[level-membership-for-cardiovascular-category]
Acquired Heart Disease
Structure of Coronary Arteries
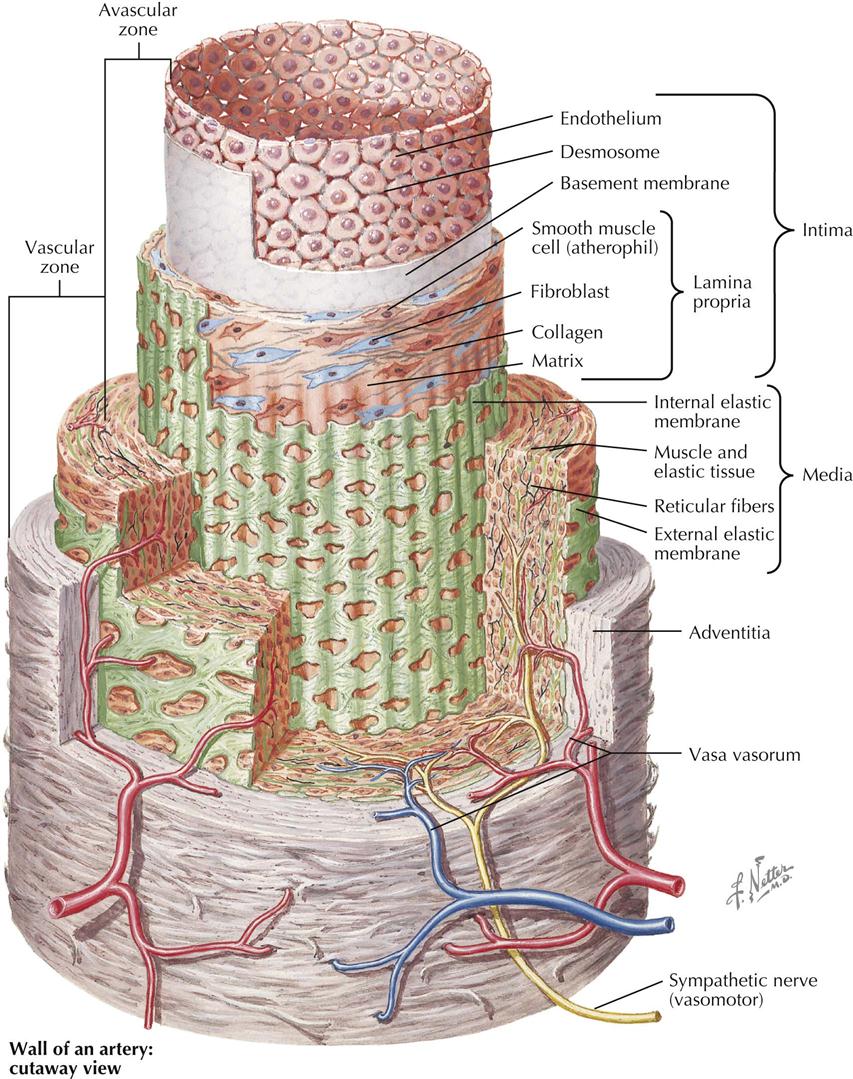
The coronary arteries are susceptible to atherosclerosis as well as its complications, particularly intravascular thrombosis, often resulting in myocardial infarction. Atherosclerosis is a form of arteriosclerosis characterized by initial involvement of the inner layer of the arterial wall, or intima. The intimal localization of early atheromas helps to differentiate the atherosclerosis type from other forms of arteriosclerosis, such as Mönckeberg’s medial sclerosis or periarteritis nodosa, that primarily involve the muscular or adventitial layers.
Recent histochemical, organ culture, and electron microscopy studies found further evidence of the complexity of the arterial wall. As Plate 6-1 shows, a medium-sized muscular artery, such as a main coronary vessel, consists of a series of concentric tubes or coaxial coats of differentiated cellular and extracellular components in three layers: tunica intima or intima, tunica media or muscular, and tunica adventitia. In the intima the innermost cell layer in direct contact with the bloodstream consists of a sheet of polygonal endothelial cells, usually less than 1 micron (µ) thick, except at the site of the cell nucleus, and elongated in a direction parallel to the vessel’s axis. Many of these endothelial cells have pinocytotic vesicles of caveolae in their cytoplasm, abundant mitochondria, well-developed granular endoplasmic reticulum, and Golgi complexes. Microscopically, the areas of contact between endothelial cells vary from simple mutual contact of the cell membranes to well-defined intercellular bridges, or desmosomes, corresponding to the so-called intercellular cement lines described in earlier light microscopy studies. A distinct basement membrane separates these endothelial cells from the subendothelial space, which varies in thickness according to age of the patient as well as size of the artery. In fetal life and shortly after birth the endothelium in the coronary arteries lies in direct contact with the internal elastic membrane, or lamina, without the subendothelial space, which appears by the end of the first decade of life.
In adulthood the intima consists of a matrix of ground substance containing small amounts of acid mucopolysaccharides and elastic and collagen fibers separating scattered intimal cells, or intimacytes. Because of morphologic and cytochemical difficulties in classifying intimacytes, their response to in vitro incorporation of serum lipids is used to identify them. Some intimacytes, or atherophils, incorporate lipid rapidly and show ultrastructural characteristics of modified smooth muscle cells, with typical bundles of myofilaments in their cytoplasm, pinocytotic vesicles, and parts of limiting basement membrane on the cell surface. On the other hand, cells called fibrophils are spindle shaped and have fingerlike cytoplasmic projections, with no basement membrane and few pinocytotic vesicles. Occasionally, large mononuclear ovoid cells appear, containing cytoplasmic inclusions with “single-unit” acid phosphatase–positive granules or lysosomes resembling macrophages.
The lamina propria is separated from the underlying smooth muscle cells of the media usually by a well-developed internal elastic membrane of tenacious matrix containing fibrils about 500 angstroms (Å) in diameter with abundant fenestrations. This internal elastic membrane is usually wavy in cross sections, and the fenestrations are oval or round openings extending across its surface. The underlying tunica media is characterized by concentric layers of smooth muscle cells 10 to 25 µ in length and oriented transversely to the artery’s main axis. Individual smooth muscle cells are surrounded by a network of collagenous and elastic fibers, which continue without transition into both internal and external elastic membranes. The external elastic membrane separates the media from the adventitia and is characterized by the presence of loosely packed collagen and elastic fibers. Small blood vessels or vasa vasorum are also found, as well as both sympathetic and parasympathetic nerve fibers from the autonomic nervous system.
These morphologic and functional characteristics of the coronary arteries are modified early in the presence of the intimal changes characteristic of atherosclerosis. The most important clinical difference between coronary and aortic atherosclerosis is the high incidence of acute thrombosis in coronary vessels, with infarction resulting in irreversible damage to the underlying myocardium.
Pathogenesis of Atherosclerosis
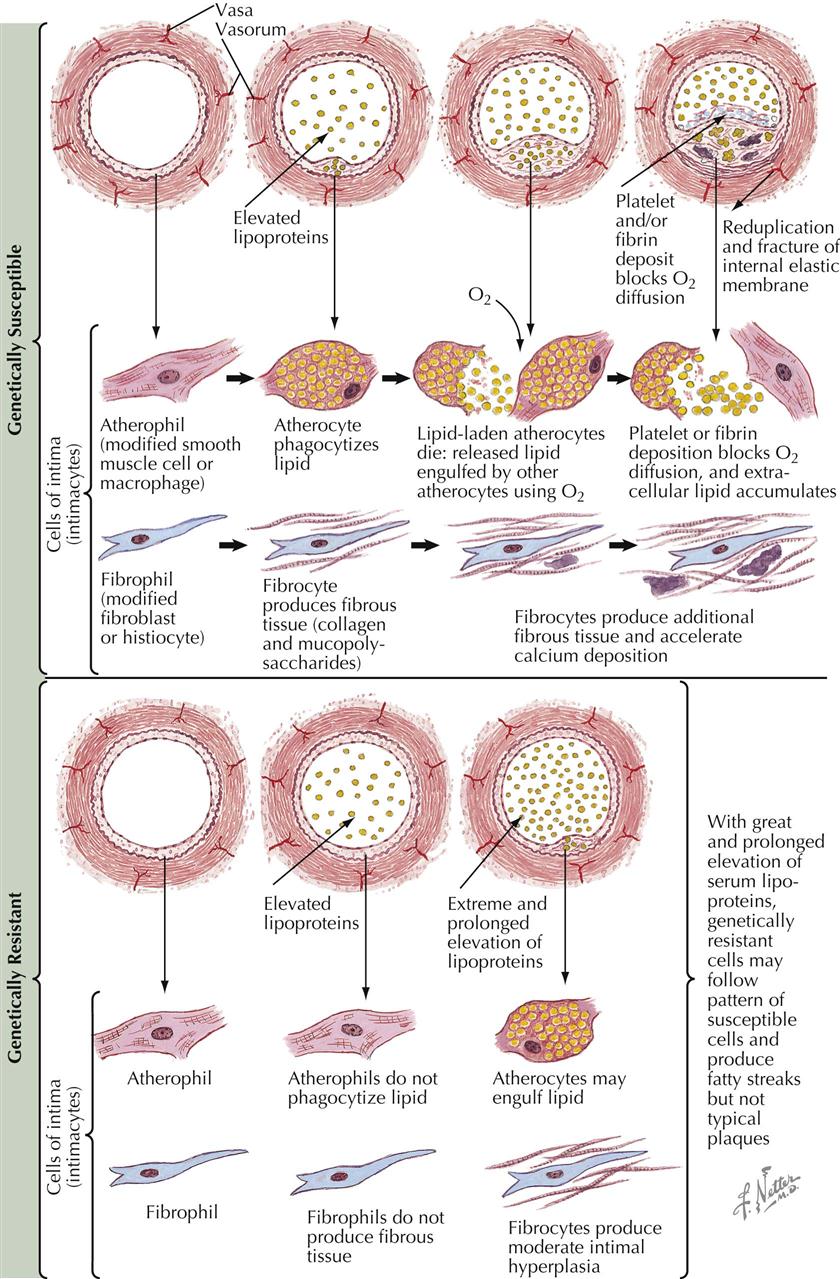
Histologic and functional characteristics of the intimal lining of the arteries help in understanding the histogenesis of spontaneous atheromas, often initiated shortly after birth, particularly the presence of different cell types in a ground substance matrix and the absence of a direct blood supply. The well-developed atherosclerotic plaque resulting from inflammatory and reparative processes is a complex lesion containing extracellular deposits of calcium salts, blood components, cholesterol crystals, and acid mucopolysaccharides. The initial changes, however, seem to occur at the cellular level, often accompanied by an abnormal intracellular storage of lipids, particularly cholesterol esters, fatty acids, and lipoprotein complexes. These findings on microscopy have strengthened the thesis that lipid infiltration from the bloodstream may be a significant factor in the growth of the atheromatous plaque. Furthermore, recognition by cytochemical techniques makes lipids helpful indicators of abnormal cell behavior, independent of their role in the etiology of atheroma.
Because of the frequency and severity of atherosclerosis, short-term organ culture techniques for the isolation of arterial intimal cells (intimacytes) identify susceptible cell populations, based on their response to incorporation of homologous serum lipids in vitro. Intimacytes from arteries, with and without histologic evidence of atherosclerosis, show two types of atherophils: genetically highly susceptible cells and genetically resistant cells to intracellular lipid accumulation.
In the presence of increased perfusion rates of plasma lipoproteins caused by local hemodynamic changes, hypertension, elevated serum lipids, or changes in endothelial surface permeability (trauma, fibrin deposition, platelet aggregation), genetically susceptible atherophils will be transformed into lipid-laden atherocytes. The secondary release of intracellular lipids from these cells will induce surrounding atherophils to incorporate them, creating a self-feeding process with cytologic changes that increase metabolic requirements, including oxygen consumption rates, which result in increased permeability of the cell membrane to lipids if not met. These events set up a cycle favoring further expansion of focal atherosclerotic changes. Collagen fibers are laid down by fibrophils, which are then transformed into fibrocytes surrounded by acid mucopolysaccharide deposits. This eventually results in a characteristic intimal hyperplasia after ground substance changes. These histologic lesions are self-perpetuating, resulting in the replacement of intimacytes by an acellular area of arterial intima that stimulates further surface involvement of the vascular wall in this interplay among deposition, inflammatory response, and scarring, with the eventual production of a typical atherosclerotic plaque.
In contrast, genetically resistant atherophils, even in the presence of elevated serum-lipid levels, do not respond (or respond minimally) to an increased perfusion of plasma lipids. If some atherocytes appear, they are few in number, and only superficial fatty streaks are developed, resulting in sparse intimal elevations with few fibrophils and no well-organized plaque. This type of cytologic response also seems less susceptible to the clinical complications of atherosclerosis, particularly thrombosis or rupture, because of the limited involvement of the vascular wall.
This approach to the pathogenesis of atherosclerosis emphasizes the clinical significance of the earliest possible identification of patients whose arteries are susceptible to atheroma, to modify or prevent environmental conditions that favor the acceleration of atherogenesis in the arterial intima cells. Local factors are also important in identification of the lesions, suggesting possible therapeutic approaches for their arrest or inhibition.
Risk Factors in Etiology of Atherosclerosis
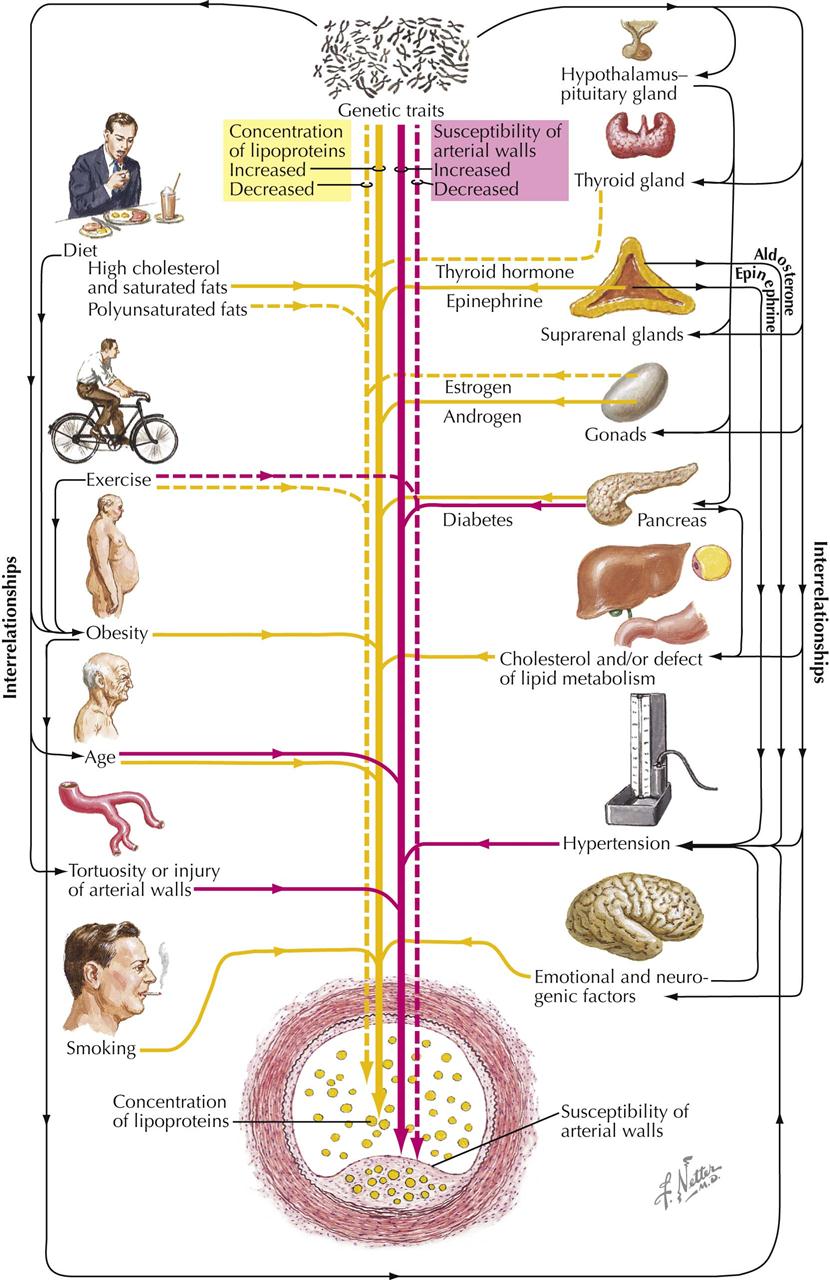
In terms of mortality, the most serious problem facing more developed countries is atherosclerosis of the cardiac and cerebral blood vessels. There is no single cause of atherosclerosis, which is often labeled a “multifaceted disease.” Atherosclerosis reflects the culmination of many factors acting over a lifetime and usually becomes clinically manifest only when angina pectoris, sudden cardiac death, myocardial infarction (MI), transient ischemic attack (TIA), or cerebrovascular accident (CVA, stroke) occurs. Heart failure is usually caused by MI.
Patients with a family history of premature manifestations of atherosclerosis may be at high risk for development of atherosclerotic disease. Older age, male gender, hypertension, diabetes, obesity, and tobacco smoking are associated with coronary atherosclerosis. Lack of exercise may interact with other clinical factors, and an exercise program (e.g., walking) is known to diminish symptoms in patients with known clinical manifestations of atherosclerosis. Excessive saturated fat in the diet is also involved in atherogenesis. High-fat diets increase blood lipid levels, particularly low-density lipoprotein (LDL) cholesterol. Elevated triglycerides and low levels of high-density lipoprotein (HDL) are also implicated in the etiology of atherosclerosis. Elevated serum uric acid may also contribute to atherogenesis, although further study is required. Unsaturated fatty acids with two or more double bonds help reduce cholesterol blood levels when their relationship to saturated fatty acids is increased.
Current concepts of atherogenicity are complex and involve monocytes, platelets, macrophages, foam cells, endothelial cells, smooth muscle cells, oxidized LDL, and many other subcellular substances. Interested readers should refer to current literature and textbooks on the subject. In general, the physician or clinician can profile a patient potentially prone to atherosclerosis, such as a hypertensive man who smokes cigarettes, exercises little, and has elevated blood cholesterol and triglyceride levels, low HDL levels, and abnormal glucose metabolism. Currently, in addition to diet and exercise, drugs used to treat lipid abnormalities include HMG CoA reductase inhibitors (statins), nicotinic acid preparations, ezetimibe, fibric acid derivatives, and bile acid sequestrants.
Pathologic Changes in Coronary Artery Disease
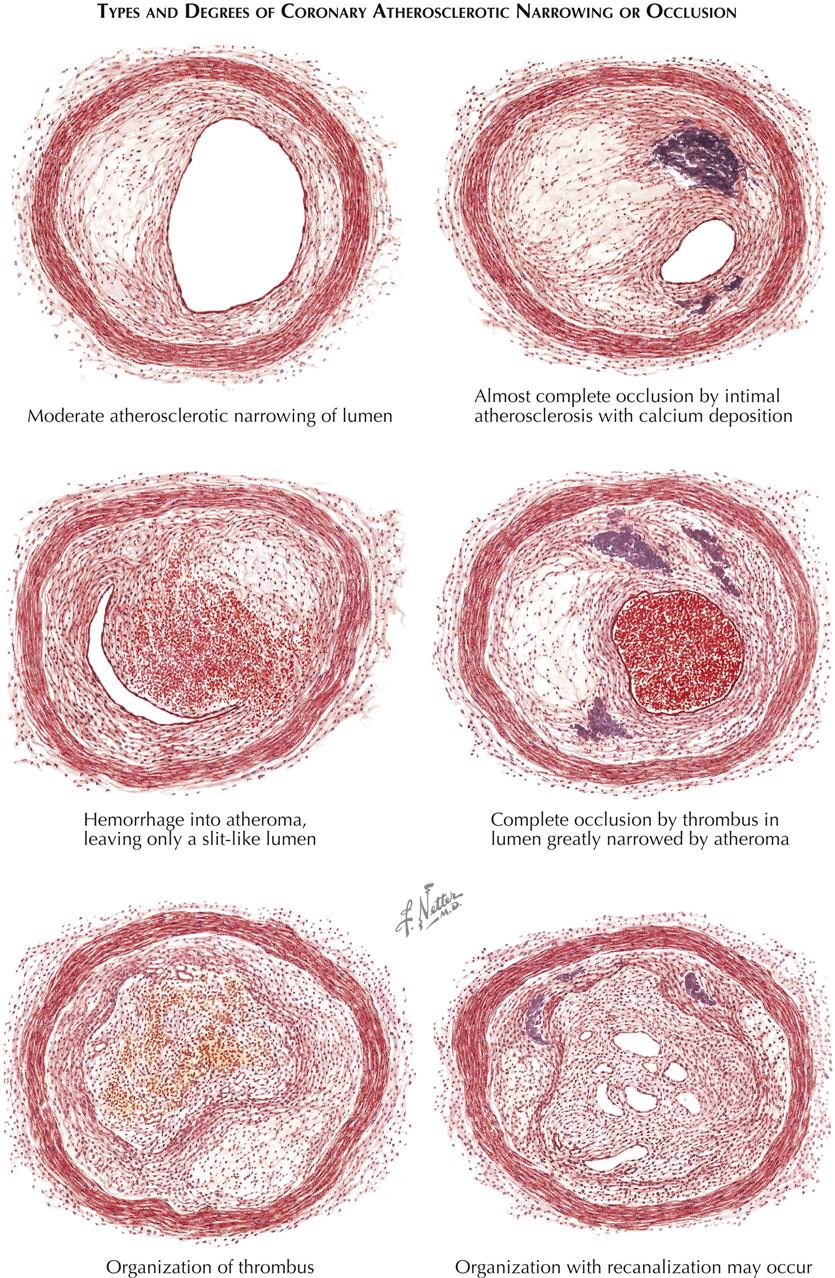
The earliest recognizable atheromatous changes in blood vessels are accumulation of aggregates of lipid-laden macrophages in the intima. The appearance of this lesion is similar to that seen in many experimental vascular lesions. Another apparently early phenomenon, difficult to correlate and possibly unrelated, is a subintimal deposit of collagen that often is primitive and thus rich in acid mucopolysaccharides. Lesions of this type shadow atherosclerosis wherever it occurs. The relationship of this vascular lesion to the type seen in Asian peoples has not been elucidated, but this disorder does occur in Asians. A further complication of this lesion is the deposition of fibrin on or within its superficial layers.
As more lipid accumulates, an atheromatous plaque with many constituents is formed. This plaque may involve only a segmental portion of the artery or its entire circumference. When the macrophages die, their lipid is released and becomes an inflammatory irritant. Fibrin accumulates, as do other blood constituents; calcium deposits form; and fibroblasts proliferate. Stenotic lesions may accumulate more surface material with deposited layers of thrombus. These lesions regress with lipid-lowering agents and antiplatelet therapy.
Even a segmental atheroma can cause atrophy of the underlying arterial wall, and the concentric type seems associated with major destruction of the lamina and parts of the media. Such changes appear secondary to interference with the nutrients permeating the intima. The stages of atheroma reveal another major feature in the lesion’s development: an inability to clear blood constituents that have permeated the media. This dynamic event is demonstrated by perfusing isolated segments of living canine arteries and observing the quantity of lipid in the vasa vasorum of the adventitia.
Hemorrhage into the lesion can result from surface blood dissecting into the plaque or from vasa vasorum bleeding. Thrombosis may occlude the artery; if the person survives, organization of the fibrin clot can progress and occasionally recanalize the artery. However, the ability of these small channels to supply any appreciable volume of blood beyond the area of obstruction is insufficient in most patients to revascularize the downstream myocardium. Rarely, an occluding thrombus completely disappears.
In addition to atheromatous and thrombus blockade of the coronary arteries, emboli can obstruct an otherwise normal coronary artery. Such emboli may consist of thrombi, valvular calcification, pieces of tumor, and occasionally even small foreign bodies. Various types of aortitis can involve the proximal vessels; syphilis of the aortic wall can occlude the ostia. The atheroma is the main disorder, however, because of its distribution. The lesions closest to the aorta (proximal) are the plaques most likely to impede flow to the cardiac microcirculation.
End-Organ Damage by Vascular Disease
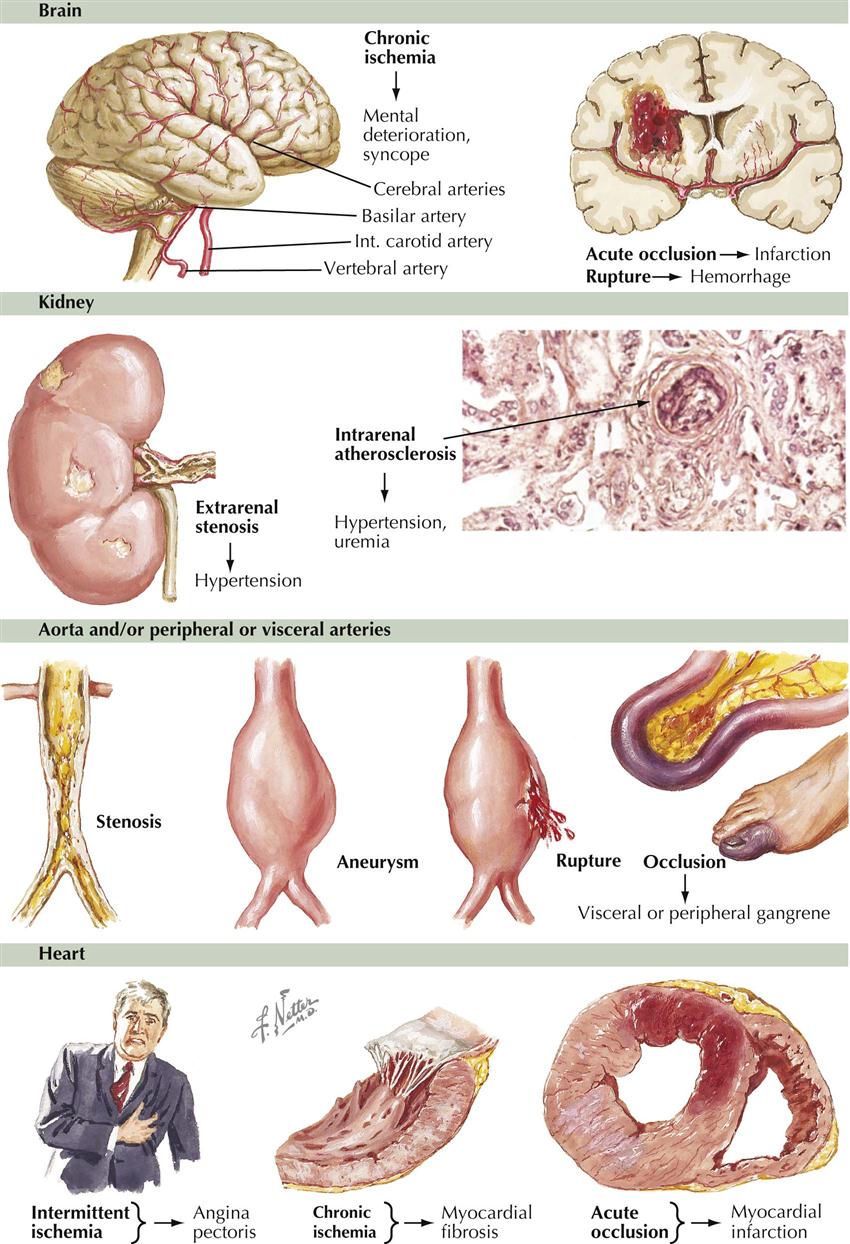
The cerebral circulation is particularly vulnerable in hypertensive patients. Stroke is the major cause of death. Hypertensive vascular disease is usually a combination of hypertension and atherosclerosis, the latter accelerated by elevated blood pressure. Infarction of the brain is more common than subarachnoid bleeding. Bleeding results from rupture of microaneurysms of the small arteries or from trauma. The coronary vessels are next most vulnerable. Angina pectoris and MI are common results of accelerated atherosclerosis, necessitating concurrent treatment of the hypertension and atherosclerosis. Many patients reduce their high blood pressure and return to normal levels, only to have an MI or die from complications of atherosclerosis. The results of the increased rate of atherogenesis are also seen in other large vessels besides the coronary arteries. Thus, aneurysms of the aorta and renal arteries are common, leading to distal embolization, dissection, and possible rupture. Reducing blood pressure has a beneficial action on these lesions. Drugs, surgery, and interventions are now available to treat the clinical effects of both hypertension and atherosclerosis, although the goal should be prevention.
Unstable Plaque Formation
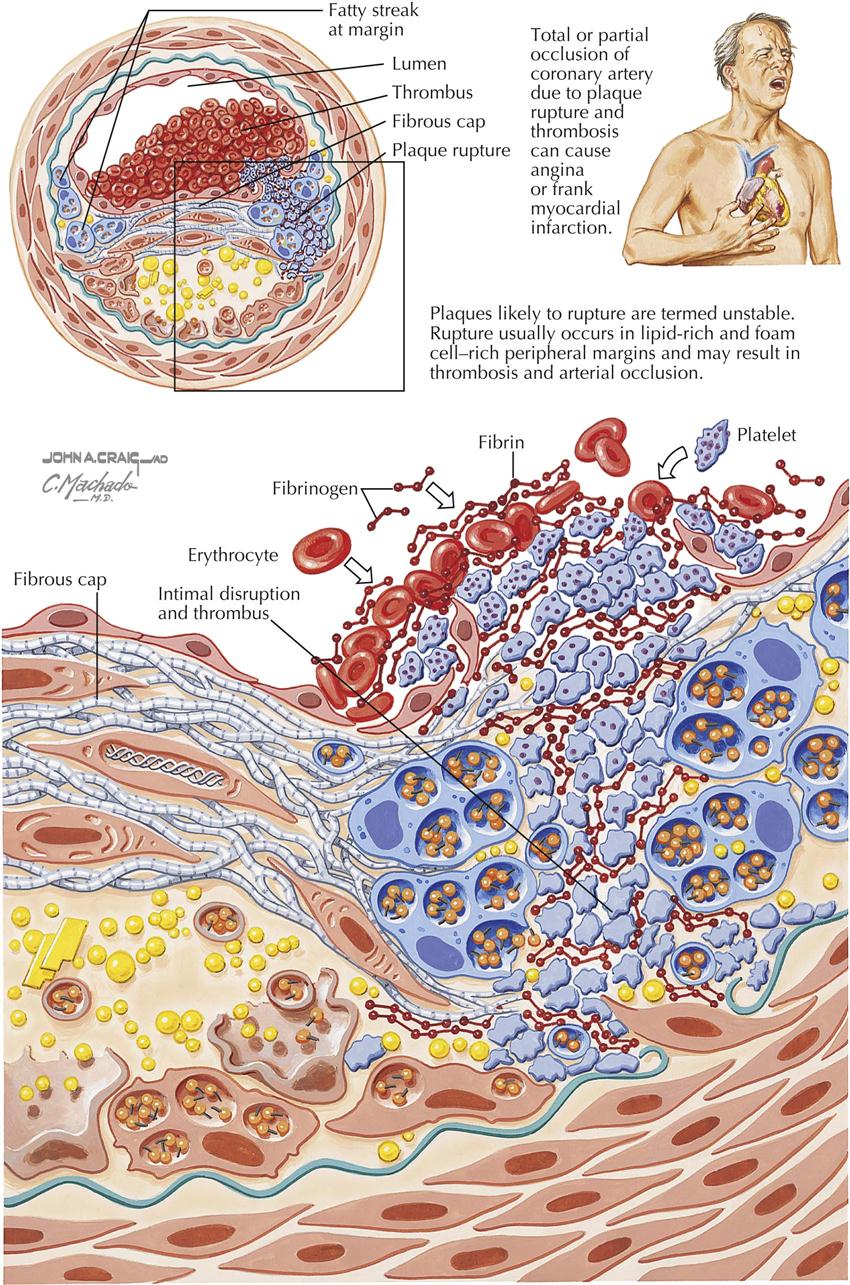
Plaques likely to rupture are called “unstable” and are the main cause of acute coronary syndromes. Plaques with a thin cap and a lipid core associated with inflammation and cap fatigue may be associated with rupture at the edges of the plaque. The plaques are rich in lipids and foam cells, found at the peripheral margins. On rupture, plaques extrude thrombogenic material, which then can trigger thrombosis on the plaque, in turn occluding the coronary artery and resulting in MI.
If the vessel only is almost occluded, a condition known as unstable angina may result. Risk factors such as cigarette smoking, hypertension, diabetes mellitus, and dyslipidemia contribute to endothelial injury, causing smooth muscle cell proliferation, inflammation, and deposition of lipid within the blood vessel wall. These events may lead to the development of unstable plaques. Other physical factors may alter plaque composition and make a plaque vulnerable to rupture; cold temperature (32°-34° C) may promote the crystallization of liquid cholesterol, and these sharp crystals may perforate the cap. Cytokines, growth factors, and oxidative stress are other factors in progression of atherosclerosis and instability.
Thrombosis related to these plaques may be caused by endothelial erosion or plaque disruption. In general, plaque erosion occurs over high-grade stenoses, whereas thrombosis related to plaque disruption is often seen in patients with few stenoses. In addition, multiple triggers of plaque disruption are probably related to increased tension, bleeding originating from the vasa vasorum, bending and twisting of the arteries, and increased systolic and pulse pressures.
Angiogenesis and Arteriogenesis
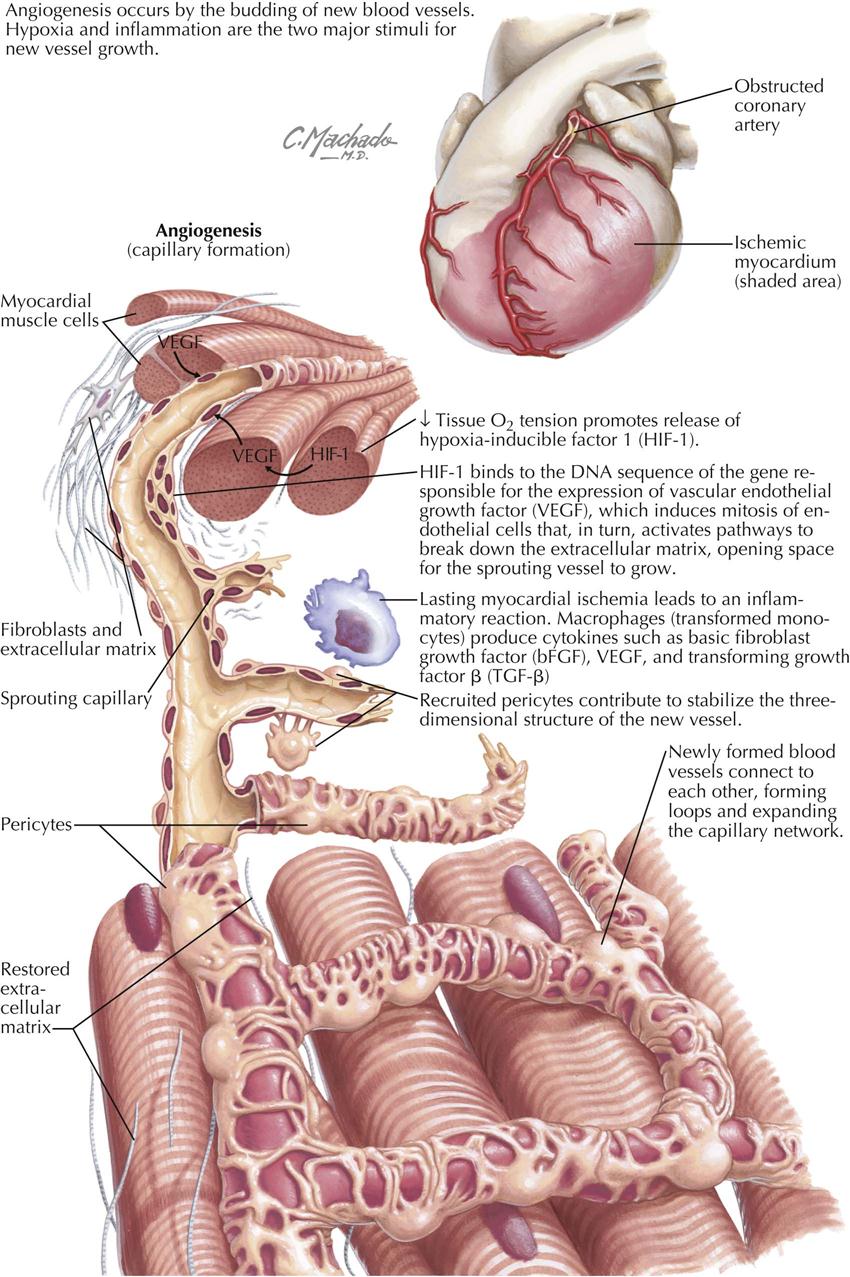
Unfortunately, the terms angiogenesis and arteriogenesis are often are confused and interpreted to have the same meaning. These processes are really two separate entities representing blood vessel changes in the heart.
Angiogenesis
Angiogenesis is essentially the budding of new blood vessels from sprouts of existing vessels in order to expand the capillary network (the microcirculation). Current investigations focus on creation of new blood vessels in areas of critical need, as in patients with large areas of infarcted myocardium surrounded by ischemic zones. The two major stimuli for new vessel growth are hypoxia, which activates hypoxia-inducible factor-1, and inflammation, which stimulates macrophages to secrete cytokines in MI patients, including basic fibroblast growth factor (bFGF) and transforming growth factor beta (TGF-β). As a result, newly formed blood vessels arise. This is particularly evident in animal models and is being tested in human trials (e.g., stem cell therapy in MI patients).
Arteriogenesis
In arteriogenesis, collateral vessels are recruited to increase blood flow to ischemic tissue. Coronary artery collateral vessels are thought to be stimulated and generated as a result of “demand ischemia” in areas of the myocardium. For example, the distal portion of a left anterior descending (LAD) coronary artery with high-grade stenosis or obstruction may receive blood from the right coronary artery (RCA) through septal collaterals or from the posterior descending coronary artery. Another potential source of collateralization is from the conus branch of the RCA, essentially one coronary artery supplying another artery, either by capillary-sized (microcirculation) collaterals or large muscular collaterals that are epicardial vessels (e.g., conus artery of RCA). This blood vessel is known to provide collateral blood flow to a severely stenotic proximal LAD artery.
Collaterals are not seen in patients without evidence of myocardial ischemia, possibly because no dormant collateral vessels are present. Also, in patients with balloon occlusion of an epicardial vessel and resultant myocardial ischemia, collateral flow through a previously dormant collateral vessel can be seen immediately. This indicates that these collateral vessels are dormant and visualized only when there is demand ischemia. Collateral vessels easily visualized at coronary angiography are categorized under arteriogenesis.
Overview of Myocardial Ischemia
In patients without myocardial ischemia, myocardial oxygen demand is balanced by myocardial oxygen supply. Determinants of myocardial oxygen supply are related to aortic diastolic pressure, left ventricular end-diastolic pressure (LVEDP), and coronary artery resistance. Determinants of myocardial oxygen demand are related to heart rate, systolic blood pressure (BP), left ventricular (LV) tension, and LVEDP. Myocardial ischemia related to balloon occlusion of a coronary artery involves a sequence of decreased coronary blood flow, as detected by perfusion scintigraphy; altered regional flow/demand ratio resulting in decreased coronary sinus oxygen saturation; LV diastolic relaxation abnormalities, detected by cardiac ultrasound; systolic contraction abnormalities, detected by cardiac ultrasound or angiography; increased LVEDP; ST-segment depression on electrocardiography; and finally angina pectoris or its equivalent symptoms (e.g., breathlessness).
At the cellular level, the current hypothesis is that myocardial ischemia increases the late sodium current, which increases the sodium content of the myocardial cells. The sodium/calcium exchanger then increases the calcium content of the cell. As a result, the LV stiffens, increasing LVEDP, which decreases endomyocardial blood flow, propagating the myocardial ischemia.
Myocardial ischemia and its usual manifestation angina pectoris result from an imbalance between myocardial oxygen supply and myocardial oxygen demand. Myocardial ischemia manifested by angina pectoris can be acute or chronic. In general, patients with an acute clinical presentation have a crescendo pattern of angina and are considered to be in an unstable or “acute coronary syndrome” state.
Although most patients with myocardial ischemia are symptomatic, 25% may be asymptomatic. However, most symptomatic patients have many episodes of asymptomatic myocardial ischemia. Laboratory manifestations of myocardial ischemia other than electrocardiographic (ECG) changes are abnormalities of perfusion, as indicated by nuclear studies, and transient regional wall motion abnormalities, usually detected by echocardiographic studies or other noninvasive imaging techniques (e.g., MRI). Clinical manifestations of myocardial ischemia are generally chest discomfort (angina pectoris), arrhythmias, and LV dysfunction in both systole and diastole.
Angina Pectoris
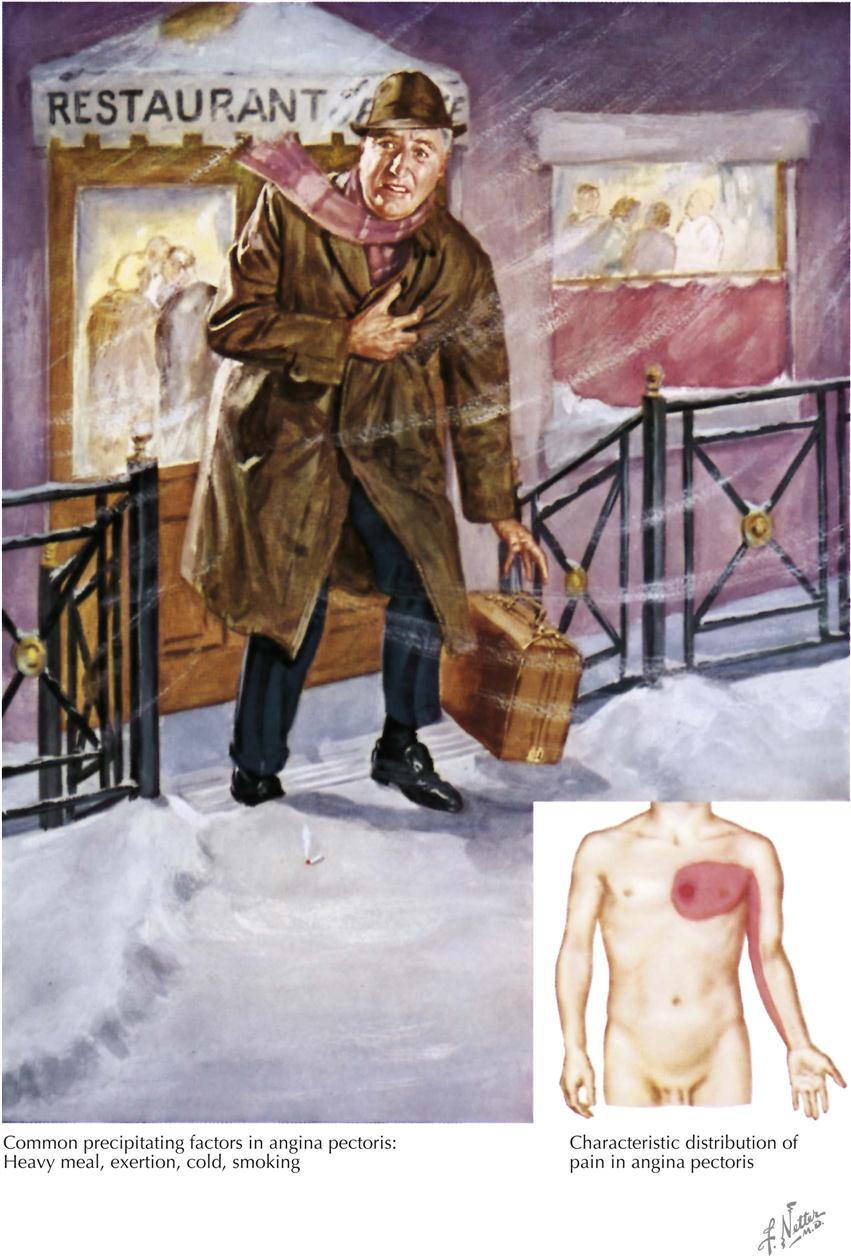
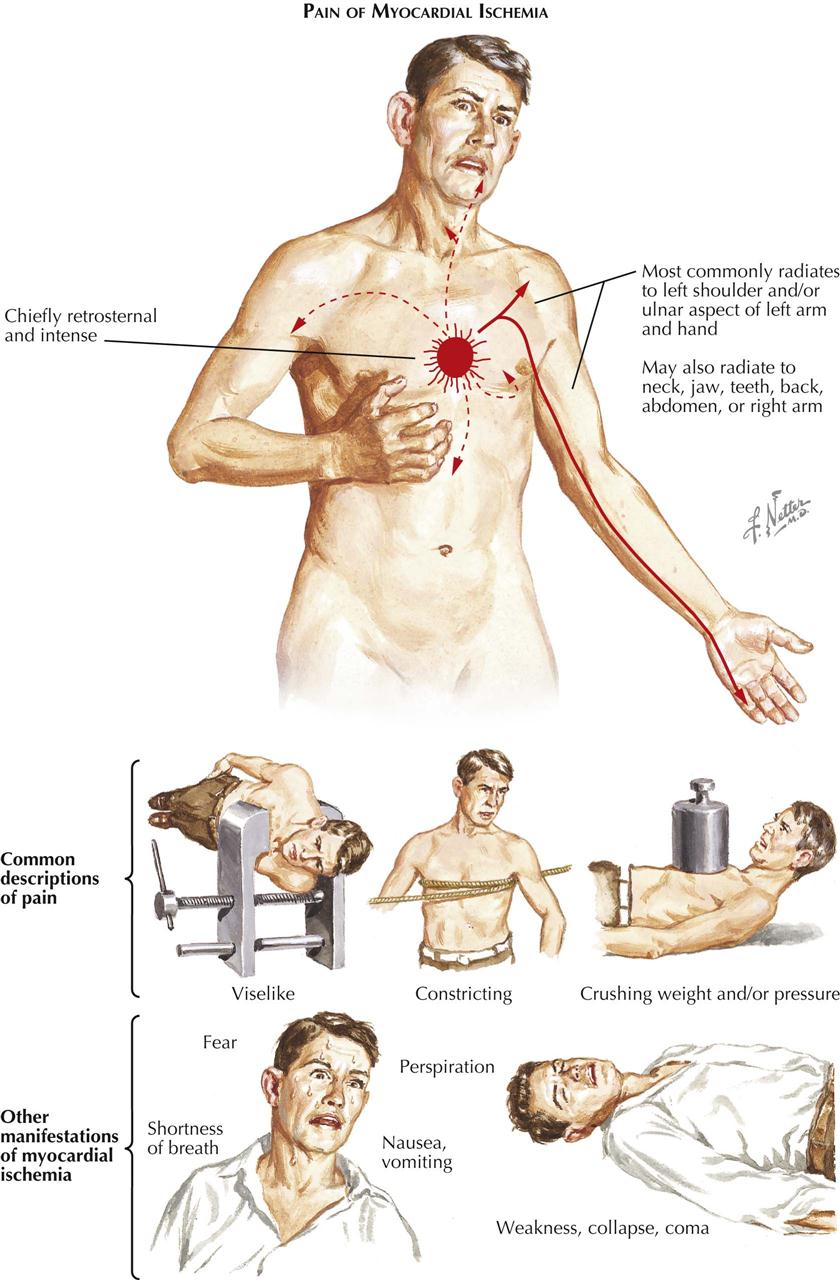
Angina pectoris (“strangling of the chest”) was first described as a serious symptom by Heberden in 1768, although he did not associate angina with coronary artery disease, as discovered later by Jenner. Angina is usually of short duration, associated with effort or emotion, and usually relieved in minutes by rest or sublingual nitroglycerin administration.
Angina pectoris varies somewhat from patient to patient, but the usual description of symptoms includes a feeling of a heavy weight, oppression, or a choking sensation under the middle of the chest and pain extending occasionally to the arms, especially the left, and almost never to the back and rarely to the neck and jaw. Many patients do not describe angina as a pain, but rather as a discomfort in the chest. It is produced, as a rule, by effort or emotion, especially in cold weather and particularly after meals or smoking. Angina rarely lasts more than 2 or 3 minutes and is relieved in a few minutes by sublingual nitroglycerin, also often used preventively, particularly if the patient knows a given situation will provoke cardiac pain. Angina must be differentiated particularly from gastrointestinal, musculoskeletal, and costochondritic origins of the discomfort.
Angina pectoris and the pain of acute MI are caused by the same disease process, but the prolonged pain in angina is associated with coronary artery occlusion and not the coronary artery stenosis of MI. The prognosis in the patient with angina pectoris varies greatly, but with medical management, many patients can continue to lead a normal life. Over months or years, their angina pectoris may diminish or disappear through natural development of collateral circulation to the ischemic zone of myocardium. However, angina is always an important symptom, and may be life threatening because of the potential for ventricular arrhythmias. Treatment of angina pectoris always includes medical therapy and often includes percutaneous coronary intervention (PCI, angioplasty/stent) or surgical bypass of a stenotic artery. Diet, blood pressure control, lipid management, nitrates, beta blockers, angiotensin-converting enzyme (ACE) inhibitors, aspirin or other antiplatelet agents, smoking cessation, and exercise are considered optimal therapy before and after a revascularization procedure.
Prevention of the underlying disease that causes angina pectoris is paramount. For those at risk, as shown by family history and high serum cholesterol, the avoidance of obesity, a diet low in animal fat, routine exercise (if no heart disease), and smoking cessation deserve first priority in any health promotion and heart disease prevention program and should start in childhood.
Detection of Myocardial Ischemia
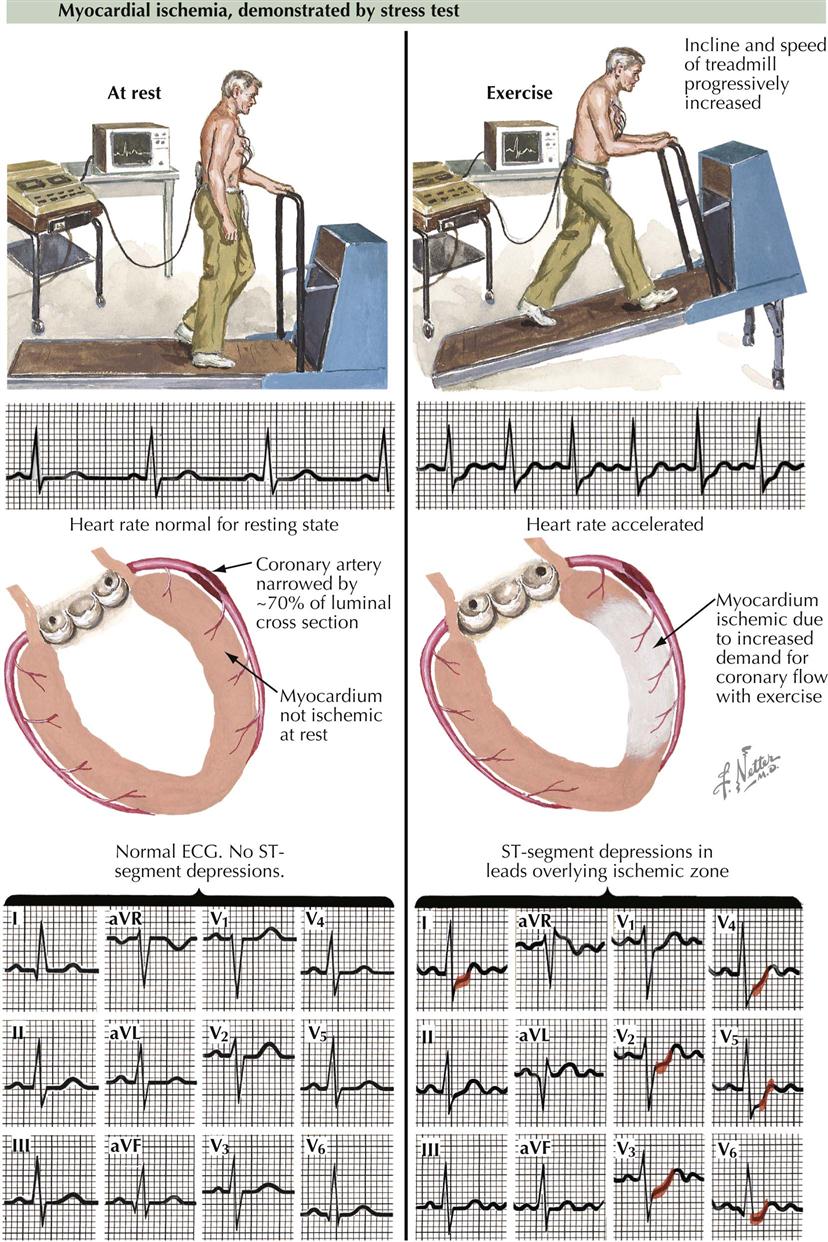
The simplest and least expensive way to diagnose the presence or absence of myocardial ischemia and to quantitate functional impairment in patients with chronic stable angina is exercise stress testing with ECG monitoring. Exercise testing is typically performed using a treadmill or bicycle to provoke chest discomfort and obtain diagnostic evidence of myocardial ischemia. It is important to record 12 leads before, during, and after exercise testing in order to detect transient changes suggesting ischemia. Under resting conditions the heart rate is usually normal and the ST segment on the electrocardiogram usually isoelectric. Thus the 12-lead ECG may be normal even if a coronary artery has 70% stenosis, the usual percentage that results in myocardial ischemia when myocardial oxygen demand is increased. The ECG is normal because the myocardium is not ischemic under these resting conditions; myocardial oxygen supply is balanced by demand, and the patient does not complain of angina pectoris or equivalents such as breathlessness. The treadmill increases speed and incline progressively, thus stressing the patient, who needs to increase heart rate and blood pressure to maintain appropriate cardiac output to meet the demand. During bicycle exercise, resistance to pedaling is increased to accomplish the same increase in heart rate and blood pressure. Patients who develop myocardial ischemia show the classic ECG changes of ST-segment depression. In the patient with 70% stenosis, under conditions of increased myocardial oxygen demand, the balance of oxygen supply and increased demand will be upset, and myocardial ischemia will result. In some patients, myocardial ischemic changes on the ECG may not be accompanied by angina pectoris, termed silent myocardial ischemia. Despite the lack of symptoms, the prognostic importance of silent or asymptomatic myocardial ischemia is the same as symptomatic disease.
Chronic stable angina is often called stable ischemic heart disease. Myocardial ischemia results when the oxygen needs of the myocardium exceed the supply of oxygen derived from coronary blood flow. The oxygen needs of the myocardium are increased by aggravating or precipitating factors such as hypertension, tachycardia, heart failure, hypermetabolic state, and sympathomimetic drugs. Myocardial oxygen demand depends on heart rate, blood pressure, and LV size, thickness, and contraction. Other factors, such as severe anemia, may result in tachycardia and increased oxygen demand.
Degree of Flow-Limiting Stenoses
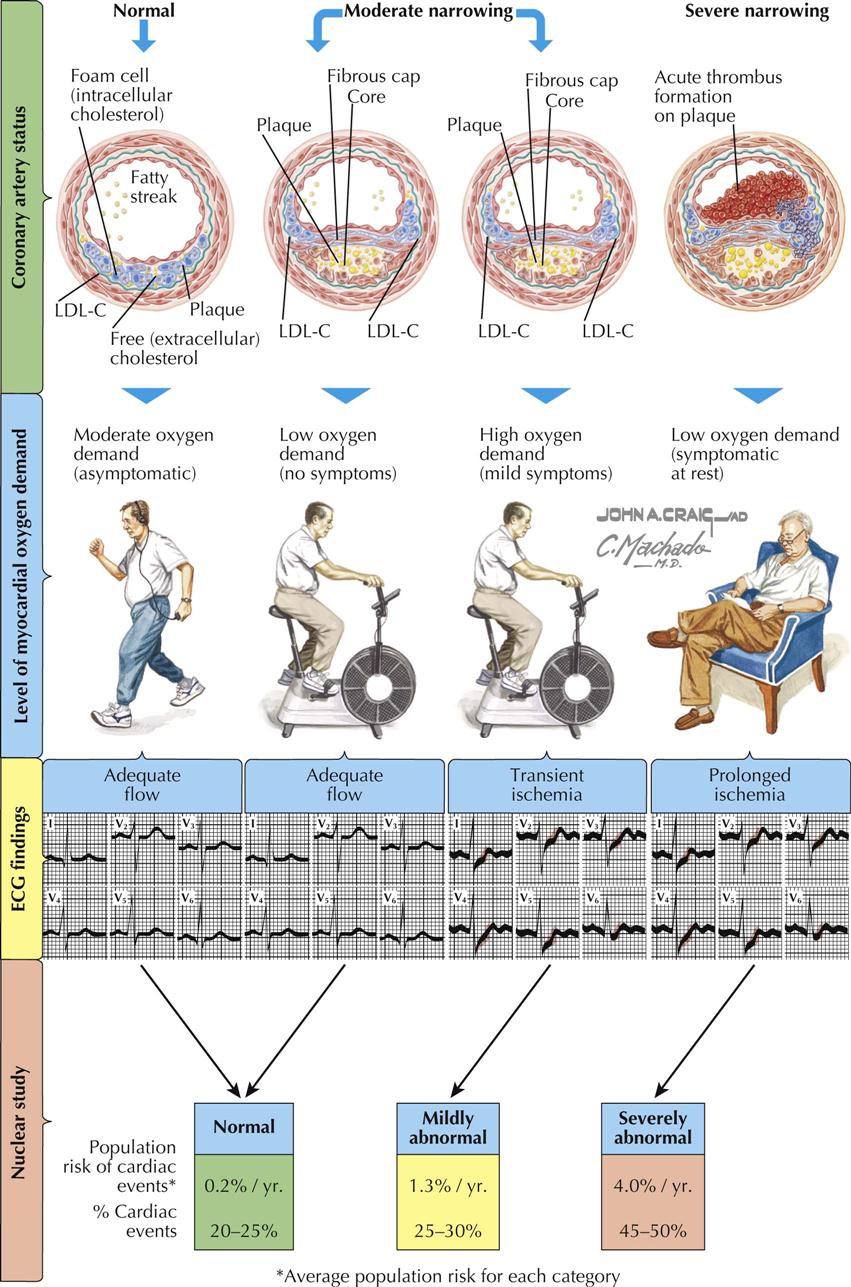
Patients with normal coronary arteries have no flow-limiting epicardial stenoses and remain asymptomatic under conditions of moderate oxygen demand, or even high oxygen demand in most cases. Patients with moderate narrowing caused by coronary atherosclerosis may also be asymptomatic or symptomatic (angina, myocardial ischemia) depending on the degree of flow-limiting stenosis to the myocardium. When flow is adequate under moderate myocardial oxygen demand, the ECG may be normal with isoelectric ST segments in all 12 leads. However, when there is high myocardial oxygen demand in this same patient with moderate coronary atherosclerosis, ECG changes in the appropriate leads (e.g., aVL, V2-V6) in patients with left-sided coronary disease demonstrate abnormal ST-segment shifts during transient ischemia and abnormal changes during prolonged ischemia at rest. In Plate 6-11, LDL-C refers to low-density cholesterol.
