Congenital Heart Disease
Physical Examination

Although many forms are not seen in adult patients, cardiologists often do see simple clues to the diagnosis of certain forms of congenital heart disease (CHD), usually acyanotic or cyanotic and postoperative. Any child with or suspected with CHD should be seen by a pediatric cardiologist at an institution with interventional pediatric cardiologists and cardiac surgeons. Adult patients with CHD should be seen and advised by a pediatric cardiologist or adult cardiologist (preferably both) at a surgical center with experts in congenital heart surgery and percutaneous procedures. Many adult patients present with arrhythmias, heart failure, or failure of the original childhood surgery. Occasionally, older patients or patients with anomalous coronary artery disease present with ischemic heart disease symptoms.
Although the history generally does not provide major clues to the diagnosis, the family history of CHD raises awareness that the offspring of these patients may also have a congenital abnormality. Diagnostic clues in the adolescent or adult patient are based on simple physical examination and electrocardiographic (ECG) and chest radiographic findings characteristic of CHD (see Plate 5-1).
Anomalies of the Great Systemic Veins
Anomalies can involve the large systemic venous trunks because of the complex embryogenesis and tremendous variability of the venous system in general. Abnormal channels almost always empty into other systemic veins and rarely cause functional changes disturbing to the patient, usually discovered incidentally at postmortem examination or during cardiovascular diagnostic or surgical procedures. Venous trunk anomalies may occur as isolated malformations but more often are associated with other cardiovascular anomalies. The presence of an anomaly, if unsuspected, may lead to troublesome or even dangerous situations when total cardiopulmonary bypass techniques are employed.
Left Superior Vena Cava
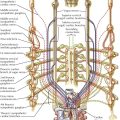
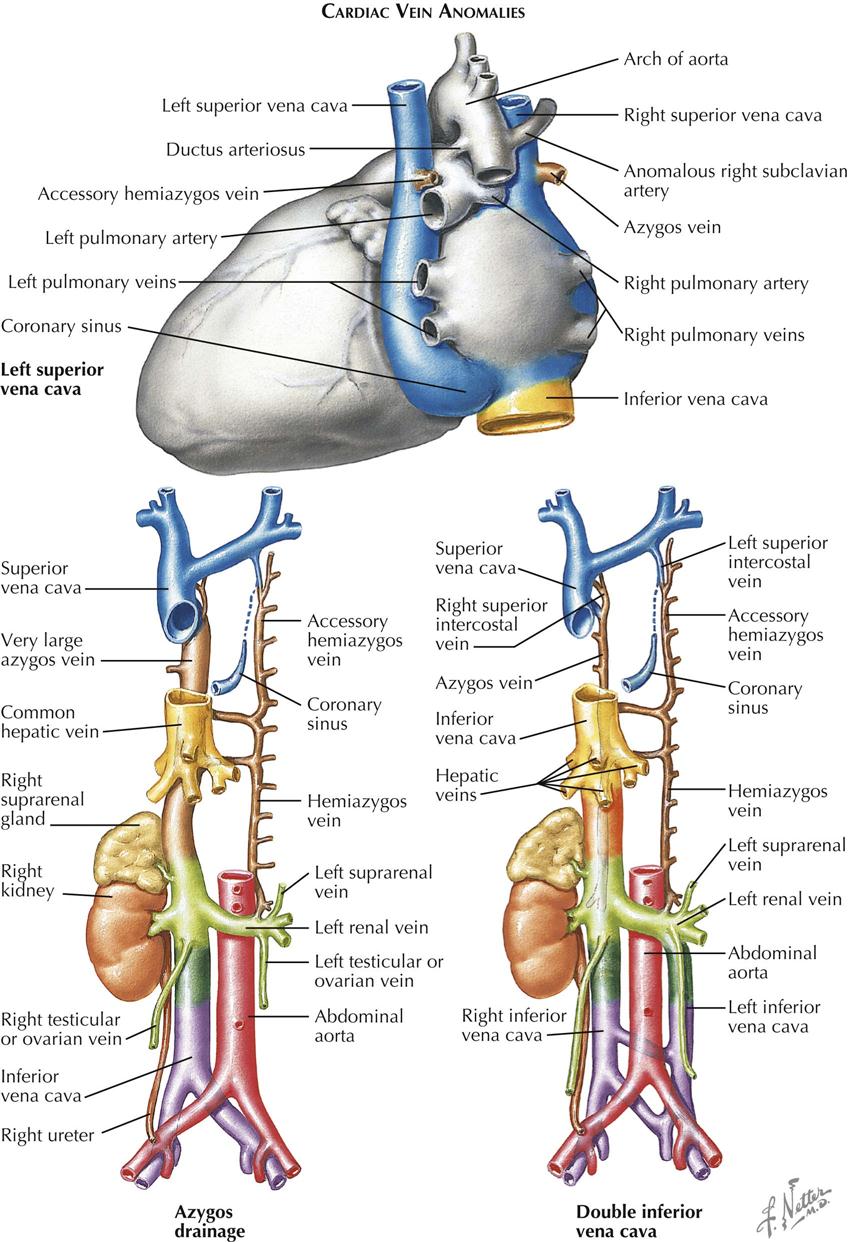
By far the most common clinically significant anomaly of the great systemic veins is persistence of the left superior vena cava (SVC). This vein, after being formed by the confluence of the left jugular and subclavian veins, descends into the chest parallel to the right superior vena cava and anterior to the left-lung hilus, usually entering the greatly dilated coronary sinus along a course normally occupied by the ligament and vein of Marshall. This topographic position is expected because embryologically, a persistent left SVC represents retention of the left anterior and common cardinal veins and the left sinus horn. Anatomically, the hemiazygos vein resembles the normal right-side azygos vein and may approximate it in size (see Plate 5-2).
The right SVC is usually present as well but may be absent. The two venae cavae may be equal in size, or one (generally the left) may be smaller than its counterpart. The left innominate vein, if present, is smaller than normal or may be more or less plexiform.
The coronary sinus ostium (coronary os) is very large because of the increased blood flow through it, detected easily by cardiac ultrasound. Occasionally, a defect is present in the wall between the sinus and left atrium (unroofed coronary sinus). Generally, such a defect results in a left-to-right shunt; that is, left atrial blood enters the coronary sinus and is carried to the right atrium. Hemodynamically, therefore, the anomaly resembles an atrial septal defect. If the defect is extremely large, particularly if the coronary os is small or atretic, the left SVC is said to “enter the left atrium.”
Patients with persistent left SVC present a typical clinical picture consisting of moderate central cyanosis without other symptoms. There is no murmur, and the heart is normal in size. The electrocardiogram (ECG) generally shows signs of left ventricular hypertrophy. Similar but more pronounced findings have been described in the rare cases of isolated drainage of the inferior vena cava into the left atrium.
Azygos Drainage of Inferior Vena Cava
Absence of the hepatic segment of the inferior vena cava (IVC) is an uncommon anomaly in which the prehepatic portion of the IVC drains into the right atrium by way of an enormously enlarged azygos vein. The hepatic veins empty into the right atrium by way of a short common stem that normally forms the most proximal part of the IVC (see Plate 5-2). Although azygos drainage of the IVC occurs rarely as an isolated lesion, usually it is associated with other serious cardiac anomalies (e.g., asplenia or polysplenia syndrome).
Double Inferior Vena Cava
Other systemic venous anomalies, including double inferior vena cava (see Plate 5-2), generally involve the IVC bed and are of more importance to general surgeons and urologists than cardiologists and cardiac surgeons. Patients with significant cardiac anomalies associated with various types of partial inversion of the thoracic or abdominal viscera are particularly prone to harbor anomalies of the great systemic venous trunks. Because such anomalies may cause difficulties at surgery, their presence or absence should be clearly established as part of the diagnostic workup, most easily through angiocardiography, computed tomography (CT), or magnetic resonance imaging (MRI).
Anomalous Pulmonary Venous Connection
In patients with anomalous pulmonary venous connection (APVC), all or some of the pulmonary veins fail to communicate with the left atrium, instead discharging blood into major systemic veins or directly into the right atrium. This discussion only considers the isolated forms of APVC. When these occur with other cardiac malformations, the clinical and hemodynamic features are usually modified or are chiefly determined by the complicating defect.
In partial anomalous pulmonary venous connection, one or more pulmonary veins empty into the proximal SVC close to the right atrium or into the sinus portion or the right atrium itself. The involved veins almost always drain all or part of the right lung; the others empty normally into the left atrium. An atrial septal defect (ASD) is generally present, especially the sinus venosus type. The clinical picture closely resembles that seen in other forms of ASD and is not discussed here.
Total Anomalous Pulmonary Venous Connection
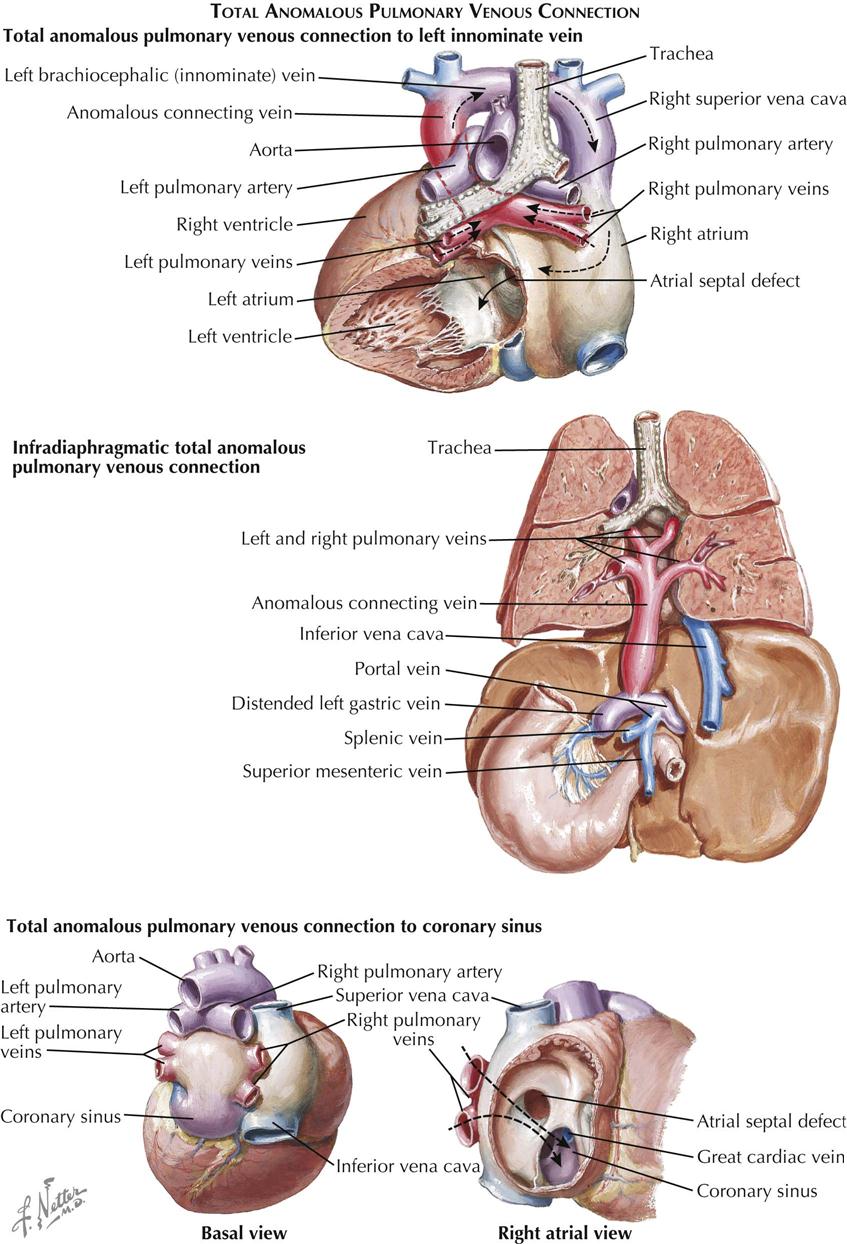
In total anomalous pulmonary venous connection (TAPVC), all the pulmonary venous blood enters the systemic venous system or the right atrium. An ASD or patent foramen ovale is always present. Types of TAPVC are distinguished by how the pulmonary venous blood enters the systemic circuit (see Plate 5-3). Embryologically, the intrapulmonary veins are derived from the venous plexus around the foregut and anastomose freely with the systemic veins early in the embryo. After the embryonic main pulmonary vein has appeared as an outgrowth of the primitive left atrium and established connections with the pulmonary venous plexus, the systemic and pulmonary venous anastomotic channels typically are obliterated. If the embryonic pulmonary vein does not develop at all or is obliterated secondarily, some of the anastomotic channels are retained, resulting in TAPVC.
TAPVC to Left Superior Vena Cava
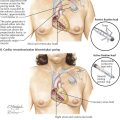
By far the most common form of TAPVC is that to a persistent left superior vena cava, illustrated as seen from behind in Plate 5-3. The right pulmonary veins converge to form a single vessel, which runs behind the small left atrium to join the left pulmonary veins. From the junction of the pulmonary veins, a large single vessel representing a persistence of the distal left SVC carries the pulmonary venous blood by way of a dilated left brachiocephalic (innominate) vein and the right SVC to the right atrium. An ASD, or more frequently a widely patent foramen ovale, allows part of the right atrial blood to enter the left atrium. Tremendous right atrial and right ventricular enlargement develops early and rapidly.
Symptoms of persistent left SVC generally appear shortly after birth, initially rapid respirations followed by dyspnea, feeding difficulties, failure to thrive, and frequent respiratory infections. As a rule, the patient has no obvious cyanosis, at least at first, reflecting the mixing of a large amount of oxygenated pulmonary venous blood with a much smaller amount of systemic venous blood. Cyanosis becomes more pronounced if bronchopneumonia or congestive heart failure is present. Failure almost always appears within the first 6 months of life, and the great majority of these infants die during their first year. For unknown reasons, a small number of patients improve, a few of whom may reach adulthood.
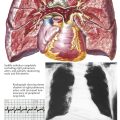
Cyanosis and clubbing of the digits are common only in older children and adults with TAPVC. The heart is enlarged. Generally there is no thrill, but a lower left parasternal “heave” is usually present. A systolic murmur of mild to moderate intensity is present at the left upper sternal border or sometimes lower. The pulmonic second sound is usually loud and often split. A diastolic tricuspid flow murmur may be present along the right lower sternal border or over the xiphoid process.
The chest radiograph features of persistent left SVC in older children and adults are characteristic. The dilated left and right superior venae cavae cause a rounded shadow in the upper mediastinum. Together with the rounded and enlarged heart shadow, a typical figure-eight or “snowman” appearance is created. The pulmonary vascularity is greatly increased. The ECG shows right-axis deviation and severe right atrial and right ventricular hypertrophy. At cardiac catheterization, the oxygen content of the right SVC blood is found to be very high, indicating a massive left-to-right (L → R) supracardiac shunt, and the blood oxygen content is almost uniform in all cardiac chambers.
The diagnosis of TAPVC to the left SVC can be confirmed easily by selective injection of a contrast medium into the pulmonary trunk. After passage of the contrast through the lungs, the anomalous veins opacify quite satisfactorily. Computed tomographic angiography (CTA) and cardiac MRI can also define the anatomic pathology of persistent left SVC.
TAPVC to Coronary Sinus
The pulmonary veins join to form a very short, wide, common vessel that empties into the hugely dilated coronary sinus in TAPVC to the coronary sinus (see Plate 5-3). The clinical picture and ECG findings are similar to those just described for left SVC. The radiographic appearance is different, in that the upper mediastinum is not widened. The right atrium may be huge. Cardiomegaly and plethora of the lung fields are seen. Cardiac catheterization fails to show the high oxygen content of the SVC; otherwise, radiograph findings are similar to those in TAPVC to the left SVC. Angiocardiography is much less helpful, and the anomalous connection to the coronary sinus may be difficult to demonstrate with certainty. CT and MRI help define the anatomic pathology.
Other types of TAPVC (e.g., to right atrium or to several different sites) are infrequently seen as isolated malformations, but also may be seen in association with other severe cardiac defects.
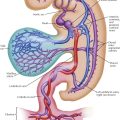
Infradiaphragmatic Type
In an unusual form of TAPVC, generally occurring as an isolated anomaly, the pulmonary veins drain into the portal venous system. This is generally referred to as infradiaphragmatic TAPVC; the other TAPVCs are classified as supradiaphragmatic. With infradiaphragmatic TAPVC the pulmonary veins join to form a single long vessel that descends in front of the esophagus and courses with it through the esophageal hiatus, to enter the proximal portal venous system, generally the left gastric vein. Usually, there is a stenotic area in the preesophageal vein just before it enters the portal venous bed. Along with the need for pulmonary venous blood to traverse the hepatic capillary bed before entering the right atrium by the hepatic veins, this stenosis causes severe pulmonary venous hypertension and is responsible for the characteristic clinical picture and laboratory findings, which are quite different from those seen in the various supradiaphragmatic TAPVCs. Plate 5-3 shows the infradiaphragmatic anomaly as seen from the back.
Severe symptoms appear soon after birth, and almost all infants with infradiaphragmatic TAPVC die within a few days or weeks. The symptoms include clear persistent cyanosis, marked dyspnea, and serious feeding difficulties. Cardiac failure becomes evident very early and is almost impossible to treat successfully. These infants are obviously very sick but show no abnormal cardiac findings. The heart is not enlarged, and a murmur, if present, is faint. The ECG is normal or near-normal. The chest radiograph is characteristic but not pathognomonic, since it is also seen in other anomalies in which pulmonary venous obstruction occurs. There is evidence of severe pulmonary venous hypertension: The hilar markings are pronounced and fuzzy, and the lungs show a reticulated appearance.
Surgical Treatment
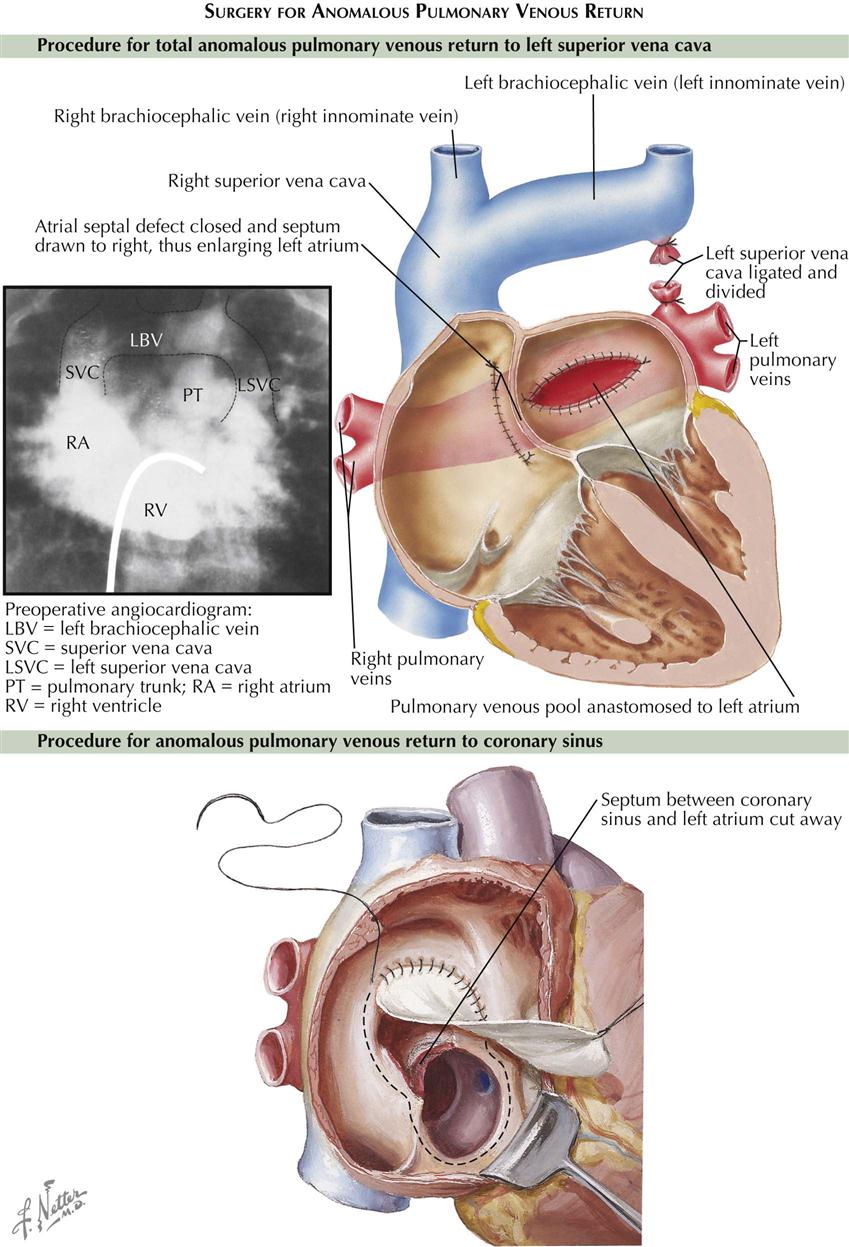
The treatment of TAPVC is surgical, to redirect pulmonary vein flow entirely to the left atrium (see Plate 5-4). In most types of total anomalous pulmonary venous return, the pulmonary veins return to a common confluence behind the left atrium. The common pulmonary vein confluence is connected to the back of the left atrium, resulting in a normal connection of pulmonary veins to left atrium. All other pulmonary vessels to the supracardiac or infracardiac areas are tied off. Any coexisting ASD is closed. Older children and adults generally have good surgical results. The repair results in normal circulation; the pulmonary veins return as normal to the left atrium, without abnormal connections or septal defects.
Anomalies of the Atria
Juxtaposition of the Atrial Appendages
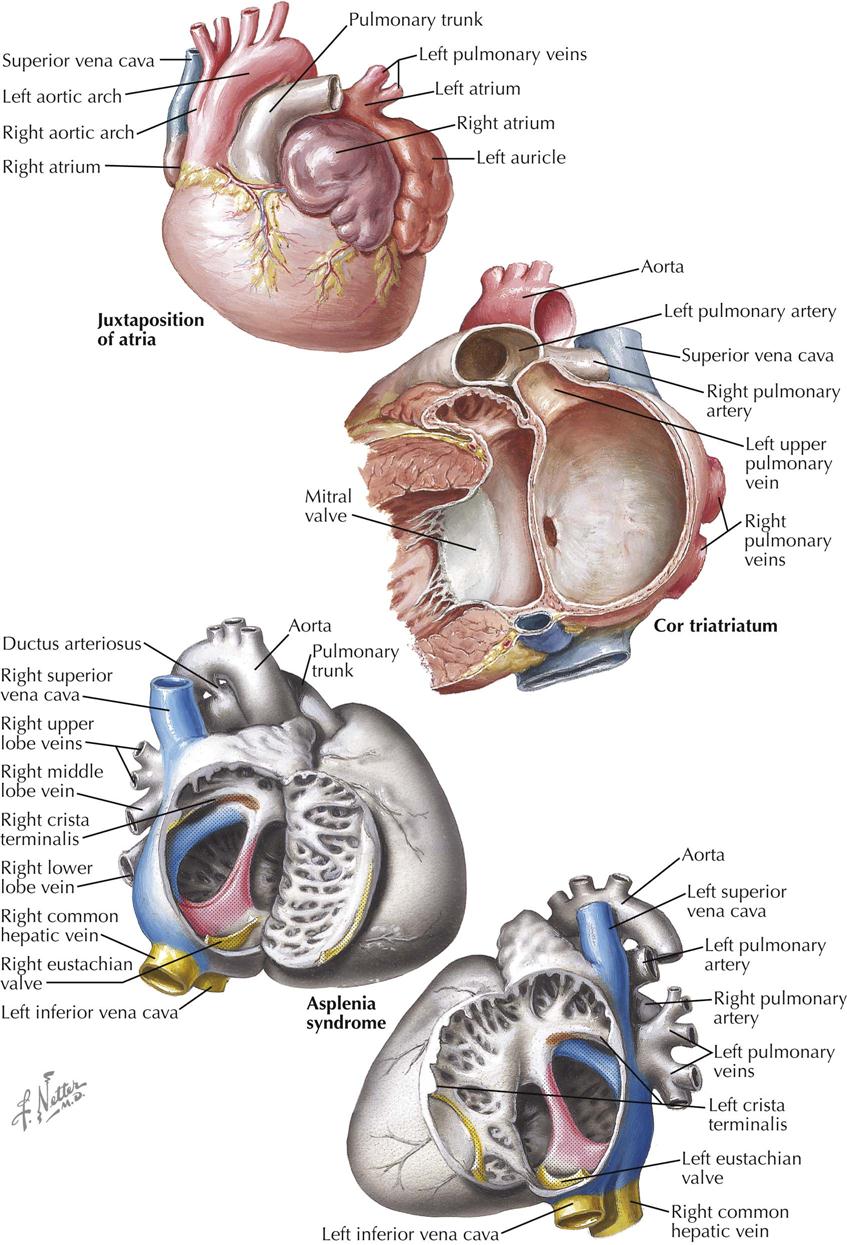
In juxtaposition of the atrial appendages (auricles), the main bodies of the atria are normally located, but there is levoposition of the right atrial appendage. Instead of being to the right of the arterial trunks, the right atrial appendage crosses behind them to appear on their left, interposing itself between the great arteries and the left atrial appendage. Juxtaposition of the atrial appendages has no functional significance because it causes no hemodynamic disturbance itself. Its presence, however, always indicates the coexistence of other major cardiac anomalies. Transposition of the great vessels and a ventricular septal defect are invariably present, and atresia of the tricuspid valve is common. Plate 5-5 also depicts a double aortic arch.
Cor Triatriatum
In the rare cor triatriatum, a fibromuscular septum divides the left atrium into a posterosuperior part receiving the pulmonary veins and an anteroinferior part giving access to the mitral valve and left atrial appendage (see Plate 5-5). Cor triatriatum is probably caused by incomplete incorporation of the embryonic common pulmonary vein into the left atrium. The original pulmonary venous ostium is represented by an opening of variable size. Rarely, the septum is imperforate, and the distal pulmonary venous compartment drains through a defect into the right atrium or an anomalous vessel into the systemic venous system. Usually the fossa or foramen ovale is located between the anteroinferior compartment and right atrium.
The severity of symptoms depends on the size of the opening between the two compartments of the left atrium. Respiratory difficulties and dyspnea may be marked, and cardiac failure develops early. If the os is very small, death occurs within the first year of life; if it is larger, symptoms appear later and closely resemble those seen in mitral stenosis, i.e., chronic cough, dyspnea, fatigability, chest pain, and hemoptysis. Cyanosis may be present, and there is marked cardiomegaly. A mild or moderate systolic murmur is usually heard, but a diastolic murmur is seldom present. The ECG usually suggests right ventricular hypertrophy because the pulmonary pressure is elevated. Cor triatriatum is easily diagnosed with transthoracic cardiac ultrasound and other imaging modalities. Surgical repair is relatively simple; the anomalous membrane is excised.
Asplenia Syndrome
Congenital absence of the spleen rarely occurs alone. Other visceral anomalies are present in most patients, about 60% of whom have the typical asplenia syndrome (see Plate 5-5). The most important feature of asplenia syndrome is the tendency for normally asymmetric organs (e.g., liver, lungs) to develop more or less symmetrically. The stomach may be located on either side or rarely in the midline. Both lungs are usually trilobed and resemble a normal right lung. The heart is generally severely malformed. A single or common ventricle is usually present and an endocardial cushion defect of the complete type often found. Transposition of the great vessels is the rule, usually associated with pulmonary stenosis. The atrial septum is reduced to a peculiar triangular band of muscle that crosses the common atrioventricular orifice. In typical cases, both the right atrium and the left atrium morphologically resemble a normal right atrium (isomerism of atria), meaning that both sinus horns have been incorporated into their corresponding atria. A coronary sinus is therefore absent. TAPVC is typically present. The great systemic veins also tend to develop symmetrically, at times with a bilateral SVC and a large vein entering on each side of the atrial floor, representing a bilateral persistence of the proximal vitelline veins. One of these drains a lobe of the liver (common hepatic vein), and the other drains the opposite lobe and the remainder of the IVC bed. The site of the viscera is impossible to determine (situs ambiguus, or heterotaxy syndrome).
