[level-membership-for-hematology-oncology-and-palliative-medicine-category]
CHAPTER 7. PHARMACOLOGY
Phyllis A. Grauer
Judicious prescribing of medication requires an understanding of the principles of pharmacology. Pharmacology is the study of the drug, the body’s effect on the drug, and the drug’s effect on the body. Variability in drug response occurs among individuals and populations, including those of different age groups and ethnic backgrounds and those with concurrent disease states and concomitant drug therapies; hence, drug dosage regimens must be individualized. These variations in response are attributed to many factors; however, they can be categorized into two major areas of study: pharmacokinetics and pharmacodynamics. Figure 7-1 illustrates the principles of pharmacokinetics and pharmacodynamics.
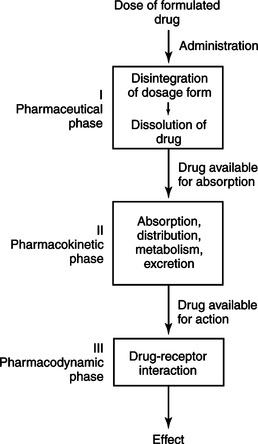 |
| Figure 7-1
(From Kuebler, K.K., Davis, M.P., & Moore, C.D. [2005]. Palliative practices: An interdisciplinary approach. St. Louis: Elsevier Mosby, Figure 4-1.)
|
Clinical pharmacokinetics examines the effects of the body on a drug, specifically absorption, distribution, metabolism, and excretion of drugs. Factors that influence these processes include the following:
▪ How quickly a drug is absorbed into the blood and how different dosages affect that absorption
▪ How the drug is distributed into organs or tissues of the body and to the site of action
▪ How the body metabolizes the drug and whether the drug is changed by the body into an active or inactive compound
▪ How long it takes the body to metabolize and eliminate half of the drug (the drug’s half-life)
▪ How long it takes the drug to be excreted from the body
Pharmacodynamics is the study of the body’s reaction to drugs. This area of pharmacology evaluates the body’s response to pharmacological, biochemical, physiological, and therapeutic effects of a drug. Basically, pharmacodynamics is the study of the activity of drugs on receptor sites within the body resulting in a clinical effect.
Of note, “pharmacogenomics” is an emerging area of study. The focus of pharmacogenomics is the identification of variations in the human genome (a total gene complement) that affect the response to medications. Advances in pharmacogenomics may permit drugs to be tailor-made for individuals and adapted to each person’s genetic makeup. Although environment, diet, age, lifestyle, and state of health all can influence a patient’s response to specific medications, understanding an individual’s genetic makeup is thought to be key in creating personalized drugs that would provide patients greater efficacy and safety. Because of the infancy and complexity of pharmacogenomics, it is beyond the scope of this chapter.
For the clinician, rational incorporation of the principles of pharmacology into therapeutic decision making will result in achieving the desired therapeutic outcome while preventing adverse drug events and promoting optimal symptom management.
UNDERSTANDING PHARMACOKINETIC PARAMETERS
Once a drug enters the circulatory system, it is distributed to tissues, reabsorbed into the bloodstream, and then eliminated from the body. Pharmacokinetic parameters define the factors that affect drug concentration within the human body over time.
Routes of Administration
A myriad of factors affect the dosage and matrix used in medications that influence the drug’s delivery to the specific site of action. These factors include the drug’s ability to penetrate barriers (i.e., the wall of the gastrointestinal tract and skin), the stability of the drug in acid environments such as the stomach (pH 2), the degree of tissue irritation when the drug is administered intramuscularly or subcutaneously, and the fraction of drug that is inactivated by the first pass through the liver. Table 7-1 describes the most common routes of administration.
| Route | Characteristics |
|---|---|
| Oral (PO) | Drug must be dispersed in solid dosage form to permeate the gastrointestinal lining and enter circulatory system. |
| Most is absorbed in small intestine, where there is less acidic environment. | |
| Rate of absorption is dependent on gastric emptying and intestinal motility. | |
| Extent of absorption is dependent on drug’s ability to permeate gastrointestinal lining and enter circulatory system. | |
| Drug enters portal circulation, passes through liver, and therefore is subject to hepatic extraction and metabolism. | |
| Drugs inactivated by acidic environment of stomach are typically enteric coated to prevent contact with stomach acid. Once the drug enters the less acidic environment of small intestine, the enteric coating dissolves, allowing drug to be dispersed and then absorbed. | |
| Extended-release drugs use various forms of pharmaceutically prepared release mechanisms so drug is released from oral dosage form over time. | |
| Sublingual (SL) | This route avoids contact with acidic stomach environment. |
| Drug is absorbed through mucosa under the tongue and enters bloodstream through numerous capillary beds. | |
| Much drug that is absorbed sublingually bypasses the liver. | |
| There is greater lipophilicity of drug and more is completely absorbed sublingually. | |
| Rectal (PR) | Rectal mucosa is fed by blood vessels that pass through liver and by blood vessels that avoid portal circulation. |
| Percent of drug absorbed through each system depends on where drug is placed in rectum. | |
| Many drugs administered rectally have erratic and often unpredictable absorption. | |
| Do not administer drugs dependent on constant serum concentration within a narrow therapeutic range (e.g., phenytoin, digoxin, warfarin). | |
| Transdermal (TD) | Skin is the body’s strongest barrier against absorption of toxins from environment into systemic circulation. |
| Few drugs will penetrate skin and be absorbed into subcutaneous capillary beds. | |
| Extent of absorption is dependent on lipophilicity and drug’s molecular structure. | |
| Amount absorbed is determined by surface area to which it is applied. | |
| Drugs administered topically for systemic absorption are best formulated in predetermined patch sizes (e.g., fentanyl TM patch). | |
| Intravenous (IV) | Drug has rapid onset of action. |
| Rate-limiting step is time it takes to reach site of action and produce therapeutic effect. | |
| Only soluble drugs are able to be administered by IV injection. | |
| Drugs administered IV are not affected by first-pass liver extraction and inactivation. | |
| Intramuscular (IM) | Rate at which a drug is absorbed from muscle into bloodstream is dependent on type of diluent used to prepare drug formulation. |
| Oil-based drugs are typically absorbed more slowly that those in aqueous solution. | |
| Drug is not affected by first-pass liver extraction and inactivation. | |
| Subcutaneous (SC) | Route is used for drugs that are not irritating to surrounding tissue and where volume of drug product administered does not typically exceed 2 ml of fluid. |
| Type of formulation used should determine how rapidly drug is absorbed into capillary walls and enters circulatory system. | |
| Drug is not affected by first-pass liver extraction and inactivation. | |
| Intraspinal | Some drugs that act on the central nervous system can be administered epidurally and intrathecally. |
| Route often allows for decreased dosage requirements and localized action, reducing intensity of adverse effects. | |
| Route can be used for opioids and other adjuvant pain medications such as local anesthetics. | |
| Inhalation (INH) | Drug is generally absorbed rapidly if particles are small. |
| Multidose inhalers require good administration technique in order to deliver drug through bronchial tree to alveolar bed for absorption. | |
| Nebulizer administration of drug is less dependent on technique and is more efficacious in patients who are weak and debilitated (although absorption is less). | |
| Topical (TOP) | Route is typically intended to exert action locally and considered to avoid systemic absorption. |
| Sites of action include skin, eyes, nose, ears, and vaginal and rectal tissues. |
It is important to note that there are several determinants associated with oral medication use that may interfere with the drug’s absorption from the gastric mucosa. These determinants include dissolution, gastric emptying time, intestinal motility, drug interactions in the gut lumen, and passage through the gut wall. Box 7-1 outlines these in further detail.
Box 7-1
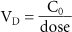
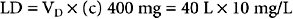
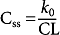
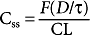
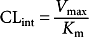
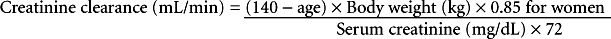
McGraw-Hill Australia Pty. Ltd.
DISSOLUTION
• Physical/chemical properties of the drug
• Crystal size and form
• Excipients (e.g., tablet fillers such as lactose)
• Dosage forms (enteric coated, sustained-release formulations)
• pH of the stomach and intestines
GASTRIC EMPTYING RATE
• Stability of the drug in an acid pH
• Solution or solid dosage forms (liquids and small particles empty more quickly)
• Presence of food or antacids
• Drugs (opioids and anticholinergics slow emptying time, metoclopramide increases emptying time)
• Disease (autonomic nervous system abnormalities such as Parkinson’s disease)
• Intestinal interactions in the gut
• Formation of complexes (tetracyclines and divalent metal compounds, e.g., Al 2+)
• Absorption (ion exchange resins, cholestyramine)
• Food (i.e., dairy products, proteins) (many antibiotics)
PASSAGE THROUGH THE GUT WALL
• Physical/chemical characteristics of the drug (quaternary ammonium compound, vancomycin)
• Metabolism by enzymes in the intestinal endothelium
Modified from Birkett, D.J. (2003). Pharmacokinetics made easy. North Ryde, Australia: McGraw-Hill Australia Pty. Ltd., Table 5-1, p. 36.
At the end of life when patients are unable to swallow and/or a parenteral route is unavailable, many drugs used for the treatment of symptoms in palliative care can be given via alternative routes of administration. Although there is a lack of published information and controlled studies regarding these alternate routes of administration, knowledge of the characteristics of the drug, including lipophilicity, molecular weight, and p Ka, can help the pharmacist predict whether a drug is likely to be absorbed via a particular route. Literature supports both benzodiazepines such as lorazepam and diazepam and certain opioids such as methadone and fentanyl as being well absorbed into the sublingual capillaries, whereas morphine is absorbed to a much lesser extent through the sublingual mucosa (Akinbi & Welty, 1999; Weinberg, Inturrisi, Reidenberg et al., 1988). The majority of the effect of morphine, as well as that of oxycodone and hydromorphone, occurs when the sublingually administered drug trickles down the esophagus and is absorbed by the gastrointestinal tract (Akinbi & Welty, 1999; Weinberg et al., 1988). Methadone administered rectally has a more rapid onset of action than does oral methadone, making it a feasible alternative when the oral and parenteral routes are not options (Dale, Sheffels, & Kharasch, 2004). Knowledge of the extent of absorption through each route of administration is imperative in order to appropriately guage the dose of medication (Katzung, 2003). The quantitative measure of the absorption of a drug into the circulation is known as the drug’s bioavailability.