Thus a normal ECG correlates with widely patent coronary arteries providing adequate flow and no evidence of ischemia. Similarly, moderate coronary artery disease (CAD) under mild stress conditions shows no ECG changes. Under high oxygen demand in patients with moderate CAD, however, transient ST-segment shifts can be found. In addition, patients with severe narrowing often have symptoms and ECG changes under resting or near-resting conditions or during trivial exercise, particularly with severe high-grade proximal stenoses (e.g., left main or proximal LAD stenosis). These patients usually have severe proximal multivessel epicardial CAD.
If done to detect myocardial perfusion abnormalities, nuclear imaging studies are usually normal if the coronary arteries have mild or no stenosis. If there is moderate stenosis with high oxygen demand, myocardial perfusion may be mildly abnormal. The treatment and prognostic implications are related to the severity and extent of the perfusion defects. For severely abnormal myocardial perfusion in patients with high-grade stenoses, aggressive medical therapy is often combined with PCI (angioplasty with stenting) or bypass surgery.
To make appropriate decisions about prognosis and therapy in the patient with symptomatic CAD, knowledge of coronary pathology must be coupled with physiologic assessment of the lesions.
Left-Sided Heart Angiography
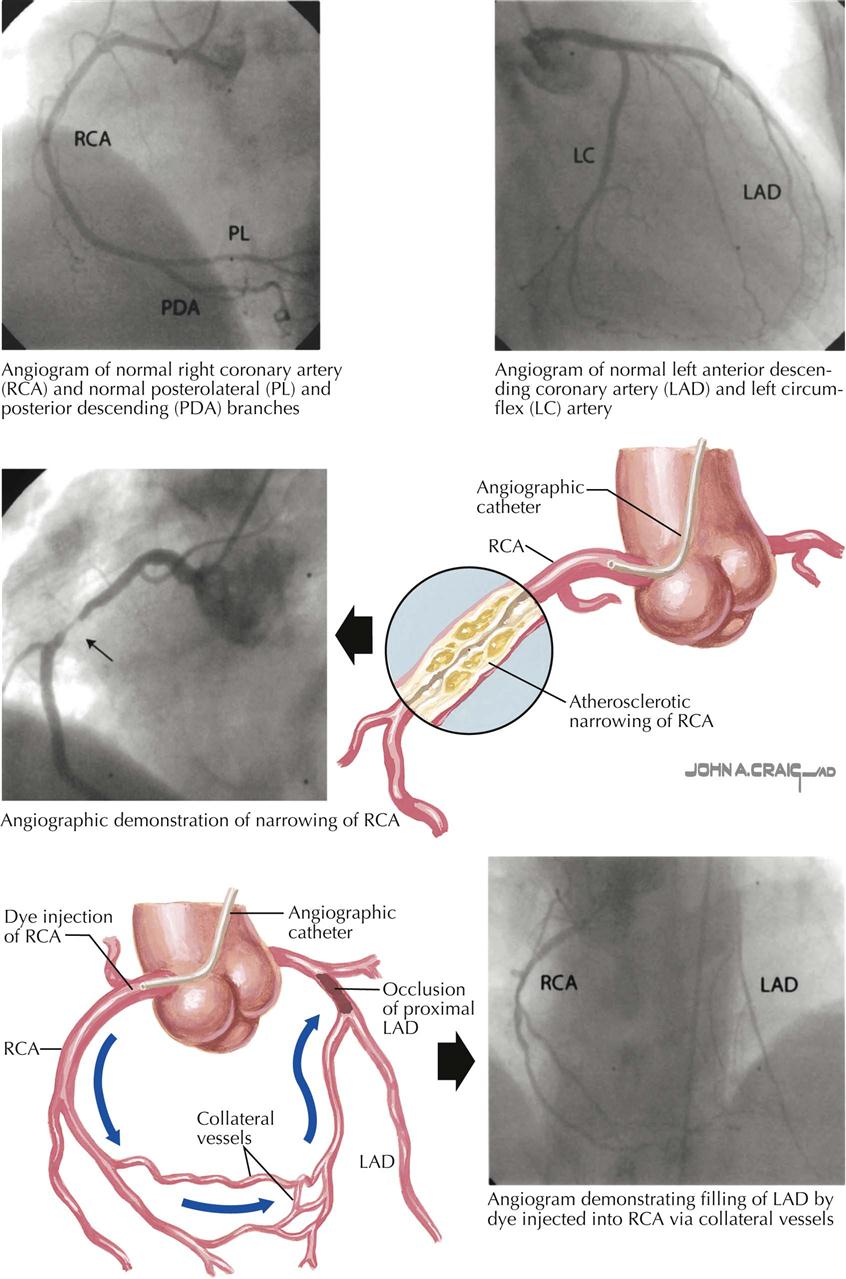
Left-sided heart catheterization and angiography includes injection of iodinated radiopaque contrast into the aorta, left ventricle, and coronary arteries to visualize the status of the aorta and its branches, presence or absence of aortic insufficiency, patency of conduit coronary bypasses, LV function and anatomy, and condition of the coronary circulation. Coronary angiography is a precise method of determining the extent, location, and severity (percent stenosis) of CAD. Multiple views of the coronary arteries are necessary to assess the significance of a specific lesion and the distribution of the right coronary, circumflex, and anterior descending arteries and the septal and diagonal distal circulation. The classic views of the right and left coronary arteries are the right anterior oblique (RAO) and left anterior oblique (LAO) projections. Multiple angulated views (caudal and cranial) are particularly important if PCI (angioplasty) is planned. One must remember that catheter-based coronary angiography is lumenography; only the vessel’s lumen is seen. The vascular wall must be visualized by intravascular ultrasound or optical coherence tomography; despite its limitations, this is a prerequisite for coronary artery surgery and in many patients allows determination of management: medical therapy (alone or plus angioplasty/stent) or bypass surgery.
Ventriculography is part of the evaluation of patients undergoing coronary angiography, to obtain the best possible assessment of ventricular function. I prefer to have ventriculography performed in the RAO and LAO views to assess the global function of the ventricle, particularly in patients with LV dysfunction.
Fractional Flow Reserve
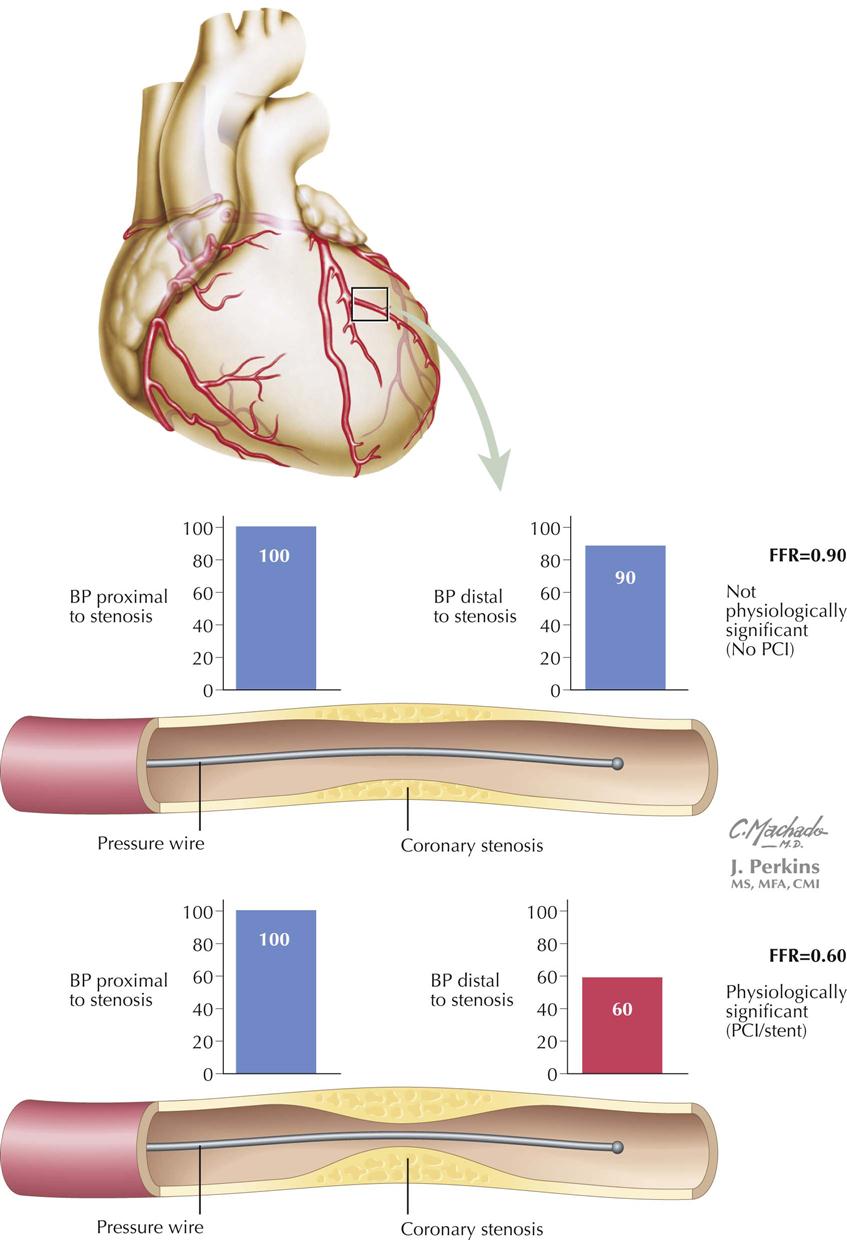
In the catheterization laboratory, interventional cardiologists can determine the physiologic significance of a specific coronary artery stenosis by passing a small wire (0.014-inch pressure guidewire) with a pressure transducer at the tip, through the stenosis in question, which allows them to measure systolic blood pressure (BP) proximal and distal to the stenosis. The systolic BP distal to the stenosis is divided by the pressure proximal to the stenosis; the resulting fraction is the percentage of BP decrease across the stenosis. Measurements are made at baseline and after infusion of adenosine, which increases coronary blood flow. For example, if BP across the stenosis drops by 50%, the fractional flow reserve (FFR) will be 0.50. The formula is FFR = Pd/Pa, where Pd is pressure distal to the stenosis and Pa is pressure proximal to the stenosis. This technique is particularly useful when the visual estimate of the clinical significance of a specific stenosis by angiography is uncertain.
If FFR is combined with intravascular coronary ultrasound, the interventional cardiologist will be able to determine the physiologic significance of the stenosis as well as the nature of the plaque being investigated, i.e., stable or unstable vulnerable plaque. FFR could also be called “functional flow reserve” because it supports that measurements reflect the pathophysiologic significance of a stenosis. The technique obviously requires cardiac catheterization and a specially designed pressure guidewire in which the pressure is measured proximal to and distal to a given epicardial coronary artery stenosis. Based on clinical trials, a cut-off point of 0.75—0.80 has been used to indicate a significant pathophysiologically important coronary artery stenosis. In my view, this measurement is equivalent to our measurements of noninvasively measured ankle-brachial index (ABI), in which a difference in pressure between the arm and leg suggests pathologic decrease in leg perfusion. An advantage is that this technique can provide immediate information for angioplasty/stent being considered for an epicardial stenotic vessel. If FFR is less than 0.75, the stenosis appears to be causing downstream myocardial ischemia, and angioplasty/stent seems appropriate. The DEFER clinical trial used FFR to determine if PCI with stenting was necessary in patients with FFR greater than 0.75. In these patients, stenting of the coronary stenosis did not influence clinical outcome. In another study, guided by FFR, fewer stents were used in patients who had FFR in the normal range. After 1 year, death, MI, or repeat revascularization occurred less in the FFR group, and hospital stay was shorter with less procedural cost.
Chronic Angina Revascularization Procedures
Revascularization involves either percutaneous coronary intervention (PCI), as first performed by Gruenzig in 1978, or coronary artery bypass graft surgery (CABG).
Percutaneous Coronary Intervention for Chronic Angina
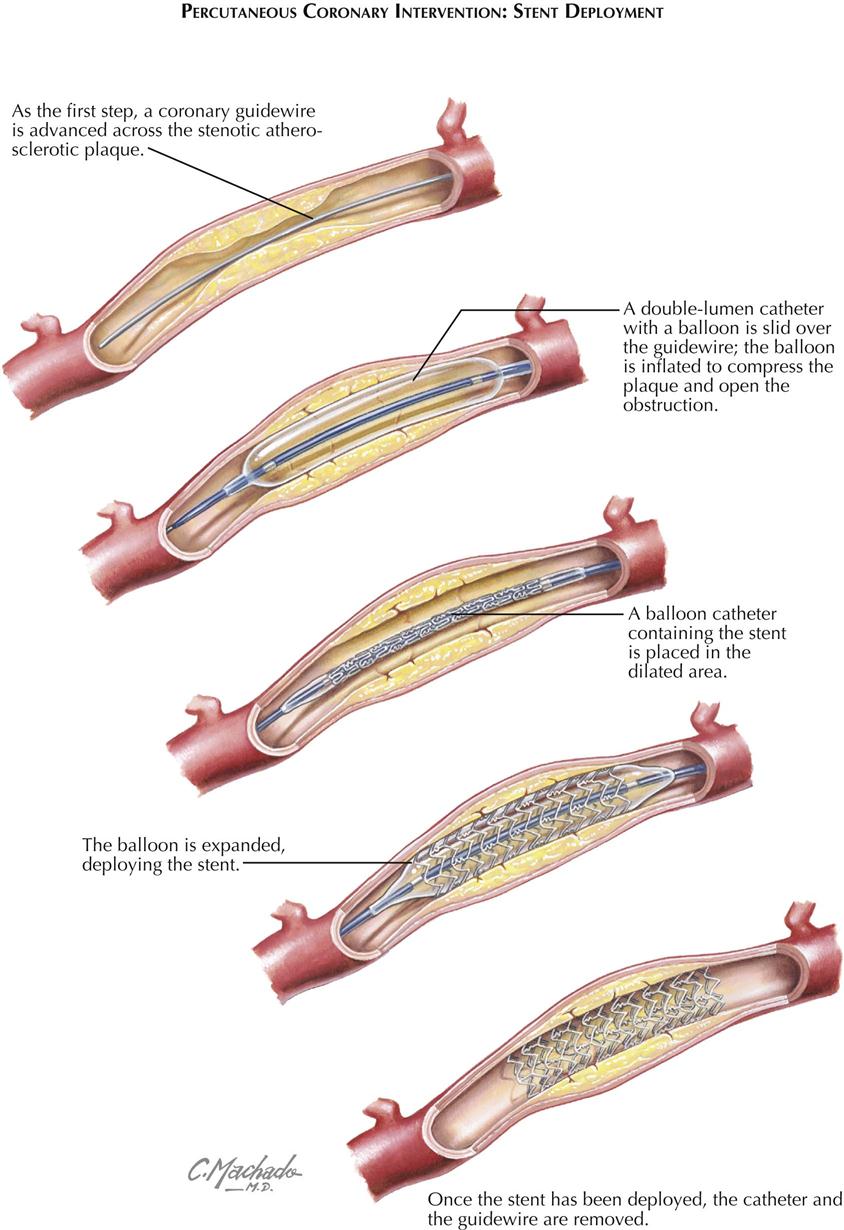
Stent Deployment
The PCI is performed through a guiding catheter introduced into the ostium of a coronary artery. A guidewire is advanced into the catheter and passed across the stenosis, and a balloon catheter is then placed at the stenosis. The balloon is inflated and the stenosis dilated. If a stent surrounds the balloon, as the balloon is inflated, the stent is deployed and forms a scaffold to maintain patency of the artery (see Plate 6-14). Stents can be bare metal (BMS) or drug eluting (DES). The balloon is deflated and removed, and the stent remains in place.
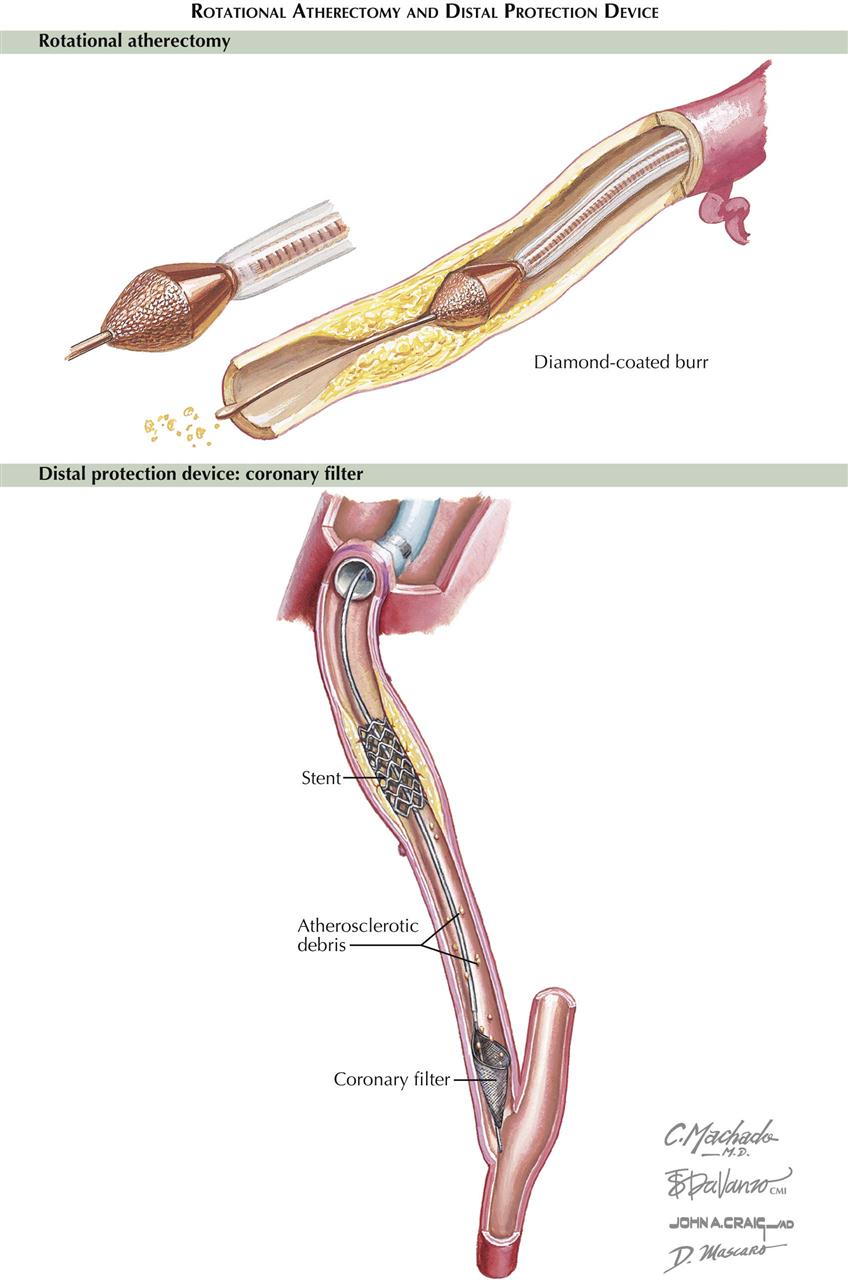
Rotational Atherectomy and Distal Protection Device
Other devices such as rotational atherectomy can be used before stenting, particularly in calcified stenoses (see Plate 6-15). A diamond-coated burr is used to fragment the plaque, widening the stenosis. Occasionally, distal protection devices are used to filter some of the detritus and atherosclerotic debris that migrate distally.
Surgery for Chronic Angina Disease
In 1968, Favaloro performed the first successful coronary artery bypass graft (CABG) at the Cleveland Clinic, where Sones had done the first selective coronary angiogram in 1958. Selective coronary angiography permitted for the first time a realistic appraisal of the patient suspected of having coronary disease because it established the presence or absence of disease, localized an existing myocardial-perfusion deficit, and assessed LV functional status. Postoperatively, Sones’ technique allowed assessment of the disease course as well as surgical success or failure. This procedure also led to percutaneous catheter-based myocardial revascularization procedures.
Before CABG was developed, several procedures were used, but none is still performed, including implantation of the internal thoracic artery directly into the myocardium (Vineberg procedure) and a Vineberg variant (Sewell procedure) in which the internal thoracic artery with a pedicle (including artery, vein, muscle, and fibrous tissue) was implanted into the myocardium.
Ventricular Aneurysmectomy
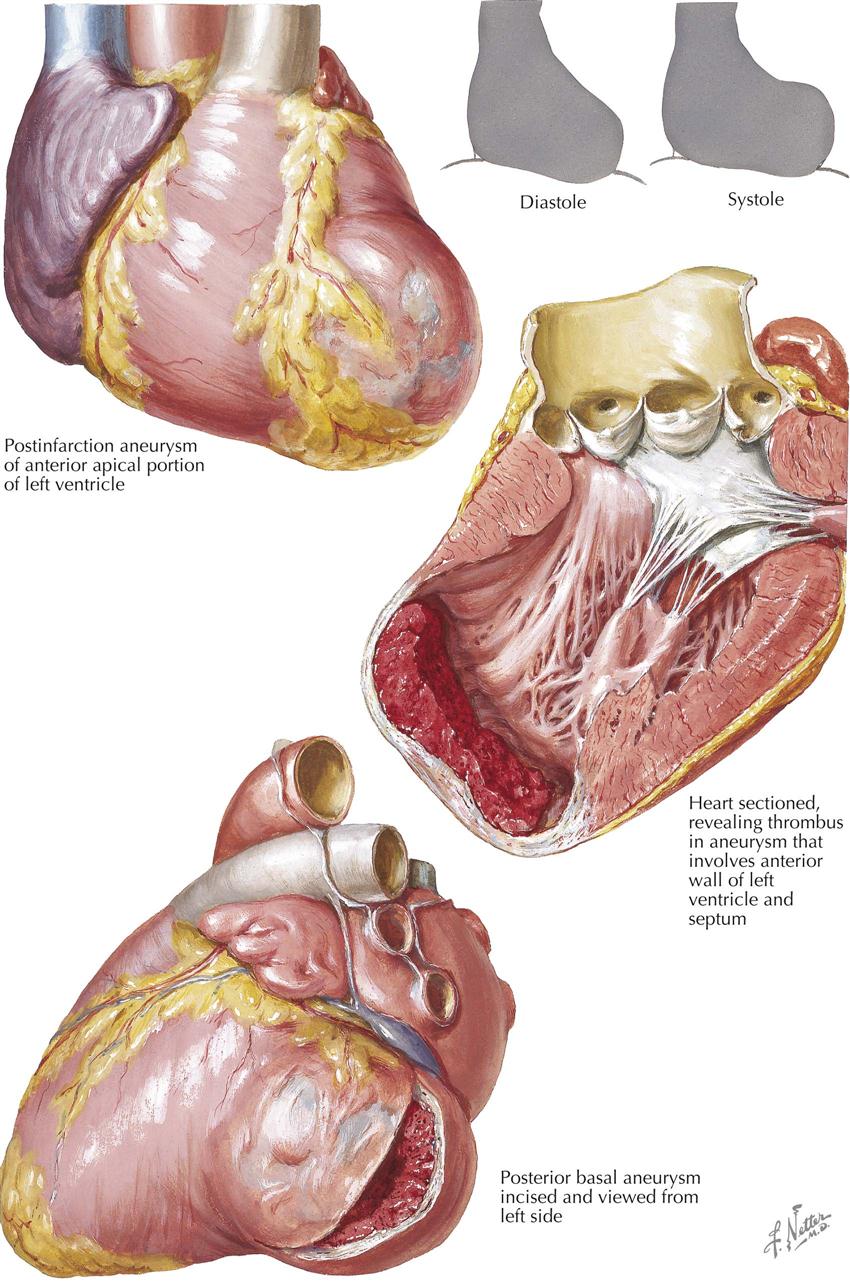
Ventricular aneurysm may occur in patients who sustain a large MI. Postinfarction aneurysm usually affects the anterior wall and the LV apex perfused by the left anterior descending branch of the left coronary artery. Aneurysm of the inferior left ventricle rarely occurs, although an infarction of sufficient magnitude to produce a ventricular aneurysm usually results in mitral incompetency secondary to papillary muscle dysfunction.
Ventricular aneurysmectomy is usually an elective procedure that is not undertaken until several months after MI, unless the patient has intractable heart failure, intractable ventricular arrhythmias, or severe mitral regurgitation not responsive to acute vasodilator therapy. Early intervention in these patients greatly increases the surgical risk.
Coronary Artery Bypass Grafts
The concept of direct coronary artery surgery uses the saphenous vein graft or an internal thoracic artery to bypass a proximally stenotic coronary artery. CABG requires target vessels either free of disease or minimally diseased. Vein graft conduits are “free grafts” and connect the ascending aorta to the coronary vessel. In contrast, internal thoracic artery conduits remain connected to the subclavian artery and connect to the coronary artery distal to the stenosis. CABG not only promotes perfusion of the distal microcirculation of the epicardial vessel, but also provides perfusion of other stenotic vessels by enhancing flow through collateral circulation.
Follow-up Studies
Angiograms months or years after CABG show patency in a high percentage of grafted vessels. However, not all saphenous vein grafts are as patent at 10 years as they were initially. The use of the internal thoracic artery as a conduit to bypass a proximal stenosis has proved to be the best CABG procedure to date. Often, bilateral internal thoracic arteries are used. Although rare, this type of conduit can stenose or thrombose, probably related to technical intraoperative problems.
Pathophysiology of Acute Coronary Syndromes
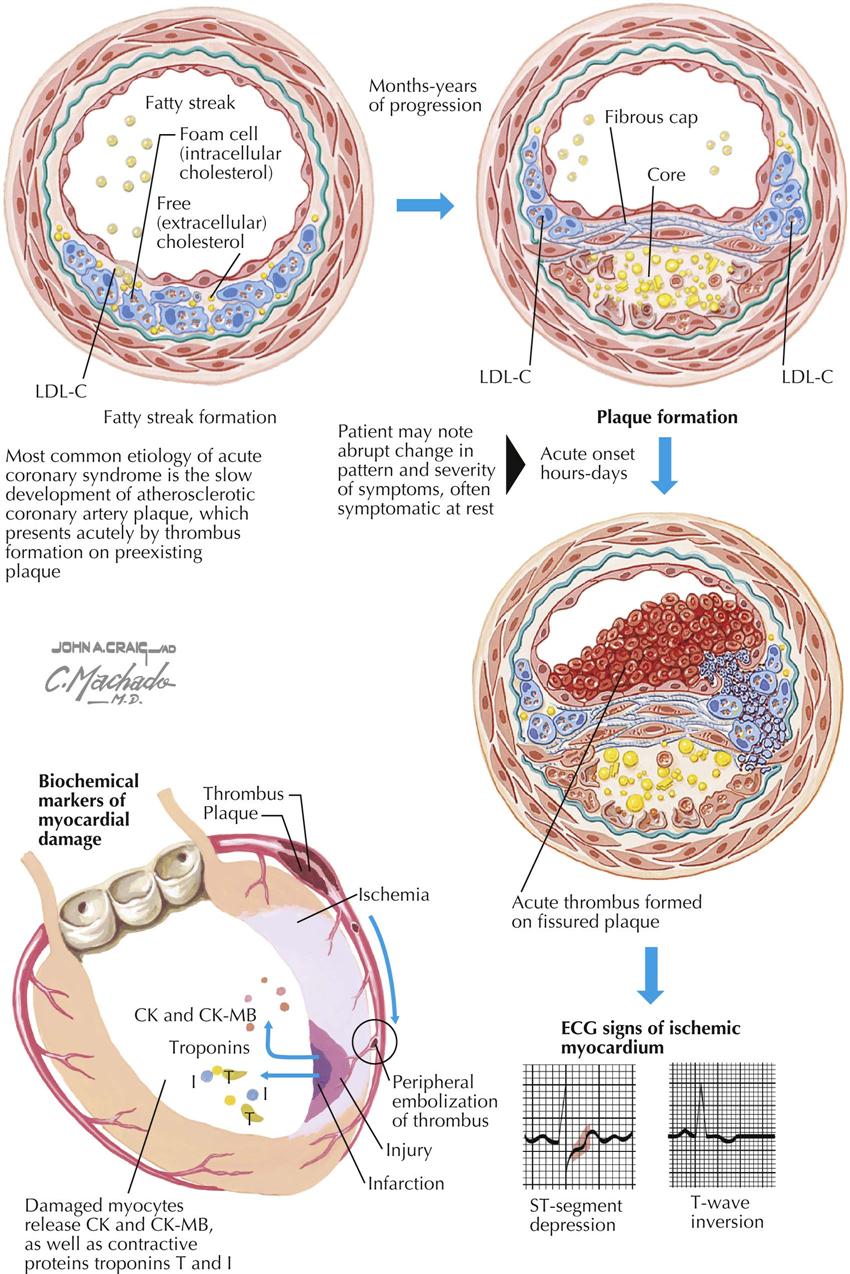
Patients with acute coronary syndrome (ACS) present in three different ways: unstable angina, non–ST-segment elevation myocardial infarction, and ST-segment elevation MI. Potential causes for the development of an ACS include extracardiac factors in the patients with severe coronary atherosclerosis, such as hypertension, tachycardia, and anemia. In 1858, Virchow proposed that injury to the inner wall of a blood vessel, possibly caused by fat, might lead to inflammation and secondary plaque formation, similar to current theory. ACS is also caused by plaque disruption resulting in transient platelet aggregation and release of vasoactive substances in diseased vessels, dynamic or intermittent coronary artery thrombosis, hemorrhagic dissection into an atheromatous plaque, and progression of coronary stenoses from plaque healing.
Acute coronary syndromes result from myocardial ischemia initiated by two primary factors: platelet and thrombus formation and resulting intense vasoconstriction from accumulation of local thromboxane A2 and serotonin, reduction of local concentrations of endothelium-derived releasing factor (EDRF), and inhibitors of platelet aggregation. These events are usually preceded by rupture or erosion of the lipid-laden plaque and involve the balance between thrombosis and thrombolysis. Patients with ACS may stabilize clinically, but this may not be associated with stabilization of the plaque. Identifying these patients requires observation for recurrent ischemia and assessment of troponin levels and possibly C-reactive protein (CRP) levels. Abnormal test results indicate increased risk for future cardiac events.
The acute coronary syndromes are not a homogeneous condition. Clinical variables evident in many patients include ECG changes, release of creatine kinase and troponins, CRP level, and differences in LV function, as well as pathology on coronary angiography. In patients with ACS, the TIMI risk score helps to predict the risk for death, MI, or recurrent ischemia at 14 days. The risks are age over 65, three risk factors for CAD, prior 50% coronary stenosis, ST-segment change on admission, angina twice in 24 hours, aspirin use within 7 days, and increased serum cardiac markers. The more risk factors present, the higher is the rate of composite endpoints in 14 days.
Myocardial Infarction—Changes in the Heart
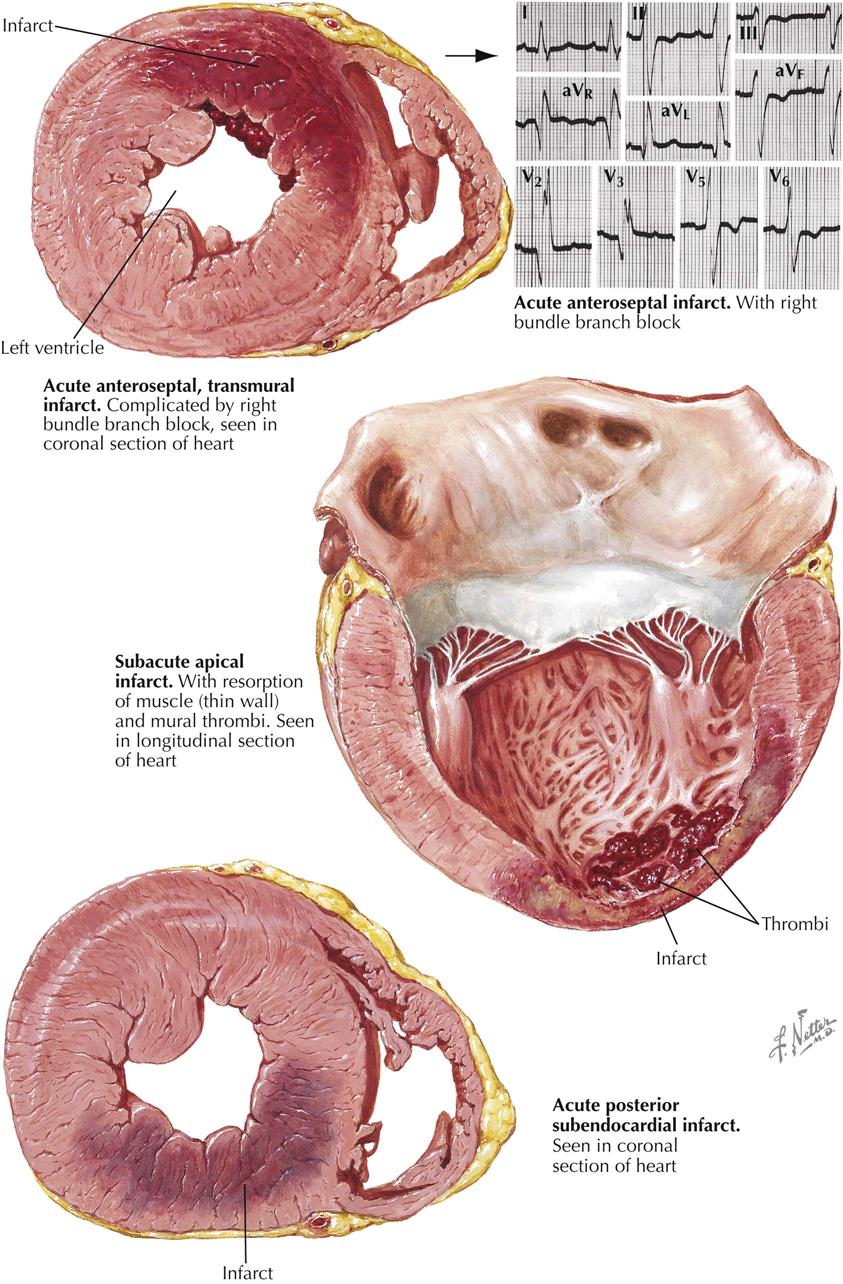
Myocardial infarction can result in an extensive panorama of changes. Death of the cardiac muscle may occur in a single zone or as multiple foci scattered diffusely throughout the heart. The latter can be found after suboptimal perfusion of the heart using a pump oxygenator, in which all the minute necrotic foci are of the same age. More often, however, the multiple minute foci can be found as destroyed zones of varying ages. In the acute phases, these microinfarcts may be extremely difficult to see and may be recognizable only microscopically as areas showing variable myofibril destruction and replacement by collagen. Chronically, these small foci may be minute white patches salted throughout the myocardium.
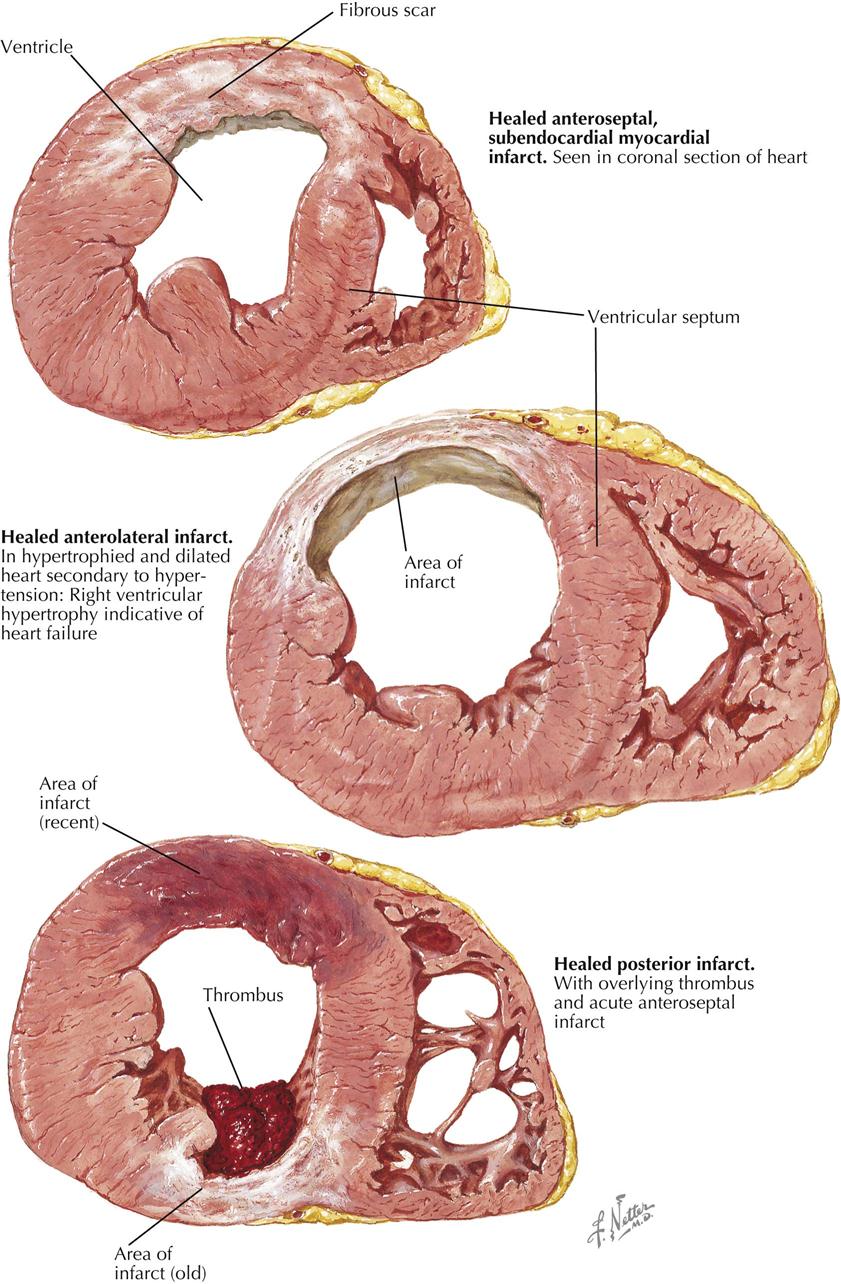
A larger myocardial infarct shows a variety of changes that depend in part on the duration of the patient’s survival after the episode. The earliest changes are associated with the resulting paralysis of vessel walls, engorgement of the capillaries, and migration of polymorphonuclear leukocytes (PMNs). Some autolytic changes then occur, and the dead muscle fibers can be recognized as showing a loss of striations and an increase in eosinophilia, fragmentation, and the gradual disappearance of nuclei. The dead muscle becomes an irritant, and more PMNs appear. Finally, the subsequent activities of repair include macrophage infiltration and replacement fibrosis. If the infarct is minute, such a sequence is of relatively short duration, but if large, remnants of necrotic muscle may be found several years later. It appears that such dead tissue has been effectively sealed away from physiologic activities.
The infarct may be named according to the area of the heart involved. Most posterior infarcts affect areas of the septum as well, and many anterior infarcts involve anterior portions of the septal wall, termed anteroseptal and posteroseptal (see Plate 6-17). A lateral infarct is found, in a small percentage of the cases. The sequence of events—muscle death, removal, and repair—may or may not result in thinning of the myocardium. Infarcts may also be classified according to the thickness of the ventricular wall involved. If all layers are affected (transmural), the term ST-segment elevation myocardial infarction (STEMI) is used, most often associated with occlusion of the coronary artery. In about 90% of these lesions, occlusion is demonstrated, as contrasted with a lesser incidence in non–ST-segment MI. In NSTEMI the zone of infarction is usually limited to the endocardial surface of the ventricle. Mural thrombosis may be a more common accompaniment of either STEMI or NSTEMI because both infarcts involve the endocardium.
A major problem with coronary artery disease is the lack of many correlations between CAD and MI in a patient who has died. Although the appearance of a lesion in the anterior descending or the main left coronary artery is a common finding in sudden cardiac death and occasionally may be the only lesion discovered, the status of the underlying cardiac muscle is not known because morphologic MI cannot be demonstrated. In about 25% of MIs encountered at autopsy—either acute or chronic but, especially, chronic—no history can be elicited as to when such a disorder occurred. In hearts with acute MI, only about half show occlusion of the coronary artery, although the other half exhibit a significant to marked stenosis of the infarcted vessel. In addition, when the immediate cause of death was believed to be CAD, no recent acute MI is found (see Plate 6-18).
Exercise may be extremely valuable in all these MI patients. Studies indicate that although the incidence of MI is not decreased, its severity is less in those who have maintained cardiac fitness.
Acute damage to areas of the conduction system, such as hemorrhage or infarction, can be demonstrated in only some cases. However, other complications are common. The immediate results of the changes in the circulatory status and constituents of the blood apparently can lead to a deposition of thrombus at numerous sites, including the coronary arteries, with a further extension of the MI. Mural thrombi form on the endocardium (probably related to healing of infarcted tissue), in the atrium (much more common on the left than the right), and possibly at all sites where previous atheromas had existed. Therefore, cerebral anoxia can result not only from the decreased cardiac output, but also from superimposed thrombosis during the early stages. Thrombosis also occurs in the systemic veins.
Embolism is a natural sequela of thrombosis, and systemic embolization occurs in an unknown number of patients with thrombi in the left side of the heart. Cerebral embolism is the most severe and damaging, although saddle embolism of the distal aorta is also serious, as is embolization in an extremity. Smaller emboli may produce any focal embolus, including splenic and renal arteries. Less often, mesenteric embolism may occur, which can be a medical disaster if not recognized early. Unfortunately, mural thrombi persist for a long period, and systemic embolization may occur at various periods following apparent recovery from the acute infarction. Pulmonary embolism is also often related to venous stasis.
As with the hypertensive heart in failure, the hypoxic heart can dilate, with resultant valvular insufficiency and subsequent regurgitation.
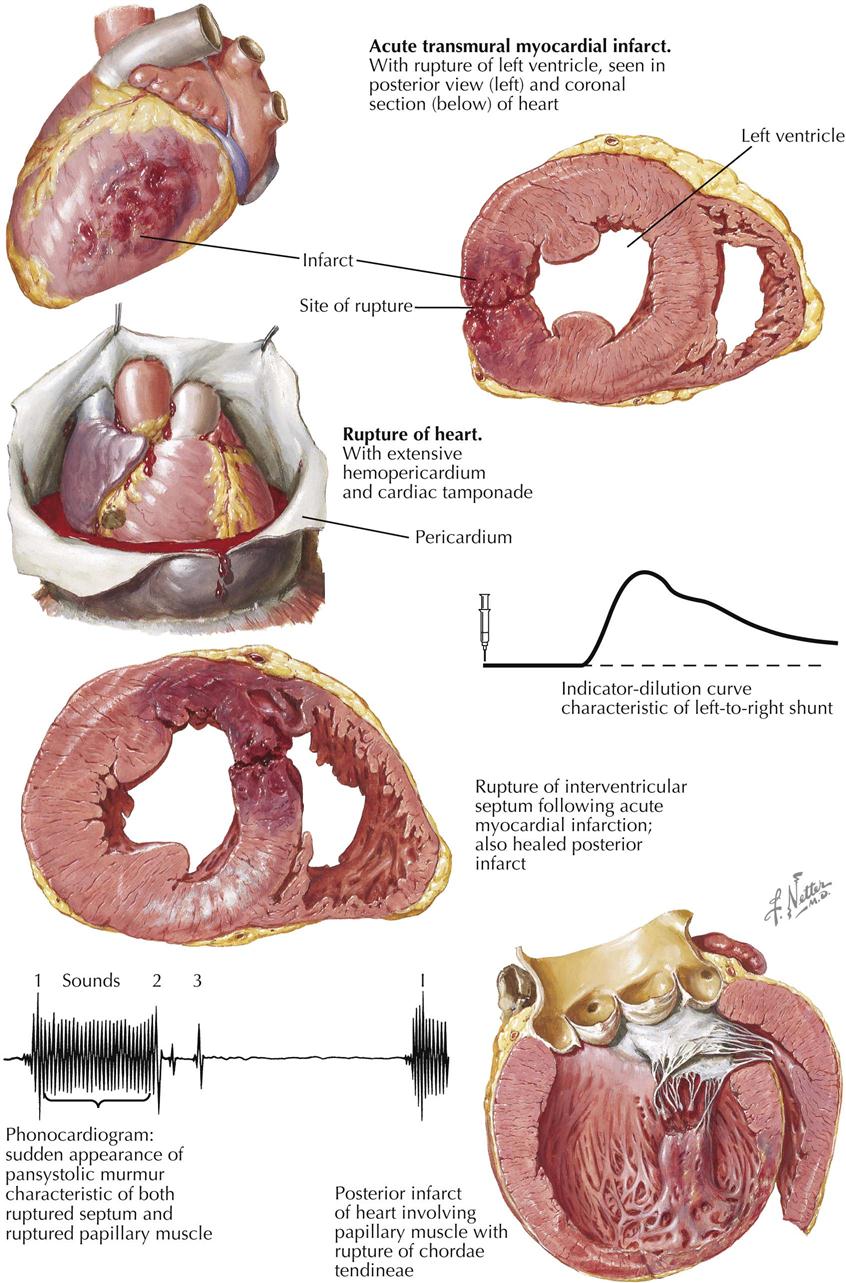
Rupture of the heart (ventricular septal defect, papillary muscle, free wall) is another variable autopsy finding (see Plate 6-19). The incidence of cardiac rupture ranges from 1% to 10% in deaths from acute MI. The area of rupture often shows a large increase in PMNs and at a much later time than such cellular elements are usually encountered; ruptures of the heart usually occurs in the first week. The classic patient prone to rupture is hypertensive, is having a first infarction, and has no visible collaterals to the infarcted artery. In addition to free rupture of the heart into the pericardial cavity and the resulting cardiac tamponade, rupture of the interventricular septum occasionally occurs. Rarely, papillary muscles rupture. The posterior papillary muscle is more likely to rupture than the anterior muscle because it does not have dual blood supply.
As a chronic complication, anterior and apical aneurysm may be more prone to mural thrombi, and thus the patient is continually exposed to the risk of embolism (see Plate 6-20).
In addition to all the complications of embolism, cardiac aneurysm, sudden death, and rupture of the heart, there is the problem of reinfarction. Some hearts demonstrate not only the zone of a previous infarct, but also areas of new infarct. The new infarct may be adjacent to the previous one or can occur in a different area of other coronary arteries.
Manifestations of Myocardial Infarction
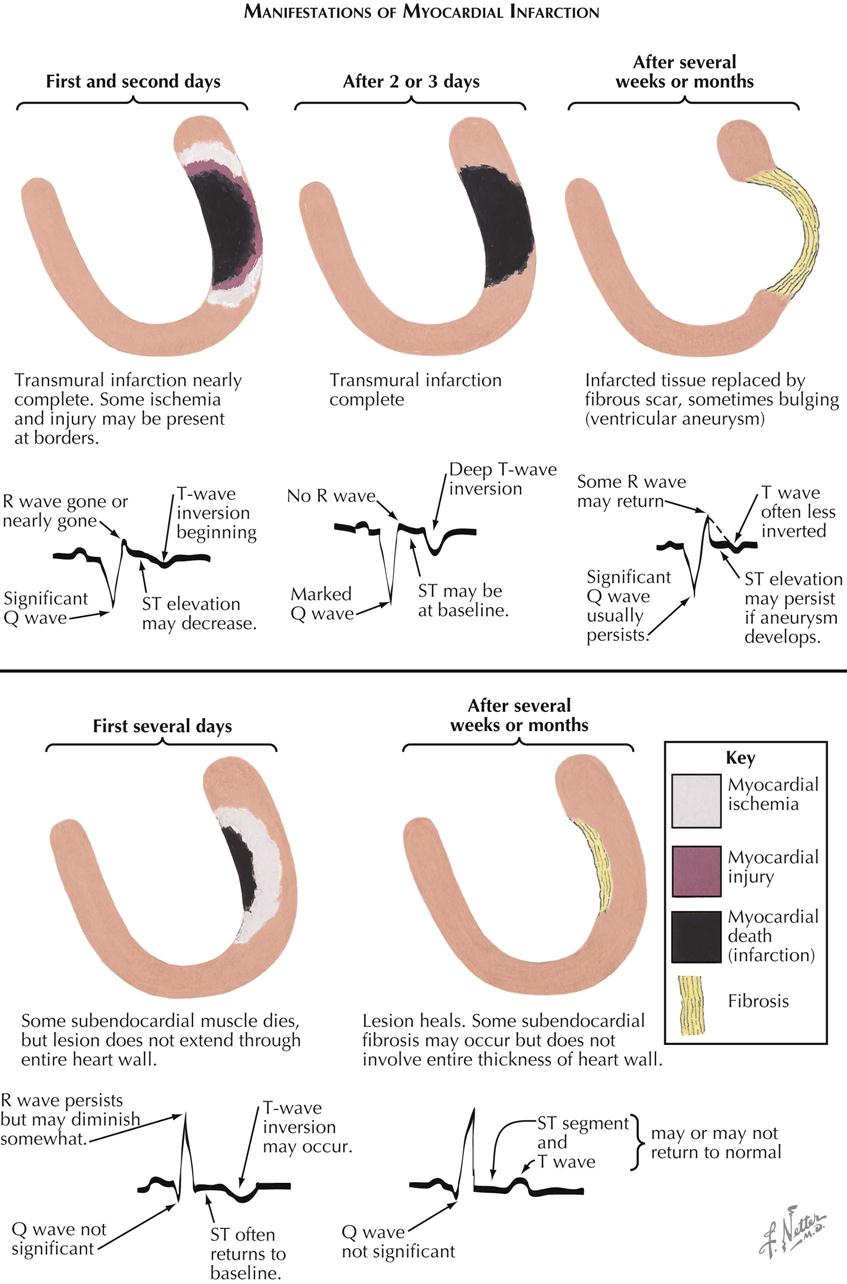
Pathologic and ECG Manifestations of Myocardial Infarction
In patients with acute MI, particularly with STEMI, pathologic changes in the heart may reveal necrosis from endocardium to the subepicardial myocardium. In the first 24 hours the ECG manifestations are variable, but usually the R wave is gone or almost gone, a Q wave is present, the ST segment is elevated, and the T wave may be inverted (see Plate 6-21). Several days later, pathology may reveal a complete infarction of the ventricular wall (transmural). The ECG continues to show Q waves, no R wave, and T-wave inversion. The ST segments may return to baseline in some cases. Several weeks or months later, the infarcted tissue may be replaced with fibrous scar, which occasionally results in a bulging ventricular aneurysm. If the aneurysm is obvious on ventriculography or cardiac ultrasound, the ST segment may be elevated. Other ECG changes usually persist (e.g., Q wave; inverted T waves, less than earlier seen), and the R wave may somewhat return.
In contrast, a patient with NSTEMI in the first 24 hours shows subendocardial necrosis, and the ECG often reveals persistent R wave, small Q wave, isoelectric ST segment, and T wave inversion. The infarcted myocardium is surrounded by a periinfarction ischemic zone. Days, weeks, or months later, the subendocardial necrosis heals, and subendocardial fibrosis replaces infarcted myocardium. This fibrosis is limited to the subendocardial area and does not extend transmurally to the epicardium. The ECG changes may or may not return to normal.
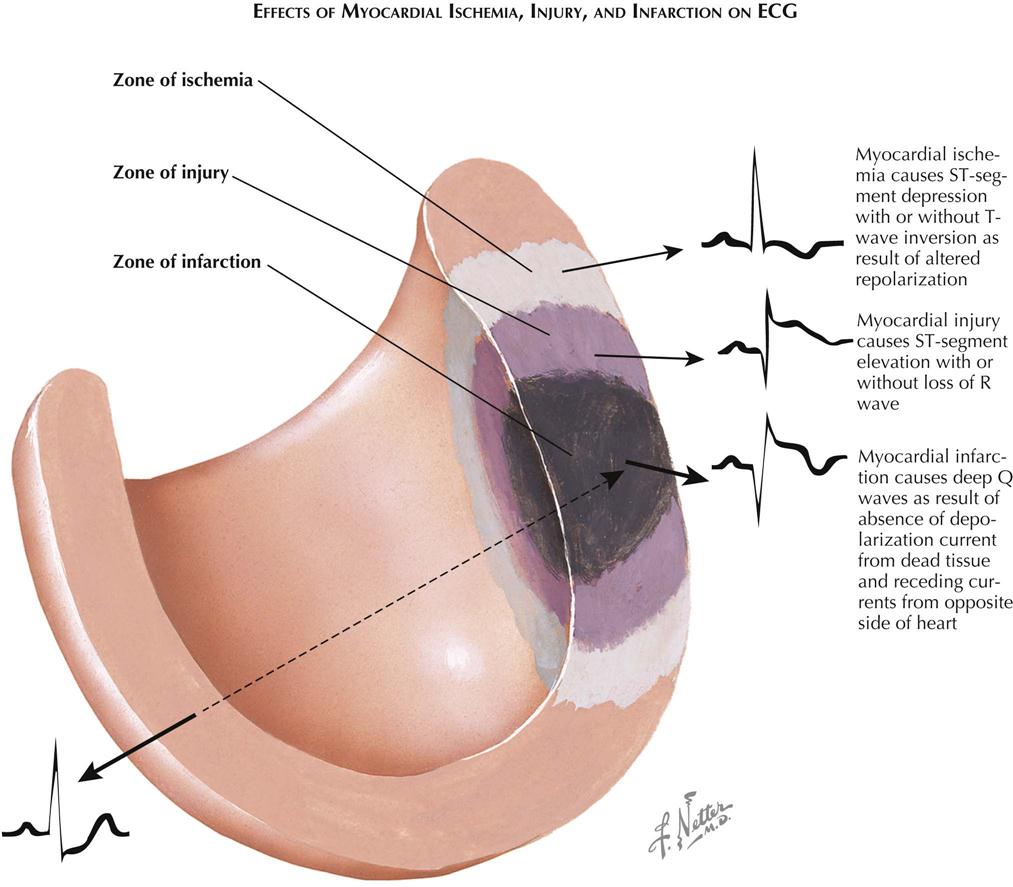
Ischemic Injury and Infarction: ECG Localization
In the myocardial region of an occluded coronary artery, ECG changes caused by ischemia, injury, and infarction can be identified (see Plate 6-22 ). ECG abnormalities are highly suggestive but not diagnostic of acute MI. In the patient who has large Q waves, ST-segment elevation, and T-wave inversion in several ECG leads, the diagnosis of an evolving MI is highly suspect; however, not all MI patients have these classic changes. Some patients present with severe chest pain and only T-wave inversion or a significant ST-segment depression, as well as elevation of cardiac markers (NSTEMI). In contrast, other patients have had obvious Q waves develop on the ECG and prominent ST-segment elevation (STEMI). All MIs begin in the subendocardial area and extend transmurally. The development of Q waves and ST-segment elevation probably relates to the transmural degree of wall involvement; the more involvement toward the epicardium, the more likely the appearance of Q waves and ST-segment elevation.
The site of the infarction can be localized in general by the ECG abnormalities. Thus, anterior and anterolateral MIs generally display ECG abnormalities of the precordial leads as well as leads I and aVL. However, all variations of these ECGs can occur, and there may be changes from V1 to V6 (including lead I) and aVL, or changes may occur only in leads I and aVL and the early precordial leads. In contrast, the diaphragmatic or inferior MI generally shows ST elevation in leads 2, 3, and aVF.
Cardiac Markers and Diagnosis of Acute Myocardial Infarction
Cardiac markers are essential for the diagnosis of acute myocardial infarction but do not appear immediately in the bloodstream if an occlusion occurs. Creatine kinase-(CK), CK isoenzymes (e.g., CK-MB), and troponin T are measurably abnormal about 4 hours after occlusion. Troponin T is the most sensitive and specific marker for acute myocardial infarction at the present time and probably is the only cardiac marker necessary to make the diagnosis of acute myocardial infarction; however, renal failure can result in the elevation of all cardiac markers in the absence of an acute myocardial infarction.