The diagnosis of asplenia syndrome should be suspected in any infant with CHD associated with some form of partial visceral heterotaxy, particularly if cyanosis is present. Howell-Jolly and Heinz bodies are typically present in the peripheral blood smear. The prognosis is poor.
Defects of the Atrial Septum
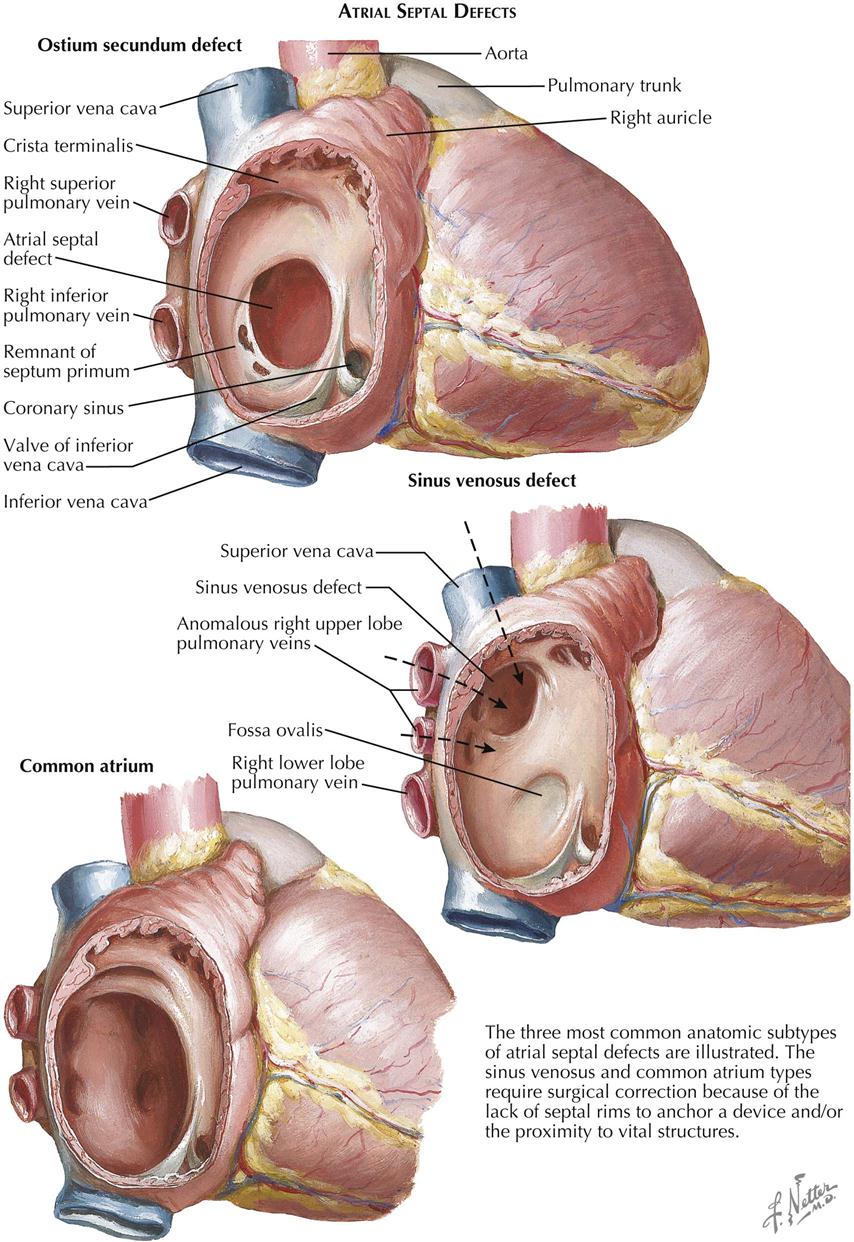
The atrial septum normally consists of two overlapping, closely adjacent components. Each forms an incomplete partition. The right-side component, corresponding to the embryonic septum secundum, is muscular and firm and has a posteroinferior oval-shaped opening, the foramen ovale. The left-side component, derived from the embryonic septum primum, is fibrous and thin and has a somewhat round opening anterosuperiorly, the ostium secundum. Together, the two components act as a one-way flap valve, allowing the flow of blood from right to left (normal before birth) but not from left to right. After birth, with the establishment of pulmonary circulation, the increased amount of blood entering the left atrium elevates the pressure in that chamber, thereby closing the flap valve. In most cases, this functional closure is eventually followed by anatomic closure; that is, the two components of the septum fuse. In the minority of cases where fusion fails, an increase in the right atrial pressure due to congenital cardiac anomalies, or any other condition that elevates right ventricular and right atrial pressure, causes the right atrial blood to flow again into the left atrium. Such a probe-patent foramen ovale, however, should not be considered a form of atrial septal defect; it causes no hemodynamic abnormalities by itself. In ASD there is an abnormal opening in the atrial septum allowing blood to flow either way; a predominantly left-to-right shunt usually exists. With associated anomalies or other conditions tending to increase right atrial pressure, the shunt is always from right to left, as in tricuspid valve atresia, or an initially left-to-right shunt reverses, as occurs after pulmonary vascular changes with pulmonary hypertension.
Ostium Secundum Defect
Of the two main types of ASD, the secundum type is more common and is one of the most frequently seen congenital cardiac anomalies (see Plate 5-6). The normal resorptive process that leads to the formation of the ostium secundum in the embryo is exaggerated, and most of the septum primum disappears. Most secundum ASDs are large, and the resulting left-to-right shunt is generally substantial, causing a several-fold increase in pulmonary blood flow. Both the right atrium and the right ventricle dilate and hypertrophy, and the pulmonary arteries enlarge considerably. Even though pulmonary venous return and therefore blood flow in the left atrium are increased, the left atrium does not enlarge because resistance to emptying into the left ventricle is higher than to the right ventricle, and thus it can readily “bleed off” through the defect into the more compliant right atrium. Systemic blood flow is generally at a low-normal rate or occasionally diminished.
The clinical features of ostium secundum ASD are not remarkable, considering the size of the defect and the magnitude of the shunt (see Plate 5-7). Only rarely are infants with ASD symptomatic; in fact, this anomaly is so well tolerated that disabling symptoms usually do not occur until adulthood, when it is the most common congenital cardiac defect. In children and young adults, the only symptoms are mild fatigability and dyspnea on exertion. Many patients are not even aware of the significance of ASDs, recognizing the defects only in retrospect after it has been corrected surgically and they have more energy and breathe more easily. Growth and development are generally normal. The heart is only slightly or moderately enlarged, and a thrill is extremely uncommon in isolated ASD. A left lower parasternal “heave” is often present, but a precordial bulge is seen only in patients with marked cardiomegaly. The murmur heard in ASD is not loud but rather systolic, medium pitched, and of the ejection type, best heard at the base to the left of the sternum. The ASD murmur is caused not by the left-to-right shunt itself but by the increased amount of blood passing through the otherwise-normal pulmonary valve. A similar mechanism is thought to cause the faint, short diastolic murmur heard in the tricuspid valve area (tricuspid flow murmur). Characteristically, the second sound at the upper left sternal border is split, and the splitting is fixed; unlike the variable splitting heard in normal children, the interval between the aortic (A2) and pulmonic (P2) components of S2 remains constant through all phases of respiration; P2 is often louder than A2. An ejection click is rare in children but may be present in adults, indicating the presence of pulmonary hypertension.
The classic chest radiograph features of secundum ASD are mild to moderate cardiomegaly, prominent right-sided heart border caused by right atrial enlargement, evidence of right ventricular (RV) enlargement, prominent pulmonary artery segment at left upper heart border caused by dilatation of the pulmonary trunk, and marked hypervascularity of the lung fields. On fluoroscopy, distinct hilar pulsations can easily be seen, called the “hilar dance.” The left atrium is never enlarged, and therefore the esophagus is not displaced posteriorly.
The ECG features are usually unmistakable. Right-axis deviation is the rule, although the axis may be normal or rarely even oriented to the left. Prominent, peaked P waves may be seen in leads II and aVF and in the right precordial leads. Most cases show an rSr′ or an rSR′ pattern over the right precordium, indicating mild to moderate RV enlargement. An rR′, an Rs, and particularly a qR pattern, not typically seen in children, indicate more severe RV hypertrophy, as seen with the development of pulmonary vascular changes and hypertension.
At cardiac catheterization, it is usually easy to enter the left atrium through the secundum defect, particularly when this is carried out from the femoral venous circulation. A distinct increase in oxygen content of the right atrium and an early opacification of the right atrium, on selective left atrial angiocardiography, demonstrate the presence of an ASD. RV and pulmonary artery pressures are normal or only slightly elevated in most children and young adults. Pulmonary hypertension may be present occasionally in early infancy or late in the disease course. A slight (10-15 mm Hg) pressure gradient across the pulmonary artery valve is common and does not, as a rule, indicate organic pulmonary valve stenosis, because it disappears after surgical closure of the defect.
Generally the clinical, chest radiograph, eECG, and cardiac ultrasound findings are so characteristic that many cardiologists do not hesitate to refer ASD patients to a surgeon, without catheterization or angiocardiographic studies. Medical treatment is often not required, but symptoms. (e.g., arrhythmias) should be treated as in any other cardiac condition. In children, cardiac failure rarely occurs, except in infants with very large defects. Bacterial endocarditis, the bane of many types of CHD, is extremely rare in uncomplicated ASD.
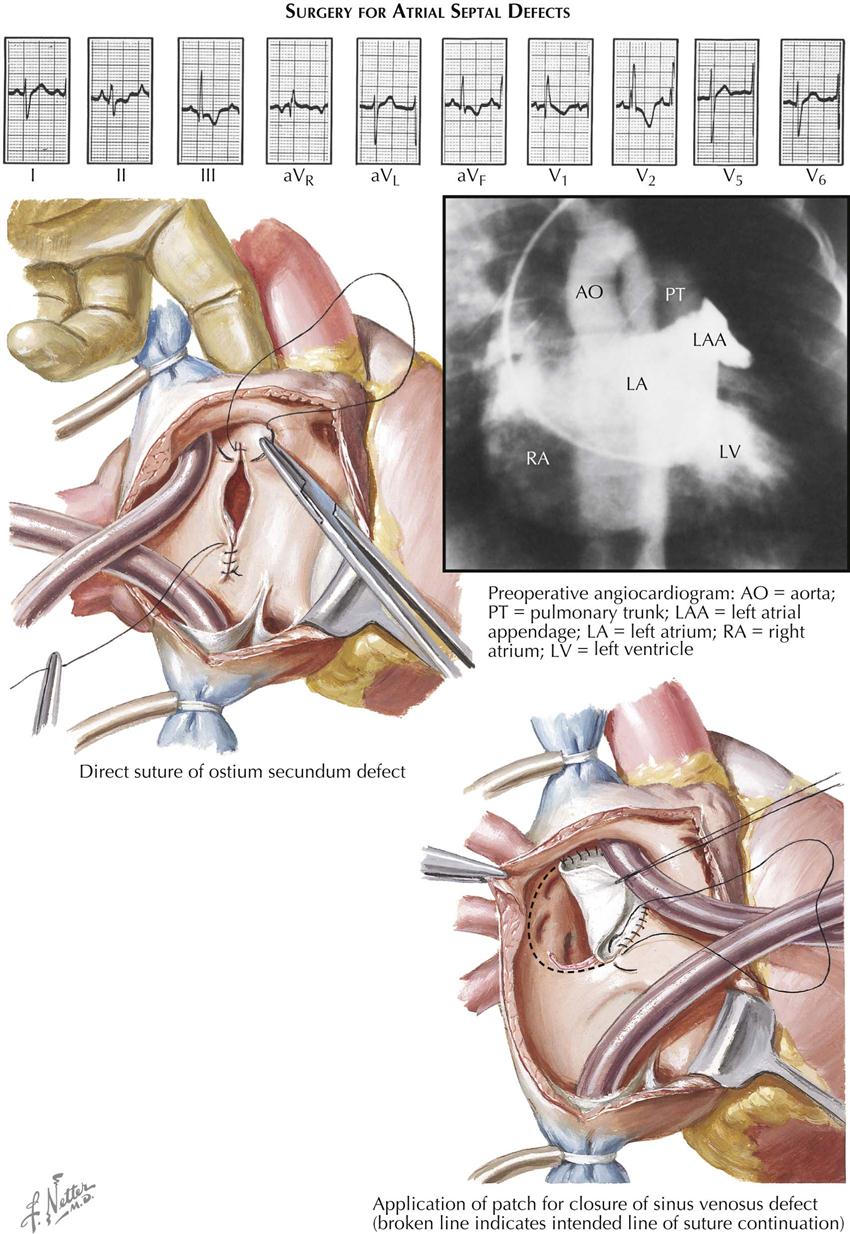
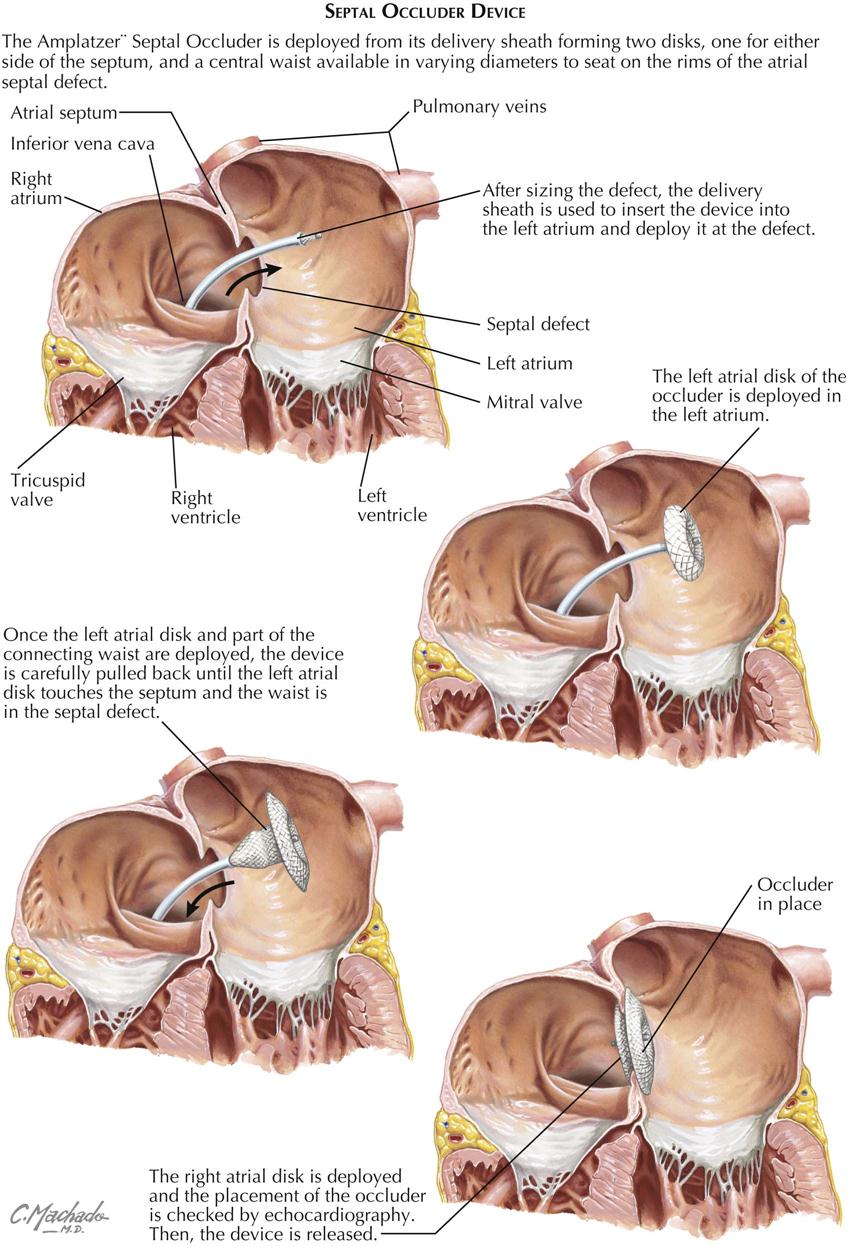
Surgical treatment ensures complete cure and should always be advised because surgery is easily done, employing cardiopulmonary bypass, and carries minimal risk. The ASD can usually be closed by direct suture. Because the defect is so well tolerated, surgery can safely be postponed until the child is 8 to 10 years or older. Percutaneous approaches using a septal occluder device (e.g., Amplatzer) have now become the transcatheter procedures of choice in patients, replacing the need for cardiopulmonary bypass (see Plate 5-8).
Common Atrium
Extremely large secundum defects, involving practically all the septum and referred to as common atrium, are seldom seen. The symptoms tend to be more pronounced, and slight arterial desaturation may be present because of easy mixing of blood at the atrial level. A prosthesis, consisting of a free pericardial graft, is usually necessary to close the defect (see Plate 5-6).
Sinus Venosus Defect
In the sinus venosus type of ASD, the region of the fossa ovalis is normal, the defect being located high in the septum at the ostium of the superior vena cava, which tends to straddle the defect (see Plate 5-6). Partial anomalous pulmonary venous return is almost always present. Such anomalous veins usually drain the right upper lobe and the middle lobe. The embryology of this much rarer kind of atrial septal defect is not clear. The clinical picture and the radiographic and electrocardiographic findings are similar to those described above.
Surgical correction of this defect may require the use of a pericardial patch to reroute the anomalous venous return to the left atrium and simultaneously close the ASD without compromising the lumen of the superior vena cava or the pulmonary veins.
“Ostium Primum” Defect
A third variety of anomalous interatrial communication is the “ostium primum” defect. Although some of its clinical features resemble other types of ASD, the ostium primum ASD differs significantly in other ways, and it is not truly a defect of the “atrial septum proper.” The primum ASD is caused by a developmental anomaly of the embryonic atrioventricular endocardial cushions (see Plates 5-9 and 5-10).
Endocardial Cushion Defects
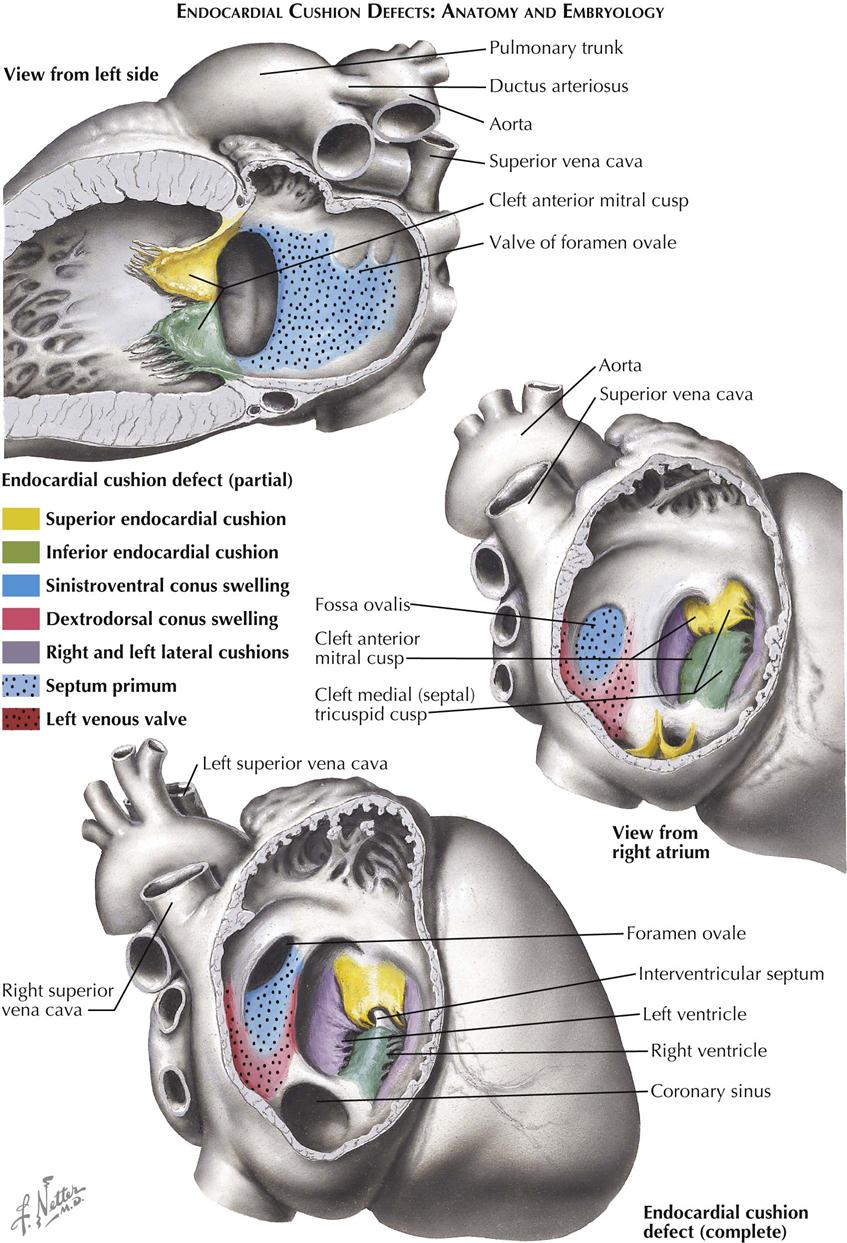
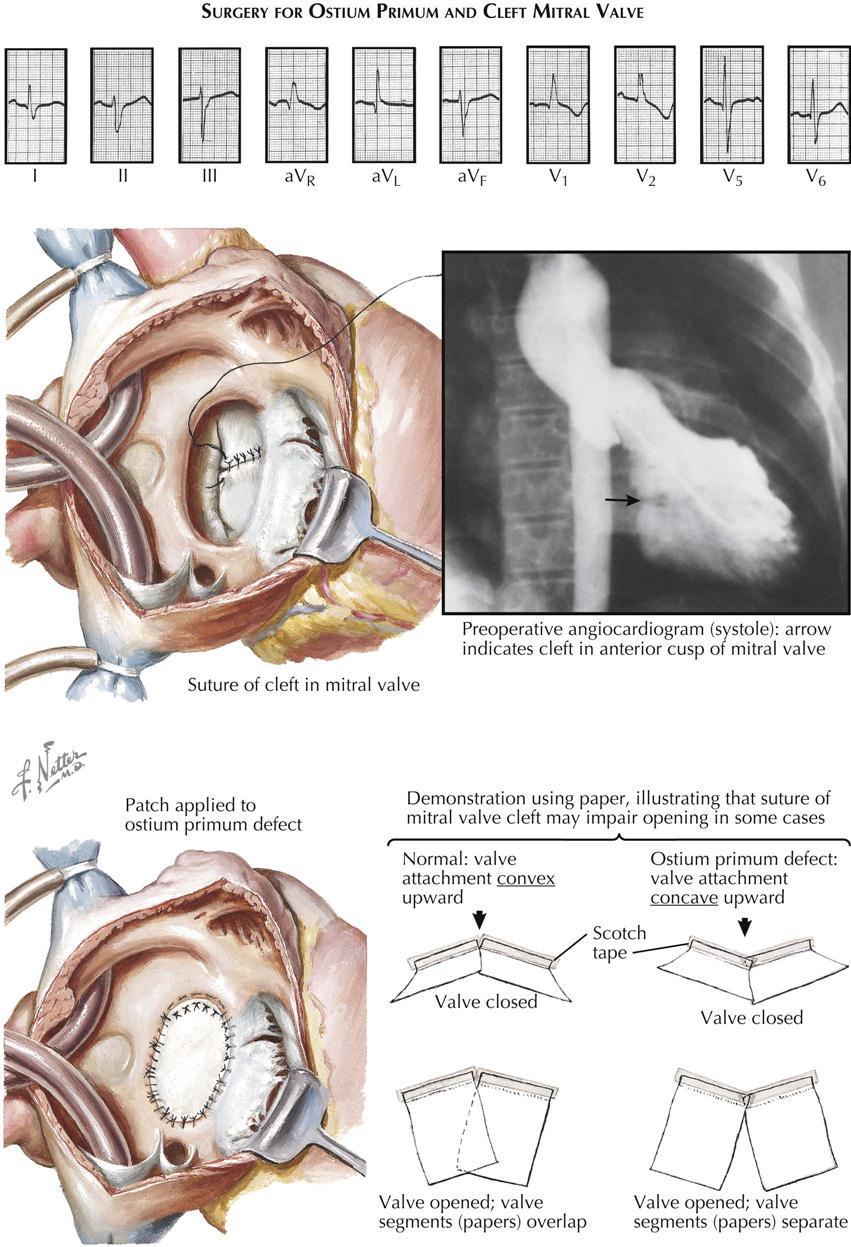
The group of anomalies known as the endocardial cushion defects (ECDs) is of interest to not only the cardiologist but the embryologist, pathologist, and surgeon as well. All ECD types are primarily caused by a developmental defect of the atrioventricular endocardial cushions. Normally, the endocardial cushions fuse with each other and bend to form an arc, the convexity of which is toward the atrial side. The atrial septum fuses with the apex of the arc, thus dividing it into two approximately equal parts. The right half contributes to the ventricular septum, the atrioventricular septum, and the medial or septal cusp of the tricuspid valve. The left half of the fused cushions forms the aortic or anterior cusp of the mitral valve.
In ECD the cushions partly fuse or do not fuse, and the arc is usually not formed (see Plate 5-9). This results in the following pathologic features characteristic of ECDs, shared by all types to varying degree:
2. The interventricular septum has a peculiar, scooped-out appearance.
3. The left ventricular outflow area is narrower and longer than normal.
4. The superior-inferior diameter of the ventricles is increased at the base.
If fusion of the cushions fails completely, the atrioventricular ostia form a large, single ostium (complete type of endocardial-cushion defect, also called persistent common atrioventricular canal), and there is a large, central septal defect that allows free communication between all four chambers. The common atrioventricular valve consists of the normal left mural (posterior) mitral valve cusp, the anterior and posterior tricuspid valve cusps, and two large cusps that cross the defect and have developed from the unfused endocardial cushions. Either cusp or both these cusps may be attached to the top of the ventricular septum by short chordae tendineae. The specimen illustrated in Plate 5-9 also has a persistent left superior vena cava.
If the cushions fuse only centrally, there is a division of the atrioventricular canal into right and left atrioventricular ostia, but the mitral valve (and often the septal cusp of the tricuspid valve) is cleft (partial ECD). Several types of ECD are distinguished, mainly depending on whether there is an interventricular or interatrial communication. The partial form, with only an interatrial communication, is known as the “ostium primum” type of ASD, as previously discussed. Again, the communication does not really correspond to the embryonic ostium primum, its position being similar to that of the atrioventricular septum of the normal heart. It must be emphasized that the atrial septum in ECDs typically is normally developed and complete, although associated ASDs do occur. The cleft mitral valve is usually incompetent. Even in cases of complete ECD, the valve may be competent.
The clinical manifestations of the ostium primum type of ECD largely resemble those seen in uncomplicated ASD. Symptoms tend to appear earlier in life, however, and growth retardation, fatigability, dyspnea, and respiratory infections are often more pronounced. Pulmonary vascular changes, resulting in right ventricular and pulmonary artery hypertension, are more common and are likely to occur earlier. A thrill is not uncommon, and auscultation again finds the systolic murmur at the left upper sternal border and the fixed splitting of S2, as in ASD. In addition, however, in just over half the cases, a high-pitched, blowing, systolic murmur of mitral insufficiency is present at or within the apex and transmitted to the axilla.