Bioavailability
The bioavailability of a drug is the fraction of the administered dose that reaches the systemic circulation. For example, the bioavailability of an intravenous injection is 100% (Birkett, 2003), whereas bioavailability will vary for other routes of administration depending on factors that affect the extent of absorption into the circulatory system and first-pass hepatic metabolism. The measurement that determines absolute bioavailability is called the area under the curve (AUC). This measurement is determined by calculating the AUC of the plasma concentration plotted over time (Birkett, 2003) (Figure 7-2).
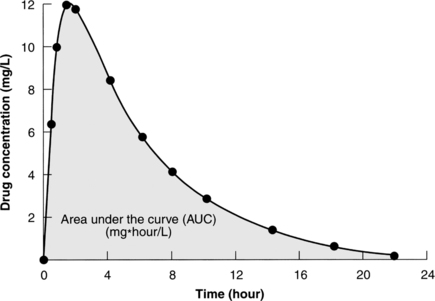 |
| Figure 7-2
(From Birkett, D.J. [2003]. Pharmacokinetics made easy. North Ryde, Australia: McGraw-Hill Australia Pty. Ltd., Figure 1-2.)
McGraw-Hill Australia
|
The bioavailability of a specific drug is the determinant of the dosage and is equivalent to the drug administered via various routes of administration. For example, chronic use of morphine administered orally has a bioavailability of around 20% to 30% of the parenteral dose (100% bioavailability) (Doyle, Hanks, Cherny et al., 2005). This means that a patient who has been receiving 10 mg of morphine intravenously will require approximately 30 mg of morphine orally to achieve the same analgesic effect as experienced from the intravenous dose (American Pain Society, 2003).
Drugs are considered bioequivalent when the extent and rate of absorption are similar and there is no difference between the therapeutic and adverse effects. A generic drug company, for example, that manufactures a drug in the same dosage form as a brand-name product will often use the AUC bioavailability data when comparing the two drugs. For a generic drug to be considered equivalent to the brand drug, the bioavailability must be similar to that of the brand product (USDHHS-FDA, 2006). The Electronic Orange Book is an online publication of the U.S. Department of Health and Human Services Food and Drug Administration that lists approved drug products and their therapeutic equivalence evaluations.
Therapeutic Range
Therapeutic range is defined as the plasma concentration that occurs between the concentration of drug needed to achieve the desired pharmacological effect and the concentration where adverse effects are observed (Figure 7-3). For some drugs, this range is narrow, and for other drugs, it is wide. The narrower the range, the more monitoring is needed to prevent adverse drug effects or clinical misadventures. Some monitoring is done based on plasma drug concentrations (e.g., gentamicin, theophylline, and digoxin). Other drug monitoring is done by measuring physiological changes that are caused by the drug (e.g., measuring international normalized ratio for Coumadin [warfarin] and thyroid-stimulating hormone for levothyroxine).
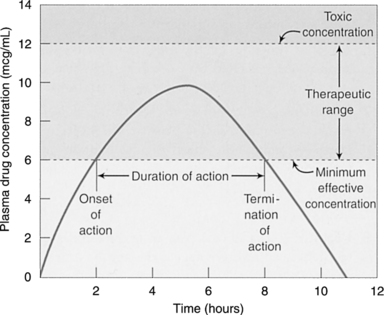 |
| Figure 7-3
(From Adams, M.P., Josephson, D.L., & Holland, L.N. [2005]. Pharmacology in nursing: A pathophysiologic approach. © 2005, pp. 53, 54. Reprinted by permission of Pearson Education, Inc., Upper Saddle River, N.J.)
Pearson Education, Inc.
|
Volume of Distribution
Once the drug is absorbed into the systemic circulation, it is distributed throughout the body. The apparent volume of distribution (V D) is a measure of where the drug goes once it is completely distributed throughout the body (Table 7-2). It is the ratio of the fraction of drug unbound to protein in the plasma to the fraction of unbound drug in the tissue, and it is expressed as liters per kilogram (L/kg). The volume of distribution is also determined by the strength of binding of the drug to plasma proteins in relationship to the strength of binding to tissue components. If a drug is highly bound by tissue and not by blood, it will allow most of the drug to be held in the tissues of the body and little will be held within the blood. In this case, the drug will have a large volume of distribution. The V D is measured by plotting the logarithm of the plasma concentration against time. The result is a straight line that can be extrapolated back to zero (Birkett, 2003). The plasma concentration at zero time (C 0) divided by the dose equals the volume of distribution:
| *Biphasic elimination: initial (terminal). |
|||
| Data from Lexi-Comp Online. (2006). Retrieved March 30, 2006, from www.lexi.com. | |||
| Drug | V D (L/kg) | V D/70 kg (L) | t½ (hr) |
|---|---|---|---|
| Morphine | 3.3 (3-4) | 230 | 2-4 (Immediate release) |
| Lorazepam | 1.3 | 91 | 12-16 |
| Diazepam | 1.1 | 77 | 20-50 |
| Chlorpromazine | 20 | 1400 | 2 (30) * |
| Haloperidol | 20 | 1400 | 20 |
| Fentanyl | 6 | 420 | 2–4 (IV), 17 (TD) |
| Methadone | 4 (1-8) | 280 | 7-59 |
| Warfarin | 7 (6-7) | 490 | 20-60 |
| Digoxin | 7 (6-7) | 490 | 37-48 |
Loading Doses
A loading dose (LD) can be used when attempting to achieve a rapid concentration of a specific drug. The volume of distribution is the pharmacokinetic parameter used when calculating an LD of a drug. For example, if a drug has a V D of 40 liters (L) and the desired concentration (c) is 10 mg/L, then a loading dose to achieve that concentration is 400 mg:
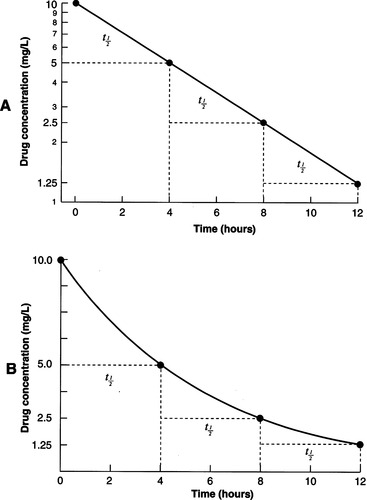
Half-Life
The half-life ( t1/2) is identified by the time it takes for the plasma concentration and the amount of drug within the body to fall by half after it has undergone absorption and distribution. Half-life is the reciprocal function of the elimination rate constant. The half-life and elimination rate constant are determined by both clearance (CL) and V D (Urso, Blardi, & Giorgi, 2002) (Figure 7-4). Half-life determines the duration of action after a single dose of a drug and the amount of time required to reach steady state with constant dosing.
 |
| Figure 7-4
(From Adams, M.P., Josephson, D.L., & Holland, L.N. [2005]. Pharmacology in nursing: A pathophysiologic approach. © 2005, pp. 53, 54. Reprinted by permission of Pearson Education, Inc., Upper Saddle River, NJ.)
Pearson Education, Inc.
|
The therapeutic concentration range, also known as the therapeutic window, is the drug concentration range where most patients will have a therapeutic effect with the least amount of adverse effects. In general, the usual therapeutic range and drug effect are proportional to the logarithm of drug concentration. Therefore, drug effect after a single dose usually declines in a linear relationship over time. Notwithstanding, a number of mechanisms result in a dissociation of the usual relationship between drug concentration and effect. Changes in volume of distribution due to dehydration of overhydration and changes in renal or liver function are examples of factors that will affect the drug concentration–and–effect relationship.
Steady State
Steady state occurs when the rate of drug administered is equal to the rate of elimination from the body. It generally takes three to five drug half-lives to reach steady state. For example, when immediate-release morphine ( t1/2 ≈4 hours) is administered routinely every 4 hours, steady state is achieved within 24 hours. For a drug with a longer half-life, such as methadone ( t1/2 ≈20 to 35 hours), steady state is achieved in approximately 6 to 10 days (Lugo, Satterfield, & Kern, 2006).
Drug concentrations are often measured as unbound plasma drug concentrations rather than as whole blood concentrations. With oral and intermittent dosing, once the steady state has been achieved, despite a fluctuation in drug doses, the amount of drug administered will equal the amount of drug eliminated. This results in an average drug concentration that is equal to an intravenous infusion (Olson, 2003).
With intermittent dosing, when the dosing interval is equal to the half-life of the drug, the result is about a two-fold fluctuation in drug concentration over the dosing interval. In cases where the drug is administered orally and at an interval that is not equal to the drug’s half-life, the degree of plasma concentration fluctuation over the dosing interval is determined by both the absorption rate and the relationship of the dosing interval to the half-life. Although a drug may be at steady state, if the dose is changed, it again will take three to five half-lives to reach a new steady state. Conversely, upon discontinuation of a dose, it takes four half-lives to eliminate 94% of the drug from the body (Birkett, 2003; Olson, 2003).
Dosing Intervals
To achieve a sustained systemic blood level of medication that has a short half-life and a narrow therapeutic window, drugs are often formulated in an oral sustained-release matrix. Morphine is an example of a drug that is available in an immediate-release form (i.e., on administration, the entire dose is available to be absorbed and distributed to the site of action) and in a sustained-release format where systemic blood levels are available for 8, 12, and 24 hours after ingestion of a single-unit dose. When morphine is administered as an immediate-release dose, it is typically dosed at an interval close to its half-life (i.e., every 4 hours) to maintain a constant therapeutic systemic level. Sustained-release morphine has the same half-life and clearance as immediate-release morphine. The sustained-release matrix formulation is designed to gradually release drug, resulting in a fraction of morphine available for absorption and distribution. Consequently, altering the sustained-release matrix through crushing, for example, will destroy the delayed absorption property and result in a bolus of the entire dosage, leading to loss of a sustained serum concentration and potentially toxicity (Olson, 2003).
Clearance
Medication clearance (CL) is the most important pharmacokinetic property of a drug. It is the measure of the efficiency of irreversible elimination of the drug from the systemic circulation and is expressed as the volume of blood cleared of unchanged drug per unit of time. Furthermore, it is the sum of drug clearance from systemic circulation, which includes the body’s vital organs. CL determines the steady-state concentration of a drug for a given dose rate.