Clinically, if the cardiac markers are normal at admission to the emergency department (ED), when the patient is having acute cardiac pain and ST-segment elevation, the MI likely has occurred recently, within 3 or 4 hours. However, if the cardiac marker is elevated, the occlusion and STEMI likely occurred more than 4 hours before ED evaluation. The onset and duration of increase in CK enzymes and troponin T after a coronary occlusion vary, but in the patient with STEMI the occlusion usually occurred recently if initial serum values are normal. If a revascularization procedure is performed early in the course of acute MI, the cardiac markers my increase much more than if a revascularization procedure had not been performed, most likely from washout of the markers from the perfused myocardium.
Recanalization of Occluded Coronary Artery in Acute Myocardial Infarction
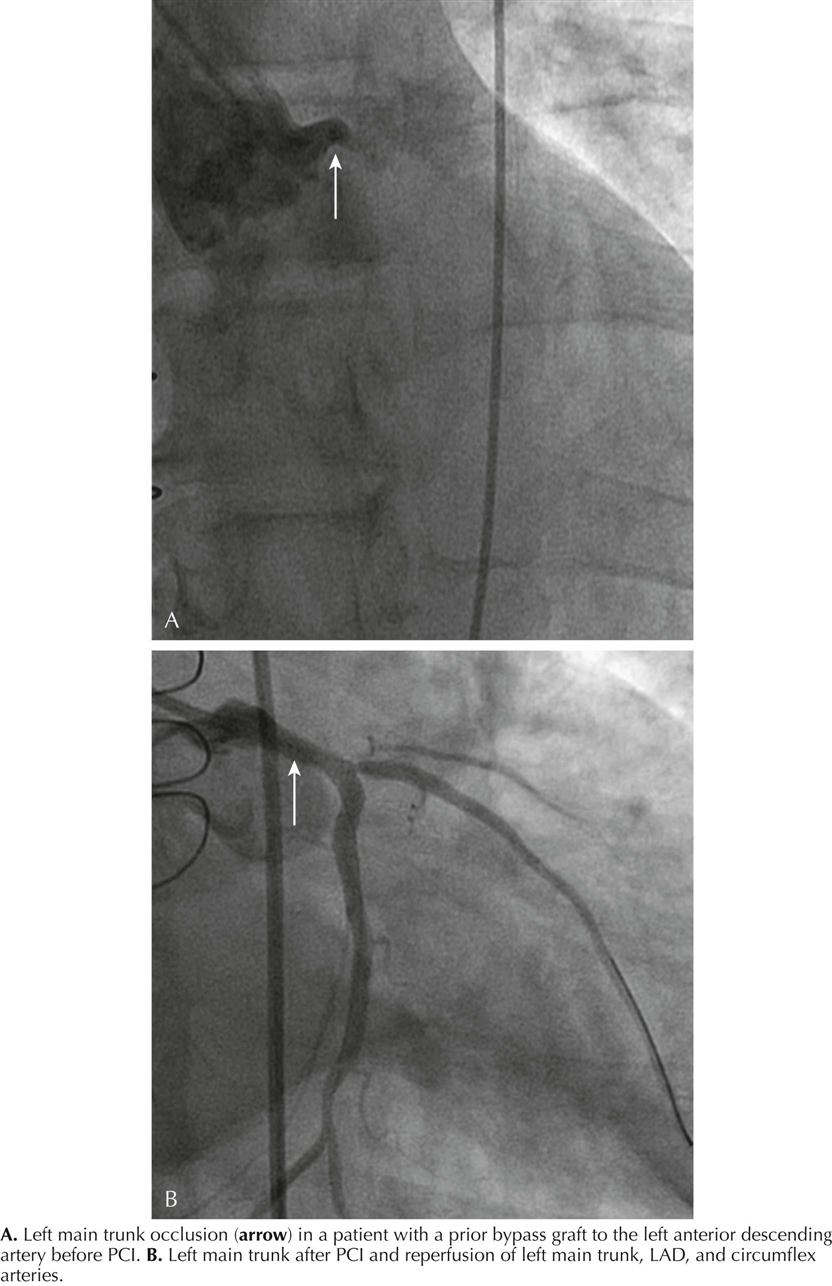
Rapid opening of the infarcted artery is generally accepted as critical to increase patient survival, preferably using PCI. However, thrombolytic therapy remains a viable option in hospitals when PCI capability is not immediately available. Percutaneous recanalization of an occluded coronary artery by balloon angioplasty and stent placement in patients with STEMI is the standard of practice. The appropriate management of acute STEMI currently is door-to-balloon (“D2B”) time—from ED arrival to passage of a guidewire into the infarcted artery. D2B time is easily and accurately measured and documented. D2B time less than 90 minutes is an acceptable goal. The rationale is that short D2B time results in less myocardial necrosis. A favorable outcome correlates well with minimal necrosis and ejection fraction (EF) greater than 40%. An unfavorable outcome correlates with a large amount of necrosis and EF less than 40%.2
Timing of Vessel Occlusion
In canine studies, MI size versus duration of coronary occlusion was measured with the circumflex coronary artery ligated. Thus the actual time of onset of coronary occlusion was identified and the time course of myocardial necrosis measured. Myocardial cells started to die about 20 minutes after onset of ischemia, with cell death complete by 6 hours.
Theoretically in the human, if mortality reduction is plotted against extent of salvage of myocardium from 0 to 3 hours, a steep reduction in percent mortality occurs, and by 3 hours the myocardial salvage is greatly reduced. After 3 hours the percent mortality reduction is similar to that found at 3 hours. Unfortunately, the exact time of vessel occlusion is not known in human MI.
Intra-Aortic Balloon Counterpulsation
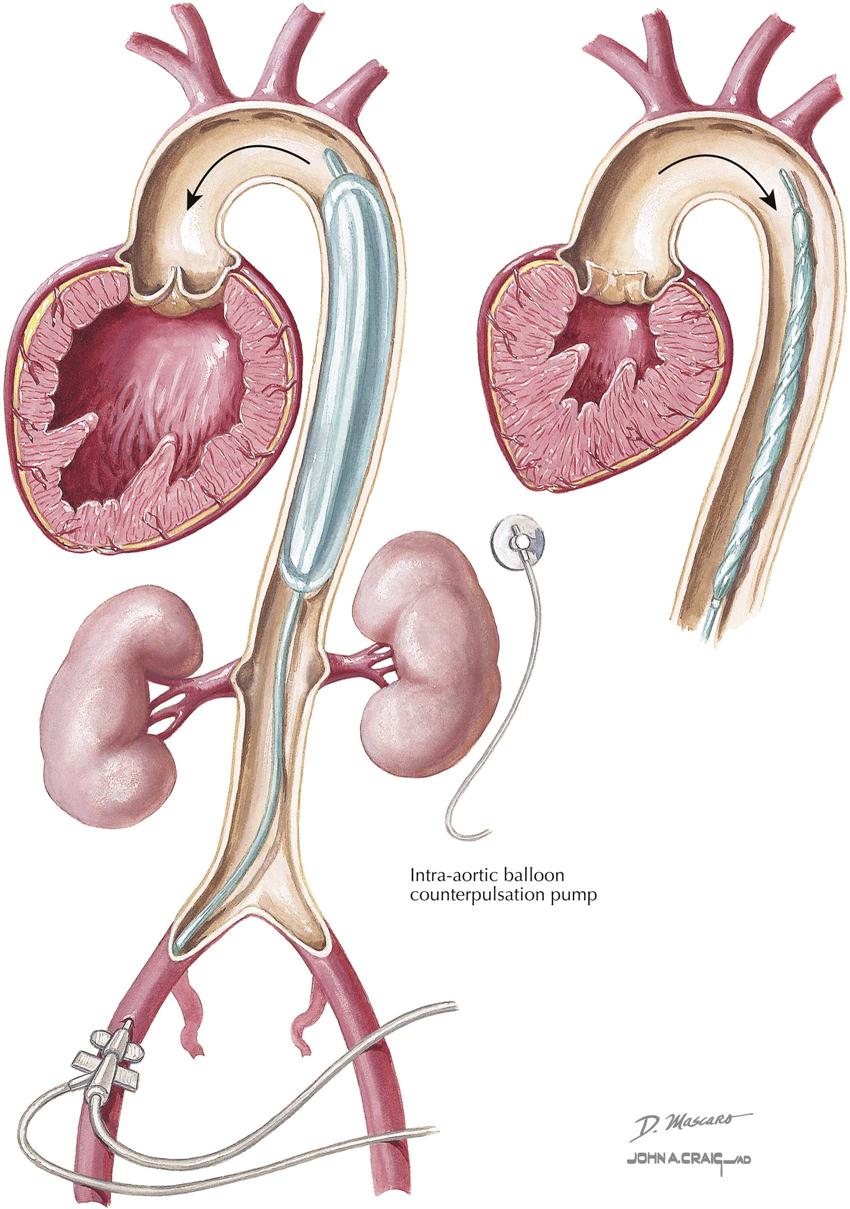
Counterpulsation is the only technique that will lower aortic systolic pressure and increase aortic diastolic pressure. To accomplish intra-aortic balloon counterpulsation (IABCP), a catheter with a cylindrical polyethylene balloon (~40 mL in adult device) is introduced into the aorta, deflated, and extended to just below the left subclavian artery and above the renal arteries, so as not to obstruct either vessel. When visualized on chest radiography or fluoroscopy, the tip of the catheter is placed at the level of the bifurcation of the right and left main bronchi, again so as not to occlude subclavian and renal arteries. Balloon expansion with helium occurs actively during diastole, and collapse of the balloon occurs actively during systole. Intra-aortic pressure is measured at the catheter tip.
Timing of balloon expansion and collapse is controlled by either ECG or pressure waveform during the cardiac cycle. When the balloon is expanded in diastole, aortic diastolic pressure increases, as does coronary perfusion pressure, which theoretically increases myocardial blood flow. When the balloon is collapsed during systole, LV afterload reduction occurs, probably a suction effect that unloads the left ventricle and increases cardiac output. When counterpulsation is working correctly, the elevated diastolic aortic pressure likely is interpreted as high blood pressure, and the baroreceptor reflexes tend to lower systolic pressure, thus decreasing myocardial oxygen demand.
The clinical indications for IABCP are cardiogenic shock of any etiology, acute mitral regurgitation that does not respond to nitroprusside, acute ventricular septal defect, and refractory acute coronary syndromes (ischemia or arrhythmias). Less clear indications relate to preprocedural use in patients considered “high risk” for a revascularization procedure such as PCI or CABG (e.g., high-grade left main coronary artery stenosis). IABCP should not be used in patients with aortic insufficiency; the increase in diastolic pressure will increase the degree of aortic insufficiency. IABCP cannot be used in patients whose aorta cannot be accessed because of severe peripheral vascular disease. Complications with IABCP are fortunately rare and include leg ischemia, aortic disruption, renal artery obstruction when the balloon is not properly placed, and bleeding.
Rheumatic Fever in Sydenham’s Chorea
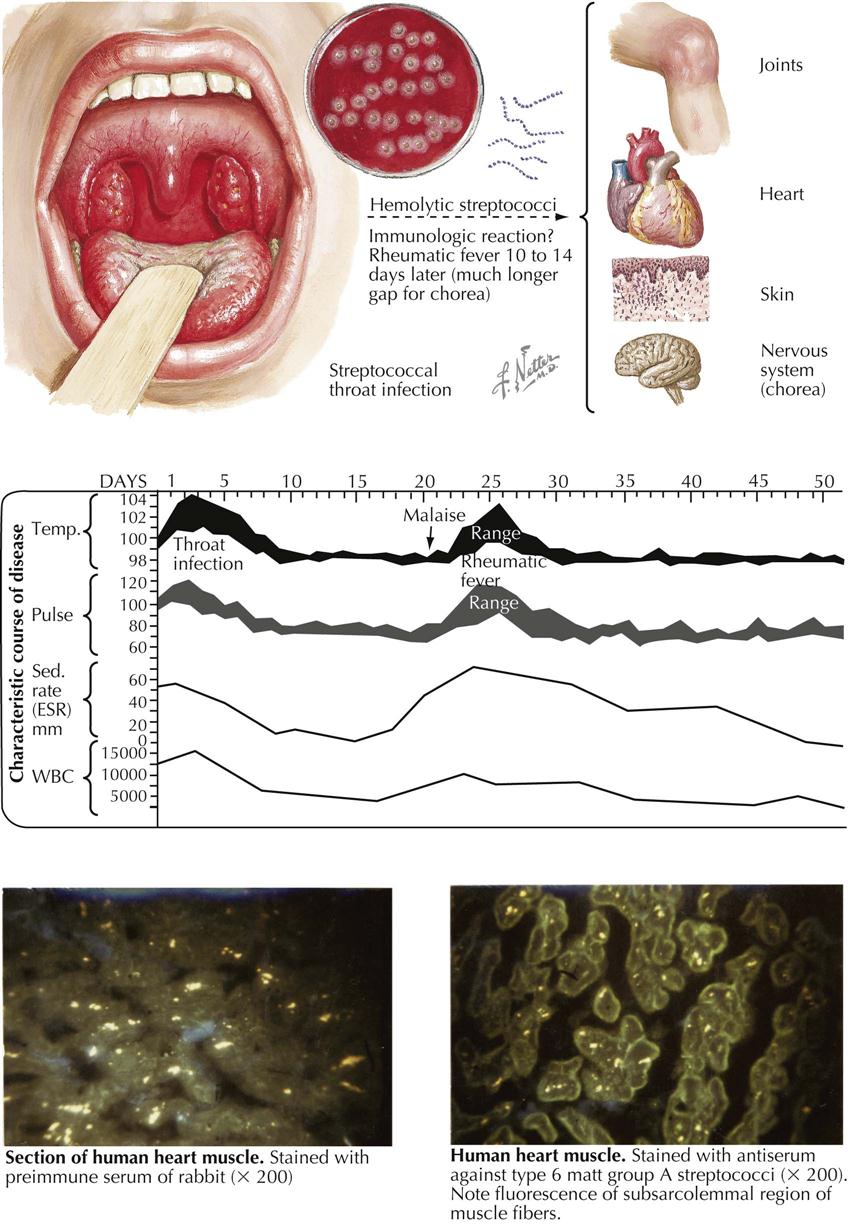
Rheumatic Fever
Although uncommon in the United States and other developed countries, rheumatic fever is common in developing countries and can affect people of all ages, except in the first years of life. The febrile onset is accompanied by severe but temporary arthritis and often by carditis affecting all three cardiac layers. The etiologic relationship of rheumatic fever to β-hemolytic streptococci is based on the following evidence:
The frequency with which rheumatic fever follows a streptococcal infection varies from 3% in epidemics to about 0.3% in sporadic infections. This discrepancy is largely caused by the greater severity of epidemic types of infection, as revealed by clinical features and the antibody titers induced. When streptococcal infections of comparable severity are studied, the incidence of subsequent rheumatic fever, even in sporadic cases, is also about 3%.
The lesions of rheumatic fever do not result from direct invasion by streptococci. When adequate precautions are taken against contamination, organisms cannot be isolated from the joints or the heart. The events relating the primary infection to these lesions are still unclear, but circulating toxins released from the organisms are unlikely to be responsible, because then the clinical manifestations of rheumatic fever would coincide with the sore throat (as do the toxic symptoms of diphtheria). Furthermore, none of the many known streptococcal products produces comparable lesions in animals. The relationship between streptococcal infection and rheumatic fever is therefore most widely regarded as immunologic, with the lesions resulting from an antigen-antibody reaction in the affected tissues. This concept was supported by observations that lesions superficially resembling those of rheumatic carditis could be produced in rabbits by the injection of massive doses of foreign serum.
A comparable massive exposure to antigen is unlikely to occur in humans. Any immunologic reaction underlying the pathogenesis of rheumatic fever is more likely to be more specific, directly involving one or more of the antigens native to the affected tissues. Many strains of β-hemolytic streptococci contain an antigen in their cell walls that cross-reacts with an antigenic component of mammalian hearts, including the human. Furthermore, animals injected with such strains of streptococci produce antibodies that react with cardiac antigens. Similar antibodies have been found in a high proportion of patients with active rheumatic fever.
However, despite the presence of firmly bound gamma globulin (presumably antibody) in the heart of many patients with rheumatic fever, circulating antibody correlates poorly with clinical severity. Cardiac damage in experimentally immunized animals is unconvincing; repeated infection in rabbits by different β-hemolytic streptococci most closely resembles human rheumatic carditis. Although the mechanism is still unclear, immunologic cross-reactivity with myocardial and other antigens is presently the most likely cause.
The clinical features of an acute attack may range from the trivial to the fulminating—from easily overlooked pallor and fatigue to an exquisitely painful arthritis with high fever, rash, and carditis sufficiently severe to lead to congestive heart failure. The temperature chart is characterized by a nonremittent type of fever with a pulse rate raised disproportionately. The arthritis is migratory, rarely lasting in any one joint for more than 1 or 2 days. Swelling is slight and accompanied by some flushing of the overlying skin. Large joints are more frequently affected than small joints, although these can become involved as well, and the clinical picture may then resemble rheumatoid arthritis.
Rheumatic Fever in Sydenham’s Chorea: Noncardiac Manifestations
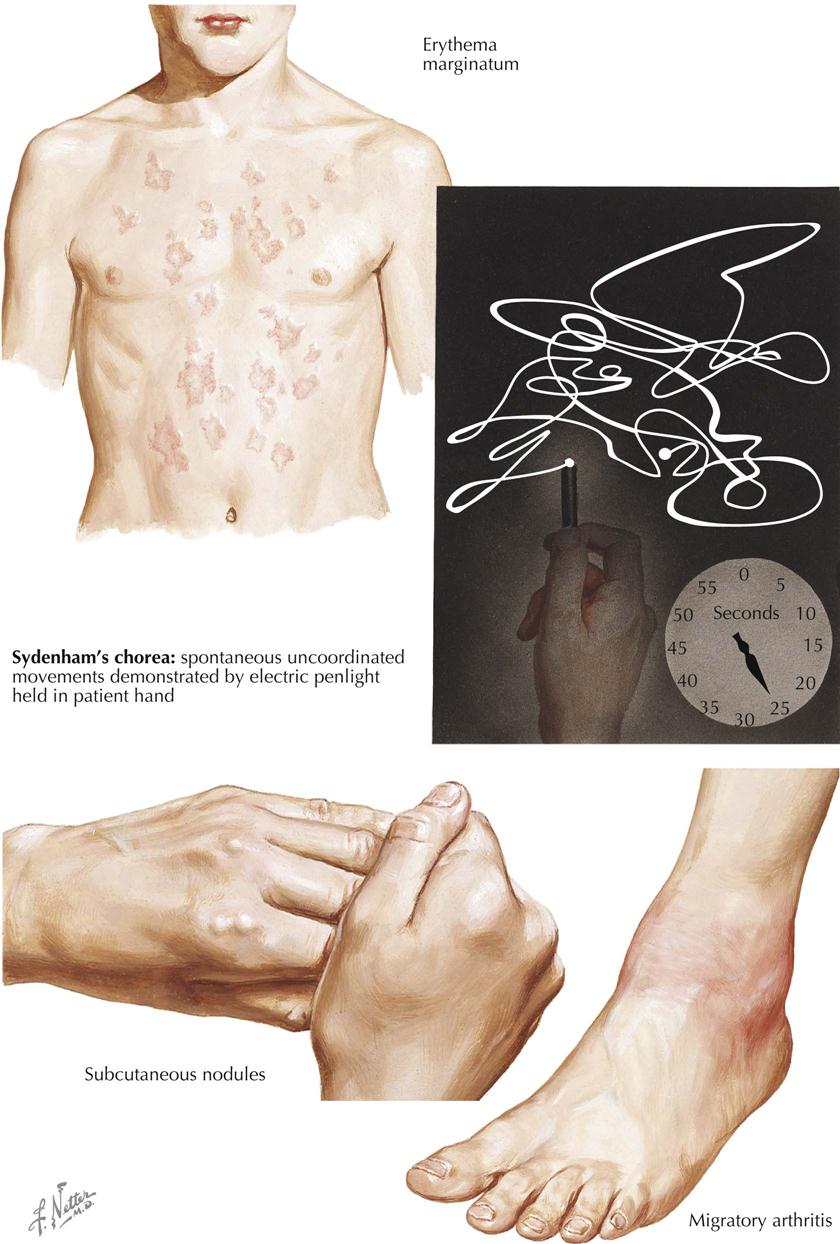
Despite the many features common to rheumatic fever and rheumatoid arthritis, notably polyarthritis and subcutaneous nodules, these are distinct nosologic entities. If differentiation is difficult in the early stages, the difference in clinical evolution rapidly distinguishes rheumatic fever from arthritis, and even in the early stages, histology of the synovium or biopsied nodule can be helpful. The lesion in rheumatic fever is capsular rather than synovial, with areas of fibrinoid necrosis accompanied by focal infiltrations of histiocytes and lymphocytes.
The joint lesions, even when severe, are entirely reversible, and permanent damage does not result. This contrasts with the cardiac lesions, which can occasionally progress to mitral stenosis and its sequelae, even when initial damage is mild. Skin rashes are uncommon but can be characteristic, especially erythema annulare, a fleeting, ring-shaped, usually flat eruption typically found on the trunk. Each lesion spreads centrifugally, leaving a slight staining in the center. Another typical feature of rheumatic fever, seen especially in severe cases, is subcutaneous nodules, mainly confined to the tissues over bony prominences, where friction combined with pressure is apparently responsible for nodule development. The nodules are most conspicuous after 2 to 3 weeks, although their impending development is often preceded by a more diffuse, boggy thickening. Histologically, a typical nodule shows an area of fibrinoid composed of parallel or interlacing bands sparsely infiltrated with histiocytes and fibroblasts. The adjacent tissue is edematous and contains groups of small vessels surrounded by similar cells. Fibrous tissue is inconspicuous.
Permanent cardiac damage correlates closely with persistent or recurrent activity of rheumatic disease, so the detection of such activity is important. The most helpful test is the erythrocyte sedimentation rate (ESR), which is almost never normal in the presence of active disease, except in patients with congestive heart failure. The converse, however—a persistently raised ESR—does not necessarily imply continued activity, especially in girls. Therefore, estimations of C-reactive protein may more accurately reflect the state of the disease activity. The greatest danger to the patient with a history of rheumatic fever is developing a new infection with β-hemolytic streptococci, because the risk of relapse is much greater than after an initial attack. The success of chemoprophylaxis has confirmed both the importance of such reinfection in the development of a relapse and the role of streptococci in rheumatic fever pathogenesis.
Environmental and genetic factors have been studied widely in an attempt to account for the considerable geographic variation in the incidence of rheumatic disease. The most impressive data are provided by Mills’ map, showing the progressive decrease in mortality from rheumatic fever as one passes from the northern to the southern latitudes of the United States. Evidence of genetic factors besides female predominance is less precise, but the increased incidence of nonsecretors of the blood group substances in patients with rheumatic fever indicates modest genetic influence. The dominant factor is environmental and related to the incidence and virulence of the local streptococci.
Sydenham’s Chorea
The rheumatic nature of Sydenham’s chorea (St. Vitus dance), first recognized just over 100 years ago, is now established by evidence similar to that for rheumatic fever, as a poststreptococcal state. This is confirmed by the frequent subsequent development of rheumatic heart disease, in the absence of any clinical evidence of rheumatic fever. The outstanding clinical features are ataxia, incoordination, and weakness, accompanied by spontaneous movements that can be most clearly depicted by the photographic record of a penlight held in the patient’s hand.
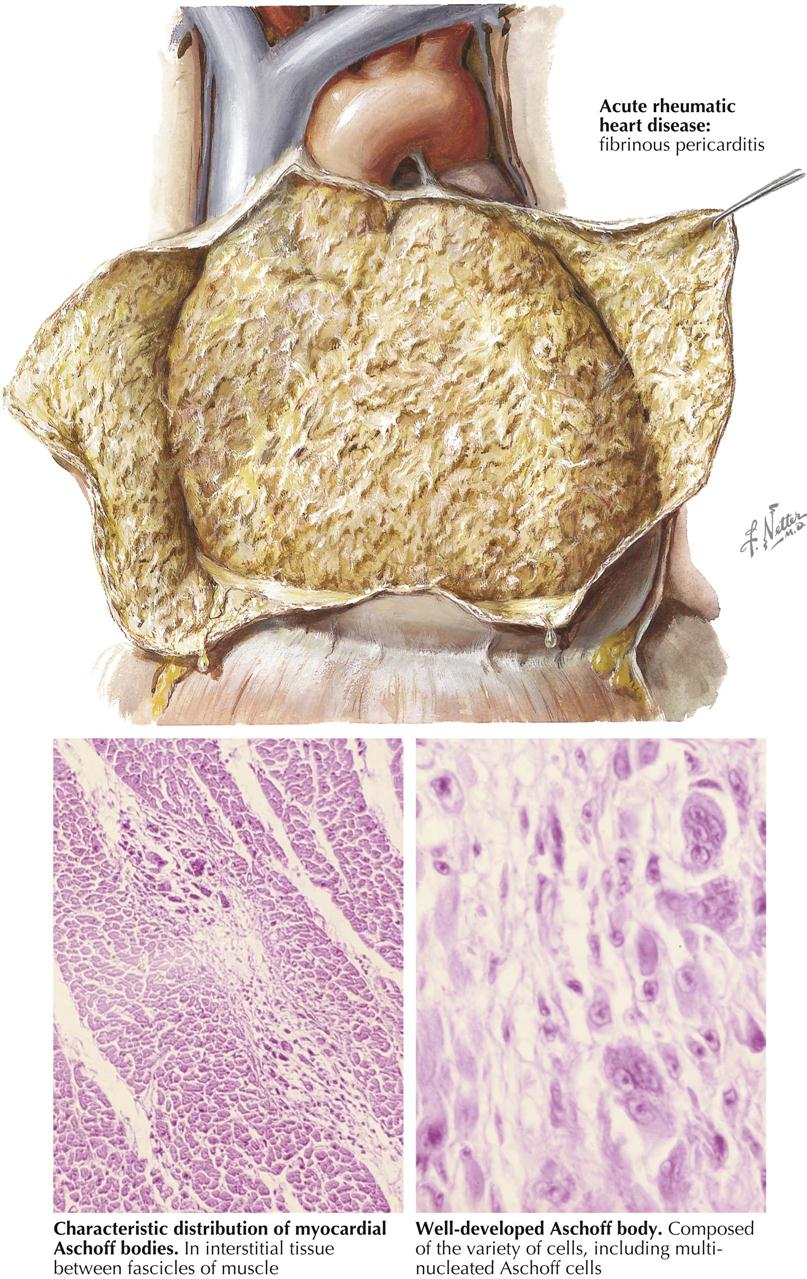
Rheumatic Heart Disease: Acute Pericarditis and Myocarditis
Rheumatic heart disease is a complication of recurrent upper-respiratory infection with group A β-hemolytic streptococci. Streptococcal products circulating from this source through the body causes a reaction in connective tissues called rheumatic inflammation. Predisposed sites include the heart, skin, and synovial membranes. In the latter two areas, inflammation usually heals by resolution, leaving no residual effects, but in the heart these recurrent respiratory infections may lead to severe deformities, usually of the valves.
Chronic rheumatic heart disease requires recurrent infections. Each attack of rheumatic inflammation goes through an active stage followed by healing. In the joints, active rheumatic involvement is characterized by migratory arthritis. In the skin, transitory subcutaneous nodules may appear. In the heart, each of the major anatomic components—the pericardium, myocardium, endocardium, and particularly the valves—may be involved.
Acute Rheumatic Pericarditis
Acute rheumatic pericarditis is characterized by variable exudation of serum and fibrin into the pericardial cavity. Large effusions of fluid may result in radiographic features of uniform cardiac enlargement and physical signs of pericardial effusion. Impairment of the inflow of blood to the heart, with resultant increased systemic venous pressure, may be evident through distention of the cervical veins. The fibrinous element of acute rheumatic pericarditis is manifested by particles of fibrin floating in the associated effusion and by the concentration of a fibrinous deposit on the visceral (epicardial) and parietal layers. The shaggy fibrin, evident when the two layers of the pericardium are separated, has given rise to the name “bread-and-butter heart” of rheumatic pericarditis. This gross feature is not specific, however, since it applies to any condition in which fibrinous pericarditis occurs. Histologically, during the active stage of rheumatic pericarditis, fibrin is deposited on the pericardial surfaces. Beneath this, capillaries and fibroblasts are mobilized and gradually enter the fibrin as granulation tissue. The pericardium shows edema and mild leukocytic infiltration. Specific Aschoff bodies, which characterize acute rheumatic myocarditis, are not usually seen in the pericardium.
Acute Rheumatic Myocarditis
The specific histologic lesion of acute rheumatic carditis is usually restricted to the myocardium and takes the form of the Aschoff body. Aschoff bodies are reactive nodules within the connective tissue and therefore are predominantly found around blood vessels of the myocardium and in other bundles of connective tissue that separate myocardial fascicles. The primary lesion appears to be an alteration of collagen, which shows a coagulation-like change with eosinophilia, a process called fibrinoid necrosis. Secondary cellular reaction to the primary process in the collagen leads to the formation of the nodule. The involved cells include nonspecific phagocytes, myocardial histiocytes, and multinucleated cells (Aschoff giant cells). In the early Aschoff body, fibroblastic proliferation is not strongly evident, but as the lesion becomes older, the phagocytic cells become less numerous and are replaced by fibroblastic cells. Toward the end of the activity of an Aschoff body, the nodule may be entirely fibrous and acellular in nature.
The characteristic scant loss of muscle in acute rheumatic carditis is paradoxical in view of the frequent occurrence of myocardial failure during this stage of the disease.
Rheumatic Heart Disease: Acute Valvular Involvement
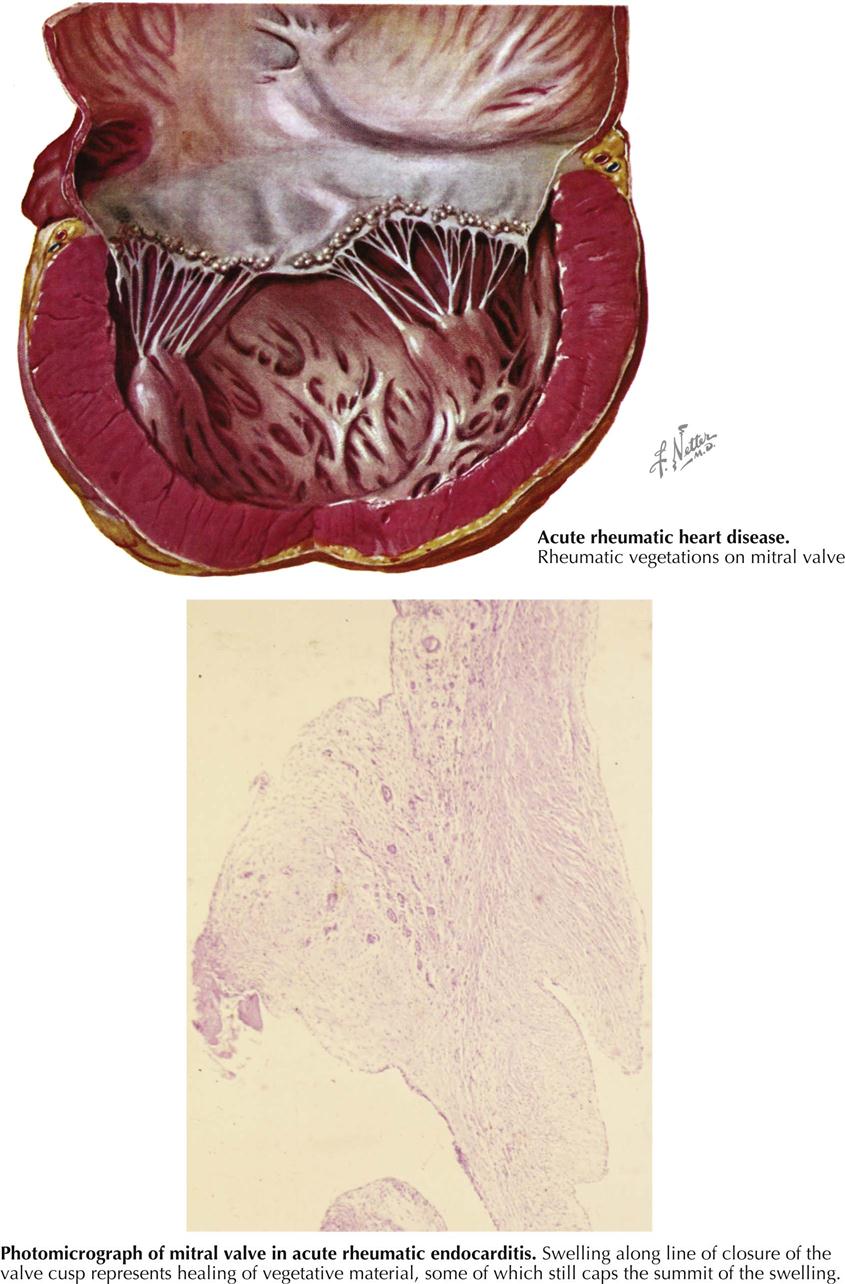
The gross changes exhibited in the cardiac valves as a manifestation of acute rheumatic involvement are highly characteristic. The mitral and aortic valves tend to be involved, whereas the tricuspid valve is affected only occasionally and the pulmonary valve, classically not at all. The primary change in the valve is edema with minimal leukocytic infiltration. A secondary erosion of the valve cusp along the line of closure occurs as a complication of closing the edematous cusp. Fibrin and platelets are then deposited along the denuded area, accounting for the row of delicate, tan, translucent beadlike lesions that usually are confined to the line of closure. From the atrioventricular valves, the lesions may extend onto the chordae tendineae. Another characteristic feature of the gross changes in rheumatic valvulitis is the absence of destructive effects on the valve tissue or chordae tendineae, with minimal deformity of the valves at this stage.
Since acute rheumatic valvulitis is almost universally associated with acute rheumatic myocarditis, the gross effects of rheumatic myocarditis in the form of ventricular dilatation are frequently evident. Mitral insufficiency may be present as a transient phenomenon during the acute stage of rheumatic carditis. The valvular disturbance is attributed primarily to the myocarditis and associated ventricular dilatation rather than intrinsic disease of the mitral valve. The involvement of the aortic valve by acute rheumatic endocarditis causes little functional change. Mitral regurgitation during acute rheumatic carditis may be associated with an area of regurgitant “jet lesions” on the posterior wall of the left atrium, called MacCallum patches, considered a focus of response to regurgitation rather than primary rheumatic mural endocarditis.
Acute rheumatic valvulitis occurring at a given time may represent either the first attack or one of several recurrent insults. The structural changes observed in a patient with acute rheumatic endocarditis will depend on whether the attack is the first event or one of several recurrent episodes. Even with a first attack, evidence may show a healing response to the inflammatory process. In such cases there is little gross alteration, except for vegetations. The response to an initial attack has certain characteristics; in the cusp beneath the vegetation, various cells (e.g., fibroblasts, macrophages) are mobilized in a palisade manner. Fibroblasts and capillaries grow into the inflamed material, replacing the fibrin. This sets the stage for the healed phase, when the sites of the valve lesions are represented by fibrous nodules. In cases where the chordae tendineae also are involved by vegetative material, healing leads to chordal thickening and sometimes adhesions between the chordae tendineae. If an attack is one of several recurrent episodes, the characteristic changes of acute rheumatic valvulitis are superimposed on residua of healed previous attacks, including cusp shortening, fibrous thickening along the line of closure, varying degrees of interadhesion between the chordae tendineae, shortening of the chordae, and vascularization of the cusps. If valvular insufficiency has resulted from earlier attacks of acute rheumatic endocarditis, an enlargement of the chambers specific for the type of such insufficiency may be observed.
Rheumatic Heart Disease: Residual Changes of Acute Rheumatic Carditis
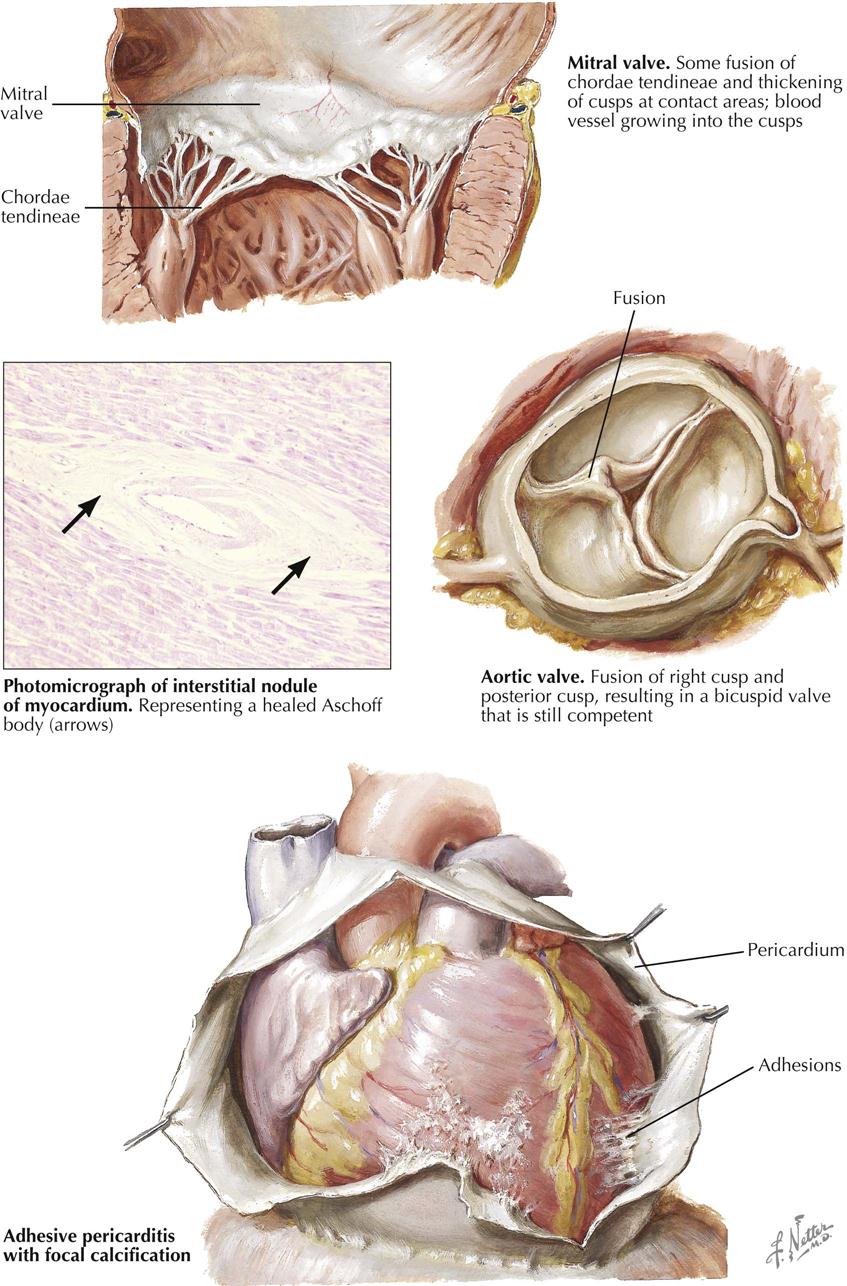
Recurrent acute rheumatic carditis may lead to severe residual changes represented by stenosis or insufficiency of one or more valves. In some patients, however, the rheumatic process leaves only minor damage, or each involved valve exhibits only minor changes. Other patients have one or two valves that may show significant deformities. Minor residual changes of acute rheumatic carditis also may be observed in the myocardium and pericardium, regardless of the specific damage to the valves.
In the mitral valve, minor residual changes are represented by fibrous thickening along the line of closure of the cusps at the site of healed lesions of acute rheumatic valvulitis. Minimal shortening of the free aspect of the cusp may occur, making the insertion of chordae tendineae appear to be directly into the free edge of the cusp. Vascularization of the mitral valve is often seen as a sign of healed rheumatic involvement. Characteristically, the vascularization involves the anterior cusp. At its base, a vessel is seen to proceed toward the free aspect of the cusp, where it arborizes. Minor degrees of chordal shortening, thickening, and interadhesion may occur in competent valves. Also, minor degrees of fusion between the cusps at their commissures may be observed. In the tricuspid valve the minor residual effects are similar to those in the mitral valve.
In the aortic valve the minor residual changes include limited degrees of fibrous thickening along the line of closure, cusp shortening, and commissural fusion. Shortening of the cusps is demonstrated in the conventional view of the opened aortic valve by a greater depth of the aortic sinuses than in the normal valve. The most common clinical observation in a patient with mitral valvulitis and stenosis is aortic regurgitation. Aortic valve commissural fusion of a degree that allows near-normal function of the valve is usually confined to one commissure. This results in one conjoined cusp about twice the width of the second cusp, which is not adherent to its neighbors. An aortic valve so altered may be termed an acquired bicuspid valve. Although such a valve may be competent and also not stenotic, it fails to open with the freedom of a normal tricuspid aortic valve and is subject to the complication of slowly becoming calcified and stenotic. Another potential complication of the minor residual effects of rheumatic disease in the valves is infective endocarditis, particularly of the mitral and the aortic valves.
In hearts with only minor residual valvular disease of rheumatic endocarditis, the myocardium is grossly normal and not hypertrophied, but small myocardial scars may exist. The latter are the residua of Aschoff bodies and are represented by avascular and acellular scars in perivascular locations. In many cases the healing of Aschoff bodies may be so complete that it is difficult to determine whether a strand of connective tissue in the myocardium is normal supporting tissue or a residual scar.
The healed effects of acute rheumatic pericarditis are rarely if ever responsible for significant disturbances of the circulation, regardless of the form that healing may take. Usually, acute rheumatic pericarditis heals by resolution, leaving a smooth-lined, nonadhesive pericardium. In other cases the fibrinous exudate is replaced focally or diffusely by fibrous adhesions. Even in uncommon instances where the entire pericardium is obliterated by fibrous adhesions, the adhesions are relatively thin and do not constitute a basis for cardiac constriction. In some cases of fibrous obliteration of the pericardial sac, focal plaques of calcium may be deposited in the adhesions, but even then there appears to be no constrictive effect.
Mitral Stenosis: Pathologic Anatomy
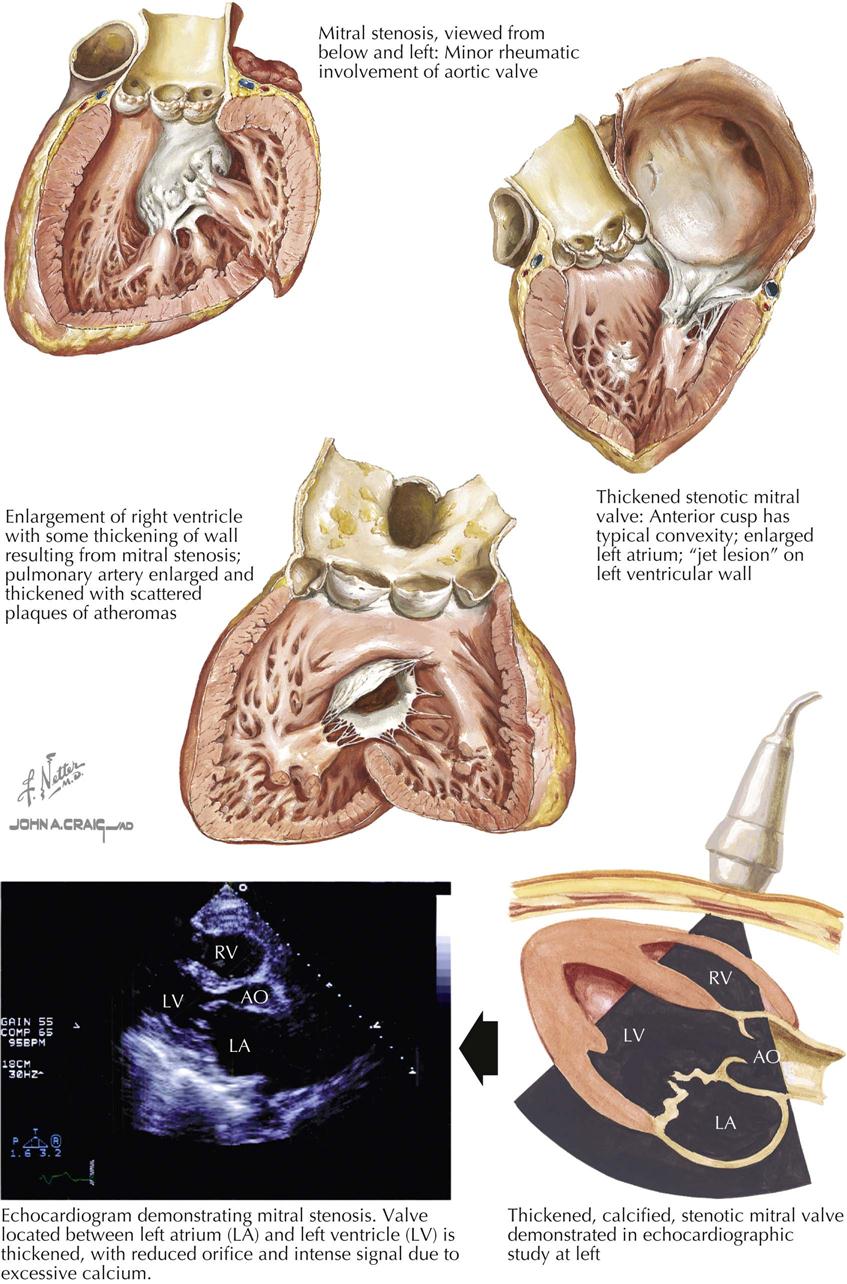
Mitral stenosis is the most common serious effect of recurrent rheumatic fever and represents the culmination of recurrent attacks: a highly deformed and obstructed mitral valve. The healing valvular lesions of acute rheumatic fever cause not only fibrous thickening of the cusps, but also and more importantly, cusp interadhesions at the commissures and chordae tendineae changes. The healing of lesions on the chordae surfaces is responsible for their fusion, obliterating the spaces between the chordae. In the normal mitral valve, blood flows through the part of the orifice between the papillary muscles and also through the spaces between the chordae, lateral to the papillary muscles. After interchordal adhesion, the secondary pathways for flow are obliterated. The chordae tendineae also undergo recurrent inflammation, causing these strands to shorten, which is responsible for the cusps being held tautly in a downward position. The chordae related to each commissure exhibit a fanlike shape. The base of the fan is attached to the cusps, with the apex to the papillary muscle. As the chordae become shortened and pull the cusps downward, they draw the base of the fan toward the apex. This tends to hold the cusps together and favors adhesion between the cusps at their commissures during healing of the valve lesions. The process of chordal shortening, with the resultant fixation of the cusps, is probably the major factor in restenosis after mitral valve commissurotomy.
The stenotic mitral valve shows a typical deformity of its anterior cusp, characterized by a convexity directed toward the atrium. In the absence of valve calcification, during left ventricular diastole when the left atrial pressure exceeds the LV pressure, the cusp at the site of this deformity buckles toward the ventricle. This buckling could be responsible for the “opening snap” at the beginning of diastole (imagine a sailboat spinnaker catching the wind), considered classic for mitral stenosis. During ventricular systole, the deformity buckles toward the atrium and impinges on the base of the posterior mitral cusp, to act as a flutter valve to prevent regurgitation through the valve.
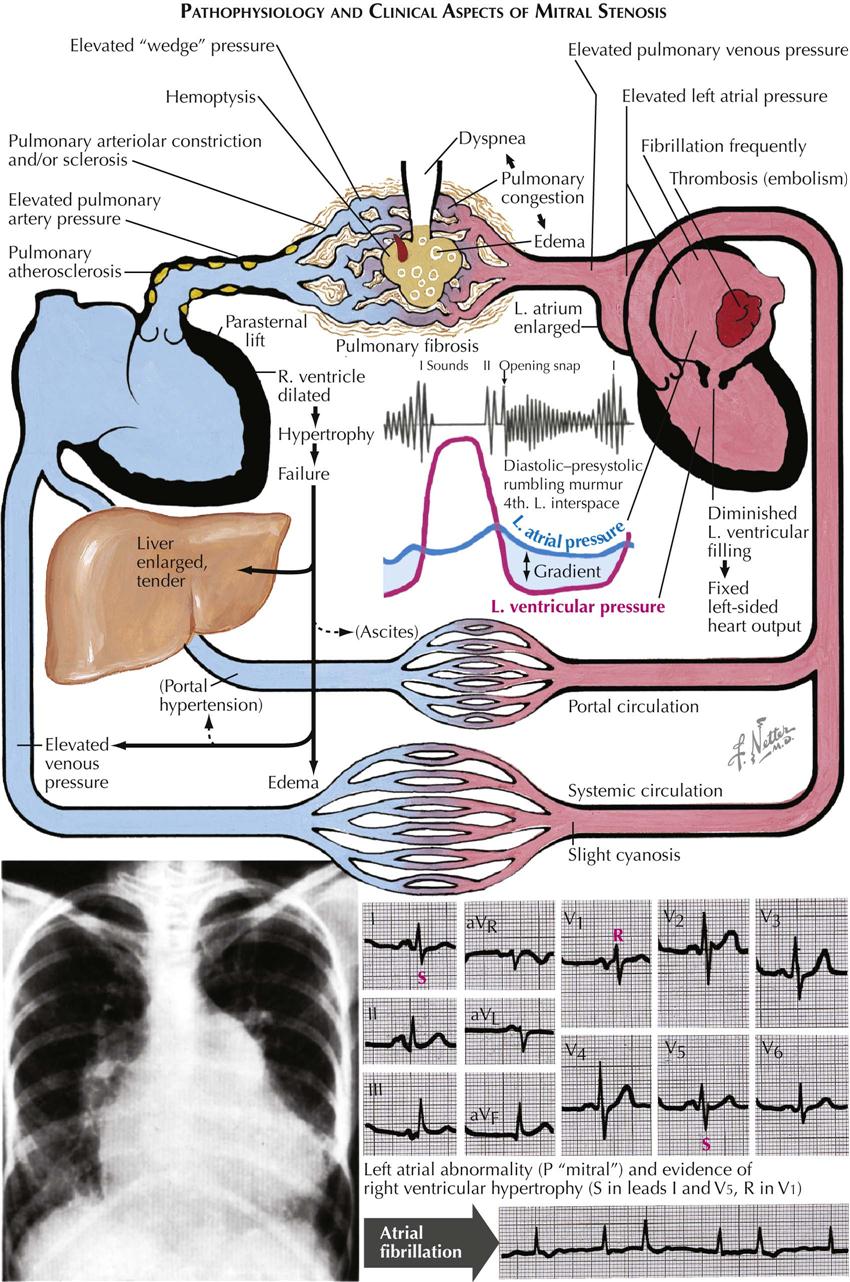
The primary functional effect of mitral stenosis is obstruction at the mitral valve.
The pressure rises in the left atrium, in the entire pulmonary vascular bed, and in the right ventricle. These functional changes result in secondary structural effects that aid in the clinical diagnosis, including enlargement of the left atrium and the main pulmonary arteries and hypertrophy of the right ventricle. The left atrial pressure exceeds the left ventricular diastolic pressure. This effect, coupled with the presence of a narrow mitral orifice, is responsible for the narrow, high-velocity stream of blood passing through the mitral orifice that creates the diastolic rumble on auscultation. At the sites where such streams strike the LV wall, “jet lesions” may develop.
In patients with established mitral stenosis, functional studies indicate diminished cardiac output, which may be fixed. Therefore the heart may not respond with an increase in output with a demand for increased tissue oxygenation. With exercise and even at rest in severe cases, there is a greater-than-normal extraction of oxygen at the systemic capillary level. This results in the characteristic increased arteriovenous oxygen difference in patients with mitral stenosis. The low cardiac output of mitral stenosis is reflected in the size of the left ventricle and the aorta. The LV cavity usually is smaller than normal, and the wall usually is normal or thinner. The diameter of the aorta may be narrower.
Mitral stenosis has classic physical findings; an opening snap is followed by a diastolic rumble, with presystolic accentuation into a loud first heart sound. These findings can be confirmed by cardiac ultrasound.
Mitral Stenosis: Secondary Anatomic Effects
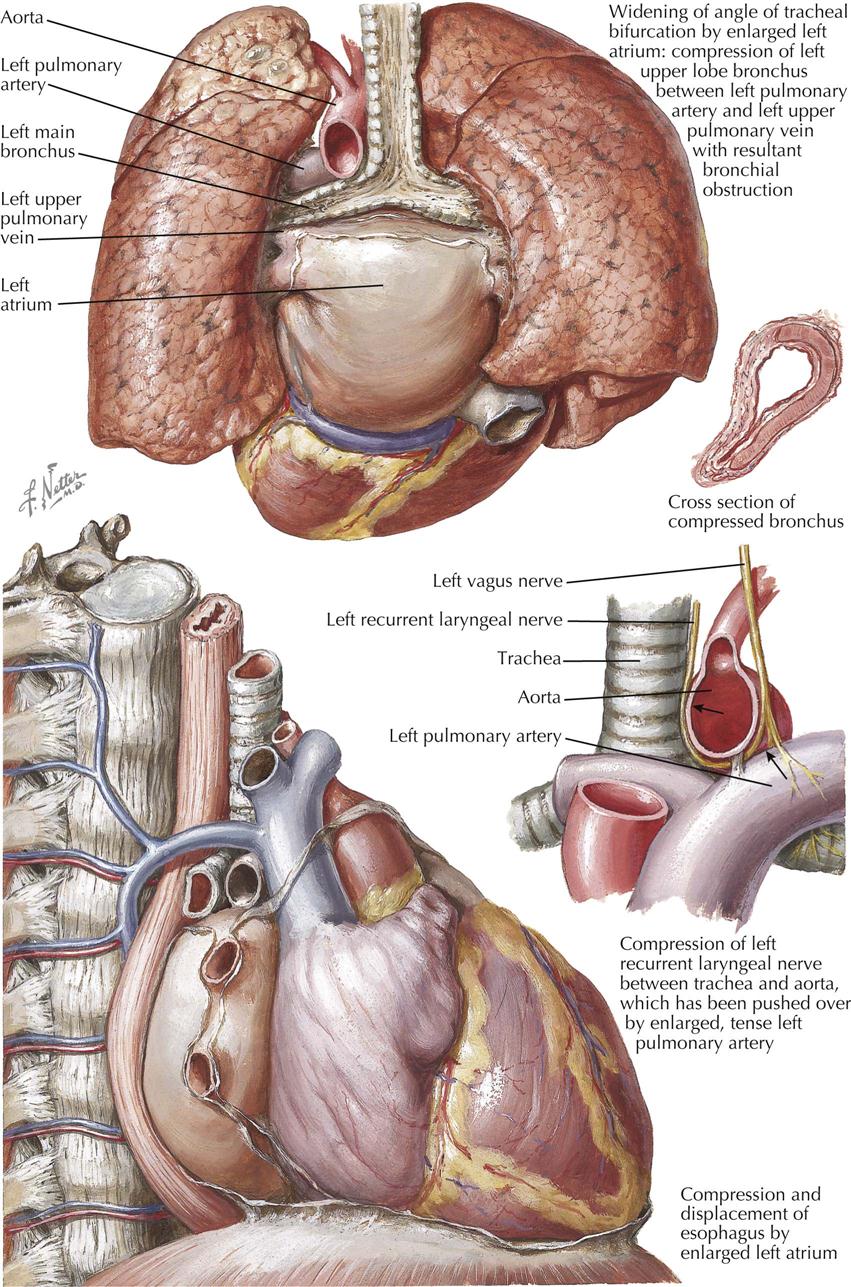
The primary functional effect of mitral stenosis is impaired flow across the mitral valve. This increases pressure in the left atrium, the entire pulmonary vascular system, and the right ventricle, causing secondary anatomic effects that include hypertrophy of the muscle in the left atrial wall and enlargement of the left atrial chamber. The wall of the right ventricle becomes hypertrophied, and the chamber may be of normal size or may be enlarged, probably a result of complicating congestive cardiac failure. Enlargement of the right ventricular chamber may in turn be responsible for dilatation of the tricuspid orifice and secondary tricuspid regurgitation. Dilatation of the major pulmonary arteries results from pulmonary hypertension, which also accentuates the second cardiac sound in the pulmonary area and contributes to atherosclerosis of the major pulmonary arteries.
The left atrium is located inferior to the tracheal bifurcation, in such a position that its superior aspect is separated from the inferior aspects of the two major bronchi by only two structures: the tracheobronchial lymph nodes and the pericardium. The tracheal bifurcation arches over the left atrium. When the left atrium becomes dilated, as in mitral stenosis, the angle of the tracheal bifurcation increases. This results mainly from an upward displacement of the left main bronchus, with the right main bronchus less affected. The close relationship between the left upper pulmonary vein and the left main bronchus may favor the latter’s upward displacement. The increased angulation (widening of angle) of the tracheal bifurcation may be identified in thoracic radiographs, and serves as a parameter in identifying left atrial enlargement.
Bronchial compression may also occur, more evident in the left main bronchus than the right. In extreme degrees of compression, the normally rounded lower aspect of the left main bronchus is represented by a sharp edge. The impaired airway resulting from bronchial compression may cause recurrent pulmonary infections. This aspect of mitral stenosis may contribute to dyspnea, which is a common symptom in patients with this valvular disease.