On chest radiography the heart tends to be somewhat larger than in ASDs. The heart may assume a configuration of left ventricular enlargement, with the apex turned down and out in the presence of significant mitral insufficiency. Even with mitral insufficiency, however, there is no left atrial enlargement unless the interatrial communication is small or absent. Other chest radiograph features are similar to those seen in primum ASD.
The ECG shows a left deviation of the QRS axis in the frontal plane, usually between 0 and −60 degrees, but sometimes more to the left. In the complete type of ECD, the QRS axis may be located in the right upper quadrant. The precordial leads are similar to those seen in ASD, but the evidence for right ventricular enlargement tends to be more pronounced, and the left precordial leads may show a pattern of left ventricular hypertrophy resulting from mitral incompetence. The left-axis deviation seen in ECD apparently is not related to possible left ventricular hypertrophy and seems to be caused by an abnormal anatomic position of the conduction system. Conduction system abnormalities such as heart block can occur.
Cardiac ultrasound can confirm the diagnosis of an ostium primum ASD and easily demonstrates mitral and tricuspid regurgitation, if present. Cardiac MRI also can define the anatomic pathophysiology. Cardiac-catheterization findings are similar to those in ASD and usually are not helpful in differentiating the two entities. Angiocardiography, on the other hand, is an extremely valuable tool because a selective left ventricular angiogram shows a configuration not observed in any other cardiac anomaly. The scooped-out ventricular septum and the long, narrow left ventricular outflow area are readily apparent during diastole, whereas during systole the two halves of the cleft mitral valve cusp are seen to bulge into the left atrium, with a notch indicating the position of the cleft. Mitral insufficiency, if present, is also readily demonstrated.
The complete type of ECD usually causes severe problems early in infancy, including repeated respiratory infections, feeding difficulties, growth retardation or serious failure to thrive, dyspnea, and congestive heart failure. Most of these children die within the first 2 years of life. Cyanosis is rare unless there is an associated obstruction of the right ventricular outflow tract, respiratory infection, or heart failure. Cardiomegaly develops rapidly after birth. In general, the larger the ventricular component, the sicker is the child; if this component is small, the clinical manifestations resemble those of the partial ostium primum type. A well-documented association exists between ECDs, particularly the complete type, and Down syndrome, which is seen in 35% to 40% of patients with a complete ECD.
The treatment of ECDs consists of open surgical correction of the malformation, always employing a cardiopulmonary bypass. Although technically much more difficult than closure of a simple ASD, the procedure now carries an acceptably low mortality rate if the anomaly is the partial type. The interatrial communication is accurately closed by employing a prosthesis of appropriate size. Direct suture should not be done in most cases, since it may cause distortion of the left atrioventricular ostium and thereby aggravate mitral insufficiency. Traditionally, the cleft in the anterior cusp of the mitral valve has been sutured, to create a more or less normal cusp and reduce insufficiency when present or prevent its development. Although suture of the cleft in cases with marked mitral incompetence seems justified, the wisdom of carrying out such a procedure in patients with a competent valve is highly debatable. In fact, suture of a competent cleft may well be contraindicated, because it will interfere with the ability of the cusp to open freely and completely and thus produce mitral stenosis (see Plate 5-10).
Correction of the complete forms of endocardial cushion defect is technically more difficult and, in some cases, impossible. In addition, children with ECD are generally smaller and more disabled.
Anomalies of Tricuspid Valve
Of the congenital tricuspid valve anomalies, only two—tricuspid valve atresia and Ebstein’s anomaly—are clinically significant. Tricuspid regurgitation and stenosis occurring as isolated lesions are extremely rare. Some forms of septal defects, such as endocardial cushion defects or ventricular septal defects, may involve the tricuspid valve’s medial cusp, rendering this cusp insufficient or allowing for a direct shunt from the left ventricle to the right atrium. Tricuspid valve stenosis usually accompanies pulmonary atresia or severe stenosis when the ventricular septum is intact. Actually, the tricuspid valve in these patients, although small and often with thickened cusps, is normally formed, and the stenosis is a secondary hypoplasia.
Tricuspid Atresia
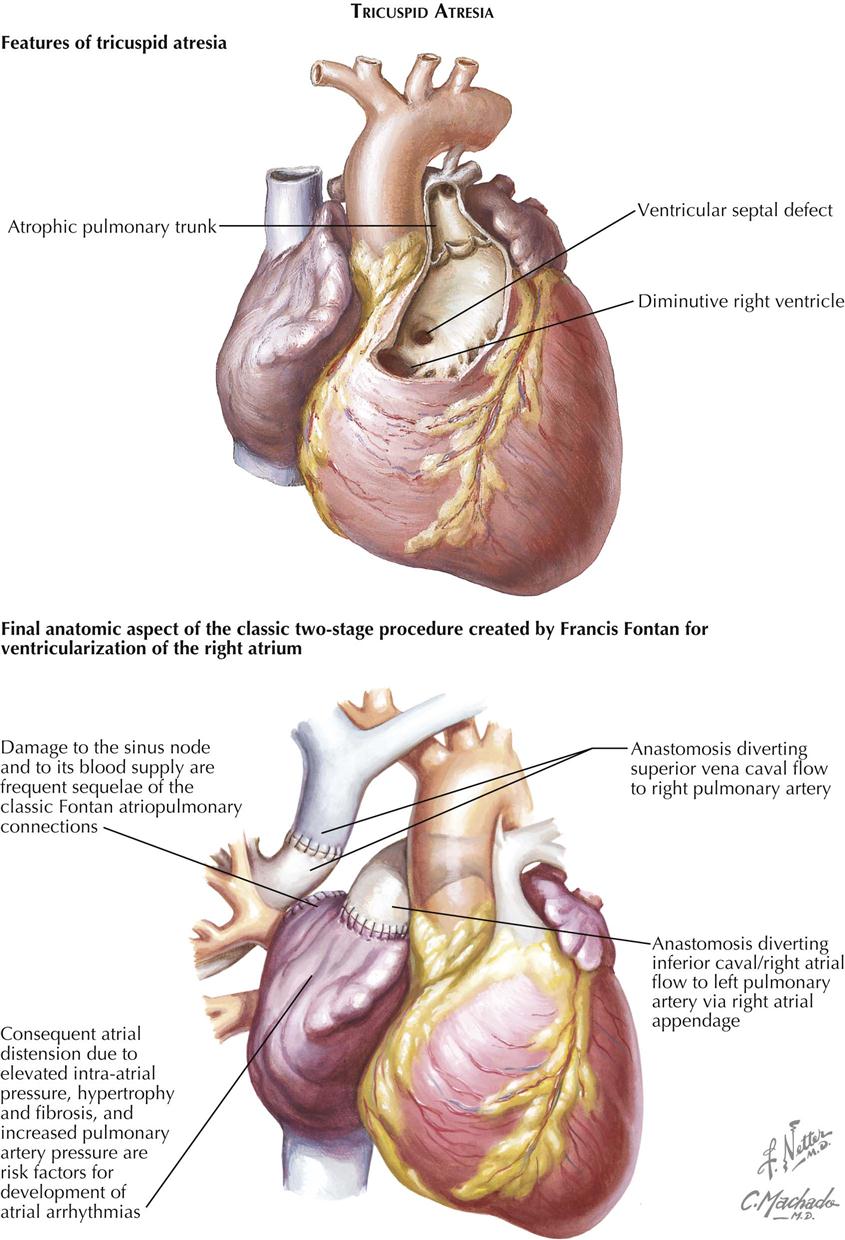
Although uncommon, tricuspid atresia is seen often enough to have considerable clinical importance. Next to transposition of the great arteries, tricuspid atresia is the most common cause of pronounced cyanosis in the neonatal period, and the degree of cyanosis is usually more marked than in cases of transposition. Only rarely is there a recognizable, small tricuspid annulus, which then forms the rim of an imperforate membrane. Usually there is only a dimple, or no indication of a tricuspid valve, in the floor of the right atrium.
Several subtypes of tricuspid atresia are distinguished based largely on whether there is an associated transposition of the great vessels (with or without pulmonary stenosis) and whether the ventricular septal defect (VSD), which is almost always present, is large or small. Of the various types, tricuspid atresia without transposition and with a relatively small VSD is by far the most common. Unfortunately, this type also carries one of the worst prognoses; the great majority of infants die in the first year and usually in weeks or months if not treated appropriately. The right atrium is dilated, and either the foramen ovale is patulous or an ASD exists. If the atrial septum is minimally patent, balloon atrial septostomy can be performed to widen a minimal patent foramen ovale or very small ASD. This allows blood to flow into the left side of the heart and thus to the systemic circulation. The mitral valve is large, as is the left ventricle. Usually there is no trace of a right ventricular inflow portion, and the infundibulum generally is present and thin walled. Although uncommon, pulmonary valve stenosis may be seen in association with tricuspid atresia.
Characteristic clinical features include the early appearance of moderate to marked cyanosis, progressing with time and increasing with crying. Cerebral hypoxic spells, similar to those seen in tetralogy of Fallot are occasionally seen, consisting of a sudden deepening of cyanosis, crying, lethargy, and at times unconsciousness. The episodes usually last only a few minutes but may lead to the death of the infant (see Plate 5-11).
Clubbing of the digits is never present at birth and takes time to develop, generally not well marked until about 3 months of age. The few children who live for any length of time usually have dyspnea on exertion (or even at rest) and fatigability. Occasionally a child may squat, but this is not a characteristic feature as in tetralogy of Fallot. Cardiomegaly is typically absent in patients of tricuspid atresia, with no precordial bulge. A systolic thrill is rare. The apical heart sounds are unremarkable; S2 at the base is normal or slightly increased and single, with P2 greatly diminished or absent as a result of the reduced pulmonary blood flow. Typically, there is a harsh systolic murmur of moderate intensity, best heard at about the third left interspace parasternally.
On chest radiography the heart is either normal in size or only very slightly enlarged. The right border of the heart is prominent because of the enlarged right atrium; the left border may have a peculiar angulated or squared-off appearance, and the pulmonary artery segment is reduced or absent. The vascularity of the lung fields is diminished. The ECG is much more helpful in arriving at a diagnosis. Left-axis deviation, left ventricular (LV) hypertrophy, and right atrial hypertrophy are invariably present. These are so typical for tricuspid atresia and so unusual in other types of cyanotic CHD that any cyanotic baby showing left-axis deviation and LV hypertrophy on the ECG, and without cardiomegaly, should be considered to have tricuspid atresia. The P waves are generally tall and peaked (often very tall), indicating right atrial enlargement. Cardiac ultrasound and MRI can easily define the anatomic pathology, including shunts, valve regurgitation, and ventricular function.
Cardiac catheterization to obtain hemodynamic data generally should not be done; it contributes little to what is already known or suspected on clinical grounds, merely adding another stressful procedure for the very sick infant to undergo. If cardiac catheterization and angiography must be done, a simple venous angiocardiogram or a selective right atrial angiocardiogram confirms the diagnosis. Opacification of the right atrium is rapidly followed by visualization of the left atrium, left ventricle, and great vessels. Generally, there is a typical, more or less triangular filling defect between the opacified right atrium and the left ventricle. Cardiac magnetic resonance angiography (MRA) with contrast can confirm the anatomy as well. This area is normally occupied by the inflow portion of the right ventricle. An LV injection in the lateral position shows the diminutive right ventricular outflow portion to be filling by way of the VSD.
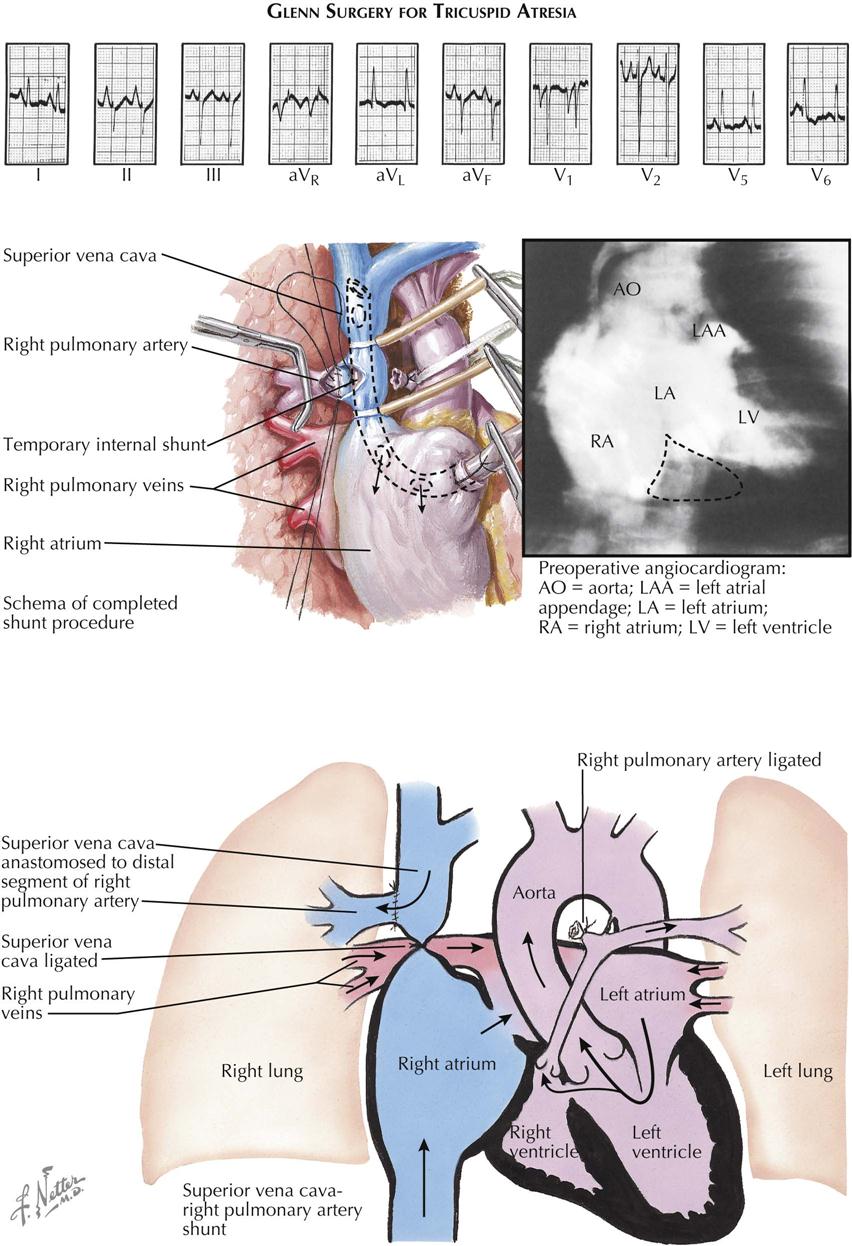
Treatment is surgical and can be only palliative. The surgery focuses on increasing pulmonary blood flow, which can also be accomplished in the newborn using prostaglandin E1. PGE1 relaxes smooth muscle in the ductus arteriosus and keeps it patent to provide temporary blood flow from the aorta to the pulmonary artery. This palliation allows time for patients with tricuspid atresia to mature to the point where a surgical procedure can be performed safely. Blalock-Taussig (subclavian to pulmonary artery anastomosis), classic Glenn (right atrium to pulmonary artery), or a Fontan (variations of vena cava to pulmonary artery) procedure provides flow to the pulmonary circulation in these patients, who depend on pulmonary blood flow for survival. These surgical procedures require the pulmonary artery pressure to be low in order for flow to enter the pulmonary circulation from the vena cava or right atrium.
A side-to-side anastomosis of the ascending aorta to the pulmonary artery can substitute for the Blalock-Taussig shunt. In the Glenn procedure, the proximal pulmonary artery is ligated, as is the superior vena cava, between the anastomotic site and the right atrium (see Plate 5-12). This operation has the considerable advantage of bringing blood directly from a large systemic vein to the right lung, thus entirely bypassing the right side of the heart. Unfortunately, it is not suitable in very small infants, who are at the highest risk and comprise the majority of cases of tricuspid atresia, because the low-pressure shunt between the small vessels has a strong tendency to thrombose, with disastrous results. This is also characteristic, but to a lesser extent, of the Blalock-Taussig operation. The Fontan procedure is another option to transfer venous blood directly into the pulmonary artery (see Plate 5-11).
Of the other (much rarer) forms of tricuspid atresia, those with a moderately large VSD and a normal to slightly increased pulmonary vascular resistance carry a much better prognosis and may not require surgery. A form that has a fairly good prognosis is that associated with transposition of the great vessels and a mild to moderate subpulmonic stenosis.
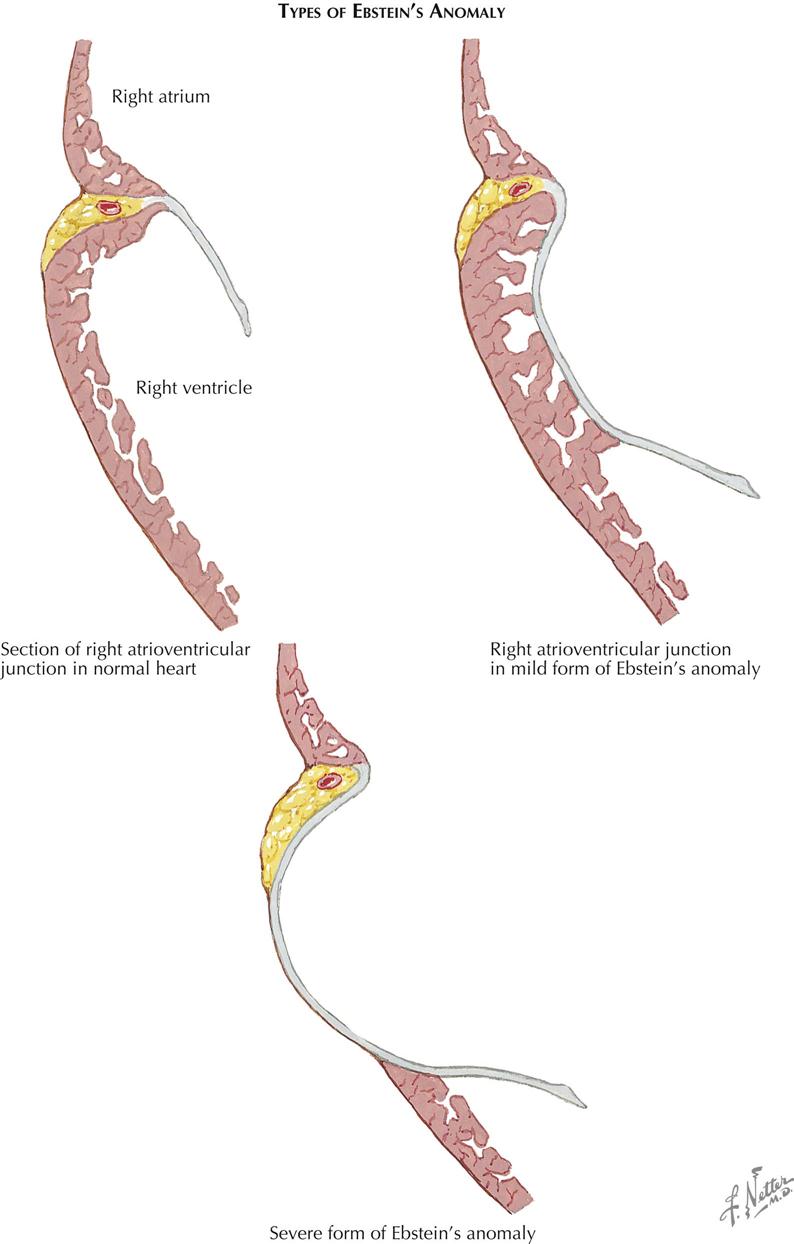
Ebstein’s Anomaly
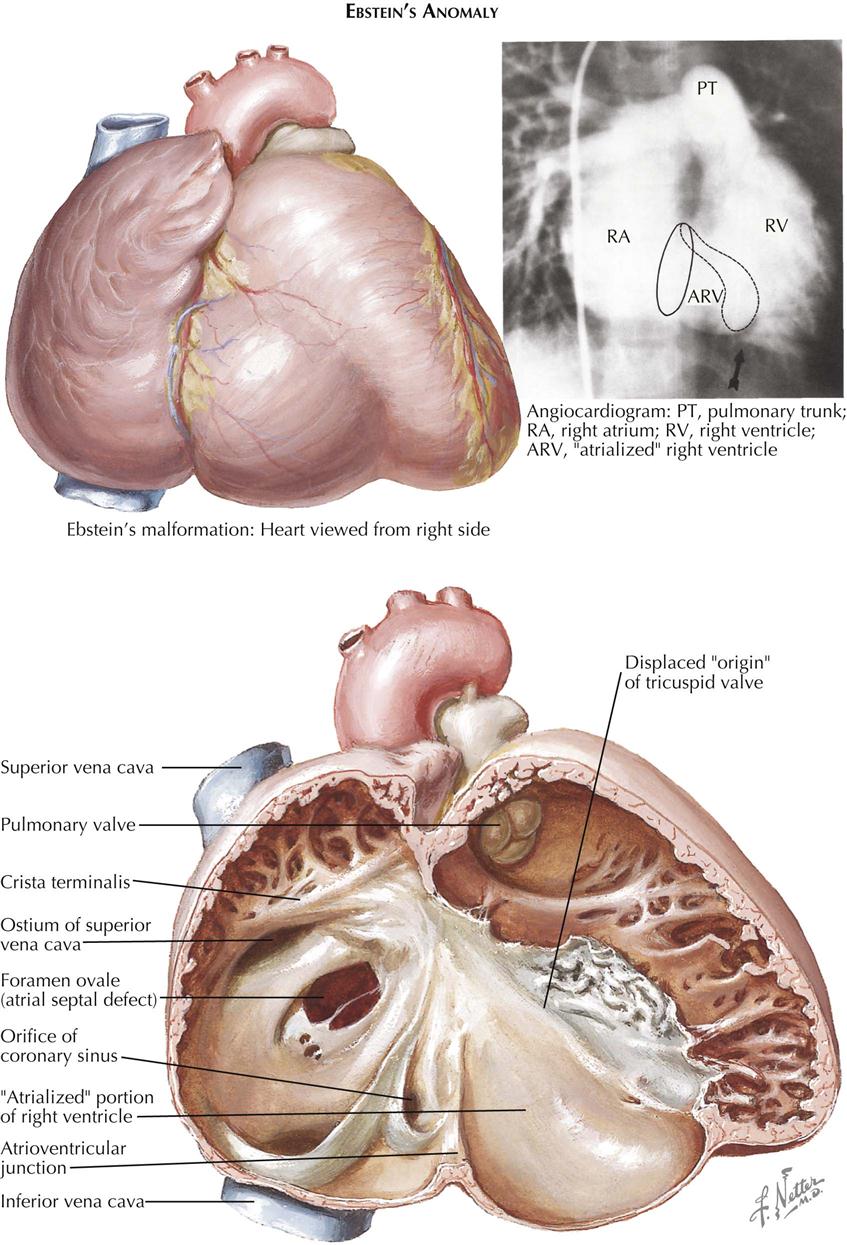
As an isolated malformation, Ebstein’s anomaly of the tricuspid valve is less common than tricuspid atresia. However, Ebstein’s malformation is of considerable clinical importance because most patients reach childhood, adolescence, or even adulthood, and therefore it must be much better tolerated than tricuspid atresia. In principle, Ebstein’s anomaly consists of a downward displacement of the tricuspid valve “origin.” The valve cusps, except for the medial two thirds of the anterior cusp, appear to originate from the right ventricular (RV) wall, often as low as the junction of the inflow and outflow portions of the right ventricle, instead of from the tricuspid annulus. The valve tissue is almost always redundant and wrinkled, and the chordae tendineae are poorly developed or absent (see Plate 5-13).
Embryologically, Ebstein’s malformation can be considered an abnormality in the undermining process of the RV wall, which normally leads to liberation of the inner layer of ventricular muscle. This process should continue until the atrioventricular junction is reached. Much of the apical portion of the valve “skirt” thus formed is normally resorbed, until only papillary muscles and narrow strands remain. The latter become fibrous (chordae tendineae), as do the valve cusps themselves. In Ebstein’s anomaly the process of undermining apparently is incomplete and does not reach the annulus. Individual cases vary greatly in this respect, and instead of cusps, chordae tendineae, and papillary muscles, there often are sheets of valve tissue with few or no chordae tendineae incorporating the papillary muscles. The anterior cusp is “liberated” very early in embryonic life, which may explain why this cusp always originates normally. The actual valve opening, located close to the crista supraventricularis, is usually much smaller than the normal tricuspid ostium, and the valve is almost always incompetent.
The downward displacement of the valve divides the right ventricle into two parts: (1) an “atrialized” part between the normal annulus and the abnormal valve origin and (2) the normal outflow portion of the right ventricle. The size of the “atrialized” portion of the right ventricle varies greatly, and its wall may be fibrous and paper thin or muscular and quite normally formed. Rarely, the valve is imperforate, or its free portion is practically nonexistent. The pulmonary valve may be stenotic or rarely atretic.
The clinical features are highly diverse, an expression of the considerable variability of the Ebstein’s pathology. In general, the larger and thinner walled the atrialized RV part, the smaller will be the remaining normally developed RV part. Also, the greater the insufficiency of the tricuspid valve, the more serious will be the hemodynamic situation. In severe cases, symptoms (cyanosis, dyspnea, feeding difficulty) may begin in the neonatal period (see Plate 5-14). The early occurrence of heart failure is an ominous sign and is usually followed by death within weeks. In milder cases, symptoms may not appear until later in childhood. Occasionally, the degree of malformation is slight and is compatible with a fairly active and normal life. Cyanosis and clubbing are usually present in older children, who tend to be underdeveloped and thin. Cyanosis in infancy often subsides temporarily, only to reappear later. Fatigue is a prominent symptom, along with exercise intolerance and dyspnea on effort. Cardiac arrhythmias are very common, usually consisting of some form of supraventricular tachycardia.
The patient with Ebstein’s anomaly almost always has considerable cardiomegaly on both the left and the right side, because of enlargement of the right atrium and the “atrialized” right ventricle, and the peripheral pulses are weak. The apical impulse is diffuse and poorly felt. A precordial bulge and thrill are unusual. S1 is of normal intensity and often is split, with the second component loud; S2 is generally normal. A loud, early diastolic S3 is heard along the left lower sternal border, and S4 may be present. A mild to moderate systolic murmur is usually present along the left lower sternal border and may be accompanied by a diastolic murmur. The systolic murmur sometimes may have a curious scratchy quality, resembling that of a pericardial friction rub.
Chest radiography shows moderate to marked cardiomegaly, and the heart is often box or funnel shaped, mainly because of tremendous right atrial enlargement and displacement and dilatation of the RV outflow tract. The pulmonary vascular markings are decreased, and the main pulmonary artery segment is small or absent. Left atrial enlargement is never seen in Ebstein’s anomaly. Rarely, the heart may be almost normal in size and shape, indicating a mild degree of malformation.
The characteristic ECG displays right-axis deviation, low voltage, and widened QRS complexes in the limb leads and the right precordial leads, and in the latter a right bundle branch-block pattern with “splintering” of the complexes. A pattern of RV hypertrophy is rarely seen, and LV hypertrophy is invariably absent. Tall, peaked P waves are usually seen in leads II, aVF, and V1 to V3, and the PR interval is usually prolonged. Wolff-Parkinson-White syndrome (see Plate 2-23) is relatively common in Ebstein’s anomaly.
Cardiac ultrasound or MRI can confirm the diagnosis of Ebstein’s anomaly. With cardiac catheterization there is a distinct tendency for arrhythmias to occur, but the procedure can be used to establish the diagnosis and determine the degree of severity. The catheter tends to coil in the right atrium and thus outlines its tremendous size. The pressure in the atrialized portion of the right ventricle is low and in general resembles that measured in the right atrium. RV pressures are normal, except in the rare case with associated pulmonary stenosis and resulting elevated RV pressure. If an electrode catheter is employed, pressure tracings and intracavitary ECGs can be recorded simultaneously. Placement of the catheter in the distal portion of the right ventricle will show typical RV pressure and ECG tracings. On pulling back into the atrialized part of the right ventricle, the former do not change significantly, whereas the pressure drops. On further withdrawal into the right atrium, the ECG complexes assume a right atrial configuration with large P waves, and the pressure tracings show no further change (see Plate 5-13).
