[level-membership-for-endocrinology-diabetes-and-metabolism-category]
Section 4 Acute metabolic complications
Hypoglycaemia
Type 1 diabetes
Iatrogenic hypoglycaemia usually results from a mismatch between:
• supply of glucose for metabolic requirements (e.g. as a result of a missed meal)
• rate of utilization (e.g. an increase due to physical exercise)
• a relative excess of insulin leading to a fall in the circulating glucose concentration.
In the Diabetes Control and Complication Trial, the overall risk of severe hypoglycaemia was increased approximately 3-fold in the intensively treated group. This was observed despite strenuous efforts to exclude patients who were thought to be at high risk of hypoglycaemia. The relation between rate of severe hypoglycaemia and mean glycated haemoglobin (HbA1c) level was inverse and curvilinear. Factors predisposing to severe hypoglycaemia are presented in Table 4.1. However, in a significant proportion of hypoglycaemic episodes, a clear predisposing factor cannot be identified even with very careful scrutiny of the circumstances.
Other causes of hypoglycaemia in insulin-treated patients are presented in Table 4.2. Their effects in individual patients are variable.
Table 4.2 Causes of recurrent hypoglycaemia in insulin-treated patients
| Changes in insulin pharmacokinetics |
The symptoms and signs of acute hypoglycaemia may be divided into two main categories:
• Autonomic (adrenergic) – arising from activation of the sympathoadrenal system (Table 4.3). During hypoglycaemia, the body normally releases adrenaline (epinephrine) and related substances. This serves two purposes:
• Neuroglycopenic – resulting from inadequate cerebral glucose delivery (Table 4.3). Specific symptoms vary by age, duration of diabetes, severity of the hypoglycaemia and the speed of the decline. The symptoms in a particular individual may be similar from episode to episode, but are not necessarily so and may be influenced by the speed at which glucose levels are dropping, as well as the previous incidence of hypoglycaemia.
Table 4.3 Autonomic, neuroglycopenic and non-specific symptoms and signs in acute hypoglycaemia
| Autonomic (adrenergic) |
| Neuroglycopenia |
| Non-specific |
Hypoglycaemia and the brain
The most serious consequence of acute hypoglycaemia is cerebral dysfunction with the risk of:
• injury (to self or others). As a result of neuroglycopenia, the patient may become irrational and aggressive. Prompt assistance from another party may then be required to avert loss of consciousness and more serious sequelae.
Clinically, hypoglycaemia may be usefully graded as follows:
Hypoglycaemia – counter-regulation
The physiological response to acute hypoglycaemia comprises:
• suppression of endogenous insulin secretion.
• activation of a hierarchy of counter-regulatory responses (Fig. 4.1).
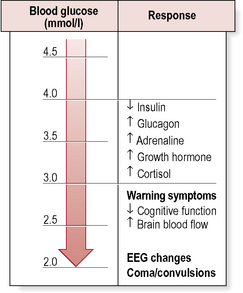
Counter-regulatory hormone responses
Nocturnal hypoglycaemia
Conventional strategies to minimize nocturnal hypoglycaemia include:
• reducing the dose of evening intermediate insulin
• moving the time of the evening injection of intermediate insulin to 2200 hours
Treatment of insulin-induced hypoglycaemia
Grade 3–4 hypoglycaemia
• Buccal glucose gel. Proprietary thick glucose gels (e.g. Glucogel, formerly known as Hypostop) or honey (with variable efficacy) can be smeared on the buccal mucosa.
• Glucagon. Parenteral glucagon (1 mg = 1 unit) can be given by intravenous, subcutaneous or, most reliably and conveniently, intramuscular injection. Glucagon is a hormone that rapidly counters the metabolic effects of insulin in the liver, causing glycogenolysis and release of glucose into the blood. Glucagon can raise the glucose by 3–6 mmol/L within minutes of administration.