Hoarseness results from paralysis of the left vocal cord and may be observed in the occasional patient with mitral stenosis. This sign must be taken as a complication of the associated pulmonary hypertension, since it is also observed in patients with other forms of cardiac disease involving pulmonary hypertension. Left vocal cord paralysis is an ultimate effect of enlargement of the major pulmonary arterial system. The aortic arch and the left pulmonary artery lie within a C-shaped angle formed by the left side of the trachea medially, the left main bronchus inferiorly, and the left upper lobe bronchus laterally. Within this confined zone, an enlarged left pulmonary artery forces the aortic arch against the left side of the trachea. In this region the left recurrent laryngeal nerve ascends after hooking around the lower aspect of the aorta. Compression of the recurrent laryngeal nerve as it courses between the trachea and aortic arch appears to explain the paralysis.
Enlargement of the left atrium is an important sign of mitral stenosis. This chamber may extend farther to the right than the right atrium, as shown on chest radiography by displacement of the esophagus and a “double” atrial shadow. The left atrium lies close to the esophagus, so the enlarged left atrium frequently causes posterior displacement of the esophagus. In extreme cases, the esophagus may also be displaced laterally, usually toward the right.
Mitral Stenosis: Secondary Pulmonary Effects
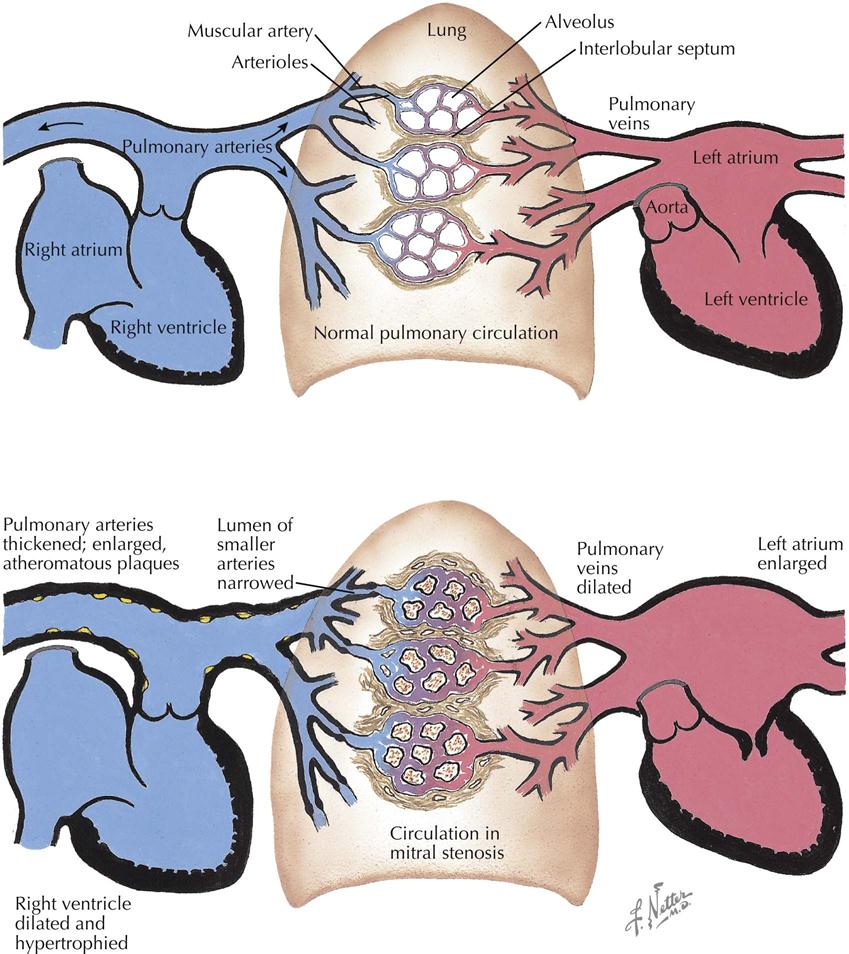
In mitral stenosis, obstruction at the diseased valve is reflected as an increase in pressure within the entire pulmonary vascular bed and right ventricle. In the normal pulmonary vascular bed, there is a low-grade differential of pressure across the arteriolar level. The small, thin-walled pulmonary arteries and arterioles are incapable of exerting high levels of vasospasm. In mitral stenosis, however, these small arterial vessels exhibit medial hypertrophy and appear to be capable of displaying considerable vasoconstriction, which is associated with greater pressure differences between pulmonary arterial and venous sides than normal. Pulmonary capillary bed pressure is determined by resistance to blood flow from the lungs (downstream stenotic mitral valve) and amount of blood flowing into the bed. If the hydrostatic pressure within the capillary bed rises to certain levels, pulmonary edema may result. The volume of flow into the capillary bed of the lung probably is determined by the degree of pulmonary arteriolar vasoconstriction, which may be seen as a protective phenomenon in mitral stenosis, guarding against flow to the degree that pulmonary edema might develop.
Although the mechanism of pulmonary arteriolar vasospasm stimulation is unknown, hypoxia may be the bottom-line cause, since oxygen does decrease pulmonary artery pressure, and when vasoconstriction is the cause, drugs such as calcium antagonists in high doses and sildenafil-like drugs also decrease pulmonary pressure in patients with evidence for pulmonary vasoconstriction. The pulmonary arteriolar bed may exert a constantly changing degree of vasoconstriction, depending on pulmonary capillary pressure.
Pulmonary arteriolar vasospasm is clinically significant in patients with tachycardia. Diastole is shortened, and in the presence of mitral stenosis, tachycardia is reflected in the retention of blood in the left atrium and in the pulmonary venous and capillary beds. Reduced blood flow into the capillary bed tends to maintain the capillary pressure at a lower level than would occur if no regulation were present. Under conditions of tachycardia, development of pulmonary edema may be ascribed to failure of the pulmonary arteriolar protective effect.
Among the structural changes in the pulmonary vascular bed is medial thickening of all classes of vessels, including the major arteries and veins. Intimal fibrous thickening may be apparent in venules, arterioles, and small arteries. These changes are nonspecific and should not be compared with the characteristic atherosclerosis, which may occur in the major pulmonary arteries. Intimal fibrous thickening of small arterial vessels is responsible for varying degrees of lumen narrowing. When present, severe changes are usually focal, with many vessels spared. This may relate to the phenomenon that pulmonary vascular disease of the degree seen in mitral stenosis does not preclude a fall in pulmonary arterial pressure if the mitral stenosis is relieved.
In addition to distention of the pulmonary capillaries, the parenchyma of the lung in mitral stenosis may show several significant alterations, including cuboidal cells lining the alveoli, fibrosis of the alveolar walls, organization of a fibrinous exudate in the alveolar spaces, and occasional spicules of bone in the alveolar spaces. Dilatation of the pulmonary lymphatics is apparent in the visceral pleura and in the interlobular septa, which probably contributes to the “straight lines” seen in the lower pulmonary lobes on chest radiographs of patients with mitral stenosis, known as Kerley B lines. This may be evidence of the assumed phenomenon in mitral stenosis that some of the fluid of the blood in the lungs is diverted to the right side of the heart by way of the lymphatics. Hemosiderosis is caused by recurrent hemorrhages from distended pulmonary alveolar capillaries, characteristically represented by the intra-alveolar accumulation of macrophages laden with iron-containing pigment. Such accumulations are responsible for the stippled appearance of the lungs in the chest radiograph and are another clinical sign of mitral stenosis.
Mitral Stenosis: Thromboembolic Complications
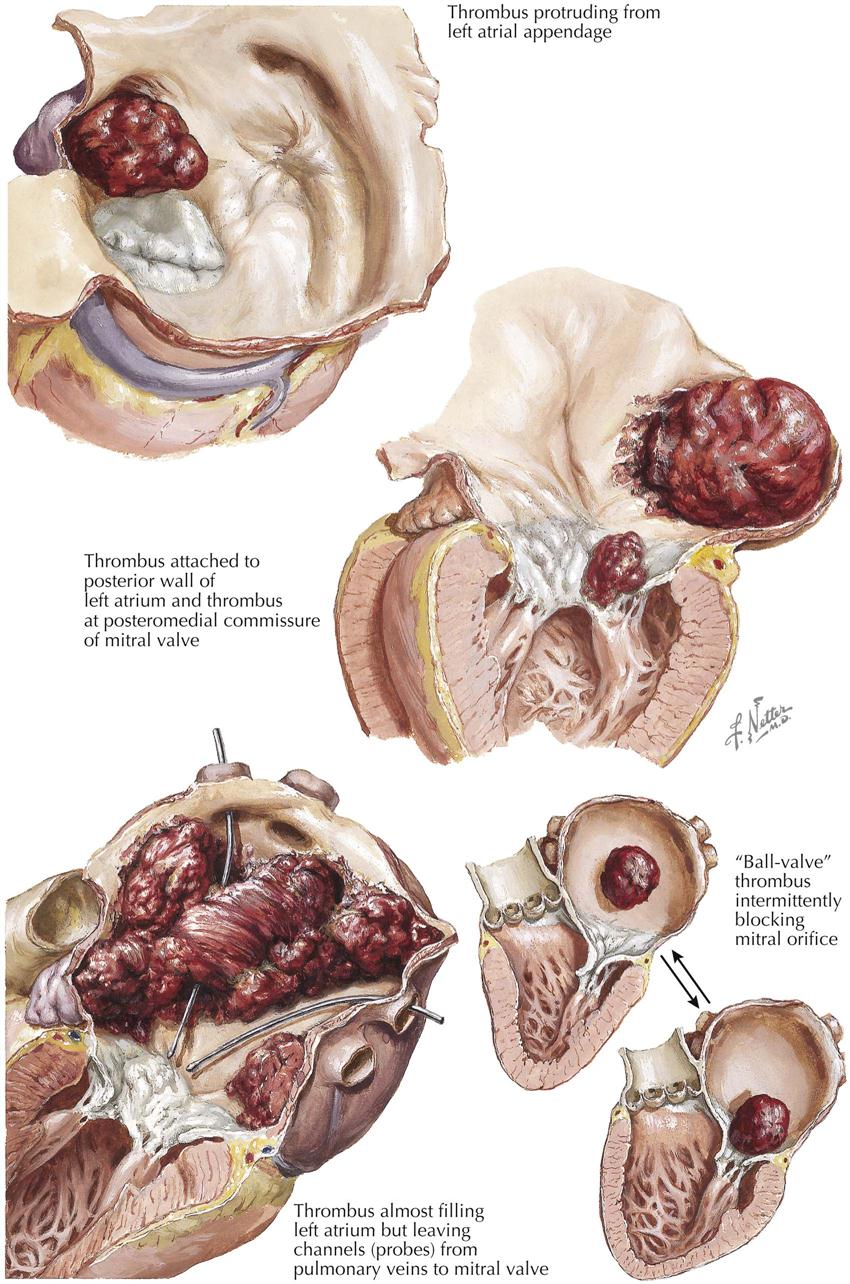
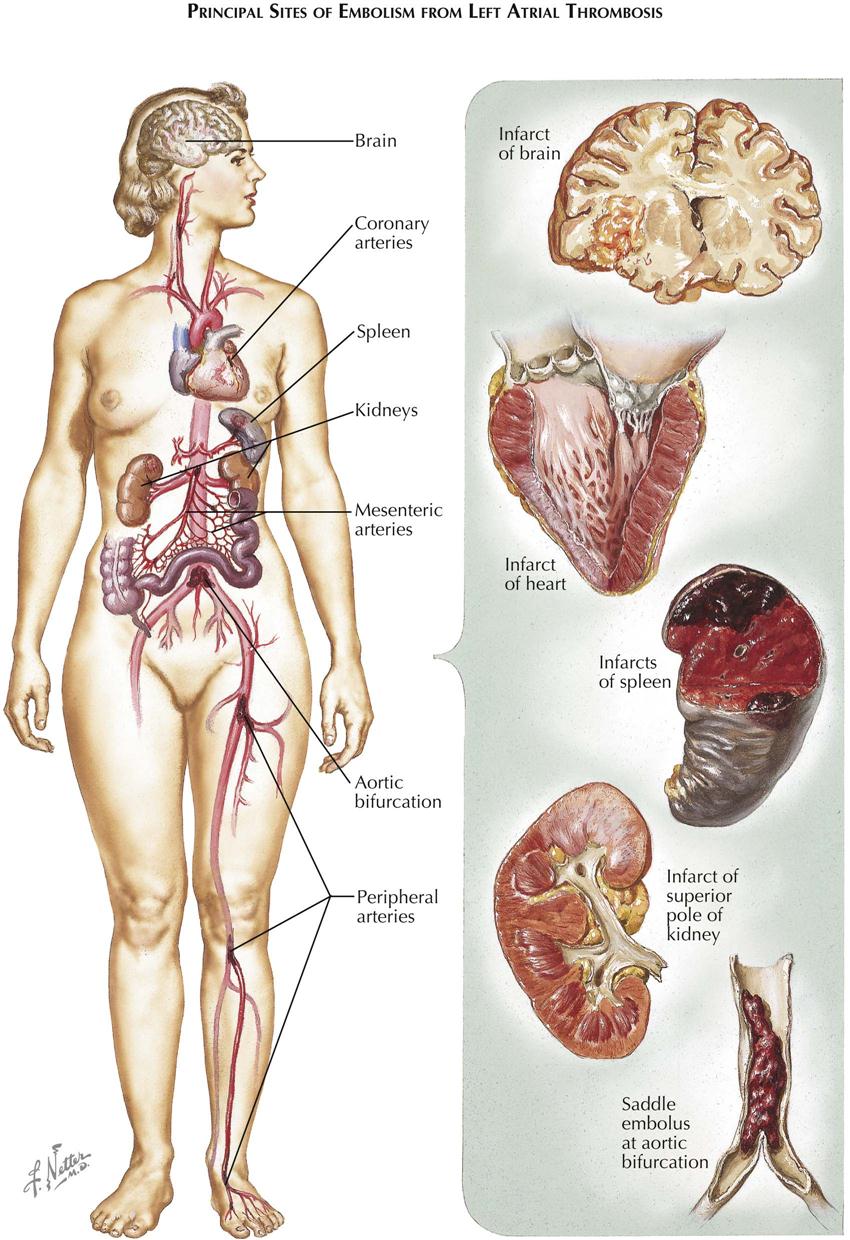
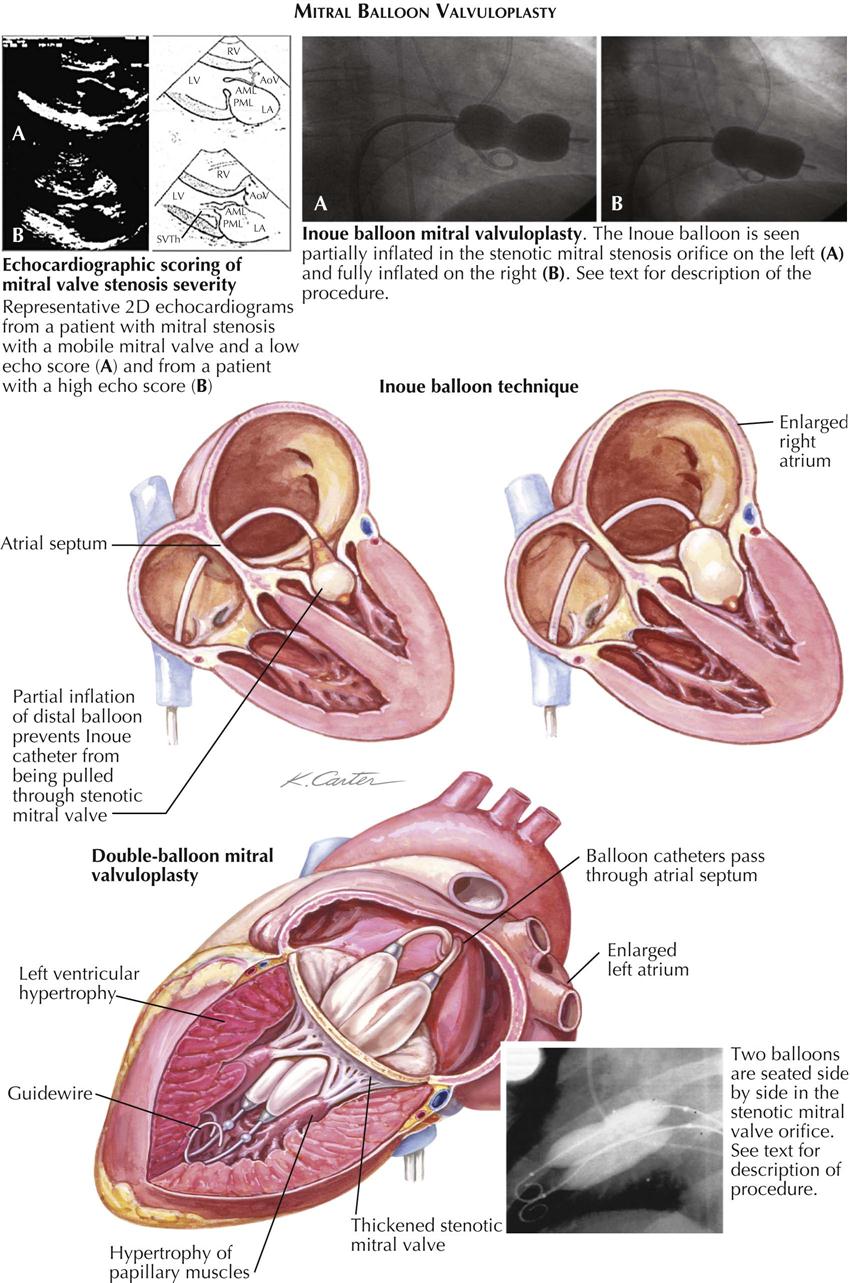
Thromboembolic complications are serious consequences of mitral stenosis and result from the common complication of left atrial thrombosis, a frequent secondary effect of which is systemic embolism. The susceptibility of the left atrium to develop thrombi in mitral stenosis relates to the incomplete emptying of the left atrium that may occur with each cardiac cycle, and in particular whether this applies when atrial fibrillation is present. In this state, left atrial thrombosis and systemic embolism are significantly more common than in patients with mitral stenosis and sinus rhythm.
Two left atrial sites are predisposed to thrombosis when mitral stenosis is present and are well visualized on transesophageal cardiac ultrasound (see Plate 6-34). These are the appendage of the mitral chamber and the posterior wall of the chamber, beginning just above the posterior cusp of the mitral valve. Thrombosis of the atrial appendage may be restricted to the appendage, or the thrombus may extend from the appendage into the main portion of the chamber. The thrombus may maintain its attachment to the wall of the main part of the left atrium and thus may be positioned for organization and firm attachment to the wall. More often, the part of the thrombus protruding from the atrial appendage in polypoid fashion into the cavity has little opportunity for attachment to the atrial wall. This type of thrombus is particularly vulnerable to fragmentation, thus serving as a basis for embolism.
Thrombi originating against the posterior wall of the left atrium are less common than thrombi of the atrial appendage. The predilection for thrombosis against the posterior wall may be associated with injury to this part of the left atrium by a jetlike stream of blood striking the wall, an expression of a minor degree of mitral insufficiency. The distribution of thrombi in some patients with mitral stenosis is so extensive that the left atrium is described as “filled” with thrombi, although there is a characteristic distribution of the thrombotic material. As the flow from the pulmonary veins through the left atrium is maintained, the process of thrombosis spares the parts of the left atrial wall involved with blood flowing from the pulmonary veins toward the mitral valve. Factors related to extensive thrombosis of the left atrium include severe mitral stenosis, older patients than usual, likelihood of left atrial wall calcification, and intractable pulmonary congestion. The severity of mitral stenosis and older patient age may in turn underlie changes in the left atrial wall, which predispose to extensive thrombosis. These include fibrous endocardial thickening and left atrial wall calcification; radiographic evidence of calcification indicates that extensively distributed thrombi are likely harbored in the left atrium. The frequent presence of intractable congestive cardiac failure in patients with extensive left atrial thrombosis also may be related to stenosis severity and older patients, possibly reflected as myocardial failure because of a longer course for the primary disease process. Extensive thrombi occupy significant space in the left atrium and thus represent an obstructive factor in addition to the primary process at the mitral valve.
Another site for thrombosis leading to embolism is the mitral valve. Thrombosis of the mitral valve tends to involve one or both commissural areas in cases of valvular calcification. This relationship suggests that the basis for thrombosis of the mitral valve is fracture of calcific valvular tissue, leaving the nonendothelialized altered valvular substance exposed to the circulation as a nucleus for thrombosis to occur. Thrombosis of the valve tends to increase the degree of the mitral stenosis.
In a special form of left atrial thrombosis, a thrombus becomes detached from its site of origin, most often the atrial appendage, and remains as a loose body in the left atrium. As this mass moves about in the left atrium, it acquires a rounded or ovoid shape. It may engage the mitral orifice but is too large to pass through the stenotic valve. Thus the mass acts as a ball valve and occludes the circulation during diastole; the patient may lose consciousness and die. Usually, however, the mass becomes dislodged from the mitral valve during systole; the circulation is reinstated, and the patient regains consciousness. In a patient with mitral stenosis, the clinical phenomenon of recurrent fainting attacks should lead to a strong suspicion of the “ball-valve” phenomenon (see Plate 6-34). A clinically similar state may be obtained in a patient with a primary myxoma of the left atrium.
In patients with mitral stenosis, embolism may occur in the pulmonary or the systemic vascular system. Pulmonary embolism is generally a complication of cardiac failure, and the usual source of emboli is the venous system of the legs, even though thrombosis of the right atrial appendage may occur, especially in patients in atrial fibrillation. Embolism of the systemic arterial system in mitral stenosis is most often a complication of thrombosis within the left atrium and less often on the stenotic mitral valve. Any of the tissues or organs supplied by the systemic arterial system may be affected (see Plate 6-35). Depending on the size of the vessel occluded, involvement of the coronary arterial system by embolism may result in sudden death, clinical evidence of STEMI, or infarction associated with nonspecific electrocardiographic manifestations (NSTEMI). Infarction of the spleen is common. In some patients with splenic infarction, the process seems silent; others may develop acute left upper abdominal pain associated with leukocytosis.
Embolism in the kidneys with renal infarction is also common. As with the spleen, some episodes may be associated with acute abdominal pain and may be confused with conditions causing an “acute surgical abdomen.” Under such circumstances, the frequent association of hematuria with renal infarction may help in the differential diagnosis. Embolism in the arteries of the gastrointestinal tract is most likely to involve the superior mesenteric artery, whereas the celiac and inferior mesenteric arteries are uncommon sites. Occlusion of the superior mesenteric artery leads to infarction of the entire small intestine (except duodenum) and right half of the colon. Embolism may occur in the branches of the extremities, but this is uncommon, at least in causing symptoms. An embolism of the aorta is more common, characteristically becoming impacted at the aortic bifurcation, a phenomenon called saddle embolism.
Mitral Balloon Valvuloplasty
Mitral balloon valvuloplasty entails passing a catheter with a special balloon from the right femoral vein to cross the interatrial septum through the foramen ovale into the left atrium and across the mitral valve (see Plate 6-36). Using the Inoue balloon, the distal portion of the balloon is inflated and pulled against the valve cusps, and then the proximal portion is dilated in order to fix the center segment of the valve orifice. Finally, the central section is inflated transiently. Another technique uses a double balloon placed side by side in the stenotic valve orifice. Mitral balloon valvuloplasty is performed in select patients with mobile mitral valves, as determined by cardiac ultrasound. The procedure usually is performed with visualization by transesophageal ultrasound and fluoroscopy and is successful, but complications can occur. The most serious complication is development of acute mitral regurgitation, usually caused by a tear in a valve leaflet or subvalvular apparatus. The transseptal puncture itself can result in complications, usually related to needle puncture of the atrium or the aorta.
Immediate results of the balloon procedure are reduction of the mitral valve gradient and relief of symptoms. Long-term follow-up data in these patients suggest that most patients are free from restenosis at 10 years; later results are less positive. Thus, individual patients need long-term clinical follow-up, including cardiac ultrasound.
Mitral Regurgitation
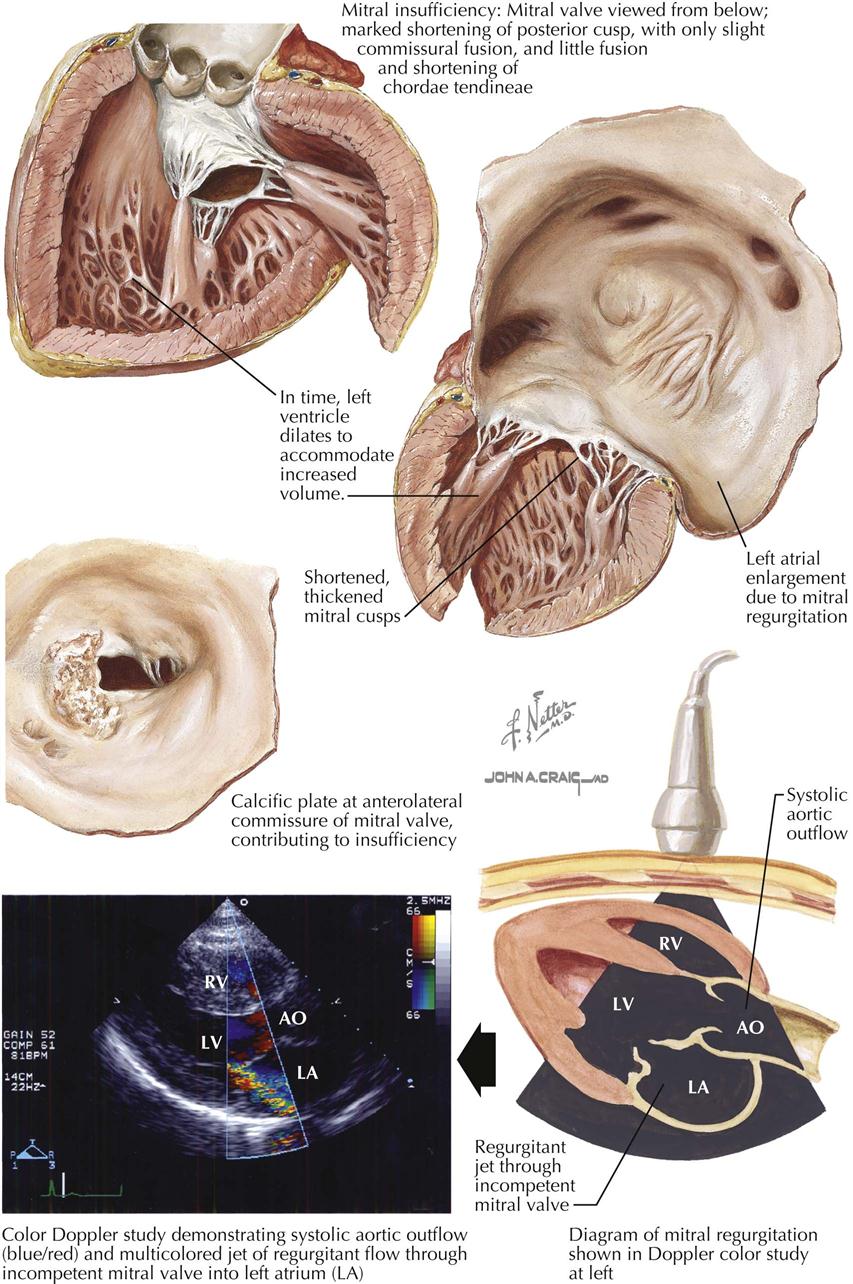
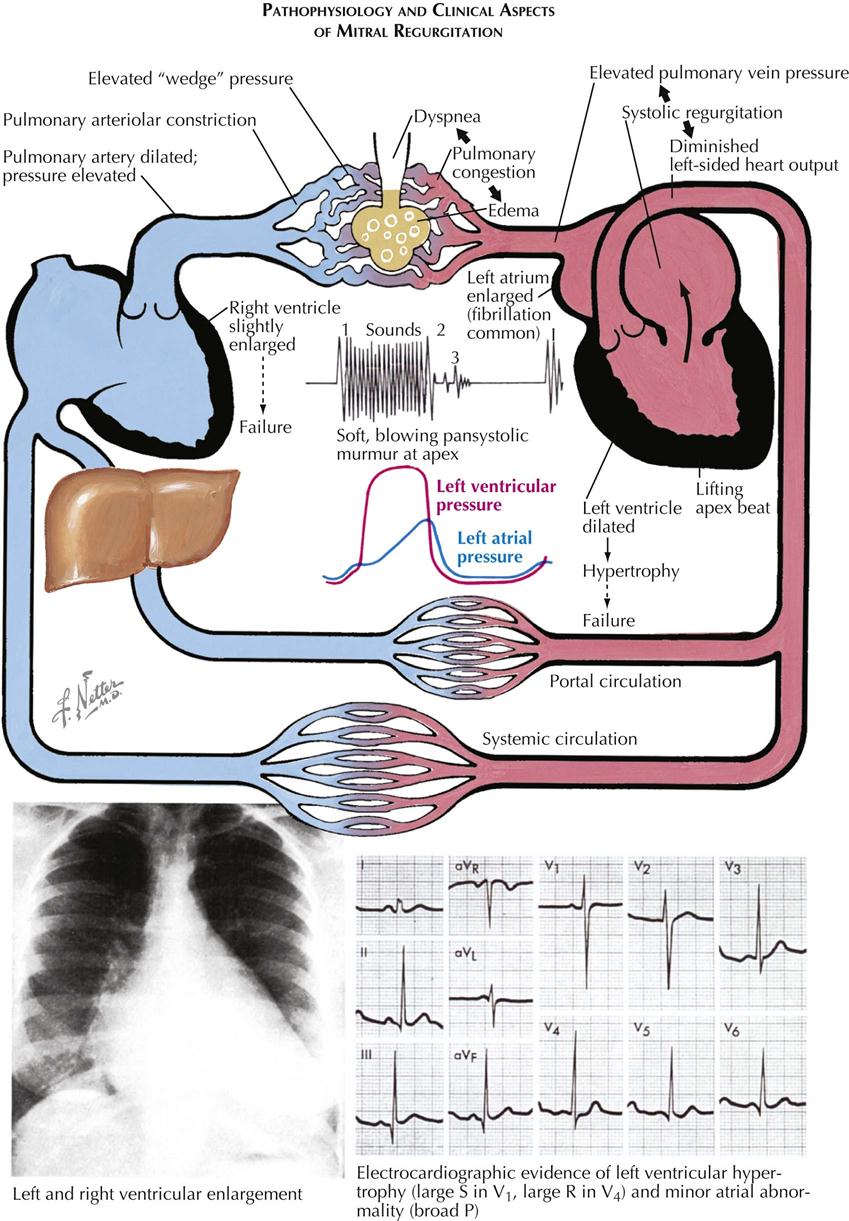
In some hearts, recurrent rheumatic mitral endocarditis results in chordal shortening and commissural fusion, changes that make the mitral valve stenotic but still competent. In other hearts, recurrent inflammation causes mitral valve incompetence. Cardiac ultrasound with color Doppler can demonstrate regurgitant flow through the incompetent mitral valve into the left atrium (see Plate 6-37). The specific anatomic changes that result in mitral insufficiency include intrinsic shortening of the cusps, commissural calcification, and left atrial enlargement. Characteristically, in patients with rheumatic mitral insufficiency, the chordae tendineae may be somewhat thickened, but no significant shortening is usually present. Intrinsic shortening is more obvious in the posterior than in the anterior mitral cusp. Evidence of anterior cusp shortening is the conversion of its free edge from a convex to concave shape. As a cusp becomes contracted, the tissue near its free end retracts, resulting in a pattern with some chordae tendineae attaching directly to the free edge of the cusp. Cusp shortening is responsible for mitral insufficiency because tissue is inadequate to guard the mitral orifice.
Some patients with mitral stenosis but no mitral regurgitation have commissural calcification. The involved commissures are held in a nearly closed position by the other changes responsible for mitral stenosis. In contrast, the mitral valve commissures are held in a nearly open position with calcification in patients who have mitral regurgitation. In fact, the calcification is responsible for fixation of the cusps in such a position so as to make them incapable of making contact with the other in the area of the calcified commissure. If both commissures are calcified, the mitral valve is converted into a fixed structure that is open throughout the cardiac cycle.
Dilatation of the left atrium is a primary effect of mitral insufficiency. Once established, however, left atrial enlargement may increase the degree of mitral valve incompetence when only one or neither commissure is fused. The aggravation of mitral insufficiency by left atrial enlargement depends primarily on the anatomic phenomenon of the left atrial wall and mitral cusps being one continuous structure. When the left atrium dilates, its posterior wall extends backward and downward, which in turn causes the posterior mitral cusp to be pulled posteriorly and away from the anterior cusp, increasing valve incompetence. In extreme cases the posterior cusp may be so displaced that it lies over the base of the left ventricle. The cusp is pulled posteriorly and downward by the left atrium while being restricted from its other end by the attached chordae tendineae. This results in fixation of the posterior cusp as it lies “hamstrung” over the LV base.
Mitral regurgitation is associated with enlargement of the LV cavity and moderate hypertrophy of the LV wall (see Plate 6-38). Enlargement of the left atrium is constant, and the degree of enlargement is usually greater than in mitral stenosis. Examples of “giant left atrium” in association with mitral valvular disease are more apt to accompany mitral insufficiency than mitral stenosis. Left atrial thrombosis occurs much less often in mitral insufficiency than in mitral stenosis, probably because of the constant washing of blood in this chamber and less likelihood that blood will be retained in the left atrium in mitral insufficiency than in mitral stenosis. The secondary effects on the esophagus, tracheal bifurcation, right ventricle, and pulmonary vascular bed are similar to those described for mitral stenosis.
On auscultation, the classic finding in mitral regurgitation is a holosystolic murmur associated with cardiomegaly, nonspecific ECG changes, left atrial enlargement, pulmonary congestion, pulmonary hypertension, right ventricular enlargement, and cardiomegaly on chest radiograph. Cardiac ultrasound using Doppler imaging will document the mitral regurgitation and provide semiquantitative assessment of the amount.
Mitral Valve Clip
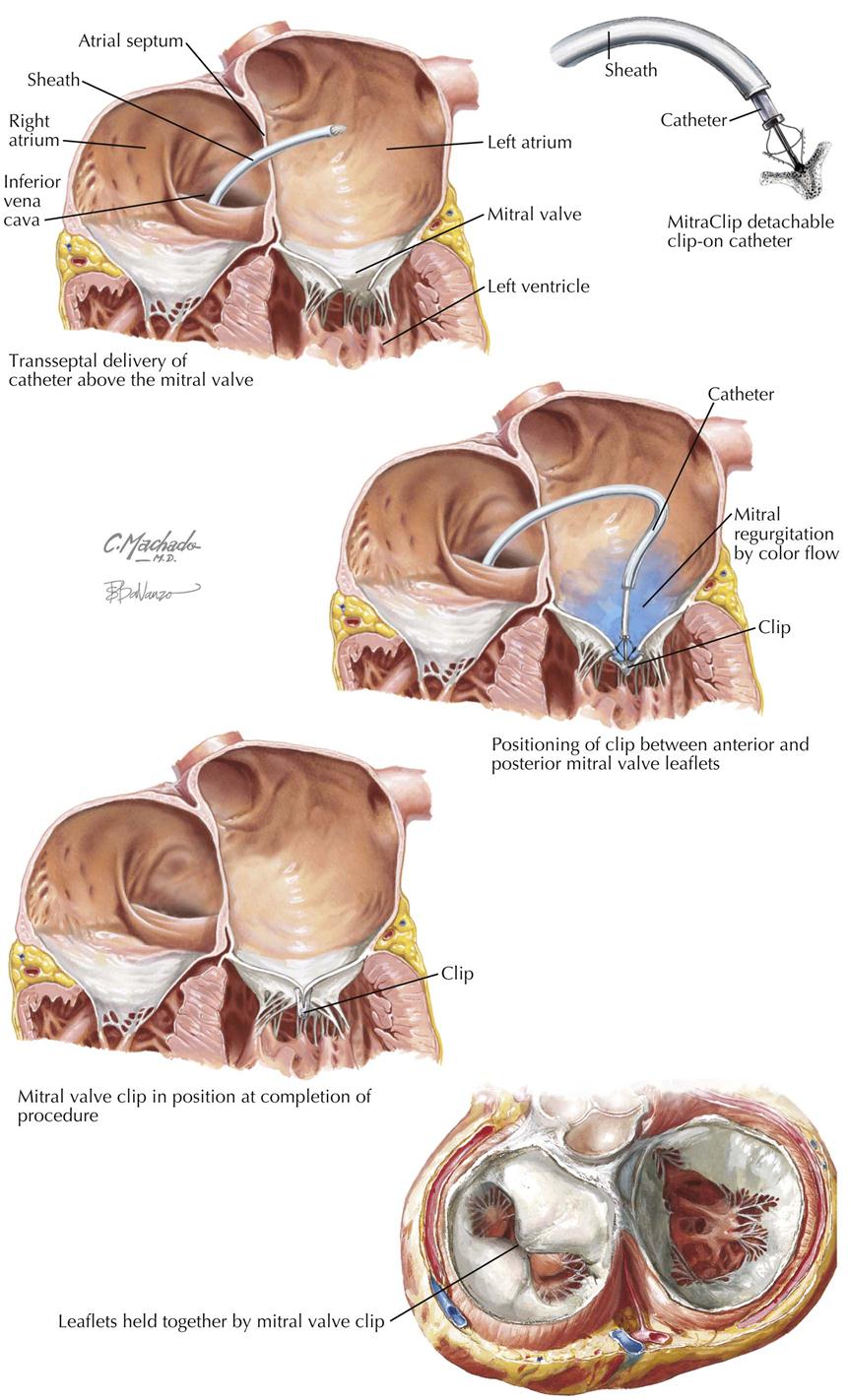
Percutaneous transseptal reduction of the mitral valve orifice of a regurgitant mitral valve is an investigative procedure. The interventional cardiologist uses a clip device to mimic the surgical procedure (Alfieri) that creates a double orifice of the mitral valve resembling a bowtie. This creates a less regurgitant orifice than the large orifice responsible for severe mitral regurgitation. The procedure consists of introducing a large sheath into the femoral vein and advancing it to the inferior vena cava, right atrium, and over a transseptal catheter into the left atrium. The clip is then passed through the sheath and positioned between the anterior and posterior mitral valve leaflets, grasping part of the commissural edges and simulating the “bowtie” surgical stitch of Alfieri. The clip is then released and left in place on the mitral valve.
A randomized controlled trial (RCT) compared the mitral valve clip (Mitra) to conventional repair or valve replacement and found that the procedure was feasible in patients with severe regurgitation (EVEREST II). The primary safety outcome was fewer blood transfusions in patients receiving the Mitra clip than in patients undergoing mitral valve surgery. The primary effective outcome favored surgery at 1 year. The mitral valve clip is unlikely to replace the usual mitral valve surgery, but in nonsurgical candidates who still require a procedure, the clip may provide an alternative therapy.
Mitral Valve Repair
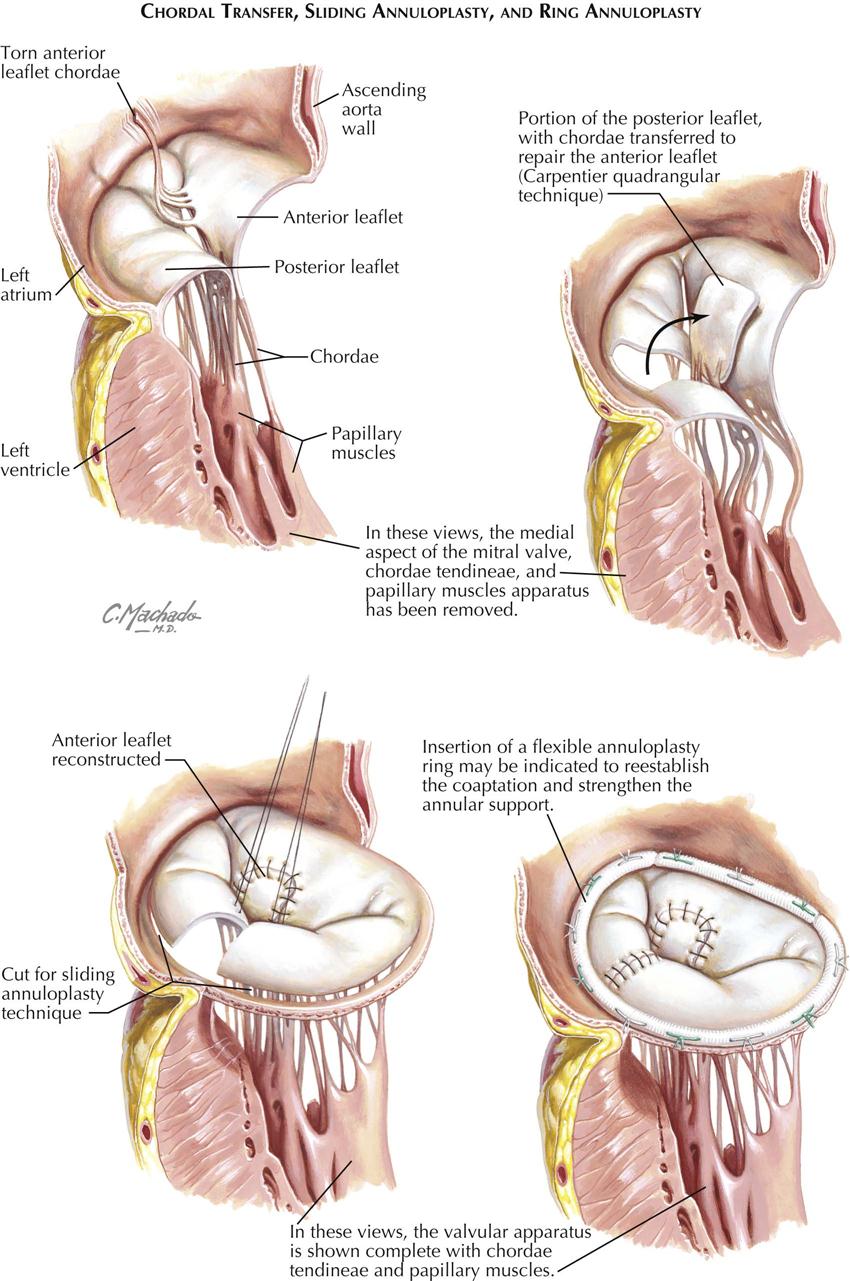
Most valve surgeons will attempt surgical repair of a regurgitant mitral valve by valve replacement or valve preservation using several evolving techniques, including the following:
Valve repair is the treatment of choice because of decreased risk of bleeding (anticoagulation not necessary unless there is another indication such as increased risk of stroke from atrial fibrillation), decreased risk of stroke (a prosthetic valve has not be used), and durability of the nonprosthetic valve (a prosthetic valve usually does not last as long as a repaired natural valve).
The cardiac surgeon must determine which procedure can be performed using criteria determined by transesophageal ultrasound and direct visualization of the valve apparatus at surgery. Approximately 90% of diseased valves from etiologies other than rheumatic can be repaired, including degenerative disease resulting in elongation or rupture of the chordal apparatus and myxomatous degeneration such as mitral valve prolapse or floppy mitral valve, as described by Barlow of South Africa.
Evidence suggests that mitral valve repair, if feasible, should be done earlier rather than later, to prevent irreversible heart damage. Severe mitral regurgitation determined by cardiac ultrasound, whether symptomatic or asymptomatic, warrants consideration of mitral valve repair, especially if the patient is in atrial fibrillation. Patients who undergo mitral valve repair must remember that they do not have an anatomically normal valve, and that endocarditis prophylaxis must be considered when certain noncardiac procedures are performed.
Mitral Valve Prolapse
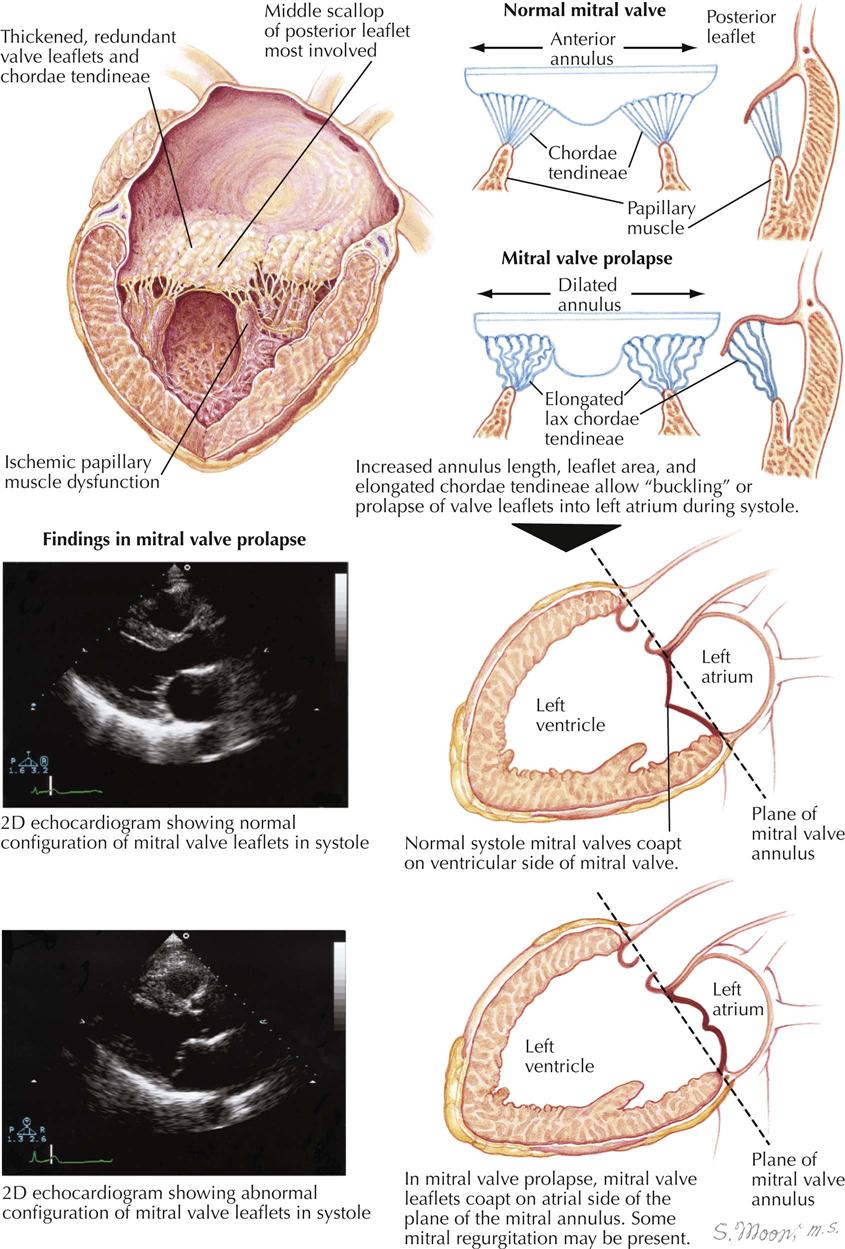
Mitral valve prolapse (MVP, ballooning mitral valve, Barlow’s syndrome) usually results from myxomatous degeneration of the valve, with thickened redundant leaflets and chordae tendineae, caused by the redundant valve tissue in ballooning or prolapse of the valve into the left atrium. If mitral insufficiency results from prolapse, physical examination reveals that the regurgitation is in late systole and is often preceded by a midsystolic click (late systolic murmur). The estimated prevalence of MVP is 2% to 3% of the population. MVP without mitral regurgitation can be determined on physical examination (isolated late systolic click) and is generally considered a benign condition. If mitral regurgitation is present, however, mortality depends on severity of the regurgitation and degree of LV dysfunction. Although infective endocarditis can occur on any valve, it is a rare event in patients with MVP without mitral regurgitation.
The diagnosis of MVP with mitral regurgitation is usually made on physical examination (late systolic click followed by a late systolic murmur) and is usually confirmed by cardiac ultrasound and Doppler imaging. Ultrasound features include an increase in the mitral annulus length, increased leaflet area, and elongated chordae tendineae. These features allow prolapse of the valve leaflets during ventricular systole. In the normal heart during systole, when the mitral valve closes, the coaptation of the valves occur on the ventricular side of the mitral valve below the plane of the mitral valve annulus. During ventricular systole in the patient with MVP, the mitral valve leaflets coapt on the atrial side of the mitral valve annulus. Occasionally, minor mitral regurgitation can be seen on Doppler ultrasound.
Aortic Stenosis: Rheumatic and Nonrheumatic Causes
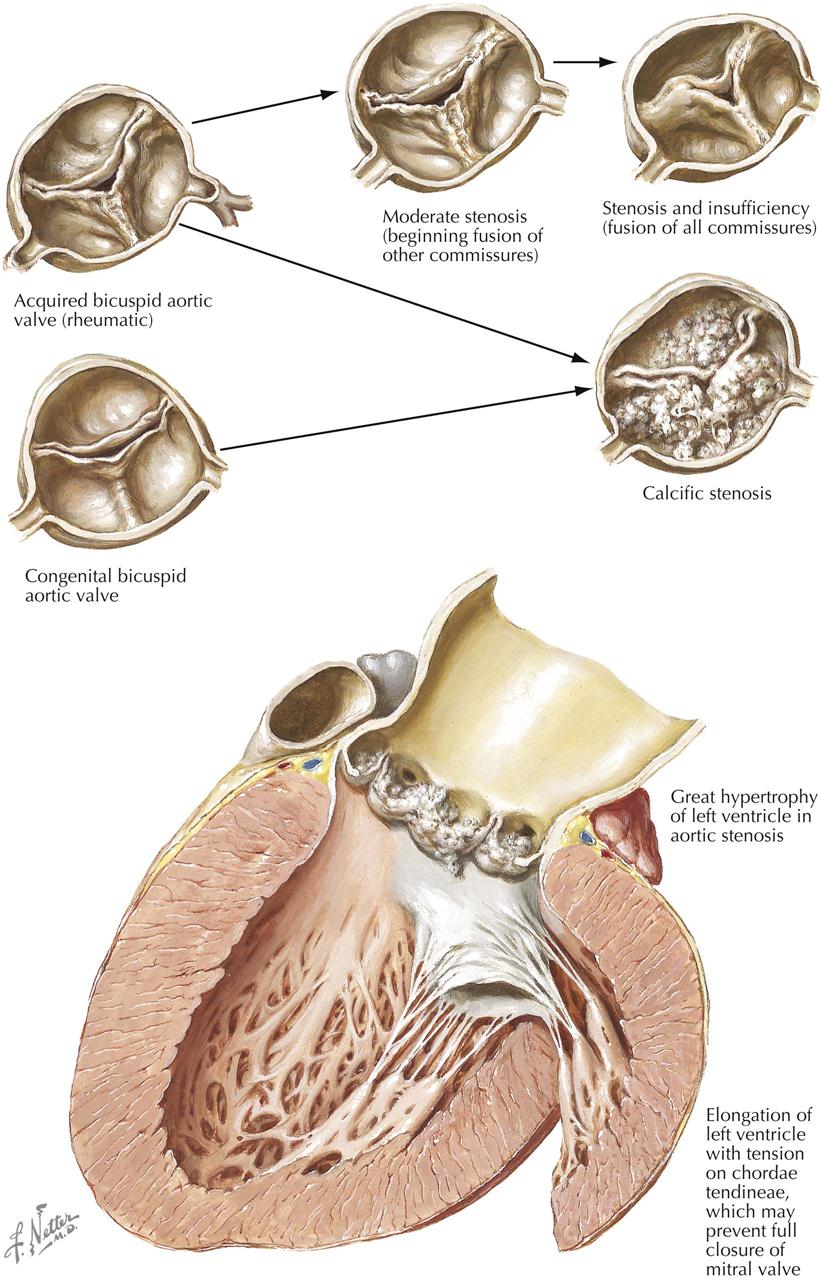
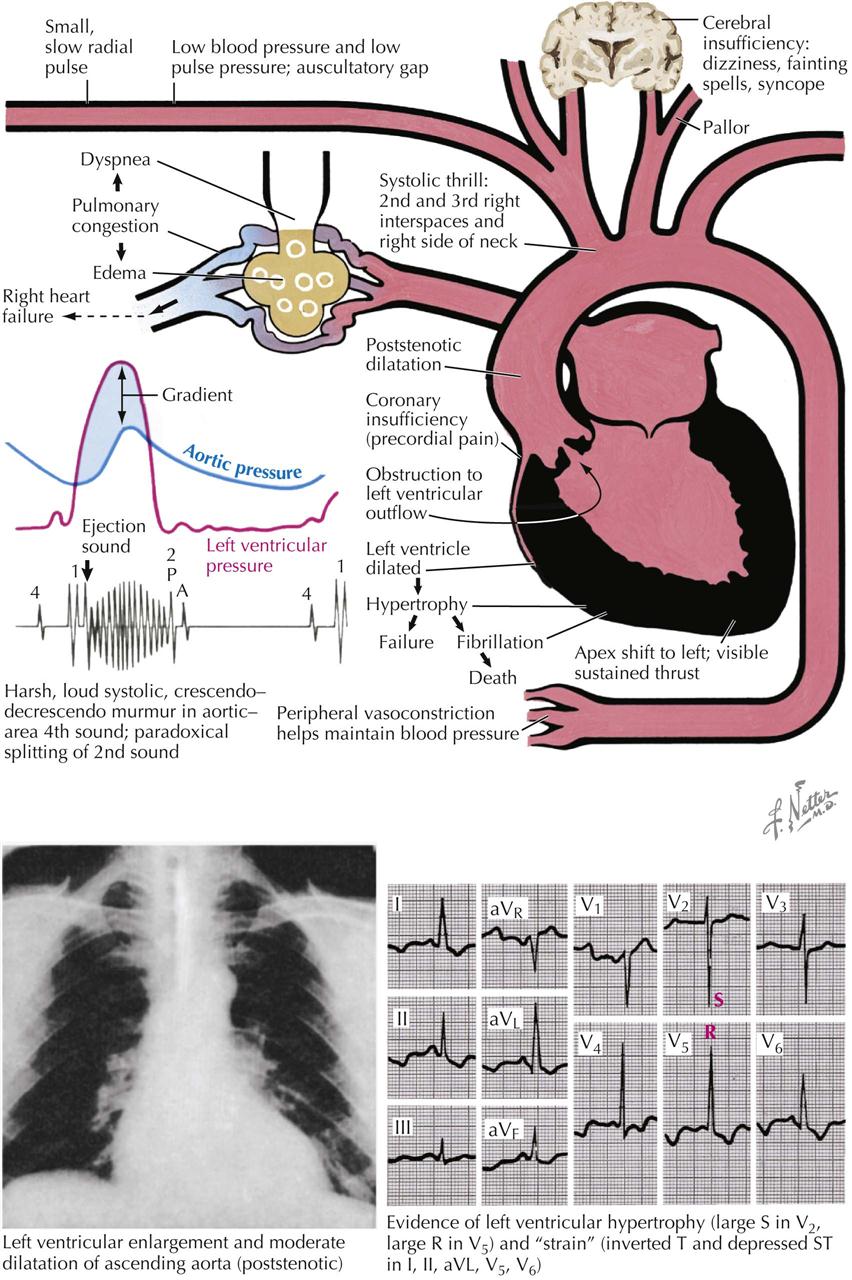
In the adult patient, obstruction at the aortic valve may be a direct result of recurrent rheumatic inflammation or a manifestation of the calcification of a bicuspid valve in the younger adult patient. In the older adult, the most common cause of aortic stenosis is progressive calcification of a trileaflet aortic valve. Diabetes, hypertension, hyperlipidemia, and chronic kidney disease may accelerate the development of calcific aortic stenosis.
In an aortic valve, various residua of rheumatic inflammation reduce the valve orifice (see Plate 6-43). The simplest type of stenosis is characterized by fusion of two of the cusps at one commissure. The resultant pattern is that of an acquired bicuspid aortic valve. The orifice of the valve, although reduced, is usually sufficiently wide so as not to cause recognizable obstruction to the egress of blood from the left ventricle. In the next stage in severity, the cusps are fused at two of the commissures. The restricted motion of the cusps by the adhesions may reduce the orifice enough to cause significant aortic stenosis. The greatest restriction of motion is achieved with fusion of adjacent cusps at each commissure. Alteration of the aortic valve in this manner is always associated with some degree of cusp shortening. Fixation of the cusps makes the valve severely stenotic, and shortening is responsible for coexistent aortic insufficiency of a degree dependent on the extent of shortening.
The second way that rheumatic inflammation leads to aortic stenosis is through the inflammatory process, which operates indirectly by first establishing an acquired bicuspid valve, which in turn may become calcified. The resultant rigidity of the cusps makes the aortic valve stenotic.
The most common type of aortic valvular stenosis is the calcific form. This process may become established on the acquired bicuspid valve or may represent a complication of a congenital bicuspid valve (see Plate 6-43 ). The most common etiology of calcific aortic valve stenosis is seen in elderly patients with a tricuspid valve. Examination of involved valves shows that such valves are often bicuspid. A congenital bicuspid valve becomes a likely candidate for calcific aortic stenosis. This concept is supported by the intrinsic features of the valve. Also, some patients with calcific aortic stenosis have a malformation associated with a high incidence of congenital bicuspid valve, such as coarctation of the aorta.
Elderly individuals typically have some calcification of the aortic cusps, and some changes may be sufficient to cause a murmur such as that heard in aortic stenosis, but the signs of LV hypertrophy are usually absent. Such patients should be considered to have aortic sclerosis. Aortic stenosis is associated with elevated LV systolic pressure greater than aortic systolic pressure. LV hypertrophy follows, giving a conical shape to the left ventricle, which extends beyond the border of the right ventricle on chest radiographs (see Plate 6-43).