However, if a drug is administered intermittently, as in the case of oral dosing, C ss is determined by
where F is the fraction of the dose absorbed into the systemic circulation, D is the dose of the drug, and τ is the dosage interval.
It is important for the clinician to note that the clearance of a drug can be altered by several factors, including liver and kidney insufficiency, changes in protein and tissue binding, and the concomitant administration of other medications.
Linear Pharmacokinetics
Once a drug is in the body, the process of elimination begins. The majority of drugs are eliminated by “first-order,” or linear, pharmacokinetics. This process of elimination is exponential or logarithmic (Figure 7-5). As an example of linear pharmacokinetics, when a dose is doubled from 200 mg/day to 400 mg/day, the patient’s serum drug concentration also doubles (Birkett, 2003).
 |
| Figure 7-5
(From Birkett, D.J. [2003]. Pharmacokinetics made easy. North Ryde, Australia: McGraw-Hill Australia Pty. Ltd., Figure 3-1.)
McGraw-Hill Australia
|
Multiple Compartment Distribution of Drugs
A drug within the systemic circulation is distributed to the body’s tissues at a rate and extent that are dependent on tissue perfusion and the ease with which it can pass through the lipid membranes of the cells. Tissues in the brain, liver, kidneys, and heart are highly perfused, whereas skeletal muscle and fat are less perfused, making distribution to these tissues much slower. The rate at which the drug is distributed from or to the site of action, in most cases, determines the onset and duration of action from its pharmacological effect (Birkett, 2003).
Distribution depends on the following four factors:
1. Blood flow: Tissues with the highest blood flow receive the drug first.
2. Protein binding: Drugs that are bound to plasma proteins do not cross lipid membranes.
3. Lipid solubility: The more lipid soluble a drug is, the more rapidly it is distributed to other tissues.
4. Degree of ionization: Only ionized (polar) drugs can cross cell membranes.
Protein Binding
To a greater or lesser extent, the majority of medications bind to plasma proteins. The major drug binding proteins in plasma are albumin, α 1-acid glycoprotein (AAG), and lipoproteins. It is the unbound drug within the bloodstream that is free to distribute throughout the tissues of the body and to the site of pharmacological activity. Unbound drug is also that portion of total plasma drug that is available for metabolism and excretion. In general, clearance of high-extraction-ratio drugs is high—a result of low protein binding; conversely, clearance of low-extraction-ratio drugs is dependent on the amount of protein binding. More highly bound drugs have a longer duration of action and a lower volume of distribution.
If a drug is highly protein bound, proportionally larger doses are required to achieve the desired therapeutic effect compared with a drug that has minimal protein binding (Birkett, 2003). Problems occur, for example, when drug B is administered and is in competition with drug A for the protein binding site. The result is an increase in the amount of free drug A. This is particularly important with drugs that are highly protein bound. If a drug is 97% bound to albumin and there is a 3% reduction in binding due to displacement by another drug, then the free drug concentration doubles from 3% to 6%. However, if a drug is 70% bound and there is a 3% reduction in binding, the increase in free drug will make little difference (70% to 73%) (Birkett, 2003).
Warfarin is an example of a drug that is highly protein bound and has a narrow therapeutic window. Displacement of warfarin from plasma protein by other drugs can be extremely dangerous and requires careful monitoring. Changes in protein binding are important when interpreting the results of therapeutic drug monitoring in medications where total rather than unbound concentrations are measured (Birkett, 2003; Davis, 2005).
Lipid Solubility
When considering the administration of diazepam in a patient who is having a seizure, the anticonvulsant effect of the diazepam occurs very quickly but wears off within a few hours. Yet the diazepam can remain in the body for several days. In this case, diazepam is rapidly absorbed into the systemic circulation where it is carried to “vessel-rich” organs—principally, the brain. After several minutes, the drug is redistributed to other tissues (fat, muscle, etc.), and the concentration within the brain decreases, allowing another seizure to possibly occur. In this setting, diazepam has been redistributed into other compartments within the body.
A graphic description of this phenomenon would be to illustrate a rapid fall in blood concentration, a plateau, and then an insidious gradual decline. The initial phase is the rapid redistribution, or alpha, phase. The plateau is the equilibrium phase (where blood concentration equals tissue concentration). The gradual decline, or beta, phase is reflective of the elimination phase, when the blood and tissue concentrations fall in tandem. This process describes a two-compartmental model (Birkett, 2003).
When the log concentration is graphed versus time and the alpha and beta elimination line can be extrapolated back to the y axis, the point where each line intersects the y axis at zero time is [c] 0, the theoretical point representing the concentration that would have existed at the start if the dose had been instantly distributed (dose/V D). From this new straight line, one can determine how long it takes for the concentration to drop by 50%—the elimination half-life. Likewise, a similar process can be performed on the alpha phase: the redistribution half-life. Log concentration is an important concept, yet the reality is that most medications are more complicated than this and require sophisticated calculations (Birkett, 2003; Katzung, 2003).
Drug Ionization
Highly ionized drugs cannot pass through lipid membranes, whereas unionized drugs can freely cross the membrane. For example, morphine is highly ionized but fentanyl is not. Consequently, fentanyl has a faster onset of action, as a result of its ability to rapidly cross lipid membranes. The degree of ionization depends on the p Ka of the drug and the pH of the local environment. The p Ka refers to the pH at which 50% of the drug is ionized. Most drugs have either a weak acid or a weak base. Acids are ionized at a high pH (i.e., in an alkaline environment). Bases are ionized within an acidic environment (low pH). For a weak acid, the more acidic the environment, the less ionized is the drug, and it can easily cross lipid membranes. Consider a drug that at an acid p Ka is 50% ionized; if two pH points are added to this (more alkaline), it becomes 90% ionized and will result in slower absorption. If the pH is reduced by two units, it becomes 10% ionized (more acidic) and can cross the lipid membrane readily. With weak bases, the opposite effect occurs (Birkett, 2003).
Local anesthetics are an example of weak bases: the closer the p Ka of the local anesthetic to the local tissue pH, the more unionized is the drug. This is why lidocaine (p Ka 7.7) has a faster onset of action than bupivacaine (p Ka 8.3). If the local tissues are alkalinized (e.g., by adding bicarbonate to the local anesthetic), then the tissue pH is brought closer to the p Ka, and the onset of action is accelerated.
DRUG CLEARANCE FROM THE BODY
The clearance of specific drugs can occur through excretion into the urine, gut, sweat, or expired air and/or through metabolic conversion into a different compound (metabolites), which often occurs in the liver. The sum of these processes equals total body clearance, and physiological clearance is determined through the following factors:
1. Blood flow to the organ that metabolizes (liver) or eliminates (kidney) the drug
2. Efficiency of the organ in extracting the drug from the bloodstream
Renal Clearance of Drugs
Renal drug clearance is identified as the net result or the addition of drug and/or its metabolites into the urine via (1) passive glomerular filtration plus (2) active tubular secretion minus (3) drug reabsorption from the urine into the blood through passive tubular reabsorption (Birkett, 2003).
Glomerular filtration occurs when drugs passively diffuse from the blood into the nephron by perfusion across the capillaries of Bowman’s capsule. Small nonionic drugs pass more readily, whereas those medications that are bound to proteins cannot pass through capillaries into the urine. The rate of glomerular filtration depends in part on blood pressure.
Tubular secretion is an active transport process (requires energy) where drugs are secreted into the nephron tubule from the efferent arteriole. This process involves carriers or transporters that bind to the drug. The size and charge of the drug are less important. There are at least two active secretion mechanisms in the proximal tubule—one for anions and one for cations. Drug interactions occur due to competition of anions for anion transporters and cations for cation transporters. Active tubular secretion is saturable, which can lead to nonlinear kinetics (see pp. 94 and 95).
Passive tubular reabsorption occurs when drugs diffuse from the nephron tubule back into the systemic circulation. This process is influenced by urine flow rate, lipid solubility, and the degree of ionization (urine pH and drug p Ka) of the drug in the tubular fluid. As with glomerular filtration, small nonionic drugs reabsorb more readily. Changing or altering the pH of the urine can influence the degree of reabsorption.
All mechanisms are reduced equally in renal dysfunction, so renal drug clearance decreases in proportion with creatinine clearance. The need to adjust drug doses in renal dysfunction depends on the fraction excreted unchanged (fe), the therapeutic index of the drug, and the presence of active or toxic metabolites. Table 7-3 illustrates the impact of renal failure and dialysis on opioid use.
| Opioid | Renal Failure | Dialysis |
|---|---|---|
| Morphine | Avoid → Glucuronide metabolites cause neuroexcitability and accumulate | Parent drug and metabolites are removed by dialysis |
| Watch for rebound as drug from central nervous system reequilibrates | ||
| Hydromorphone | Caution → Less effect of glucuronide metabolite accumulation than with morphine | Parent drug partially removed by dialysis, no data on metabolites |
| Oxycodone | Insufficient data → Anecdotal data of central nervous system depression | Avoid; no data |
| Codeine | Do not use → Active metabolite accumulates | Do not use |
| Methadone | Appears safe → In GFR >10 mL/min, some recommend decrease in dose | Parent drug is not dialyzed and metabolites are inactive |
| Fentanyl | Probably safe | Not dialyzed |
Hepatic First-Pass Clearance
Many drugs are extensively metabolized by the liver. Drugs administered orally are delivered from the gut to the portal vein in the liver. The liver extracts a portion of the administered drug, leaving less drug available to distribute to the site of action. Morphine, for example, is a drug that exhibits extensive first-pass elimination. To achieve the same therapeutic effect, the dose of oral morphine must be three times the intravenous dose (Birkett, 2003; Doyle et al., 2005).
The systemic bioavailability of a drug after oral administration is determined by the extent of absorption into the portal circulation and first-pass extraction by the liver. The extraction ratio of a drug is measured from 0 (no drug is removed on the first pass through the liver) to 1.0 (the drug is completely removed from the circulation on the first pass through the liver). Drugs that enter the liver for the first time through the portal system and are extensively and irreversibly removed by the liver have high extraction ratios (Birkett, 2003). The extent of first-pass hepatic extraction is a major determinant of the bioavailability of high–, but not low–, hepatic extraction ratio drugs and is a major source of variability in drug response for high–hepatic extraction ratio drugs. The degree of hepatic extraction determines the appropriate route of drug administration, the effects of liver disease, specific drug interactions, and the correlation between oral and intravenous doses.
