech disturbance without weakness | 1 | |
| Duration | Symptoms lasted >60 min | 2 |
| Symptoms lasted 10–60 min | 1 | |
| Symptoms lasted < 10 min | 0 |
(From Rothwell PM, Giles MF, Flassmann E, et al. A simple score (ABCD) to identify individuals at high risk of stroke after transient ischaemic attack. Lancet 2005; 366: 29–36)
Brain imaging
A head CT or MRI scan is indicated in most patients with TIA to exclude lesions that occasionally mimic TIA, such as subdural haematomas and brain tumours. CT, and more particularly MRI, may show areas of infarction which match the symptoms of an ischaemic event that, on clinical grounds, has completely resolved. CT is less sensitive than MRI in detecting posterior territory ischaemic lesions, particularly in the brain stem. In TIAs due to atrial fibrillation or another known cardiac source, brain imaging to exclude ICH is necessary prior to commencing anticoagulation. The exception is in cases of emboli from endocarditis in which anticoagulation is contraindicated owing to the increased risk of secondary ICH.
Imaging vessels
Imaging in stroke
Brain imaging
Recent studies have suggested that MRI is as accurate as CT in diagnosing acute ICH.6 This is significant, as it means that, where facilities are immediately available, CT may be bypassed in acute stroke and MRI can be used to both to exclude ICH and to scan for ischaemia/infarction with DWI. As already mentioned, other modalities such PWI and MRA/MRV may also give important diagnostic information and influence treatment decisions. However, MRI may not be feasible in a significant number of stroke patients, due either to standard contraindications to MRI or other factors such as haemodynamic instability, impaired consciousness or vomiting and agitation. In one study the proportion of patients intolerant of MRI was 1:10.
Treatment
TIAs
Ischaemic stroke
A more active approach to the acute management of ischaemic stroke is seen as having the potential to improve neurological outcomes. The ED is the place where these important treatment decisions will largely be made. Most patients with a stroke will require hospital admission for further evaluation and treatment, as well as for observation and possible rehabilitation. Studies of stroke units show that patients benefit from being under the care of physicians with expertise in stroke and a multidisciplinary team that can manage all aspects of their care.10
Intracerebral haemorrhage (ICH)
Medical management
Primary ICH is a medical emergency with a high mortality of between 35% and 50%, with half of these deaths occurring in the first 2 days. There is also a very high risk of dependency. Haematomas can expand rapidly, and there is a significant risk of early neurological deterioration and increasing intracranial pressure (ICP). Treatment of raised ICP in a setting of ICH involves a range of modalities similar to those used in head trauma. These include elevation of the head of the bed, analgesia, sedation, an osmotic diuretic such as mannitol and hypertonic saline, hyperventilation, drainage of CSF via ventricular catheter, and neuromuscular paralysis.
There is no good evidence regarding the management of hypertension in the setting of ICH sufficient to make firm recommendations. Guidelines have been published, but treatment should be individualized and take place in consultation with neurology/neurosurgery/intensive care specialists.18 Sudden falls in blood pressure and hypotension should be avoided, as they may aggravate cerebral ischaemia in the setting of raised ICP, which is often associated with ICH.
Early studies of the use of recombinant factor VIIa have shown promise when administered within 3 hours of stroke onset, showing a significant reduction in haematoma expansion and improved mortality.19 Steroids are not indicated in ICH. Anticonvulsant prophylaxis is common practice.
Surgical management
1 Executive Committee of the Asymptomatic Carotid Atherosclerosis Study. Endarterectomy for asymptomatic carotid artery stenosis. Journal of the American Medical Association. 1995;273:1421-1428.
2 North American Symptomatic Carotid Endarterectomy Trial Collaborators (NASCET). Beneficial effects of carotid endarterectomy in symptomatic patients with high grade carotid stenosis. New England Journal of Medicine. 1991;325:445-453.
3 Kouthari RU, Panciolli A, Liu T, et al. Cincinatti Pre Hospital Stroke Scale: reproducibility and validity. Annals of Emergency Medicine. 1999;33:373-378.
4 Goldstein LB, Samsa GP. Reliability of the National Institute of Health Stroke Scale: extension to non-neurologists in the context of a clinical trial. Stroke. 1997;28:307-310.
5 Rothwell PM, Giles MF, Flassmann E, et al. A simple score (ABCD) to identify individuals at high risk of stroke after transient ischaemic attack. Lancet. 2005;366:29-36.
6 Kidwell CS, Chalela JA, Saver JL, et al. Comparison of MRI and CT for detection of acute intracerebral hemorrhage. Journal of the American Medical Association. 2004;292:1823-1834.
7 The ESPRIT Study Group. Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT). Lancet. 2006;367:1665-1673.
8 CAPRIE Steering Committee. A randomized, blinded, control trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet. 1996;348:1329-1339.
9 The Stroke Prevention by Aggressive Reduction in Cholesterol levels (SPARCL) Investigators. High dose atorvastatin after stroke or transient ischaemic attack. New England Journal of Medicine. 2006;355:549-559.
10 Duffy BK, Phillips PA, Davis SM, et al. Evidence based care and outcomes of acute stroke managed in hospital specialty units. Medical Journal Australia. 2003;178:318-323.
11 International Stroke Trial Collaborative Group. The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among. 19435 patients with acute ischaemic stroke. Lancet. 1997;349:1569-1581.
12 CAST (Chinese Acute Stroke Trial) Collaborative Group. CAST: randomised placebo controlled trial of early aspirin use in. 20000 patients with acute ischaemic stroke. Lancet. 1997;349:1641-1649.
13 Hoffman J. Tissue plasminogen activator (tPA) for acute ischaemic stroke: why has so much been made of so little? Medical Journal Australia. 2003;179:333-334.
14 National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group (NINDS). Tissue plasminogen activator for acute ischaemic stroke. New England Journal of Medicine. 1995;333:1581-1587.
15 Albers GW. Intravenous tissue-type plasminogen activator for treatment of acute stroke: the Standard Treatment with Alteplase to Reverse Stroke (STARS) study. Journal of the American Medical Association. 2000;83:1145-1150.
16 Katzan IL, Furlan AJ, Lloyd LE, et al. Use of tissue type plasminogen activator for acute ischaemic stroke: the Cleveland Area Experience. Journal of the American Medical Association. 2000;283:1511-1518.
17 Wahlgren N, Ahmed N, Davalos A, et al. Thrombolysis with alteplase for acute ischaemic stroke in the Safe Implementation of Thrombolysis in Stroke (SITS-MOST): an observational study. Lancet. 2007;369:275-282.
18 Broderick JP, Connolly S, Feldman E, et al. AHA/ASA Guidelines for the management of spontaneous intracerebral hemorrhage in adults. ICH. Stroke. 2007;38:2001.
19 Mayer S, Brun MC, Begtiup K, et al. Recombinant Factor 7a for acute ICH. New England Journal of Medicine. 2005;352:777-785.
20 Vahedi K, Hofmijer J, Juettler E, et al. Early decompressive surgery in malignant infarction of the middle cerebral artery: a pooled analysis of three randomised controlled trials. Lancet Neurology. 2007;6:215-222.
Rothwell PM. Atherothrombosis and ischaemic stroke. British Medical Journal. 2007;334:379-381.
Alberts MJ, Latchaw RE, Selman WR, et al. Brain Attack Coalition. Recommendations for comprehensive stroke centres: a consensus statement of the Brain Attack Coalition. Stroke. 2005;36:1597-1616.
Libman RB, Wirkowski E, Alvir J. Conditions that mimic stroke in the emergency department. Archives of Neurology. 1995;52:1119-1122.
Schriger DL, Kalafut M, Starkman S, et al. Cranial computed tomography interpretation in acute stroke. Physician accuracy in determining eligibility for thrombolytic therapy. Journal of the American Medical Association. 1998;279:1293-1297.
Adams HPJr, Zoppo G, Alberts MJ, et al. AHA/ASA Guidelines for the early management of adults with ischaemic stroke. Stroke. 2007;38:1655.
on behalf of the MATCH InvestigatorsDiener H, Bogousslavsky J, Brass LM, et al. Acetylsalicilic acid on a background of clopidogrel in high risk patients randomized after recent stroke or transient ischaemic attack: The Match trial results. Lancet. 2004;364:331-337.
Sacco RL, Adams R, Albers G, et al. AHA/ASA Guidelines for the prevention of stroke with ischaemic stroke or transient ischaemic attack. Stroke. 2006;37:577.
Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of a score to predict very early stroke risk after transient ischaemic attack. Lancet. 2007;369:283-292.