Glucagon comes in a glucagon emergency rescue kit, which includes tiny vials containing 1 mg, the standard adult dose. The glucagon in the vial is a lyophilized pellet: it must be reconstituted with 1 ml sterile water, which is included in the ‘kit’. Relatives or friends can learn how to reconstitute and administer glucagon, and this should bolster confidence when dealing with a patient prone to recurrent, severe hypoglycaemia. If glucagon is ineffective within 10–15 min, intravenous dextrose should be administered.
• Intravenous glucose. Administer 25 mL 50% dextrose (or 100 mL 20% dextrose) into a large vein. If a person is unconscious, has had seizures, or has an altered mental status and cannot receive oral glucose gel or tablets, emergency personnel should establish a peripheral or central intravenous line and administer a solution containing dextrose and saline. Dextrose 25% and 50% are heavily necrotic owing to their hyperosmolarity, and should be given only through a patent intravenous line – any infiltration can cause tissue necrosis. Thrombophlebitis is also a well-recognized complication of intravenous delivery.
Behavioural problems specific to type 1 diabetes
Idiopathic ‘brittle diabetes’
Considerable difficulties in glycaemic control can occur as a result of a number of problems including delayed gastric emptying (gastroparesis) (see p. 18), drug interactions (e.g. β-blockers, steroids) and problems with insulin absorption.
Type 2 diabetes
Management
Treatment of hypoglycaemia in type 2 diabetes
• Intravenous dextrose. This remains the mainstay of therapy; it is well recognized that continuous infusions of 5% or 10% dextrose may be required for several days.
• Glucagon. This is less likely to work in type 2 diabetes as hepatic glycogen stores are usually very low when the patient presents with hypoglycaemia. In addition, glucagon also stimulates endogenous insulin release.
• Diazoxide. This drug antagonizes the actions of sulphonylureas by opening adenosine triphosphate (ATP)-sensitive potassium channels in the membranes of islet β-cells. However, the drug is associated with adverse cardiovascular effects, including tachycardia and orthostatic hypotension, which may be hazardous particularly in elderly patients with a compromised vasculature or impaired baroreceptor reflexes.
• Octreotide. This has been shown to be an effective and well-tolerated agent for the prevention of sulphonylurea-induced hypoglycaemia under controlled experimental conditions. However, clinical experience for this indication remains very limited.
• Mannitol and dexamethasone have been recommended for cerebral oedema; however, the evidence base remains poor.
Diabetic ketoacidosis
Diagnosis
Investigations
Diabetic ketoacidosis may be diagnosed when the combination of hyperglycaemia, acidosis and ketones on urinalysis is demonstrated (Table 4.4). Arterial blood gas measurement is usually performed to confirm the acidosis. Subsequent measurements taken to monitor progress may be taken from a vein, as there is little difference between the arterial and the venous pH.
Table 4.4 Initial management of a patient with suspected diabetic ketoacidosis
| History |
| Initially, brief and relevant, including: |
Venous blood is withdrawn for laboratory measurement of:
An arterial blood sample (corrected for hypothermia) is taken for:
Repeat laboratory measurement of blood glucose, electrolytes, urea, gases at 2 and 6 h
BUN, blood urea nitrogen; DKA, diabetic ketoacidosis; ECG, electrocardiography; PCO2, partial pressure of carbon dioxide; PO2, partial pressure of oxygen.
Criteria
• Mild: blood pH mildly decreased to between 7.25 and 7.30 (normal 7.35–7.45); serum bicarbonate decreased to 15–18 mmol/L (normal above 20 mmol/L); the patient is alert
• Moderate: pH 7.00–7.25, bicarbonate 10–15 mmol/L; mild drowsiness may be present
• Severe: pH below 7.00, bicarbonate below 10 mmol/L; stupor or coma may occur.