Factors Affecting Hepatic Clearance of Drugs
Hepatic extraction of drugs is determined by the following three parameters (Table 7-4):
| Dependence on | ||||
|---|---|---|---|---|
| Extent of Liver Clearance | Examples of Drugs | Blood Flow (≈90 L/hr) | Intrinsic Clearance (0 to 1.0) | Protein Binding (0% to 100%) |
| High | Propranolol | Greatly dependent | Little to no dependence | Little to no dependence |
| Lidocaine | ||||
| Morphine | ||||
| Intermediate | Acetaminophen | Moderately dependent | Moderately dependent | Moderately dependent |
| Desipramine | ||||
| Nortriptyline | ||||
| Low | Warfarin | Little to no dependence | Greatly dependent | Greatly dependent |
| Diazepam | ||||
| Carbamazepine | ||||
1. Fraction of drug unbound in plasma
2. Intrinsic activity of the metabolizing liver enzymes
3. Liver blood flow (normal liver blood flow = 90 L/min)
DRUG METABOLISM
The majority of medications require biotransformation or metabolism before they can be excreted from the human body. In general, this metabolism results in a drug that is more polar and water soluble. Metabolism can render a drug either active or inactive, and in some cases, drug metabolism will transform an inactive or less active drug (prodrug) into a more active medication. An example can be found when codeine is metabolized into morphine, which is a more potent opioid agonist than the parent drug, codeine. Most metabolism occurs in the liver, although metabolism can occur in other tissues (kidneys, brain, skin, blood, lungs, and gastrointestinal tract). Medications are usually transformed by two types of metabolic reactions: phase I and phase II reactions. Phase I (nonsynthetic) reactions convert the parent drug through oxidation, reduction, or hydrolysis to a more polar form. Phase II (synthetic) reactions conjugate the drug with a group such as glucuronic acid, glycine glutamine, sulfate, methyl, or acetate to create a hydrophilic conjugate that is able to be renally excreted (Olson, 2003).
Morphine is extensively metabolized by phase II glucuronidation to morphine 3-glucuronide (M3G) (55%) and morphine 6-glucuronide (M6G) (10%). Although M6G has been shown to have analgesic activity, M3G has no beneficial action and is thought to be responsible for neurotoxic side effects (hyperalgesia, allodynia, and myoclonus), particularly in high doses and in patients with renal insufficiency (Davis, Varga, Dickerson et al., 2003; Dean, 2004). There appears to be significant individual variability in the degree of glucuronidation that would explain why some patients can tolerate higher doses of morphine than others without experiencing adverse side effects (Holthe, Klepstad, Zahlsen et al., 2002).
Ten percent of oxycodone is metabolized by phase I O-demethylation via the CYP450 enzyme CYP2D6 to oxymorphone and the inactive metabolite noroxycodone by N-demethylation. Although oxymorphone is 14 times as potent as oxycodone, the contribution of oxymorphone to analgesia is considered to be of no consequence. Renal insufficiency does decrease elimination of oxycodone and its metabolites, but the impact is less significant than that of morphine (Davis et al., 2003).
Drug metabolism is determined by the affinity of the drug for the enzyme (Km) and the velocity of the reaction (Vmax). The equation for enzyme drug reaction is
The velocity (Vmax) of the conversion of drug to metabolite depends on the quantity of the enzyme, the enzyme structure, and drug interactions (Birkett, 2003).
CYTOCHROME P450 ENZYMES
The cytochrome system is responsible for the majority of drug metabolism. Variations in drug clearance (up to 10-fold) can occur between individuals as a result of genetic enzyme polymorphisms that regulate enzyme activity in the cytochrome P450 (CYP) enzyme system. The most predominant cytochrome enzymes involved in drug metabolism include CYP3A4, CYP2D6, CYP1A2, CYP2C9, and CYP2C19. These enzymes account for 90% of drug metabolism. Based on liver mass, the fractions of total cytochrome isoforms are approximately 30% CYP3A4, 20% CYP2C9, 13% CYP1A2, and 1% to 5% CYP2D6 (Birkett, 2003; Ingelman-Sundberg, 2001). A vast number of medications are metabolized by a handful of cytochrome enzymes. These enzymes have wide drug specificity. In addition, some drugs are metabolized by several different cytochrome enzymes. Many drugs that are frequently used in palliative care are affected by the CYP450 enzymes and result in potentially dangerous drug interactions. Methadone, for example, is metabolized primarily by CYP3A4 but also by CYP2D6, CYP1A2, CYP2C9, and CYP2C19. Several drugs can activate or induce enzyme metabolism. When this occurs, substrate drugs that are metabolized by the induced enzyme are metabolized more rapidly, resulting in a decrease in blood levels in the body and possible reduction in the therapeutic effect. Cigarette smoke can also induce CYP1A2. A patient who is stabilized on methadone and quits smoking may experience symptoms of methadone toxicity (Davis & Walsh, 2001). Likewise, enzymes can be inhibited by competitive (reversible) or noncompetitive (irreversible) drug binding to the enzyme active site, thereby preventing further drug metabolism. Noncompetitive inhibition can be reversed by a generation of a new enzyme. The tricyclic antidepressant desipramine competitively inhibits CYP2D6, resulting in increased desipramine plasma levels when administered with methadone (Davis & Walsh, 2001). A substrate drug that is metabolized by an enzyme that is inhibited by another drug will result in decreased metabolism leading to higher serum drug concentrations and possible toxicity. Table 7-5 lists drugs that are substrates, inducers, and inhibitors of CYP enzymes.
| CYP1A2 | |
| Substrates | Amitriptyline, caffeine, clomipramine, clozapine, cyclobenzapine, imipramine, methadone, metoclopramide, mirtazapine, olanzapine, propranolol, riluzole, R-warfarin, theophylline, zileuton |
| Inducers | Amobarbital, butabarbital, charcoal-broiled beef, cigarette smoke, cruciferous vegetables, mephobarbital, mexiletine, omeprazole, pentobarbital, phenobarbital, phenytoin, secobarbital |
| Inhibitors | Cimetidine, ciprofloxacin, diltiazem, enoxacin, erythromycin, fluoxetine, fluvoxamine, grapefruit juice, mexiletine, norfloxacin, paroxetine, sertraline, verapamil, zileutin |
| CYP2C9 | |
| Substrates | Amitriptyline, carvedilol, celecoxib, diclofenac, flurbiprofen, fluvastatin, glimepiride, ibuprofen, imipramine, irbesartan, losartan, montelukast, naproxen, phenytoin, piroxicam, tolbutamide, torsemide, S-warfarin, zarfirlukast |
| Inducers | Butabarbital, carbamazepine, mephobarbital, pentobarbital, phenobarbital, rifampin, rifapentine, secobarbital |
| Inhibitors | Amiodarone, cimetidine, fluconazole, fluvastatin, metronidazole, miconazole, ritonavir, sulfamethoxazole, trimethoprim, zafirlukast, zileutin |
| CYP2C19 | |
| Substrates | Amitriptyline, citalopram, clomipramine, diazepam, imipramine, lansoprazole, mephenytoin, omeprazole, pentamidine, phenytoin, propranolol, R-warfarin |
| Inducers | Phenytoin, rifampin |
| Inhibitors | Felbamate, fluoxetine, fluvoxamine, ketoconazole, omeprazole |
| CYP2D6 | |
| Substrates | Amitriptyline, captopril, carvedilol, chlorpromazine, clomipramine, clozapine, codeine, desipramine, dextromethorphan, dihydrocodeine, diphenhydramine, encainide, flecanide, fluoxetine, haloperidol, hydrocodone, hydromorphone, imipramine, loratadine, maprotiline, methadone, metoprolol, mexiletine, mirtazapine, nortriptyline, ondansetron, oxycodone, paroxetine, perphenazine, propafenone, propranolol, risperidone, ritonavir, sertraline, thioridazine, timolol, tolterodine, tramadol, trazodone |
| Inducers | Bromocriptine, cimetidine, clarithromycin, cyclosporine, danazol, diltiazem, ergotamine, erythromycin, ethinyl estradiol, fluconazole, fluoxetine, fluvoxamine, gestodene, grapefruit juice, indanivir, itraconazole, ketoconazole, miconazole, midazolam, nefazodone, nicardipine, nifedipine, omeprazole, paroxetine, progesterone, propoxyphene, quinidine, ritonavir, sertraline, testosterone, troleandomycin, valproic acid, verapamil, zafirlukast, zileutin |
| Inhibitors | Amiodarone, cimetidine, diltiazem, fluoxetine, fluvoxamine, haloperidol, paroxetine, propafenone, quinidine, sertraline, thioridazine, tramadol, tricyclic antidepressants |
| CYP3A4 | |
| Substrates | Acetaminophen, alfentanil, alprazolam, amitriptyline, amlodipine, amprenavir, astemizole, atorvastatin, buspirone, carbamazepine, cerivastatin, citalopram, clarithromycin, codeine, cyclosporine, dapsone, delavirdine, dexamethasone, diazepam, diltiazem, disopyramide, donepezil, efavirenz, erythromycin, ethinyl estradiol, felodipine, fentanyl, finasteride, haloperidol, imipramine, indinavir, isradipine, itraconazole, ketoconazole, lansoprazole, lidocaine, loratadine, losartan, lovastatin, methadone, midazolam, mirtazapine, montelukast, nefazodone, nelfinavir, nicardipine, nifedipine, nimodipine, nisoldipine, pimozide, prednisone, propafenone, quetiapine, quinidine, quinine, repaglinide, rifabutin, ritonavir, saquinavir, sertraline, sibutramine, sildenafil, simvastatin, sufentanil, tacrolimus, tamoxifen, terfenadine, testosterone, theophylline, tolterodine, toremifene, triazolam, troleandomycin, valproic acid, verapamil, R-warfarin, zileuton, zolpidem |
| Inducers | Amobarbital, butabarbital, carbamazepine, clarithromycin, dexamethasone, ethosuximide, isoniazid, nafcillin, pentobarbital, phenobarbital, phenytoin, primidone, rifabutin, rifampin, rifapentine, troglitazone |
| Inhibitors | Cyclosporine, cimetidine, diltiazem, erythromycin, fluconazole, fluoxetine, grapefruit juice, ketoconazole, midazolam, nifedipine, paroxetine, progesterone, propoxyphene, sertraline, testosterone |
Interpatient difference in the degree of activity of CYP enzymes is extensive. Age and ethnicity also factor into these variations. Individual genetic influence and polymorphism are major topics of research (Ingelman-Sundberg, 2001).