On selective right atrial angiocardiography, contrast successively opacifies the smooth-walled right atrium, the atrialized RV part, and often (after some delay) the trabeculated RV outflow portion. The diaphragmatic right border of the heart may have a trilobed, scalloped appearance. Injection into the RV outflow outlines the “incomplete” right ventricle and regurgitation across the tricuspid valve.
Medical treatment is indicated mainly in patients with Ebstein’s anomaly who have congestive heart failure or paroxysmal supraventricular tachycardia. Therapy consists of the usual anticongestive measures: digitalis, diuretics, oxygen, sedation, bed rest, and reduced salt intake. Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) and beta-blocker therapy should be considered in heart failure patients, as well as antiarrhythmics to control the tachycardia.
Surgical treatment is complicated and usually palliative. The choice of procedure depends on whatever pathology is present. In general, surgery should be advised only for patients with Ebstein’s anomaly who are symptomatic and incapacitated. Rehabilitation of incapacitated patients has been achieved by prosthetic replacement of the anomalous valve and, in suitable cases, by resection or plication of the nonfunctioning atrialized part of the right ventricle. An ASD, if present, should probably be closed at the same time, to achieve the full benefit of the procedure. If the tricuspid valve is replaced, long-term anticoagulation must be achieved.
Anomalies of the Ventricular Septum
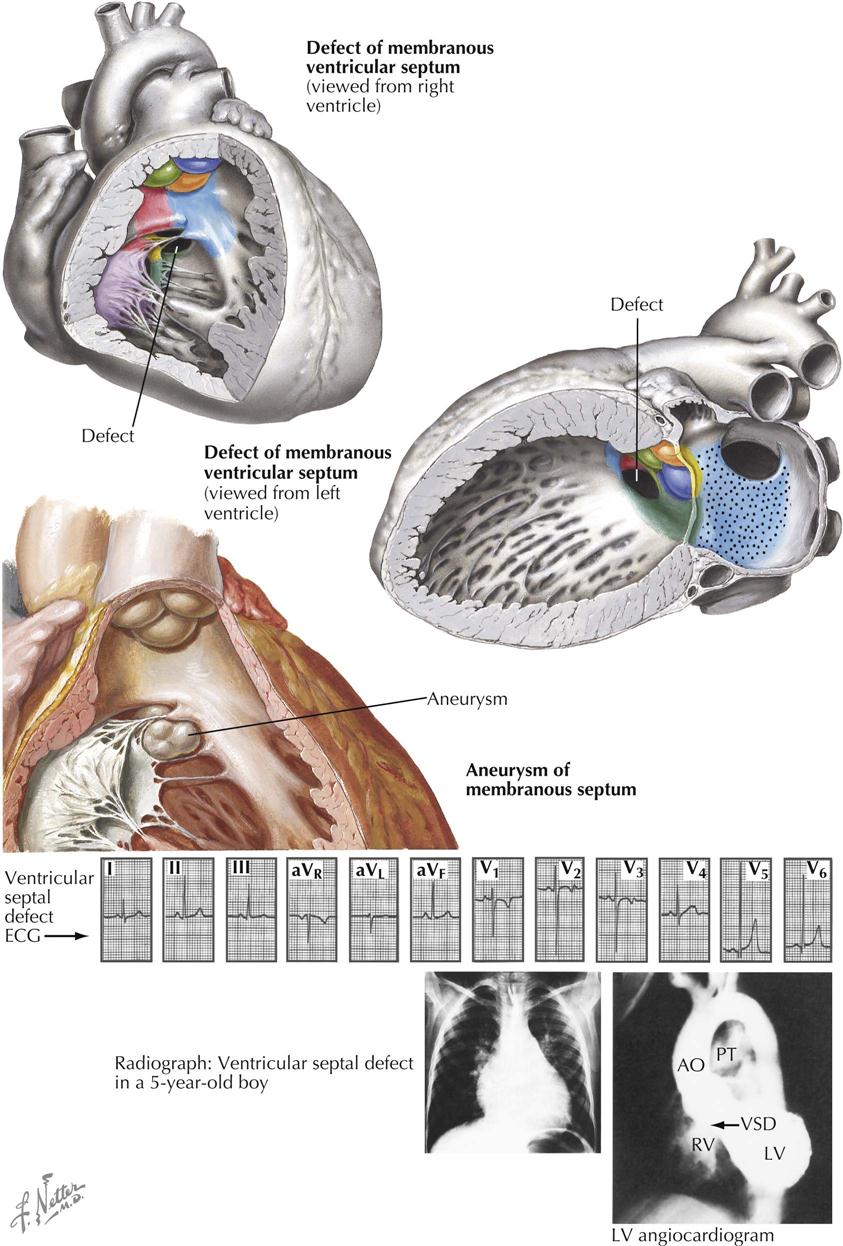
Ventricular Septal Defects (“Membranous”)
Of the anomalies involving ventricular septal defects, those located beneath the aortic valve, the membranous ventricular septal defects, are by far the most common. Not only are these defects frequently seen in association with other cardiac anomalies, but even when occurring as isolated lesions, the membranous VSDs constitute the most important and also the most common type of congenital heart disease. This is not surprising considering the complex embryologic history of the subaortic portion of the ventricular septum. This is the last part of the septum to close, a closure effected by the fusion of components from the embryonic muscular septum, endocardial cushions, and conal swellings. Anomalous development of any one or several of these contributors will lead to a defect of the ventricular septum. Therefore, although located in the same general area, membranous VSDs may vary considerably in position and size. Some are found immediately beneath the right and posterior aortic valve cusps; these probably are caused mainly by deficiency of the conus septum and, because of a lack of support for the aortic valve cusps, may lead to prolapse of one or both cusps, causing aortic regurgitation. Other membranous VSDs mainly caused by deficiency of the right limb of the endocardial cushions, or failure of otherwise normally developed endocardial cushions to fuse with the ventricular septum and conus septum, are located a few millimeters away from the aortic valve, leaving a rim of muscular or fibrous tissue. All these defects are located in the general area where the membranous septum is found in the normal heart, and thus are usually rather loosely referred to as “membranous septal defects.”
Small Defects and Shunts
The clinical features vary, as might be expected in an anomaly with a diverse pathologic anatomy. Children who have small defects and shunts are well developed and asymptomatic, and the ECG is normal. Chest radiographs also are generally normal, although occasionally the vascular pattern may be slightly increased, with evidence of some left atrial enlargement. Such cases are referred to as having “maladie de Roger.” A harsh systolic murmur, often well localized and, at times, quite loud, is heard best over the lower left parasternal area or (sometimes) somewhat higher up. A thrill may be palpable. Treatment is generally not indicated unless the anomaly is complicated by endocarditis, which, fortunately, occurs only rarely.
Large Defects
Large VSDs may cause symptoms in early infancy. Growth failure is usual in such cases; weight gain may be distressingly slow, and the children are pale, delicate-looking, and scrawny. Feeding difficulties, respiratory infections, and congestive failure are common, and the infants may spend more time in the hospital than at home. There is cardiomegaly, and a loud, harsh, holosystolic murmur audible over the left lower sternum, accompanied by a thrill, is almost invariably present. An apical diastolic rumble, ascribed to torrential blood flow across the mitral valve, is often also heard.
The chest radiograph shows cardiomegaly, mainly caused by biventricular and left atrial enlargement; marked hypervascularity of the lungs, with a prominent pulmonary trunk and main pulmonary arteries; and a relatively small aorta. ECG generally reveals right-axis deviation and evidence of biventricular enlargement. This often takes the form of large, biphasic QRS complexes in the midprecordial leads (Katz-Wachtel phenomenon). Cardiac ultrasound and MRI easily confirm the diagnosis of VSD and can define its anatomic pathology and location.
Cardiac catheterization readily demonstrates a marked increase in the oxygen content of the RV blood samples, and the catheter may enter the left ventricle or aorta through the VSD, especially when the catheter is advanced from the superior vena cava. Catheters advanced from the inferior vena cava generally do not cross the VSD. The RV and pulmonary artery pressures are elevated and may reach systemic levels. The pulmonary hypertension is caused in part by some increase in pulmonary vascular resistance, but mostly by the greatly increased pulmonary blood flow, which may be several times that of the systemic blood flow. The injection of a radiopaque medium selectively into the pulmonary trunk, after passage through the lungs, demonstrates the interventricular shunt. A selective left ventricular angiogram will give even clearer pictures of the shunt.
Therapeutically, infants with large VSDs may present a serious problem. Every effort should be made to carry them through the first year, after which many improve greatly, probably because of the relative decrease in size of the VSD. If medical treatment is unsuccessful, a pulmonary banding procedure may be done, or in some cases a percutaneous approach may be used to close the VSD with a device. With banding, a plasticlike band is placed around the pulmonary trunk, just above the valve, and tightened until the diameter of the vessel is reduced by about two thirds, and the pressure distally has dropped closer to normal. Usually, there is a concomitant rise in aortic pressure, indicating a more favorable pulmonary/systemic blood flow ratio. The surgical results may be excellent, although many failures occur as well. In any case, banding is a temporary procedure followed by closure of the defect later, at which time the band is removed.
Moderate-Sized Defects
Fortunately, most children with moderately sized VSDs do not have the stormy infancy previously described, although respiratory infections are prevalent and many patients are small for their age. Dyspnea on exertion is also common. Congestive heart failure occurs rarely in older children, however, and the physician should always consider the possibility of a complicating lesion, such as prolapse of an aortic valve cusp causing aortic regurgitation, or bacterial endocarditis. A harsh, rather loud, holosystolic murmur accompanied by a thrill is generally best heard along the lower left sternal border. An apical diastolic murmur of moderate intensity (mitral flow murmur) is usually audible at the apex.
Clinical examination and chest radiography usually show moderate cardiomegaly; the pulmonary vasculature is distinctly increased, and the left atrium is enlarged. The ECG typically shows a normal or right-axis deviation with a pattern of so-called left ventricular diastolic overloading, consisting of deep Q waves, very tall R waves, and often tall, peaked T waves in the left precordial leads. Evidence for biventricular enlargement is also common. Cardiac ultrasound and MRI can define the size and position of the VSD. Cardiac-catheterization findings are similar to those described above; however, the right ventricular and pulmonary-artery pressures are generally only slightly or moderately elevated and show little tendency to rise during childhood. A left ventricular angiogram is easily done in this age group, and it will clearly demonstrate the size and position of the defect.
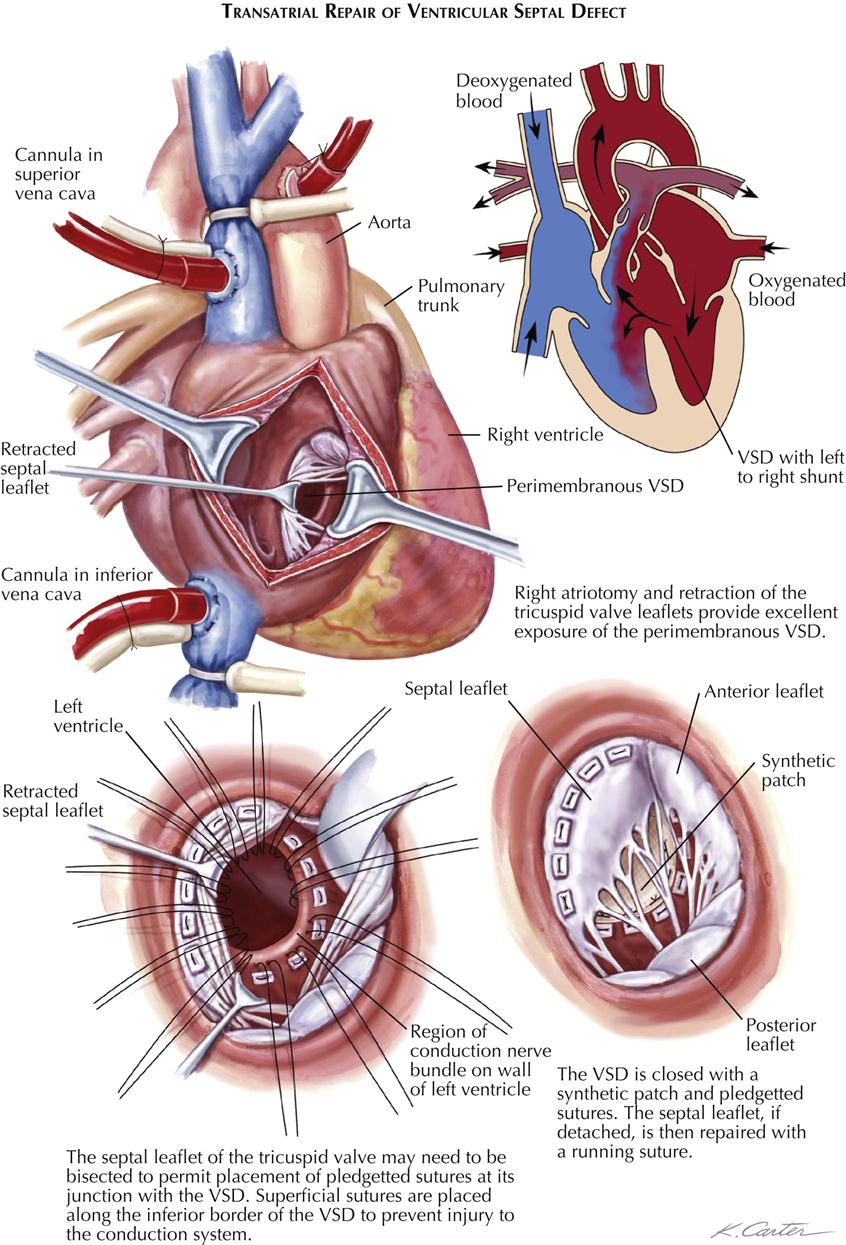
Treatment of patients with moderate-sized VSDs is surgical and consists of closure of the defect transatrially, using direct suture or a prosthesis and cardiopulmonary bypass (see Plate 5-17). The surgical risk is low, but heart block caused by injury to the atrioventricular bundle can occur infrequently.
Pulmonary Hypertension
Some patients with VSDs either always have had, or develop as young adults, marked pulmonary hypertension because of vascular changes in the lungs. The pulmonary vascular resistance equals or exceeds systemic vascular resistance, and the shunt across the defect is (or becomes) bidirectional or mainly from right to left, causing cyanosis and digital clubbing. In some patients a murmur is barely audible, an expression of the presence of equal pressures in the two ventricles and minimal net shunt. P2 is loud and snapping, and the pulmonary valve may become incompetent, resulting in a diastolic murmur at the left upper sternal border.
Chest radiography reveals little or no cardiomegaly. The pulmonary artery and the branches are usually dilated and the lung fields are clear. ECG reveals right axis deviation and RV hypertrophy.
Prostaglandins, endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and activators of soluble guanylate cyclase are used to decrease pulmonary artery pressure in many VSD patients with hypertension, especially those with some evidence of pulmonary artery constriction. Surgical closure of the defect carries a prohibitive mortality (~100%) and is contraindicated. Lung transplant may be the only surgical procedure to reduce symptoms and prolong life.
Aneurysm of Membranous Septum
Aneurysms of the membranous septum are being diagnosed with increasing frequency as selective left ventricular angiocardiography, echocardiography, CT, and MRI are increasingly done. The aneurysm may be intact or may contain one or more perforations (see Plate 5-15). The aneurysm itself produces no symptoms, unless large enough to cause RV outflow obstruction, or unless an aortic cusp prolapses into it; both are rare complications.
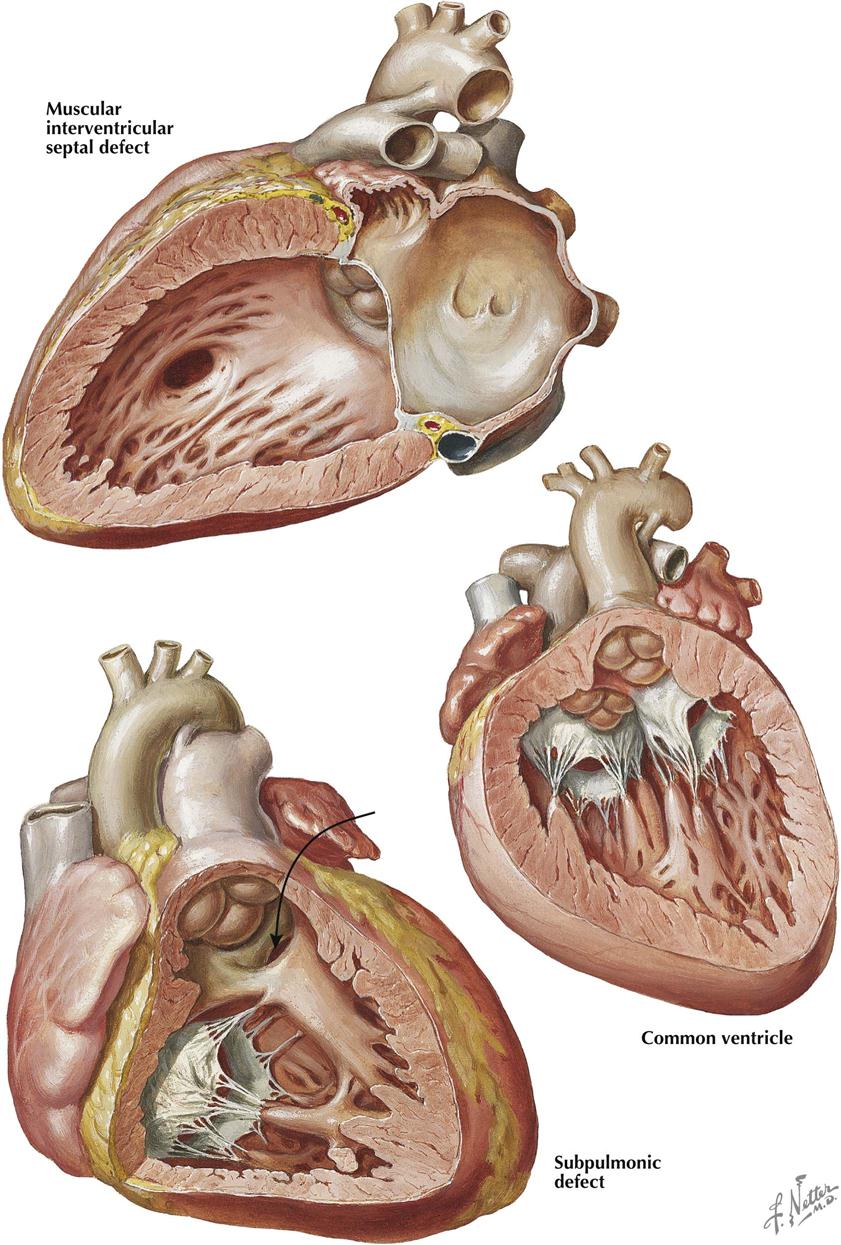
Muscular Interventricular Septal Defects
Defects of the muscular interventricular septum may occur anywhere in the septum (see Plate 5-16). These defects may be single or multiple and any size. If located in the trabeculated apical part of the septum, the defect may go undetected. Some defects have a “Swiss cheese” appearance. In the adult population without CHD, ventricular septal rupture secondary to acute myocardial infarction can produce comparable signs and symptoms as the congenital variety, and the prognosis is poor. The symptoms and signs depend on the combined size of the defects as well as the degree of ventricular dysfunction. Treatment is surgical or with a percutaneous closure device.
A special form of muscular interventricular septal defect is located beneath the two arterial valves and is caused by malalignment of the truncus and conus septa, which do not meet each other and therefore cannot fuse. The truncus septum is deviated to the left, and the pulmonary artery overrides the anteriorly located defect. The murmur tends to be located somewhat higher than usual and may sound superficial.
Common Ventricle
In a common ventricle the entire septum is absent except for a low muscular ridge, usually present along the posteroinferior ventricular wall (see Plate 5-16). Both atrioventricular valves enter the common chamber, and both structurally resemble the normal mitral valve. The two posterior papillary muscles, together with the low muscular ridge, may form a single muscle mass. The two great arteries are transposed, and both may originate from the common chamber, or one (usually the aorta) may spring from a small outflow chamber separated from the main ventricular body by a muscular septum–like ridge. Associated pulmonary stenosis often occurs and, if not too severe, generally improves the prognosis. Ventricular inversion is common and is present in the specimen illustrated here.
The clinical features depend largely on the presence of pulmonary stenosis; patients with stenosis present similar to those with tetralogy of Fallot (see Plate 5-18). If there is no stenosis, the symptoms and signs are those of a large VSD, except that the thrill and the loud systolic murmur are not present. A systolic murmur at the base is probably caused by a large pulmonary blood flow across the normal pulmonary valve. As in VSD, an apical diastolic rumble may be heard. Vascular changes in the lung develop early, resulting in a high resistance to blood flow and pulmonary hypertension.
On chest radiography the heart is normal in size if pulmonary stenosis is present, and the pulmonary vasculature is diminished. In patients without pulmonary stenosis, cardiomegaly may be present, associated with an increase in the pulmonary vasculature. In patients with severe pulmonary hypertension as a result of the intrapulmonary vascular changes, cardiomegaly is mild or absent; the hilar vessels are large, but peripherally the markings are diminished. There is no characteristic ECG findings associated with the common ventricle, and the QRS axis and precordial lead patterns vary greatly. Selective angiocardiography establishes the diagnosis, as does the cardiac echocardiography, MRI, or CTA.
At present, treatment of common ventricle can be only palliative; correction is not possible for anatomic and hemodynamic reasons. Patients with mild to moderate pulmonary stenosis require no surgical treatment and may do well for many years. Patients with severe pulmonary stenosis may have a Blalock-Taussig shunt or an anastomosis from vena cava to pulmonary artery. In young children without stenosis, a pulmonary artery banding procedure may be considered.
Anomalies of Right Ventricular Outflow Tract
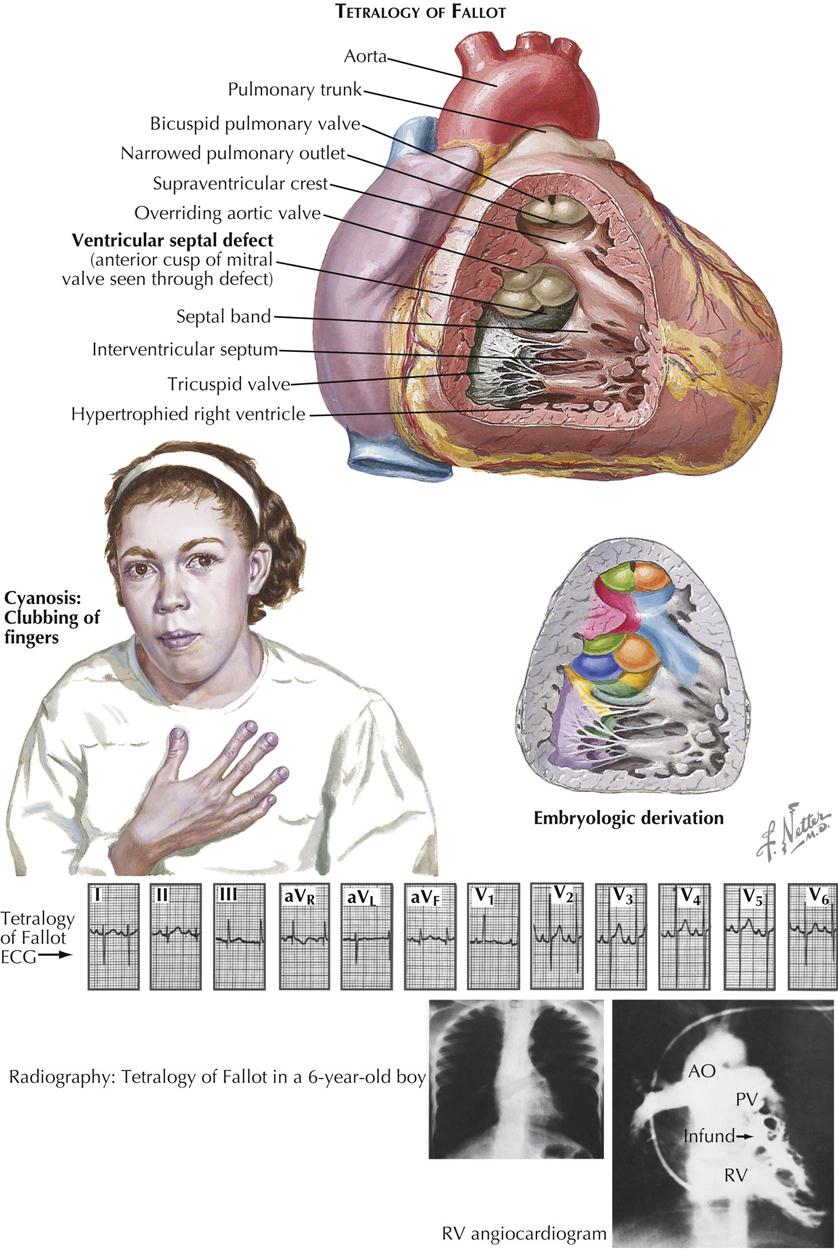
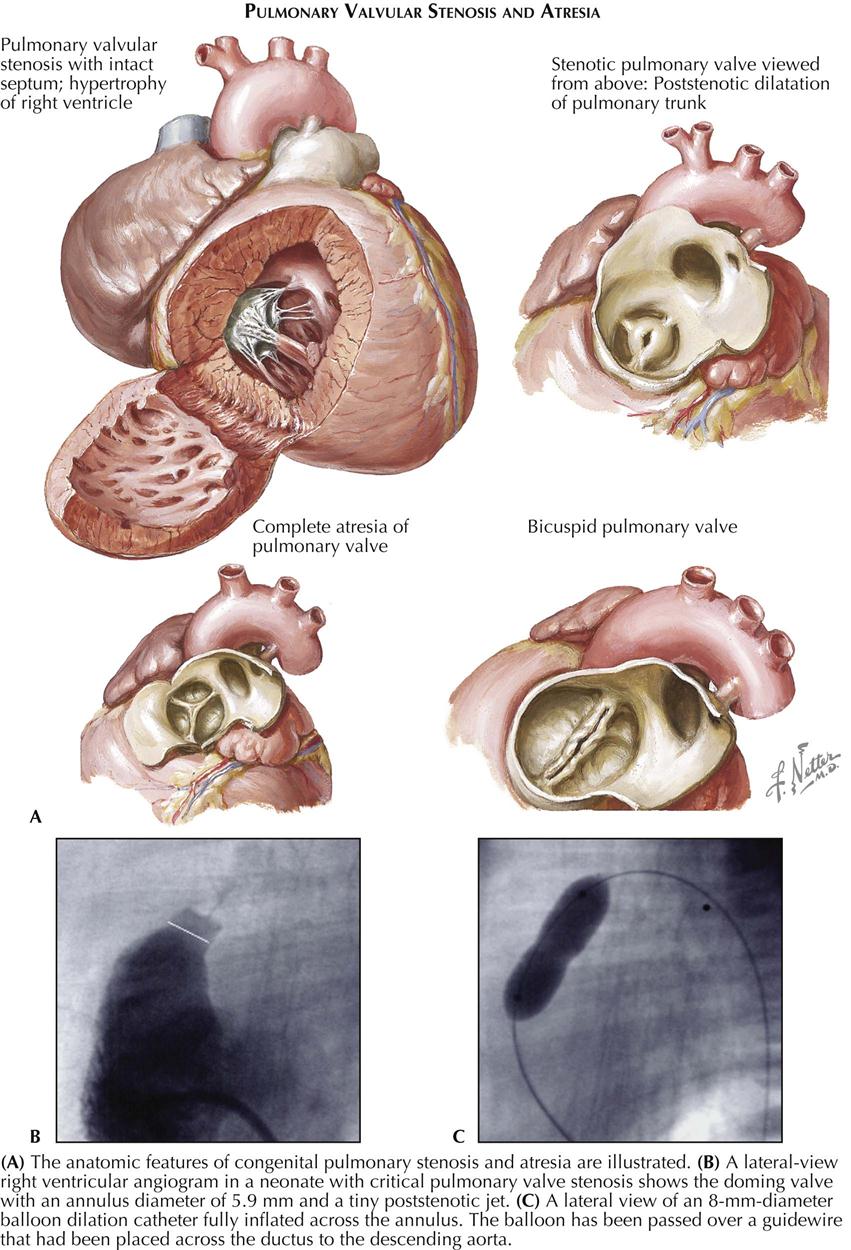
Tetralogy of Fallot
Tetralogy of Fallot is by far the most common form of cyanotic congenital heart disease that is compatible with life. Patients reaching adulthood are uncommon but not rare.