Metabolic acidosis has a number of potential serious pathological effects:
• exert a negative inotropic effect on cardiac muscle
• exacerbate systemic hypotension via peripheral vasodilatation
• increase the risk of ventricular arrhythmias
DKA usually presents with an anion gap acidosis (anion gap typically 25–35 mmol/L). A wide variety of acid–base disturbances have been reported. Some causes are listed shown in Table 4.5. The anion gap is increased when plasma:
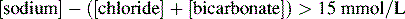
Management
Hyperglycaemia
The main aims in the treatment of diabetic ketoacidosis are replacing the lost fluids and electrolytes while suppressing the high blood sugars and ketone production with insulin (Table 4.6). Admission to an intensive care unit or similar high-dependency area for close observation may be necessary.
• A urinary catheter should be inserted if the patient is oliguric.
• Patients with hyperkalaemia should have a cardiac monitor in order to monitor for peaked T waves.
• Patients who are drowsy or have a reduced conscious level should have a nasogastric tube inserted to avoid aspiration.
Table 4.6 Guidelines for the management of severe diabetic ketoacidosis in adults
| Fluids and electrolytes |
| Volumes:
Fluids: • Isotonic (‘normal’) saline (150 mmol/L) is routine • Hypotonic (‘half-normal’) saline (75 mmol/L) if serum sodium exceeds 150 mmol/L (no more than 1–2 L – consider 5% dextrose with increased insulin if marked hypernatraemia) • 5% dextrose 1 L 4–6-hourly when blood glucose has fallen to 15 mmol/L (severely dehydrated patients may require simultaneous saline infusion) • Sodium bicarbonate (100 mL) as 1.26% solution* (or 8.4% solution if large vein cannulated) if pH < 7.0 (with extra potassium – see text) Potassium: |
• A bolus of 20 units intravenously may be given while an insulin infusion is being prepared
• Give 6 units/h initially until blood glucose level has fallen to 15 mmol/L. Thereafter, adjust rate (1–4 units/h usually) during dextrose infusion to maintain blood glucose concentration at 5–10 mmol/L until patient is eating again
• Search for and treat precipitating cause (e.g. infection, myocardial infarction)
• Hypotension usually responds to adequate fluid replacement
• CVP monitoring in elderly patients or if cardiac disease present
• Pass nasogastric tube if conscious level impaired to avoid aspiration of gastric contents
• Urinary catheter if conscious level impaired or no urine passed within 4 h of start of therapy
• Continuous ECG monitoring may warn of hyperkalaemia or hypokalaemia (plasma potassium should be measured at 0, 2 and 6 h – or more often as indicated)
• Adult respiratory distress syndrome (ARDS) – mechanical ventilation (100% oxygen, IPPV); avoid fluid overload
• Mannitol (up to 1 g/kg intravenously) if cerebral oedema suspected. Dexamethasone as alternative (induces insulin resistance). Consider cranial CT to exclude other pathology (e.g. cerebral haemorrhage, venous sinus thrombosis)
• Treat specific thromboembolic complications if they occur
• Meticulously updated clinical and biochemical record using a purpose-designed flow chart
CT, computed tomography; CVP, central venous pressure; DKA, diabetic ketoacidosis; ECG, electrocardiography; IPPV, intermittent positive-pressure ventilation.
* 1.26% sodium bicarbonate (NaHCO3) = 12.6 g NaHCO3, 150 mmol each of Na+ and HCO3−/L.
Fluid replacement
Despite a proportionally greater loss of body water, plasma sodium concentrations are usually normal or low (Table 4.7). However, in DKA, plasma electrolyte concentrations may be falsely depressed by grossly raised plasma lipid concentrations. Plasma should therefore be inspected for turbidity.
Table 4.7 Average electrolyte deficits in adults with diabetic ketoacidosis
| Electrolyte | Deficit (mmol) |
|---|---|
| Sodium | 500 |
| Chloride | 350 |
| Potassium | 300–1000 |
| Calcium | 50–100 |
| Phosphate | 50–100 |
| Magnesium | 25–50 |
The amount of fluid depends on the estimated degree of dehydration (Table 4.8). If dehydration is so severe as to cause hypovolaemic shock or a depressed level of consciousness, rapid infusion of saline is recommended to restore circulating volume. When the circulating volume has been restored, this should be followed by gradual correction of interstitial and intracellular fluid deficits. Overenthusiastic fluid replacement may lead to respiratory distress syndrome and/or cerebral oedema.