8.3 Subarachnoid haemorrhage
Pathology and epidemiology
SAH is the presence of extravasated blood within the subarachnoid space. The incidence is 5–7 per 100 000 patient-years, but is significantly higher (around 20 per 100 000) in Japan and Finland, for reasons that are unclear. Although incidence increases with age, about half of those affected are under 55, the condition being most common in the 40–60 age group. Excluding head trauma, which remains the most common cause, non-traumatic or spontaneous SAH results from rupture of a cerebral aneurysm in approximately 85% of cases, non-aneurysmal perimesencephalic haemorrhage in 10%, and the remaining 5% from other rare causes including rupture of mycotic aneurysms, intracranial arterial dissection, aterio-venous malformations, vasculities, central venous thrombosis, bleeding diatheses, tumours and drugs such as cocaine, amphetamines and anticoagulants.
Clinical features
History
The history is critical to the diagnosis of SAH:
Examination
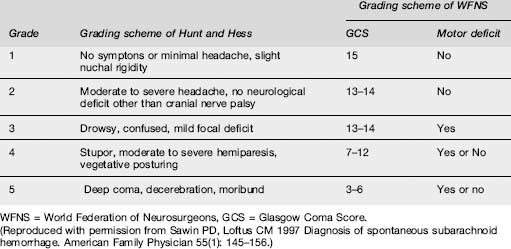
Patients are categorized into clinical grades from I to V, according to their conscious state and neurological deficit. Two grading schemes, that of Hunt and Hess and that of the World Federation of Neurosurgeons, which is preferred, are depicted in Table 8.3.1. The higher the score, the worse the prognosis.
Investigations
Imaging
A brain CT scan without contrast is the initial investigation of choice. In the first 24 hours after haemorrhage it can demonstrate the presence of subarachnoid blood in more than 95% of cases. (Fig. 8.3.1). The sensitivity, however, decreases with time owing to the rapid clearance of blood, with only 80% of scans positive at 3 days and 50% positive at 1 week. CT will also demonstrate the site and extent of the haemorrhage, indicate the possible location of the aneurysm, and demonstrate the presence of hydrocephalus and other pathological changes.
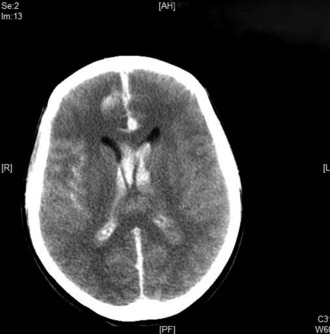
Lumbar puncture
It is important to measure the opening pressure when performing a lumbar puncture, as CSF pressure may be elevated in SAH or in other conditions such as intracranial venous thrombosis or pseudotumour cerebri, or low in spontaneous intracranial hypotension.
Complications
Early complications
Management
General measures
Conclusion
Al-Shahi R, White PM, Davenport RJ, et al. Subarachnoid haemorrhage. British Medical Journal. 2006;333:235-240.
de Gans K, Nieuwkamp DJ, Rinkel GJ, et al. Timing of aneurysmal surgery in subarachnoid hemorrhage: a systematic review of the literature. Neurosurgery. 2002;50:336-340.
Dorhout Mees SM, Rinkel GJE, Vermeulen M, van Gijn J. Calcium antagonists for aneurysmal subarachnoid haemorrhage, 1999. Cochrane Database of Systematic Reviews, (4): CD000277. DOI: 10.1002/14651858.CD000277.pub3
Dorhout Mees SM, van den Bergh WM, et al. Antiplatelet therapy for aneurysmal subarachnoid haemorrhage, 2007. Cochrane Database of Systematic Reviews (4): CD006184. DOI: 10.1002/14651858.CD006184.pub2
Edlow JA, Caplan LR. Primary care: Avoiding pitfalls in the diagnosis of subarachnoid hemorrhage. New England Journal of Medicine. 2000;342:29-36.
Naval NS, Stevens RD, Mirski MA. Controversies in the management of subarachnoid haemorrhage. Critical Care Medicine. 2006;34:511-524.
Roos YBWEM, Rinkel GJE, Vermeulen M, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage, 1998. Cochrane Database of Systematic Reviews (4): CD001245. DOI: 10.1002/14651858.CD001245
Sawin PD, Loftus CM. Diagnosis of spontaneous subarachnoid hemorrhage. American Family Physician. 1997;55:145-156.
Suarez JI, Tarr RW, Selman WR. Current concepts: Aneurysmal subarachnoid haemorrhage. New England Medical Journal. 2006;354:387-398.
van Gijn J, Kerr RS, Rinkel GJE. Subarachoid haemorrhage. Lancet. 2007;369:306-318.
8.4 Altered conscious state
Pathophysiology
Numerous scales have been proposed to define consciousness but the one that has found universal acceptance is the Glasgow Coma (or Responsiveness) Scale (GCS) (Table 8.4.1). Initially described in 1974 for the assessment of traumatic head injuries, 25 years of experience have shown that the scale can also be used in non-traumatic situations to provide a structured assessment of an individual’s conscious state at various points in time, and also to monitor progress. Trends provided by repeated measurements of the GCS give clinicians an objective measure to monitor a patient’s deterioration or improvement in response to therapy. In quantifying and standardizing the various responses, the GCS has enabled clinicians worldwide to compare data and therapies. In the spectrum from full awareness to unrousable, coma or unconsciousness is arbitrarily defined as a GCS ≤8.
| The GCS is scored between 3 and 15, 3 being the worst and 15 the best. It is composed of three parameters: Best Eye Response, Best Verbal Response, Best Motor Response, as given below. |
Differential diagnoses
There are several well known mnemonics to assist in remembering the rather diverse list. Some are listed in Table 8.4.2. However, the long list of apparently disparate causes can be divided pathophysiologically into structural insults and metabolic insults.
| T | Trauma |
| I | Infection |
| P | Psychogenic |
| (P) | (Porphyria) |
| S | Seizure |
| Syncope | |
| Space-occupying lesion | |
| A | Alcohol and other toxins |
| E | Endocrinopathy |
| Encephalopathy | |
| Electrolyte disturbances | |
| I | Insulin – Diabetes |
| O | Oxygen: Hypoxia of any cause |
| Opiates | |
| U | Uraemia including Hypertension |
| C | erebral |
| O | verdose |
| M | etabolic |
| A | sphyxia and other A ssociations |
Metabolic insults are usually due to systemic pathology that affects primarily the forebrain, although direct depression of the brain stem may also occur. There are seldom lateralizing signs. The solution to the problem is the correction of the underlying metabolic impairment. Naturally, as in all clinical practice, there are no absolute distinctions. Uncorrected, any of the metabolic causes can eventually cause cerebral oedema and herniation, leading thence to brainstem compression with lateralizing signs and death. Table 8.4.3 lists the more common and important causes of an altered conscious state.
Table 8.4.3 Causes of alteration in conscious state
Clinical assessment
Primary survey
A bedside glucose determination may identify clinical or biochemical hypoglycaemia, which should be treated with glucose. There is no evidence that 50 mL of intravenous 50% dextrose will cause harm even in an already hyperglycaemic patient, and a case could be made for routinely administering glucose to any patient with an altered conscious state if a bedside glucose estimation is not readily available.
Secondary survey
Examination
Pupillary findings and eye signs may also be useful to differentiate metabolic and structural insults, and more importantly to detect incipient uncal herniation. Intact oculocephalic reflexes and preservation of the ‘doll’s eyes’ response indicates an intact medial longitudinal fasciculus and by default an intact brain stem, suggesting a metabolic cause for coma (Table 8.4.4). There are four pairs of nuclei governing ocular movements, and they are spread between the superior and inferior midbrain and the pons. The pattern of ocular movement dysfunction can be used to pinpoint the site of a brainstem lesion (Table 8.4.4). Likewise, specific testing of the oculovestibular reflex and the cranial nerve examination can be used to precisely locate a brainstem lesion but is of limited use in the emergency setting except as a predictor of herniation (Table 8.4.5).
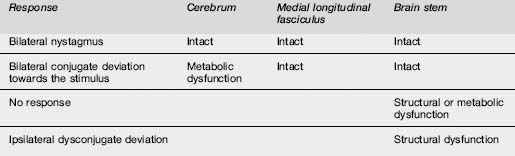
Table 8.4.5 Patterns of dysfunction in various parameters determined by the site of the structural or metabolic insult
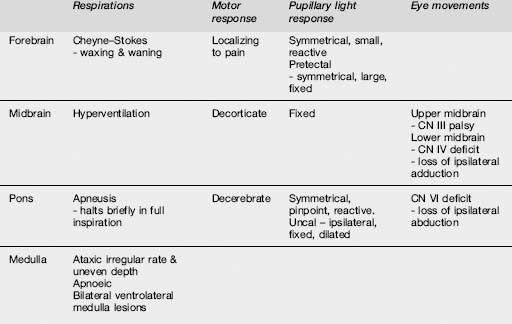
More generally, skin examination may reveal needle tracks suggestive of drug use or a meningococcal rash. Mucosal changes such as cyanosis or the cherry-red glow of carbon monoxide poisoning can be diagnostic. Cardiac monitoring and cardiovascular examination should identify rhythm disturbances, the murmurs of endocarditis and valvular disease, or evidence of shock from myocardial ischaemia or infarction. Respiratory patterns may aid in identifying the site of the lesion (Table 8.4.5). Abdominal examination may detect organomegaly, ascites, bruits or pulsatile masses.