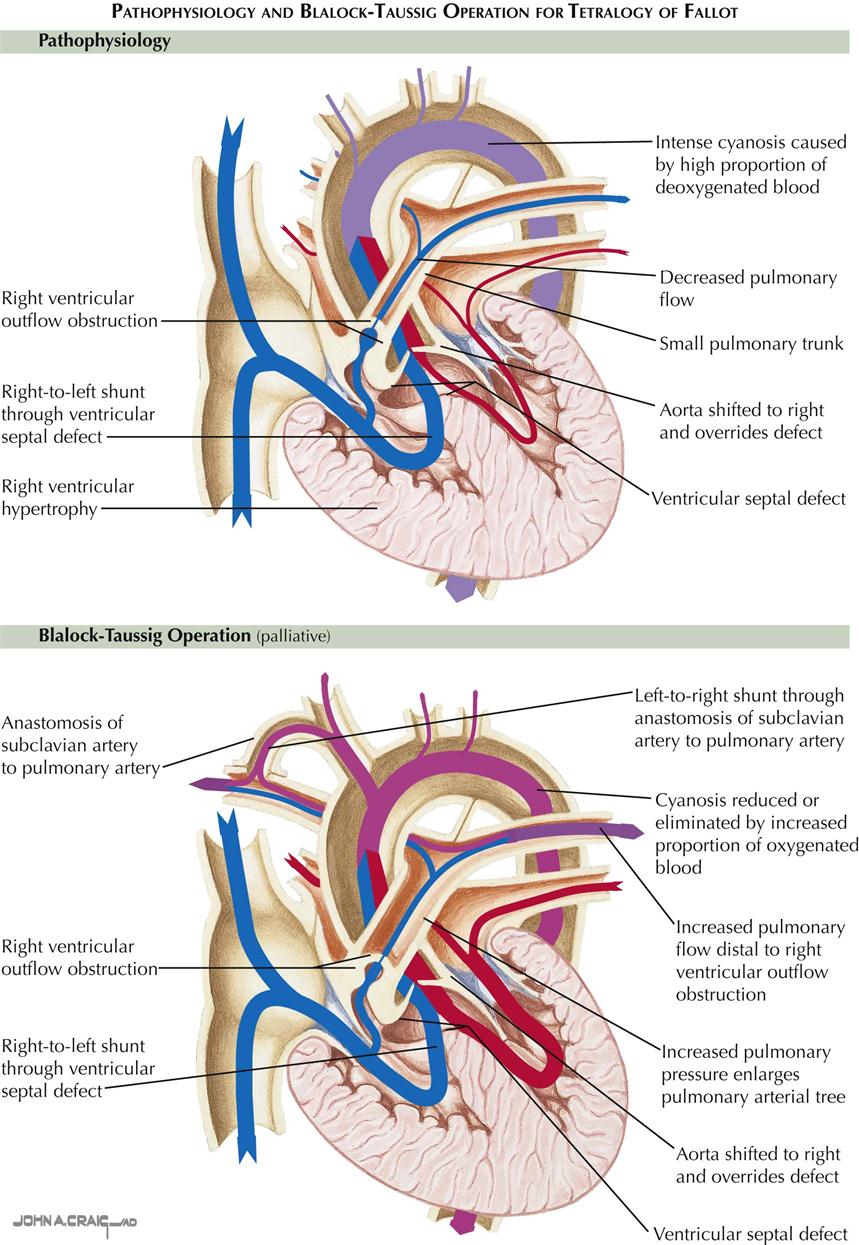
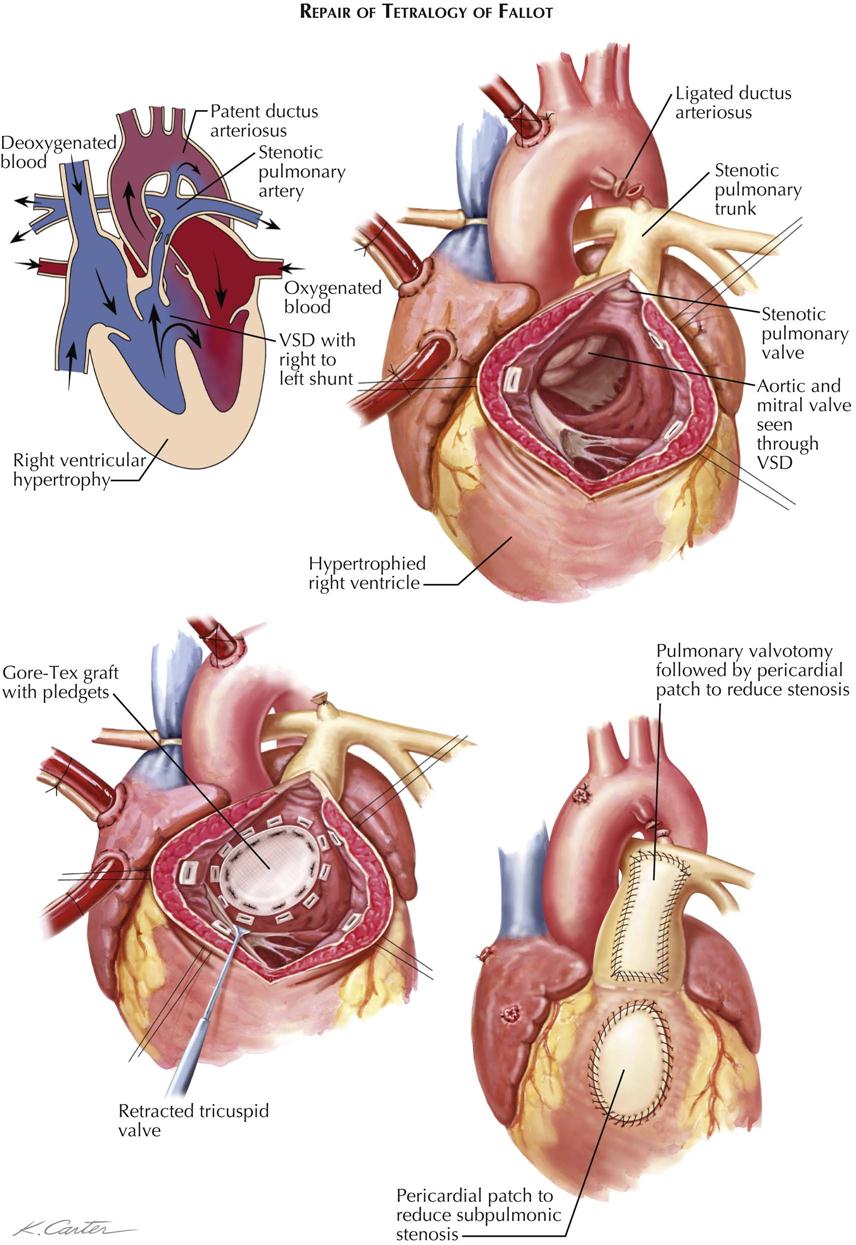
Classically, as described by Fallot, the four abnormalities that constitute the complex are right ventricular outflow tract stenosis (or atresia), ventricular septal defect, aortic straddling of the VSD (seems to originate from both ventricles), and RV hypertrophy (see Plate 5-18). Anatomically, there is classically RV infundibular stenosis, but the pulmonary valve, although frequently bicuspid, is stenotic in only about 40% of patients. Nonstenotic valves may be hypoplastic, as part of a general hypoplasia of the pulmonary trunk. Stenotic valves may be bicuspid, tricuspid, or dome shaped, without well-defined cusps. The degree of infundibular stenosis ranges from complete atresia to barely detectable. The VSD is usually large, offering little or no resistance to blood flow, and involving not only the area of the membranous septum but also the adjacent, more anterior portions of the ventricular septum. The aortic straddling of the VSD, although variable, is always unmistakable, and the aorta often appears to originate primarily from the right ventricle. The right ventricle is always hypertrophied, reflecting the high RV pressure, which is identical to left ventricular pressure.
From a developmental point of view, tetralogy of Fallot is a simple anomaly caused by a single embryologic error (see Plate 5-18). The conus septum is located too far anteriorly, particularly in its lower part, dividing the conus into a smaller, anterior, RV part (thus the infundibular stenosis) and a larger posterior part. The conus cannot form the crista supraventricularis and participate in closure of the interventricular septum, which in turn makes it impossible for the aortic valve to seat itself in its normal position, with its free edge so far removed from the tricuspid valve that the aortic valve cannot contribute to the formation of the tricuspid valve. This is why the medial papillary muscle is absent and the tricuspid valve is abnormally formed in tetralogy of Fallot. The truncus septum is also usually displaced anteriorly, accounting at least in part for the small pulmonary trunk and disproportionately large ascending aorta.
The clinical picture depends mainly on the degree of RV outflow obstruction, which usually is moderate, at least initially. The shunt across the ventricular septum is mainly from left to right (acyanotic tetralogy of Fallot). Therefore, many children with tetralogy are not clinically cyanotic during the first few months of life but become so as they grow and as the stenosis worsens. More venous blood enters the aorta directly from the right ventricle, and the pulmonary blood flow decreases in a relative sense. At first, cyanosis is obvious only with exertion or crying, but generally within the first few years of life the children become cyanotic even at rest, and clubbing of the fingers and toes develops (see Plate 5-18). Occasionally, the infundibular stenosis is so mild that cyanosis never develops (“pink” tetralogy). These children, particularly in infancy, may behave more like patients who have a VSD with large left-to-right shunts.
At the other extreme are cases where the infundibulum and/or pulmonary valve are atretic or severely stenotic. Such infants are generally cyanotic from birth, although the severity of the condition may be masked by a short-lived patency of the ductus arteriosus, which temporarily maintains good pulmonary circulation. Unfortunately, however, the ductus usually closes within the first 2 weeks of life, often causing rapid deterioration of the infant’s condition, so that surgical intervention to increase pulmonary blood flow becomes imperative.
A worrisome phenomenon can occur in young children with tetralogy of Fallot, the hypoxic episode or “blue spell.” A period of crying suddenly leads to a marked increase in cyanosis, dyspnea, and unconsciousness, sometimes with convulsions. Such episodes may occur only occasionally or as often as several times a day and may last for minutes or hours. The spells tend to be associated with bowel movements or feeding and to occur early in the day, although the episodes can happen at any time for no apparent reason. Hypoxic events are serious and may be fatal. The hypoxia results from a sudden spasm of the RV infundibulum and a corresponding decrease in pulmonary blood flow. Although more common in frankly cyanotic infants, hypoxic episodes may also occur in the less severe forms of tetralogy.
Squatting is characteristically assumed by children of walking age with cyanotic tetralogy of Fallot. Squatting usually follows some degree of physical exertion, which may simply be walking. The posture rapidly restores arterial oxygen saturation by an incompletely understood mechanism, although the increased arterial vascular resistance during squatting may decrease right-to-left shunting. Dyspnea and hyperpnea on exertion are common, as in all forms of cyanotic CHD. These children typically are underdeveloped with clubbing of the fingers and toes, except in the first few months of life. There is usually no prominence of the left side of the chest, and a thrill is palpable at the lower left sternal border in most patients.
On auscultation, S1 is normal, and A2 is loud but P2 is diminished or absent. In acyanotic or mildly cyanotic forms of tetralogy of Fallot, P2 may be present, and S2 may then be widely split. The systolic murmur is usually loud and of the stenotic crescendo-decrescendo type, ending before or at aortic closure. In general, the more severe the tetralogy, the shorter is the murmur; occasionally, no murmur is heard at all. Likewise, during a hypoxic episode, the murmur may be less obvious or may disappear altogether, only to return on recovery.
The chest radiograph characteristically shows the heart to be normal in size (see Plate 5-18). The apex is elevated, and the pulmonary segment is small or concave, the boot-shaped heart (“coeur in sabot”). The aortic arch is prominent, and the pulmonary vasculature may be diminished. The aortic arch is located on the right side in about 2.5% of cases. The ECG characteristically shows right-axis deviation and RV hypertrophy of the systolic-overload type, with tall R waves in the right precordial leads. Transition is usually early and rather sudden in V2 or V3 and is an expression of the heart’s small size. Additional evidence of left ventricular (LV) hypertrophy may be present in the left precordial leads in pink tetralogy, when the ventricular shunt is mainly or exclusively from left to right (see Plate 5-18).
On cardiac catheterization the pressures in the ventricles are usually equal and at systemic levels; RV and LV pressure tracings are identical and of normal configuration. Evidence of bidirectional shunting across the VSD shows the right-to-left component is usually dominant. Arterial oxygen saturation varies considerably among patients. The aorta is often entered easily from the right ventricle, when the catheter is advanced from either the SVC or IVC and pulmonary artery pressure is low.
Angiocardiography is especially valuable to the surgeon in delineating the anatomy of RV outflow and determining the size and position of the pulmonary artery. Cardiac ultrasound and MRA are also helpful. The prognosis depends on the severity of RV outflow obstruction. Infants who are cyanotic at or shortly after birth seldom survive the first year, unless surgery is performed. Those with lesser forms of tetralogy of Fallot may live for many years and although handicapped in many ways, are usually alert and do well intellectually. The more common and serious complications are bacterial endocarditis, cerebral vascular accident (CVA, stroke) caused by thrombosis or severe hypoxia, and brain abscess. Central nervous system symptoms appearing in patients older than 2 years with cyanotic CHD almost always indicate the presence of a brain abscess. It is extremely rare in infants. On the other hand, CVA seldom occurs in patients older than 2 years. The etiology of brain abscesses and cerebral thrombosis may be related to shunting of blood from the venous system to the arterial system (see Plate 5-18).
Treatment of patients with tetralogy of Fallot is both medical and surgical. Heart failure is extremely uncommon beyond infancy but may occur in babies and should be treated in the usual manner. Hypoxic spells are often dramatically relieved by the administration of morphine and oxygen and squatting. Cyanotic children with normal or near-normal hemoglobin levels are usually anemic and should be given iron therapy until hemoglobin has increased to 15 to 17 g/dL (gm%). Once popular in patients with high hematocrit levels, venesection is now known to increase the symptoms and signs of hypoxia. Venesection should be done in small increments with caution, and then only in symptomatic patients with extremely high hematocrit (≥80 mL/dL).
Blalock-Taussig and Brock Operations
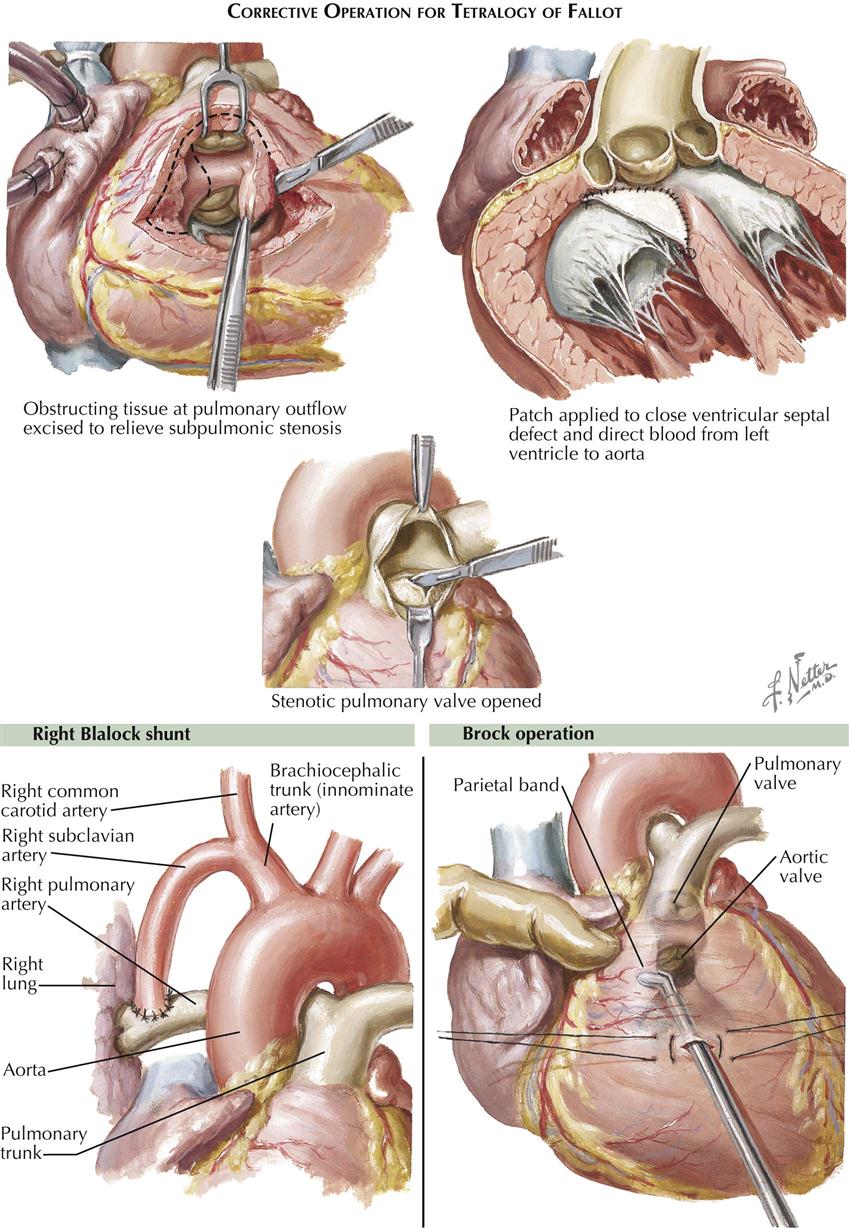
Surgical treatment is much more important in tetralogy of Fallot. Any cyanotic infant or child too young for correction but with significant symptoms should have the benefit of some type of palliative procedure. An end-to-side subclavian artery–pulmonary artery shunt, the Blalock-Taussig operation, is the procedure of choice in patients after infancy (see Plate 5-19). Technically straightforward, the surgery carries minimal risk of creating too large a shunt, which might lead to heart failure. In infants, who have small vessels, the Blalock-Taussig procedure is much less satisfactory; the shunt often thromboses immediately, or the infant rapidly outgrows it, necessitating a second operation on the opposite side. Creation of a direct side-to-side ascending aorta–pulmonary artery shunt may be preferable, the Waterston (Waterston-Cooley) operation. The older Potts procedure, with anastomosis from descending aorta to pulmonary artery, was found to be so difficult and dangerous to undo when done at a later age that it has been abandoned.
An alternate procedure that may be done in young infants, and that may be preferable to some surgeons for older children as well, is the Brock operation (see Plate 5-20). This consists of removing at least some of the stenosing infundibular muscle tissue through an RV stab wound, cutting the pulmonary valve as well if it is stenotic. The Brock procedure is performed blindly, is difficult to do adequately, and carries risks, particularly to the aortic valve.
The results of correction of tetralogy of Fallot are dramatic. The RV (pulmonary) outflow obstruction must be adequately relieved, which involves excision of as much obstructing muscle tissue as possible and possibly the use of an outflow patch to widen the infundibulum and at times the pulmonary root and trunk as well. The VSD is closed, employing a second patch of appropriate size (see Plate 5-21).
In the most severe forms of tetralogy of Fallot, in which the patient has atresia or near-atresia of the RV outflow tract, correction may not be possible. A permanent superior vena cava–right pulmonary artery shunt often provides excellent palliative therapy. A conduit from the right ventricle to the pulmonary artery (Rastelli procedure) may also improve oxygenation.
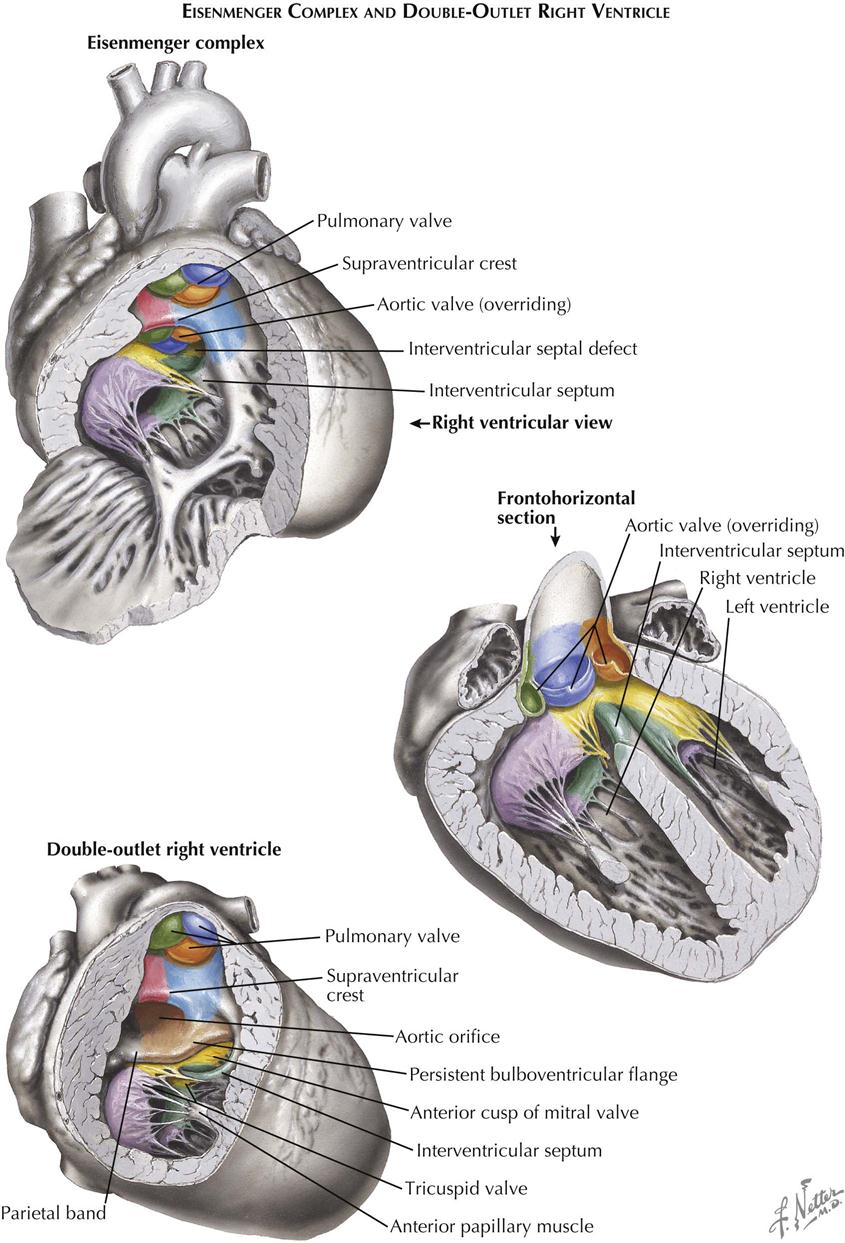
Eisenmenger Complex
Patients with Eisenmenger complex have a large ventricular septal defect similar to that seen in tetralogy of Fallot, and the aorta straddles the VSD (see Plate 5-22). However, the crista supraventricularis is not significantly displaced, although hypoplastic and occasionally nearly absent. As in tetralogy, the tricuspid valve is abnormally formed in this anomaly. There is no RV outflow obstruction, so the pulmonary artery is large. The anomaly is rare, and the term “Eisenmenger syndrome” arose to designate situations where a left-to-right shunt, regardless of its level, changed gradually to a predominantly right-to-left shunt, with severe pulmonary hypertension caused by pulmonary vascular changes and a concomitant rise in pulmonary vascular resistance (PVR). Pathologically, the Eisenmenger complex may be differentiated from a simple VSD by noting the anomalous tricuspid valve. In a simple VSD, the tricuspid is normally formed, and a medial papillary muscle is present. At times, differentiation of the Eisenmenger complex from acyanotic tetralogy of Fallot is difficult, since in tetralogy, as in all forms of tetralogy, the crista may not only be displaced but also be hypoplastic. Embryologically, the anomaly is caused by hypoplasia of the conus septum.
In young children the clinical picture of Eisenmenger complex is that of a VSD with a large left-to-right shunt and RV and pulmonary-artery hypertension. As might be expected, P2 is very loud, and an ejection click is usually present. Pulmonary vascular changes develop early, and older children become increasingly cyanotic.
On chest radiography, cardiomegaly and increased pulmonary vascularity are seen in young children. In older patients the heart is only slightly enlarged or normal, and the vascular pattern of the peripheral lung fields is attenuated, while the main pulmonary arteries remain large. Initially the ECG typically shows biventricular enlargement, in older patients resembling more and more the pattern seen in tetralogy of Fallot. At cardiac catheterization, the right ventricular and pulmonary-artery pressures are high and equal to the systemic pressure, and, even in acyanotic patients with a predominant left-to-right shunt, some systemic arterial desaturation may be detectable by oximetry. Angiocardiography occasionally demonstrates the hypoplastic crista.
Pulmonary banding should be carried out early, in an effort to prevent the development of pulmonary hypertension and increased PVR. Once a right-to-left shunt is established, surgery is contraindicated, and in the severely symptomatic patient, lung transplantation should be considered.
Double-Outlet Right Ventricle
In the double-outlet right ventricle, both the aorta and the pulmonary artery originate from the right ventricle (see Plate 5-22). The pulmonary artery is normally located; the aorta arises from the ventricle to the right of and posterior to the pulmonary artery. Pulmonary valvular with infundibular stenosis is common and usually severe. A muscular band of varying width derived from the bulboventricular flange separates the aortic valve from the mitral valve. A VSD is always present, forming the sole outlet for the left ventricle. Embryologically, double-outlet right ventricle probably results from a persistent bulboventricular flange, which keeps the aortic and mitral valves separated and prevents the normal transfer of the aorta to the left ventricle. The conus septum cannot develop in its normal relation to the right atrioventricular valve, so it cannot contribute to the formation of the A-V valve.
Clinical and chest radiograph findings for double-outlet right ventricle are similar to Eisenmenger complex. The ECG shows biventricular or predominantly RV enlargement, and the QRS axis usually is oriented to the right and superiorly. When pulmonary stenosis is present, the double outlet may be mistaken for tetralogy of Fallot. Cardiac catheterization demonstrates systemic RV pressures and may show mild systemic arterial desaturation, even in acyanotic children. The aortic blood always has higher oxygen saturation than the pulmonary-artery blood. Angiocardiographically, the aorta is far to the right, the aortic valve is too “high,” and the aorta and pulmonary artery are almost in the same frontal plane. These features can be seen with cardiac ultrasound, CTA, and MRI.
Treatment for double-outlet right ventricle is surgical. As in Eisenmenger complex, pulmonary banding should be done early in patients without pulmonary stenosis. Correction is feasible (in those who do not have prohibitive PVR changes) by connecting the VSD and the adjacent right-side aorta by a half-shell–shaped patch. Occasionally, enlargement of the VSD will be required. Where pulmonary with infundibular stenosis is present, outflow tract enlargement with a prosthesis is usually also necessary, because of not only the severity of the pulmonary stenosis, but also the obstructive effect of the anterior bulge of the subjacent tunnel patch. A major right coronary conal artery is often present and should be taken into account in placing the ventriculotomy; not knowing its position may be dangerous in some patients with pulmonary stenosis.