• Use sodium chloride 0.9% (see example, Table 4.9) – infusion rates will vary between patients; remember the risk of possible cardiac failure in elderly patients.
• If hypotension (systolic blood pressure < 100 mmHg) or signs of poor organ perfusion are present, consider use of colloid if increased rate of sodium chloride 0.9% does not restore circulating volume.
• Measure U&Es, venous bicarbonate and laboratory glucose at the end of hour 2 and hour 4.
Table 4.8 Assessment of hydration (over-estimation of dehydration is dangerous)
| Degree of dehydration | Clinical signs |
|---|---|
| Mild, 3% | Only just clinically detectable |
| Moderate, 5% | Dry mucous membranes, reduced skin turgor |
| Severe, 8% | As above, plus sunken eyes, poor capillary return |
| + Shock | May be severely ill with poor perfusion, thready rapid pulse (reduced blood pressure is not likely and is a very late sign) |
Table 4.9 Initial fluid replacement
| Fluid | Rate (mL/h) | Time (h) |
|---|---|---|
| 0.9% sodium chloride 1 L | 1000 | 1 |
| 0.9% sodium chloride 1 L with potassium chloride | 500 | 2 |
| 0.9% sodium chloride 1 L with potassium chloride | 500 | 2 |
| 0.9% sodium chloride 1 L with potassium chloride | 250 | 4 |
| 0.9% sodium chloride 1 L with potassium chloride | 250 | 4 |
| 0.9% sodium chloride 1 L with potassium chloride | 250 | 4 |
| 0.9% sodium chloride 1 L with potassium chloride | 125 | 8 |
| Total | 7 L | 25 |
Insulin
The aims of insulin treatment in DKA are:
• inhibition of lipolysis (and hence ketogenesis)
• inhibition of hepatic glucose production
• enhanced disposal of glucose and ketone bodies by peripheral tissues.
• Transfer to subcutaneous insulin. The first subcutaneous injection should comprise or include an appropriate dose of short-acting insulin. Precise recommendations are difficult to make. The subcutaneous insulin should continue to be administered for 30–60 min before the intravenous infusion is terminated to allow time for absorption of insulin from the subcutaneous depot, thereby avoiding transient insulinopenia.
• Intramuscular insulin. If intravenous insulin administration is impracticable for any reason, an intramuscular regimen can be used as a safe alternative. This begins with a bolus of 20 units short-acting insulin, followed by 6 units/h into the deltoid until blood glucose level has reached 15 mmol/L. Insufficient rehydration may cause erratic absorption of intramuscular injections, resulting in apparent insulin resistance.
Potassium
There should be no potassium in the first litre unless known to be <3.5 mmol/L. Thereafter, add dosages below to each 1 L of fluid, as shown in Table 4.10.
| Plasma potassium (mmol/L) | Potassium added (mmol)/L fluid) |
|---|---|
| < 3.5 | 40* |
| 3.5–5.5 | 20 |
| > 5.5 | 0 |
* Severe hypokalaemia may require more aggressive replacement. Must be given in 1 L fluid; avoid infusion rates of potassium > 20 mmol/h.
Hypokalaemia increases the risk of dangerous irregularities in the heart rate.
Cerebral oedema
Cerebral oedema, which is the most dangerous complication of DKA, is probably the result of a number of factors (Table 4.11). Some authorities maintain that it is the result of over-vigorous fluid replacement, but the complication may develop before treatment has been commenced. It is more likely to occur in those with more severe DKA, in children, and in the first episode of DKA. Likely factors in the development of cerebral oedema are dehydration, acidosis, increased level of inflammation and coagulation. Together these factors lead to decreased blood flow to parts of the brain, which may then swell once fluid replacement is commenced. The swelling of brain tissue leads to raised intracranial pressure, which is reflected in a rising blood pressure and a falling heart rate, and ultimately herniation, where the swollen brain compresses vital structures in the brain stem, leading to death.