NONLINEAR PHARMACOKINETICS
In linear pharmacokinetics, doubling the dose of drug given usually results in doubled serum drug concentration at a steady state, because the drug elimination rate is proportional to the drug concentration in the blood. There are some drugs that do not follow this rule. Instead of the serum concentration of the drug increasing proportionally with an increase in the amount of drug given, the serum concentration may be either more or less than expected.
When the drug-metabolizing enzymes or renal active secretion processes become saturated, a larger-than-expected increase in drug concentration (both total and unbound) may be seen with increased dose of drug. After the point of saturation is reached, even a small increase in dosage rate can result in a large increase in drug concentration and consequent toxicity. This is known as Michaelis-Menten kinetics (Birkett, 2003).
When the dosage of a low-clearance drug is increased, saturation of protein-binding sites can cause a less-than-expected increase in total drug concentration. Although the concentration of unbound drug increases because there are no protein-binding sites available for the drug, clearance of the drug (based on the unbound portion of the drug) increases, resulting in a less-than-expected change in the overall steady-state concentration of drug (Birkett, 2003).
Therapeutic Drug Monitoring
Some drugs require therapeutic drug monitoring of serum concentration to maintain a target concentration range. By maintaining the drug concentration within these parameters, optimal therapeutic affect will be achieved and drug toxicity can be avoided. However, drug concentrations need to be interpreted in the context of the clinical features of the patient. Interpreting drug concentration information and making drug regimen adjustments depend on obtaining accurate information about both the timing and the handling of the serum sample and about patient-specific factors that influence drug disposition. Very few drugs that are used for symptom control in palliative care require monitoring of serum concentrations. However, those drugs that remain necessary for patient comfort and have narrow therapeutic ranges, such as digoxin, theophylline, and phenytoin, may need to be monitored as the patient’s condition changes (e.g., weight loss or dehydration) to avoid toxicity.
PHARMACODYNAMICS
Drugs exert their pharmacodynamic action at three different levels: cellular, organism or individual, and population levels (Olson, 2003).
Cellular Pharmacodynamics
At the cellular level, drugs produce their action at specific receptor sites. Receptors are typically proteins or glycoproteins that reside on the cell surface, on an organelle within the cell, or in the cytoplasm of the cell in finite numbers. Hence, the effect of the drug at the therapeutic dose plateaus either before or at the point of receptor saturation. Once a drug binds to the receptor, one of several actions is likely to occur:
1. Ion channel is opened or closed (e.g., calcium or sodium channels).
2. Biochemical messengers called “second messengers” are activated. These secondary messengers initiate a series of chemical reactions within the cell, which transfer the signal stimulated by the drug. These messengers include compounds such as cyclic adenosine monophosphate, cyclic guanosine monophosphate, calcium, and inositol phosphates.
3. Normal cellular function, such as DNA synthesis, bacterial cell wall production, and protein synthesis, is physically changed.
4. A cellular function is “turned on” or “turned off” (activated or deactivated) (Olson, 2003).
Furthermore, drug response is dependent on both the affinity of the drug for its receptor and the drug’s efficacy once it is bound to the receptor.
The drug’s affinity to a receptor refers to the strength with which it binds to a receptor. At steady state, equilibrium exists between the amount of drug that is bound to receptors and drug that is free. Drugs can dissociate from a receptor after they have been bound to the receptor. The dissociation constant ( KD) of a drug is the measure of its affinity for a particular receptor and is the concentration of drug required to achieve a 50% receptor occupancy. The higher the affinity of a drug to a receptor, the less likely it is to dissociate from the receptor. Several classifications are used to identify receptor activity and include the following.
Agonists
Drugs that alter the physiology of a cell are referred to as agonists. Typically, the change in cell function does not occur until a minimum number of receptors are occupied. Agonists can further be classified based on their degree of activity at the receptor level (Olson, 2003).
▪ Strong agonist. If a maximal effect is exerted when only a small fraction of receptors on the cell are occupied, the drug is considered a strong agonist. Morphine is an example of a strong opioid agonist.
▪ Weak agonist. Conversely, if a greater number of receptors must be occupied in order to produce the same effect as a strong agonist, the drug is considered to be a weak agonist. Propoxyphene is considered to be a weak opioid agonist.
▪ Partial agonist. If all the receptors are occupied by a drug, yet the maximum desired effect is still not achieved, the drug is considered to be a partial agonist. An example of a partial opioid agonist is butorphanol.
Antagonists
Antagonists inhibit or block responses caused by agonists (Olson, 2003).
▪ Competitive antagonists. Drugs that compete with agonists for the receptor site are known as competitive antagonists. While the receptor site is occupied by an antagonist, an agonist cannot bind to the receptor because it appears to have less affinity for the receptor than does the antagonist. As the dose of the agonist increases, it can be overcome by the antagonist when binding to any additional receptor sites and reclaim the receptor site from the antagonist at equilibrium. Therefore, competitive antagonists are considered to be surmountable. Naloxone is an example of a competitive opioid antagonist.
▪ Noncompetitive antagonists. Drugs that bind to sites other than the receptor sites where an agonist binds are known as noncompetitive antagonists. Once an antagonist is bound to a receptor site, it changes the configuration of the agonist receptor site and renders it unrecognizable by the agonist. This type of antagonism is insurmountable. Angiotensin II receptor blockers are examples of noncompetitive antagonists of angiotensin II.
▪ Irreversible antagonist. These agents are nonequilibrium competitive antagonists that compete for the same receptor site that an agonist does; however, they are insurmountable. That is, they cannot be displaced by increased doses of an agonist, as is the case with competitive antagonist agents. Phenoxybenzamine is an irreversible α 1-antagonist.
Antagonism of therapeutic effect is a result from processes that are unrelated to receptor binding sites and is further described.
▪ Physiological antagonism. Antagonism of therapeutic activity that can occur when two drugs have unrelated mechanisms of action but produce opposite effects. The cholinergic agent urecholine decreases urinary retention, while oxybutynin, an anticholinergic drug, causes urinary retention and therefore physiologically antagonizes the action of urecholine.
▪ Antagonism by neutralization. Drugs that bind to one another when administered at the same time and render both drugs inactive can cause antagonism by neutralization. Aluminum hydroxide, for example, when administered with tetracycline, inactivates tetracycline.
ORGANISM PHARMACODYNAMICS
Unlike receptor site pharmacodynamics, organism pharmacodynamics identifies the observations that are made regarding the activity of drugs in an individual or organism. Various terms are used to reflect observations at this level (Olson, 2003):
▪ Efficacy refers to the degree of drug that is able to produce a maximum effect. For example, an antilipidemic drug (drug A) may decrease low-density lipoproteins (LDL) by 30% at the maximum dose, whereas another antilipidemic agent (drug B) lowers LDL by only 20% at the maximum dose. In this case, drug A is more efficacious in lowering LDL than drug B (Olson, 2003).
▪ Potency signifies the amount of drug required to produce a given response. When comparing parenterally administered opioid analgesics, hydromorphone 1.5 mg produces the same amount of analgesia as morphine 10 mg. Therefore, hydromorphone is more potent than morphine (Olson, 2003).
POPULATION PHARMACODYNAMICS
Several definitions are used to describe the behavior of drugs and how they interface within the general patient population. These definitions are further described (Olson, 2003).
▪ Effective concentration 50% (EC 50) is the concentration of drug that produces the desired therapeutic effect in 50% of the population. In general, drug doses should range between EC 20 and EC 80.
▪ Lethal dose 50% (LD 50) is the concentration of drug that causes death in 50% of the population.
▪ Therapeutic index is calculated by dividing the LD 50 by the EC 50. It is used to identify the measure of safety of a specific drug.
▪ Margin of safety is the margin or difference between the therapeutic and lethal dose of a specific drug.
PHARMACODYNAMIC AND PHARMACOKINETIC PROPERTIES
Age
The incidence of adverse drug reactions (ADRs) is directly related to the number of medications a patient is taking and the presence of concomitant diseases. What is considered a “therapeutic burden” can occur at any age; however, the presence of older age and the physiological changes that occur in the body (i.e., reduced organ function) can increase the risk of medication misadventures (Abernethy, 1999). The geriatric population is at risk due to multiple disease states and their impact on the pharmacokinetic and pharmacodynamic responses to drug therapy. The rate of ADRs in older adults (older than 65 years) is two to three times higher than that of younger adults. It is estimated that 20% of all hospital admissions of geriatric patients are due to unrecognized ADRs (Turnheim, 2003, 2004).
The effect of aging on pharmacokinetic parameters varies. In general, both aging and pharmacokinetic properties determine the extent of absorption after oral administration of medication is unchanged by the aging process. However, transdermal administration of medication may be altered in older patients. In order to achieve the optimal systemic effects from transdermal medication, the drug must diffuse across the stratum corneum and enter the microcirculation within the dermal layers. There are alterations in skin features that affect transdermal delivery in the aging, such as drying of the stratum corneum, changes in lipid composition of the skin, and decrease in dermal capillaries. The chemical composition of the drug determines the effect age will have on absorption. Lipophilic drugs tend to be absorbed to a lesser extent in elderly than in younger patients. Fentanyl is an example of a lipophilic drug that has shown to have decreased absorption in the elderly (Cusack, 2004; Turnheim, 2003).
As patients age, their body fat tends to increase while lean body mass decreases. Frequently, elderly patients become more dehydrated due to decreased fluid intake, diuretic medications, and reduced appetite. Changes in plasma protein in the elderly are small but can be more pronounced if the patient is severely ill or malnourished. All these factors can alter the volume of distribution of drugs. Alternately, changes in transport mechanisms in tissues such as the small intestine and the kidney can also affect drug distribution and elimination (Cusack, 2004).
Liver function and its composition incur minimal changes with age, even though the liver size and blood flow decrease. Synthetic phase 2 metabolism (glucuronidation, acetylation, and sulfation) has not been shown to be altered in the elderly. However, phase 1 CYP metabolism may vary with age. Genetic polymorphism of enzymes may be one explanation for these changes (Cusack, 2004). Alteration in renal drug elimination does occur in the elderly. Glomerular filtration changes with aging are thought to be due to structural changes within the nephron. The Cockcroft-Gault formula for determining creatinine clearance includes age in calculation along with gender, serum creatinine, and body weight as follows:
Age can also alter renal tubular function. Renal tubular transport of organic bases does not appear to be changed, but renal tubular organic acid transport declines with age (Cusack, 2004).