A special form of double-outlet right ventricle, in which the VSD is located anteriorly beneath the pulmonary valve, is known as the Taussig-Bing complex. The pulmonary valve overrides the defect, and at cardiac catheterization the saturation of the pulmonary artery blood is higher than that of the aorta. Corrective surgery consists of completing the transposition of the pulmonary artery to the left ventricle by patch closure of the VSD, and intraatrial venous transposition, as described by Mustard. Although the result is a hemodynamic improvement, whether the right ventricle can sustain the systemic circulation for a normal life span remains conjectural. Again, the degree of PVR must be considered in selecting patients for surgery.
Right Ventricular Outflow Obstruction with Intact Ventricular Septum: Pulmonary Stenosis
Right ventricular outflow obstruction with intact ventricular septum is usually caused by stenosis of the pulmonary valve. “Pure” subvalvular (infundibular) stenosis is rare and may be the result of an abnormality in the architecture of the RV outflow musculature, or part of the myocardial-dysplasia syndrome and associated with hypertrophic cardiomyopathy.
The pathologic anatomy of the pulmonary artery root and valve varies in isolated or pure pulmonary valvular stenosis (see Plate 5-23). Typically, the valve is more or less dome or cone shaped, with the valve ostium located at the apex of the dome. Rudimentary fused commissures are present near the base of the dome, and the sinuses of Valsalva are hypoplastic. In other cases the valve cusps are fairly normal but thickened, and the commissures are fused for a variable distance or, occasionally, completely obliterated (pulmonary valve atresia). The valve may be bicuspid or tricuspid. A bicuspid (but not stenotic) pulmonary valve causes little or no functional disturbance and has minimal clinical significance, but it may be prone to endocarditis. Unlike the aortic valve, calcification does not occur in the pulmonary valve later in life. Even in relatively mild cases of pulmonary stenosis, RV hypertrophy is present. If the stenosis is severe, the hypertrophy becomes immense, and the tricuspid valve is often hypoplastic and thickened and may be incompetent.
The clinical features vary considerably depending on the degree of stenosis. Children with mild or moderate pulmonary stenosis are well developed, are not cyanotic, and are asymptomatic, except perhaps for fatigue and dyspnea on exertion. In those with severe stenosis, cyanosis is common and usually caused by a right-to-left shunt at the atrial level through a patent foramen ovale. A tricuspid regurgitant murmur may be present in these children, which accounts for increased right atrial pressure and thus allows a right-to-left shunt through a patent foramen ovale. Heart failure is rare but may occur, generally in infancy. A forceful precordial heartbeat may be palpable, and a thrill is usually felt at the base, to the left of the sternum, in the suprasternal notch. In the same areas a typically loud, diamond-shaped systolic murmur is audible, often preceded by an ejection click if the stenosis is mild or moderate. If present, both A2 and P2 of S2 are clearly split in proportion to the severity of the stenosis. Good correlation exists between ECG findings and degree of stenosis. The ECG may be normal in mild cases, but usually clear evidence of RV hypertrophy is present. In general, the more severe the stenosis, the more the QRS axis shifts to the right, and the taller the R waves become in the right precordial leads. The T waves are usually inverted in the right precordial leads but may be upright, even if the corresponding QRS complex looks unimpressive. In severe cases the sequence of R and S waves may be reversed in the precordial leads.
On chest radiography the heart is normal or only slightly enlarged, except in severe cases of considerable enlargement, particularly in the presence of right-sided heart failure. The vasculature is normal or somewhat diminished, and there is poststenotic dilatation of the pulmonary trunk and left pulmonary artery, except when the stenosis is subvalvular or extreme. At cardiac catheterization the RV pressure is elevated, up to 200 mm Hg or more in severe cases; pulmonary artery pressure is normal or decreased. The arterial blood is desaturated in severe cases in which a right-to-left shunt is present at the atrial level. Selective RV angiocardiography is helpful in outlining the particular anatomy present, as is cardiac ultrasound and MRI.
No treatment is indicated in patients with mild pulmonary stenosis. In the more severe cases, the treatment is surgical and consists of relieving the obstruction, employing open cardiopulmonary bypass procedures. Closed transventricular pulmonary valvotomy is indicated as an emergency procedure in infants with severe stenosis who are cyanotic, have syncopal episodes, or are in heart failure. In infants it is advisable to relieve the stenosis, at least partially, by the transventricular approach, if RV pressure is 100 mm Hg or higher. Catheter-based balloon valvuloplasty is a preferred option, and the results are excellent. Even if no clear symptoms are present, this is done in an effort to prevent the development of massive RV hypertrophy, which may make the surgery difficult and hazardous in older children.
Anomalies of Left Ventricular Outflow Tract
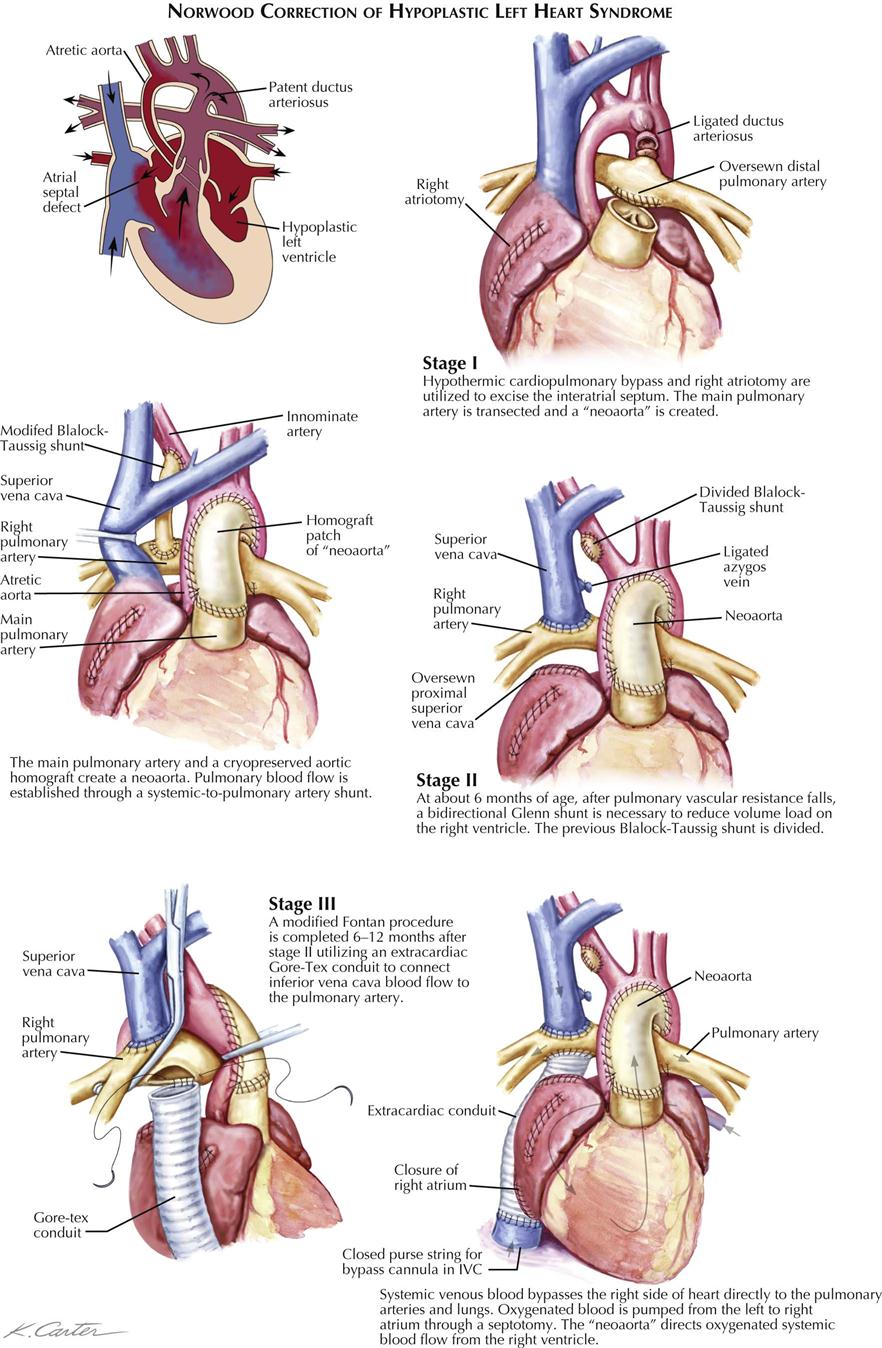
Bicuspid Aortic Valve
Aortic valve anomalies are common. A bicuspid (but not stenotic) aortic valve causes no symptoms in children or young adults, and the only finding is a systolic murmur in the aortic area (see Plate 5-24). Later in life, however, such valves almost invariably become thickened and calcified, with resultant significant stenosis. In a bicuspid aortic valve, the two cusps are unequal in size, with the larger cusp approximately equally divided by an abortive raphe. A truly bicuspid valve, equally divided, is uncommon.
Aortic Valvular Stenosis
Stenotic aortic valves are usually also bicuspid (see Plate 5-24). A developed tricuspid valve with partially fused commissures is occasionally seen. Poststenotic dilatation of the ascending aorta is common. Stenotic aortic valves calcify later in life. The clinical picture varies with the degree of stenosis. Severe aortic stenosis may cause cardiac failure and death early in infancy. More often, however, aortic stenosis is well tolerated, and both children and young adults are usually asymptomatic and well developed. Symptoms consist of fatigue, chest pains, and syncope. Cardiac failure is rare in children. Obvious cardiomegaly is uncommon, but a precordial or apical “slow-rising heave” is usually present. A loud, typically harsh systolic ejection murmur, preceded by an ejection click and often accompanied by a thrill, is best heard at the second interspace, to the right of the sternum, and is transmitted well to the apex and along the carotid arteries. A suprasternal thrill is always palpable.
Chest radiography shows that the heart is normal or mildly enlarged, with a rounded left border in most cases. A poststenotic dilatation of the ascending aorta may be present. The ECG is normal in mild and moderately severe cases. Normal radiographs and ECGs do not necessarily indicate mild aortic stenosis. On the other hand, clear ECG evidence of LV hypertrophy generally indicates significant obstruction. The vectorcardiogram is more sensitive in aortic stenosis because the QRS forces in the horizontal plane are found to be directed far posteriorly. The only accurate means of evaluating the severity of aortic stenosis is cardiac catheterization to determine the pressure gradient across the aortic valve. Cardiac Doppler ultrasound can assess severity of aortic stenosis by measuring velocity across the aortic valve and along with angiocardiography is useful in distinguishing valvular aortic stenosis from subvalvular LV outflow obstruction. Cardiac ultrasound, CTA, and MRI are also helpful.
Imaging will also indicate whether the aortic root is hypoplastic, making surgery more difficult, unless aortic replacement or reconstruction is done.
The prognosis during childhood and adolescence is good. Sudden death, the only major complication other than bacterial endocarditis, does occur, but its incidence is minimal in patients with aortic stenosis and normal ECG. Nevertheless, aortic stenosis of other than a mild degree is the only congenital cardiac anomaly in which the patient’s activities, particularly participation in competitive sports, should be restricted. Surgery is reserved for patients who are symptomatic, show deterioration on the ECG, or have high-pressure gradients across the aortic valve.
Aortic Atresia
In patients with aortic atresia, the aortic valve is completely atretic or severely stenotic, and the ascending aorta is extremely hypoplastic (see Plate 5-24). The aorta serves merely to bring blood to the coronary arteries. The aorta is always fed by a widely patent ductus arteriosus. The ventricular septum is generally intact, and the left ventricle is diminutive or occasionally absent. Its endocardium is fibroelastic, thickened, and pearly white, resembling enamel. The mitral valve usually is tiny but normally formed; rarely it is atretic or absent. Left atrial blood shunts across an ASD, or more often across a foramen ovale whose valve has prolapsed to the right into the right atrium.
Prognosis is poor, and unless a Norwood procedure is performed, the baby will die within weeks of birth. The surgery can be divided into three main stages (see Plate 5-26).
Fibrous Subaortic Stenosis
Fibrous subaortic stenosis is an uncommon anomaly in which a partial or complete ring of fibrous tissue is located just below the aortic valve (see Plate 5-25). This tissue may extend into the base of the aortic sinuses of Valsalva, and the aortic valve cusps may be abnormally formed. Occasionally, the aortic valve is stenotic or incompetent. The fibrous ring causes a subaortic stenosis of varying degree. The embryology of this anomaly is not clear but probably results from malformation of the proximal extremity of the truncus septum where it joins the conus septum.
Clinical, radiograph, and ECG findings for fibrous subaortic stenosis are similar to those for the more common valvular aortic stenosis. Diagnosis can be confirmed by cardiac ultrasound. Cardiac catheterization allows entry into the left ventricle retrograde from the aorta. The ventricular pressure is elevated to a varying degree, depending on the degree of stenosis. On drawing the catheter back into the aorta, the ventricular systolic pressure drops sharply as soon as the subaortic ring has been passed and maintains a ventricular pressure pattern; no further decrease is usually seen on withdrawal beyond the valve, although an aortic pressure pattern is seen. Thus the infundibular nature of the stenosis is demonstrated. Selective LV angiocardiography can visualize the stenotic ring, as can cardiac ultrasound, MRI, and CT.
The treatment of the patient with fibrous subaortic stenosis is surgical and consists of transaortic removal of the ring during cardiopulmonary bypass. The ring is predominantly a crescent across the anterior two thirds of the outflow tract, and thus damage to the aortic cusp of the mitral valve must be avoided (see Plate 5-24). Because the aortic valve cusps are usually normal in fibrous subaortic stenosis, the long-term surgical result is more favorable than in valvular aortic stenosis.
Hypertrophic Cardiomyopathy
In hypertrophic cardiomyopathy (HCM), no discrete stenosis of the subvalvular region is demonstrable anatomically. The anomaly represents one manifestation of a condition characterized by enormous hypertrophy of the ventricular musculature. Most frequently, this involves the left ventricular wall, particularly the septum (asymmetric septal hypertrophy, ASH) but the right ventricle may also be hypertrophied, as in the specimen shown in Plate 5-25, which depicts the anterosuperior half of the heart as seen from below and behind. The cause of the severe ventricular hypertrophy and whether the condition represents a true anatomic anomaly remain unknown. Several members in a family may be affected.
Clinically, patients with HCM are asymptomatic initially, and most live into late childhood or early adult life. Sudden death is common, particularly in the adolescent during strenuous athletic competition. The clinical picture resembles other forms of aortic stenosis, with important differences. Usually there is a systolic murmur, heard best at the lower left sternal border rather than in the aortic area and possibly related to mitral insufficiency. No ejection click occurs. Mitral insufficiency may be an associated finding and tends to be maximal rather late in systole. The peripheral pulses are usually easily palpable and may seem brisk, because the initial phase of ventricular ejection is normal, giving the typical rapid rise in arterial pressure. With further contraction, the aortic subvalvular area is suddenly narrowed severely (systolic anterior motion of anterior leaflet of mitral valve, SAM), and the arterial pressure drops while the proximal ventricular pressure rises. Finally, as the outflow part of the left ventricle relaxes again, the remainder of the blood can be discharged into the aorta. This causes a second hump in the arterial-pressure tracing.
Chest radiography shows that the heart is mildly to moderately enlarged, with a rounded left border. The pulmonary vasculature is normal. The ECG shows LV hypertrophy, even when minimal or no pressure gradient is demonstrable in the aortic subvalvular area. If present at cardiac catheterization or during cardiac Doppler ultrasound, a pressure gradient may be localized in the body of the ventricle, at the level of the apices of the hypertrophied papillary muscles, or in the subaortic area. The pressure gradient may vary considerably in severity from day to day and is increased, or induced when not present initially, by exercise or by the infusion of isoproterenol, inhalation of amyl nitrite, or other arterial vasodilators. Angiocardiographically, or with cardiac ultrasound, MRI, or CTA, the left ventricle is thick walled with a peculiar appearance in systole. Mitral incompetence is typically present.
Surgery with the Morrow procedure (septal myectomy) is the reference standard of management of symptomatic HCM. Alcohol septal ablation has become an alternative nonsurgical therapy with excellent results in most patients, producing a local myocardial infarction in the hypertrophied septum. When the subaortic pressure gradient is relieved by resection or ablation of the hypertrophied septal muscle mass, the patients have greatly reduced or eliminated symptoms.
Supravalvular aortic stenosis is an interesting form of aortic obstruction in which there is a waistlike narrowing of the ascending aorta just above the aortic valve. The ascending (or even entire) aorta may be hypoplastic, and the sinuses of Valsalva often are aneurysmal. Coarctation and hypoplasia of the pulmonary arteries are common. The lesion is often part of a syndrome; the other manifestations are mental retardation, peculiar facies with wide nasal bridge, abnormally formed ears, recessed chin, and narrow jaw with irregularly placed teeth, and hypercalcemia in early childhood. The diagnosis can be established best by aortography, cardiac ultrasound, MRI, and CTA.
Surgical treatment consists of incising the ascending aorta longitudinally across the narrow area, followed by interposition of a prosthetic patch or replacement of the ascending aorta.
Transposition of the Great Vessels
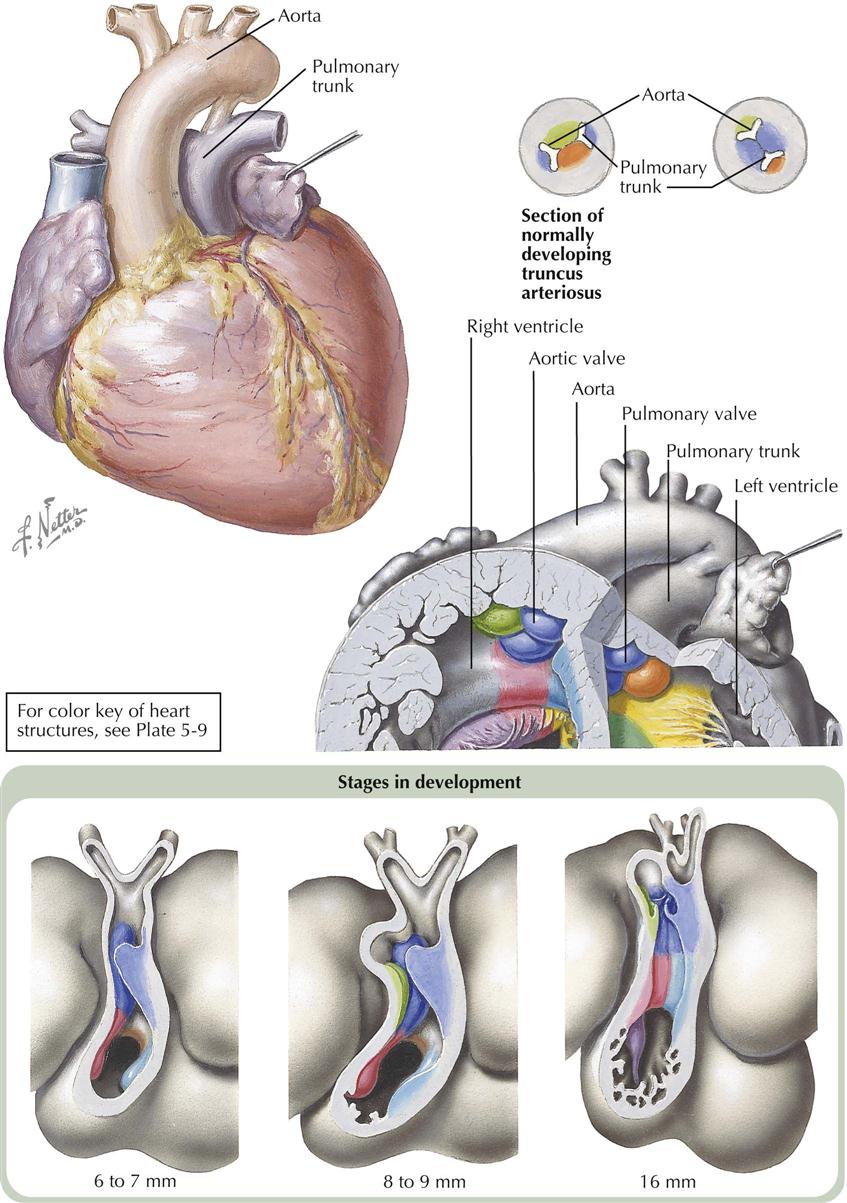
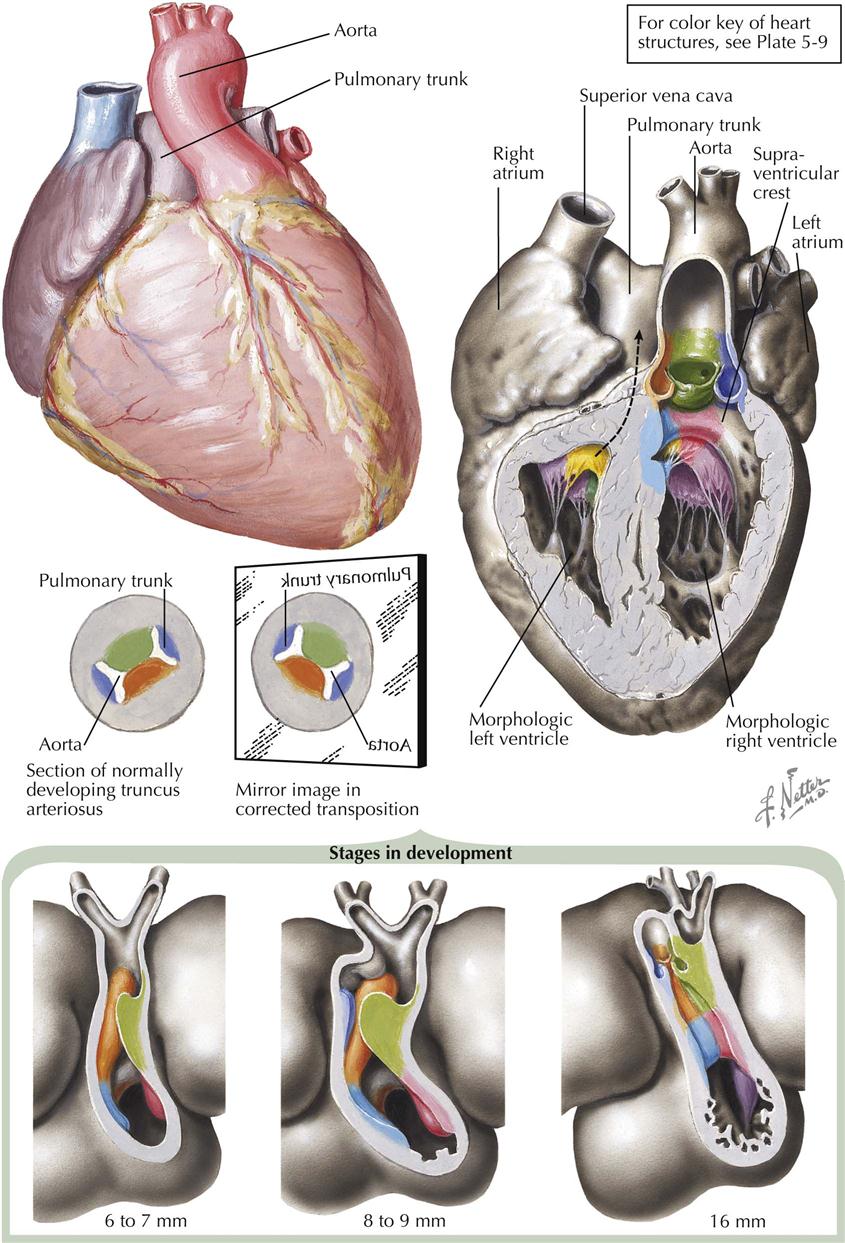
An abnormal anteroposterior relationship of the two arterial trunks, with one or both vessels arising from the wrong ventricle, is extremely common and often forms a component of complex cardiac anomalies. In simple, complete transposition of the great vessels, the aorta arises anteriorly from the right ventricle and the pulmonary trunk arises posteriorly from the left ventricle, with the two arterial trunks running parallel to each other. This discussion here considers only the complete type of transposition, without associated anomalies other than a septal defect, patent ductus arteriosus, or pulmonary stenosis (see Plate 5-27).
The anteroposterior relationship between the aorta and the pulmonary artery varies, but most often in transposition of the great vessels, the pulmonary artery lies posterior and to the left of the aorta. In uncomplicated cases, the ventricles are normally formed. The aortic valve, however, lies slightly more to the right of the pulmonary valve than in a normal heart. In less than half the cases, the ventricular septum is intact, and no other anomalies are present.
The great morphologic similarity of hearts that have isolated transposition of the great vessels suggests that the anomaly is simple, that is, caused by a single embryologic error. Furthermore, with normally formed ventricles, the error probably occurs in the truncus arteriosus. Two pairs of truncus swellings usually develop. Of these, the major pair executes the partitioning of the truncus arteriosus, and the intercalated valve swellings merely form a pair of arterial cusps. Transposition may result if the wrong truncus swellings become the major pair; the pulmonary and aortic intercalated valve swellings form the truncus septum and align themselves, respectively, with the sinistroventral and dextrodorsal conus swellings. The result is that the aorta arises from the right ventricle anteriorly, and the pulmonary artery arises from the left ventricle posteriorly. The conus septum develops normally, and therefore its derivatives—the crista supraventricularis, medial part of tricuspid valve, and medial papillary muscle—are normal.
Transposition of the great vessels, the most frequent cause of cardiac failure in early infancy, particularly with an intact septum, shows the following clinical features. If a VSD is present, failure occurs usually after a few weeks or months. If pulmonary stenosis is also present, it may be delayed much longer or may occur only late, as a terminal event. Cardiomegaly is absent at birth but is usually already marked within the first 2 weeks of life. The anteroposterior diameter of the chest is increased, and a left precordial bulge is common. Cyanosis may be present from birth or may appear within the first few days or weeks of life. It appears earlier, is more intense, and progresses more rapidly if associated anomalies, which provide for mixing between the circulations, are absent. Conversely, children with large ASD or VSD may not become cyanotic for many months or even, in exceptional cases, for a few years. Cyanosis increases on crying, but this intensification usually is not as pronounced as in cardiac anomalies where venoarterial shunting is associated with diminished pulmonary blood flow (e.g., tetralogy of Fallot). Although the birth weight is usually normal, weight gain is poor, and infants who survive for some time become progressively more underweight. Dyspnea and rapid, shallow respirations are usually present.
The second heart sound at the base is loud because of the proximity of the aortic valve to the chest wall. S2 may appear single because of poor transmission of P2 resulting from the far-posterior location of the pulmonary valve. In cases with an intact ventricular septum, a murmur is usually absent or, if present, is not loud. If a VSD is present, a murmur is almost always audible and may be quite loud, but often not harsh or pansystolic. A diastolic mitral flow murmur may be present. In patients with associated pulmonary stenosis, a moderately loud systolic murmur, often accompanied by a thrill, is audible at the base.
The chest radiographic findings are usually typical. The heart is enlarged and characteristically egg shaped, and the upper mediastinum is narrow because the aorta and pulmonary artery lie almost in the same sagittal plane. The pulmonary vasculature is increased, with corresponding left atrial enlargement. In cases with pulmonary stenosis, there is little or no cardiomegaly, and the vasculature is normal or diminished. The ECG typically shows right-axis deviation, right atrial hypertrophy, and RV hypertrophy. In the newborn period, it may be difficult to detect ECG abnormalities unless qR patterns are present in the right precordial leads. Additional evidence for LV hypertrophy is usually seen in patients with a large VSD or patent ductus arteriosus.