| Complication | Clinical findings |
|---|---|
| Cerebral oedema | This is unpredictable, occurs more frequently in younger children and newly diagnosed diabetics, and has a mortality rate of around 25%. Producing a slow correction of the metabolic abnormalities reduces the risk |
| Hypokalaemia | This is preventable with careful monitoring and management |
| Aspiration pneumonia | Use a nasogastric tube in semiconscious or unconscious patients |
| Gastric stasis | A succussion splash may be evident on abdominal examination |
| Adult respiratory distress syndrome | ARDS has been reported in patients with DKA, usually in patients under 50 years of age. Clinical features include dyspnoea, tachypnoea, central cyanosis and non-specific chest signs. Arterial hypoxia is characteristic and chest X-ray reveals bilateral pulmonary infiltrates. Management involves respiratory support with IPPV and avoidance of fluid overload |
| Thromboembolism | Thromboembolic complications can cause mortality in DKA as a consequence of:
DIC has also been reported as a rare complication. The role of routine anticoagulation has not been clearly established in DKA and in the absence of other risk factors is not generally recommended |
DIC, disseminated intravascular coagulation; DKA, diabetic ketoacidosis; IPPV, intermittent positive pressure ventilation.
Counselling and follow-up
All patients with DKA should be referred to the diabetes team on the day after admission.
Diabetic hyperosmolar non-ketotic syndrome – hyperosmolar hyperglycaemic syndrome
Diabetic HONK syndrome is a characterized by:
• marked hyperglycaemia (plasma glucose often in excess of 50 mmol/L)
• profound dehydration with pre-renal uraemia
• depression of the level of consciousness; coma is well recognized.
Diagnosis
• Urinalysis. This will reveal heavy glycosuria and a negative or perhaps ‘trace’ reaction with Ketostix.
• Blood chemistry. The diagnosis is confirmed by a markedly raised plasma glucose concentration. Pre-renal uraemia, a raised haematocrit and a mild leukocytosis are common. Depression of consciousness generally occurs when plasma osmolality exceeds around 340 mosmol/L. Although there is considerable interindividual variation, alternative causes of an impaired level of consciousness should be considered in patients whose osmolality is less marked (Table 4.12).
| Plasma osmolality (the osmotic pressure exerted by a fluid across a membrane) can be measured in the laboratory (e.g. by freezing point depression) and estimated using the formula: Plasma osmolality (mosmol/L) = 2 × (plasma Na+ + plasma K+) + plasma glucose + plasma urea (where Na+, K+, glucose and urea are in mmol/L) The values for Na+ and K+ are doubled to allow for their associated Cl− anions. |
Treatment
• Initial therapy. Intravenous rehydration, electrolyte replacement and insulin therapy are usually similar to that recommended for DKA. Controversy surrounds the choice of isotonic or hypotonic saline for initial rehydration. It is recommended that isotonic saline (150 mmol/L) is used in preference to hypotonic saline (75 mmol/L) unless plasma sodium exceeds 150 mmol/L. A rise in plasma sodium is frequently observed as blood glucose falls with treatment, and water moves back into the intracellular compartment. The rise in sodium may also be partially explained by the reciprocal relationship that exists between plasma glucose and sodium concentrations.
• Thromboembolic complications. Despite the high frequency of thromboembolic complications in patients with HONK, the role of routine anticoagulation remains uncertain. Neurological signs usually reverse when hyperglycaemia is controlled; epilepsy also responds to insulin and rehydration, but often not to specific antiepileptic drugs in the presence of hyperosmolality.
• Subsequent antidiabetic treatment. Insulin treatment is usually required for the first few months. However, these patients generally secrete significant quantities of endogenous insulin, allowing successful long-term treatment with oral hypoglycaemic agents. Possible precipitating factors (e.g. thiazide diuretics, glucose drinks) should be carefully avoided in the future.