Pharmacodynamic changes that occur with age impact the effects of drugs in the elderly patient. These changes occur both at the receptor level and as a result of homeostatic changes. Elderly patients tend to have a decrease in dopamine D 2 receptors. This can lead to an increased incidence of extrapyramidal symptoms from dopamine antagonist drugs. Downregulation of β-adrenoceptors from increased norepinephrine release results in a reduced response from β-adrenergic agonists. Likewise, β-blockers produce less antihypertensive activity, probably due to lower renin levels in the geriatric patient. Decreased cholinergic neurons and receptors in the elderly most likely explain why anticholinergic drugs can cause increased confusion (Turnheim, 2003).
Drug-induced orthostatic hypotension occurs at a 5% to 33% incidence in the elderly population. Homeostatic mechanisms for counterregulating blood pressure in response to changes in position progressively decrease in the aging patient. This is potentiated by drugs that decrease either blood pressure or vascular volume. The result is an increased risk of falls (Turnheim, 2004).
Elderly patients are often sensitive to centrally acting drugs; therefore, it is important for the clinician to consider reducing initial doses by 50% (Turnheim, 2003). This heightened sensitivity is a direct result of a reduction in brain size and a decrease in the number of neurons and synapses (Turnheim, 2003).
An increased awareness of physiological changes that occur with aging should alert the clinician to use caution when determining which drug and what dose to use in the elderly patient. Typically, when adjusting medication doses in the elderly, it is prudent to “start low and titrate slowly.”
Gender
Females have a larger volume of distribution of lipophilic drugs compared with men, because women typically have a higher percentage of body fat (Craft, 2003). Reduced activity of CYP3A and CYP2D6 in women leads to an increase in bioavailability of drugs that are metabolized by these enzymes. Therefore, clearance of drugs like oxycodone, desipramine, sertraline, and oxycodone (CYP2D6) can be reduced compared with males, so dosages for women may be less (Davis et al., 2003; Schwartz, 2003). Men tend to have greater renal function, such as glomerular filtration and tubular secretion, than women relative to corrected body size (Schwartz, 2003). Glucuronidation (influencing, among others, acetaminophen) is diminished in females (Schwartz, 2003). Gender-related differences in drug metabolism may lead to ADRs and drug-drug interactions. Although these gender differences are of note (10% to 30% difference), other factors such as age and disease states have greater influence on pharmacokinetics than gender (Schwartz, 2003).
Ethnicity
Whites, Asians, and African Americans have varying amounts of CYP2D6, CYP2C9, and CYP2C19, resulting in variations in drug interactions and rates of metabolism of drugs metabolized by these families of enzymes (Davis & Walsh, 2001). A number of palliative medications are influenced by this, such as diazepam, phenytoin, and warfarin (Bjornsson, Callaghan, Einolf et al., 2003).
Smoking and Alcohol Consumption
Cigarette smoking increases the metabolism of specific drugs that are substrates of CYP1A2, whereas alcohol increases metabolism of drugs that are substrates of CYP2E1. If changes occur in use or amount of alcohol or cigarettes, affected drug dosages may need to be adjusted. Methadone is an example of a drug where metabolism is influenced by the chronic use of alcohol and cigarette use (Davis & Walsh, 2001). If a patient discontinues the use of alcohol or cigarettes while prescribed methadone, it is likely that the patient will experience increased side effects (e.g., sedation) from an elevation in methadone levels. If this occurs in a terminally ill patient, it is important that the clinician does not mistakenly attribute this as the dying process.
Disease State
Disease states can dramatically influence the pharmacokinetics and pharmacodynamics of drugs. Depending on the disease state, any number of circumstances can occur. In patients who are malnourished (e.g., a patient with a malignancy), drugs that are highly protein bound will have an increase in free drug resulting in possible toxicity. Diabetic patients may experience an exacerbation of hyperglycemia due to use of corticosteroids. Drugs that are primarily metabolized by the liver (e.g., acetaminophen, morphine) should be used cautiously or avoided completely in patients who have end-stage liver failure. Oxycodone elimination half-life increases threefold (13.9 hours) in end-stage liver disease, necessitating reduced doses or extended dose intervals (Davis, Walsh, LeGrand et al., 2002). Extensive edema or ascites, for example, may result in third spacing of hydrophilic drugs, such as digoxin, leading to decreased serum concentrations and increased half-life. Renal failure has a profound effect on most opioids. Table 7-6 lists considerations in opioid dosing as it relates to renal failure.
| Morphine | Methadone | Oxycodone | Hydromorphone | ||||
|---|---|---|---|---|---|---|---|
| GFR (mL/min) | Dosage (% of Normal) | GFR (mL/min) | Dosage (% of Normal) | GFR (mL/min) | Dosage (% of Normal) | GFR (mL/min) | Dosage (% of Normal) |
| 20-50 | 75 | 20-50 | 100 | >60 | 100 | >60 | 100 |
| 10-20 | 50 | 10-20 | 100 | 30-60 | ? | 30-60 | 50 |
| 10 | 25 | 10 | 59 | <30 | ? Reduce | <30 | 25 |
POLYPHARMACY
Polypharmacy has many definitions. Most commonly, it is defined in the outpatient setting as the concomitant ingestion of four or more medications. Perhaps a more appropriate definition is the prescription, administration, or use of more medications than are clinically indicated in a given patient. This definition avoids enumeration of medications and recognizes that unnecessary adverse events can be the result of one unnecessary medication. Polypharmacy has distinct subgroups. Appropriate polypharmacy is when a patient is on several medications but each is appropriately indicated. In this case, decreasing the number of medications would not be beneficial. Inappropriate polypharmacy is when a patient is taking more medications than are necessary (Rollason & Vogt, 2003). The prevalence of polypharmacy increases with age. Two-thirds of persons over the age of 70 take between two and four medications, and one-fifth take five or more medications. The incidence of ADRs increases exponentially as the number of medications taken increases. The risk of drug-drug interactions approaches 100% when patients take eight or more medications (Rollason & Vogt, 2003). These interactions can include increased or decreased efficacy due to synergistic effects or competitive activity. Failure to recognize significant drug interactions may result in undertreatment or overdosing (Bernard & Bruera, 2000). The clinician should consider ongoing and careful monitoring for effectiveness and appropriateness of prescribed medications.
PRINCIPLES OF GOOD PRESCRIBING
When considering prescribing a medication for a patient
▪ Identify patient-related pharmacokinetic and pharmacodynamic factors.
▪ Identify drug-related pharmacokinetic and pharmacodynamic factors.
▪ Identify desired therapeutic outcome.
▪ Anticipate adverse effects and drug-drug interactions.
▪ Implement monitoring strategies.
REFERENCES
Abernethy, D.R., Aging effects on drug disposition and effect, Geriatr Nephrol Urol 9 (1999) 15–19.
Adams, P.A.; Josephson, D.L.; Holland, L.N., Pharmacology for nurses: A pathophysiologic approach. ( 2005)Pearson Prentice Hall, Upper Saddle River, N.J.
Akinbi, M.S.; Welty, T.E., Benzodiazepines in the home treatment of acute seizures, Ann Pharmacother 33 (1999) 99–102.
American Pain Society, Principles of Analgesic Use in the Treatment of Acute Pain and Cancer Pain. ( 2003)American Pain Society, Glenville, Ill..
Bernard, S.A.; Bruera, E., Drug interactions in palliative care, J Clin Oncol 18 (2000) 1780–1799.
Birkett, D.J., Pharmacokinetics made easy. ( 2003)McGraw-Hill Australia Pty. Ltd, North Ryde, Australia.
Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; et al.Pharmaceutical Research and Manufacturers of America Drug Metabolism/Clinical Pharmacology Technical Working Groups, The conduct of in vitro and in vivo drug-drug interaction studies: A PhRMA perspective, J Clin Pharmacol 43 (2003) 443–469.
Craft, R.M., Sex differences in drug- and non-drug-induced analgesia, Life Sci 72 (2003) 2675–2688.
Cusack, B.J., Pharmacokinetics in older persons, Am J Geriatr Pharmacother 2 (2004) 274–302.
Dale, O.; Sheffels, P.; Kharasch, E.D., Bioavailabilities of rectal and oral methadone in healthy subjects, Br J Clin Pharmacol 58 (2004) 156–162.
Davis, M., Pharmacology, In: (Editors: Kuebler, K.K.; Davis, M.P.; Moore, C.D.) Palliative practices: An interdisciplinary approach ( 2005)Elsevier Mosby, St. Louis.
Davis, M.P.; Varga, J.; Dickerson, D.; et al., Normal-release and controlled-release oxycodone: Pharmacokinetics, pharmacodynamics, and controversy, Support Care Cancer 11 (2003) 84–92.
Davis, M.P.; Walsh, D., Methadone for relief of cancer pain: A review of pharmacokinetics, pharmacodynamics, drug interactions and protocols of administration, Support Care Cancer 9 (2001) 73–83.
Davis, M.P.; Walsh, D.; LeGrand, S.; et al., Symptom control in cancer patients: The clinical pharmacology and therapeutic role of suppositories and rectal suspensions, Support Care Cancer 10 (2002) 117–138.
Dean, M., Opioids in renal failure and dialysis patients, J Pain Symptom Manage 28 (2004) 497–504.
Doyle, D.; Hanks, G.; Cherny, N.; et al., Oxford textbook of palliative medicine. 3rd ed. ( 2005)Oxford University Press, New York.
Holthe, M.; Klepstad, P.; Zahlsen, K.; et al., Morphine glucuronide-to-morphine plasma ratios are unaffected by the UGT2B7, H268Y and UGT1A1*28 polymorphisms in cancer patients on chronic morphine therapy, Eur J Clin Pharmacol 58 (2002) 353–356.
Ingelman-Sundberg, M., Genetic susceptibility to adverse effects of drugs and environmental toxicant: The role of the CYP family of enzymes, Mutat Res 482 (2001) 11–19.
Katzung, B.G., Basic & clinical pharmacology. 9th ed. ( 2003)Lange Medical Books/McGraw-Hill, San Francisco, Calif..