Investigations
Specific laboratory testing
Venom detection kits can be used in specific clinical situations, and evidence of systemic envenomation can be screened for with other tests, such as coagulation profiles and creatinine kinase.
Management
The algorithm in Figure 8.4.1 is aimed at correcting immediate life-threatening pathology and then identifying and treating reversible structural and metabolic causes.
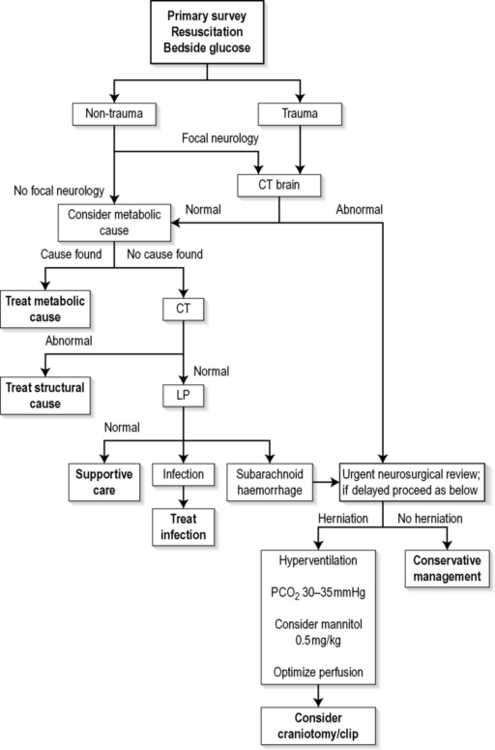
Following initial resuscitation, it is important to identify patients in whom trauma is known or suspected. These have a higher risk of skull fractures and focal intracranial pathology, and are more likely to have increased intracranial pressure requiring urgent imaging and subsequent neurosurgical consultation and definitive management. The same pathway is required for patients who have a non-traumatic cause for coma but who have lateralizing signs suggesting a focal intracranial lesion. Evidence of brainstem herniation is a neurosurgical emergency. A CT scan is helpful in diagnosing the cause of cerebral herniation and should be obtained expeditiously. Neurosurgical consultation can be arranged concurrently so as not to impede smooth transit to theatre for those requiring urgent craniotomy. Cerebral resuscitation is continued concurrently, with relative hyperventilation to maintain a PCO2 of 30–35 mmHg. The role of mannitol is still controversial, but it may be used in consultation with the neurosurgical team. The diuretic effect may, however, add to haemodynamic compromise and secondary neurological embarrassment.
Prognosis
Hasbun R, Abrahams J, Jekel J, Quagliarello VJ. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. New England Journal of Medicine. 2001;345(24):1727-1732.
Hoffman JR, Schriger DL, Luo JS. The empiric use of naloxone in patients with altered mental status: A reappraisal. Annals of Emergency Medicine. 1991;20:246-252.
Hoffman JR, Schriger DL, Votey SR, et al. The empiric use of hypertonic glucose in patients with altered mental status: A reappraisal. Annals of Emergency Medicine. 1992;21:20-24.
Hoffman DS, Goldfrank LR. The poisoned patient with altered consciousness: Controversies in the use of a ‘coma cocktail’. Journal of the American Medical Association. 1995;274:562-569.
Kelly AM, Kerr D, Dietze P, et al. A randomised trial of intranasal versus intramuscular naloxone in prehospital treatment for suspected opioid overdose. Medical Journal Association. 2005;182:24-27.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet. 1974;2:81-84.
Teasdale G, Jennett B. Aspects of coma after head injury. Lancet. 1977;1:878-881.
8.5 Seizures
Introduction
The terms ‘seizure’, ‘convulsion’ and ‘fit’ are often used both interchangeably and incorrectly. A seizure is an episode of abnormal neurological function caused by an abnormal electrical discharge of brain neurons. The seizure is also referred to as an ictus or ictal period. A convulsion is an episode of excessive and abnormal motor activity. Seizures can occur without convulsions, and convulsions can be caused by other conditions. The term ‘fit’ is best avoided in medical terminology, but is a useful term for non-medical personnel.
Seizures are common. It has been estimated that up to 10% of the population will have at least one seizure in their lifetime, and 1–3% of the population will develop epilepsy.1 A single seizure may be a reaction to an underlying disorder, part of an established epileptic disorder, or an isolated event with no associated pathology. The challenge is to rapidly identify and treat life-threatening conditions as well as to identify benign conditions that require no further investigation or treatment.
The manifestations of epileptic disorders are extremely varied. Two international classifications have been developed: the International Classification of Epileptic Seizures, and the International Classification of Epilepsy and Epileptic Syndromes.2,3 The former divides epileptic seizures into two major categories: partial and generalized. Partial epileptic seizures are further classified according to the impairment or the preservation of consciousness into simple partial and complex partial seizures. Either condition may secondarily generalize into tonic–clonic seizures. Generalized seizures can be divided into convulsive and non-convulsive types.
Convulsive seizures are generalized tonic–clonic seizures or grand mal seizures. Non-convulsive generalized seizures include absence seizures (previously termed petit mal seizures), myoclonic, tonic and atonic seizures. Under the International Classification, epilepsy and epileptic syndromes are initially classified according to their corresponding types of seizure into localization related and generalized disorders. Each disorder can be further classified according to its relationship to aetiological or predisposing factors into symptomatic, cryptogenic or idiopathic types.3 Different seizure types are associated with differing aetiological and prognostic factors. The details of the classification systems are not as important in emergency medicine as the concept of recognizing the different seizure types and being aware of the accepted terminology when discussing and referring cases.
Given the high frequency of this condition in emergency departments (ED) it is important to have a management strategy formulated in advance. One such approach has been developed by the American College of Emergency Physicians.4 The four main management concepts are as follows:
First seizures
| Hypoxia |
| Hypoglycaemia |
| Head trauma |
| Meningitis and encephalitis, including HIV disease |
| Metabolic, including hyponatraemia, hypocalcaemia, hyperthyroidism, uraemia and eclampsia |
| Drug overdose, including alcohol, tricyclics, theophylline, cocaine, amphetamine and isoniazid |
| Drug withdrawal, including alcohol, benzodiazepines, narcotics, cocaine and anticonvulsants |
| Cerebral tumour or stroke |
(Reproduced with permission from Brown AF, Wilkes GJ. Emergency department management of status epilepticus. Emergency Medicine 1994; 6: 49–61)
A complete physical and neurological examination is mandatory. Evidence of alcohol and drug ingestion and head trauma is particularly important. A comprehensive medication history may include agents known to reduce the seizure threshold in susceptible individuals, e.g. tramadol, selective serotonin reuptake inhibitors.5 A careful mental state examination in seemingly alert patients may reveal evidence of a resolving post-ictal state or of underlying encephalopathy. All patients not fully alert should not be assumed to simply be in a post-ictal state until other causes are excluded. Of particular importance is any evidence of underlying illness, such as fever, nuchal rigidity (meningitis) or cardiac murmurs (endocarditis). Needle tracks, evidence of chronic liver disease, dysmorphic features and marks such as café-au-lait spots (neurofibromatosis) are important aetiological clues. Complications such as tongue biting, broken teeth and peripheral injuries are not uncommon in generalized seizures. Stress fractures can occur, particularly in the elderly, and posterior dislocation of the shoulder is an uncommon but significant and easily overlooked finding.
The investigations necessary following an uncomplicated seizure are minimal. Although it is common practice to order a variety of tests, such as electrolytes, blood sugar level and full blood count, these are rarely of benefit in the fully recovered patient. Elevated neutrophil counts in blood and CSF may be seen as a result of a generalized seizure in the absence of an infectious disorder. Although electrolyte abnormalities may cause seizures they are unlikely to be the cause if the patient has recovered. A serum prolactin level at 20 and 60 minutes post seizure may be helpful if the diagnosis is in doubt. Patients with an abnormal physical or neurological examination should be managed according to clinical findings and the results of laboratory and radiological investigations. Findings suggestive of meningitis, encephalitis or subarachnoid haemorrhage are indications for cranial CT scan and lumbar puncture.
