CASE 7
Paul is 4 years of age and suffers from recurrent gram-negative bacterial infections, all of which have eventually cleared with long-term antibiotic therapy. Although he has been hospitalized three times for pneumonia (Chlamydia pneumoniae), there is no evidence for increased susceptibility to viral or fungal infections, and all of his childhood immunizations are up to date. An infectious disease specialist at a local tertiary care center was unable to establish an etiology for the problem. Further exploration of the family tree revealed that both male and female relatives had presented with a similar clinical history of recurrent infectious illnesses. Blood cell count and differential, performed on several occasions, indicated normal numbers of B cells, T cells, neutrophils, and monocytes. Serum immunoglobulin levels were also shown to be normal for all isotypes, as were antibody titers for various vaccine antigens. Other culture assays, performed to investigate cytokine production (from T cells) after stimulation with known T cell polyclonal activators, were normal. How would you proceed?
QUESTIONS FOR GROUP DISCUSSION
RECOMMENDED APPROACH
Implications/Analysis of Family History
An analysis of Paul’s family history revealed that both males and females had presented with similar symptoms, indicating that the recurrent infections are not likely caused by a defective gene encoded on the X chromosome. The familial nature of the problem, however, suggests a genetic basis for the recurrent infections.
Implications/Analysis of Laboratory Investigation
Paul’s initial blood work consisting of a complete blood cell count and differential was normal, so this is not a problem in T cell or B cell development (see Case 1). As predicted, T cell numbers/function were normal, as were serum immunoglobulin levels, indicating that B cell isotype switch mechanisms are intact.
Assessment of B Cells
Although assessment of serum immunoglobulins has typically been performed using serum electrophoresis, immunoelectrophoresis, or immunofixation, most laboratories use rate nephelometry, which provides a rapid and quantitative measure (see Case 5). Whereas B cell function can be analyzed using in vitro assays, antibody titers after immunization with protein antigens (e.g., tetanus toxoid) and polysaccharide (e.g., Haemophilus influenzae type b) are used initially to determine B cell responses. If these antibody titers (and serum immunoglobulins) are low, then the more advanced in vitro tests are performed.
Assessment of Phagocytes
Given that the infections are primarily bacterial, and that the B cell arm of the immune system is normal, one would suspect a phagocytic or complement dysfunction. Analysis of the respiratory burst (see Case 6) to rule out chronic granulomatous disease as a possibility should be performed.
Altered Cell Trafficking
In the presence of normal cell numbers and function, recurrent infection could be caused by the inability of immune cells to get to the site of infection or the lack of antigen presentation to T cells. However, a defect in antigen presentation would also result in viral and fungal infections, ruling out defective antigen presentation as a likely explanation. Altered cell trafficking is also not high on the list of possible explanations because there is no evidence for the classic presentation of necrotic skin lesions. To rule out this disorder, you should measure the expression of CD18, the common chain in β2 integrins. (See Additional Laboratory Tests.)
Additional Laboratory Tests
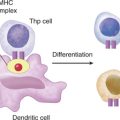
Flow cytometric analysis for the expression of CD18, a cell surface adhesion molecule, was within the normal range, ruling out the immunodeficiency disorder leukocyte adhesion defect (see Case 8) as a possible cause of Paul’s infections. Tests to assess total complement function (CH50 and AP50) were normal (see Case 11). We are faced with the fact that recurrent bacterial and fungal infections are occurring despite the presence of an intact and normal immune system and normal trafficking. Therefore, a defect in antigen recognition is one of the few remaining candidates to account for Paul’s problem with infections and so we should look at antigen recognition by dendritic cells and macrophages, cells of the innate immune system.
DIAGNOSIS
On the basis of the just-described tests, the diagnosis was a lack of TLR4 on the cell surface.
ETIOLOGY: LACK OF TLR4
Role of Other TLRs
Other TLRs that play an important role in innate immunity include TLR2, which binds peptidoglycan and lipoteichoic acid of Gram-positive bacteria, and lipoproteins present in several pathogens. In contrast, TLR9 binds CpG motif of bacterial oligonucleotides. Interaction of TLRs with their cognate ligands results in the intracellular activation of NFκB and altered gene transcription (see Case 10). Many TLRs are expressed at the cell surface, whereas several (TLR7, TLR8) are apparently expressed only intracellularly (likely detecting viral targets and/or altered expression of host molecules associated with “danger”). There is speculation as to why it is necessary for dendritic cells to express, in addition, this PRR repertoire (given that general inflammation associated with infection [viral/bacterial] is thought to result in sufficient dendritic cell activation [with cytokine production] to “drive” subsequent T cell activation). One explanation may be that in addition to providing a further activation signal to dendritic cells (measured by, for instance, the increased CD40 expression on these cells engaging PRRs), the different PRRs provide some additional information about the nature of the immunologic insult recognized (e.g., bacterial/viral) and thus help “mold” the resulting protective response.
The discovery of TLRs, their cognate ligands, and signaling transduction pathways has opened the door to a myriad of possibilities for therapeutic intervention in which signaling via TLRs is either suppressed or enhanced. The suppression or downregulation of TLR4 by administering pharmacologic agents that compete for binding with lipopolysaccharide would offer hope for patients with septic shock. Alternatively, enhancing signaling via one TLR versus another could drive antigen-dependent T cell development toward a Th1 or Th2 response, depending on the needs of the patient.