Lugo, R.A.; Satterfield, K.L.; Kern, S.E., Pharmacokinetics of methadone, J Pain Palliat Care Pharmacother 19 (2006) 13–24.
Olson, J., Clinical pharmacology made ridiculously simple. 2nd ed. ( 2003)MedMaster, Miami, Fla..
Rollason, V.; Vogt, N., Reduction of polypharmacy in the elderly, Drugs Aging 20 (2003) 817–832.
Schwartz, J.B., The influence of sex on pharmacokinetics, Clin Pharmacokinet 42 (2003) 107–121.
Turnheim, K., When drug therapy gets old: Pharmacokinetics and pharmacodynamics in the elderly, Exp Gerontol 38 (2003) 843–853.
Turnheim, K., Drug therapy in the elderly, Exp Gerontol 39 (2004) 1731–1738.
Urso, R.; Blardi, P.; Giorgi, G., A short introduction to pharmacokinetics, Eur Rev Med Pharmacol Sci 6 (2002) 33–44.
U.S. Department of Health and Human Services Food and Drug Administration, Electronic orange book, Retrieved February 10, 2006, from www.fda.gov/cder/orange/default.htm ( 2006).
Weinberg, D.S.; Inturrisi, C.E.; Reidenberg, B.; et al., Sublingual absorption of selected opioid analgesics, Clin Pharmacol Ther 44 (1988) 335–342.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
CHAPTER 7. PHARMACOLOGY
Phyllis A. Grauer
Judicious prescribing of medication requires an understanding of the principles of pharmacology. Pharmacology is the study of the drug, the body’s effect on the drug, and the drug’s effect on the body. Variability in drug response occurs among individuals and populations, including those of different age groups and ethnic backgrounds and those with concurrent disease states and concomitant drug therapies; hence, drug dosage regimens must be individualized. These variations in response are attributed to many factors; however, they can be categorized into two major areas of study: pharmacokinetics and pharmacodynamics. Figure 7-1 illustrates the principles of pharmacokinetics and pharmacodynamics.
|
|
| Figure 7-1
(From Kuebler, K.K., Davis, M.P., & Moore, C.D. [2005]. Palliative practices: An interdisciplinary approach. St. Louis: Elsevier Mosby, Figure 4-1.)
|
Clinical pharmacokinetics examines the effects of the body on a drug, specifically absorption, distribution, metabolism, and excretion of drugs. Factors that influence these processes include the following:
▪ How quickly a drug is absorbed into the blood and how different dosages affect that absorption
▪ How the drug is distributed into organs or tissues of the body and to the site of action
▪ How the body metabolizes the drug and whether the drug is changed by the body into an active or inactive compound
▪ How long it takes the body to metabolize and eliminate half of the drug (the drug’s half-life)
▪ How long it takes the drug to be excreted from the body
Pharmacodynamics is the study of the body’s reaction to drugs. This area of pharmacology evaluates the body’s response to pharmacological, biochemical, physiological, and therapeutic effects of a drug. Basically, pharmacodynamics is the study of the activity of drugs on receptor sites within the body resulting in a clinical effect.
Of note, “pharmacogenomics” is an emerging area of study. The focus of pharmacogenomics is the identification of variations in the human genome (a total gene complement) that affect the response to medications. Advances in pharmacogenomics may permit drugs to be tailor-made for individuals and adapted to each person’s genetic makeup. Although environment, diet, age, lifestyle, and state of health all can influence a patient’s response to specific medications, understanding an individual’s genetic makeup is thought to be key in creating personalized drugs that would provide patients greater efficacy and safety. Because of the infancy and complexity of pharmacogenomics, it is beyond the scope of this chapter.
For the clinician, rational incorporation of the principles of pharmacology into therapeutic decision making will result in achieving the desired therapeutic outcome while preventing adverse drug events and promoting optimal symptom management.
UNDERSTANDING PHARMACOKINETIC PARAMETERS
Once a drug enters the circulatory system, it is distributed to tissues, reabsorbed into the bloodstream, and then eliminated from the body. Pharmacokinetic parameters define the factors that affect drug concentration within the human body over time.
Routes of Administration
A myriad of factors affect the dosage and matrix used in medications that influence the drug’s delivery to the specific site of action. These factors include the drug’s ability to penetrate barriers (i.e., the wall of the gastrointestinal tract and skin), the stability of the drug in acid environments such as the stomach (pH 2), the degree of tissue irritation when the drug is administered intramuscularly or subcutaneously, and the fraction of drug that is inactivated by the first pass through the liver. Table 7-1 describes the most common routes of administration.
| Route | Characteristics |
|---|---|
| Oral (PO) | Drug must be dispersed in solid dosage form to permeate the gastrointestinal lining and enter circulatory system. |
| Most is absorbed in small intestine, where there is less acidic environment. | |
| Rate of absorption is dependent on gastric emptying and intestinal motility. | |
| Extent of absorption is dependent on drug’s ability to permeate gastrointestinal lining and enter circulatory system. | |
| Drug enters portal circulation, passes through liver, and therefore is subject to hepatic extraction and metabolism. | |
| Drugs inactivated by acidic environment of stomach are typically enteric coated to prevent contact with stomach acid. Once the drug enters the less acidic environment of small intestine, the enteric coating dissolves, allowing drug to be dispersed and then absorbed. | |
| Extended-release drugs use various forms of pharmaceutically prepared release mechanisms so drug is released from oral dosage form over time. | |
| Sublingual (SL) | This route avoids contact with acidic stomach environment. |
| Drug is absorbed through mucosa under the tongue and enters bloodstream through numerous capillary beds. | |
| Much drug that is absorbed sublingually bypasses the liver. | |
| There is greater lipophilicity of drug and more is completely absorbed sublingually. | |
| Rectal (PR) | Rectal mucosa is fed by blood vessels that pass through liver and by blood vessels that avoid portal circulation. |
| Percent of drug absorbed through each system depends on where drug is placed in rectum. | |
| Many drugs administered rectally have erratic and often unpredictable absorption. | |
| Do not administer drugs dependent on constant serum concentration within a narrow therapeutic range (e.g., phenytoin, digoxin, warfarin). | |
| Transdermal (TD) | Skin is the body’s strongest barrier against absorption of toxins from environment into systemic circulation. |
| Few drugs will penetrate skin and be absorbed into subcutaneous capillary beds. | |
| Extent of absorption is dependent on lipophilicity and drug’s molecular structure. | |
| Amount absorbed is determined by surface area to which it is applied. | |
| Drugs administered topically for systemic absorption are best formulated in predetermined patch sizes (e.g., fentanyl TM patch). | |
| Intravenous (IV) | Drug has rapid onset of action. |
| Rate-limiting step is time it takes to reach site of action and produce therapeutic effect. | |
| Only soluble drugs are able to be administered by IV injection. | |
| Drugs administered IV are not affected by first-pass liver extraction and inactivation. | |
| Intramuscular (IM) | Rate at which a drug is absorbed from muscle into bloodstream is dependent on type of diluent used to prepare drug formulation. |
| Oil-based drugs are typically absorbed more slowly that those in aqueous solution. | |
| Drug is not affected by first-pass liver extraction and inactivation. | |
| Subcutaneous (SC) | Route is used for drugs that are not irritating to surrounding tissue and where volume of drug product administered does not typically exceed 2 ml of fluid. |
| Type of formulation used should determine how rapidly drug is absorbed into capillary walls and enters circulatory system. | |
| Drug is not affected by first-pass liver extraction and inactivation. | |
| Intraspinal | Some drugs that act on the central nervous system can be administered epidurally and intrathecally. |
| Route often allows for decreased dosage requirements and localized action, reducing intensity of adverse effects. | |
| Route can be used for opioids and other adjuvant pain medications such as local anesthetics. | |
| Inhalation (INH) | Drug is generally absorbed rapidly if particles are small. |
| Multidose inhalers require good administration technique in order to deliver drug through bronchial tree to alveolar bed for absorption. | |
| Nebulizer administration of drug is less dependent on technique and is more efficacious in patients who are weak and debilitated (although absorption is less). | |
| Topical (TOP) | Route is typically intended to exert action locally and considered to avoid systemic absorption. |
| Sites of action include skin, eyes, nose, ears, and vaginal and rectal tissues. |
It is important to note that there are several determinants associated with oral medication use that may interfere with the drug’s absorption from the gastric mucosa. These determinants include dissolution, gastric emptying time, intestinal motility, drug interactions in the gut lumen, and passage through the gut wall. Box 7-1 outlines these in further detail.
Box 7-1
McGraw-Hill Australia Pty. Ltd.
DISSOLUTION
• Physical/chemical properties of the drug
• Crystal size and form
• Excipients (e.g., tablet fillers such as lactose)
• Dosage forms (enteric coated, sustained-release formulations)
• pH of the stomach and intestines
GASTRIC EMPTYING RATE
• Stability of the drug in an acid pH
• Solution or solid dosage forms (liquids and small particles empty more quickly)
• Presence of food or antacids
• Drugs (opioids and anticholinergics slow emptying time, metoclopramide increases emptying time)
• Disease (autonomic nervous system abnormalities such as Parkinson’s disease)
• Intestinal interactions in the gut
• Formation of complexes (tetracyclines and divalent metal compounds, e.g., Al 2+)
• Absorption (ion exchange resins, cholestyramine)
• Food (i.e., dairy products, proteins) (many antibiotics)
PASSAGE THROUGH THE GUT WALL
• Physical/chemical characteristics of the drug (quaternary ammonium compound, vancomycin)
• Metabolism by enzymes in the intestinal endothelium
Modified from Birkett, D.J. (2003). Pharmacokinetics made easy. North Ryde, Australia: McGraw-Hill Australia Pty. Ltd., Table 5-1, p. 36.