There are no clear guidelines to the routine need for or urgency of neuroimaging following a single uncomplicated seizure. Patients with focal neurological signs, those who do not recover to a normal examination, and those with a history of head trauma or intracranial pathology should all undergo cranial CT as soon as possible. The dilemma arises in patients with complete recovery and no focal signs. The incidence of abnormalities on CT in this group of patients is less than 1%.6 The decision as to whether and when to scan patients in this group will be determined largely by local factors. Generally, a contrast CT (more sensitive for subtle lesions) is performed on an outpatient basis prior to review. MRI is more sensitive than CT for infarcts, tumours, inflammatory lesions and vascular lesions, but cost and availability limit its use as a primary investigative modality.
Status epilepticus
Status epilepticus (SE) may be defined as ‘two or more seizures without full recovery of consciousness between seizures, or recurrent epileptic seizures for more than 30 minutes’.7
Status epilepticus has been reported to account for 1–8% of all hospital admissions for epilepsy, 3.5% of admissions to neurological intensive care, and 0.13% of all visits to a university hospital ED. It is more common at the extremes of age, with over 50% of all cases occurring in children and a disproportionately high incidence in those over 60 years of age. SE is also more frequent in the mentally handicapped and in those with structural cerebral pathology, especially of the frontal lobes. Four to 16% of adults and 10–25% of children with known epilepsy will have at least one episode of SE. However, SE occurs most commonly in patients with no previous history of epilepsy.8
Many compensatory physiological changes accompany seizures. As the duration is increased these mechanisms begin to fail, with an increased risk of permanent damage. Brain damage resulting from prolonged SE is believed to be caused by excitatory amino acid neurotransmitters such as glutamate and aspartate. These lead to an influx of calcium into neuronal cytoplasm and an osmotolysis with cell destruction. Continuing seizure activity itself contributes substantially to neuronal damage, which is further exacerbated by hypoxia, hypoglycaemia, lactic acidosis and hyperpyrexia. When seizures continue for over 60 minutes, the risk of neuronal injury increases despite optimal delivery of oxygen and glucose. The longer an episode of SE continues, the more refractory to treatment it becomes, and the more likely it is to result in permanent neuronal damage. Mortality increases from 2.7% with seizure duration under 1 hour, to 32% with duration beyond this.8 Generalized convulsive SE is therefore a medical emergency.
The principal pharmacological agents used are benzodiazepines and phenytoin. The benzodiazepines used vary between countries, with little clinical evidence to support any particular one. In Australasian centres midazolam is preferred, in increments of 1–2 mg i.v. If i.v. access cannot be rapidly secured, midazolam i.m. at a dose of 0.2 mg/kg will terminate most seizures.9 Alternatives to midazolam are diazepam and clonazepam. Diazepam can be administered rectally if necessary, and this technique can be taught to parents with high-risk children. However, onset of action by this route in adults is slow and unpredictable. All benzodiazepines share the disadvantages of respiratory depression, hypotension, and a short duration of clinical effect.
Phenytoin is usually used as a second-line agent in a dose of 15–20 mg/kg at a rate of no more than 50 mg/min. Rapid administration is associated with bradyarrhythmias and hypotension. The common practice of administering 1 g is inadequate for most adults. The effect of phenytoin does not commence until 40% of the dose has been administered; for this reason it should be commenced at the same time that i.v. benzodiazepines are given. Most people on anticonvulsants who present in SE have negligible drug levels, and the side effects from a full loading dose on top of a therapeutic level are minimal. The full loading dose should therefore be given even when the patient is known to be on therapy.10
The most common causes of failure to control seizures are:
When benzodiazepines and phenytoin are ineffective, expert advice should be sought. Drugs that may be used in the control of SE are summarized in Table 8.5.2. Inhalational or barbiturate anaesthesia can also be used. Both require expert airway control, and in some cases inotropic support. Management in an intensive care unit is mandatory.
| Drug | Bolus (i.v. unless stated otherwise) | Maintenance infusion |
|---|---|---|
| Midazolam | 0.02–0.1 mg/kg 0.15–0.3 mg/kg i.m. | 0.05–0.4 mg/kg/h |
| Phenytoin | 15–20 mg/kg at up to 50 mg/min, followed by further 5 mg/kg | N/A |
| Phenobarbitone | 10–20 mg/kg at 60–100 mg/min | 1–4 mg/kg/day |
| Thiopentone | 5 mg/kg | 1–3 mg/kg/h |
| Pentobarbitone (USA only) | 5 mg/kg at 25 mg/min | 0.5–3 mg/kg/h |
| Propofol | 2 mg/kg | 5–10 mg/kg/h |
| Lignocaine | 2 mg/kg | 3–6 mg/kg/h |
| Chlormethiazole | 0.8% solution, 40–100 mL over 10 minute | 0.8% solution 0.5–4 mL/min |
| Paraldehyde | 0.15 mL/kg i.m. or 0.3–0.5 mL/kg rectally diluted 1:1 with vegetable oil |
(Modified with permission from Brown AF, Wilkes GJ. Emergency department management of status epilepticus. Emergency Medicine 1994; 6: 49–61)
Non-convulsive seizures
Non-convulsive seizures may be partial (focal) or generalized. Complex partial seizures and focal seizures account for approximately one-third of all seizures, whereas primary generalized non-convulsive seizures (absence seizures) account for 6%.11
Non-convulsive status epilepticus (NCS) accounts for at least 25% of all cases of SE and is diagnosed more frequently when actively considered. Absence seizures rarely result in complete unresponsiveness, and patients may appear relatively normal to unfamiliar observers. NCS may precede or follow convulsive seizures and may easily create the perception of a cerebral vascular or psychiatric event. The longest reported episode of absence status is 60 days, and that of complex partial status 28 days.12
Treatment of non-convulsive seizures in the acute setting is the same as for convulsive seizures.12 The event is terminated with benzodiazepines in most instances, and should be followed by a search for precipitating causes. An estimated 50% of patients with simple partial seizures have abnormal CT scans.12 Long-term seizure control uses different agents from those used for convulsive seizures, highlighting the importance of involving a neurological service when planning follow-up.12
Pseudoseizures
Pseudoseizures are more common in women, less common after 35 years of age, and rare in patients over 50.13 They may be associated with a conversion disorder, malingering, Munchausen syndrome or Munchausen syndrome by proxy. Patients with conversion disorder differ from malingerers by being unaware of the psychiatric cause of their actions.
Pseudoseizures typically last more than 5 minutes, compared to neurogenic seizures which usually terminate within 1–2 minutes. Multiple patterns of seizures tend to occur in individual patients, and post-ictal periods are either very brief or absent. Patients with recall of events during what appears to be a generalized convulsive seizure are likely to have had a psychogenic seizure. Extremity movement out of phase from one side to the other and head turning from side to side typify pseudoseizures. Forward pelvic thrusting occurs in 44% of patients with pseudoseizures and is highly suggestive of the diagnosis.14
The most definitive means of differentiating pseudoseizures is by ictal EEG or video-EEG monitoring. Unfortunately, this is of little value in the ED. Blood gas determinations demonstrate a degree of acidaemia in neurogenic tonic–clonic seizures, but not in patients with pseudoseizures. Pulse oximetry will detect a fall in SaO2 during neurogenic but not pseudoseizures. Serum prolactin levels rise and peak 15–20 minutes after generalized tonic–clonic seizures, and then fall with a half-life of 22 minutes. The levels do not consistently rise with partial seizures, and remain normal with pseudoseizures.15
Alcohol-related seizures
Seizures represent 0.7% of ED visits, and alcohol contributes to approximately 50% of these. 16 The majority of alcohol-related seizures occur as part of the alcohol withdrawal syndrome.17
Drug-related seizures
Some medications are also associated with lowering seizure threshold in susceptible individuals. Tramadol in particular has been increasingly prescribed for analgesia in recent times and associated with new-onset seizures at normal therapeutic doses.5 A complete medication history is therefore essential.
Post-traumatic seizures
Post-traumatic epilepsy develops in 10–15% of serious head injury survivors.18 More than half will have their first seizure within 1 year. Significant risk factors are central parietal injury, dural penetration, hemiplegia, missile wounds and intracerebral haematomas.19 Early treatment with phenytoin for severe head injuries reduces the incidence of seizures in the first week only.20
Seizures and pregnancy
In previously diagnosed epileptics there is an increased risk of seizures during pregnancy of 17%.21 Anticonvulsant levels are influenced by reduced protein binding, increased drug binding and reduced absorption of varying degrees. The final effect on free drug levels is unpredictable and is most variable around the time of delivery.22 Careful clinical monitoring is essential, and monitoring of free drug levels rather than total serum levels may be necessary in selected patients. Anticonvulsants also interfere with the metabolism of vitamins D, K and folic acid. Supplementation is advisable.
Isolated simple seizures place both mother and fetus at increased danger of injury, but are otherwise generally well tolerated. Generalized seizures during labour cause transient fetal hypoxia and bradycardia of uncertain significance. Generalized convulsive SE is life-threatening to both mother and fetus at any stage of pregnancy.