In patients with aortic stenosis, coronary artery disease may be present to the same extent as seen in the general population. Areas of MI may be observed in those with normal coronary arteries but is usually associated with significant degrees of coronary artery disease. In an occasional case, coronary arterial narrowing results from embolism, the source of which may be calcific material in the diseased aortic valve or emboli from infective endocarditis of the aortic valve.
The pathophysiology and clinical features of aortic stenosis include a pressure gradient across the aortic valve resulting in LV outflow obstruction, LV hypertrophy, increased LVEDP, LV enlargement on ECG, and a harsh, loud, systolic crescendo-decrescendo murmur in the aortic area. Pulmonary congestion can occur if left atrial pressure is elevated, which in turn increases pulmonary capillary wedge pressure. The carotid pulse is narrow and rises slowly (pulsus parvus and tardus). A systolic thrill can be palpated in the carotid arteries. Chest radiography can reveal dilatation of the ascending aorta.
Aortic Regurgitation: Pathology
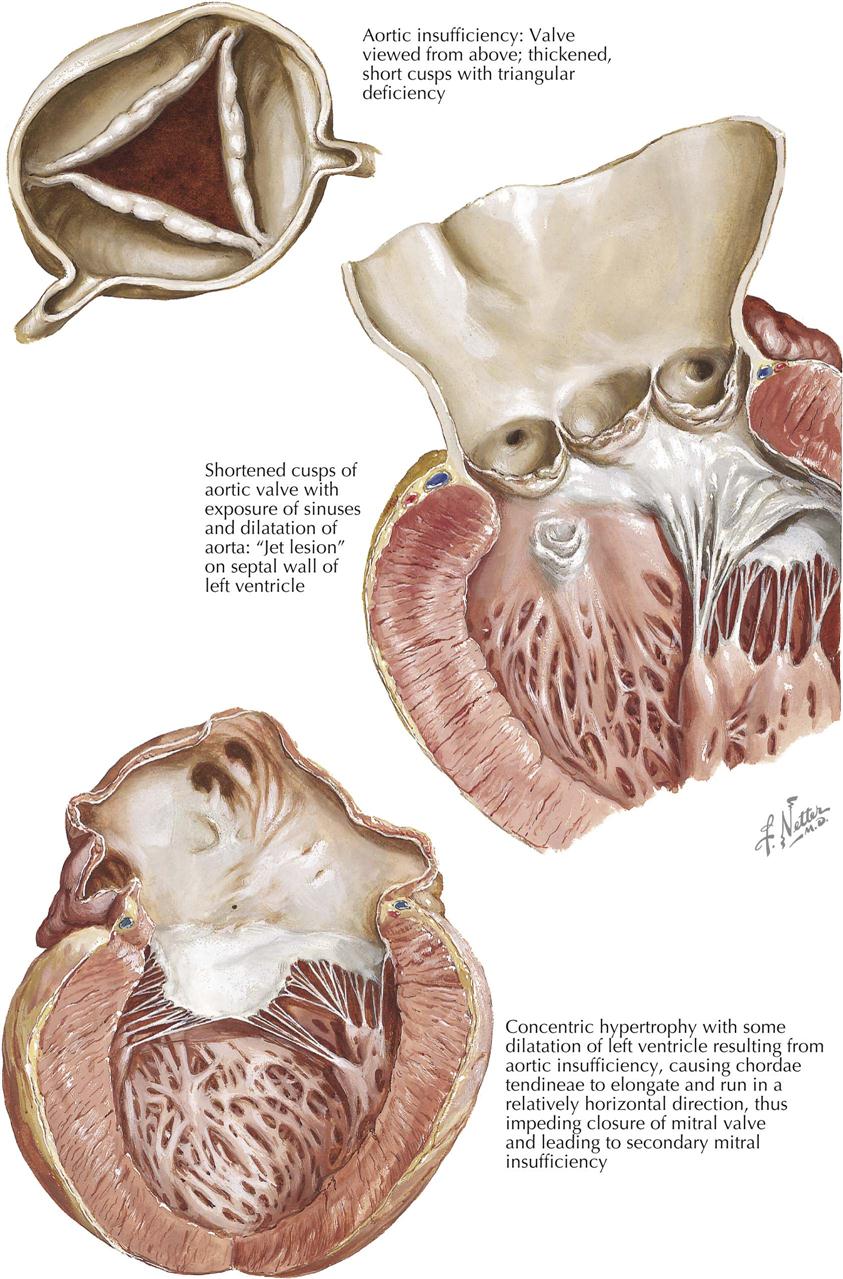
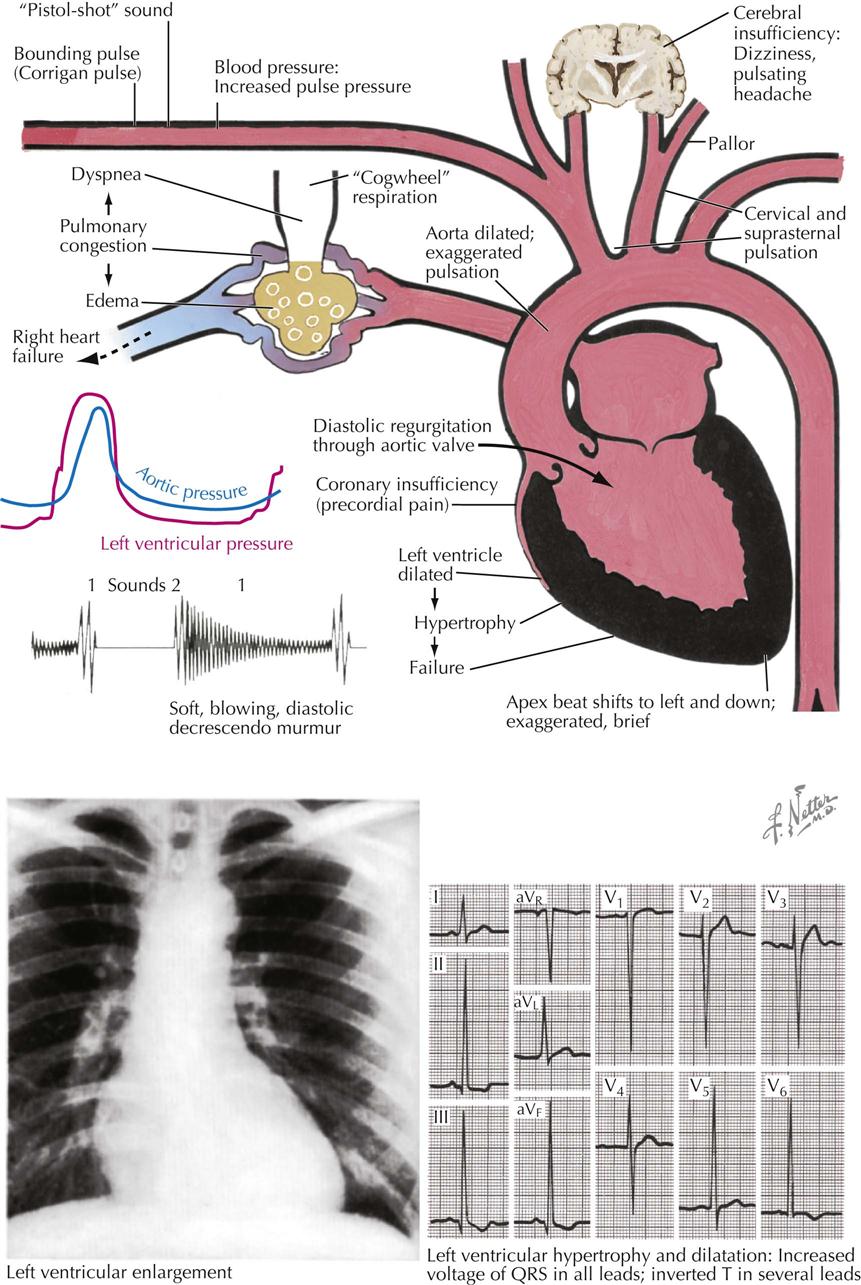
If recurrent rheumatic inflammation affects the aortic cusps but spares the commissures, isolated aortic regurgitation may result. This functional change is derived from shortening of the scarred cusps. As each cusp is shortened, some of its “extra length,” which makes the valve competent, is lost. With enough deformation the cusps become too short to make the valve competent. The incompetent orifice of the valve is then represented by a triangular opening bounded by the affected cusps (see Plate 6-44).
Shortening of the cusps involves not only their width but also their length. Thus, when the left ventricle and the incompetent aortic valve are opened in the conventional pathologic dissection, more aortic sinuses are evident than with a normal aortic valve.
Secondary signs of aortic regurgitation include widening or dilatation of the ascending aorta and alterations in the left ventricle. LV changes include hypertrophy and regurgitant “jet lesions” on the wall of the subaortic region. The jet lesions are responses to the trauma of the regurgitant stream striking the wall. These may be present on the septal wall of the outflow area or on the ventricular aspect of the anterior mitral valve leaflet. Characteristically, the fibrous tissue of the jet lesion is oriented in a cuspid pattern, the open parts of the cusps facing the source of the regurgitant stream. Jet lesions in the subaortic area provide important anatomic evidence for aortic regurgitation, and their location may correspond to the site of origin of the diastolic murmur of aortic regurgitation. The LV dilatation and hypertrophy of aortic regurgitation may be extreme; in the heaviest hearts observed pathologically, aortic regurgitation is a common underlying problem. Along with thickening of the wall, the cavity becomes enlarged in both lateral and downward directions. The LV changes may be responsible for the development of secondary mitral regurgitation for the following reasons:
The LV hypertrophy accompanying aortic regurgitation or aortic stenosis may be seen as a basis for increased resistance to LV filling. This manifestation is expressed functionally as elevated LVEDP, which in turn may be considered an obstruction to pulmonary venous flow and thus comparable to mitral stenosis. Left atrial, pulmonary capillary, and pulmonary arterial pressures are increased., and the parenchyma may simulate qualitatively that in mitral stenosis, although the degree of change is usually less in aortic valvular disease. The right ventricular hypertrophy accompanying aortic stenosis or aortic insufficiency may be derived similar to the right ventricular hypertrophy of mitral stenosis.
Clinical manifestations also include diastolic regurgitation of blood through the aortic valve, which may result in inadequate filling of the coronary arteries, premature closure of the mitral valve (Austin-Flint murmur), mitral regurgitation, and pulmonary edema. A blowing, decrescendo diastolic murmur is heard best in the third left intercostal space (Erbs area). Because of the aortic regurgitation, there can be a wide pulse pressure, bounding carotid and other artery pulsation, LV enlargement on physical examination and chest radiography, and LV hypertrophy on the ECG. A systolic murmur is usually audible, related to the large volume of blood flowing across a diseased valve in systole (see Plate 6-45).
Transcutaneous Aortic Valve Replacement
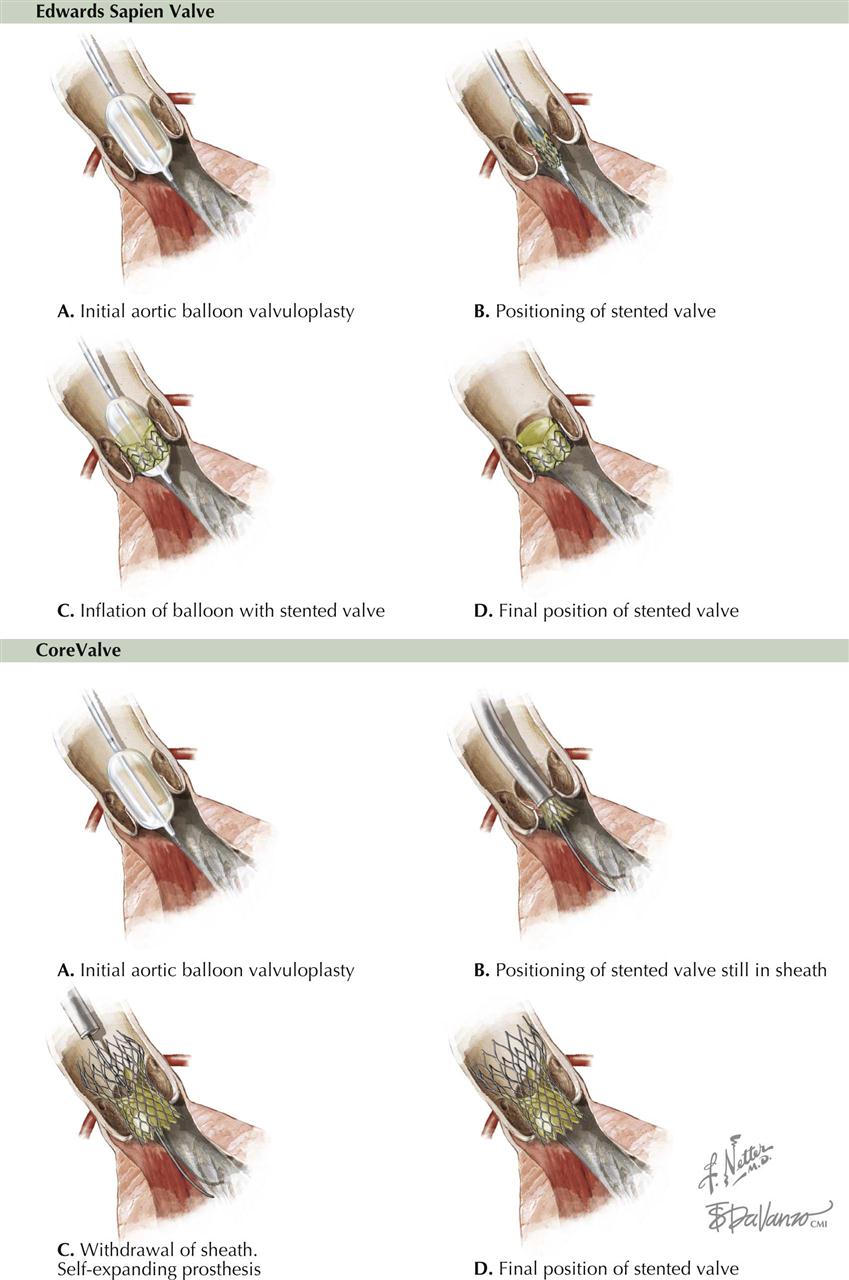
Cribier performed the first transcutaneous aortic valve replacement (TAVR) in 2002, with an estimated 80,000 implanted worldwide since then. Catheter-based TAVR uses a transfemoral, transapical, transsubclavian, transaortic, or even transcarotid approach.
Currently, two devices are available in the United States (more in Europe) for implantation in appropriate patients: a self-expanding trileaflet porcine pericardial valve mounted on a metal frame (CoreValve, Medtronic) and a trileaflet valve of bovine pericardium mounted on a metal stent with expandable balloon (Sapien, Edwards Laboratories). Both are deployed after intra-aortic balloon valvuloplasty. The valve must be positioned precisely and oriented correctly to avoid interference with the coronary artery orifices. Either valve is generally used in high-risk patients with severe aortic stenosis, and except for a higher incidence of stroke than with surgical aortic valve replacement, outcome results are comparable over the short term. Although TAVR outcomes continue to improve, concerns remain with respect to vascular injury, stroke, paravalvular regurgitation, and valve durability. However, with ongoing refinement of transcatheter valve systems, techniques, and patient selection, TAVR likely will be used in a broad population of patients.
Surgical aortic valve replacement (SAVR) has long been the mainstay of therapy for severe aortic stenosis. Randomized controlled trials by interventional cardiologists and cardiac surgeons and ongoing surveillance will play an important role as rigorous clinical evaluation begins for minimally invasive therapies for structural heart disease. In high-risk patients with severe aortic stenosis, transcatheter and surgical procedures for aortic valve replacement have similar rates of survival at 1 year, although there are important differences in risks associated with the procedure. The transcatheter procedure was associated with a higher risk of stroke than the surgical replacement (5.5% vs. 2.4% after 30 days; 8.3% vs. 4.3% after 1 year), although this difference was not statistically significant at 2 years.
Cystic Medial Necrosis of Aorta
Cystic medial necrosis (also called Erdheim disease) of the aorta is another cause of aortic regurgitation. In some patients the aortic valve may be competent, while other manifestations, such as mitral regurgitation or dissection of the aorta, may occur. Cystic medial necrosis is seen in elastic arteries and is characterized histologically by deposits within the media of amorphous basophilic accumulations, or microcysts, and accounting for the “cystic” necrosis. Initially tiny isolated lesions, the microcysts tend to coalesce and in extreme cases replace broad areas of the media. While the microcysts are small, the elastic laminae of the aorta may be intact. In the presence of coalesced microcysts, however, the elastic laminae in a given area are interrupted, and such fibers then recoil. The histologic effect is that multiple areas of the media are devoid of elastic fibers. The overall gross effect of this process is an increase in the diameter of the aorta in the involved segments. In the aorta the greatest effect of cystic medial necrosis is evident from the root of the vessel distally to include the entire ascending aorta and varying extents of the arch.
Among patients with significant cystic medial necrosis of the aorta, variation occurs as to body habitus. In some this is normal, and the enlargement of the aorta is called idiopathic dilatation of the aorta. Others exhibit distinct characteristics of body habitus and other effects which, collectively, are called Marfan syndrome (see Plate 6-47). These patients characteristically are unusually tall and have correspondingly long bones of the arms, legs, feet, and hands. The arm span exceeds the total body height. These patients also have a high-arched palate, dislocation of the optic lenses, and a tendency toward emphysema. Biochemical abnormality may be detected by high urinary hydroxyproline levels.
Patients with extensive cystic medial necrosis of the aorta, including those with arachnodactyly, often have cardiovascular disease. Although certain congenital malformations of the heart have been identified, these are not common, and the cardiovascular association may assist in making a diagnosis. Lesions involving the aorta, the aortic valve, the atrioventricular valves, and the pulmonary trunk appear to have a direct association with cystic medial necrosis.
The aortic valve effect is a common manifestation and results in aortic regurgitation. This functional abnormality may develop in several ways, most simply through extensive dilatation of the aortic root, including each sinus of Valsalva. This process may be the sole cause of aortic valve incompetence. Some patients show extreme enlargement with prolapse of the aortic cusps, which compounds the effect of aortic dilatation in causing aortic regurgitation.
Cystic Medial Necrosis of Aorta: Surgical Management
The aorta that harbors significant degrees of cystic medial necrosis tends to rupture. This complication takes three major forms. First, aortic rupture causes a simple hemorrhage, resulting in exsanguination or cardiac tamponade from hemopericardium. Second, the rupture results in a classic aortic dissection, with complications that may include coronary artery occlusion or occlusion of peripheral vessels. Third, the tear does not extend through the entire wall of the aorta and results in a localized intravascular hematoma. If present in the ascending aorta, this hematoma may distort the aortic valve sufficiently to initiate or exacerbate aortic regurgitation. Regardless of whether a localized dissection or an intramural hematoma is present, the ascending aorta dilated by cystic medial necrosis may cause some alteration in the shape of the heart on chest radiographs. Because the aortic origin is within the cardiac outline, however, major degrees of aortic dilatation may go undetected unless ultrasound or CT images of the aorta are obtained.
The atrioventricular (A-V) valves of patients with cystic medial necrosis of the aorta may show changes indicating the weakness of connective tissues. Characteristically, the valve substance between the insertion of the chordae tendineae tends to balloon up toward the atrium (myxomatous degeneration, mitral valve prolapse), and there may also be elongation of the chordae. These changes may account for incompetence of either the mitral or the tricuspid A-V valve, although the mitral valve is affected more often. In the pulmonary artery, cystic medial necrosis manifests as dilatation of the involved vessels. So-called idiopathic dilatation of the pulmonary trunk may be a manifestation of cystic medial necrosis in this vessel.
Medical management of Marfan aortic disease consists of beta-adrenergic blockade to decrease the force and velocity of contraction of the left ventricle on the proximal aorta. The rationale is that beta-blocker therapy reduces the force and velocity of aortic ejection on the weakened tissue of the aorta and thus slows the progression of aortic dilatation. Non-dihydropyridine calcium channel antagonists may give similar results.
Surgical therapy of Marfan syndrome is considered when the risk of aortic dissection is apparent with progressive enlargement of the aortic root (see Plate 4-48). Two methods are used. With the traditional method, the surgeon replaces the aorta with a graft and the aortic valve with a mechanical valve. With the valve-sparing method, the ascending aorta is replaced with a graft and the valve reimplanted (see also Plate 6-54).
Surgical Management of Abdominal and Thoracic Aortic Dissection and Aneurysms
Surgical management of other types of aortic dissections and aneurysms are typically deferred until risk of rupture outweighs the risk of repair. Currently, endovascular therapy of dissection or aneurysm repair is often used by vascular surgeons or cardiac surgeons to treat an abdominal or thoracic aortic aneurysm or dissection. With this procedure, a large stent is introduced into the aorta percutaneously.
Syphilitic Aortic Disease
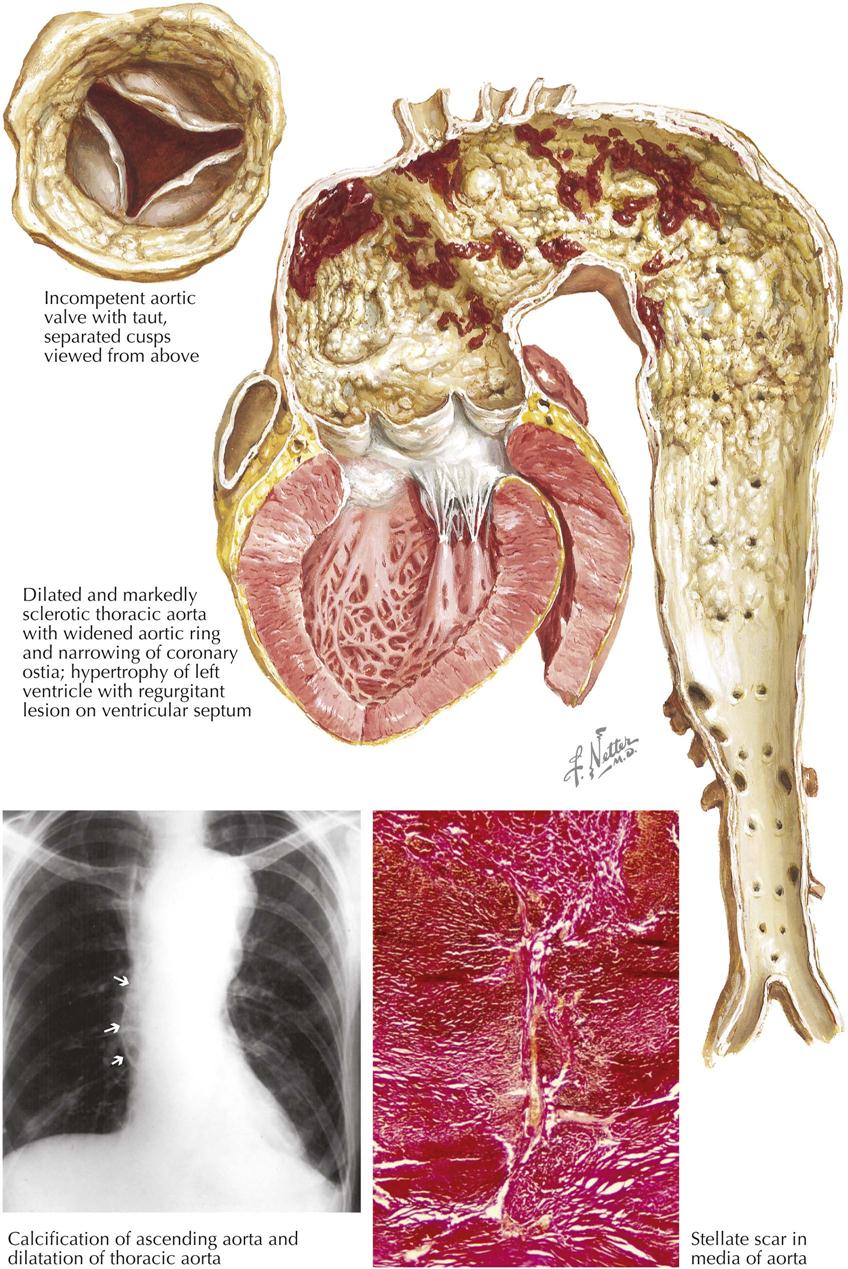
Syphilis of the aorta is a classic cause of aortic valve regurgitation. In rheumatic heart disease or infective endocarditis, changes in the valve cusps represent the primary causes of valvular incompetence. In syphilis, aortic insufficiency results primarily from changes in the aortic wall rather than in the cusps. Moreover, syphilitic heart disease has another manifestation; this relates to coronary artery obstruction because this is an aortic vasculitis.
The primary affected site is the media of the thoracic aorta, which shows many microscopic foci where tissue has been lost and replaced by delicate stellate scars. Lymphocytes and plasma cells may be present in the scars. In addition, alterations may occur within the adventitial and intimal layers. The adventitia shows fibrous thickening and lymphocytic and plasma cell infiltration. Also, the vasa vasorum may show nonspecific fibrous proliferation of the intima, with corresponding degrees of narrowing of the lumen. This change has led investigators to determine whether the medial change of syphilis is an effect of direct infection of the media or a result of faulty aortic nutrition, secondary to the alterations in the vasa vasorum.
Certain gross characteristics are displayed by the aorta, which is the end organ in syphilitic disease. Clinical identification of these features provides circumstantial evidence for a syphilitic background in the patient with aortic regurgitation.
The combination of two features of syphilitic heart disease—widening of the affected portion of the aorta and localization to the thoracic portion of the vessel—results in a characteristic appearance of the syphilitic aorta. Beginning at the root of the aorta and extending for a variable distance along the thoracic portion, the vessel is widened. In the classic example, widening extends to the level of the diaphragm, where because of a lack of dilatation of the abdominal portion, the descending aorta assumes a funnel shape as it becomes continuous with the abdominal segment. Grossly, another feature usually becomes apparent: a strong tendency for the involved portion to show diffuse atherosclerosis. This lesion may display focal calcification and may be ulcerated. Calcification in the secondary atherosclerotic lesion of the thoracic aorta involving the ascending aorta may be observed in chest radiographs. This process, together with evidence of dilatation of the ascending aorta, presents strong but not specific evidence of syphilitic aortitis. The complicating process of atherosclerosis may result in one of the classic features of syphilitic aortic disease, narrowing of the coronary arterial ostia. The same process in the aorta may also compromise the lumen of the branches arising from the arch.
In addition to the effects of secondary atherosclerosis, the syphilitic aorta may show complications of the medial disease beyond simple uniform dilatation. These include formation of saccular aneurysms in any segment of the thoracic aorta, except in the wall of the aortic sinuses of Valsalva, which appears to be spared any significant effect. Rupture of the thoracic aorta into a serous cavity or an adjacent vessel, such as the superior vena cava or pulmonary artery, may be a complication of a saccular aneurysm or simple dilatation.
In the specific area of the ascending aorta, the process of widening may be responsible for aortic regurgitation. As the aorta widens, each aortic valve cusp may be pulled away from its neighbor at the commissure, resulting in the classic feature of commissural separation. More important, as related to complicating aortic regurgitation, the widening of the aorta causes undue tension on the individual cusps. The cusps become bowed, and their length shortens relative to the size of the aortic orifice. A triangular deficiency in the aortic valve appears, and aortic regurgitation ensues. Once the valve becomes incompetent, secondary fibrotic changes may occur in the free extremities of the cusps. The aortic valve incompetence causes LV hypertrophy. Also, at the impact site of the regurgitant stream, the LV outflow area may show regurgitant “jet lesions.”
Syphilitic aortic disease has become a rare disease in developed countries since the advent of penicillin use after World War II.
Prosthetic Valve Surgery: First and Second Generation
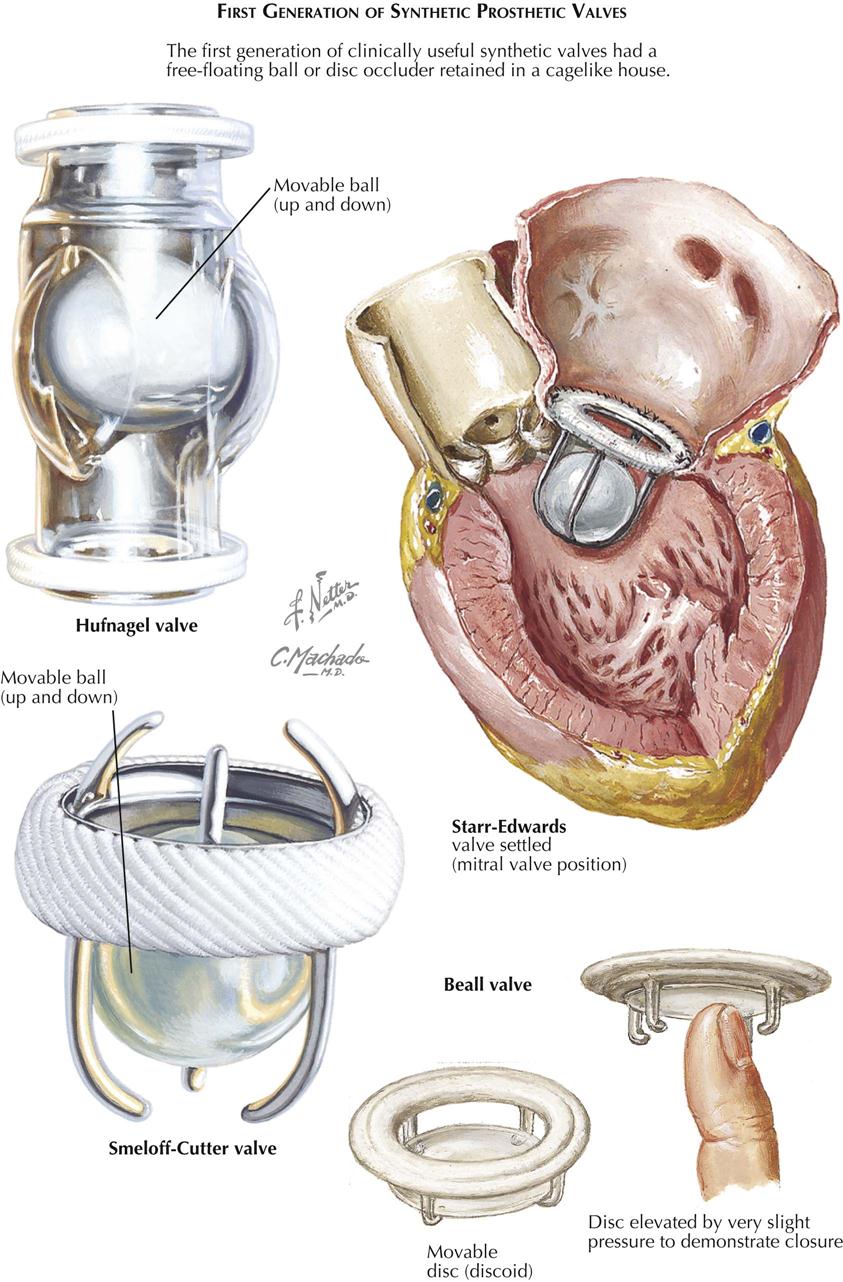
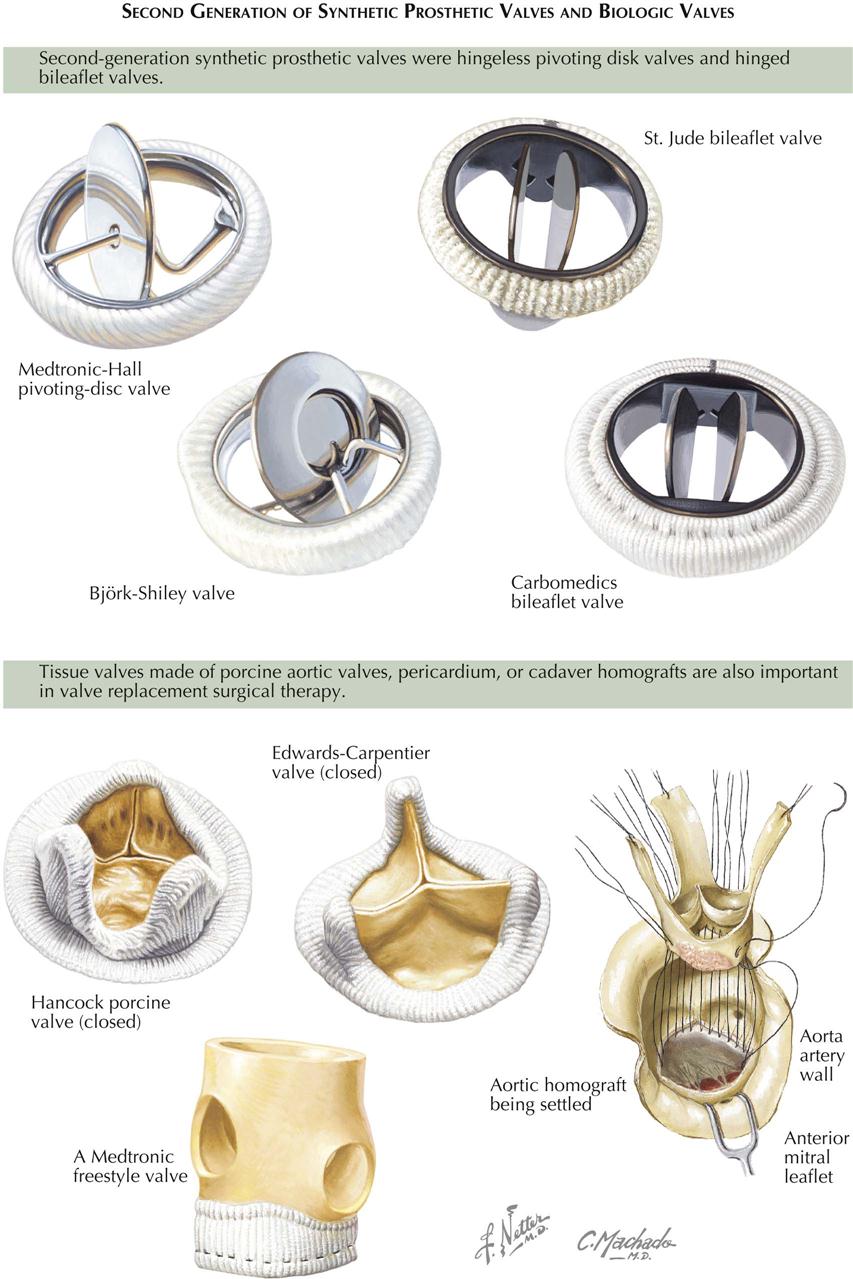
Surgery for Acquired Heart Disease
Effective surgery for acquired heart disease began about 1897 when Rehn first successfully sutured a wound of the heart. Surgical procedures for acquired valvular disease were initiated with Cutler’s pioneering effort (1920), which converted mitral stenosis into mitral insufficiency by using a valvulotome inserted transventricularly. Souttar had an early success (1925) with a transatrial mitral commissurotomy for mitral stenosis. Contributions by Bailey and Harken to closed mitral operations aided in the meteoric development and popularization of cardiac surgery.
A true breakthrough in curative efforts was achieved when the techniques of cardiopulmonary bypass were introduced in 1954, when a direct surgical approach to all cardiac valves first became feasible. These benefits and advantages are so demonstrably sound that, although closed mitral commissurotomy still is suitable in some patients, open-heart techniques are preferable for the correction of valvular pathology in most patients.
With the introduction of direct-vision procedures, it was hoped that restoration of valvular function could be accomplished with the natural valvular structures. Therefore, for both calcific aortic and mitral stenoses, debridement and mobilization of fixed valve cusps were attempted. Unfortunately, success was usually incomplete and transient. Somewhat better results have been obtained following plication of a dilated insufficient valvular annulus of the type present in some cases of mitral or tricuspid insufficiency. Also, plication of valve cusps, in the management of certain forms of ruptured chordae tendineae, has functioned acceptably. The overall immediate and long-term accomplishments of procedures for acquired valve disease were substantially amplified by the introduction and (shortly thereafter) ready availability of prosthetic devices, which permitted total valvular replacement. To be realistic, however, it must be emphasized that none of the presently available prosthetic valves achieves the physiologic and mechanical features of a normal human heart valve.
First-Generation Prosthetic Valves
First-generation prosthetic heart valves were designed as mechanical occluders (see Plate 6-50). Second-generation prosthetic valves were either mechanical or bioprosthetic. The mechanical valves were initially pivoting hingeless and somewhat later hinged leaflet valves were developed. Bioprosthetic valves are either porcine or human valves, and valves made from pericardium (porcine or bovine). First-generation prosthetic valves were free floating (e.g., caged disks or balls). In 1952, Hufnagel designed and implanted the Hufnagel valve, a movable ball up and down inserted in the descending aorta, primarily used to prevent blood from returning back into the heart of patients with severe aortic regurgitation.
In 1960 Albert Starr implanted the Starr-Edwards valve, a free-floating ball in a cage. Until early this century, the Starr-Edwards valve was used often in patients with aortic and mitral disease who needed replacement. Anticoagulation was needed whether the valve was placed in the aorta or mitral position. Introduced in 1964, the Smeloff-Cutter valve, a mechanical valve prosthesis, was hemodynamically similar to the Starr-Edwards prosthesis. The Smeloff-Cutter valve was a double-cage ball prosthesis and represented a major design difference from the Starr-Edwards valve. The Smeloff-Cutter was used in the aortic position with less risk of thrombosis than the Starr-Edwards, particularly in select patients who could not be anticoagulated. Both these mechanical valves allowed many patients to lead long and useful lives and could be implanted in the aortic or mitral position. These valves were discontinued as evolution of valve technology resulted in other mechanical valves.
Arthur Beall invented a free-floating disc valve with a very low profile (Beall valve) that could be used in the mitral position, particularly in patients with a small LV cavity (see Plate 6-50 ). The low profile seemed to be an advantage of the Beall valve. The free-floating disc valve evolved between 1960 and 1962 primarily for the atrioventricular position. Initially it uniformly produced essentially normal hemodynamics and excellent clinical results, without embolization. It does not impinge on the ventricular septum or obstruct the LV outflow tract. The extremely lightweight polypropylene disc allows for a rapid response with minimal pressure variations. Closure of the valve is complete on initiation of systole.
The replacement of mitral and tricuspid valves presented a particularly difficult problem because of several factors: (1) low pressure in the atrium and the resultant tendency toward thrombus formation with systemic embolization; (2) overgrowth of tissue on the metallic or plastic parts, which results in valvular dysfunction, necrosis of tissue, and embolization; (3) inertia of the valvular mechanism; (4) occlusion of a portion of the mechanism, with regurgitation before complete closure and dislodgement of the sewing ring produced by the large mass striking the areas of suture; (5) incomplete ventricular filling because of the large size of the occluding portion of the mechanism, thus requiring use of a valve of small orifice size; and (6) obstruction to the LV outflow area because of the angulation.
The Beall valve was used extensively worldwide from 1965 to 1970 but was replaced by other mechanical valves, such as the pivoting hingeless valves.
Second-Generation Prosthetic Valves
Second-generation mechanical prosthetic valves were initially pivoting, hingeless, tilting-disc valves (e.g., Medtronic-Hall, Björk-Shiley) that could be used in the aortic or mitral position (see Plate 6-51). These valves consisted of a circular occluder with excursion limited by metal struts. The disc provided two orifices, one large and one somewhat smaller, when in the open position. Both valves do require anticoagulation.
The tilting-disc valves were used for many years until mechanical hinged leaflet valves were developed (St. Jude and Carbometrics bileaflet valves). This type of bileaflet mechanical valve replaced all the previous mechanical valves and is now the most favored type of mechanical valve. These valves have struts attached to the valve ring that contain two semicircular leaflets. The bileaflet valves provide more blood flow than either caged-ball or tilting-disc mechanical valves. In fact, the bileaflet valves have almost a square centimeter opening greater than other mechanical valves. Thrombogenicity remains a problem, but these valves are less thrombogenic than previous versions of mechanical valves. The bileaflet valves do require chronic anticoagulation, but these valves can last 20 years or longer, provided the patient adheres to a rigid anticoagulation protocol, principally with warfarin. Patients must also be aware that these valves can also cause hemolysis, but that is not a common problem.
A new mechanical valve under development is the ONYX bileaflet valve made of Pyrolite carbon. This valve may require less anticoagulation than previous mechanical valves. Long-term results compared to other bileaflet valves will be needed for further evaluation.
Biologic Valves
Bioprosthetic or biologic valves (tissue valves) are made from animal aortic valves (e.g., porcine aortic valves) or biologic tissue (e.g., bovine or equine pericardium). These types of biologic valve are called xenografts. When used to form the valve leaflets, pericardium is sewn into a circular metal frame. The valve leaflets are extremely flexible, and patients do not need lifelong anticoagulation with warfarin. These bioprostheses include the Edwards Laboratories Magna bovine pericardial valve, the Mosaic-Medtronic porcine aortic valve, Medtronic freestyle valve, Hancock porcine valve, and the Carpentier valve (see Plate 6-51).
Human valves, called homografts, are obtained from human cadavers, usually within 12 hours after death of the donor. The normal aortic valve is removed with its own support structure in place. The valves are sterilized by the freeze-irradiation method. When implanted in patients, homografts require no anticoagulation. Human valves have minimal residual gradient across the valve and the best hemodynamic performance for the patient. The human valve is often used in patients whose aortic valve is damaged by infective endocarditis. Long-term results are excellent, and replacement of the valve because of deterioration at 10 years is about 10%.
Surgery for Acquired Heart Disease (Valvar Replacement)
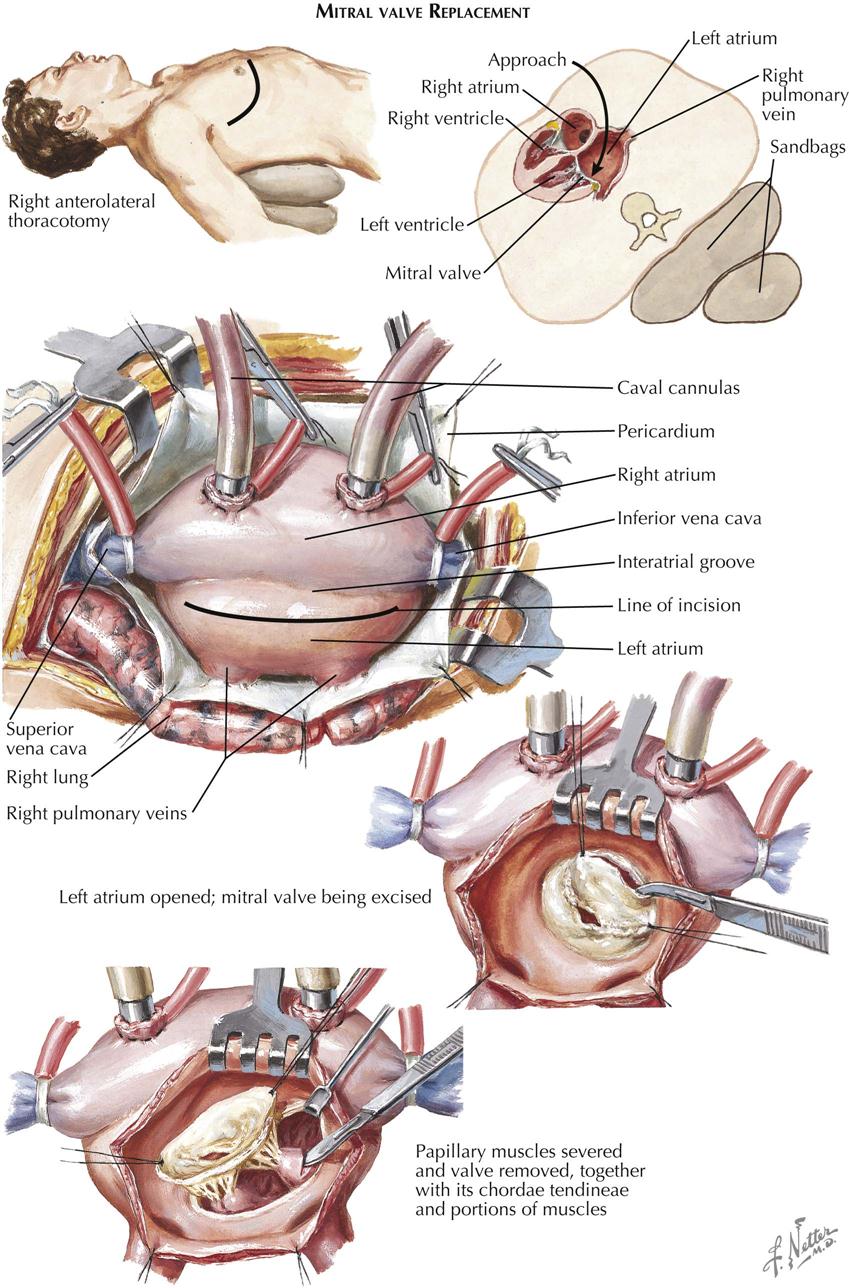
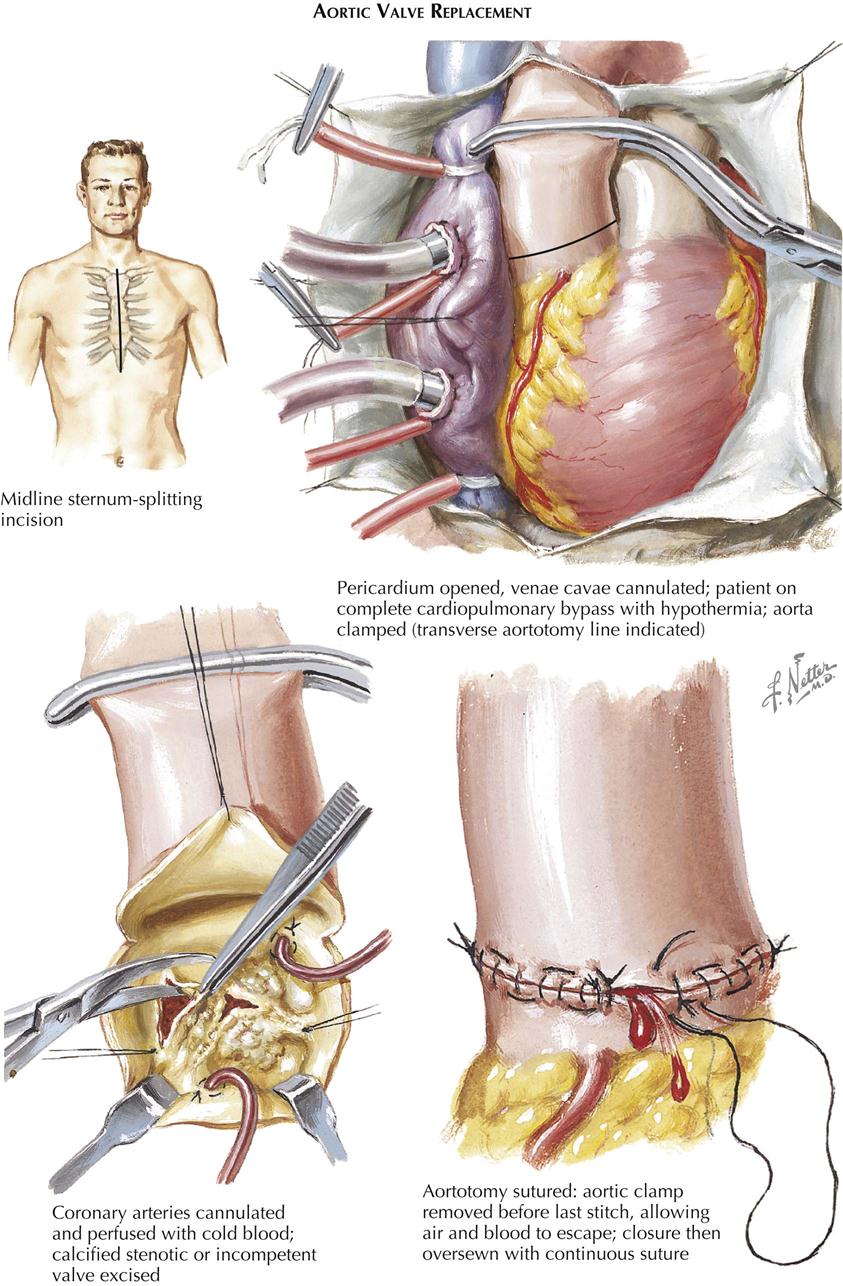
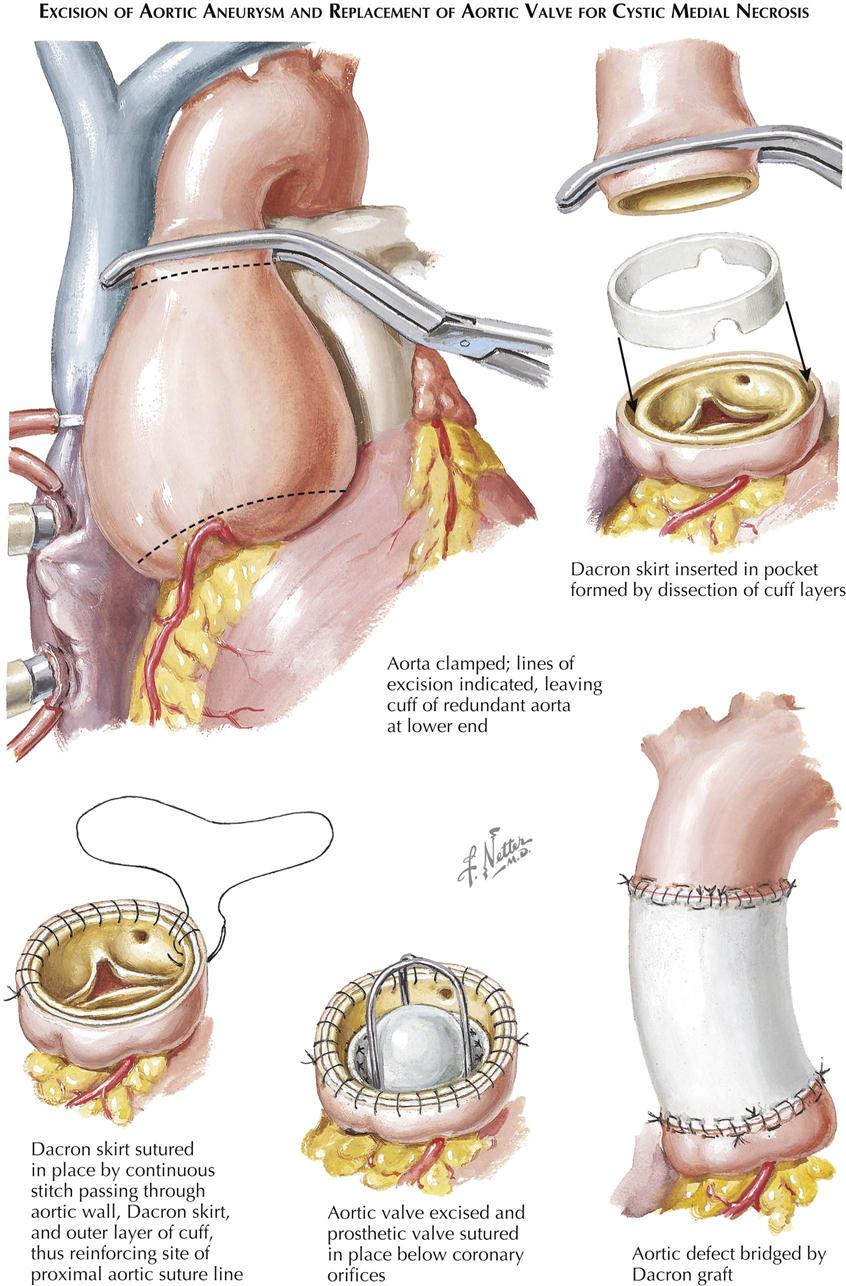
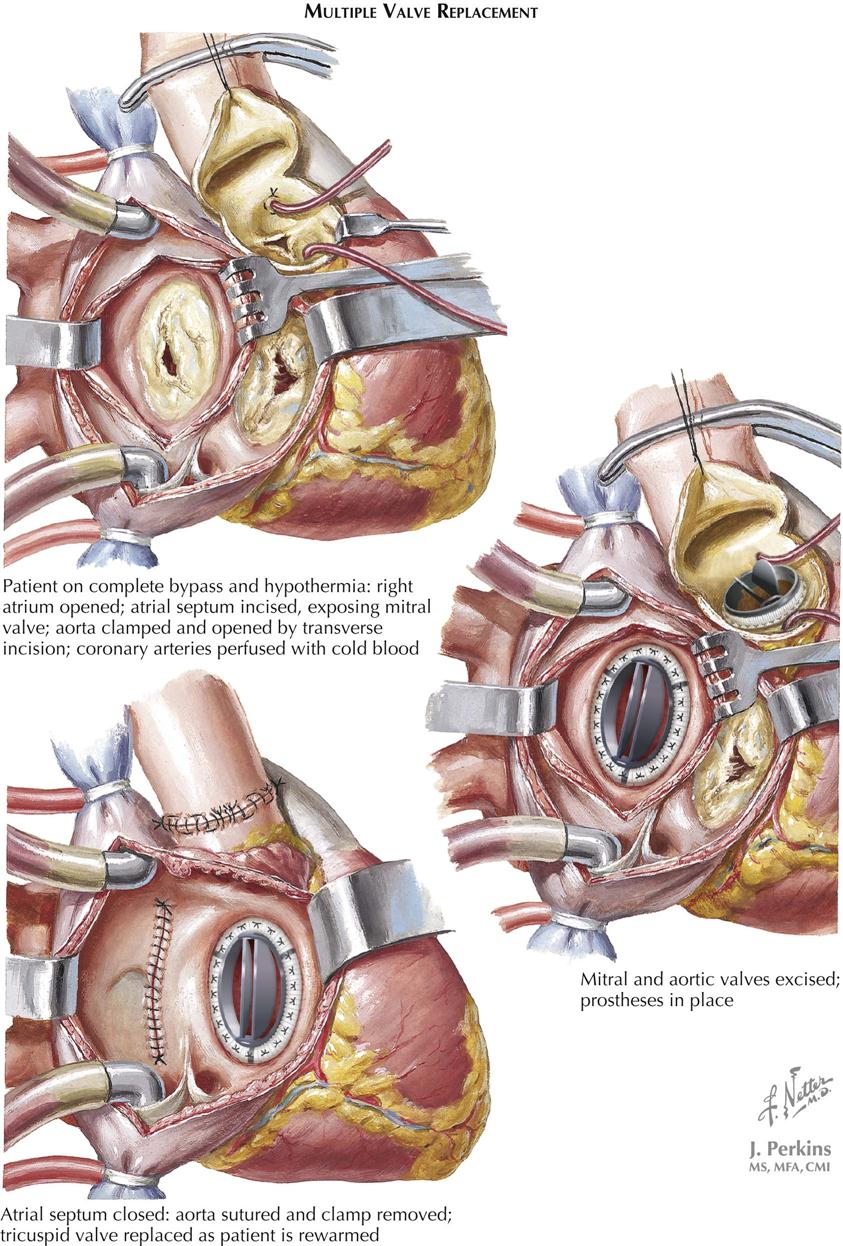
See previous discussion, “Surgery for Acquired Heart Disease,” in Plate 6-51.
Critical goals for improved prosthetic heart valve design are superior flow characteristics, development of prosthetic materials which arouse less adverse reactions in soft tissues or blood, and the elimination of mechanical breakdown. The caged ball valve of Starr and Edwards was used in the aortic, mitral, and tricuspid areas.
Mitral Valve Replacement
Acquired calcific mitral stenosis is the most common valvular lesion resulting from rheumatic fever and four times more common in women than men. Patients in New York Heart Association (NYHA) classes II and III are appropriate candidates for surgical relief of mitral stenosis (see Plate 6-52). Those in NYHA Class IV, although operable, clearly have a higher surgical risk. Mitral commissurotomy (balloon valvuloplasty or surgery) is still indicated for patients with pure mitral stenosis who have pliable mitral valves detected by physical examination and cardiac ultrasound. Coexisting heart failure is treated, whenever possible, until compensation has been achieved. However, a few patients with severe right-sided heart failure, acute pulmonary edema, or severe hemoptysis may fail to respond to all forms of medical management and may require emergency mitral valvotomy. Rheumatic activity and bacterial endocarditis are relative temporal contraindications for surgery. On the other hand, neither pregnancy nor older age represents an absolute contraindication.
When surgery is indicated, a mitral valve procedure under direct vision is the method of choice for managing significant mitral insufficiency, whether alone or with mitral stenosis. Cardiopulmonary bypass (CPB) is also the best technique if the mitral valve is heavily calcified or mitral disease coexists with aortic or tricuspid valve problems. Also, CPB should be used primarily in most patients with a history of peripheral embolization. Certain patients in NYHA Class II, and virtually all in NYHA III and IV are candidates, with less risk for patients in classes II and III. Preferably, all patients will come to operation restored to cardiac compensation by an adequate period of appropriate treatment. However, in isolated NYHA IV patients, mitral valve replacement may represent the sole remaining resource, and therefore the operation must be undertaken, despite residual cardiac failure.