Although the diagnosis usually can be made clinically, confirmation by angiocardiography, cardiac ultrasound, MRI, or CTA may be required, especially when the picture is obscured by associated anomalies. A peripheral venous angiocardiogram is easily done and indicated in extremely sick infants, but selective angiocardiography is more precise and preferred.
The prognosis in simple transposition of the great vessels is extremely poor, and the great majority of infants die within the first 3 months of life. It is considerably better if an ASD or VSD with pulmonary stenosis is also present.
Surgical Treatment of Transposition of Great Vessels
Medical treatment is useful only in combating cardiac failure, and surgery is usually performed early. In infants this consists of the creation of an ASD, the Blalock-Hanlon procedure, which provides satisfactory palliation in patients who survive (see Plate 5-28). Also, an ASD can be created “medically” using the Rashkind procedure. A balloon-tipped catheter is introduced by the femoral vein and passed through the foramen ovale into the left atrium. After inflation of the balloon with dilute radiopaque fluid, the catheter is withdrawn forcefully, thus tearing the thin valve of the foramen ovale (see Plate 5-29). This procedure can be carried out even in small, sick infants, and it may be lifesaving.
Various methods of anatomic, or at least functional, correction have been devised. The Mustard operation (atrial switch) uses cardiopulmonary bypass to remove the atrial septum and suture a pericardial graft into the atrium so that the pulmonary venous blood is directed toward the right (systemic) ventricle and the systemic venous blood toward the left (pulmonary) ventricle. Atrial switch is most successful in children older than 2 years in whom the ventricular septum is intact and the only associated lesion is a naturally occurring or artificially created ASD. The Mustard procedure provided good palliative results but now is infrequently used because of problems developing during adolescence or early adulthood.
The Jatene procedure (arterial switch) is now often used to treat transposition of the great vessels (see Plate 5-29). If pulmonary stenosis or VSD is present correction is much less satisfactory, and the surgical mortality is high. Children with transposition and VSD rapidly become inoperable; the early development of pulmonary vascular changes results in a high PVR. Relief of the stenosis of the posteriorly positioned pulmonary-artery valve is technically difficult and often is impossible to perform satisfactorily. In patients with a VSD and marked pulmonary stenosis, the creation of a palliative Blalock-Taussig shunt is preferred (see Plate 5-20, bottom left).
Transposition of Great Vessels with Inversion of Ventricles (Corrected Transposition of Great Vessels)
As in simple, complete transposition of the great vessels, the ascending aorta is situated anterior and parallel to the pulmonary trunk but arises anteriorly from the left-side ventricle, and the pulmonary trunk originates posteriorly from the right-side ventricle (see Plate 5-30). Thus the transposition is at least functionally corrected; that is, the aorta receives arterial blood and the pulmonary artery receives venous blood. In addition to the reversed anteroposterior relationship of the great vessels, the left-right relationship of the ventricles is also reversed. The right-side ventricle morphologically resembles a normal left ventricle, and its atrioventricular valve is a mitral valve. (The valves always follow the ventricle.) The left-side ventricle structurally resembles a right ventricle and contains a tricuspid valve. The morphology and position of the atria are normal.
The left A-V valve is usually abnormal and incompetent. Ebstein’s anomaly of the left-side tricuspid valve is common. Other associated defects are preexcitation of the ventricle (Wolff-Parkinson-White syndrome), VSD, pulmonary stenosis, and double-inlet (right-side) left ventricle with a rudimentary left-side (RV) outflow chamber from which the aorta arises.
Corrected transposition can be seen as caused by a single embryologic error. If very early the cardiac tube bends to the left rather than the right, and the bulboventricular loop internally develops normally, but in mirror image, then all structures derived from the bulboventricular part of the heart (e.g., A-V valves, ventricles, proximal great arteries) will become inverted. Only the intrapericardial, freely movable part of the embryonic heart can participate in the inversion; the fixed, extrapericardial parts (atria, sinus venosus, truncoaortic sac) cannot. Therefore, the atria develop and are located normally. Development of the truncoaortic sac itself proceeds normally; because the inverted truncus arteriosus is partitioned in mirror image, the end result is transposition of the great vessels, with the aorta arising anteriorly from a left-side right ventricle and the pulmonary trunk posteriorly from a right-side left ventricle.
The clinical features of transposition with ventricular inversion are determined largely by the character and severity of associated anomalies. Conduction disturbances, with varying degrees of heart block, are common and may occur in the absence of VSD or other gross defects. In the rare uncomplicated cases, S2 at the base to the left of the sternum is loud because of the anterior location of the aortic valve. A low-grade systolic murmur of uncertain origin may be audible at the base. Other auscultatory findings may also vary.
Chest radiographs may show features suggesting corrected transposition. The vascular pedicle may be narrow, as in simple complete transposition. Of more significance is an indentation of the left side of a barium-filled esophagus, caused by the enlarged, posteriorly located pulmonary trunk. The left upper heart border may be unusually straight or even convex because of the anterior and leftward position of the ascending aorta. Other radiographic features vary greatly and are determined by the associated lesions. The ECG often shows differing amounts of heart block, and the presence of congenital complete heart block in an otherwise asymptomatic child should suggest corrected transposition. Reversal of the initial ventricular activation is evident in the ECG by the absence of a Q wave in leads I, aVL, and the left precordial, and a qR or QS pattern in the right precordial leads. Associated defects will modify the ECG.
At cardiac catheterization the diagnosis may be suspected because of the unusually medial and posterior position of the tip of the venous catheter if it can be made to enter the pulmonary trunk, and the anterior and far-leftward position of the arterial catheter if it is passed into the ascending aorta. Angiocardiography is more valuable than cardiac catheterization and easily establishes the diagnosis as does cardiac ultrasound, CMR, and CT angiography.
The prognosis of the very rare, uncomplicated transposition cases without conduction disturbances should be good. In complicated cases, prognosis depends on the severity of the associated anomalies, which also determine the type of procedure if surgery is indicated.
Anomalies of the Truncus Septum
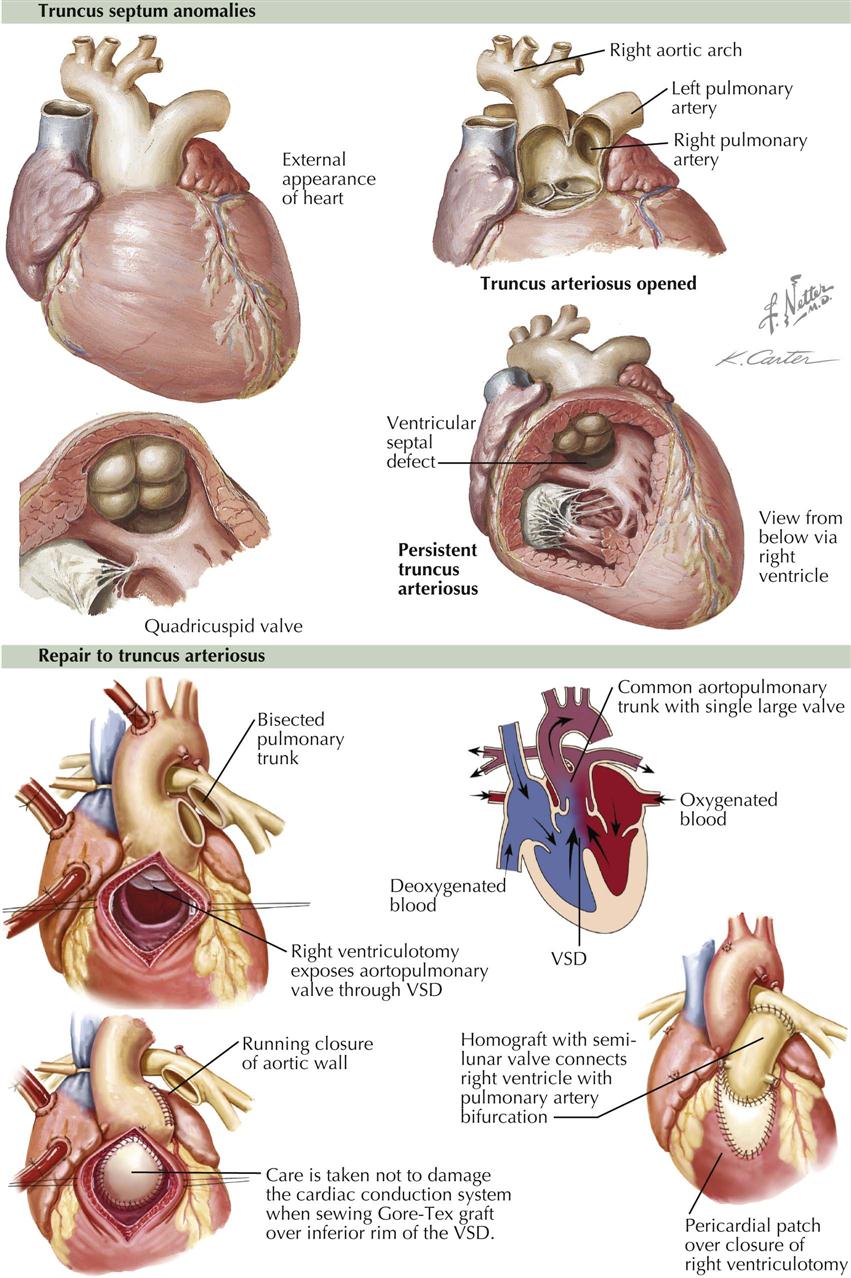
In truncus septum anomalies, a large single vessel arising from the heart gives off the coronary and pulmonary arteries and the aortic arch with its usual branches. The truncal valve is generally tricuspid but may be quadricuspid or bicuspid. A large VSD is always present and located anteriorly. Several types of truncus septum anomaly are known, as determined by how the pulmonary arteries arise from the common trunk. In the most common form, a short main stem left coronary artery bifurcates into a right and a left pulmonary artery. More rarely, these arteries arise independently from the trunk, or the pulmonary arteries, as such, are absent (see Plate 5-31).
The clinical features of the most common truncus septum defect depend largely on the pulmonary vascular bed. If PVR in this bed is high, pulmonary blood flow will be the same as or less than systemic flow. The child is cyanotic and has polycythemia, finger clubbing, dyspnea on exertion, and easy fatigability. If PVR is still low (usually in infants and very young children), pulmonary blood flow is greatly increased. Cyanosis is only mild or absent, but the infants are dyspneic and have feeding difficulties, frequent respiratory infections, and growth failure. Congestive heart failure is common. With the development of a high PVR in the few surviving children, the left-to-right shunt gradually diminishes, and the heart decreases in size. The patient’s general condition improves, but cyanosis appears and generally is progressive. Some patients remain almost acyanotic for many years, whereas others are among the most deeply cyanotic individuals ever seen.
A systolic murmur is best heard at the third or fourth intercostal space to the left of the sternum and is preceded by an ejection click. S1 is normal; S2 is very loud and may be followed by a diastolic murmur, which is usually caused by incompetence of the truncal valve. A continuous machinery murmur, so characteristic of patent ductus arteriosus, is unusual.
The chest radiograph findings vary. In children with large left-to-right shunts, the heart is large, at times with an upturned apex, and the vascularity is much increased. The left upper heart border is usually concave, and the aortic knob is large. As the magnitude of this shunt diminishes with increased PVR, the cardiomegaly and pulmonary plethora also decrease. The ECG shows a normal or, more commonly, a right axis deviation and either right ventricular hypertrophy or in a case with large left to right shunts, biventricular or rarely only left ventricular hypertrophy, tall peaked P waves are common. Retrograde aortic angiography can establish the diagnosis.
Truncus Arteriosus Repair
If left untreated, truncus arteriosus can be fatal. Surgery to repair truncus arteriosus is generally successful, especially if the repair occurs before 2 months of age (see Plate 5-31). Surgery is required to close the VSD with a patch and separate systemic from pulmonary blood flow. This prevents pulmonary hypertension and damage to the lung. The pulmonary arteries are then disconnected from the single great vessel, and a conduit with a valve is placed from the right ventricle to these pulmonary arteries (Rastelli repair).
Aorticopulmonary Septal Defect
This is a rare congenital anomaly, usually characterized by the presence of a large defect between the ascending aorta and the pulmonary trunk (see Plate 5-31). Initially, the clinical features are those of a large left-to-right shunt at the ventricular or arterial level and are roughly intermediate in severity between those of persistent truncus arteriosus and patent ductus arteriosus. The ECG usually shows a pattern of biventricular hypertrophy, at least in childhood, and the chest radiography resemble those seen in a large patent ductus arteriosus. Retrograde aortography is diagnostic. The prognosis without surgical treatment, though more favorable than that of truncus arteriosus, is still rather poor, and the mortality during the first year of life is significant. Fortunately, surgical repair of the defect, employing a cardiopulmonary bypass, can be carried out rather easily, but it should be done at an early age, before a marked increase in PVR renders the patient inoperable.
Anomalous Left Coronary Artery and Aneurysm of Sinus of Valsalva
The anomalous origin of both coronary arteries from the pulmonary artery is extremely rare and is not compatible with postnatal life. A similar, equally rare anomaly involving the right coronary artery causes no symptoms.
Anomalous Left Coronary Artery
More common but still infrequently seen is the anomalous origin of the left coronary artery (LCA) from the pulmonary artery (see Plate 5-32). Since pressures in the aorta and pulmonary artery are equal before birth, and oxygen saturation of the blood in these vessels is much the same, the anomaly is of no consequence prenatally.
After birth, with the normally occurring fall in pulmonary artery pressure, perfusion of the anomalous LCA is greatly reduced, resulting in myocardial ischemia. Given time, the normally present but small intercoronary anastomoses expand. The normal right coronary artery and its branches become dilated and tortuous, but the LCA remains small and thin walled. The potential benefit for the LV myocardium from the developing large intercoronary anastomoses, however, is largely lost because of stealing of the blood into the low-pressure pulmonary artery, with minimal blood reaching the myocardium itself. The left ventricle dilates greatly, and its myocardium becomes fibrotic, particularly in its anterolateral and apical parts. The endocardium is thickened and fibroelastotic, and calcifications may be present.
In infants the main clinical features of anomalous LCA are congestive heart failure with episodes of distress, characterized by pallor, restlessness, slight cyanosis, dyspnea, and sweating. The attacks may be precipitated by feeding or straining and are thought to be ischemic. Between episodes, the child is happy and asymptomatic until the onset of congestive heart failure. The heart is considerably enlarged, but any murmur is insignificant. Symptoms usually do not appear until the babies are 4 to 6 weeks old, and most die within the next few weeks. Occasionally the child’s condition improves, and the cardiomegaly recedes. A minority of cases are asymptomatic in early childhood and may present themselves later with signs of mitral insufficiency. Sudden death is common.
There are no characteristic radiographic findings with anomalous LCA. There is general cardiomegaly in infants, and the pulmonary vasculature is normal, or evidence may indicate pulmonary venous congestion. In older patients the heart is normal in size or only moderately enlarged. The ECG in symptomatic infants typically shows the pattern of anterolateral myocardial infarction and usually LV hypertrophy. In older children and adults the ECG generally indicates only LV hypertrophy. Cardiac catheterization is not helpful, but aortography and selective coronary angiography confirms the diagnosis.
Treatment is surgical: ligation of the defective artery at its origin to prevent further runoff (“coronary steal”). Current surgical procedures establish revascularization by creating a coronary artery system using either a left subclavian artery–coronary artery anastomosis, a saphenous vein bypass graft, aortopulmonary window and intrapulmonary tunnel extending from anomalous ostium to the window, or direct reimplantation. By establishing a patent coronary artery system, most patients experience normalization of LV systolic function, thereby improving long-term survival.
Aneurysm of Aortic Sinus of Valsalva
An aortic sinus of Valsalva aneurysm is caused by a congenital weakness of the bottom of the right coronary sinus or less often the noncoronary sinus (see Plate 5-32). The aneurysm itself does not usually cause symptoms. Rarely, conduction disturbances are present, including complete heart block. Rupture of a congenital aortic sinus aneurysm, occurring usually in young adults, is into a cardiac chamber, generally the right ventricle or right atrium. The sudden onset of an often large aortocardiac shunt may precipitate congestive heart failure or may be rapidly fatal. Therefore the clinical features are often dramatic: dyspnea, chest pain, bounding pulses, and a machinery murmur accompanied by a thrill over the lower precordial area.
Chest roentgenograms are normal in intact aneurysms; after rupture, cardiomegaly usually develops rapidly, and the vasculature is increased. The ECG is not specific. Retrograde aortography establishes the diagnosis of aortic sinus aneurysm.
Anomalous Coronary Arteries Seen in Adult Patients
In general, the coronary artery anomalies considered benign in terms of development of major adverse coronary cardiac events include (1) separate origin of the left anterior descending and circumflex arteries from the left sinus of Valsalva, (2) ectopic origin of the circumflex artery from the right sinus of Valsalva, (3) ectopic coronary origin from the posterior sinus of Valsalva, (4) anomalous coronary origin from the ascending aorta, (5) absent circumflex artery, (6) intracoronary communications, and (7) small coronary artery fistula. Coronary anomalies associated with serious sequelae (e.g., myocardial ischemia, myocardial infarction, syncope, cardiac arrhythmias, congestive heart failure, sudden death) include ectopic coronary origin from the pulmonary artery, ectopic coronary origin from the opposite aortic sinus, single coronary artery, and large coronary fistula.
Adults with Coronary Artery Anomalies
Predicting adverse events in patients with coronary anomalies is at best an educated guess. Since adults with anomalies are survivors, the anomalies are most often incidental findings in symptomatic patients, usually caused by coronary artery disease (CAD) found in other vessels (e.g., origin of circumflex coronary from right sinus of Valsalva). Other patients may have CAD with anomalous right coronary artery (RCA) of origin from the pulmonary artery or huge congenital coronary artery aneurysms. A few patients have had coronary stenosis in an anomalous coronary artery, which then could undergo angioplasty.
The most common congenital anomaly seen in adult patients is an anomalous circumflex coronary artery, which comes from the right sinus of Valsalva as a separate orifice or arises off a branch of the RCA. This anomalous circumflex vessel typically is retrocardiac and therefore probably benign. At coronary angiography in the catheterization laboratory, it may be difficult to determine if the artery is anterior to the great vessels, posterior to the great vessels, between the great vessels, or intramural within the aorta; CT coronary angiography can easily determine this.
As a general rule, any artery originating from either sinus of Valsalva and passing between the great vessels or taking an intramural course through the aorta may be at risk and may cause sudden cardiac death. The potential for poor outcomes from an anomalous coronary artery thus depends on the course of the anomalous vessel. Compression can occur (Laplace’s law), or if the artery takes off at an acute angle, the ostia of the anomalous coronary artery can be severely narrowed.
Again, it is often challenging to diagnose the position of the anomaly during catheter-based coronary angiography, because it is difficult to see the relationship to the great vessels. CT angiography allows evaluation of not just artery caliber and lumen but also course and relationship to adjacent structures, as well as the takeoff angle of the anomalous vessel from the sinus of Valsalva.
Potentially Serious Coronary Anomalies
An anomalous RCA that comes off either the left sinus of Valsalva or branches of the single LCA can be retroaortic, but in the vast majority of patients, it tracks between the aortic and pulmonary artery or even may track intramurally in the aorta. The latter two courses may result in major adverse cardiac events, although data are sparse. In contrast, an anomalous LCA originating from the right sinus of Valsalva or branches of a single RCA can be retroaortic, anterior, or intramural. In most patients, however, the anomalous LCA courses between the aorta and pulmonary artery and thus may be more dangerous than other coronary arteries of ectopic origin.
Anomalies of Aortic Arch System
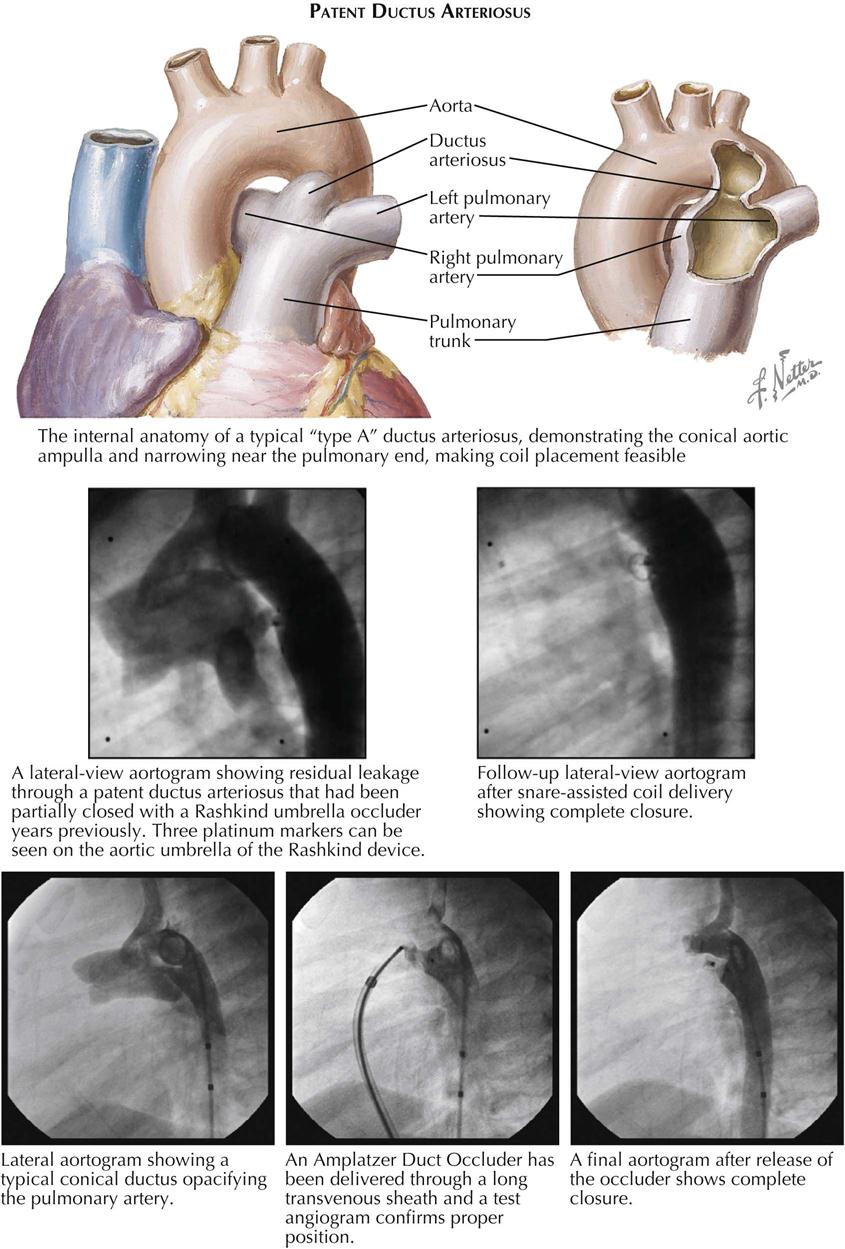
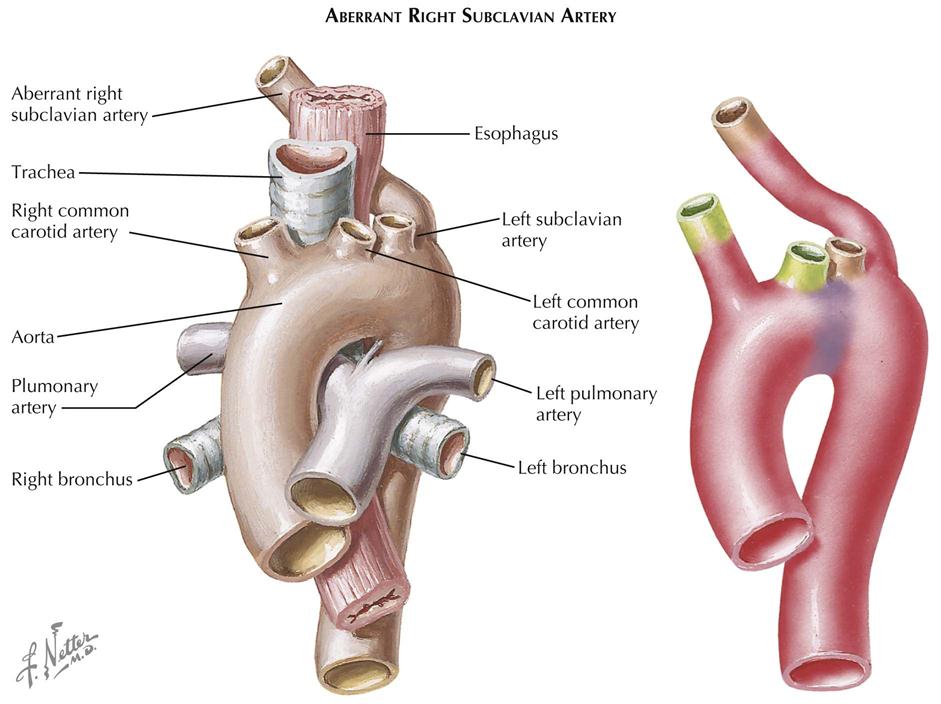
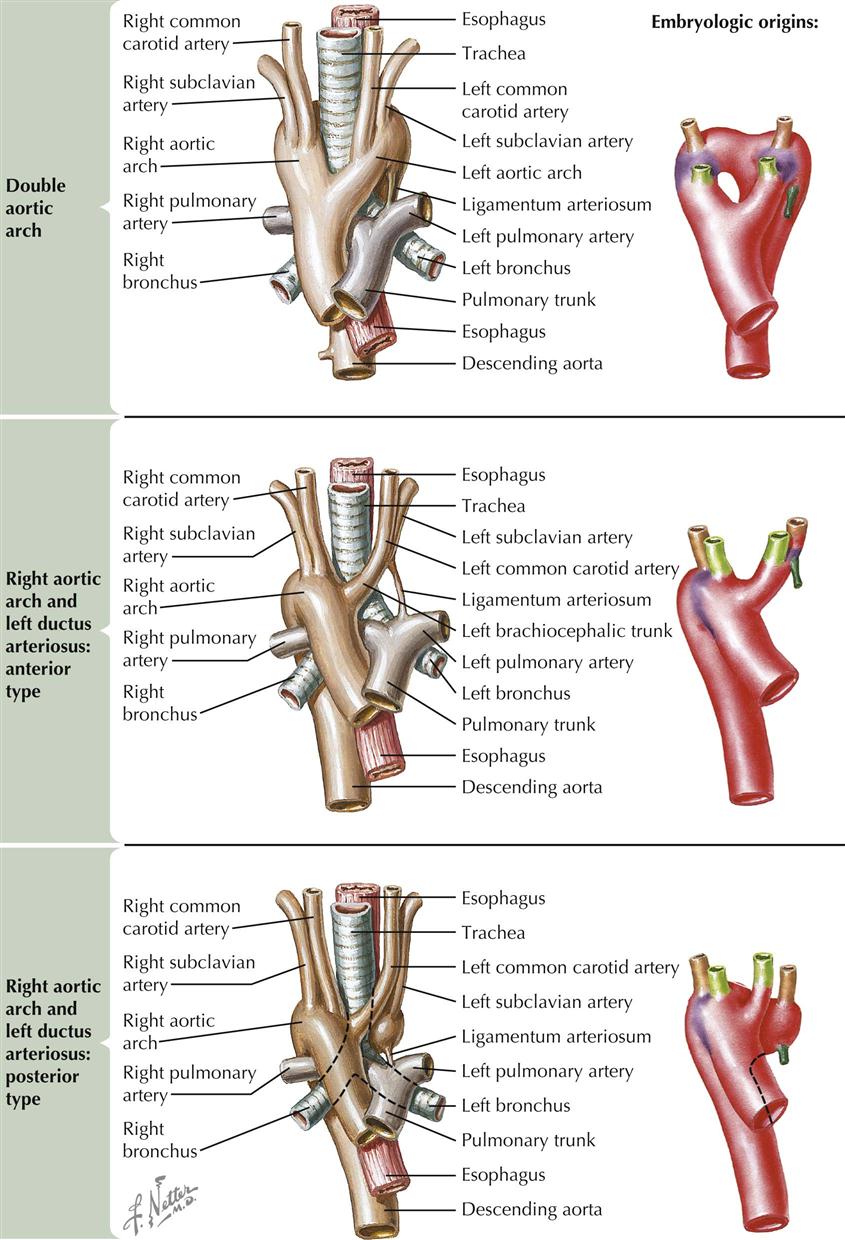
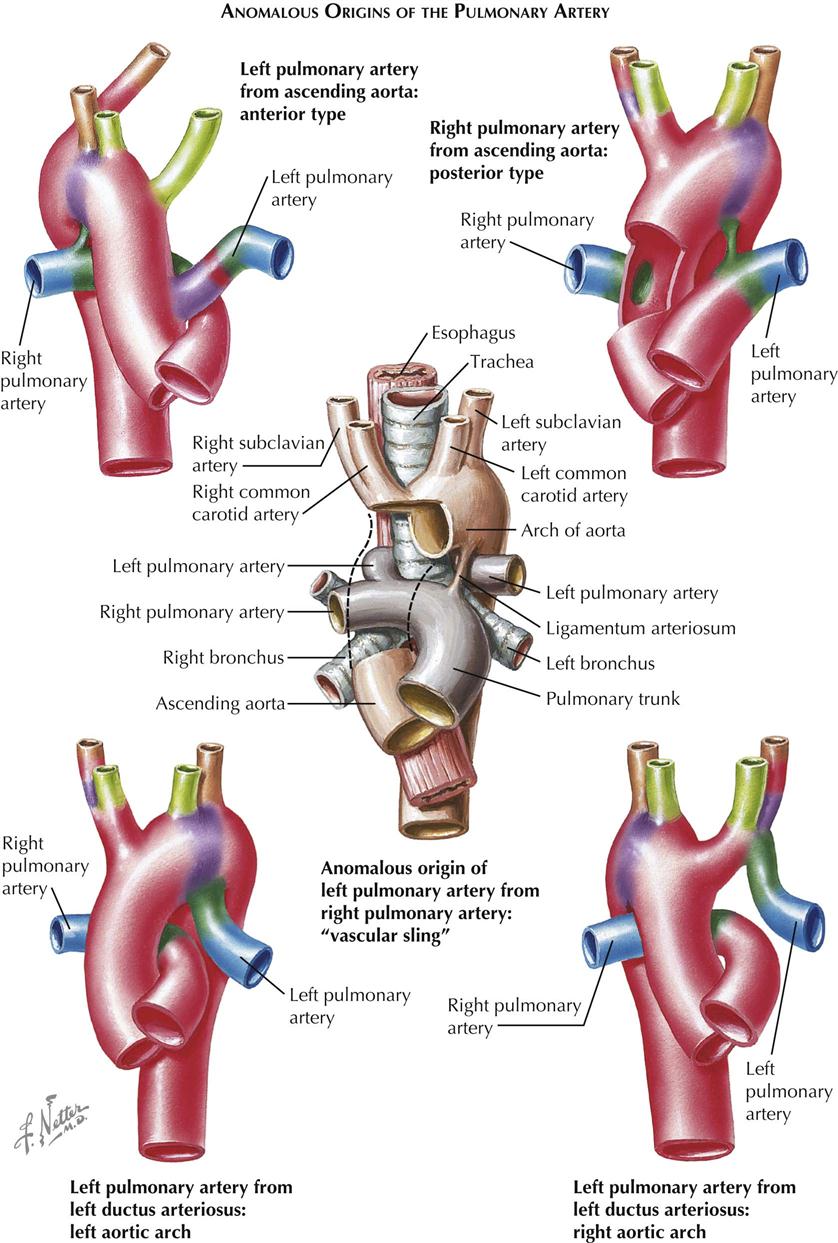
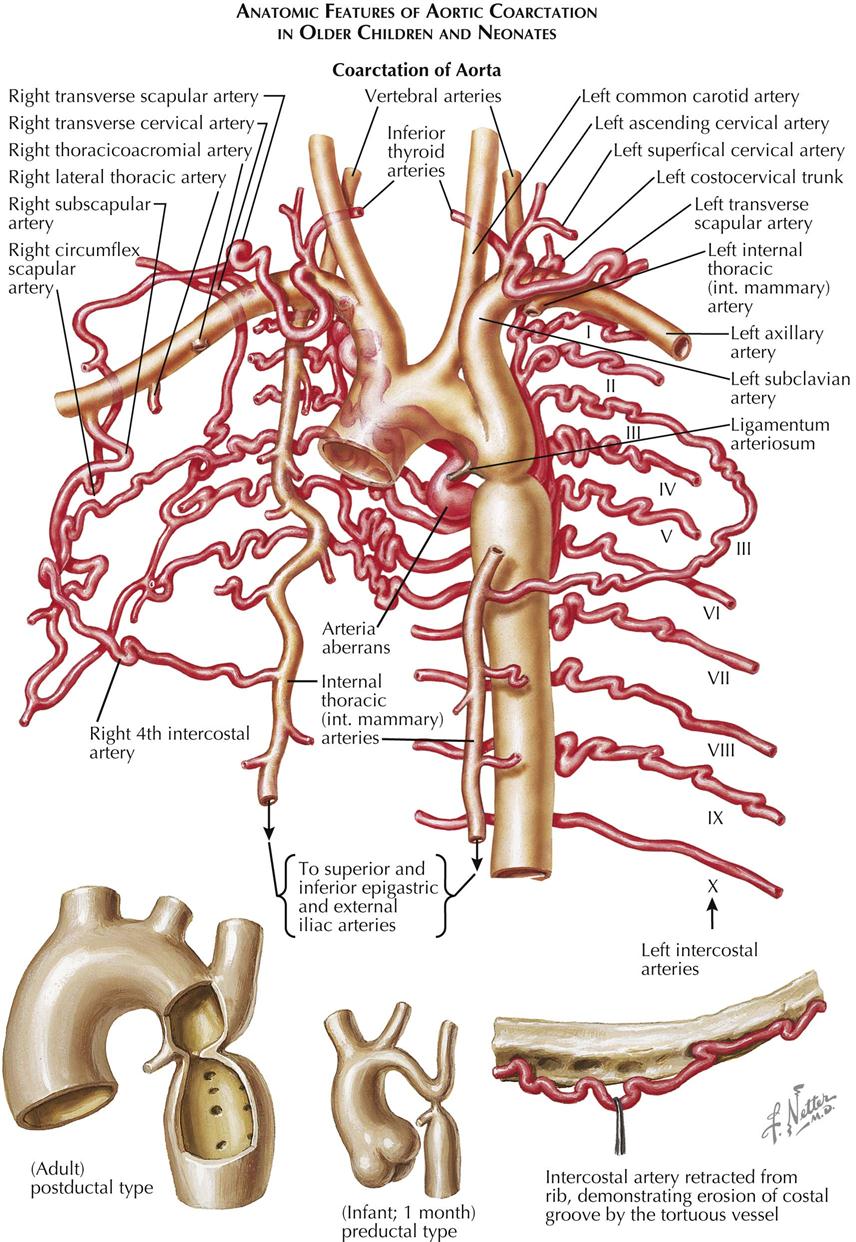
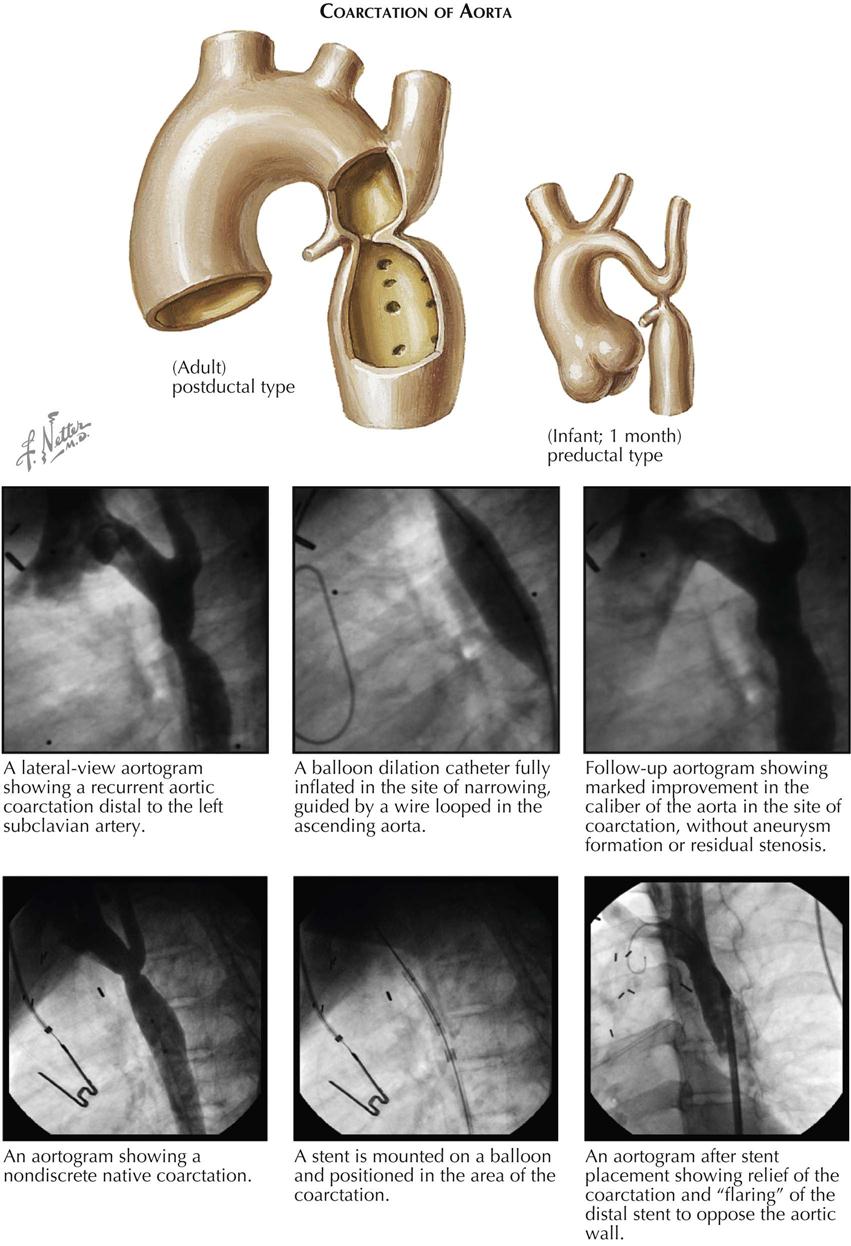
Patent Ductus Arteriosus
As an isolated anomaly, patent ductus arteriosus (PDA) is one of the most common and most benign types of congenital heart disease. The anomaly represents a continued patency of a channel—the ductus arteriosus—that connects the origin of the left pulmonary artery to the aorta and, in fetal life, allows most of the right ventricular blood to bypass the nonfunctioning lungs. After birth and with onset of respiration, its usefulness ends, and the ductus normally closes, at least functionally, within hours after birth. It is not known why, in some babies, the ductus arteriosus remains patent. PDA is a common cardiovascular anomaly in the rubella syndrome, which occurs in children whose mothers had German measles during the first 2 months of pregnancy. PDA is often associated with other cardiac anomalies and in some patients (e.g., with aortic atresia) is always present (see Plates 5-24 and 5-26).