All of the anticonvulsants cross the placenta and are potentially teratogenic. The risk of malformation in children is increased from 3.4% in the general population to 3.7% in epileptic mothers.23 In general, the types of malformation associated are not drug specific, apart from the increased risk of neural tube defects associated with valproate and carbamazepine. Prenatal screening for such defects is advised in patients who become pregnant while taking these agents. The risk from uncontrolled seizures greatly outweighs the risk from prophylactic medication in patients with good seizure control.3,24
Eclampsia is the occurrence of seizures in patients with pregnancy-induced toxaemia occurring after the 20th week of pregnancy, and consists of a triad of hypertension, oedema and proteinuria. One in 300 women with pre-eclampsia progresses to eclampsia. Seizures are typically brief, self-terminating, usually preceded by headache and visual disturbances, and tend to occur without warning.25 Treatment is directed at controlling the seizures and hypertension, and expedient delivery of the baby. Magnesium sulphate is effective in seizure control and is associated with a better outcome for both mother and baby than standard anticonvulsant and antihypertensive therapy. 26–29 The mechanism of action is unclear.25
Management of SE in pregnancy includes consideration of eclampsia, positioning in the left lateral position, and assessment and monitoring of fetal wellbeing. Urgent control of seizures is essential for both mother and baby. Phenobarbital may reduce the incidence of intraventricular haemorrhage in premature infants, and should be considered in place of phenytoin in this circumstance.30 Early involvement of obstetric and neurology services is essential.
1 Engel JJr, Starkman S. Overview of seizures. Emergency Medicine Clinics of North America. 1994;12(4):895-923.
2 Mosewich RK, So EL. A clinical approach to the classification of seizures and epileptic syndromes [see Comments]. Mayo Clinic Proceedings. 1996;71(4):405-414.
3 Cavasos JE, et al. Seizures and Epilepsy: Overview and Classification 2005. http://www.eMedicine.com, August 2007. Accessed
4 American College of Emergency Physicians. Clinical policy for the initial approach to patients presenting with a chief complaint of seizure, who are not in status epilepticus. Annals of Emergency Medicine. 1993;22(5):875-883.
5 Labate A, Newton MR, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
6 Reinus WR, Wippold FJD, Erickson KK. Seizure patient selection for emergency computed tomography. Annals of Emergency Medicine. 1993;22(8):1298-1303.
7 Treiman DM. Electroclinical features of status epilepticus. Journal of Clinical Neurophysiology. 1995;12(4):343-362.
8 Brown AF, Wilkes GJ. Emergency department management of status epilepticus. Emergency Medicine. 1994;6:49-61.
9 McDonagh TJ, Jelinek GA, Galvin GM. Intramuscular midazolam rapidly terminates seizures in children and adults. Emergency Medicine. 1992;4:77-81..
10 Lowenstein DH, Alldredge BK. Status epilepticus. New England Journal of Medicine. 1998;338(14):970-976.
11 Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993;34(3):453-468.
12 Jagoda A. Nonconvulsive seizures. Emergency Medical Clinics of North America. 1994;12(4):963-971.
13 Riggio S. Psychogenic seizures. Emergency Medicine Clinics of North America. 1994;12(4):1001-1012.
14 Gates JR, Ramani V, Whalen S, et al. Ictal characteristics of pseudoseizures. Archives of Neurology. 1985;42(12):1183-1187.
15 Dana-Haeri J, Trimble MR. Prolactin and gonadotrophin changes following partial seizures in epileptic patients with and without psychopathology. Biology Psychiatry. 1984;19(3):329-336.
16 Morris JC, Victor M. Alcohol withdrawal seizures. Emergency Medicine Clinics of North America. 1987;5(4):827-839.
17 Krumholz A, Grufferman S, Orr ST, et al. Seizures and seizure care in an emergency department. Epilepsia. 1989;30(2):175-181.
18 Dugan EM, Howell JM. Posttraumatic seizures. Emergency Medicine Clinics of North America. 1994;12(4):1081-1107.
19 Feeney DM, Walker AE. The prediction of posttraumatic epilepsy. A mathematical approach. Archives of Neurology. 1979;36(1):8-12.
20 Temkin NR, Haglund MM, Winn HR. Causes, prevention, and treatment of post–traumatic epilepsy. New Horizons. 1995;3(3):518-522.
21 Shuster EA Seizures in pregnancy. Emergency Medicine Clinics of North America. 1994;12(4):1013-1025.
22 Yerby MS, Friel PN, McCormick K. Antiepileptic drug disposition during pregnancy. Neurology. 1992;42(4 Suppl 5):12-16.
23 Stanley FJ, Priscott PK, Johnston R, et al. Congenital malformations in infants of mothers with diabetes and epilepsy in Western Australia, 1980–1982. Medical Journal of Australia. 1985;143(10):440-442.
24 Yerby MS. Risks of pregnancy in women with epilepsy. Epilepsia. 1992;33(Suppl 1):S23-26. discussion S26–27
25 Sibai BM. Medical disorders in pregnancy, including hypertensive diseases. Current Opinion in Obstetrics and Gynaecology. 1991;3(1):28-40.
26 The Eclampsia Trial Collaborative Group. Which anticonvulsant for women with eclampsia? Evidence from the Collaborative Eclampsia Trial [published erratum appears in Lancet 346(8969): 258]. Lancet. 1995;345(8963):1455-1463.
27 Lucas MJ, Leveno KJ, Cunningham FG. A comparison of magnesium sulfate with phenytoin for the prevention of eclampsia [see Comments]. New England Journal of Medicine. 1995;333(4):201-205.
28 Duggan K, Macdonald G. Comparative study of different anticonvulsants in eclampsia. Journal of Obstetric and Gynaecological Research. 1997;23(3):289-293.
29 Jagoda A, Riggio S. Emergency department approach to managing seizures in pregnancy. Annals of Emergency Medicine. 1991;20(1):80-85.
30 Morales WJ. Antenatal therapy to minimize neonatal intraventricular hemorrhage. Clinical Obstetrics and Gynaecology. 1991;34(2):328-335.
8.6 Syncope and vertigo
Introduction
Syncope and vertigo are relatively common symptoms. They are often described by patients using the term ‘dizziness’; however, it is essential to differentiate between the two. Syncope and vertigo both represent a significant diagnostic challenge and it is important to risk-stratify patients accurately to distinguish between potentially life-threatening and benign causes.
Syncope
Syncope as a presenting symptom represents about 1–1.5% of all emergency department (ED) attendances.1 It is a symptom, not a diagnosis. It is defined as a loss of consciousness induced by the temporarily insufficient flow of blood to the brain. Patients recover spontaneously, without therapeutic intervention or prolonged confusion.
The causes of syncope are summarized in Table 8.6.1. The most common cause in all age groups is neurally mediated syncope, also known as neurocardiogenic or vasovagal syncope.2 Orthostatic hypotension and cardiac causes are the next most common.3
| Neurally mediated | Cardiac |
| Vasovagal/neurocardiogenic | Structural valvular disease such as aortic stenosis |
| Situational: cough, micturition, defaecation | Unstable angina |
| Carotid sinus syndrome | Myocardial infarction |
| Bradyarrhythmias such as sinus node disease, AV block | |
| Tachyarrhythmias such as VT, SVT and torsadesde pointes | |
| Pacemaker/defibrillator dysfunction | |
| Pulmonary hypertension | |
| Pulmonary embolus | |
| Aortic dissection | |
| Orthostatic hypotension | Neurological |
| Dehydration | Vertebrobasilar transient ischaemic attack |
| Vasodilatation | Subclavian steal |
| Migraines | |
| Medication | Psychiatric |
| Antihypertensives | |
| β-Blockers | |
| Cardiac glycosides | |
| Diuretics | |
| Antiarrhythmics | |
| Antiparkinsonian drugs | |
| Nitrates | |
| Alcohol |
Clinical features
Orthostatic hypotension occurs when the patient moves from a lying position to a sitting or standing position. If the required autonomic changes fail to compensate adequately, even healthy individuals will experience lightheadedness or blurring of their vision, and possibly a loss of consciousness. The most vulnerable people are those with blunted or impaired autonomic reflexes, such as the elderly, those on certain medications (particularly vasodilators, antihypertensive agents and β-blockers) and those who are relatively volume depleted due to heat, excessive fluid losses or inadequate oral intake.
Cardiac syncope is more likely to present with an absent or brief prodrome. Sudden unexplained loss of consciousness should raise suspicion for a cardiac arrhythmia, particularly in the high-risk patient. Both tachycardia and bradycardia can be responsible. A syncopal event while supine is of particular concern, and a predictor for a cardiac cause.4 Those that occur during exertion should prompt a search for structural heart disease, in particular aortic stenosis.