Prosthetic-valve replacement is almost always necessary in severely calcific mitral stenosis. This is not necessarily true for most patients with acquired mitral insufficiency who should have mitral valve repair. An exceptional case of ruptured papillary muscles or chordae tendineae can be realistically corrected by suture approximation of the cusps. Pericardial patch closure of a perforated valve cusp, secondary to bacterial endocarditis, does not require prosthetic replacement. Ring annuloplasty of the mitral annulus, to reduce the size of the valve’s orifice and improve the approximation of the cusps, usually accompanies mitral valve repair in patients with mitral regurgitation.
Aortic Valve Replacement
Prosthetic aortic-valve replacement has an increasingly significant role in the management of patients with aortic stenosis or insufficiency in whom clinical manifestations of cardiac decompensation, syncope, and angina regularly affect their life activities. This role prevails because patients with hemodynamic evidence of a serious compromise of cardiac function, particularly those with significantly elevated LVEDP, are unlikely to be benefited long by procedures other than aortic-valve replacement. All potential candidates should undergo right- and left-sided heart catheterization (and angiography of the aorta and left ventricle), to evaluate hemodynamic function more precisely. The diseased valve cusps are carefully excised, and calcium is removed from the valve’s annulus or even from the LV outflow area, preserving a small valvular remnant on the wall. Particular care must be exercised to ensure that the upper rim of the valvular prosthesis lies well below the coronary ostia (see Plate 6-53).
Aortic Valve Regurgitation
Aortic valve regurgitation can result from not only intrinsic aortic valve disease but also lesions of the ascending aorta. Syphilitic aortic disease, aortic dissection, weakness of aortic media (Marfan syndrome), senile or idiopathic aortic dilatation, and traumatic nonspecific inflammatory aortic disease may lead to aortic regurgitation because of dilatation of the aortic annulus or structural fatigue of the tissue supporting the aortic cusps. Regurgitation may be associated with idiopathic cystic medial necrosis, producing aneurysmal dilatation of the ascending aorta (see Plate 6-54). The aortic valve is usually tricuspid and contains no calcifications. Aortic regurgitation is secondary to dilatation of the annulus, thereby increasing the area of the valvular orifice. The cusps are thinned out, and the free edges of the valve cusps may be rolled. The ascending aortic wall proper is dilated and thinned. Disruption and fragmentation of the elastic fibers occur. At times, early aortic dissection with limited extravasations within the media is observed. These patients may exhibit classic signs and symptoms of aortic regurgitation. Simple anteroposterior and lateral chest radiographs show marked dilatation of the ascending aorta. Left-sided heart catheterization and angiocardiography usually confirm the evidence for the diagnosis of massive aortic regurgitation in the presence of a widely dilated ascending aorta.
Surgical Technique
After clinical, ultrasound, and hemodynamic evaluation, surgical correction of the aortic lesion includes prosthetic valvular replacement, resection of the ascending aortic aneurysm, and replacement with a prosthetic graft (see Plate 6-54 ). Through a midline sternotomy, the patient is placed on CPB and moderate systemic hypothermia induced. The aorta is cross-clamped distal to its point of dilatation, usually immediately proximal to the takeoff of the innominate artery. The proximal line of resection is about 1 cm above the level of the coronary arterial takeoffs. The aneurysm is completely excised, and the coronary arteries are cannulated and perfused continuously throughout the procedure. Dissecting hematoma within the proximal aortic cuff may be encountered. To obliterate this and particularly to ensure a firmer base for suturing the graft to the diseased and thinned aortic wall, a synthetic polyester (Dacron) skirt is fashioned and positioned within the split residual aortic root. This Dacron cylinder is sutured in place by continuous stitches, passing successively through the inner aortic wall, the polyester skirt, and the outer aortic layer. This procedure reinforces the proximal aorta. Once this has been accomplished, the aortic valve is resected and replaced by a prosthesis, which may be a composite conduit and valve. Prosthetic valvular replacement is necessary because these secondarily deformed aortic valves are usually not suitable candidates for reconstructive procedures.
Multiple-Valve Replacement
Significant involvement of more than one cardiac valve is more complicated to evaluate quantitatively and to manage surgically (see Plate 6-55). Aortic and mitral valve disease is the most common combination and requires particularly careful study. The best decision in the more complex cases is based on the physical findings, cardiac ultrasound, and left- and right-sided catheterization, and angiocardiographic findings. These data usually provide appropriate qualitative and quantitative estimations of the overall state of valvular disease.
Decisions involving the tricuspid valve are less precisely based on derivable data. In general, however, when serious organic involvement of the tricuspid valve is present, prosthetic replacement is required. On the other hand, functional tricuspid insufficiency, most often in mitral stenosis, usually improves after correction of the primary problem. In many patients the tricuspid valve can be restored to competency with a ring valvuloplasty, especially if no structural valve damage is present. Thus, experience can only partly resolve the dilemma of whether to intervene surgically. Patients who show little or no improvement in signs and symptoms of tricuspid-valve disease, despite prolonged intense medical treatment for multivalve-induced cardiac failure, usually require prosthetic tricuspid replacement. The same holds true for patients who, in the presence of normal or near-normal pulmonary artery pressure (at catheterization), have significant structural damage to the tricuspid valve resulting in tricuspid regurgitation. Surgery provides an additional opportunity for evaluating tricuspid-valve function, using intraoperative transesophageal ultrasound while the heart is beating, as well as direct inspection within the atrium after instituting CPB. In general, if the tricuspid valve seems to be structurally damaged, it should be replaced rather than repaired, or a semirigid valvuloplasty ring used.
Insertion of Trileaflet Aortic Valve
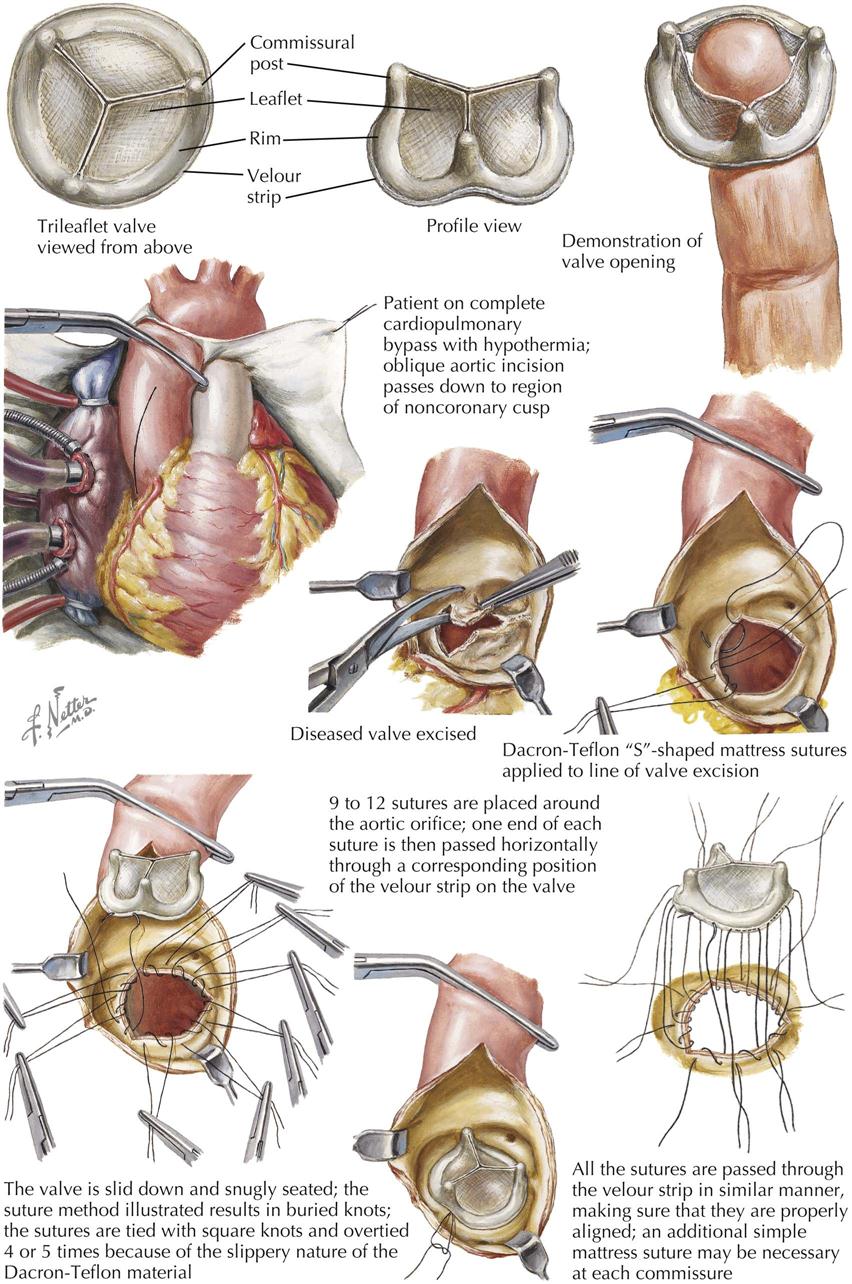
The basic concept of an artificial valvular mechanism that would function permanently and reliably in the human body was first introduced in 1952. Since that time, many variations of the original ball valve have been evolved. The free-floating discoid valve was a later type, first used clinically in 1962. Although all these variations produced progressively better results, all had an inherent problem as well: the central space was occupied by the occlusive portion of the valve mechanism. All such valves produced an alteration in the direction of blood flow, introduced a resistance imposed by the ball or disc in this area of high-pressure and high-velocity flow, and depended on availability of a marginal or restricted space at the periphery of the occlusive part of the mechanism in the plane of maximum travel.
The trileaflet tissue valve embodies the principle of self-suspended flexible leaflets (cusps) and has certain features not previously used in mechanical prosthetic valves. The first leaflet valves were made of polypropylene covered with silicone rubber. Polypropylene is extremely resistant to flexion fatigue, and no other currently available plastic material approaches it in flex life. The three leaflets were suspended from the base within the aortic sinuses, without attachment to the aortic wall at their commissure. This makes closure independent of changes in the aortic diameter and configuration, since these may vary widely from individual to individual and under different physiologic conditions. The leaflets are very pliable, and the valve has an extremely low opening pressure. Subsequent trileaflet valves are biologic tissue, either a porcine or bovine valve or an equine pericardial valve.
The proximal orifice of the trileaflet aortic valve is equal to the size of the ventricular outflow area. The profile is sufficiently low to permit the entire valve to lie within the sinuses of Valsalva. The base of the valve follows a scalloped contour that duplicates the configuration of the normal aortic cusp attachments to the aortic root. This was the first valve requiring only secure attachment of its base to the aortic annulus; once this was achieved, valve opening and closure were ensured. Trileaflet aortic valves are lightweight, and its low resistance to blood flow permits fixation with minimal technical difficulty. Accumulation of clots with subsequent embolism has been a serious problem in previous types of mechanical valvular prostheses and remains so in current mechanical valves that require chronic anticoagulation.
Trileaflet valve sizes must be selected appropriate to the aortic root. Because of the wide valve orifice and the low resistance to flow, care should be taken not to attempt the insertion of an oversized valve; even the smaller trileaflet valves provide an orifice approaching normal. Accurate sizing simplifies insertion.
Aortic Valve Biologic Grafts
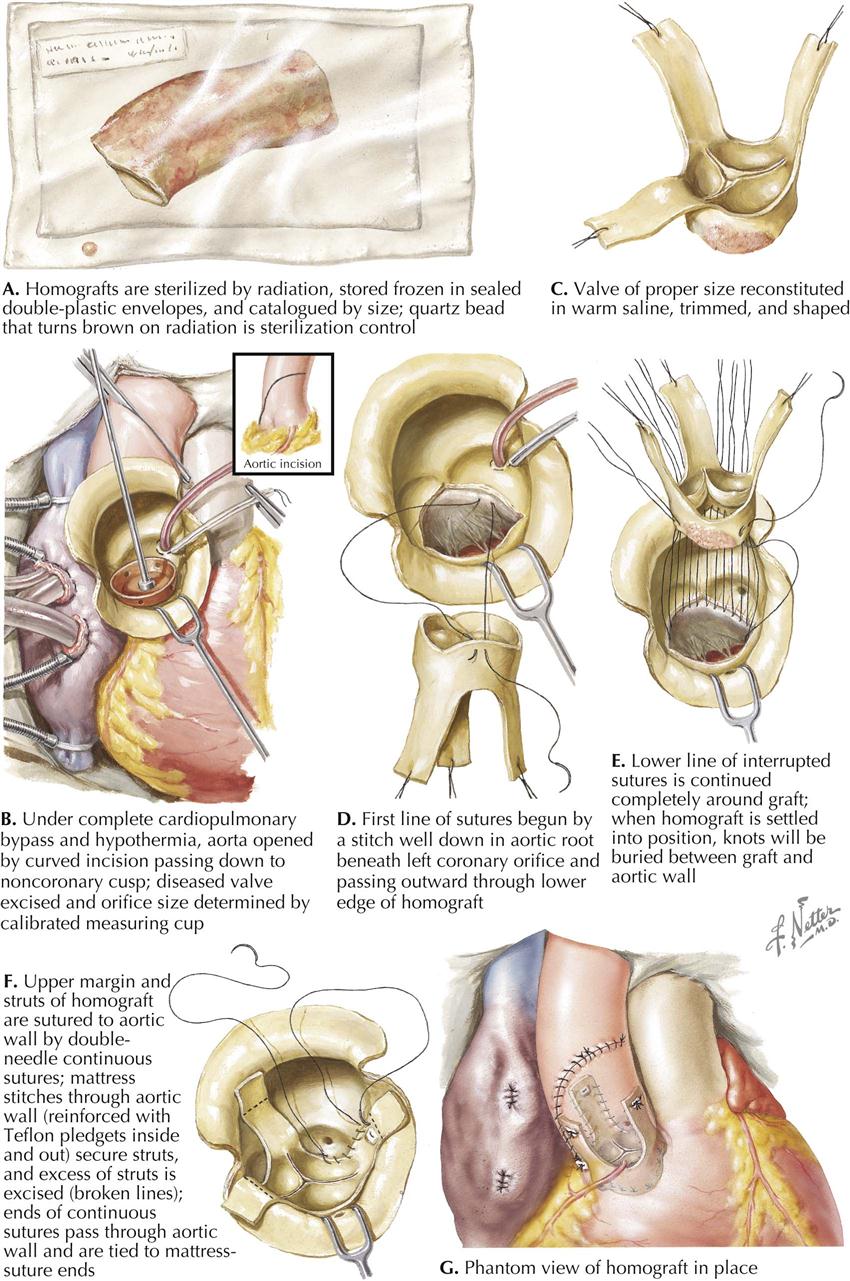
Aortic valve replacement using homograft or heterograft valves has been evaluated clinically over many years. The advantages of these nonviable tissue grafts are the absence of late peripheral arterial emboli and elimination of postoperative long-term anticoagulation therapy.
The technique of harvesting, preparing, sterilizing, and storing these homograft valves has an influence on their structure and function. Laboratory tests have shown that the freeze-irradiation method of sterilization and storage of the aortic valve homografts causes the least change in their gross morphology and tensile strength. The principal feature of this technique is irradiation of the graft in the frozen state to bactericidal energy levels of 2 to 2.2 megarads in a 5- to 6-second exposure. The packaged sterilized valve (available from valve bank) uses a discolored quartz bead as an indicator of radiation exposure (see Plate 6-57, A). The aortic valves are obtained unsterile from autopsy specimens less than 12 hours after death. All valves are trimmed of fat and cardiac muscle to reduce the bulk of muscle that may become interposed between the graft and the host. Careful dissection of the aortic root is required to preserve the aortic sinuses. The internal diameter of the valve is measured and recorded before packaging and freezing the graft to −50° C.
A graft is selected equal in size to the internal diameter of the patient’s aortic root following excision of the diseased aortic valve (Plate 6-57, B). The 2- to 3-mm wall thickness of the graft ensures a replacement that is somewhat greater in diameter than the aortic root implant site. The valve selected is reconstituted in warm saline solution under sterile conditions (Plate 6-57, C). Excess muscle and mitral valve tissue are trimmed from the homograft valve, leaving a 2-mm rim below the sinuses of Valsalva. Parts of the homograft aortic wall are excised into the sinuses to accommodate the host’s coronary artery orifices. The struts of the aortic wall remaining on the homograft valve will be used later, during implantation, to provide added support for the cusp commissures.
The graft is positioned so that the mitral valve remnant lies below the left coronary orifice to reduce bulk and to ensure placement well below the lumen of the coronary artery. Sutures are then passed through the base of the homograft from inside out, thus placing the suture line between the host aorta and the graft (Plate 6-57, D-F).
The technique of biologic valve implantation is more precise and time-consuming than that required for the artificial prosthesis. Correct positioning and suturing of these grafts are essential for proper valve function postoperatively. The operative mortality and clinical results of aortic homograft valve replacement compare favorably with those reported after insertion of the ball-valve prosthesis. The absence of late peripheral arterial emboli and lack of the need for postoperative anticoagulation therapy are the strongest arguments for the use of the homografts. Postoperative aortic diastolic murmurs have been reported, usually not of hemodynamic significance.
A satisfactory size relationship must exist between the host aortic root and the homograft, and the homograft valve selected must be large enough in diameter to remain sufficient when the aortic ring is distended by systemic pressure. The graft size selected is based on the internal diameter of the aortic root of the recipient; thus the wall thickness of the graft, 2 to 3 mm, represents the excess area available when the aortic root dilates. Tailoring the aortic root helps reduce the diameter of large aortic bases to accommodate the homograft.
Clinical follow-up indicates the aortic homograft valve functions satisfactorily, with no evidence of breakdown or restenosis. Late peripheral arterial emboli are rare, and again, no postoperative anticoagulation is required. Although changes in the design of artificial prostheses will ultimately solve the thromboembolism problem, at present the aortic homograft remains far superior in this regard.
Tricuspid Stenosis and Regurgitation and Multivalvular Disease
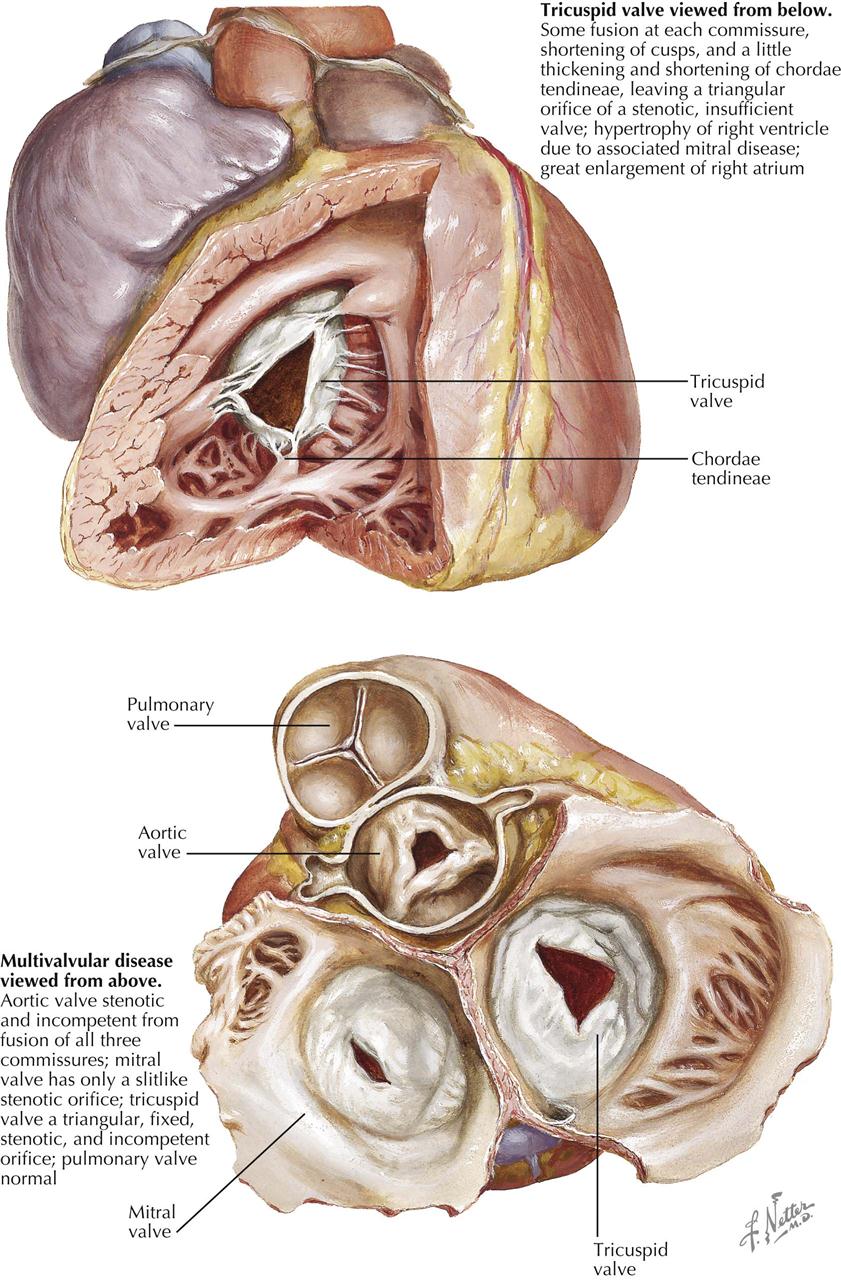
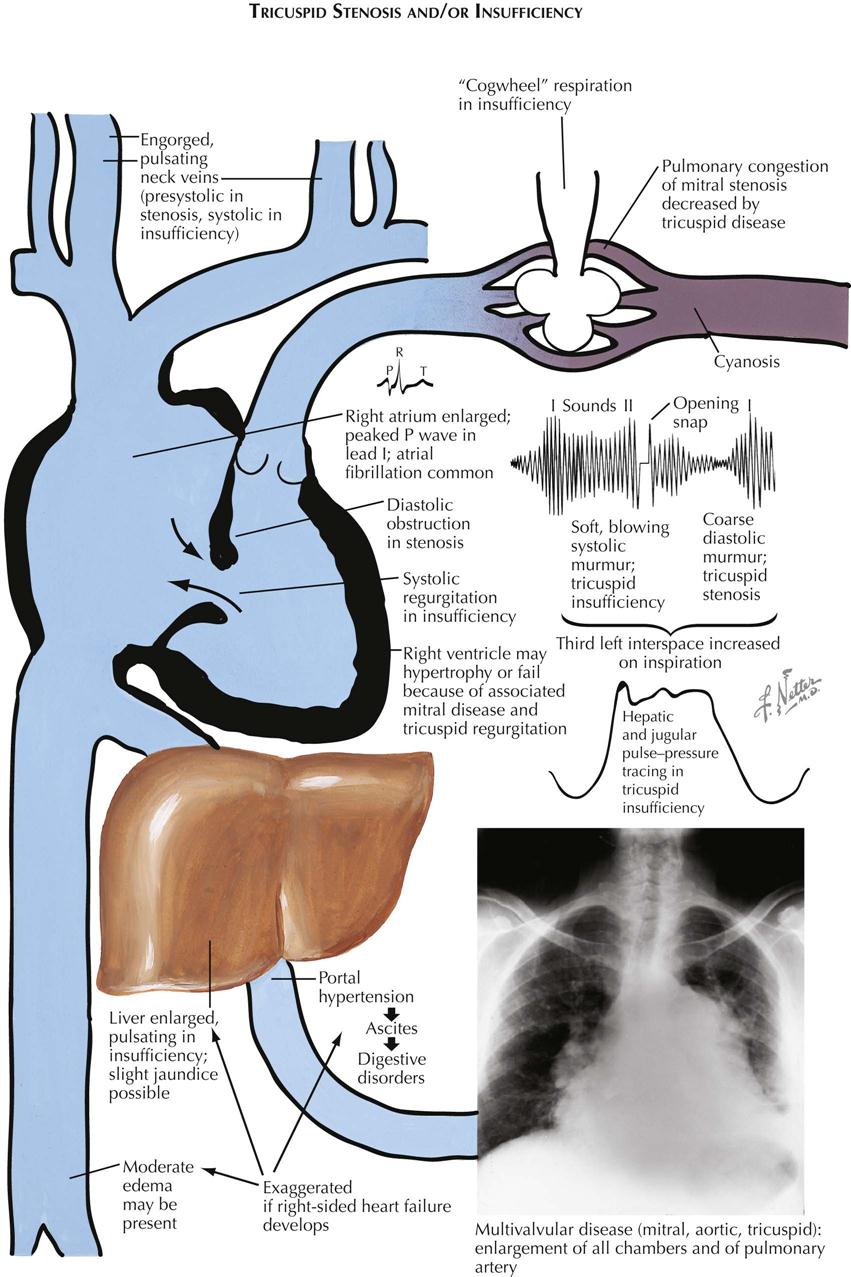
Stenosis and Regurgitation of Triscuspid Valve
Rheumatic deformity of the tricuspid valve is characterized principally by fusion at each of the commissures and shortening of the cusps; chordal changes are usually minimal. These changes are responsible for reduced caliber of the orifice and incompetence of the valve. The usual coexistence of stenosis and insufficiency in the rheumatic tricuspid valve contrasts with involvement of the mitral valve, often manifested as stenosis or insufficiency. One characteristic effect of rheumatic tricuspid valve disease is an enlarged right atrium (see Plate 6-58). Chronic rheumatic involvement of the tricuspid valve is rarely if ever an isolated phenomenon; the mitral valve is also involved and in some cases the aortic valve as well.
In patients with mitral stenosis and evidence of tricuspid regurgitation, it remains to be determined if the regurgitation represents intrinsic involvement of the tricuspid valve or is secondary to the mitral stenosis promoting right ventricular failure. Certain features favor secondary tricuspid insufficiency in the patient with known mitral stenosis; congestive heart failure is usually evident. In intrinsic rheumatic disease of the tricuspid valve, stenosis and insufficiency usually coexist, whereas in secondary tricuspid insufficiency, stenosis is absent. Therefore, patients with no signs of tricuspid stenosis must be seen as having secondary rather than primary insufficiency.
Multivalvular Disease
Involvement of more than one valve by deforming rheumatic disease generally consists of disease of the mitral and aortic valves while the tricuspid valve is essentially unaffected. The combination of aortic and mitral disease usually takes the form of predominant mitral stenosis and predominant aortic regurgitation but some aortic stenosis. Both severe primary aortic regurgitation and severe primary mitral regurgitation are rarely seen together, possibly because their coexistence tends to be a lethal combination.
When three valves are involved, the pulmonary valve is usually spared of major disease, but the tricuspid valve is affected. In trivalvular rheumatic disease, the lesions of the aortic and mitral valves are generally similar to those observed when only the left-sided heart valves are affected. The tricuspid valve shows the characteristic features of intrinsic rheumatic disease (see Plate 6-58). When multiple valves are involved, the effects on the chambers vary with both the state of compensation of the heart and the dominance of the disease in the various valves. Involvement of the tricuspid valve is universally associated with enlargement of the right atrium. The mitral valvular disease results in right ventricular hypertrophy. When congestive heart failure is present, regardless of the state of the tricuspid valve, the right ventricle is enlarged. In the compensated heart with mitral valve disease and a normal tricuspid valve, the right ventricle may be of normal size. The pulmonary artery and the left atrium are enlarged because of the high pressure in the left atrium (and thus the pulmonary artery) secondary to the mitral disease.
In patients with multivalvular disease in whom the aortic valve also participates with the mitral valve, the left ventricle is hypertrophied, but considerably less than observed in patients with isolated aortic valvular disease. The mitral valve disease tends to be associated with a low cardiac output, so the effects of aortic valvular disease, especially aortic stenosis, are minimized.
Amyloidosis
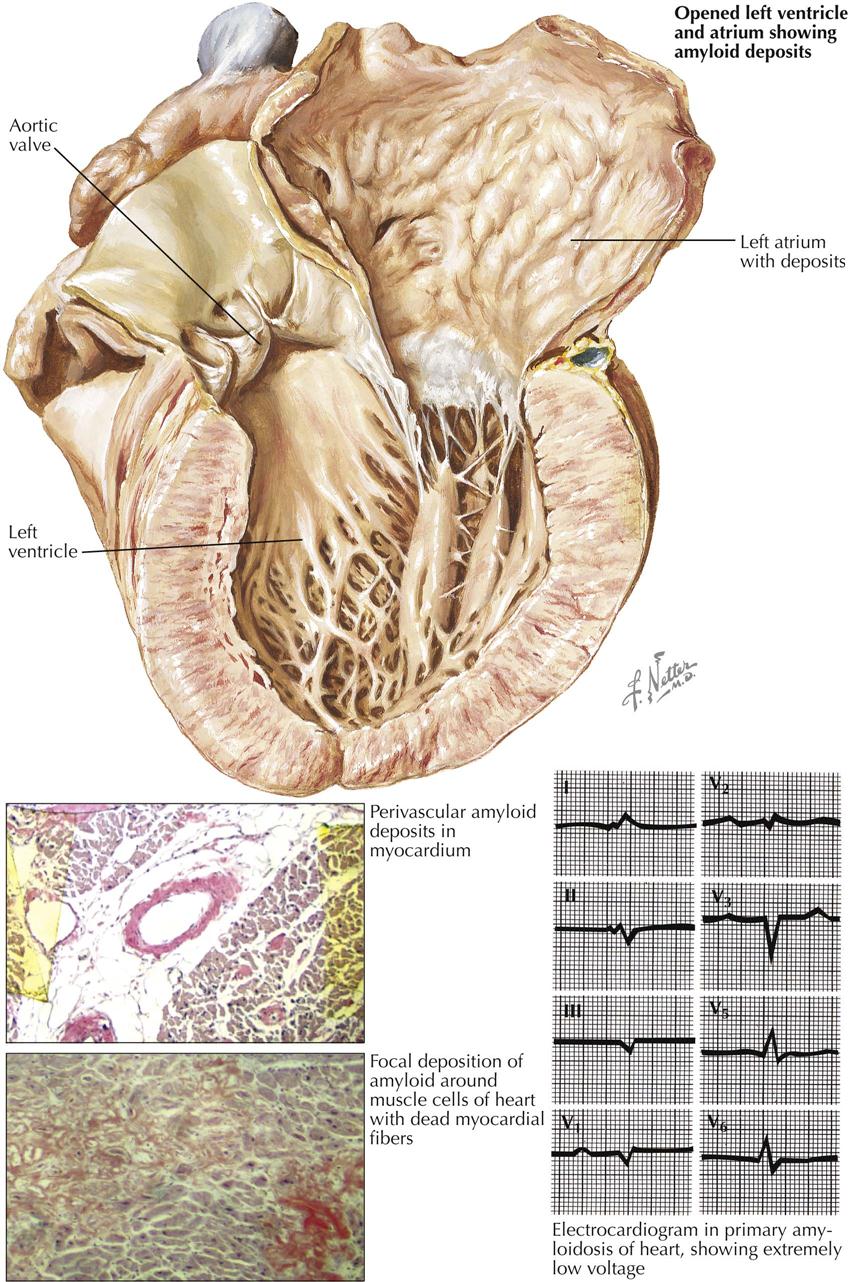
Accumulation of insoluble amyloid results in a restrictive infiltrative cardiomyopathy that may occur under four circumstances: (1) as part of primary systemic amyloidosis, (2) as part of amyloidosis complicating multiple myeloma, (3) as part of secondary systemic amyloidosis, and (4) as a localized phenomenon in elderly persons. In primary systemic amyloidosis the liver, spleen, and kidneys are usually found without amyloid deposits. Of 99 patients with primary amyloidosis, 92 showed severe involvement of the heart. Patients with multiple myeloma often develop amyloidosis of the heart.
The clinician should consider the diagnosis of amyloidosis of the heart when chronic, intractable heart failure develops in a patient age 50 or older, particularly if the heart failure remains unexplained, even in the presence of the usual symptoms and signs. The heart typically is enlarged, but hypertension is absent. The excursions of the heart are slow, and noncharacteristic murmurs may be heard. Murmurs typical of valvular disease can be detected only if the valves are affected by depositions of amyloid. The ECG indicates a low voltage (see Plate 6-60). Disturbances of the atrioventricular conduction may be caused by amyloidosis of the bundle of His and the bundle branches. Cardiac ultrasound with Doppler imaging often reveals a restrictive cardiomyopathy. Cardiac muscle also may “sparkle” on the echocardiogram. Cardiac biopsy and staining with Congo red confirm the diagnosis of amyloidosis of the heart by demonstrating apple-green birefringence under polarized light. Electron microscopic studies of homogeneous amyloid demonstrate delicate fibers thought to be responsible for the effect of the green birefringence. Biopsies of the skin, rectal mucosa, or myocardium may help establish the diagnosis. On ultrasound the amyloid heart is characterized by biventricular thickening, biatrial enlargement, restrictive hemodynamics, and a small cavity with a relatively preserved ejection fraction.
On cardiac biopsy, microscopic homogeneous deposits of amyloid are translucent and refract the light strongly (see Plate 6-60). Sections of myocardium stained with Congo red exhibit a network of homogeneous bands surrounding the bundles of myocardial fibers and separating these. The myocardial fibers are compressed and often are atrophied to very flat fibers with pinlike nuclei. The interstices are diffusely widened but not infiltrated by inflammatory cells. The pseudomyocardial hypertrophy explains the insufficiency of both heart chambers. Rarely, as a result of obstruction of a coronary artery by nodular deposits of amyloid, infarction or disseminated necroses of the myocardium may occur. When amyloidosis complicates multiple myeloma (20% of cases), the distribution of amyloidosis resembles that in primary systemic amyloidosis. Cardiac involvement is common.
Secondary typical or systemic amyloidosis occurs in diseases with chronic inflammation and severe tissue necrosis, especially chronic tuberculosis of the lungs or other organs. Other primary underlying diseases are bronchiectasis, empyema of the pleural cavities, chronic osteomyelitis, and tumors, as well as leprosy, syphilis, and echinococcosis (hydatid disease). The incidence of secondary amyloidosis has decreased as a result of antibiotic therapy. In systemic amyloidosis the liver, spleen, and kidneys are affected predominantly, but the heart also may be involved. The depositions of amyloid are much less extensive than in primary systemic amyloidosis of the heart and demonstrate a different pattern. Amyloid is usually deposited in the media of the arteries and veins and in the capillaries between the endothelial cells and the basement membrane. The lumen of the capillaries remains patent, as in the arteries and veins, and thus the complications are not so serious as in primary systemic amyloidosis of the heart.
Deposits of amyloid may be found in the blood vessels and in relation to myocardial fibers of the myocardium of elderly persons without underlying disease. The incidence of presenile or senile amyloidosis, which also may involve the brain, is low in patients under age 60. However, the heart can be affected in patients over 80.
Septic Myocarditis
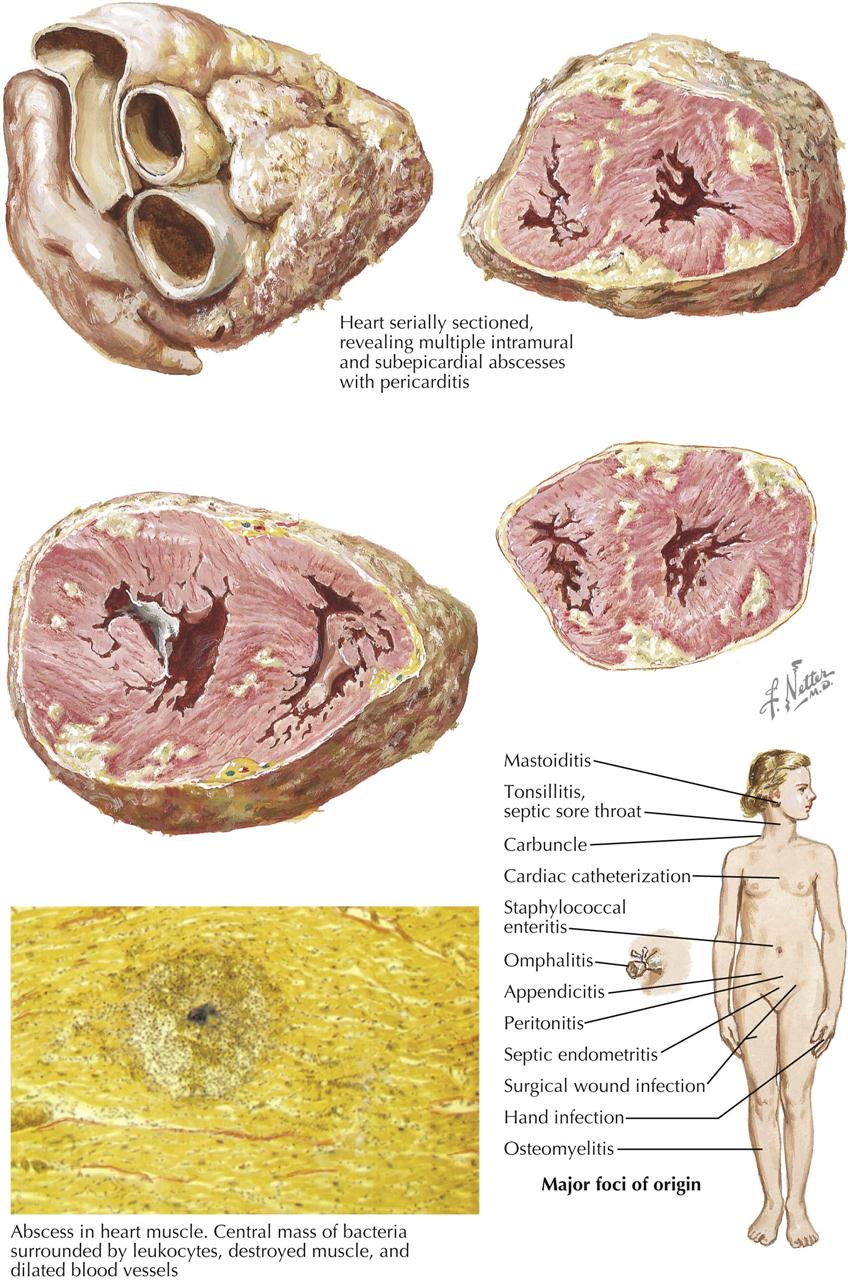
Septic or suppurative myocarditis can lead to myocardial abscesses and other complications, but it is now rarely seen since the introduction of antibiotics, not disappearing completely because of the development of antibiotic-resistant strains of bacteria. The number of known major foci permitting the spread of infection by bacteremia (which precedes suppurative myocarditis) increases with the use of new diagnostic and therapeutic methods, such as indwelling intravascular catheters.
Any virulent bacteria are capable of causing acute septic myocarditis. Newborns, nursing mothers, and patients with previous viral infection are predisposed, as are diabetic and severely burned patients. In some patients the process is associated with or a complication of bacterial endocarditis; in others the valves are not affected. The first nonspecific signs of myocarditis in septicemia are fever, leukocytosis, and shock. Circumscribed areas of necrotic and damaged myocardium may lead to ECG changes. Murmurs can be heard only in patients who have developed endocarditis or pericarditis. Frequently, in the patient with acute bacteremia, no macroscopic changes are visible in the heart because the rapid course of the septicemia does not allow the tissues to react against the spreading bacteria. Clinically, signs of sepsis predominate (e.g., shock).
Diagnosis and treatment of septic myocarditis depend on culture of the myocardial strains. Dense colonies of bacteria within the dilated capillaries and veins are the earliest detectable finding in microscopic sections of the myocardium (see Plate 6-61). In later stages these are small abscesses showing abundant bacteria in their centers, with a circular wall of numerous PMNs. The surrounding myocardial fibers can reveal homogeneous cytoplasm with unstained or pyknotic nuclei. The entire focus is surrounded by a hemorrhagic marginal zone.
When the heart is sectioned at autopsy, myocardial abscesses are often visible as small yellow points or stripes, measuring 1 or 2 mm, beneath the thin endocardium of the right ventricle (see Plate 6-61). The irregular and bizarre formation of the abscesses is usually determined by the anatomic course of the muscle bundles. Subendocardially located abscesses can perforate into the ventricular lumen and cause bacterial endocarditis. Subepicardial abscesses can involve the pericardium as well. Frequently, the focus often can be detected easily, such as after perforation into the pericardial sac, but in a few cases it may be impossible to find. Therefore, primary hematogenous spread cannot always be ruled out with certainty.
Microorganisms (viruses, rickettsiae, bacteria, protozoa) or their toxins can produce myocarditis. The morphologic findings alone do not clarify the etiology of myocarditis. Focal or diffuse myocarditis may develop without clinical manifestations. Therefore, to diagnose myocarditis, all clinical information and bacteriologic and virologic examination results should be evaluated together.
Diphtheritic and Viral Myocarditis
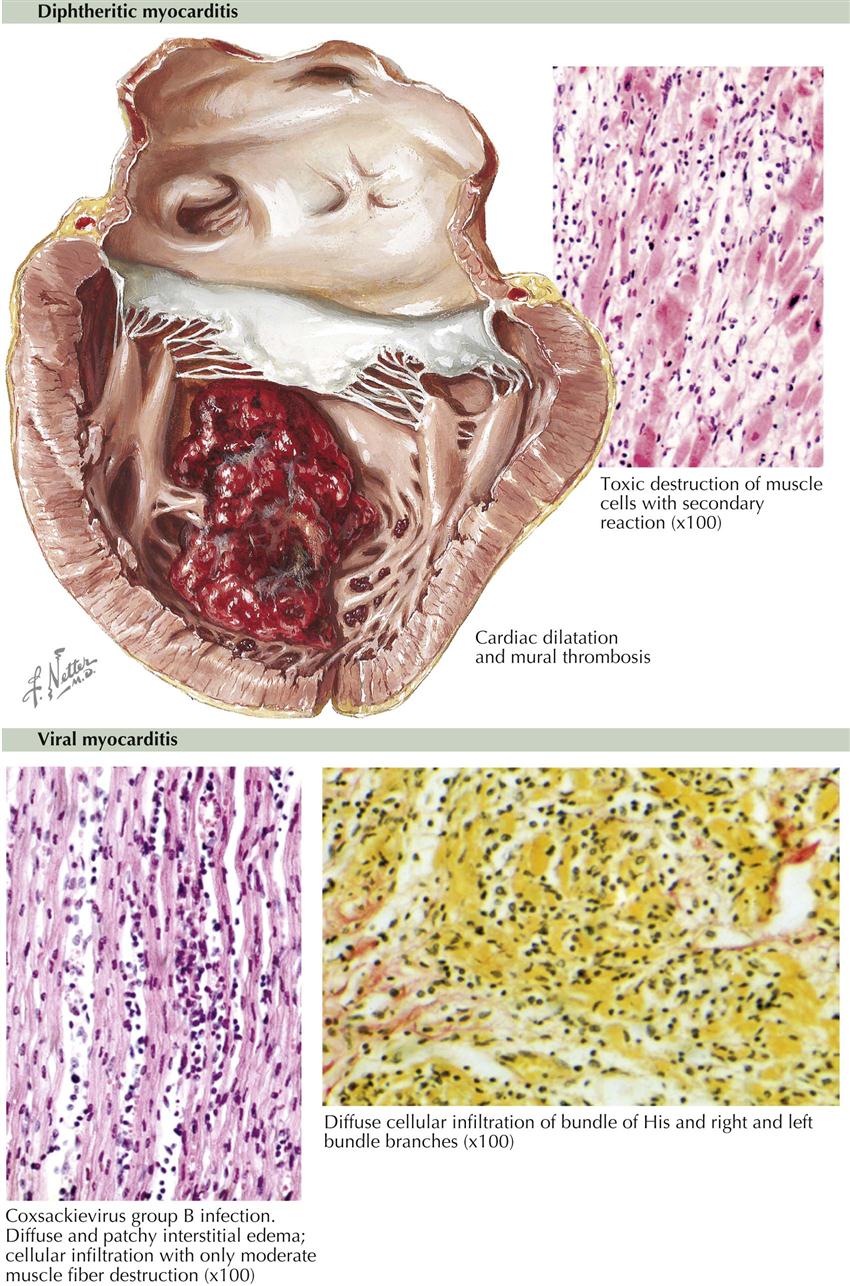
Diphtheritic Myocarditis
In diphtheria of the upper respiratory tract or the result of wound diphtheria, myocarditis is caused by the powerful exotoxins of Corynebacterium diphtheriae (Klebs-Löffler bacillus). This type of toxic myocarditis occurred frequently before the introduction of active immunization, and even now small epidemics may occur outside the United States.
In acute diphtheria the heart is flabby, and the myocardial fibers have a boiled appearance. Both ventricles are dilated, and small mural thrombi may be found between the trabeculae (see Plate 6-62). The left ventricle, particularly the apex, may be filled with large ball thrombi. Decreasing blood pressure, arrhythmias, and complete heart block may precede acute heart failure. Microscopically, the myocardial fibers are swollen and demonstrate a dustlike fatty degeneration. The interstices are edematous. In subacute cases the myocardial fibers show granular or hyaline necrosis. PMNs and mobilized histiocytes are distributed throughout the interstitial connective tissues.
The secondary inflammatory reactions are frequently arranged focally but also may be diffuse. In the surrounding areas of toxic necrosis, circulatory disturbances also may develop, leading to further necrosis. The protracted shock finally affects the coronary blood supply in such a manner that additional disseminated hypoxic necrosis occurs. After several weeks the necrotic myocardial fibers will be organized and replaced by fine reticular fibrosis. The extent of such diffuse fibrosis can often be determined only by quantitative methods, measuring the components of connective tissue and myocardial fibers. Fibrosis and even scarring can be so severe that death results from chronic heart failure.
Viral Myocarditis
The incidence of viral myocarditis has increased. Known causative organisms in humans are coxsackievirus group, adenovirus, parvovirus, echovirus, Epstein-Barr virus, German measles, and human immunodeficiency virus. The etiology can be ascertained only if virologic examination is successful. Epidemiologic studies can provide only hints about the possible organism. Newborns, infants, children, and adolescents are predisposed. Myocarditis can be preceded by meningitis, encephalitis, pleurisy, hepatitis, and pericarditis, as well as gastrointestinal symptoms. After 3 to 10 days, fever, tachycardia, cyanosis, and finally signs of cardiac failure may develop. The ECG reveals severe changes suggesting damaged myocardium (e.g., increased troponin T) or alterations of the conduction system. Radiography shows an enlarged heart. Cardiac ultrasound usually reveals diminished global or regional ventricular function.
Except for dilatation of both ventricles, the myocardium appears macroscopically unchanged. Several tissue blocks should be sampled for microscopic examination because viral myocarditis affects the myocardium focally, and large areas of the heart may be spared. The histologic findings also depend largely on the stage of the inflammatory process. The first stage of viral invasion into the myocardial fiber can only be studied experimentally. During the next stage, disturbances of cell metabolism, a few or several myocardial fibers may undergo necrosis. In the third stage, interstitial accumulations of lymphocytes, plasma cells, and histiocytes are the predominant features. These third-stage changes are observed most frequently in sections of the myocardium and the conduction system at autopsy. The diffuse interstitial fibrosis of the fourth stage does not differ from the fibrosis caused by toxic myocarditis. The extensive scarring may cause chronic congestive heart failure.
Myocarditis in Sarcoidosis and Scleroderma
Sarcoidosis
Among the interstitial myocarditides, sarcoidosis (Besnier-Boeck-Schaumann disease) is second in incidence to rheumatic myocarditis. In sarcoidosis the lungs, liver, spleen, and lymph nodes are involved much more frequently and extensively than the heart (see Plate 6-63). However, the heart may be affected in 20% of all cases, and death may result from myocarditis in 6%. Sarcoidosis of the lungs can lead to pulmonary hypertension and thus right-sided heart failure and cor pulmonale without myocarditic changes.
Myocarditic granulomas are found particularly in the LV myocardium but may also be present in other areas of the heart. Some granulomas measure up to 4 mm in diameter and are recognizable grossly. Granulomas of the right atrium may cause arrhythmias and sinus tachycardia. Granulomas involving the bundle of His and the atrioventricular (A-V) node can lead to abnormalities in atrioventricular conduction (sarcoid of A-V node), including complete A-V block. Therefore, death may occur during Morgagni-Adams-Stokes attacks. At first the myocarditic foci consist of perivascular accumulations of histiocytes. Later, granulomas develop and demonstrate a few Langhans giant cells (see Plate 6-63). Finally, granulomas may be replaced by hyaline scars. The recurrent course of the disease may result in diffuse fibrosis of the myocardium. However, the granulomatous lesions are nonspecific. Similar changes can occur in tuberculosis, brucellosis, and tularemia. Therefore, the histologic findings can be interpreted only within the context of the clinical situation.
The etiology of sarcoidosis is unknown. Some experts hypothesize that cardiac sarcoid is related to an abnormal immune response to environmental factors (e.g., pine pollen) or infectious agents (e.g., mycobacterium) in a genetically susceptible individual. Sarcoidosis shows a special predilection for black patients of African descent. More than one case of sarcoidosis can be observed in families or in siblings, for reasons still undetermined.
Scleroderma
An interstitial myocarditis can also occur in scleroderma. Myocardial fibrosis may be caused by impaired perfusion secondary to arteriolar and microvascular disease. The focal myocardial lesions consist mainly of lymphocytes and histiocytes (see Plate 6-63). In later stages, perivascular interstitial fibrosis progresses, and scarring extends diffusely throughout the myocardium. Similar changes are seen in systemic lupus erythematosus and dermatomyositis. Therefore the diagnosis can be made only after a review of all clinical data. The etiology of scleroderma (systemic sclerosis) is unknown. Initially, the disease is characterized by edema and erythema of the skin, followed by hardening and atrophy. In addition to cardiac involvement, other internal organs are often affected, including the lungs, kidneys, and alimentary tract.
Idiopathic Myocarditis
Giant Cell Myocarditis
Among the isolated myocarditides of unknown origin granulomatous myocarditis with giant cells should be distinguished. However, giant cell myocarditis can only be diagnosed by endomyocardial biopsy. Animal studies suggest that the disease is mediated by T lymphocytes.
In granulomatous myocarditis the enlarged heart shows dilatation of both ventricles and atria, and usually weighs 500 g or more. The cut surface of the myocardium has a dim, gray, patchy appearance. The focal lesions are not surrounded by hemorrhagic marginal zones. The coronary arteries are delicate. The typical gross findings often facilitate the diagnosis. Microscopically, the focal granulomatous lesions are composed of dense infiltrates of lymphocytes, plasma cells, and histiocytes, together with giant cells of the Langerhans type. As remnants of myocardial fibers, giant cells with numerous nuclei may also be present. Other areas of the myocardium show fibrosis. Some cases of giant cell myocarditis also reveal granulomas in other organs, suggesting an association with sarcoidosis. However, as long as the etiology of granulomatous myocarditis with giant cells remains unclear, final classification is still unresolved, although the disease is thought to be caused by T lymphocytes.
The diagnosis of giant cell myocarditis often can be made at cardiac biopsy and at autopsy. Clinical examination reveals an enlarged heart, with passive congestion of the lungs and abdominal organs. Usually the body temperature is not elevated, but in a few cases it may reach septic levels. The ECG shows abnormalities indicating myocardial damage or conduction system disturbances. A diagnosis of giant cell myocarditis is justified only if other causes of cardiac failure (e.g., rheumatic heart disease, idiopathic cardiac hypertrophy, amyloidosis) can be excluded.
Acute Isolated (Fiedler) Myocarditis
Isolated eosinophilic myocarditis (Fiedler myocarditis) reveals necroses of myocardial fibers associated with dense interstitial infiltrates of eosinophilic leukocytes, as well as a few lymphocytes and plasma cells. Some believe that viruses cause acute isolated (idiopathic) myocarditis, and others that certain features suggest allergic reactions in the etiology. However, etiology and pathogenesis remain unknown. Parietal thromboses may develop over foci located directly beneath the endocardium and may spread as extensively, as in parietal fibroplastic endocarditis. Some patients with eosinophilic myocarditis also develop vascular lesions, as in pericarditis.
Idiopathic myocarditis usually takes a rapid, fatal course and often is called pernicious myocarditis. Thus, endomyocardial biopsy should be performed urgently and if positive, high-dose steroids administered.
Endomyocardial Fibrosis
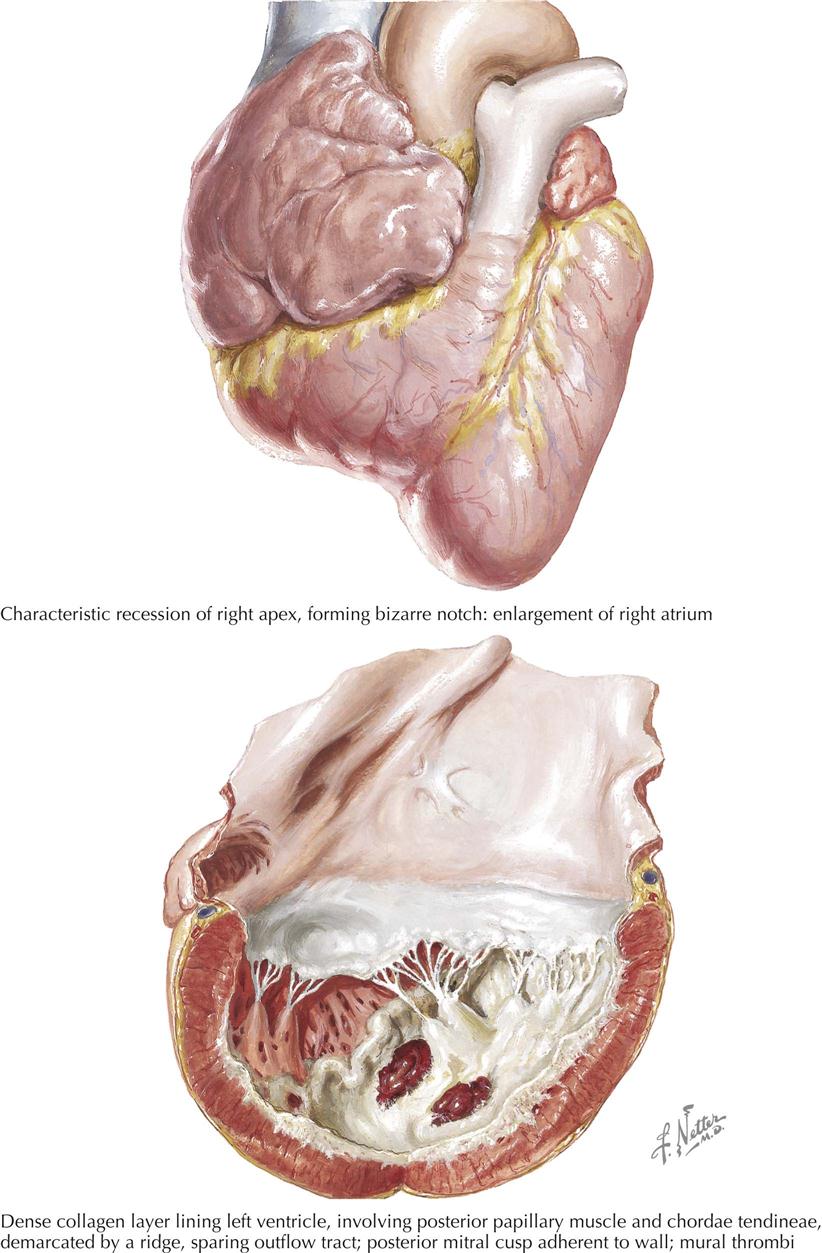
Endomyocardial fibrosis is common in many tropical countries, although cases are seen in temperate regions as well. The pathologic lesion is a scar replacing the endocardium and subjacent myocardium, which is confined to the ventricular inflow areas. A raised firm ridge marks the junction of the inflow and outflow areas, best seen in the left ventricle. The unscarred part of the endocardium may show a mild opacification from elastomyofibrosis. Lesions may be focally distributed in the inflow area, and the areas of preference are the apex and the site behind the posterior cusp of the A-V valve; the cusp adheres to the mural endocardium. In more severe cases the posterior cusp is lost in a mass of fibrous tissue extending from atrium to apex and partly up the septal and anterior walls, engulfing the papillary muscle and rendering the A-V valve incompetent. The A-V valves exhibit no specific lesions, and the semilunar valves are entirely unaffected.
On the right side, the ventricular configuration results in filling and obliterating the ventricular cavity with a mass of thrombus and organizing fibrous tissue. This can be recognized externally by a severe recession of the right apex or less often by a recession higher on the ventricular wall. The effective cavity is reduced to a shallow saucer, and fibrous tissue extends upward toward the pulmonary valve; the endocardium immediately below the valve is normal or opacified by elastomyofibrosis. The right atrium may be enormously dilated, similar to a rubber balloon.
In both ventricles the endocardium may be covered by thrombi. Although it may detach and produce a massive embolus in rare cases, the thrombus is usually firmly attached and does not loosen, so infarctions are uncommon. Beneath the thickened endocardium, there are small blood lagoons—dilated thebesian veins—and from these and the endocardial scar tissue, tongues of fibrous tissue extend into the inner third or half of the myocardium, but never involve its full thickness. The major coronary vessels are normal; no changes are seen in the minor vessels except for an occasional small focus of inflammatory cells and, in late stages when fibrosis is severe, an obliterative arteritis. Severe calcification may develop in the valve or mural endocardium, which is important radiologically, indicating that the constriction is endocardial, not pericardial, although a large pericardial effusion may be present. No constant extracardiac lesions are seen except for those of congestion. The heart weight may be increased but is often reduced, and despite the voluminous atria, the ventricles often are small and shrunken.
The cause of endocardial myofibrosis is unknown, and the early lesions have not yet been identified. Most cases are seen only when endocardial scarring is advanced. The disease may be biventricular, and the clinical manifestations may change depending on which ventricle is most diseased. If the left ventricle alone is affected, myofibrosis runs a rapid course with severe pulmonary hypertension. If the right side alone is involved, progress is much slower. Patients have high central venous pressure and may show exophthalmos. They have no peripheral edema but severe, extremely tense ascites with high-protein fluid. The liver is exceptionally large.