The clinical features of uncomplicated PDA are characteristic in the majority of cases. The ductus arteriosus is rather small, and there are few or no symptoms in early childhood. Growth and development are normal. The heart is normal in size or may be enlarged slightly to moderately, depending on the magnitude of the shunt across the ductus arteriosus. A thrill is often palpable over the left upper sternal border, usually systolic in time, but may continue into diastole. There is a characteristic machinery or fistulous-type murmur that starts shortly after S1, increases in intensity, and decreases again after the end of systole. The characteristic features of the murmur result from aortic pressure being higher than pulmonary artery pressure during all phases of the cardiac cycle, because PVR is considerably lower than resistance in the systemic vascular bed. The easy and rapid runoff of blood during diastole into the low-resistance pulmonary vascular bed results in a wide pulse pressure and explains the bounding peripheral pulses typically present in children with PDA. In premature infants, intravenous indomethacin may help close a PDA by stimulating the muscles inside the ductus to constrict.
Some infants with a large ductus arteriosus may become symptomatic early in life or may even progress to congestive heart failure (see Plate 5-34). The symptoms and physical findings resemble those seen in babies with a large VSD and massive left-to-right shunt.
The chest radiographic findings are similar to those in VSD, but the aortic arch, instead of being small, is usually large. The ECG is normal in almost half the cases; the remainder show LV hypertrophy of the volume-overload type or, in a minority of cases, biventricular hypertrophy. The clinical findings are generally so characteristic that cardiac catheterization and angiocardiographic studies are not necessary. When in doubt, aortograms reveal the ductus arteriosus. Treatment is simple and consists of surgical division of the PDA or percutaneous occlusion. Surgical repair is usually recommended for infants younger than 6 months who have large defects that are causing symptoms. PDA may also be repaired by a cardiac catheterization procedure using occluder devices. Closure is contraindicated once the patient has become cyanotic because of shunt reversal.
Aberrant Right Subclavian Artery
An anomalous right subclavian artery, originating from the descending aortic arch as a last branch and crossing behind the esophagus to the right arm, is common both as an isolated anomaly and in association with other defects. The anomaly can be explained by postulating a disappearance of the right fourth aortic arch, which normally forms the most-proximal part of the right subclavian artery, and persistence of the normally disappearing right dorsal aorta (see Plate 5-35).
An anomalous right subclavian artery can be detected radiographically because it causes a posterior and oblique indentation of the barium-filled esophagus at the level of the fourth thoracic vertebra. This anomaly has been blamed for causing dysphagia lusoria, but rarely does so. Usually it is an incidental finding.
In an embryo with crown-to-rump (C-R) length of 7 to 8 mm, the first two pairs of aortic arches have disappeared, as such. The fifth pair, never well developed in humans, has had only a fleeting existence. The remaining third, fourth, and sixth pairs of arches are well developed, originate from the truncoaortic sac, and encircle the developing esophagus and tracheobronchial tree to join the right and left dorsal aortas. All these aortic arches are normally retained, except for the distal portion of the right sixth arch, which has already disappeared in a 14-mm embryo. The establishment of a normal aortic arch system involves the involution of three additional vessel segments.
The encirclement of the trachea and esophagus by the aortic arch system in an early embryo does not constrict these structures because of the wide opening in the arterial ring. With further development, however, the arteries shorten and widen and the ring becomes tighter, leading to compression of the esophagus and the trachea unless the ring system is opened. To do this, only the distal portion of one of the dorsal aortas needs to disappear. If the distal right dorsal aorta persists as a major channel, a vascular ring can be prevented only if both the distal right sixth arch and the right dorsal aortic segment between the right fourth and sixth arches disappear. This simply results in an anomalous right subclavian artery. However, if only the right distal sixth arch vanishes, a double aortic arch is formed. If only the segment of dorsal aorta between the fourth and sixth arches is removed, the result will be a right-side ductus of the posterior type.
Double Aortic Arch
In patients with double aortic arch the two arches may be equal in size but usually are not (see Plate 5-36). Most often the right-side arch is dominant, regardless of whether the descending aorta courses to the right or more frequently to the left of the spine. At times the left arch is atretic. In most cases the ductus (ligamentum) arteriosus is on the left side, but it may be on the right. Rarely there is a bilateral ductus arteriosus. Left and right common carotid and subclavian arteries arise from their corresponding arches.
The clinical symptoms of double aortic arch reflect obstruction of the trachea and esophagus, and the severity depends on the tightness of the vascular ring. Symptoms generally are present in childhood, often in early infancy, and occasionally patients are asymptomatic and the ring is discovered incidentally. Wheezing, cough, inspiratory stridor, repeated respiratory infections, and aspiration pneumonia are common problems. Extension of the head and back tends to relieve respiratory difficulties, and infants often spontaneously assume such a position of hyperextension. Dysphagia varies in severity, often increasing or appearing when the child begins to take solid foods. Chest radiographic examination, including esophagogram, is important to demonstrate the ring’s constriction of both the esophagus and the trachea.
Surgery is indicated in symptomatic-patients and at times must be done as an emergency procedure. It consists of division of the smaller or atretic arch. If present, a ductus arteriosus or ligament on the same side also should be divided. Otherwise, the patient will be left with a vascular ring of a different type, formed by the right arch, left ductus arteriosus, and pulmonary artery.
Right Aortic Arch with Left-Side Contralateral Ductus Arteriosus
This condition may or may not cause symptoms. If the ductus originates from the bifurcation of the (here left-side) innominate artery, no ring is formed. However, if the ductus arteriosus arises posteriorly from a diverticulum (representing the left dorsal aorta) of the descending arch, a ring is formed by the right arch, left ductus arteriosus, and pulmonary artery (see Plate 5-36). Symptoms of right aortic arch with left-side contralateral ductus arteriosus are similar to those already described for aortic arch anomalies but generally appear later and tend to be less severe. This type of anomaly can also occur in mirror-image fashion in patients with a left aortic arch. Treatment is simple and consists of division of the ductus arteriosus or ligament.
Other Aortic Arch Anomalies
Other variants of aortic arch anomalies involving the third or fourth arches occur, but as long as no constricting ring is formed, these defects only occasionally cause symptoms and are of little clinical importance. Anomalies of the aortic-arch system that mainly involve the sixth arches are uncommon and usually referred to as cases of “absent” left or right pulmonary artery, which is misleading. The embryonic pulmonary arteries proper arise as branches of the sixth arches and actually have appeared before the ventral and dorsal primordia of the sixth arches have joined each other to complete these arches. In most cases of “absent” pulmonary artery, the derivatives of the (intrapulmonary) embryonic pulmonary arteries are present, even though they may have lost contact with the pulmonary trunk and receive their blood supply from a systemic arterial source. True, complete absence of a pulmonary artery and its terminal branches, with the lung supplied solely by anomalous systemic arteries arising from the aorta beyond the arch, is rare. Cases of “absent” pulmonary artery fall into one of two categories: either the distal pulmonary artery of the involved lung is continuous with a large artery originating from the ascending aorta, or it is supplied by a ductus arteriosus.
Pulmonary Artery Arising From Ascending Aorta
This anomaly may be of the anterior or posterior type. The pathogenesis of these two types is entirely different. As indicated in Plate 5-37, the anterior type of pulmonary artery consists of the left fourth arch, a segment of the dorsal aorta, distal part of the left sixth arch, and the left embryonic pulmonary artery. It is always contralateral to the aortic arch and can occur with either a right or a left aortic arch, and the subclavian artery contralateral to the aortic arch can be expected to arise anomalously from the descending aortic arch. The posterior type of artery consists of the proximal portion of the sixth arch and the embryonic pulmonary artery. Apparently, at the time of partitioning of the truncus and truncoaortic sac, it was left “stranded.” It may be expected to be located always on the right side (in situs solitus individuals) with either a right or a left aortic arch, and the branching pattern of the aortic arch vessels is normal, unless another independent anomaly of these vessels is present. A pulmonary artery arising from the ascending aorta is generally large. The clinical features are similar to those seen in other conditions with anomalous communications at the arterial level and depend on the magnitude of the shunt. The diagnosis of pulmonary artery from ascending aorta can best be established by angiocardiography.
Pulmonary Artery Arising From Ductus Arteriosus
If the proximal segment of one of the sixth arches disappears early, the corresponding embryonic pulmonary artery will be supplied instead by the distal sixth arch segment, that is, the ductus arteriosus (see Plate 5-37). Such a ductus arteriosus originates from the aortic arch if the arch is on the same side, or from the innominate artery if the aortic arch is on the opposite side. The ductus arteriosus tends to close, and a large left-to-right shunt therefore is not usually present. In many cases this duct obliterates completely, and the involved lung then is supplied by bronchial artery collaterals.
Left Pulmonary Arising From Right Pulmonary Artery (“Vascular Sling”)
An anomalous left pulmonary artery arising from the right pulmonary artery is an interesting malformation. The vessel always comes off the posterior aspect of the right pulmonary artery at the level of the right main bronchus and carina, running between the trachea and the esophagus to the left lung. The ductus arteriosus is located normally on the left side. Other cardiovascular anomalies may be associated. In some cases, tracheobronchial anomalies have been reported, including complete tracheal rings or a right upper lobe bronchus arising independently from the trachea some distance above the carina (“bronchus suis”). No dysphagia is present, but severe respiratory difficulties are usually manifest at an early age. Inspiratory stridor and expiratory stridor generally are pronounced, and emphysema or atelectasis and pneumonitis of the right upper lobe, or even of the entire right lung, are usual. Lateral chest radiography may show an ovoid mass separating the barium-filled esophagus and air-filled lower trachea. Although not pathognomonic, this finding should strongly suggest the presence of a “vascular sling.” A pulmonary artery angiogram establishes the diagnosis (see Plate 5-37). Treatment is surgical and consists of detaching the anomalous vessel from the right pulmonary artery, followed by reimplantation into the main pulmonary artery. The prognosis without treatment is poor; most infants succumb within the first year of life.
Coarctation of Aorta
Coarctation of the aorta is a congenital narrowing of the descending aorta that usually occurs in the area of the ductus arteriosus, which may be patent (see Plate 5-38). If the coarctation is located proximal to the ductus arteriosus, it is called preductal; if located distal, the coarctation is termed postductal. Preductal coarctation is usually associated with other intracardiac anomalies, and the ductus arteriosus is often widely patent (PDA). The preductal type is most often seen in infants. Postductal coarctation is usually not associated with other intracardiac defects, except for aortic valve anomalies, and is the type usually found in older children and adults.
The coarctation may be mild but usually is tight or even atretic, and the distal aorta receives most of its blood through collaterals. These collaterals are generally extremely abundant, particularly in older children and adults. The main collateral routes are from branches of the subclavian arteries (internal thoracic, transverse scapular, transverse cervical) and the intercostal arteries. Other collateral routes are formed by cervical vessels at the thoracic inlet, the vertebral and anterior spinal arteries. Aortic valve anomalies are common in coarctation of aorta, occurring in about 80% or more of patients.
The clinical features of coarctation are characteristic in most patients. Symptoms are typically absent in childhood, and growth and development are normal (see Plate 5-39). As a rule, coarctation of aorta is diagnosed indirectly and incidentally because the femoral pulses are diminished or absent, a chest radiograph shows rib notching, or the patient has hypertension. Some patients complain of coldness of the feet or headaches. In older adults, symptoms are mainly caused by long-standing hypertension. Bacterial endocarditis, generally of an anomalous aortic valve and rarely at the site of coarctation, may occur at any age beyond early childhood. Rupture of the aorta and intracranial hemorrhage are most often seen in young adults. Some patients develop difficulty in infancy leading to severe congestive heart failure. Associated cardiac defects may be responsible for the early decompensation, but this is not always the case. The most important findings are absent or greatly diminished femoral pulses; this is a pathognomonic sign in children and young adults because other conditions causing obstruction of the lower aorta are rare in these age groups. Hypertension in the proximal aortic bed usually is absent in young children but becomes increasingly common and severe with advancing age. Other findings are systolic murmur and visible or palpable pulsations around the scapulae and in the axillae and intercostal spaces.
Chest radiography shows a normal or enlarged heart (if no significant aortic-valve disease is present); notching of the ribs in older children and adults, caused by erosion of the ribs by the tortuous dilated intercostal arteries; and often a notch in the left border of the upper descending aortic shadow, indicating the site of the coarctation. An esophagogram may outline the opposite (also notched) border of the coarcted aortic segment. MRI can also define the coarctation. In uncomplicated cases the ECG is either normal or more often shows LV hypertrophy. Cardiac catheterization is not essential. If information is desired concerning the status of the aortic valve or coronary arteries, or if other cardiac anomalies are suspected, cardiac catheterization is best postponed until after the coarctation has been corrected surgically or by percutaneous balloon dilatation and stent deployment (see Plate 5-39). Open-heart surgical techniques to repair aortic coarctation include resection with end-to-end anastomosis, patch aortoplasty, left subclavian flap angioplasty, and bypass graft repair. Balloon angioplasty/stent is also an option for initially treating aortic coarctation or for treating recoarctation after surgery (see Plate 5-39).
Endocardial Fibroelastosis and Glycogen Storage Disease
Endocardial fibroelastosis typically is a disease of early childhood, although it has occasionally been described in older children and even in young adults. The pathologic features are characteristic (see Plate 5-40). The left ventricle is hugely dilated and may be almost globular. The interior is coated by a shiny white layer representing the thickened endocardium, and the trabeculae are coarse and relatively few in number. The mitral and aortic valves may be thickened, rather stiff, and occasionally incompetent. Endocardial fibroelastosis also occurs in association with severe cardiac malformations, such as aortic stenosis or atresia, congenital mitral stenosis, and coarctation of aorta. Fibroelastosis is also seen in various forms of acquired valvar disease, presumably as a secondary lesion. Its distribution in these patients is variable and spotty.
The clinical picture is somewhat characteristic. The age of onset is generally between 2 and 7 months but can be earlier or later. Before onset, asymptomatic and perfectly healthy infants appear to develop symptoms of a cold, followed by irritability, refusal to feed, tachypnea, dyspnea, fever, and cough. Signs of cardiac failure, as evidenced by hepatomegaly, abdominal pain, generalized edema with puffy eyelids, and often the appearance of a systolic murmur, reveal the problem to be a more serious one. The heart can be greatly enlarged, with a gallop rhythm and tachycardia.
Chest radiography and cardiac ultrasound confirm the heart’s size, with evidence of pulmonary venous congestion, with or without pneumonitis. The left upper lobe may be overinflated because of compression of the left main bronchus. The ECG shows left-axis deviation or more frequently a normal axis and almost always evidence of lv hypertrophy with tall qR patterns and inverted T waves in the left precordial leads. Additional RV hypertrophy may be present in infants with marked failure, but this disappears after successful treatment. Arrhythmias and conduction disturbances are common. Cardiac catheterization and angiocardiography are usually not necessary, unless associated defects are suspected.
The treatment of endocardial fibroelastosis is the same as for chronic cardiac failure; its acute exacerbations are often precipitated by respiratory infections. Early and prolonged treatment with digoxin may be helpful; digoxin cessation may result in acute cardiac failure, Other therapeutic measures for acute failure and exacerbations of failure may be required, and precipitating factors such as infection and anemia demand immediate attention. Warfarin therapy should be considered if patients have thrombotic/embolic complications.
Glycogen Storage Disease
Excessive deposition of glycogen in the tissues is caused by a hereditary error in carbohydrate metabolism. It is transmitted as an autosomal recessive gene and often occurs in siblings. Several types are distinguished, and the enzyme defect of most of these is known. In the cardiac variety, first described by Pompe, glycogen is deposited in abnormal amounts in all tissues but especially in the heart, which is enormously enlarged and globular, with tremendously thick and solid walls (see Plate 5-40). Microscopically, large central vacuoles in the myocardial fibers, with a thin surrounding shell of cytoplasm, give the sections a peculiar lace appearance. Glycogen can be demonstrated with various histochemical staining techniques. Clinically, the patient has marked cardiomegaly and muscular weakness. Congestive heart failure appears early in infancy. There may be macroglossia, also from abnormal glycogen deposition in the tongue muscle. Chest radiography reveals marked cardiomegaly, and the ECG characteristically shows a short PR interval, with or without Wolff-Parkinson-White syndrome, and LV hypertrophy. The prognosis is poor, and death usually occurs within the first year of life. However, enzyme replacement therapy in infantile-onset patients has been shown to decrease heart size, maintain normal heart function, and reduce glycogen accumulation. Alglucosidase alfa has received U.S. Food and Drug Administration (FDA) approval for the treatment of infants and children with glycogen storage disease.