Risk stratification
Most of the published literature on assessment of patients presenting to EDs with syncope has focused on identifying risk factors for mortality or adverse cardiac outcome. Colivicchi et al.5 developed the OESIL score, based on four high-risk factors identified in a multicentre Italian study aimed at predicting mortality at a year. These were age over 65 years, a history of cardiovascular disease (which encompasses ischaemic heart disease, congestive cardiac failure, cerebrovascular disease and peripheral vascular disease), an abnormal ECG (including signs of ischaemia, arrhythmias, prolonged QT interval, AV block or bundle branch block) and absence of the typical prodrome. Martin6 derived a similar group of risk factors in a cohort of syncope patients and then validated these prospectively. More recently, Quinn et al.7,8 devised and then validated the San Francisco Syncope Rule (SFSR), where five factors were used to predict serious short-term and longer-term outcomes. These factors are:
Distilling these factors, patients with syncope can be divided into high- and low-risk groups as shown in Table 8.6.2. Low-risk patients can be safely discharged for outpatient follow-up, but controversy over high-risk patients remains. It is likely that there is a significant proportion of patients in the high-risk group who are actually intermediate risk, and given further evaluation in the ED or a short-stay unit could also be safely discharged; however, it is more difficult to identify this subset.
| High risk | Low risk |
|---|---|
| Chest pain consistent with IHD | Age < 45 years |
| History of congestive cardiac failure | Otherwise healthy |
| History of ventricular arrhythmias | Normal ECG |
| Pacemaker/defibrillator dysfunction | Normal cardiovascular exam |
| Abnormal ECG (findings such as prolonged QTc interval, conduction abnormalities, acute ischaemia) | Prodrome (consistent with neurally mediated syncope or orthostatic hypotension) |
| Exertional syncope/valvular heart disease | |
| Age > 60 years |
A number of projects have attempted to further define risk groups or assess risk stratification approaches. The Risk Stratification of Syncope in the Emergency department (ROSE pilot)9 compared the performance of the OESIL score, SFSR and the Edinburgh Royal Infirmary ED Syncope Guidelines and found that although the SFSR showed the best sensitivity for detecting adverse events, this was at the expense of increased hospital admissions. Similarly, an Australian validation study found that the SFSR was fairly sensitive but that it would have increased admissions by 9% if all high risk patients were admitted.10 The Syncope Evaluation in the Emergency Department Study (SEEDS)11 randomized patients deemed to be intermediate risk to either conventional assessment or assessment in a specialized syncope unit. It reported that a specialized unit increased the diagnostic yield and reduced the need for inpatient hospital admission.
Clinical investigations
The only two mandatory investigations are a 12-lead ECG and blood glucose. These should add enough information to the clinical findings to stratify the patient as high or low risk for an adverse outcome. Research has found that a serum troponin taken at least 4 hours after a syncopal event is not a sensitive predictor of an adverse cardiac outcome.12
Treatment
Patients who are deemed high risk for a cardiac cause need continuous cardiac monitoring for at least 24 hours and admission for further evaluation. This may include echocardiography to identify structural heart problems and to quantify an ejection fraction, or electrophysiological studies.
Prognosis
Syncope in a patient with underlying heart disease implies a poor prognosis, with data suggesting that a third will die within a year of the episode.13 Overall, those with syncope on a background of congestive cardiac failure are at the highest risk for an adverse outcome.1 In the absence of underlying heart disease, syncope is not associated with excess mortality.2
Vertigo
Vertigo is defined as the disabling sensation in which the affected individual feels that he himself or his surroundings are in a state of constant movement. It has a reported 1-year incidence of 1.4%.14 Like syncope, it is a symptom not a diagnosis, and has as many causes. The difficulty is that whereas many of the causes of vertigo are benign, it may be a symptom of serious neurological conditions such as vertebrobasilar stroke.
Aetiology
The causes of vertigo may be divided into peripheral and central (Table 8.6.3).
| Peripheral | Central |
|---|---|
| Benign paroxysmal positional vertigo (BPPV) | Cerebellar haemorrhage and infarction |
| Vestibular neuritis | Vertebrobasilar insufficiency |
| Acute labyrinthitis | Neoplasms |
| Ménière’s disease | Multiple sclerosis |
| Ototoxicitiy | Wallenberg’s syndrome (lateral medullary syndrome) |
| Eighth-nerve lesions such as acoustic neuromas | Migrainous vertigo |
| Cerebellopontine angle tumours | |
| Post-traumatic vertigo |
Clinical features
As previously described, vertigo may be central or peripheral in origin. Peripheral vertigo tends to be more intense and associated with nausea, vomiting, diaphoresis and auditory symptoms such as tinnitus or hearing loss (although hearing loss can rarely occur with vascular insufficiency in the posterior cerebral circulation, as the auditory apparatus is supplied via the anterior inferior cerebellar artery or the posterior inferior cerebellar artery). There may also be a history of ear trauma, barotrauma, ear infection or generalized illness. The onset of the vertigo tends to be subacute, coming on over minutes to hours. Central vertigo tends to be less severe and associated with neurological symptoms and signs such as headache, weakness of the limbs, ataxia, incoordination and dysarthria. These symptoms may be the harbinger of more serious causes, such as cerebellar lesions or demyelinating diseases (Table 8.6.4).
| Peripheral | Central | |
|---|---|---|
| Onset | Acute | Gradual |
| Severity | Severe | Less intense |
| Duration, pattern | Paroxysmal, intermittent; minutes to days | Constant; usually weeks to months |
| Positional | Yes | No |
| Associated nausea | Frequent | Infrequent |
| Nystagmus | Rotatory – vertical, horizontal | Vertical |
| Fatigue of symptoms, signs | Yes | No |
| Hearing loss/tinnitus | May occur | Not usually |
| CNS symptoms, signs | No | Usually |
Clinical investigations
Dynamic manoeuvres can be both diagnostic and therapeutic. The Dix–Hallpike test15 can diagnose benign paroxysmal positional vertigo (BPPV). It should not be performed on patients with carotid bruits, and patients must be warned that the test may provoke severe symptoms.
Initially, the patient should be seated upright, close enough to the head of the bed so that when they are supine the head will be able to extend back a further 30–45°. To test the right posterior semicircular canal, the head is initially rotated 30–45° to the right. Keeping the head in this position, the patient is quickly brought to the horizontal position with the head placed 30–45° below the level of the bed. A positive test is indicated by rotatory nystagmus towards the affected ear. The test is then repeated on the left side.
Treatment
Treatment depends on the cause. Benign paroxysmal positional vertigo (BPPV) has the classic history of position-induced vertigo lasting only seconds. If BPPV is suspected, the Dix–Hallpike test is performed to identify the affected ear. The Epley manoeuvre15 or ‘canalith repositioning manoeuvre’ aims to move any unwanted particles out of the semicircular canals and thus ease the symptoms for which they are responsible. The steps of this manoeuvre are:
Vestibular neuritis is unilateral and thought to be caused by a viral infection or inflammation. Episodes are acute in onset and may be severe, lasting for days, usually associated with nausea and vomiting. The sense of perpetual movement is present even with the eyes closed, and is made worse by movement of the head. Symptomatic treatment, with medications such as antihistamines, antiemetics and benzodiazepines, is often all that is indicated. If nausea and vomiting are severe, intravenous fluid therapy may be needed. There are some reports of trials using steroids for vestibular neuritis, but this treatment remains unproven.16
Ménière’s disease has the classic triad of vertigo, sensorineural hearing loss and tinnitus. Attacks last from minutes to hours, and may recur with increasing frequency as the disease progresses. It is caused by dilatation of the endolymphatic system due to excessive production or problems with reabsorption of the endolymph (endolymphatic hydrops). Medical management traditionally involves salt restriction and diuretics, although a Cochrane Review has questioned the efficacy of this.17
Vertebrobasilar insufficiency can produce vertigo, often accompanied by unsteadiness and visual changes. Symptoms may be provoked by head position and often include headache. Importantly, however, patients with cerebellar infarction occasionally present with vertigo without other symptoms or signs of neurological impairment.18 Treatment involves addressing cardiovascular risk factors as well as antiplatelet therapy.
1 American College of Emergency Physicians. Clinical Policy: Critical issues in the evaluation and management of adult patients presenting to the emergency department with syncope. Annals of Emergency Medicine. 2007;49:431-444.
2 Strickberger SA, Benson DW, Biaggioni I, et al. AHA/ACCF Scientific Statement on the Evaluation of Syncope. Circulation. 2006;113:316-327.
3 Linzer M, Yang EH, Estes M, et al. Diagnosing syncope Part 1: Value of history, physical examination and electrocardiography. Clinical Efficacy Assessment project of the American College of Physicians. Annals of Internal Medicine. 1997;126:989-996.