Differentiation from constrictive pericarditis may be extremely difficult in some cases, but diagnosis of endomyocardial fibrosis has been greatly facilitated by cardiac ultrasound and angiocardiography. The posterior A-V valve cusp is severely damaged regardless of the ventricle affected, with resulting mitral and tricuspid incompetence and usually stenosis. The early lesion may be a pyrexial form of carditis, followed shortly by evidence of progressive cardiac damage. Whatever the cause, the pathogenesis is destruction of the endocardium and underlying myocardium of the ventricular-inflow areas, with the formation of scar tissue. Elastic tissue in the affected areas is almost entirely lost, and elastosis is infrequently seen, except at the edges of the scar. No significant diagnostic blood changes have been found.
Löffler’s Endocarditis
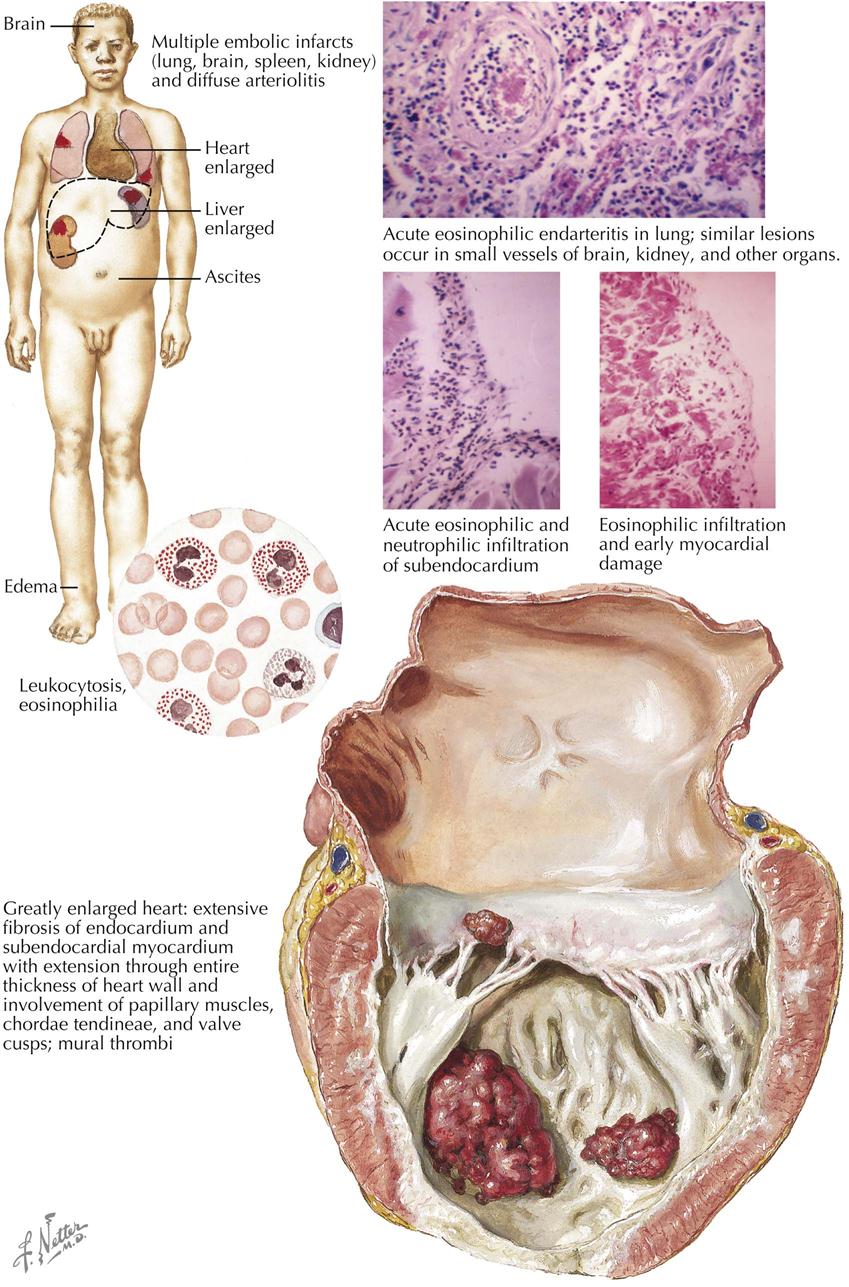
Löffler’s endocarditis, or eosinophilic endomyocardial disease, is an uncommon but distinctive form of disease in which the heart is the organ mainly affected by eosinophilic arteritis, probably allergic. Men are affected most, and the disease can occurs between ages 7 and 65. The disease has worldwide distribution, but no specific cause of Löffler’s endocarditis is known. At autopsy the changes of congestive heart failure are seen, usually with considerable effusion into the serous cavities. Fresh or older infarcts are often found in the brain, kidneys, and other organs. The heart may be of normal size but is usually enlarged, either generally or only on one side, most often the left. In acute cases, minimal change may be observed, except marked pallor of the endocardium and subjacent myocardium. However, electron microscopy shows swelling and necrosis of the endothelial cells, an eosinophilic infiltration of both the endocardium and the thebesian veins, focal necrosis of myocardial fibers with an eosinophilic infiltration, and an eosinophilic arteritis of the small coronary arteries with an occasional focus of fibrinoid necrosis. A similar eosinophilic arteritis is found in the small arteries of the brain, kidneys, lungs, and striated muscle, but the main coronary arteries are not involved.
More advanced cases will show extensive lesions. The endocardium is thickened and scarred, gray-white in color, and covered with mural thrombi, both atrial and ventricular, in varying stages of organization and attachment. Scar tissue may cover the papillary muscle and shorten and thicken the chordae tendineae, thus distorting the A-V valve, which itself usually exhibits valvulitis with vegetations. The semilunar valves may also show these lesions. The myocardium has areas of necrosis, sometimes with frank hemorrhage, organization, and scarring; the lesions often involve the full thickness of the ventricular myocardium. Between the myocardium and the thickened endocardium, small blood lagoons may be detected in dilated thebesian veins. A great eosinophilic infiltration usually is present in the necrotic and fibrosing areas of the myocardium. The cells are also abundant in the thickened endocardium. The original endocardium is largely destroyed, replaced by scar tissue with some elastosis. In more chronic cases, tissue eosinophilia may diminish or disappear, the arteries show only an obliterative arteritis, and severe endomyocardial scarring is found, usually maximal at the apex and spreading to involve the endocardial inflow and outflow tracts.
The basic arteritis may result in cerebral, abdominal, or renal manifestations initially, or arthralgias, muscle pains, and in some cases polyneuritis. The symptoms may present acutely with a fulminant course, or slowly with a progressive evolution. In a special patient subgroup, the endocarditis develops after several years of attacks of respiratory disease with bronchospasm. Cardiac decompensation develops, often with major embolic accidents. Pyrexia is common. Radiology may demonstrate cardiac enlargement, and ECG evidence may suggest necrosis, especially of the subendocardial myocardium, and probably confirmed by cardiac MRI. Mitral insufficiency may develop. High blood eosinophil count is most important in diagnosis, with levels as high as 130,000 cells/mm3 reported. The eosinophilia may not be marked or even present at the onset, and it frequently diminishes and may disappear terminally or in the chronic stages, when the disease may closely mimic all the signs and symptoms of a constrictive pericarditis.
The important points in distinguishing Löffler’s endocarditis are blood and tissue eosinophilia (in the endocardium and myocardium particularly), eosinophilic arteritis, endocardial scarring which may involve the inflow or outflow areas, valvulitis, and involvement of the whole thickness of the ventricular myocardium.
Becker’s Disease
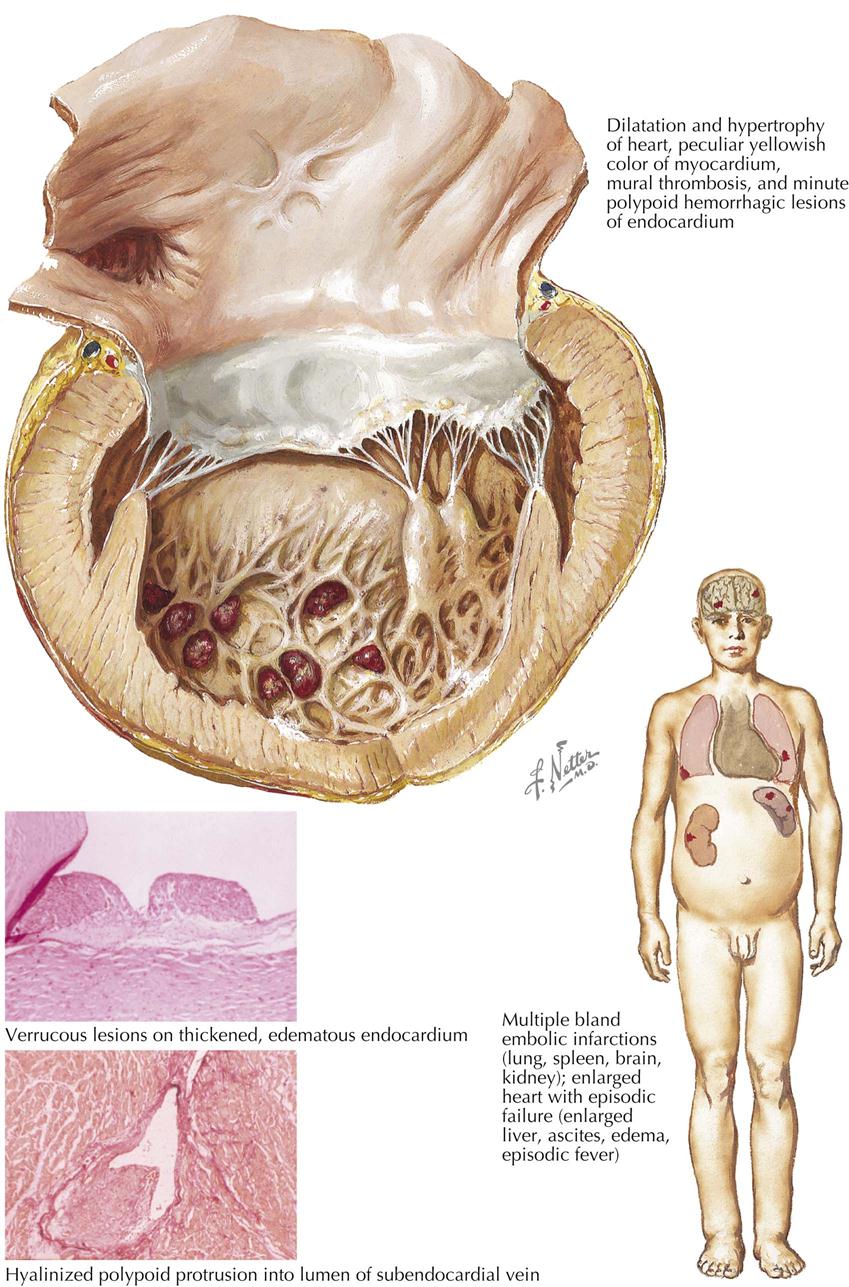
Becker’s disease occurs in all races in South Africa but is found in other areas as well. It appears to be a specific type of heart disease, the basic lesion a form of verrucous angiitis that especially affects the subendocardial blood vessels. The lesions found at autopsy reflect the acuteness or chronicity of Becker’s disease. At any stage that death occurs, severe lesions of congestive heart failure with large effusions in the serous cavities are found, as well as multiple infarctions in the lungs and often in the brain, spleen, kidneys, and other organs as well. The heart is greatly dilated and usually considerably heavier. Mural thrombi are present in the ventricles and may be small or may cover two thirds of the mural endocardium, interfering mechanically with A-V valve function. The heart valves show no specific lesions, and the main coronary arteries are normal. In acute cases the endocardium not covered by thrombus is neither thickened nor opaque, although a fine surface deposition of fibrin may be seen. Small, dark spots may be visible, representing hemorrhagic polyps on the endocardial surface. In acute cases, pallor of the myocardium under the mural thrombi is seen, and in the later stages, small areas of granulation tissue and streaks of fibrous tissue are visible in the inner third of the myocardium. In more chronic cases, irregular plaques of fibroelastotic endocardial thickening are found, scattered irregularly over the inflow and outflow tracts, but most evident at the apex. The hearts often are stiff and rubbery with a curious tan or yellowish color.
Microscopically, progressive changes are recognizable. In the acute stage of Becker’s disease, the endocardium is thickened with a serous and mucinous edema, and it may be acellular or infiltrated with inflammatory cells, wholly polymorphonuclear or mixed in type. Fibrin is diffusely or focally deposited on the endocardial surface, and, where damage is severe, the thrombus accretes. The focal fibrin deposits may form polyps, and hemorrhaging polyps may produce the black spots. The deposited fibrin and elastic-tissue proliferation organize into the fibroelastotic plaques of the late stages. A verrucous angiitis of the subendocardial vessels commences with ectatic dilatation and deposits of fibrin on the walls. These may develop into narrow-stalked polyps, which break loose and produce microembolic lesions, or the fibrin deposits may be organized into a fibrinofibrous cushion, rich in capillary tissue, to one side of the vessel, or occasionally occluding it. Similar changes are found in pulmonary blood vessels. In the acute stages the myofibers show little change, and the loss of striation, nuclear hyperchromatism, and fragmentation appear to be anoxic. Interstitial edema may be marked, and the changes are confined to the inner third of the myocardium. In the chronic stages, some interstitial fibrosis may develop. Small perivascular inflammatory cell infiltrates may be seen, but usually minimal myocardial inflammatory-cell infiltration is seen.
The cause of Becker’s disease is unknown. It may develop acutely, with severe congestive heart failure, and the serous effusions are typically out of proportion to the severity of the cardiac failure. Abdominal pain from acute liver congestion may be intense. There is often a fever with PMN leukocytosis, but no eosinophilia with tachycardia or cardiomegaly. Radiology reveals only an enlarged and flaccid heart. ECG changes are not distinctive, and low-voltage patterns are usual. Embolic episodes, large or small, are common. The patient may progress to death in cardiac failure, unresponsive to all treatment, or may recover completely. After a variable interval, relapse occurs, which may result in recovery or death. The episodes may be repeated over many months, with the intervals of improvement progressively shorter and periods of congestive failure progressively protracted. Treatment at all stages is disappointing, because Becker’s disease is refractory to all therapy. Ultimately, death occurs, often rapidly, in a final episode of heart failure.
Pathologically, the cardiac lesions in Becker’s disease can be recognized by the enlargement and stiffness of the heart, the fibroelastotic patches, and the distinctive verrucous angiitis of the subendocardial blood vessels.
Beriberi
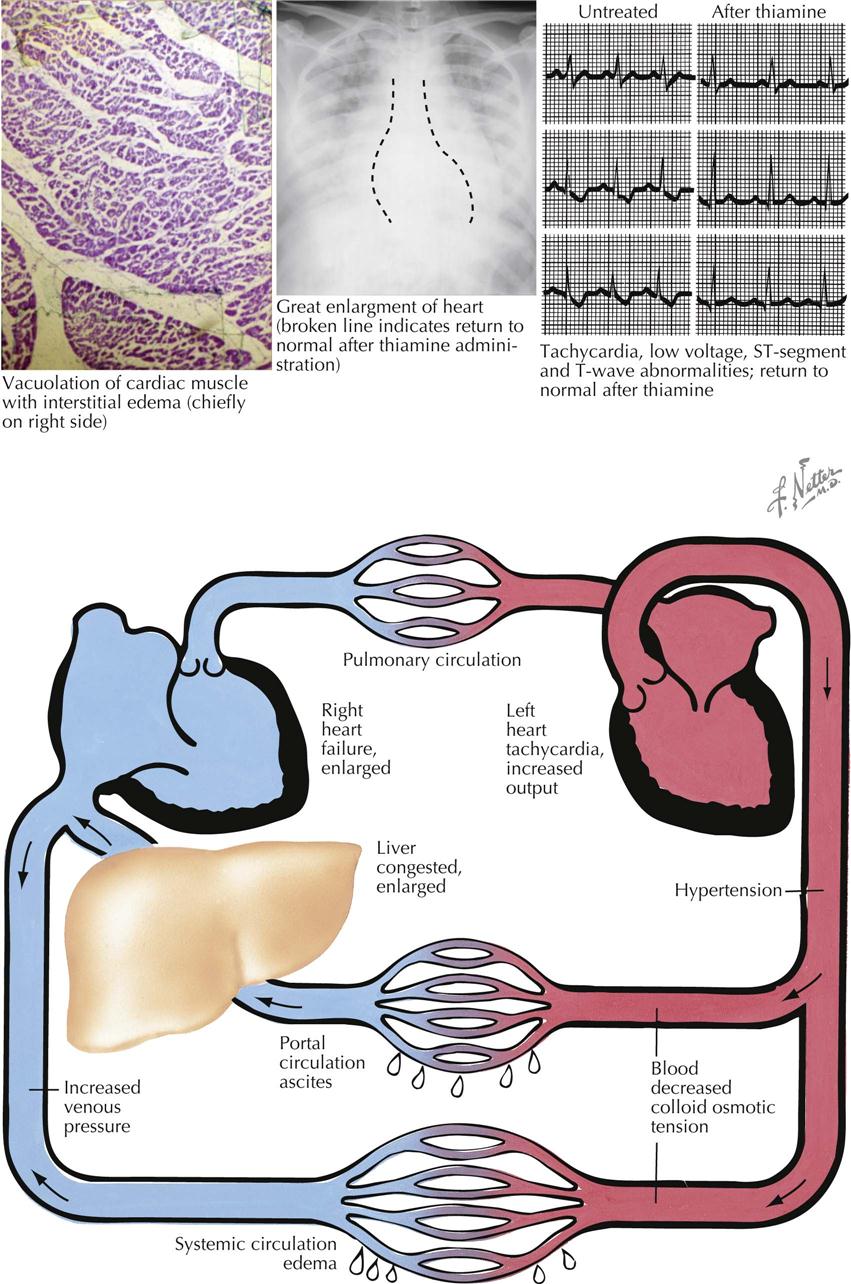
The term beriberi should be confined to the lesions that result from a dietary deficiency of thiamine. Cardiovascular beriberi is seen as a fulminant disease of small, breast-fed infants, reflecting severe thiamine deficiency in the mothers. It has a seasonal incidence, as does adult cardiovascular beriberi, in regions where the disease is caused by a diet deficient in a simple staple such as husked rice. Small epidemics may occur in prisons, camps, and fishing boats. Beriberi is sometimes seen as an acute disease in alcoholic patients with a restricted diet. The crucial feature of all these types is that they respond to adequate thiamine quickly, except in the most severe terminal cases. The response is rapid and dramatic.
The circulatory rate in cardiovascular beriberi is extremely rapid, with bounding pulse and increased blood pressure. “Pistol shot” sounds are heard over the extremities, and the hands and feet are warm despite peripheral edema of rapid onset; ascites may occur, possibly with polyneuritis. The heart must increase its workload to meet the demand of this hyperkinetic circulation, and the rapid return of blood to the heart leads to disproportionate right-sided dilatation and hypertrophy. The basic change apparently is in the peripheral circulation, with a great opening of arteriovenous anastomoses, especially in striated muscles. The initial effect of thiamine seems to be on the peripheral circulation; the workload on the heart is abruptly diminished, and rapid alteration in heart size and rate results. These changes are so rapid that thiamine administration can be used as a therapeutic test for cardiovascular beriberi. When terminal, death results from the sudden failure of the heart to meet the sustained call for high output. The terminal failure may be sudden, with severe pulmonary edema.
It has been impossible thus far to define specific changes caused by thiamine deficiency in human hearts with cardiovascular beriberi. The hearts invariably exhibit a degree of right-sided dilatation and hypertrophy, but the microscopic findings may show only myofiber hypertrophy, with enlargement and hyperchromatism of the nuclei, which are cigar shaped or blunt ended. Watery vacuolation of the myofibers and interstitial edema have often been recorded, along with fatty infiltration and varying degrees of myocardial fibrosis. These changes seem to reflect the patient’s general nutritional status rather than specific lesions of thiamine deficiency. These lesions do not resemble those found in animal models of thiamine deficiency. The animals die suddenly, showing focal patchy necrosis of muscle cells with an inflammatory infiltrate. Lesions of this type are not reported in humans. The lesions seen in animals are similar to those induced by extreme potassium deficiency, but a combination of thiamine and potassium deficiency does not produce the lesions.
In Western countries, a high-output cardiovascular disease seen in alcoholic patients may respond completely to thiamine administration. Some patients taking thiamine may have their status changed from high-output failure to low-output failure, in all probability the result of an alcoholic cardiomyopathy.
Nutritional deficiencies in humans only rarely are caused by lack of a single essential dietary element. Thiamine deficiency may occur in otherwise well-nourished individuals; most patients have diets typically low in protein and fat but high in carbohydrates, thus developing multiple deficiencies. Since its administration is so effective in cardiovascular beriberi, thiamine should be given as quickly as possible. If improvement does not follow, or if only some cardiovascular modifications are altered, the remaining cardiovascular lesions are from some cause other than thiamine deficiency. In the light of current views on the metabolic derangements in thiamine deficiency, it seems unlikely that a state of chronic beriberi heart disease could exist.
Cardiomyopathies
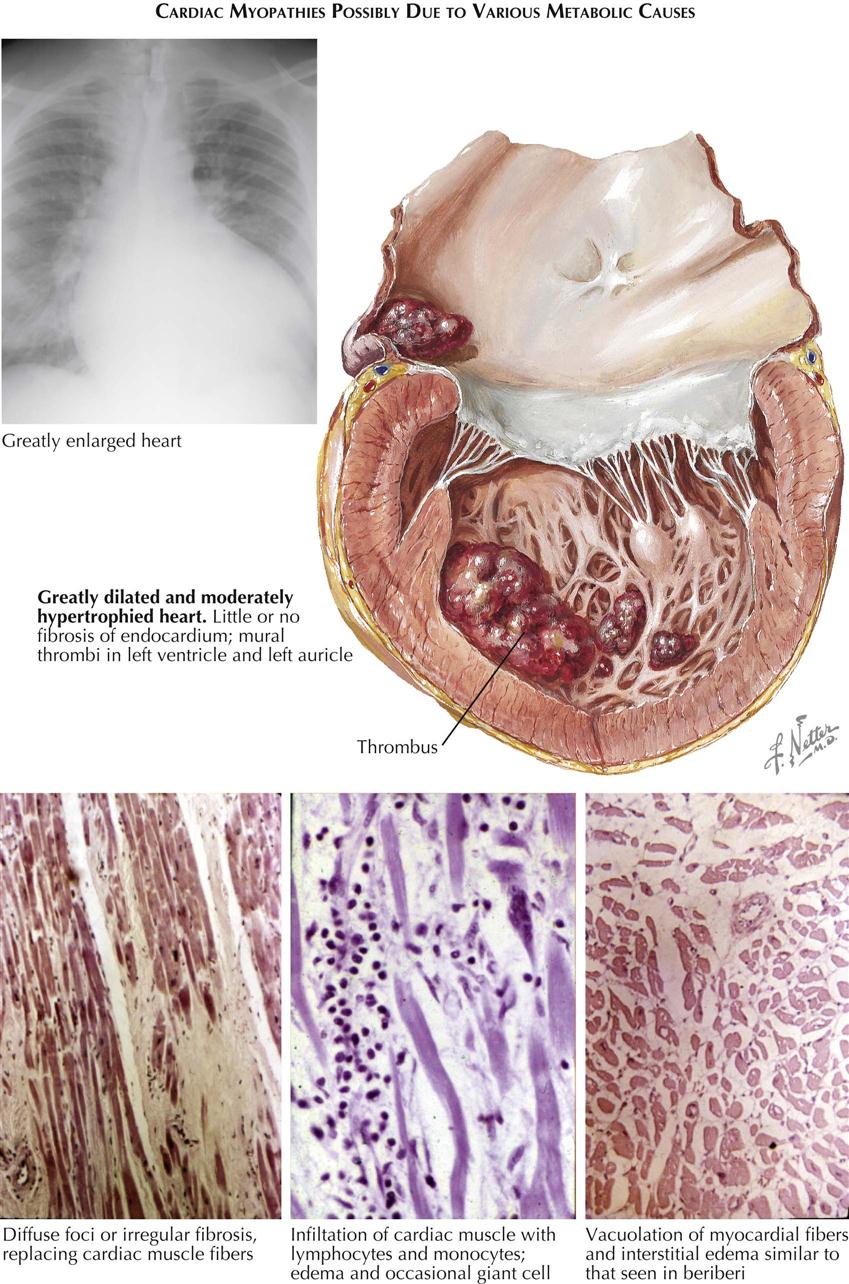
In any cardiac clinic, an occasional patient is seen who exhibits evidence of cardiac disease, typically with a greatly enlarged heart but with no evidence of valvular disorder, coronary-artery disease, or hypertension. The term nonischemic cardiomyopathy is often used with these patients. Clues to the cause and nature of the disease may be found in the family history, a past episode of infections, or the evidence of accompanying endocrine disturbance, a generalized vasculitis, or some form of systemic disease. Hemochromatosis, amyloidosis, glycogen storage disease, and nervous system disorders must be excluded and evidence of a persisting or past episode of bacterial, viral, or protozoal disease evaluated.
The cause of the cardiomegaly is still obscure in some patients, including women later in pregnancy or in the postpartum state, called peripartum cardiomyopathy. There is a sizable group of patients worldwide with “idiopathic cardiomegaly.” Many are suspected to have malnutrition or a history of alcoholism (often difficult to elicit in detail), which indicates thiamine deficiency, but administration often produces no improvement. Some patients may recover spontaneously, only to relapse later with a later pregnancy. In some the cardiomyopathy may progress rapidly and relentlessly, or slow deterioration may occur. Physicians should be alert to epidemics, particularly with beer drinkers’ cardiomyopathy. In many cases, alcohol is the most important factor, although similar conditions also affect those who do not drink alcohol.
Radiologic and ultrasonic examination is usually unhelpful other than in demonstrating a grossly enlarged heart, which typically continues to increase in size. The ECG usually is abnormal, but the abnormalities are nonspecific (e.g., left bundle branch block, T-wave changes). Cardiac ultrasound and MRI confirm the poor contraction. Ultrasound can easily define chamber dilatation and decreased ejection fraction. Unfortunately, if the cause was not found in life, it cannot be satisfactorily established at autopsy. Evidence of congestive heart failure is found with an enlarged heart, compounded by varying degrees of hypertrophy and dilatation. Some degree of coronary arteriosclerosis may be found, especially in communities where beriberi is common, but this is usually insufficient to explain the ventricular derangement. The coronary vessels are often normal, or may be widely patent. The myocardium is often soft and flabby, or may be stiffer than normal and exhibit pallor, with evidence of myocardial fibrosis. The endocardium may be normal or covered in part by mural thrombi, or may be patchily thickened and opaque.
The histologic changes in cardiomyopathy may also be indeterminate, and some patients may show only hypertrophy of myocardial fibers, with enlarged, hyperchromatic, blunt-ended or cigar-shaped nuclei. Watery vacuolation of myofibers may occur, perhaps with some accumulation of lipid droplets. The myofibers may be separated by interstitial edema. Diffuse fibrosis or focal scars may be present. Cellular infiltration of variable intensity is seen; the cell types vary but typically include an infiltrate of lymphocytes and monocytes and occasionally a giant cell. Histochemical studies may show greatly diminished numbers of oxidative enzymes.
Study of the endocardial lesions, especially areas of endocardial thickening, may help elucidate the pathogenetic mechanisms in cardiomyopathy. Considerable endocardial smooth muscle hypertrophy indicates previous acute cardiac dilatation. Pathologic study should include carefully oriented blocks and exact mapping of lesions, as well as careful correlation with any extracardiac lesions, to identify the causes of these presumed metabolic cardiomyopathies. However, meticulous clinical history taking into the patient’s background and circumstances surrounding onset of cardiomyopathy are most important. As a rule, only symptomatic relief can be given.
Acquired Immunodeficiency Syndrome and the Heart
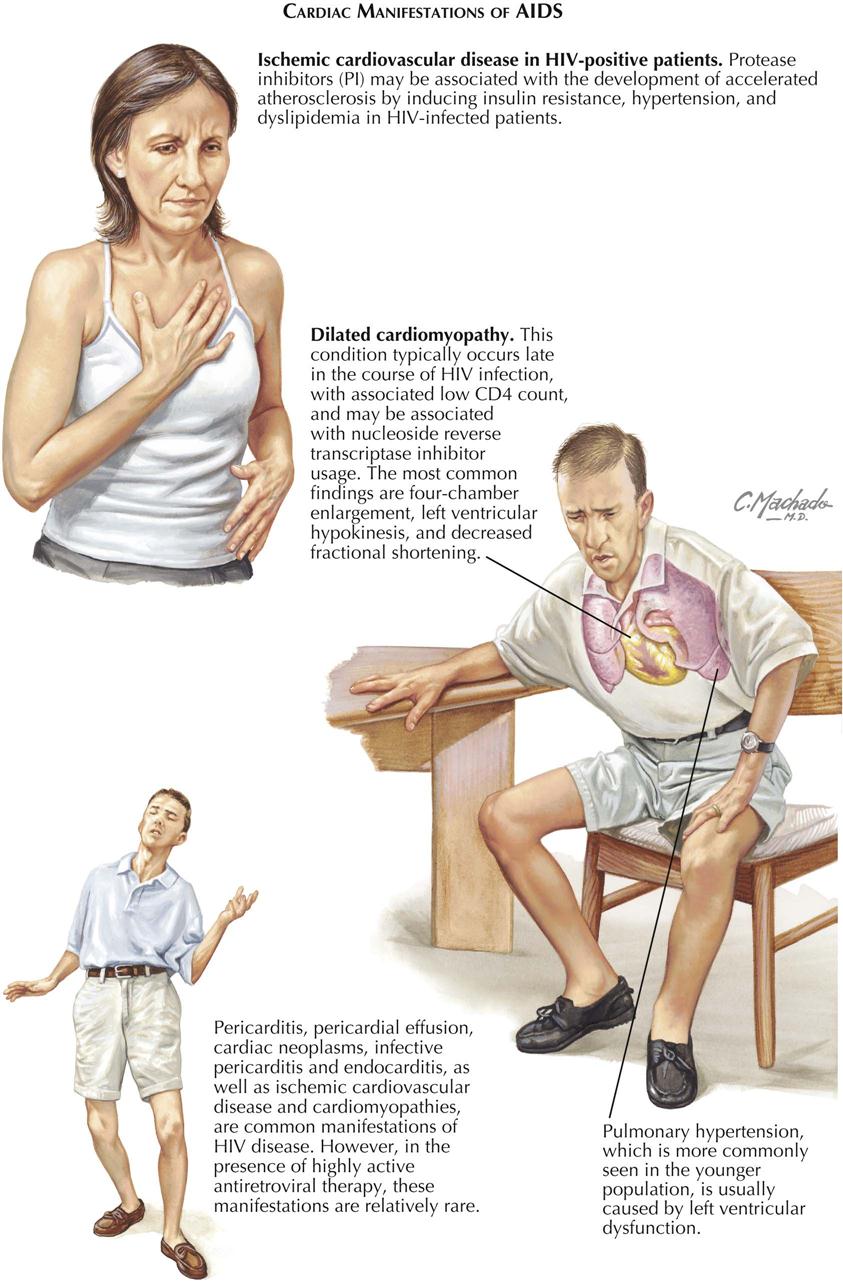
Patients with human immunodeficiency virus (HIV) are living much longer because of highly active antiretroviral therapy (HAART). As a result of the chronicity of HIV infection, however, patients can develop multiple cardiovascular manifestations, including hypertension, pericarditis, cancer (rare), endocarditis, and pulmonary hypertension. An estimated 20% or more of patients with HIV infection at autopsy have some evidence of heart disease (see Plate 6-70).
Drugs used to treat HIV infection (e.g., protease inhibitors) can increase the risk for coronary artery disease and development of accelerated coronary atherosclerosis. The mechanism probably relates to the induction of insulin resistance as well the development of systemic hypertension and hypercholesterolemia. Patients who develop heart disease should have treatment based on the pathogenesis and etiology, similar to patients without HIV infection, although drug interactions in patients with HIV may require adjustments in therapy. Despite this caveat, any modifiable risk factor (e.g., lipid abnormalities, hypertension, smoking, diabetes, heart failure) need aggressive therapy. In any patient with HIV, opportunistic infections such as tuberculosis can complicate cardiac management, and thus prophylaxis must be considered based on the patient’s level of immunity.
Other conditions such as dilated cardiomyopathy can occur, usually late in the disease course. HIV infection can directly affect the myocardium, and the decreased immunity makes the heart more susceptible to infection than in the non-HIV patient. The heart failure caused by cardiomyopathy is usually associated with a low CD4 count and may be associated with reverse transcriptase inhibitor use. The typical cardiac findings in these heart failure patients are marked LV dysfunction, manifested by global hypokinesis and decreased ejection fraction. Drug use (cocaine, amphetamines) can contribute to the cardiac failure of HIV cardiomyopathy patients. Pulmonary hypertension can result from the elevated LVEDP in these heart failure patients.
In the modern era of HAART, patients with HIV are living longer, and cardiac disease has become a growing problem; pericarditis, pericardial effusions from tuberculosis, Kaposi’s sarcoma, or lymphoma can occur. The pericardial effusions can be readily detected by cardiac ultrasound and other noninvasive modalities. Rarely, cardiac tamponade can occur. Cardiac cancers, infective endocarditis, and ischemic heart disease are also relatively rare complications of HIV. Infections, however, including fungal (e.g., Candida, histoplasmosis, cryptococcosis, aspergillosis), viral (e.g., herpes simplex, cytomegalovirus), bacterial (e.g., tuberculosis), and parasitic (e.g., toxoplasmosis), can cause myocarditis in HIV patients. Conduction disturbance can result from lymphomatous infiltration. The cardiologist and infectious disease specialist must work together to meet the therapeutic challenge of cardiovascular disease in HIV patients.
Substance Abuse and the Heart
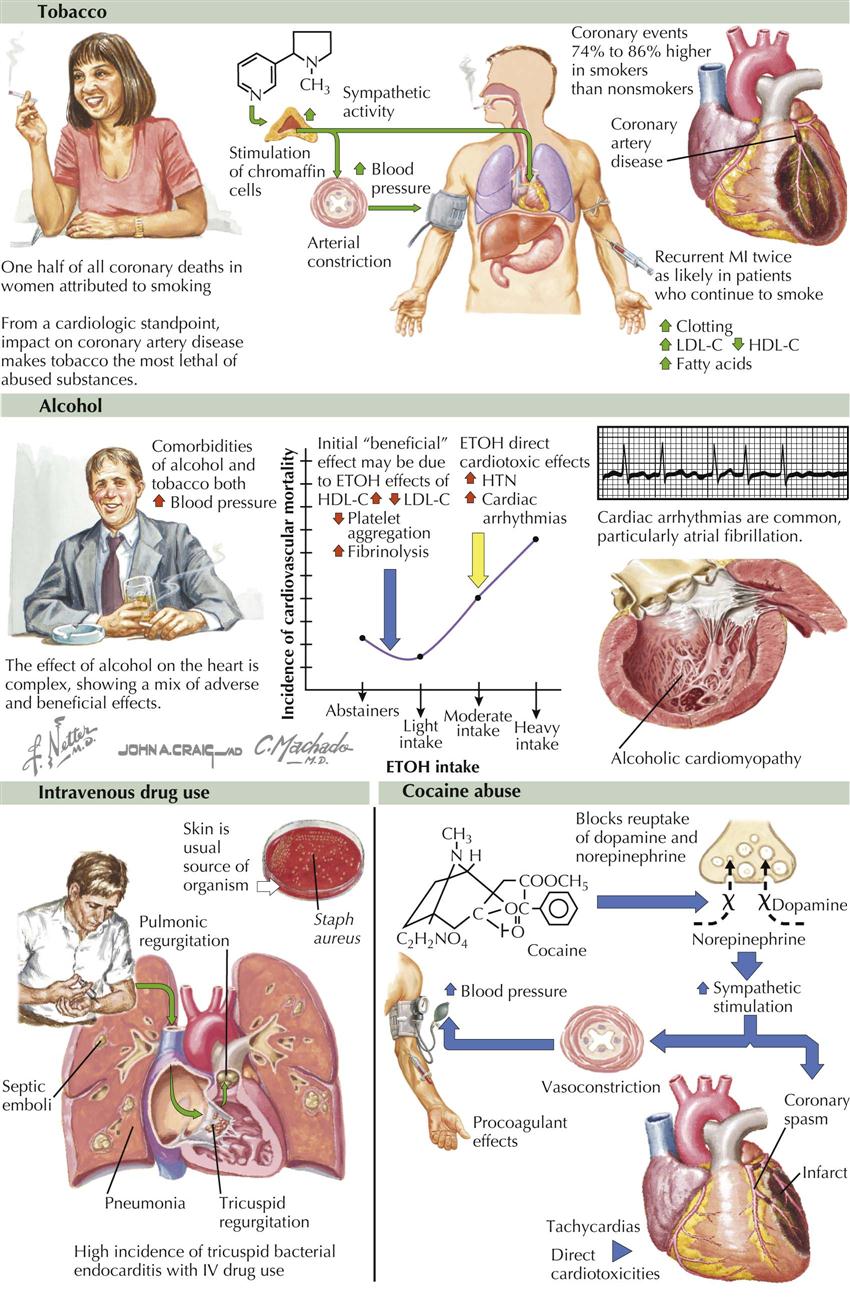
The combination of addiction and physical harm makes intravenous heroin probably the most dangerous abused drug, followed by cocaine, barbiturates, and amphetamines. Any use of injected drugs can transmit bacteria (most often Staphylococcus aureus) from the skin into the bloodstream. This can result in septic emboli to the lung, pneumonia, and infective endocarditis of any valve, especially tricuspid and pulmonary.
Tobacco and Alcohol
Tobacco and alcohol are extremely addicting but usually less harmful in the short term than heroin and cocaine. Half of all coronary deaths in women are attributed to cigarette smoking. Coronary events, such as myocardial infarction, are approximately 80% higher in tobacco smokers than nonsmokers. Recurrent MI is twice as likely to occur in patients who continue to smoke than in those who stop. Tobacco smoking increases sympathetic activity and catecholamines, constricts blood vessels, and increases blood pressure.
The effect of alcohol (ETOH) on the heart has been shown to have adverse and beneficial effects. Alcohol and smoking can both raise blood pressure. The initial beneficial effect may be related to increased high-density lipoprotein cholesterol (HDL-C), decreased low-density lipoprotein cholesterol (LDL-C), decreased platelet aggregation, and increased fibrinolysis. However, alcohol (ETOH) has direct cardiotoxic effects (e.g., alcoholic cardiomyopathy) because it can increase blood pressure and increase cardiac arrhythmias, e.g. atrial fibrillation (see Plate 6-71; HTN, hypertension). Although there is some evidence that light intake of alcohol is beneficial, it is not recommended that a person who does not drink alcohol should start, since even low levels of alcohol may increase blood pressure.
Cannabis is less addicting than tobacco or alcohol, although it may be harmful related to judgment errors. As of 2013, marijuana was available in select patients for medical use in 20 U.S. states.
Cocaine
A common illegal “street drug,” cocaine, as with amphetamines, acts by multiple mechanisms on brain catecholaminergic neurons; its reinforcing effects may involve inhibition of dopamine uptake. By blocking the reuptake of dopamine and norepinephrine, the latter increases sympathetic stimulation of blood vessels, increases blood pressure, has procoagulant effects, and may induce tachycardias and coronary artery spasm, which may lead to MI. Cocaine also has direct cardiotoxicity; in the United States, 15,000 people die directly from cocaine use or from judgment errors (e.g., motor vehicle crashes). Several cardiovascular complications are closely related to cocaine use. Chest pain syndromes may result in MI. Other adverse cardiac events are related to hypertension and include aortic dissection, stroke, nonfatal arrhythmias such as atrial fibrillation, and fatal dysrhythmias such as ventricular tachycardia and fibrillation. On occasion, patients present with heart failure symptoms related to pulmonary edema.
Pericardial Disease
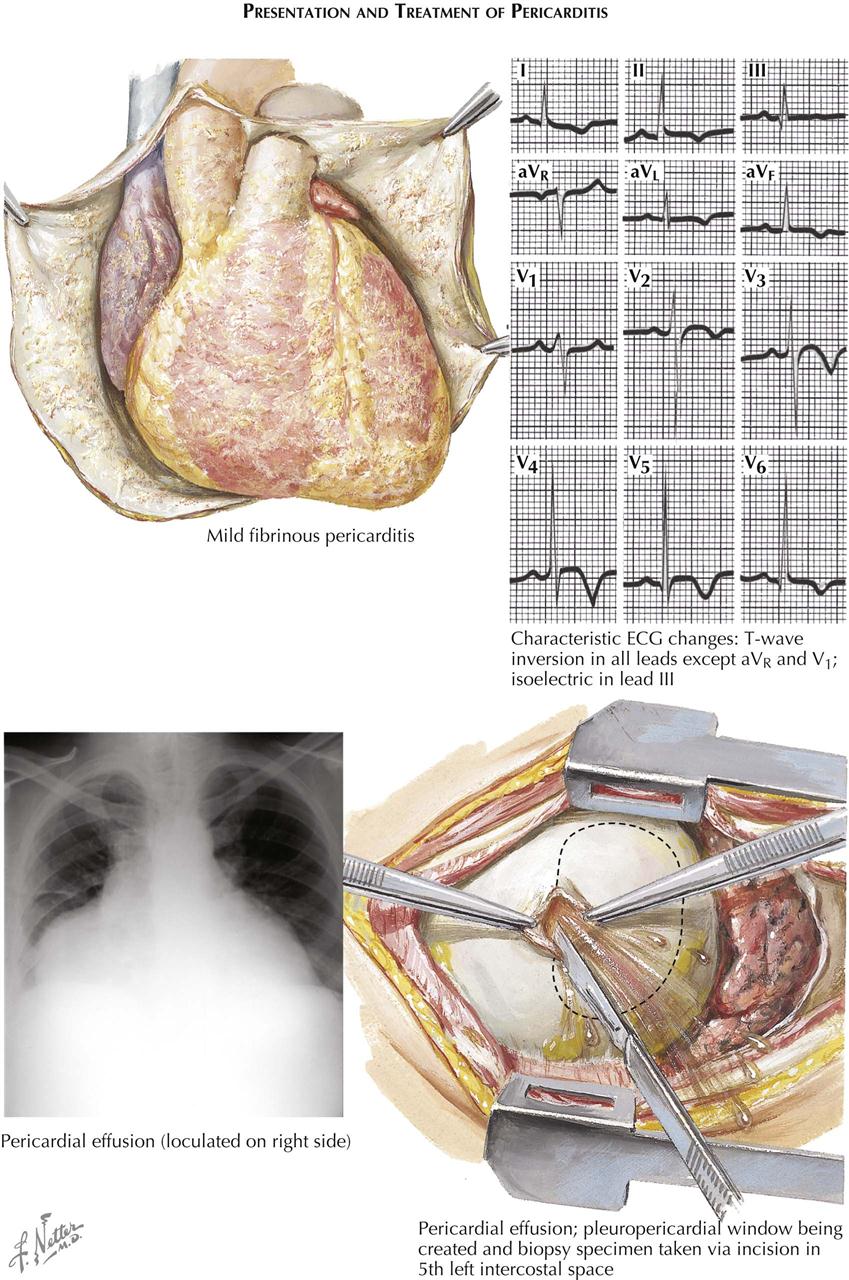
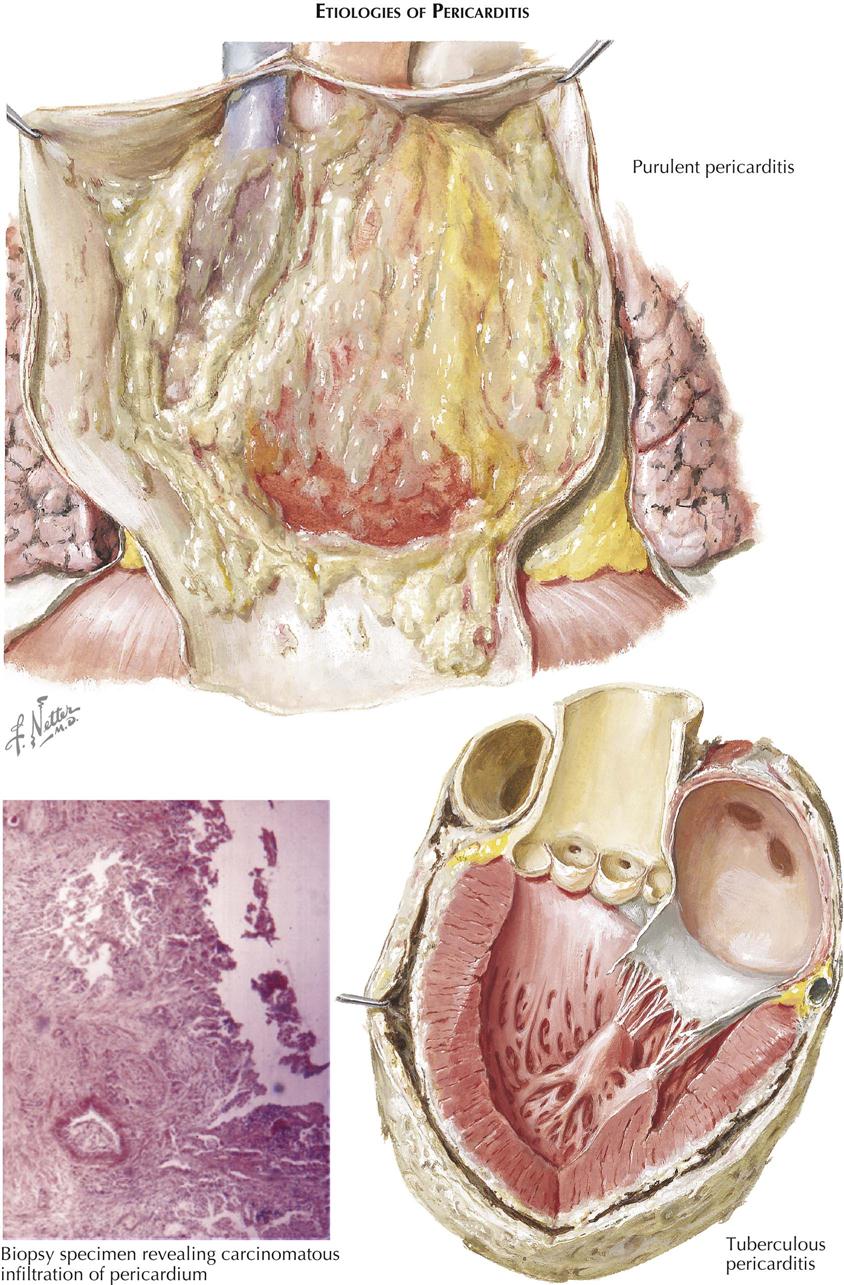
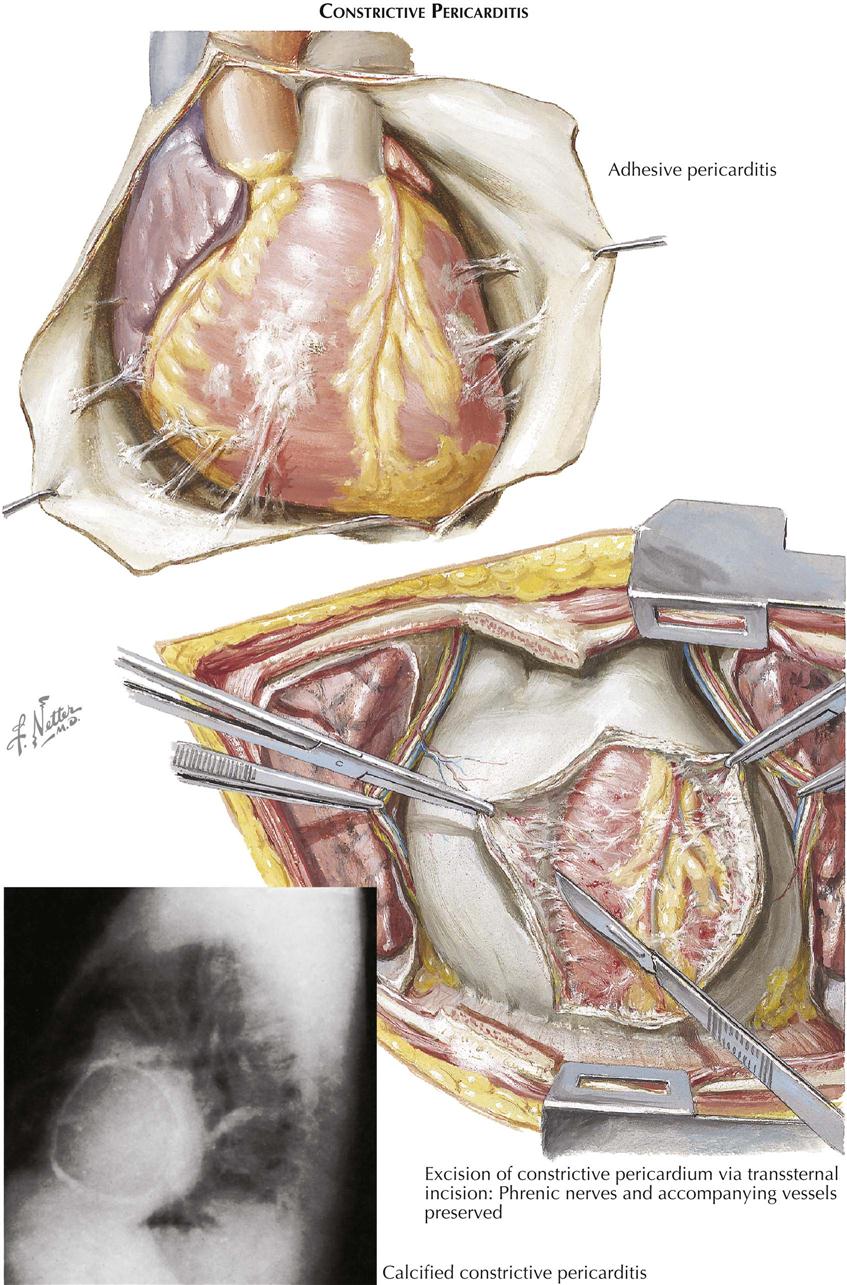
Pericarditis can be acute, subacute, or chronic inflammation of the pericardium. Identifying the cause of pericarditis is important because therapy depends on the underlying process; thus the best way to classify pericarditis is by etiology. Pericarditis can result from infection (viral, bacterial, parasitic), systemic autoimmune diseases (lupus, rheumatoid arthritis), immune processes (rheumatic fever, post-MI), myocardial disease (acute MI, myocarditis), metabolic disorders (uremia, myxedema), trauma, direct injury (penetrating thoracic/esophageal perforation, cardiac perforation from device), blunt trauma to chest (steering wheel injury), mediastinal radiation, and cancer (primary tumors, secondary metastases/melanoma).
The incidence of pericarditis depends on several factors, including flulike epidemics (viral pericarditis), areas of the world in which trauma is common (war zones, etc.) and areas of the world in which infectious disease or rheumatic fever is still very prevalent (Africa, India, etc). Benign viral or idiopathic pericarditis tends to recur. Bacteriologic studies show a viral origin in some patients; however, many cases are not caused by a viral infection but probably result from an autoimmune reaction, because recurrences can be suppressed by therapy with nonsteroidal antiinflammatory drugs (NSAIDs), aspirin, and colchicine.
Acute Pericarditis
Acute pericarditis may occur as serous, serosanguineous, purulent, and bloody forms. Serous pericarditis is the type usually seen in rheumatic fever and benign nonspecific or viral pericarditis. Purulent pericarditis usually results from bacterial infection with such organisms as pneumococcus, staphylococcus, or streptococcus, but any bacterium can be infective. Serosanguineous pericarditis must be distinguished from hemorrhage into the pericardium and is most often noted when the etiology is a malignancy or pericardial injury. Adhesive pericarditis may develop, rarely after the acute serous or serosanguineous forms and more likely after purulent or bloody pericarditis (see Plate 6-74). The chronic effusive adhesive form is also seen in tuberculous pericarditis, often with pericardial thickening. Constriction of one or both cardiac tracts (inflow and outflow) can result from chronic adhesive pericarditis and may require pericardiectomy.
The onset of pericarditis usually is accompanied by chest pains, although pain is not a prominent symptom and may even be absent in some cases, especially when pericardial effusion appears early, which probably prevents the parietal and visceral pericardia from rubbing together. The pain usually is substernal and may radiate to the left shoulder and arm or the neck. The character of the pain may closely simulate that of the onset of acute MI, thus presenting a difficult diagnostic problem. Classically, the pain of pericarditis is relieved by sitting up. If there is pleural involvement, the pain may be increased by deep inspiration. With the development of effusion, dyspnea becomes the more prominent symptom (see Plate 6-72). This is probably caused by mechanical pressure on the lungs, compressing not only the alveolar structure but also the smaller bronchial branches. If cardiac tamponade occurs, physiologic changes include distention of neck veins from increased right atrial pressure. Associated symptoms depend on the etiology of pericarditis. In acute inflammatory conditions, chills, fever, and sweating often are present.
The most important physical sign of acute pericarditis is a pericardial friction rub, usually heard best just to the left of the sternum and fairly well localized. This to-and-fro scratching sound seems to be closer to the ear than other cardiac sounds or murmurs. In pericarditis caused by inflammatory conditions or MI, the rub may be heard for only a short time, but it is usually persistent when pericarditis results from neoplasm or uremia. The friction rub may be heard in the presence of considerable pericardial effusion, even as much as 1 liter, because of the uneven accumulation of fluid in the pericardium.
In the presence of significant effusion, the heart sounds will diminish in intensity, and on percussion the area of cardiac dullness will be increased. An area of dullness with bronchial breathing may be noted below the angle of the left scapula (Ewart’s sign). Occasionally, there may be so much pericardial fluid that by greatly increasing intrapericardial pressure, it interferes with cardiac function (cardiac tamponade). Venous pressure rises, heart rate increases, pulse slows, and pulse amplitude may decrease during inspiration (paradoxical pulse). Blood pressure may fall as much as 20 mm Hg at the end of inspiration, an exaggeration of the normal drop in blood pressure. The rapid formation of pericardial effusion is important in determining whether tamponade will occur. A small, rapidly developing effusion may cause tamponade, whereas a large effusion developing over several weeks, as in tuberculous pericardial effusion, or effusion related to hypothyroidism, will not cause tamponade.
Serial chest radiographs in particular demonstrate an enlarging cardiac silhouette suggesting pericardial effusion. However, the fluid volume must be at least 250 mL before a significant change in the cardiac shadow permits a definitive diagnosis. The cardiac silhouette, depending on the amount of fluid present, will assume a pear-shaped or water-bottle form.
The ECG is important in the diagnosis of pericarditis, which may be confused with acute myocardial infarction (see Plate 6-72). Classically, elevation of the ST segment usually occurs in most of the 12 leads, whereas in MI, depending on the location of the infarction, the precordial leads or inferior leads show changes (ST elevation). The shape of the ST segment may also be similar to that seen in MI. In pericarditis the PR segments may be depressed in lead II and elevated in lead aVR . The moat common ECG abnormality is nonspecific T-wave changes. The ECG changes probably reflect inflammation of the epimyocardium, since the pericardium itself is electrically inert. Two-dimensional cardiac ultrasound readily determines the presence and location of pericardial effusion and is required if pericardiocentesis is planned.
Purulent Pericarditis
Purulent pericarditis may not be recognized because of being part of the general disease (see Plate 6-73). However, ECG changes suggesting pericarditis (and epimyocarditis), cardiac ultrasound evidence of pericardial effusion, or ultrasonic evidence of diminished cardiac pulsations may call attention to the purulent condition. A pericardial rub is also helpful, but it is not diagnostic for purulent pericarditis. Antibiotics and early drainage by pericardiocentesis or surgery are indicated, and when used early, these measures substantially reduce mortality, otherwise close to 100%.
Tuberculous Pericarditis
Tuberculous pericarditis is a complication usually resulting from extension of a contiguous tuberculous process in the lungs or the lymph nodes (see Plate 6-73). It starts as a fibrinous reaction, with or without effusion (see Plate 6-72), and then the pericardium thickens as a result of tubercles, caseation, and fibrosis. The thickened, adherent pericardium assumes a “shaggy” appearance. Constrictive effusive pericarditis may occur in this early period, or later if pericardial calcification results. Treatment in the early stages before constriction is by conventional long-term antitubercular therapy (as determined by TB specialist). When constrictive physiology is present, pericardiectomy is required (see Plate 6-74).
Nonspecific Pericarditis
The recurrent nonspecific type of pericarditis, such as associated with autoimmune reactions, postcommissurotomy pericarditis, and post-MI pericarditis, is often benefited by antiinflammatory therapy (steroids, NSAIDs, aspirin, colchicine). Such treatment merely suppresses the signs and symptoms and therefore is usually continued indefinitely.
In the patient with pericardial effusion, pericardiocentesis is required only when tamponade threatens, or for diagnostic purposes, as in suspected malignancy or bacterial infection. A surgical approach removes fluid through a “pericardial window” into the left pleural space, and pericardial biopsy can be obtained (see Plate 6-72). When a pericardial window is created, fluid in the pericardial space passes into the left pleural cavity, from where it is absorbed. Alternatively, a catheter can be inserted into the pericardial space to facilitate pericardial drainage to a collecting system.