At the end of life when patients are unable to swallow and/or a parenteral route is unavailable, many drugs used for the treatment of symptoms in palliative care can be given via alternative routes of administration. Although there is a lack of published information and controlled studies regarding these alternate routes of administration, knowledge of the characteristics of the drug, including lipophilicity, molecular weight, and p Ka, can help the pharmacist predict whether a drug is likely to be absorbed via a particular route. Literature supports both benzodiazepines such as lorazepam and diazepam and certain opioids such as methadone and fentanyl as being well absorbed into the sublingual capillaries, whereas morphine is absorbed to a much lesser extent through the sublingual mucosa (Akinbi & Welty, 1999; Weinberg, Inturrisi, Reidenberg et al., 1988). The majority of the effect of morphine, as well as that of oxycodone and hydromorphone, occurs when the sublingually administered drug trickles down the esophagus and is absorbed by the gastrointestinal tract (Akinbi & Welty, 1999; Weinberg et al., 1988). Methadone administered rectally has a more rapid onset of action than does oral methadone, making it a feasible alternative when the oral and parenteral routes are not options (Dale, Sheffels, & Kharasch, 2004). Knowledge of the extent of absorption through each route of administration is imperative in order to appropriately guage the dose of medication (Katzung, 2003). The quantitative measure of the absorption of a drug into the circulation is known as the drug’s bioavailability.
Bioavailability
The bioavailability of a drug is the fraction of the administered dose that reaches the systemic circulation. For example, the bioavailability of an intravenous injection is 100% (Birkett, 2003), whereas bioavailability will vary for other routes of administration depending on factors that affect the extent of absorption into the circulatory system and first-pass hepatic metabolism. The measurement that determines absolute bioavailability is called the area under the curve (AUC). This measurement is determined by calculating the AUC of the plasma concentration plotted over time (Birkett, 2003) (Figure 7-2).
|
|
| Figure 7-2
(From Birkett, D.J. [2003]. Pharmacokinetics made easy. North Ryde, Australia: McGraw-Hill Australia Pty. Ltd., Figure 1-2.)
McGraw-Hill Australia
|
The bioavailability of a specific drug is the determinant of the dosage and is equivalent to the drug administered via various routes of administration. For example, chronic use of morphine administered orally has a bioavailability of around 20% to 30% of the parenteral dose (100% bioavailability) (Doyle, Hanks, Cherny et al., 2005). This means that a patient who has been receiving 10 mg of morphine intravenously will require approximately 30 mg of morphine orally to achieve the same analgesic effect as experienced from the intravenous dose (American Pain Society, 2003).
Drugs are considered bioequivalent when the extent and rate of absorption are similar and there is no difference between the therapeutic and adverse effects. A generic drug company, for example, that manufactures a drug in the same dosage form as a brand-name product will often use the AUC bioavailability data when comparing the two drugs. For a generic drug to be considered equivalent to the brand drug, the bioavailability must be similar to that of the brand product (USDHHS-FDA, 2006). The Electronic Orange Book is an online publication of the U.S. Department of Health and Human Services Food and Drug Administration that lists approved drug products and their therapeutic equivalence evaluations.
Therapeutic Range
Therapeutic range is defined as the plasma concentration that occurs between the concentration of drug needed to achieve the desired pharmacological effect and the concentration where adverse effects are observed (Figure 7-3). For some drugs, this range is narrow, and for other drugs, it is wide. The narrower the range, the more monitoring is needed to prevent adverse drug effects or clinical misadventures. Some monitoring is done based on plasma drug concentrations (e.g., gentamicin, theophylline, and digoxin). Other drug monitoring is done by measuring physiological changes that are caused by the drug (e.g., measuring international normalized ratio for Coumadin [warfarin] and thyroid-stimulating hormone for levothyroxine).
|
|
| Figure 7-3
(From Adams, M.P., Josephson, D.L., & Holland, L.N. [2005]. Pharmacology in nursing: A pathophysiologic approach. © 2005, pp. 53, 54. Reprinted by permission of Pearson Education, Inc., Upper Saddle River, N.J.)
Pearson Education, Inc.
|
Volume of Distribution
Once the drug is absorbed into the systemic circulation, it is distributed throughout the body. The apparent volume of distribution (V D) is a measure of where the drug goes once it is completely distributed throughout the body (Table 7-2). It is the ratio of the fraction of drug unbound to protein in the plasma to the fraction of unbound drug in the tissue, and it is expressed as liters per kilogram (L/kg). The volume of distribution is also determined by the strength of binding of the drug to plasma proteins in relationship to the strength of binding to tissue components. If a drug is highly bound by tissue and not by blood, it will allow most of the drug to be held in the tissues of the body and little will be held within the blood. In this case, the drug will have a large volume of distribution. The V D is measured by plotting the logarithm of the plasma concentration against time. The result is a straight line that can be extrapolated back to zero (Birkett, 2003). The plasma concentration at zero time (C 0) divided by the dose equals the volume of distribution:
| *Biphasic elimination: initial (terminal). |
|||
| Data from Lexi-Comp Online. (2006). Retrieved March 30, 2006, from www.lexi.com. | |||
| Drug | V D (L/kg) | V D/70 kg (L) | t½ (hr) |
|---|---|---|---|
| Morphine | 3.3 (3-4) | 230 | 2-4 (Immediate release) |
| Lorazepam | 1.3 | 91 | 12-16 |
| Diazepam | 1.1 | 77 | 20-50 |
| Chlorpromazine | 20 | 1400 | 2 (30) * |
| Haloperidol | 20 | 1400 | 20 |
| Fentanyl | 6 | 420 | 2–4 (IV), 17 (TD) |
| Methadone | 4 (1-8) | 280 | 7-59 |
| Warfarin | 7 (6-7) | 490 | 20-60 |
| Digoxin | 7 (6-7) | 490 | 37-48 |
Loading Doses
A loading dose (LD) can be used when attempting to achieve a rapid concentration of a specific drug. The volume of distribution is the pharmacokinetic parameter used when calculating an LD of a drug. For example, if a drug has a V D of 40 liters (L) and the desired concentration (c) is 10 mg/L, then a loading dose to achieve that concentration is 400 mg:
Half-Life
The half-life ( t1/2) is identified by the time it takes for the plasma concentration and the amount of drug within the body to fall by half after it has undergone absorption and distribution. Half-life is the reciprocal function of the elimination rate constant. The half-life and elimination rate constant are determined by both clearance (CL) and V D (Urso, Blardi, & Giorgi, 2002) (Figure 7-4). Half-life determines the duration of action after a single dose of a drug and the amount of time required to reach steady state with constant dosing.
|
|
| Figure 7-4
(From Adams, M.P., Josephson, D.L., & Holland, L.N. [2005]. Pharmacology in nursing: A pathophysiologic approach. © 2005, pp. 53, 54. Reprinted by permission of Pearson Education, Inc., Upper Saddle River, NJ.)
Pearson Education, Inc.
|
The therapeutic concentration range, also known as the therapeutic window, is the drug concentration range where most patients will have a therapeutic effect with the least amount of adverse effects. In general, the usual therapeutic range and drug effect are proportional to the logarithm of drug concentration. Therefore, drug effect after a single dose usually declines in a linear relationship over time. Notwithstanding, a number of mechanisms result in a dissociation of the usual relationship between drug concentration and effect. Changes in volume of distribution due to dehydration of overhydration and changes in renal or liver function are examples of factors that will affect the drug concentration–and–effect relationship.
Steady State
Steady state occurs when the rate of drug administered is equal to the rate of elimination from the body. It generally takes three to five drug half-lives to reach steady state. For example, when immediate-release morphine ( t1/2 ≈4 hours) is administered routinely every 4 hours, steady state is achieved within 24 hours. For a drug with a longer half-life, such as methadone ( t1/2 ≈20 to 35 hours), steady state is achieved in approximately 6 to 10 days (Lugo, Satterfield, & Kern, 2006).
Drug concentrations are often measured as unbound plasma drug concentrations rather than as whole blood concentrations. With oral and intermittent dosing, once the steady state has been achieved, despite a fluctuation in drug doses, the amount of drug administered will equal the amount of drug eliminated. This results in an average drug concentration that is equal to an intravenous infusion (Olson, 2003).
With intermittent dosing, when the dosing interval is equal to the half-life of the drug, the result is about a two-fold fluctuation in drug concentration over the dosing interval. In cases where the drug is administered orally and at an interval that is not equal to the drug’s half-life, the degree of plasma concentration fluctuation over the dosing interval is determined by both the absorption rate and the relationship of the dosing interval to the half-life. Although a drug may be at steady state, if the dose is changed, it again will take three to five half-lives to reach a new steady state. Conversely, upon discontinuation of a dose, it takes four half-lives to eliminate 94% of the drug from the body (Birkett, 2003; Olson, 2003).
Dosing Intervals
To achieve a sustained systemic blood level of medication that has a short half-life and a narrow therapeutic window, drugs are often formulated in an oral sustained-release matrix. Morphine is an example of a drug that is available in an immediate-release form (i.e., on administration, the entire dose is available to be absorbed and distributed to the site of action) and in a sustained-release format where systemic blood levels are available for 8, 12, and 24 hours after ingestion of a single-unit dose. When morphine is administered as an immediate-release dose, it is typically dosed at an interval close to its half-life (i.e., every 4 hours) to maintain a constant therapeutic systemic level. Sustained-release morphine has the same half-life and clearance as immediate-release morphine. The sustained-release matrix formulation is designed to gradually release drug, resulting in a fraction of morphine available for absorption and distribution. Consequently, altering the sustained-release matrix through crushing, for example, will destroy the delayed absorption property and result in a bolus of the entire dosage, leading to loss of a sustained serum concentration and potentially toxicity (Olson, 2003).
Clearance
Medication clearance (CL) is the most important pharmacokinetic property of a drug. It is the measure of the efficiency of irreversible elimination of the drug from the systemic circulation and is expressed as the volume of blood cleared of unchanged drug per unit of time. Furthermore, it is the sum of drug clearance from systemic circulation, which includes the body’s vital organs. CL determines the steady-state concentration of a drug for a given dose rate.
However, if a drug is administered intermittently, as in the case of oral dosing, C ss is determined by
where F is the fraction of the dose absorbed into the systemic circulation, D is the dose of the drug, and τ is the dosage interval.
It is important for the clinician to note that the clearance of a drug can be altered by several factors, including liver and kidney insufficiency, changes in protein and tissue binding, and the concomitant administration of other medications.
Linear Pharmacokinetics
Once a drug is in the body, the process of elimination begins. The majority of drugs are eliminated by “first-order,” or linear, pharmacokinetics. This process of elimination is exponential or logarithmic (Figure 7-5). As an example of linear pharmacokinetics, when a dose is doubled from 200 mg/day to 400 mg/day, the patient’s serum drug concentration also doubles (Birkett, 2003).
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading. Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