4 Jhanjee R, van Dijk JG, Sakaguchi S, et al. Syncope in adults: terminology, classification and diagnostic strategy. Pacing and Clinical Electrophysiology. 2006;29:1160-1169.
5 Colivicchi F, Ammirati F, Melina D, et al. Development and prospective validation of a risk stratification system for patients with syncope in the emergency department. European Heart Journal. 2003;24:811-819.
6 Martin TP, Hanusa BH, Kapoor WN. Risk stratification of patients with syncope. Annals of Emergency Medicine. 1997;29:459-466.
7 Quinn JV, Stiell IG, McDermott DA, et al. Derivation of the San Francisco Syncope Rule to predict patients with short-term serious outcomes. Annals of Emergency Medicine. 2004;43:224-232.
8 Quinn JV, Stiell IG, McDermott DA, et al. Prospective validation of the San Francisco Syncope Rule to predict patients with short-term serious outcomes. Annals of Emergency Medicine. 2006;47:448-454.
9 Reed MJ, Newby DE, Coull AJ, et al. Risk Stratification of Syncope in the Emergency Department (ROSE) pilot study: A comparison of existing Syncope guidelines. Emergency Medicine Journal. 2007;24:270-275.
10 Cosgriff T, Kelly AM, Kerr D. External validation of the San Francisco Syncope Rule in the Australian context. Canadian Journal of Emergency Medicine. 2007;9:157-161.
11 Shen WK, Decker WW, Smars PA, et al. Syncope evaluation in the Emergency Department (SEEDS). Circulation. 2004;110:3636-3645.
12 Hing R, Harris R. Relative utility of serum troponin and the OESIL score in syncope. Emergency Medicine of Australasia. 2005;17:31-38.
13 Crane SD. Risk stratification of patients with syncope in an accident and emergency department. Emergency Medicine Journal. 2002;19:23-27.
14 Neuhauser HK, von Brevern HM, Radtke A, et al. Epidemiology of vestibular vertigo: a neurotological study of the general population. Neurology. 2005;65:898-904.
15 Tintinalli J, Kelen G, Stapczynski S, editors. Emergency medicine. A comprehensive study guide, 6th edn. American College of Emergency Physicians. 2003, 1402-1405.
16 Strupp M, Zingler VC, Arbuso V, et al. Methylprednisolone, valaciclovir, or the combination for vestibular neuritis. New England Journal of Medicine. 2004;351:354-361.
17 Seemungal BM. Neuro-otological emergencies. Current Opinion in Neurology. 2007;20:32-39.
18 Lee H, Yi HA, Cho YW, et al. Nodulus infarction mimicking peripheral vestibulopathy. Neurology. 2003;60:1700-1702.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
section 8 Neurology
8.1 Headache
Introduction
Headache is a common ailment that is often due to a combination of physical and psychological factors. The vast majority are benign and self-limiting and are managed by patients in the community. Only a very small proportion of patients experiencing headache attend emergency departments (ED) for treatment. The challenges are to distinguish potentially life-threatening causes from the more benign, and to effectively manage the pain of headache.
Pathophysiology
The structures in the head capable of producing headache are limited. They include:
The pathological processes that may cause headache are:
The pathophysiological causes of headache are summarized in Table 8.1.1.
| Extracranial | Intracranial | |
|---|---|---|
| Tension/traction | Muscular headache | Intracranial tumour |
| ‘Tension headache’ | Cerebral abscess | |
| Intracranial haematoma | ||
| Vascular | Migraine | Severe hypertension |
| Inflammatory | Temporal arteritis | Meningitis |
| Sinusitis | Subarachnoid haemorrhage | |
| Otitis media | ||
| Mastoiditis | ||
| Tooth abscess | ||
| Neuralgia |
Assessment
Headache patterns
Some headaches have ‘classic’ clinical features: these are listed in Table 8.1.2. It must be remembered that, as with all diseases, there is a spectrum of presenting features and the absence of the classic features does not rule out a particular diagnosis. Every patient must be assessed on their merits and, if symptoms persist without reasonable explanation, further investigation should be undertaken.
Table 8.1.2 Classic clinical complexes and cause of headache
| Preceded by an aura | |
|
| Throbbing unilateral headache, nausea | Migraine | |
| Family history | ||
| Sudden onset | |
|
| Severe occipital headache; ‘like a blow’ | Subarachnoid haemorrhage | |
| Worst headache ever | ||
| Throbbing/constant frontal headache | |
|
| Worse with cough, leaning forward | Sinusitis | |
| Recent URTI | ||
| Pain on percussion of sinuses | ||
| Paroxysmal, fleeting pain | |
|
| Distribution of a nerve | Neuralgia | |
| Trigger manoeuvres cause pain | ||
| Hyperalgesia of nerve distribution | ||
| Unilateral with superimposed stabbing | |
|
| Claudication on chewing | Temporal arteritis | |
| Associated malaise, myalgia | ||
| Tender artery with reduced pulsation | ||
| Persistent, deep-seated headache | |
|
| Increasing duration and intensity | Tumour: primary or secondary | |
| Worse in morning | ||
| Aching in character | ||
| Acute, generalized headache | |
|
| Fever, nausea and vomiting | Meningitis | |
| Altered level of consciousness | ||
| Neck stiffness +/– rash | ||
| Unilateral, aching, related to eye | |
|
| Nausea and vomiting | Glaucoma | |
| Raised intraocular pressure | ||
| Aching, facial region | |
|
| Worse at night | Dental cause | |
| Tooth sensitive to heat, pressure |
Tension headache
The pathological basis of tension headaches remains unclear, but increased tension of the neck or cranial muscles is a prominent feature. A family history of headaches is common, and there is an association with an injury in childhood or adolescence. The most common precipitants are stress and alteration in sleep patterns.
Migraine
Treatment
The effectiveness of commonly used agents is summarized in Table 8.1.3. Dosing and administration are summarized in Table 8.1.4. At present the most effective agents seem to be the phenothiazines (chlorpromazine, prochlorperazine, droperidol and possibly haloperidol) and the triptans, each of which has achieved > 70% efficacy in a number of studies. Note that triptans are contraindicated in patients with a history of ischaemic heart disease, uncontrolled hypertension or with the concomitant use of ergot preparations.
| Agent | Drug dosing/administration |
|---|---|
| Chlorpromazine i.m. | 12.5 mg intravenously, repeated every 20 minutes as needed to a maximum dose of 37.5 mg, accompanied by 1 L normal saline over 1 hour to avoid hypotension OR 25 mg in 1 L normal saline over 1 hour, repeated if necessary |
| Droperidol (i.m. or i.v.) | 2.5 mg |
| Prochlorperazine (i.m. or i.v.) | 10 mg/12.5 mg (depending on packaging) |
| Sumatriptan (s.c., i.n.) | 6 mg SC, 20 mg i.n. |
| Metoclopramide (i.v.) | 10–20 mg |
| Ketorolac (i.m. or i.v.) | 30 mg i.v.; 60 mg i.m. |
| Tramadol (i.m.) | 100 mg |
Pethidine is not indicated for the treatment of migraine. Its reported effectiveness is only 56%, it has a high rate of rebound headache and it carries a risk of dependence. In two small RCTs haloperidol administered as 5 mg in 500 mL normal saline was reported to give significant pain relief in more than 80% of patients. Lignocaine (lidocaine) has been shown to be no more effective than placebo. The data on dihydroergotamine are difficult to interpret because it is often used in combination with other agents, e.g. metoclopramide; however, it has also been shown to be less effective than chlorpromazine and sumatriptan in acute treatment, and to have a high rate of unpleasant side effects. There are insufficient data to assess the effectiveness of CGRP receptor antagonists. Sodium valproate has also shown moderate effectiveness in small studies, but there are insufficient data to draw a valid conclusion. The efficacy of intravenous magnesium sulphate (1 or 2 mg) remains unclear. It was shown in a small placebo-controlled trial to be effective, but in another study the combination of magnesium with metoclopramide was less effective than metoclopramide and placebo.
Trigeminal neuralgia
Australian and New Zealand College of Anaesthetists and Faculty of Pain Medicine. Acute pain management: scientific evidence, 2nd edn. Canberra: National Health and Medical Council (Australia), 2005.
Friedman BW, Greenwald P, Bania TC, et al. Randomized trial of IV dexamethasone for acute migraine in the emergency department. Neurology. 2007. (Epub ahead of print)
Kelly AM. Specific pain syndromes: Headache. In: Mace S, Ducharme J, Murphy M, editors. Pain management and procedural sedation in the emergency department. New York: McGraw-Hill, 2006.
Kelly AM, Kerr D, Clooney M. Impact of oral dexamethasone versus placebo after ED treatment of migraine with phenothiazines on the rate of recurrent headache: a randomized controlled trial. Emergency Medicine Journal. 2008;25:26-29.