Acute Cor Pulmonale and Pulmonary Embolism
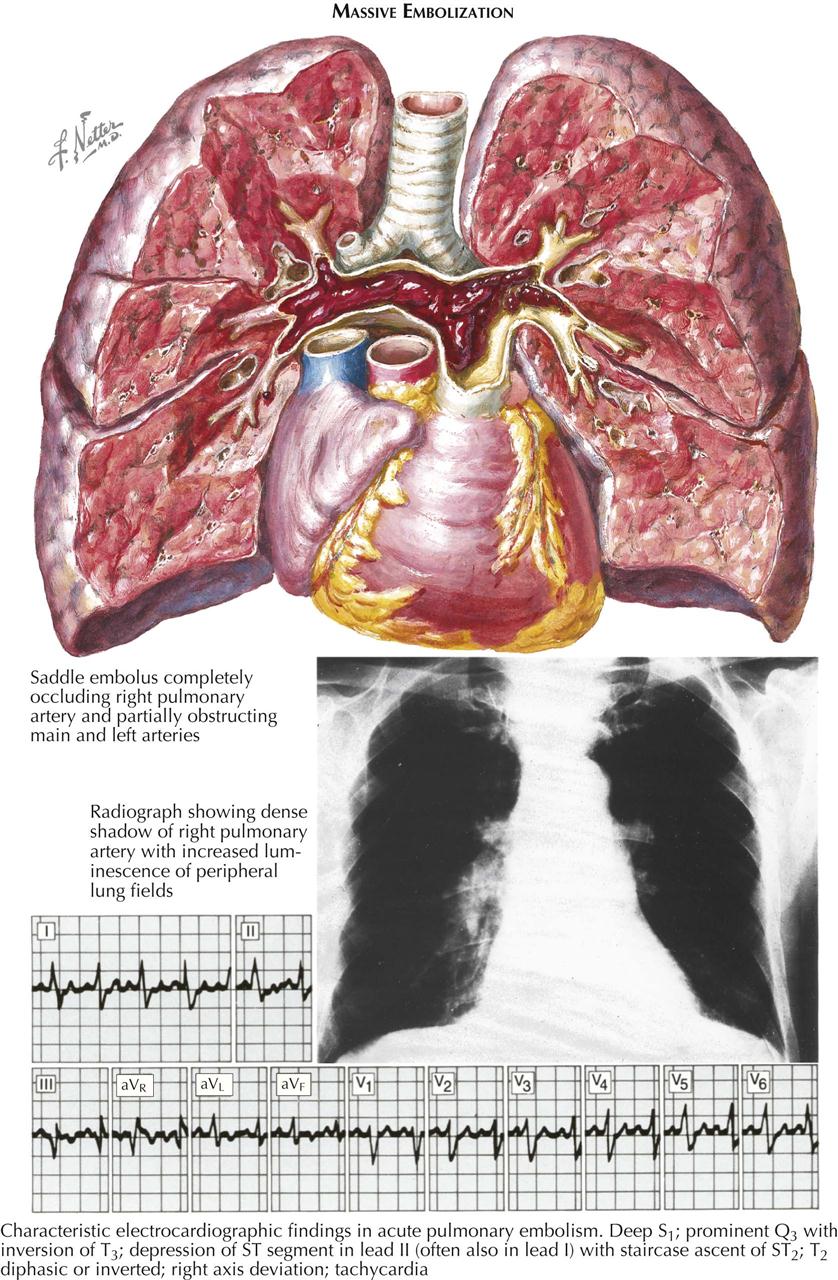
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Acquired Heart Disease
Structure of Coronary Arteries
The coronary arteries are susceptible to atherosclerosis as well as its complications, particularly intravascular thrombosis, often resulting in myocardial infarction. Atherosclerosis is a form of arteriosclerosis characterized by initial involvement of the inner layer of the arterial wall, or intima. The intimal localization of early atheromas helps to differentiate the atherosclerosis type from other forms of arteriosclerosis, such as Mönckeberg’s medial sclerosis or periarteritis nodosa, that primarily involve the muscular or adventitial layers.
Recent histochemical, organ culture, and electron microscopy studies found further evidence of the complexity of the arterial wall. As Plate 6-1 shows, a medium-sized muscular artery, such as a main coronary vessel, consists of a series of concentric tubes or coaxial coats of differentiated cellular and extracellular components in three layers: tunica intima or intima, tunica media or muscular, and tunica adventitia. In the intima the innermost cell layer in direct contact with the bloodstream consists of a sheet of polygonal endothelial cells, usually less than 1 micron (µ) thick, except at the site of the cell nucleus, and elongated in a direction parallel to the vessel’s axis. Many of these endothelial cells have pinocytotic vesicles of caveolae in their cytoplasm, abundant mitochondria, well-developed granular endoplasmic reticulum, and Golgi complexes. Microscopically, the areas of contact between endothelial cells vary from simple mutual contact of the cell membranes to well-defined intercellular bridges, or desmosomes, corresponding to the so-called intercellular cement lines described in earlier light microscopy studies. A distinct basement membrane separates these endothelial cells from the subendothelial space, which varies in thickness according to age of the patient as well as size of the artery. In fetal life and shortly after birth the endothelium in the coronary arteries lies in direct contact with the internal elastic membrane, or lamina, without the subendothelial space, which appears by the end of the first decade of life.
In adulthood the intima consists of a matrix of ground substance containing small amounts of acid mucopolysaccharides and elastic and collagen fibers separating scattered intimal cells, or intimacytes. Because of morphologic and cytochemical difficulties in classifying intimacytes, their response to in vitro incorporation of serum lipids is used to identify them. Some intimacytes, or atherophils, incorporate lipid rapidly and show ultrastructural characteristics of modified smooth muscle cells, with typical bundles of myofilaments in their cytoplasm, pinocytotic vesicles, and parts of limiting basement membrane on the cell surface. On the other hand, cells called fibrophils are spindle shaped and have fingerlike cytoplasmic projections, with no basement membrane and few pinocytotic vesicles. Occasionally, large mononuclear ovoid cells appear, containing cytoplasmic inclusions with “single-unit” acid phosphatase–positive granules or lysosomes resembling macrophages.
The lamina propria is separated from the underlying smooth muscle cells of the media usually by a well-developed internal elastic membrane of tenacious matrix containing fibrils about 500 angstroms (Å) in diameter with abundant fenestrations. This internal elastic membrane is usually wavy in cross sections, and the fenestrations are oval or round openings extending across its surface. The underlying tunica media is characterized by concentric layers of smooth muscle cells 10 to 25 µ in length and oriented transversely to the artery’s main axis. Individual smooth muscle cells are surrounded by a network of collagenous and elastic fibers, which continue without transition into both internal and external elastic membranes. The external elastic membrane separates the media from the adventitia and is characterized by the presence of loosely packed collagen and elastic fibers. Small blood vessels or vasa vasorum are also found, as well as both sympathetic and parasympathetic nerve fibers from the autonomic nervous system.
These morphologic and functional characteristics of the coronary arteries are modified early in the presence of the intimal changes characteristic of atherosclerosis. The most important clinical difference between coronary and aortic atherosclerosis is the high incidence of acute thrombosis in coronary vessels, with infarction resulting in irreversible damage to the underlying myocardium.
Pathogenesis of Atherosclerosis
Histologic and functional characteristics of the intimal lining of the arteries help in understanding the histogenesis of spontaneous atheromas, often initiated shortly after birth, particularly the presence of different cell types in a ground substance matrix and the absence of a direct blood supply. The well-developed atherosclerotic plaque resulting from inflammatory and reparative processes is a complex lesion containing extracellular deposits of calcium salts, blood components, cholesterol crystals, and acid mucopolysaccharides. The initial changes, however, seem to occur at the cellular level, often accompanied by an abnormal intracellular storage of lipids, particularly cholesterol esters, fatty acids, and lipoprotein complexes. These findings on microscopy have strengthened the thesis that lipid infiltration from the bloodstream may be a significant factor in the growth of the atheromatous plaque. Furthermore, recognition by cytochemical techniques makes lipids helpful indicators of abnormal cell behavior, independent of their role in the etiology of atheroma.
Because of the frequency and severity of atherosclerosis, short-term organ culture techniques for the isolation of arterial intimal cells (intimacytes) identify susceptible cell populations, based on their response to incorporation of homologous serum lipids in vitro. Intimacytes from arteries, with and without histologic evidence of atherosclerosis, show two types of atherophils: genetically highly susceptible cells and genetically resistant cells to intracellular lipid accumulation.
In the presence of increased perfusion rates of plasma lipoproteins caused by local hemodynamic changes, hypertension, elevated serum lipids, or changes in endothelial surface permeability (trauma, fibrin deposition, platelet aggregation), genetically susceptible atherophils will be transformed into lipid-laden atherocytes. The secondary release of intracellular lipids from these cells will induce surrounding atherophils to incorporate them, creating a self-feeding process with cytologic changes that increase metabolic requirements, including oxygen consumption rates, which result in increased permeability of the cell membrane to lipids if not met. These events set up a cycle favoring further expansion of focal atherosclerotic changes. Collagen fibers are laid down by fibrophils, which are then transformed into fibrocytes surrounded by acid mucopolysaccharide deposits. This eventually results in a characteristic intimal hyperplasia after ground substance changes. These histologic lesions are self-perpetuating, resulting in the replacement of intimacytes by an acellular area of arterial intima that stimulates further surface involvement of the vascular wall in this interplay among deposition, inflammatory response, and scarring, with the eventual production of a typical atherosclerotic plaque.
In contrast, genetically resistant atherophils, even in the presence of elevated serum-lipid levels, do not respond (or respond minimally) to an increased perfusion of plasma lipids. If some atherocytes appear, they are few in number, and only superficial fatty streaks are developed, resulting in sparse intimal elevations with few fibrophils and no well-organized plaque. This type of cytologic response also seems less susceptible to the clinical complications of atherosclerosis, particularly thrombosis or rupture, because of the limited involvement of the vascular wall.
This approach to the pathogenesis of atherosclerosis emphasizes the clinical significance of the earliest possible identification of patients whose arteries are susceptible to atheroma, to modify or prevent environmental conditions that favor the acceleration of atherogenesis in the arterial intima cells. Local factors are also important in identification of the lesions, suggesting possible therapeutic approaches for their arrest or inhibition.
Risk Factors in Etiology of Atherosclerosis
In terms of mortality, the most serious problem facing more developed countries is atherosclerosis of the cardiac and cerebral blood vessels. There is no single cause of atherosclerosis, which is often labeled a “multifaceted disease.” Atherosclerosis reflects the culmination of many factors acting over a lifetime and usually becomes clinically manifest only when angina pectoris, sudden cardiac death, myocardial infarction (MI), transient ischemic attack (TIA), or cerebrovascular accident (CVA, stroke) occurs. Heart failure is usually caused by MI.
Patients with a family history of premature manifestations of atherosclerosis may be at high risk for development of atherosclerotic disease. Older age, male gender, hypertension, diabetes, obesity, and tobacco smoking are associated with coronary atherosclerosis. Lack of exercise may interact with other clinical factors, and an exercise program (e.g., walking) is known to diminish symptoms in patients with known clinical manifestations of atherosclerosis. Excessive saturated fat in the diet is also involved in atherogenesis. High-fat diets increase blood lipid levels, particularly low-density lipoprotein (LDL) cholesterol. Elevated triglycerides and low levels of high-density lipoprotein (HDL) are also implicated in the etiology of atherosclerosis. Elevated serum uric acid may also contribute to atherogenesis, although further study is required. Unsaturated fatty acids with two or more double bonds help reduce cholesterol blood levels when their relationship to saturated fatty acids is increased.
Current concepts of atherogenicity are complex and involve monocytes, platelets, macrophages, foam cells, endothelial cells, smooth muscle cells, oxidized LDL, and many other subcellular substances. Interested readers should refer to current literature and textbooks on the subject. In general, the physician or clinician can profile a patient potentially prone to atherosclerosis, such as a hypertensive man who smokes cigarettes, exercises little, and has elevated blood cholesterol and triglyceride levels, low HDL levels, and abnormal glucose metabolism. Currently, in addition to diet and exercise, drugs used to treat lipid abnormalities include HMG CoA reductase inhibitors (statins), nicotinic acid preparations, ezetimibe, fibric acid derivatives, and bile acid sequestrants.
Pathologic Changes in Coronary Artery Disease
The earliest recognizable atheromatous changes in blood vessels are accumulation of aggregates of lipid-laden macrophages in the intima. The appearance of this lesion is similar to that seen in many experimental vascular lesions. Another apparently early phenomenon, difficult to correlate and possibly unrelated, is a subintimal deposit of collagen that often is primitive and thus rich in acid mucopolysaccharides. Lesions of this type shadow atherosclerosis wherever it occurs. The relationship of this vascular lesion to the type seen in Asian peoples has not been elucidated, but this disorder does occur in Asians. A further complication of this lesion is the deposition of fibrin on or within its superficial layers.
As more lipid accumulates, an atheromatous plaque with many constituents is formed. This plaque may involve only a segmental portion of the artery or its entire circumference. When the macrophages die, their lipid is released and becomes an inflammatory irritant. Fibrin accumulates, as do other blood constituents; calcium deposits form; and fibroblasts proliferate. Stenotic lesions may accumulate more surface material with deposited layers of thrombus. These lesions regress with lipid-lowering agents and antiplatelet therapy.
Even a segmental atheroma can cause atrophy of the underlying arterial wall, and the concentric type seems associated with major destruction of the lamina and parts of the media. Such changes appear secondary to interference with the nutrients permeating the intima. The stages of atheroma reveal another major feature in the lesion’s development: an inability to clear blood constituents that have permeated the media. This dynamic event is demonstrated by perfusing isolated segments of living canine arteries and observing the quantity of lipid in the vasa vasorum of the adventitia.
Hemorrhage into the lesion can result from surface blood dissecting into the plaque or from vasa vasorum bleeding. Thrombosis may occlude the artery; if the person survives, organization of the fibrin clot can progress and occasionally recanalize the artery. However, the ability of these small channels to supply any appreciable volume of blood beyond the area of obstruction is insufficient in most patients to revascularize the downstream myocardium. Rarely, an occluding thrombus completely disappears.
In addition to atheromatous and thrombus blockade of the coronary arteries, emboli can obstruct an otherwise normal coronary artery. Such emboli may consist of thrombi, valvular calcification, pieces of tumor, and occasionally even small foreign bodies. Various types of aortitis can involve the proximal vessels; syphilis of the aortic wall can occlude the ostia. The atheroma is the main disorder, however, because of its distribution. The lesions closest to the aorta (proximal) are the plaques most likely to impede flow to the cardiac microcirculation.
End-Organ Damage by Vascular Disease
The cerebral circulation is particularly vulnerable in hypertensive patients. Stroke is the major cause of death. Hypertensive vascular disease is usually a combination of hypertension and atherosclerosis, the latter accelerated by elevated blood pressure. Infarction of the brain is more common than subarachnoid bleeding. Bleeding results from rupture of microaneurysms of the small arteries or from trauma. The coronary vessels are next most vulnerable. Angina pectoris and MI are common results of accelerated atherosclerosis, necessitating concurrent treatment of the hypertension and atherosclerosis. Many patients reduce their high blood pressure and return to normal levels, only to have an MI or die from complications of atherosclerosis. The results of the increased rate of atherogenesis are also seen in other large vessels besides the coronary arteries. Thus, aneurysms of the aorta and renal arteries are common, leading to distal embolization, dissection, and possible rupture. Reducing blood pressure has a beneficial action on these lesions. Drugs, surgery, and interventions are now available to treat the clinical effects of both hypertension and atherosclerosis, although the goal should be prevention.
Unstable Plaque Formation
Plaques likely to rupture are called “unstable” and are the main cause of acute coronary syndromes. Plaques with a thin cap and a lipid core associated with inflammation and cap fatigue may be associated with rupture at the edges of the plaque. The plaques are rich in lipids and foam cells, found at the peripheral margins. On rupture, plaques extrude thrombogenic material, which then can trigger thrombosis on the plaque, in turn occluding the coronary artery and resulting in MI.
If the vessel only is almost occluded, a condition known as unstable angina may result. Risk factors such as cigarette smoking, hypertension, diabetes mellitus, and dyslipidemia contribute to endothelial injury, causing smooth muscle cell proliferation, inflammation, and deposition of lipid within the blood vessel wall. These events may lead to the development of unstable plaques. Other physical factors may alter plaque composition and make a plaque vulnerable to rupture; cold temperature (32°-34° C) may promote the crystallization of liquid cholesterol, and these sharp crystals may perforate the cap. Cytokines, growth factors, and oxidative stress are other factors in progression of atherosclerosis and instability.
Thrombosis related to these plaques may be caused by endothelial erosion or plaque disruption. In general, plaque erosion occurs over high-grade stenoses, whereas thrombosis related to plaque disruption is often seen in patients with few stenoses. In addition, multiple triggers of plaque disruption are probably related to increased tension, bleeding originating from the vasa vasorum, bending and twisting of the arteries, and increased systolic and pulse pressures.
Angiogenesis and Arteriogenesis
Unfortunately, the terms angiogenesis and arteriogenesis are often are confused and interpreted to have the same meaning. These processes are really two separate entities representing blood vessel changes in the heart.
Angiogenesis
Angiogenesis is essentially the budding of new blood vessels from sprouts of existing vessels in order to expand the capillary network (the microcirculation). Current investigations focus on creation of new blood vessels in areas of critical need, as in patients with large areas of infarcted myocardium surrounded by ischemic zones. The two major stimuli for new vessel growth are hypoxia, which activates hypoxia-inducible factor-1, and inflammation, which stimulates macrophages to secrete cytokines in MI patients, including basic fibroblast growth factor (bFGF) and transforming growth factor beta (TGF-β). As a result, newly formed blood vessels arise. This is particularly evident in animal models and is being tested in human trials (e.g., stem cell therapy in MI patients).
Arteriogenesis
In arteriogenesis, collateral vessels are recruited to increase blood flow to ischemic tissue. Coronary artery collateral vessels are thought to be stimulated and generated as a result of “demand ischemia” in areas of the myocardium. For example, the distal portion of a left anterior descending (LAD) coronary artery with high-grade stenosis or obstruction may receive blood from the right coronary artery (RCA) through septal collaterals or from the posterior descending coronary artery. Another potential source of collateralization is from the conus branch of the RCA, essentially one coronary artery supplying another artery, either by capillary-sized (microcirculation) collaterals or large muscular collaterals that are epicardial vessels (e.g., conus artery of RCA). This blood vessel is known to provide collateral blood flow to a severely stenotic proximal LAD artery.
Collaterals are not seen in patients without evidence of myocardial ischemia, possibly because no dormant collateral vessels are present. Also, in patients with balloon occlusion of an epicardial vessel and resultant myocardial ischemia, collateral flow through a previously dormant collateral vessel can be seen immediately. This indicates that these collateral vessels are dormant and visualized only when there is demand ischemia. Collateral vessels easily visualized at coronary angiography are categorized under arteriogenesis.
Overview of Myocardial Ischemia
In patients without myocardial ischemia, myocardial oxygen demand is balanced by myocardial oxygen supply. Determinants of myocardial oxygen supply are related to aortic diastolic pressure, left ventricular end-diastolic pressure (LVEDP), and coronary artery resistance. Determinants of myocardial oxygen demand are related to heart rate, systolic blood pressure (BP), left ventricular (LV) tension, and LVEDP. Myocardial ischemia related to balloon occlusion of a coronary artery involves a sequence of decreased coronary blood flow, as detected by perfusion scintigraphy; altered regional flow/demand ratio resulting in decreased coronary sinus oxygen saturation; LV diastolic relaxation abnormalities, detected by cardiac ultrasound; systolic contraction abnormalities, detected by cardiac ultrasound or angiography; increased LVEDP; ST-segment depression on electrocardiography; and finally angina pectoris or its equivalent symptoms (e.g., breathlessness).
At the cellular level, the current hypothesis is that myocardial ischemia increases the late sodium current, which increases the sodium content of the myocardial cells. The sodium/calcium exchanger then increases the calcium content of the cell. As a result, the LV stiffens, increasing LVEDP, which decreases endomyocardial blood flow, propagating the myocardial ischemia.
Myocardial ischemia and its usual manifestation angina pectoris result from an imbalance between myocardial oxygen supply and myocardial oxygen demand. Myocardial ischemia manifested by angina pectoris can be acute or chronic. In general, patients with an acute clinical presentation have a crescendo pattern of angina and are considered to be in an unstable or “acute coronary syndrome” state.
Although most patients with myocardial ischemia are symptomatic, 25% may be asymptomatic. However, most symptomatic patients have many episodes of asymptomatic myocardial ischemia. Laboratory manifestations of myocardial ischemia other than electrocardiographic (ECG) changes are abnormalities of perfusion, as indicated by nuclear studies, and transient regional wall motion abnormalities, usually detected by echocardiographic studies or other noninvasive imaging techniques (e.g., MRI). Clinical manifestations of myocardial ischemia are generally chest discomfort (angina pectoris), arrhythmias, and LV dysfunction in both systole and diastole.
Angina Pectoris
Angina pectoris (“strangling of the chest”) was first described as a serious symptom by Heberden in 1768, although he did not associate angina with coronary artery disease, as discovered later by Jenner. Angina is usually of short duration, associated with effort or emotion, and usually relieved in minutes by rest or sublingual nitroglycerin administration.
Angina pectoris varies somewhat from patient to patient, but the usual description of symptoms includes a feeling of a heavy weight, oppression, or a choking sensation under the middle of the chest and pain extending occasionally to the arms, especially the left, and almost never to the back and rarely to the neck and jaw. Many patients do not describe angina as a pain, but rather as a discomfort in the chest. It is produced, as a rule, by effort or emotion, especially in cold weather and particularly after meals or smoking. Angina rarely lasts more than 2 or 3 minutes and is relieved in a few minutes by sublingual nitroglycerin, also often used preventively, particularly if the patient knows a given situation will provoke cardiac pain. Angina must be differentiated particularly from gastrointestinal, musculoskeletal, and costochondritic origins of the discomfort.
Angina pectoris and the pain of acute MI are caused by the same disease process, but the prolonged pain in angina is associated with coronary artery occlusion and not the coronary artery stenosis of MI. The prognosis in the patient with angina pectoris varies greatly, but with medical management, many patients can continue to lead a normal life. Over months or years, their angina pectoris may diminish or disappear through natural development of collateral circulation to the ischemic zone of myocardium. However, angina is always an important symptom, and may be life threatening because of the potential for ventricular arrhythmias. Treatment of angina pectoris always includes medical therapy and often includes percutaneous coronary intervention (PCI, angioplasty/stent) or surgical bypass of a stenotic artery. Diet, blood pressure control, lipid management, nitrates, beta blockers, angiotensin-converting enzyme (ACE) inhibitors, aspirin or other antiplatelet agents, smoking cessation, and exercise are considered optimal therapy before and after a revascularization procedure.
Prevention of the underlying disease that causes angina pectoris is paramount. For those at risk, as shown by family history and high serum cholesterol, the avoidance of obesity, a diet low in animal fat, routine exercise (if no heart disease), and smoking cessation deserve first priority in any health promotion and heart disease prevention program and should start in childhood.
Detection of Myocardial Ischemia
The simplest and least expensive way to diagnose the presence or absence of myocardial ischemia and to quantitate functional impairment in patients with chronic stable angina is exercise stress testing with ECG monitoring. Exercise testing is typically performed using a treadmill or bicycle to provoke chest discomfort and obtain diagnostic evidence of myocardial ischemia. It is important to record 12 leads before, during, and after exercise testing in order to detect transient changes suggesting ischemia. Under resting conditions the heart rate is usually normal and the ST segment on the electrocardiogram usually isoelectric. Thus the 12-lead ECG may be normal even if a coronary artery has 70% stenosis, the usual percentage that results in myocardial ischemia when myocardial oxygen demand is increased. The ECG is normal because the myocardium is not ischemic under these resting conditions; myocardial oxygen supply is balanced by demand, and the patient does not complain of angina pectoris or equivalents such as breathlessness. The treadmill increases speed and incline progressively, thus stressing the patient, who needs to increase heart rate and blood pressure to maintain appropriate cardiac output to meet the demand. During bicycle exercise, resistance to pedaling is increased to accomplish the same increase in heart rate and blood pressure. Patients who develop myocardial ischemia show the classic ECG changes of ST-segment depression. In the patient with 70% stenosis, under conditions of increased myocardial oxygen demand, the balance of oxygen supply and increased demand will be upset, and myocardial ischemia will result. In some patients, myocardial ischemic changes on the ECG may not be accompanied by angina pectoris, termed silent myocardial ischemia. Despite the lack of symptoms, the prognostic importance of silent or asymptomatic myocardial ischemia is the same as symptomatic disease.
Chronic stable angina is often called stable ischemic heart disease. Myocardial ischemia results when the oxygen needs of the myocardium exceed the supply of oxygen derived from coronary blood flow. The oxygen needs of the myocardium are increased by aggravating or precipitating factors such as hypertension, tachycardia, heart failure, hypermetabolic state, and sympathomimetic drugs. Myocardial oxygen demand depends on heart rate, blood pressure, and LV size, thickness, and contraction. Other factors, such as severe anemia, may result in tachycardia and increased oxygen demand.
Degree of Flow-Limiting Stenoses
Patients with normal coronary arteries have no flow-limiting epicardial stenoses and remain asymptomatic under conditions of moderate oxygen demand, or even high oxygen demand in most cases. Patients with moderate narrowing caused by coronary atherosclerosis may also be asymptomatic or symptomatic (angina, myocardial ischemia) depending on the degree of flow-limiting stenosis to the myocardium. When flow is adequate under moderate myocardial oxygen demand, the ECG may be normal with isoelectric ST segments in all 12 leads. However, when there is high myocardial oxygen demand in this same patient with moderate coronary atherosclerosis, ECG changes in the appropriate leads (e.g., aVL, V2-V6) in patients with left-sided coronary disease demonstrate abnormal ST-segment shifts during transient ischemia and abnormal changes during prolonged ischemia at rest. In Plate 6-11, LDL-C refers to low-density cholesterol.
Thus a normal ECG correlates with widely patent coronary arteries providing adequate flow and no evidence of ischemia. Similarly, moderate coronary artery disease (CAD) under mild stress conditions shows no ECG changes. Under high oxygen demand in patients with moderate CAD, however, transient ST-segment shifts can be found. In addition, patients with severe narrowing often have symptoms and ECG changes under resting or near-resting conditions or during trivial exercise, particularly with severe high-grade proximal stenoses (e.g., left main or proximal LAD stenosis). These patients usually have severe proximal multivessel epicardial CAD.
If done to detect myocardial perfusion abnormalities, nuclear imaging studies are usually normal if the coronary arteries have mild or no stenosis. If there is moderate stenosis with high oxygen demand, myocardial perfusion may be mildly abnormal. The treatment and prognostic implications are related to the severity and extent of the perfusion defects. For severely abnormal myocardial perfusion in patients with high-grade stenoses, aggressive medical therapy is often combined with PCI (angioplasty with stenting) or bypass surgery.
To make appropriate decisions about prognosis and therapy in the patient with symptomatic CAD, knowledge of coronary pathology must be coupled with physiologic assessment of the lesions.
Left-Sided Heart Angiography
Left-sided heart catheterization and angiography includes injection of iodinated radiopaque contrast into the aorta, left ventricle, and coronary arteries to visualize the status of the aorta and its branches, presence or absence of aortic insufficiency, patency of conduit coronary bypasses, LV function and anatomy, and condition of the coronary circulation. Coronary angiography is a precise method of determining the extent, location, and severity (percent stenosis) of CAD. Multiple views of the coronary arteries are necessary to assess the significance of a specific lesion and the distribution of the right coronary, circumflex, and anterior descending arteries and the septal and diagonal distal circulation. The classic views of the right and left coronary arteries are the right anterior oblique (RAO) and left anterior oblique (LAO) projections. Multiple angulated views (caudal and cranial) are particularly important if PCI (angioplasty) is planned. One must remember that catheter-based coronary angiography is lumenography; only the vessel’s lumen is seen. The vascular wall must be visualized by intravascular ultrasound or optical coherence tomography; despite its limitations, this is a prerequisite for coronary artery surgery and in many patients allows determination of management: medical therapy (alone or plus angioplasty/stent) or bypass surgery.
Ventriculography is part of the evaluation of patients undergoing coronary angiography, to obtain the best possible assessment of ventricular function. I prefer to have ventriculography performed in the RAO and LAO views to assess the global function of the ventricle, particularly in patients with LV dysfunction.
Fractional Flow Reserve
In the catheterization laboratory, interventional cardiologists can determine the physiologic significance of a specific coronary artery stenosis by passing a small wire (0.014-inch pressure guidewire) with a pressure transducer at the tip, through the stenosis in question, which allows them to measure systolic blood pressure (BP) proximal and distal to the stenosis. The systolic BP distal to the stenosis is divided by the pressure proximal to the stenosis; the resulting fraction is the percentage of BP decrease across the stenosis. Measurements are made at baseline and after infusion of adenosine, which increases coronary blood flow. For example, if BP across the stenosis drops by 50%, the fractional flow reserve (FFR) will be 0.50. The formula is FFR = Pd/Pa, where Pd is pressure distal to the stenosis and Pa is pressure proximal to the stenosis. This technique is particularly useful when the visual estimate of the clinical significance of a specific stenosis by angiography is uncertain.
If FFR is combined with intravascular coronary ultrasound, the interventional cardiologist will be able to determine the physiologic significance of the stenosis as well as the nature of the plaque being investigated, i.e., stable or unstable vulnerable plaque. FFR could also be called “functional flow reserve” because it supports that measurements reflect the pathophysiologic significance of a stenosis. The technique obviously requires cardiac catheterization and a specially designed pressure guidewire in which the pressure is measured proximal to and distal to a given epicardial coronary artery stenosis. Based on clinical trials, a cut-off point of 0.75—0.80 has been used to indicate a significant pathophysiologically important coronary artery stenosis. In my view, this measurement is equivalent to our measurements of noninvasively measured ankle-brachial index (ABI), in which a difference in pressure between the arm and leg suggests pathologic decrease in leg perfusion. An advantage is that this technique can provide immediate information for angioplasty/stent being considered for an epicardial stenotic vessel. If FFR is less than 0.75, the stenosis appears to be causing downstream myocardial ischemia, and angioplasty/stent seems appropriate. The DEFER clinical trial used FFR to determine if PCI with stenting was necessary in patients with FFR greater than 0.75. In these patients, stenting of the coronary stenosis did not influence clinical outcome. In another study, guided by FFR, fewer stents were used in patients who had FFR in the normal range. After 1 year, death, MI, or repeat revascularization occurred less in the FFR group, and hospital stay was shorter with less procedural cost.
Chronic Angina Revascularization Procedures
Revascularization involves either percutaneous coronary intervention (PCI), as first performed by Gruenzig in 1978, or coronary artery bypass graft surgery (CABG).
Percutaneous Coronary Intervention for Chronic Angina
Stent Deployment
The PCI is performed through a guiding catheter introduced into the ostium of a coronary artery. A guidewire is advanced into the catheter and passed across the stenosis, and a balloon catheter is then placed at the stenosis. The balloon is inflated and the stenosis dilated. If a stent surrounds the balloon, as the balloon is inflated, the stent is deployed and forms a scaffold to maintain patency of the artery (see Plate 6-14). Stents can be bare metal (BMS) or drug eluting (DES). The balloon is deflated and removed, and the stent remains in place.
Rotational Atherectomy and Distal Protection Device
Other devices such as rotational atherectomy can be used before stenting, particularly in calcified stenoses (see Plate 6-15). A diamond-coated burr is used to fragment the plaque, widening the stenosis. Occasionally, distal protection devices are used to filter some of the detritus and atherosclerotic debris that migrate distally.
Surgery for Chronic Angina Disease
In 1968, Favaloro performed the first successful coronary artery bypass graft (CABG) at the Cleveland Clinic, where Sones had done the first selective coronary angiogram in 1958. Selective coronary angiography permitted for the first time a realistic appraisal of the patient suspected of having coronary disease because it established the presence or absence of disease, localized an existing myocardial-perfusion deficit, and assessed LV functional status. Postoperatively, Sones’ technique allowed assessment of the disease course as well as surgical success or failure. This procedure also led to percutaneous catheter-based myocardial revascularization procedures.
Before CABG was developed, several procedures were used, but none is still performed, including implantation of the internal thoracic artery directly into the myocardium (Vineberg procedure) and a Vineberg variant (Sewell procedure) in which the internal thoracic artery with a pedicle (including artery, vein, muscle, and fibrous tissue) was implanted into the myocardium.
Ventricular Aneurysmectomy
Ventricular aneurysm may occur in patients who sustain a large MI. Postinfarction aneurysm usually affects the anterior wall and the LV apex perfused by the left anterior descending branch of the left coronary artery. Aneurysm of the inferior left ventricle rarely occurs, although an infarction of sufficient magnitude to produce a ventricular aneurysm usually results in mitral incompetency secondary to papillary muscle dysfunction.
Ventricular aneurysmectomy is usually an elective procedure that is not undertaken until several months after MI, unless the patient has intractable heart failure, intractable ventricular arrhythmias, or severe mitral regurgitation not responsive to acute vasodilator therapy. Early intervention in these patients greatly increases the surgical risk.
Coronary Artery Bypass Grafts
The concept of direct coronary artery surgery uses the saphenous vein graft or an internal thoracic artery to bypass a proximally stenotic coronary artery. CABG requires target vessels either free of disease or minimally diseased. Vein graft conduits are “free grafts” and connect the ascending aorta to the coronary vessel. In contrast, internal thoracic artery conduits remain connected to the subclavian artery and connect to the coronary artery distal to the stenosis. CABG not only promotes perfusion of the distal microcirculation of the epicardial vessel, but also provides perfusion of other stenotic vessels by enhancing flow through collateral circulation.
Follow-up Studies
Angiograms months or years after CABG show patency in a high percentage of grafted vessels. However, not all saphenous vein grafts are as patent at 10 years as they were initially. The use of the internal thoracic artery as a conduit to bypass a proximal stenosis has proved to be the best CABG procedure to date. Often, bilateral internal thoracic arteries are used. Although rare, this type of conduit can stenose or thrombose, probably related to technical intraoperative problems.
Pathophysiology of Acute Coronary Syndromes
Patients with acute coronary syndrome (ACS) present in three different ways: unstable angina, non–ST-segment elevation myocardial infarction, and ST-segment elevation MI. Potential causes for the development of an ACS include extracardiac factors in the patients with severe coronary atherosclerosis, such as hypertension, tachycardia, and anemia. In 1858, Virchow proposed that injury to the inner wall of a blood vessel, possibly caused by fat, might lead to inflammation and secondary plaque formation, similar to current theory. ACS is also caused by plaque disruption resulting in transient platelet aggregation and release of vasoactive substances in diseased vessels, dynamic or intermittent coronary artery thrombosis, hemorrhagic dissection into an atheromatous plaque, and progression of coronary stenoses from plaque healing.
Acute coronary syndromes result from myocardial ischemia initiated by two primary factors: platelet and thrombus formation and resulting intense vasoconstriction from accumulation of local thromboxane A2 and serotonin, reduction of local concentrations of endothelium-derived releasing factor (EDRF), and inhibitors of platelet aggregation. These events are usually preceded by rupture or erosion of the lipid-laden plaque and involve the balance between thrombosis and thrombolysis. Patients with ACS may stabilize clinically, but this may not be associated with stabilization of the plaque. Identifying these patients requires observation for recurrent ischemia and assessment of troponin levels and possibly C-reactive protein (CRP) levels. Abnormal test results indicate increased risk for future cardiac events.
The acute coronary syndromes are not a homogeneous condition. Clinical variables evident in many patients include ECG changes, release of creatine kinase and troponins, CRP level, and differences in LV function, as well as pathology on coronary angiography. In patients with ACS, the TIMI risk score helps to predict the risk for death, MI, or recurrent ischemia at 14 days. The risks are age over 65, three risk factors for CAD, prior 50% coronary stenosis, ST-segment change on admission, angina twice in 24 hours, aspirin use within 7 days, and increased serum cardiac markers. The more risk factors present, the higher is the rate of composite endpoints in 14 days.
Myocardial Infarction—Changes in the Heart
Myocardial infarction can result in an extensive panorama of changes. Death of the cardiac muscle may occur in a single zone or as multiple foci scattered diffusely throughout the heart. The latter can be found after suboptimal perfusion of the heart using a pump oxygenator, in which all the minute necrotic foci are of the same age. More often, however, the multiple minute foci can be found as destroyed zones of varying ages. In the acute phases, these microinfarcts may be extremely difficult to see and may be recognizable only microscopically as areas showing variable myofibril destruction and replacement by collagen. Chronically, these small foci may be minute white patches salted throughout the myocardium.
A larger myocardial infarct shows a variety of changes that depend in part on the duration of the patient’s survival after the episode. The earliest changes are associated with the resulting paralysis of vessel walls, engorgement of the capillaries, and migration of polymorphonuclear leukocytes (PMNs). Some autolytic changes then occur, and the dead muscle fibers can be recognized as showing a loss of striations and an increase in eosinophilia, fragmentation, and the gradual disappearance of nuclei. The dead muscle becomes an irritant, and more PMNs appear. Finally, the subsequent activities of repair include macrophage infiltration and replacement fibrosis. If the infarct is minute, such a sequence is of relatively short duration, but if large, remnants of necrotic muscle may be found several years later. It appears that such dead tissue has been effectively sealed away from physiologic activities.
The infarct may be named according to the area of the heart involved. Most posterior infarcts affect areas of the septum as well, and many anterior infarcts involve anterior portions of the septal wall, termed anteroseptal and posteroseptal (see Plate 6-17). A lateral infarct is found, in a small percentage of the cases. The sequence of events—muscle death, removal, and repair—may or may not result in thinning of the myocardium. Infarcts may also be classified according to the thickness of the ventricular wall involved. If all layers are affected (transmural), the term ST-segment elevation myocardial infarction (STEMI) is used, most often associated with occlusion of the coronary artery. In about 90% of these lesions, occlusion is demonstrated, as contrasted with a lesser incidence in non–ST-segment MI. In NSTEMI the zone of infarction is usually limited to the endocardial surface of the ventricle. Mural thrombosis may be a more common accompaniment of either STEMI or NSTEMI because both infarcts involve the endocardium.
A major problem with coronary artery disease is the lack of many correlations between CAD and MI in a patient who has died. Although the appearance of a lesion in the anterior descending or the main left coronary artery is a common finding in sudden cardiac death and occasionally may be the only lesion discovered, the status of the underlying cardiac muscle is not known because morphologic MI cannot be demonstrated. In about 25% of MIs encountered at autopsy—either acute or chronic but, especially, chronic—no history can be elicited as to when such a disorder occurred. In hearts with acute MI, only about half show occlusion of the coronary artery, although the other half exhibit a significant to marked stenosis of the infarcted vessel. In addition, when the immediate cause of death was believed to be CAD, no recent acute MI is found (see Plate 6-18).
Exercise may be extremely valuable in all these MI patients. Studies indicate that although the incidence of MI is not decreased, its severity is less in those who have maintained cardiac fitness.
Acute damage to areas of the conduction system, such as hemorrhage or infarction, can be demonstrated in only some cases. However, other complications are common. The immediate results of the changes in the circulatory status and constituents of the blood apparently can lead to a deposition of thrombus at numerous sites, including the coronary arteries, with a further extension of the MI. Mural thrombi form on the endocardium (probably related to healing of infarcted tissue), in the atrium (much more common on the left than the right), and possibly at all sites where previous atheromas had existed. Therefore, cerebral anoxia can result not only from the decreased cardiac output, but also from superimposed thrombosis during the early stages. Thrombosis also occurs in the systemic veins.
Embolism is a natural sequela of thrombosis, and systemic embolization occurs in an unknown number of patients with thrombi in the left side of the heart. Cerebral embolism is the most severe and damaging, although saddle embolism of the distal aorta is also serious, as is embolization in an extremity. Smaller emboli may produce any focal embolus, including splenic and renal arteries. Less often, mesenteric embolism may occur, which can be a medical disaster if not recognized early. Unfortunately, mural thrombi persist for a long period, and systemic embolization may occur at various periods following apparent recovery from the acute infarction. Pulmonary embolism is also often related to venous stasis.
As with the hypertensive heart in failure, the hypoxic heart can dilate, with resultant valvular insufficiency and subsequent regurgitation.
Rupture of the heart (ventricular septal defect, papillary muscle, free wall) is another variable autopsy finding (see Plate 6-19). The incidence of cardiac rupture ranges from 1% to 10% in deaths from acute MI. The area of rupture often shows a large increase in PMNs and at a much later time than such cellular elements are usually encountered; ruptures of the heart usually occurs in the first week. The classic patient prone to rupture is hypertensive, is having a first infarction, and has no visible collaterals to the infarcted artery. In addition to free rupture of the heart into the pericardial cavity and the resulting cardiac tamponade, rupture of the interventricular septum occasionally occurs. Rarely, papillary muscles rupture. The posterior papillary muscle is more likely to rupture than the anterior muscle because it does not have dual blood supply.
As a chronic complication, anterior and apical aneurysm may be more prone to mural thrombi, and thus the patient is continually exposed to the risk of embolism (see Plate 6-20).
In addition to all the complications of embolism, cardiac aneurysm, sudden death, and rupture of the heart, there is the problem of reinfarction. Some hearts demonstrate not only the zone of a previous infarct, but also areas of new infarct. The new infarct may be adjacent to the previous one or can occur in a different area of other coronary arteries.
Manifestations of Myocardial Infarction
Pathologic and ECG Manifestations of Myocardial Infarction
In patients with acute MI, particularly with STEMI, pathologic changes in the heart may reveal necrosis from endocardium to the subepicardial myocardium. In the first 24 hours the ECG manifestations are variable, but usually the R wave is gone or almost gone, a Q wave is present, the ST segment is elevated, and the T wave may be inverted (see Plate 6-21). Several days later, pathology may reveal a complete infarction of the ventricular wall (transmural). The ECG continues to show Q waves, no R wave, and T-wave inversion. The ST segments may return to baseline in some cases. Several weeks or months later, the infarcted tissue may be replaced with fibrous scar, which occasionally results in a bulging ventricular aneurysm. If the aneurysm is obvious on ventriculography or cardiac ultrasound, the ST segment may be elevated. Other ECG changes usually persist (e.g., Q wave; inverted T waves, less than earlier seen), and the R wave may somewhat return.
In contrast, a patient with NSTEMI in the first 24 hours shows subendocardial necrosis, and the ECG often reveals persistent R wave, small Q wave, isoelectric ST segment, and T wave inversion. The infarcted myocardium is surrounded by a periinfarction ischemic zone. Days, weeks, or months later, the subendocardial necrosis heals, and subendocardial fibrosis replaces infarcted myocardium. This fibrosis is limited to the subendocardial area and does not extend transmurally to the epicardium. The ECG changes may or may not return to normal.
Ischemic Injury and Infarction: ECG Localization
In the myocardial region of an occluded coronary artery, ECG changes caused by ischemia, injury, and infarction can be identified (see Plate 6-22 ). ECG abnormalities are highly suggestive but not diagnostic of acute MI. In the patient who has large Q waves, ST-segment elevation, and T-wave inversion in several ECG leads, the diagnosis of an evolving MI is highly suspect; however, not all MI patients have these classic changes. Some patients present with severe chest pain and only T-wave inversion or a significant ST-segment depression, as well as elevation of cardiac markers (NSTEMI). In contrast, other patients have had obvious Q waves develop on the ECG and prominent ST-segment elevation (STEMI). All MIs begin in the subendocardial area and extend transmurally. The development of Q waves and ST-segment elevation probably relates to the transmural degree of wall involvement; the more involvement toward the epicardium, the more likely the appearance of Q waves and ST-segment elevation.
The site of the infarction can be localized in general by the ECG abnormalities. Thus, anterior and anterolateral MIs generally display ECG abnormalities of the precordial leads as well as leads I and aVL. However, all variations of these ECGs can occur, and there may be changes from V1 to V6 (including lead I) and aVL, or changes may occur only in leads I and aVL and the early precordial leads. In contrast, the diaphragmatic or inferior MI generally shows ST elevation in leads 2, 3, and aVF.
Cardiac Markers and Diagnosis of Acute Myocardial Infarction
Cardiac markers are essential for the diagnosis of acute myocardial infarction but do not appear immediately in the bloodstream if an occlusion occurs. Creatine kinase-(CK), CK isoenzymes (e.g., CK-MB), and troponin T are measurably abnormal about 4 hours after occlusion. Troponin T is the most sensitive and specific marker for acute myocardial infarction at the present time and probably is the only cardiac marker necessary to make the diagnosis of acute myocardial infarction; however, renal failure can result in the elevation of all cardiac markers in the absence of an acute myocardial infarction.
Clinically, if the cardiac markers are normal at admission to the emergency department (ED), when the patient is having acute cardiac pain and ST-segment elevation, the MI likely has occurred recently, within 3 or 4 hours. However, if the cardiac marker is elevated, the occlusion and STEMI likely occurred more than 4 hours before ED evaluation. The onset and duration of increase in CK enzymes and troponin T after a coronary occlusion vary, but in the patient with STEMI the occlusion usually occurred recently if initial serum values are normal. If a revascularization procedure is performed early in the course of acute MI, the cardiac markers my increase much more than if a revascularization procedure had not been performed, most likely from washout of the markers from the perfused myocardium.
Recanalization of Occluded Coronary Artery in Acute Myocardial Infarction
Rapid opening of the infarcted artery is generally accepted as critical to increase patient survival, preferably using PCI. However, thrombolytic therapy remains a viable option in hospitals when PCI capability is not immediately available. Percutaneous recanalization of an occluded coronary artery by balloon angioplasty and stent placement in patients with STEMI is the standard of practice. The appropriate management of acute STEMI currently is door-to-balloon (“D2B”) time—from ED arrival to passage of a guidewire into the infarcted artery. D2B time is easily and accurately measured and documented. D2B time less than 90 minutes is an acceptable goal. The rationale is that short D2B time results in less myocardial necrosis. A favorable outcome correlates well with minimal necrosis and ejection fraction (EF) greater than 40%. An unfavorable outcome correlates with a large amount of necrosis and EF less than 40%.2
Timing of Vessel Occlusion
In canine studies, MI size versus duration of coronary occlusion was measured with the circumflex coronary artery ligated. Thus the actual time of onset of coronary occlusion was identified and the time course of myocardial necrosis measured. Myocardial cells started to die about 20 minutes after onset of ischemia, with cell death complete by 6 hours.
Theoretically in the human, if mortality reduction is plotted against extent of salvage of myocardium from 0 to 3 hours, a steep reduction in percent mortality occurs, and by 3 hours the myocardial salvage is greatly reduced. After 3 hours the percent mortality reduction is similar to that found at 3 hours. Unfortunately, the exact time of vessel occlusion is not known in human MI.
Intra-Aortic Balloon Counterpulsation
Counterpulsation is the only technique that will lower aortic systolic pressure and increase aortic diastolic pressure. To accomplish intra-aortic balloon counterpulsation (IABCP), a catheter with a cylindrical polyethylene balloon (~40 mL in adult device) is introduced into the aorta, deflated, and extended to just below the left subclavian artery and above the renal arteries, so as not to obstruct either vessel. When visualized on chest radiography or fluoroscopy, the tip of the catheter is placed at the level of the bifurcation of the right and left main bronchi, again so as not to occlude subclavian and renal arteries. Balloon expansion with helium occurs actively during diastole, and collapse of the balloon occurs actively during systole. Intra-aortic pressure is measured at the catheter tip.
Timing of balloon expansion and collapse is controlled by either ECG or pressure waveform during the cardiac cycle. When the balloon is expanded in diastole, aortic diastolic pressure increases, as does coronary perfusion pressure, which theoretically increases myocardial blood flow. When the balloon is collapsed during systole, LV afterload reduction occurs, probably a suction effect that unloads the left ventricle and increases cardiac output. When counterpulsation is working correctly, the elevated diastolic aortic pressure likely is interpreted as high blood pressure, and the baroreceptor reflexes tend to lower systolic pressure, thus decreasing myocardial oxygen demand.
The clinical indications for IABCP are cardiogenic shock of any etiology, acute mitral regurgitation that does not respond to nitroprusside, acute ventricular septal defect, and refractory acute coronary syndromes (ischemia or arrhythmias). Less clear indications relate to preprocedural use in patients considered “high risk” for a revascularization procedure such as PCI or CABG (e.g., high-grade left main coronary artery stenosis). IABCP should not be used in patients with aortic insufficiency; the increase in diastolic pressure will increase the degree of aortic insufficiency. IABCP cannot be used in patients whose aorta cannot be accessed because of severe peripheral vascular disease. Complications with IABCP are fortunately rare and include leg ischemia, aortic disruption, renal artery obstruction when the balloon is not properly placed, and bleeding.
Rheumatic Fever in Sydenham’s Chorea
Rheumatic Fever
Although uncommon in the United States and other developed countries, rheumatic fever is common in developing countries and can affect people of all ages, except in the first years of life. The febrile onset is accompanied by severe but temporary arthritis and often by carditis affecting all three cardiac layers. The etiologic relationship of rheumatic fever to β-hemolytic streptococci is based on the following evidence:
The frequency with which rheumatic fever follows a streptococcal infection varies from 3% in epidemics to about 0.3% in sporadic infections. This discrepancy is largely caused by the greater severity of epidemic types of infection, as revealed by clinical features and the antibody titers induced. When streptococcal infections of comparable severity are studied, the incidence of subsequent rheumatic fever, even in sporadic cases, is also about 3%.
The lesions of rheumatic fever do not result from direct invasion by streptococci. When adequate precautions are taken against contamination, organisms cannot be isolated from the joints or the heart. The events relating the primary infection to these lesions are still unclear, but circulating toxins released from the organisms are unlikely to be responsible, because then the clinical manifestations of rheumatic fever would coincide with the sore throat (as do the toxic symptoms of diphtheria). Furthermore, none of the many known streptococcal products produces comparable lesions in animals. The relationship between streptococcal infection and rheumatic fever is therefore most widely regarded as immunologic, with the lesions resulting from an antigen-antibody reaction in the affected tissues. This concept was supported by observations that lesions superficially resembling those of rheumatic carditis could be produced in rabbits by the injection of massive doses of foreign serum.
A comparable massive exposure to antigen is unlikely to occur in humans. Any immunologic reaction underlying the pathogenesis of rheumatic fever is more likely to be more specific, directly involving one or more of the antigens native to the affected tissues. Many strains of β-hemolytic streptococci contain an antigen in their cell walls that cross-reacts with an antigenic component of mammalian hearts, including the human. Furthermore, animals injected with such strains of streptococci produce antibodies that react with cardiac antigens. Similar antibodies have been found in a high proportion of patients with active rheumatic fever.
However, despite the presence of firmly bound gamma globulin (presumably antibody) in the heart of many patients with rheumatic fever, circulating antibody correlates poorly with clinical severity. Cardiac damage in experimentally immunized animals is unconvincing; repeated infection in rabbits by different β-hemolytic streptococci most closely resembles human rheumatic carditis. Although the mechanism is still unclear, immunologic cross-reactivity with myocardial and other antigens is presently the most likely cause.
The clinical features of an acute attack may range from the trivial to the fulminating—from easily overlooked pallor and fatigue to an exquisitely painful arthritis with high fever, rash, and carditis sufficiently severe to lead to congestive heart failure. The temperature chart is characterized by a nonremittent type of fever with a pulse rate raised disproportionately. The arthritis is migratory, rarely lasting in any one joint for more than 1 or 2 days. Swelling is slight and accompanied by some flushing of the overlying skin. Large joints are more frequently affected than small joints, although these can become involved as well, and the clinical picture may then resemble rheumatoid arthritis.
Rheumatic Fever in Sydenham’s Chorea: Noncardiac Manifestations
Despite the many features common to rheumatic fever and rheumatoid arthritis, notably polyarthritis and subcutaneous nodules, these are distinct nosologic entities. If differentiation is difficult in the early stages, the difference in clinical evolution rapidly distinguishes rheumatic fever from arthritis, and even in the early stages, histology of the synovium or biopsied nodule can be helpful. The lesion in rheumatic fever is capsular rather than synovial, with areas of fibrinoid necrosis accompanied by focal infiltrations of histiocytes and lymphocytes.
The joint lesions, even when severe, are entirely reversible, and permanent damage does not result. This contrasts with the cardiac lesions, which can occasionally progress to mitral stenosis and its sequelae, even when initial damage is mild. Skin rashes are uncommon but can be characteristic, especially erythema annulare, a fleeting, ring-shaped, usually flat eruption typically found on the trunk. Each lesion spreads centrifugally, leaving a slight staining in the center. Another typical feature of rheumatic fever, seen especially in severe cases, is subcutaneous nodules, mainly confined to the tissues over bony prominences, where friction combined with pressure is apparently responsible for nodule development. The nodules are most conspicuous after 2 to 3 weeks, although their impending development is often preceded by a more diffuse, boggy thickening. Histologically, a typical nodule shows an area of fibrinoid composed of parallel or interlacing bands sparsely infiltrated with histiocytes and fibroblasts. The adjacent tissue is edematous and contains groups of small vessels surrounded by similar cells. Fibrous tissue is inconspicuous.
Permanent cardiac damage correlates closely with persistent or recurrent activity of rheumatic disease, so the detection of such activity is important. The most helpful test is the erythrocyte sedimentation rate (ESR), which is almost never normal in the presence of active disease, except in patients with congestive heart failure. The converse, however—a persistently raised ESR—does not necessarily imply continued activity, especially in girls. Therefore, estimations of C-reactive protein may more accurately reflect the state of the disease activity. The greatest danger to the patient with a history of rheumatic fever is developing a new infection with β-hemolytic streptococci, because the risk of relapse is much greater than after an initial attack. The success of chemoprophylaxis has confirmed both the importance of such reinfection in the development of a relapse and the role of streptococci in rheumatic fever pathogenesis.
Environmental and genetic factors have been studied widely in an attempt to account for the considerable geographic variation in the incidence of rheumatic disease. The most impressive data are provided by Mills’ map, showing the progressive decrease in mortality from rheumatic fever as one passes from the northern to the southern latitudes of the United States. Evidence of genetic factors besides female predominance is less precise, but the increased incidence of nonsecretors of the blood group substances in patients with rheumatic fever indicates modest genetic influence. The dominant factor is environmental and related to the incidence and virulence of the local streptococci.
Sydenham’s Chorea
The rheumatic nature of Sydenham’s chorea (St. Vitus dance), first recognized just over 100 years ago, is now established by evidence similar to that for rheumatic fever, as a poststreptococcal state. This is confirmed by the frequent subsequent development of rheumatic heart disease, in the absence of any clinical evidence of rheumatic fever. The outstanding clinical features are ataxia, incoordination, and weakness, accompanied by spontaneous movements that can be most clearly depicted by the photographic record of a penlight held in the patient’s hand.
Rheumatic Heart Disease: Acute Pericarditis and Myocarditis
Rheumatic heart disease is a complication of recurrent upper-respiratory infection with group A β-hemolytic streptococci. Streptococcal products circulating from this source through the body causes a reaction in connective tissues called rheumatic inflammation. Predisposed sites include the heart, skin, and synovial membranes. In the latter two areas, inflammation usually heals by resolution, leaving no residual effects, but in the heart these recurrent respiratory infections may lead to severe deformities, usually of the valves.
Chronic rheumatic heart disease requires recurrent infections. Each attack of rheumatic inflammation goes through an active stage followed by healing. In the joints, active rheumatic involvement is characterized by migratory arthritis. In the skin, transitory subcutaneous nodules may appear. In the heart, each of the major anatomic components—the pericardium, myocardium, endocardium, and particularly the valves—may be involved.
Acute Rheumatic Pericarditis
Acute rheumatic pericarditis is characterized by variable exudation of serum and fibrin into the pericardial cavity. Large effusions of fluid may result in radiographic features of uniform cardiac enlargement and physical signs of pericardial effusion. Impairment of the inflow of blood to the heart, with resultant increased systemic venous pressure, may be evident through distention of the cervical veins. The fibrinous element of acute rheumatic pericarditis is manifested by particles of fibrin floating in the associated effusion and by the concentration of a fibrinous deposit on the visceral (epicardial) and parietal layers. The shaggy fibrin, evident when the two layers of the pericardium are separated, has given rise to the name “bread-and-butter heart” of rheumatic pericarditis. This gross feature is not specific, however, since it applies to any condition in which fibrinous pericarditis occurs. Histologically, during the active stage of rheumatic pericarditis, fibrin is deposited on the pericardial surfaces. Beneath this, capillaries and fibroblasts are mobilized and gradually enter the fibrin as granulation tissue. The pericardium shows edema and mild leukocytic infiltration. Specific Aschoff bodies, which characterize acute rheumatic myocarditis, are not usually seen in the pericardium.
Acute Rheumatic Myocarditis
The specific histologic lesion of acute rheumatic carditis is usually restricted to the myocardium and takes the form of the Aschoff body. Aschoff bodies are reactive nodules within the connective tissue and therefore are predominantly found around blood vessels of the myocardium and in other bundles of connective tissue that separate myocardial fascicles. The primary lesion appears to be an alteration of collagen, which shows a coagulation-like change with eosinophilia, a process called fibrinoid necrosis.
[/not-level-membership-for-cardiovascular-category]