8.2 Stroke and transient ischaemic attacks
Pathophysiology
Ischaemic strokes
These are the results of several pathological processes (Table 8.2.1):
| Ischaemic stroke |
Haemorrhagic stroke
Haemorrhagic stroke is the result of vessel rupture into the surrounding intracerebral tissue or subarachnoid space. Subarachnoid haemorrhage is the subject of a separate chapter in this book (see Chapter 8.3). The neurological defect associated with an intracerebral haemorrhage is the consequence of direct brain injury, secondary occlusion of nearby vessels, reduced cerebral perfusion caused by associated raised intracranial pressure, and cerebral herniation. The causes of intracerebral haemorrhage (ICH) include:
Prevention
This particularly applies to ischaemic strokes. Non-modifiable risk factors for stroke include:
Primary prevention
Hypertension is the most important modifiable risk factor. The benefit of antihypertensive treatment in stroke prevention has been well shown. The other major risk factors for atherosclerosis and its complications – diabetes, smoking and hypercholesterolaemia – often contribute to increased stroke risk. These should be managed according to standard guidelines. The most important cardiac risk factor for TIA and stroke is atrial fibrillation, both chronic and paroxysmal. Warfarin is recommended to prevent cardioembolism, except in unsuitable patients. Those with contraindications to warfarin should initially receive aspirin. Other major cardiac risk factors include endocarditis, mitral stenosis, prosthetic heart valves, recent myocardial infarction and left ventricular aneurysm. Less common risk factors include atrial myxoma, a patent foramen ovale and cardiomyopathies.
A carotid bruit or carotid stenosis found in an otherwise asymptomatic patient is associated with an increased stroke risk. However, the role of carotid endarterectomy in these patients is controversial. In a highly selected patient group, the asymptomatic carotid atherosclerosis study (ACAS)1 showed a small but significant benefit in reduction of stroke or death at 5 years following surgery for angiographically proven stenosis >60% compared to medical therapy. The benefit was much lower than that achieved in symptomatic carotid stenosis shown in the North American Symptomatic Carotid Endarterectomy Study (NASCET 2),2 and can only be achieved with low perioperative mortality and stroke rates.
Ischaemic stroke syndromes
The symptoms and signs of stroke or TIA correspond to the area of the brain affected by ischaemia or haemorrhage (Table 8.2.2).
Anterior circulation ischaemia
The anterior circulation supplies blood to 80% of the brain and consists of the ICA and its branches, principally the ophthalmic, middle cerebral and anterior cerebral arteries. Hence this system supplies the optic nerve, retina, frontoparietal and most of the temporal lobes. Ischaemic injury involving the anterior cerebral circulation commonly has its origins in atherothrombotic disease of the ICA. Atherosclerosis of this artery usually affects the proximal 2 cm, just distal to the division of the common carotid artery. Advanced lesions may be the source of embolism to other parts of the anterior circulation, or cause severe stenosis with resultant hypoperfusion distally if there is inadequate collateral supply via the Circle of Willis. This is usually manifest by signs and symptoms in the middle cerebral artery (MCA) territory (Table 8.2.3). Less commonly, lesions of the intracranial ICA and MCA may cause similar clinical features.
| Homonymous hemianopia |
| Contralateral hemiplegia affecting face and arm more than leg |
| Contralateral hemisensory loss |
| Dysphasias with dominant hemispheric involvement (usually left) |
| Spatial neglect and dressing apraxia with non-dominant hemispheric involvement. |
Embolism to the ophthalmic artery or its branches causes monocular visual symptoms of blurring, loss of vision and field defects. When transient, this is referred to as amaurosis fugax, or transient monocular blindness.
Posterior circulation ischaemia
Specific brainstem syndromes include:
Pre-hospital care
A pre-hospital evaluation tool that has been developed and validated is the Cincinatti Prehospital Stroke Scale or FAST: F –facial movements, A –arm movements, S – speech, and T – test.3 Pre-hospital personnel who identify patients with acute onset of neurological deficits identified using this simple scale can then notify the ED in order to mobilize appropriate staff and forewarn Radiology, so as to expedite assessment and imaging, particularly if thrombolysis is being considered.
Clinical evaluation in the ED
Examination
Central nervous system
This includes assessing the level of consciousness, pupil size and reactivity, extent of neurological deficit, presence of neck stiffness and funduscopy for signs of papilloedema and retinal haemorrhage. Quantifying the neurological deficit using a stroke scale such as the 42-point National Institute of Health Stroke Scale (NIHSS)4 is useful in the initial assessment, and also for monitoring progress in a more objective way than clinical description alone. Strokes with a NIHSS score >22 are classified as severe.
In the case of TIA all clinical signs may have resolved. The average TIA lasts less than 15 minutes.
Differential diagnosis (Table 8.2.4)
| Intracranial space-occupying lesion |
| Subdural haematoma |
| Brain tumour |
| Brain abscess |
| Postictal neurological deficit – Todd’s paresis |
| Head injury |
| Encephalitis |
| Metabolic or drug-induced encephalopathy |
| Hypoglycaemia, hyponatraemia etc. |
| Wernicke–Korsakoff syndrome |
| Drug toxicity |
| Hypertensive encephalopathy |
| Multiple sclerosis |
| Migraine |
| Peripheral nerve lesions |
| Functional |
Complications of stroke
Investigations
General
TIAs and non-disabling strokes should be evaluated similarly in order to promptly diagnose and manage a potentially treatable process that might lead to a subsequent major stroke. The risk of a stroke following a TIA is now appreciated to be much higher than previously thought, and may be as high as 30% in the first week. The ABCD stroke risk score from TIA has been developed and validated to evaluate the risk of a stroke in the first 7 days following a TIA.5 This has the potential to guide the urgency of investigations, such as carotid ultrasound, required to determine the underlying causes of the TIA. The scoring system is outlined in Table 8.2.5. In patients with an ABCD score <4 there is minimal short-term risk of stroke. With scores of 4, 5 and 6 the risk is 2.2%, 16.5% and 35%, respectively. Other patient groups are at increased risk of stroke independent of the ABCD scoring system. These include patients with diabetes, multiple TIAs within a short period, and patients with a probable or proven cardioembolic source. Diabetes has been incorporated in the recently published ABCD2 scoring system (see Further Reading).
| ABCD | Risk factor | Score |
|---|---|---|
| Age | Below 60 | 0 |
| Above 60 | 1 | |
| Blood pressure | BP > systolic 140 mmHg, and/or diastolic 90 mmHg | 1 |
| Clinical | Unilateral weakness of face, arm, hand or leg | 2 |
| Speech disturbance without weakness | 1 | |
| Duration | Symptoms lasted >60 min | 2 |
| Symptoms lasted 10–60 min | 1 | |
| Symptoms lasted < 10 min | 0 |
(From Rothwell PM, Giles MF, Flassmann E, et al. A simple score (ABCD) to identify individuals at high risk of stroke after transient ischaemic attack. Lancet 2005; 366: 29–36)
Brain imaging
A head CT or MRI scan is indicated in most patients with TIA to exclude lesions that occasionally mimic TIA, such as subdural haematomas and brain tumours. CT, and more particularly MRI, may show areas of infarction which match the symptoms of an ischaemic event that, on clinical grounds, has completely resolved. CT is less sensitive than MRI in detecting posterior territory ischaemic lesions, particularly in the brain stem. In TIAs due to atrial fibrillation or another known cardiac source, brain imaging to exclude ICH is necessary prior to commencing anticoagulation. The exception is in cases of emboli from endocarditis in which anticoagulation is contraindicated owing to the increased risk of secondary ICH.
Imaging vessels
Imaging in stroke
Brain imaging
Recent studies have suggested that MRI is as accurate as CT in diagnosing acute ICH.6 This is significant, as it means that, where facilities are immediately available, CT may be bypassed in acute stroke and MRI can be used to both to exclude ICH and to scan for ischaemia/infarction with DWI. As already mentioned, other modalities such PWI and MRA/MRV may also give important diagnostic information and influence treatment decisions. However, MRI may not be feasible in a significant number of stroke patients, due either to standard contraindications to MRI or other factors such as haemodynamic instability, impaired consciousness or vomiting and agitation. In one study the proportion of patients intolerant of MRI was 1:10.
Treatment
TIAs
Ischaemic stroke
A more active approach to the acute management of ischaemic stroke is seen as having the potential to improve neurological outcomes. The ED is the place where these important treatment decisions will largely be made. Most patients with a stroke will require hospital admission for further evaluation and treatment, as well as for observation and possible rehabilitation. Studies of stroke units show that patients benefit from being under the care of physicians with expertise in stroke and a multidisciplinary team that can manage all aspects of their care.10
[/not-level-membership-for-emergency-medicine-category]