Lactic acidosis
Causes
There are several different causes of lactic acidosis including:
Treatment and prognosis
• Intravenous bicarbonate. The role of bicarbonate is contentious. In one double-blind, placebo-controlled trial of intravenous administration of sodium bicarbonate, no improvement in cardiac haemodynamics occurred, although significant improvement in the arterial partial pressure of carbon dioxide (PaCO2) occurred. Animal models of lactic acidosis have shown that intravenous administration of bicarbonate may increase lactate production (particularly by the splanchnic bed), decrease portal vein flow, lower intracellular pH in muscle and liver, lower arterial pH, and worsen the cardiac output.
• Sodium dichloroacetate. Dichloroacetate is the most potent stimulus of pyruvate dehydrogenase, the rate-limiting enzyme for the aerobic oxidation of glucose, pyruvate and lactate. Dichloroacetate may inhibit glycolysis and, thereby, lactate production. Dichloroacetate also exerts a positive inotropic effect that has been attributed to improvement in myocardial glucose use and high-energy phosphate production. Data from animal studies and one placebo-controlled double-blind clinical trial showed that dichloroacetate was superior to placebo in improving the acid–base status of the patients; however, the magnitude of change was small and did not alter haemodynamics or survival.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
Section 4 Acute metabolic complications
Hypoglycaemia
Type 1 diabetes
Iatrogenic hypoglycaemia usually results from a mismatch between:
• supply of glucose for metabolic requirements (e.g. as a result of a missed meal)
• rate of utilization (e.g. an increase due to physical exercise)
• a relative excess of insulin leading to a fall in the circulating glucose concentration.
In the Diabetes Control and Complication Trial, the overall risk of severe hypoglycaemia was increased approximately 3-fold in the intensively treated group. This was observed despite strenuous efforts to exclude patients who were thought to be at high risk of hypoglycaemia. The relation between rate of severe hypoglycaemia and mean glycated haemoglobin (HbA1c) level was inverse and curvilinear. Factors predisposing to severe hypoglycaemia are presented in Table 4.1. However, in a significant proportion of hypoglycaemic episodes, a clear predisposing factor cannot be identified even with very careful scrutiny of the circumstances.
Other causes of hypoglycaemia in insulin-treated patients are presented in Table 4.2. Their effects in individual patients are variable.
Table 4.2 Causes of recurrent hypoglycaemia in insulin-treated patients
| Changes in insulin pharmacokinetics |
The symptoms and signs of acute hypoglycaemia may be divided into two main categories:
• Autonomic (adrenergic) – arising from activation of the sympathoadrenal system (Table 4.3). During hypoglycaemia, the body normally releases adrenaline (epinephrine) and related substances. This serves two purposes:
• Neuroglycopenic – resulting from inadequate cerebral glucose delivery (Table 4.3). Specific symptoms vary by age, duration of diabetes, severity of the hypoglycaemia and the speed of the decline. The symptoms in a particular individual may be similar from episode to episode, but are not necessarily so and may be influenced by the speed at which glucose levels are dropping, as well as the previous incidence of hypoglycaemia.
Table 4.3 Autonomic, neuroglycopenic and non-specific symptoms and signs in acute hypoglycaemia
| Autonomic (adrenergic) |
| Neuroglycopenia |
| Non-specific |
Hypoglycaemia and the brain
The most serious consequence of acute hypoglycaemia is cerebral dysfunction with the risk of:
• injury (to self or others). As a result of neuroglycopenia, the patient may become irrational and aggressive. Prompt assistance from another party may then be required to avert loss of consciousness and more serious sequelae.
Clinically, hypoglycaemia may be usefully graded as follows:
Hypoglycaemia – counter-regulation
The physiological response to acute hypoglycaemia comprises:
• suppression of endogenous insulin secretion.
• activation of a hierarchy of counter-regulatory responses (Fig. 4.1).
Counter-regulatory hormone responses
Nocturnal hypoglycaemia
Conventional strategies to minimize nocturnal hypoglycaemia include:
• reducing the dose of evening intermediate insulin
• moving the time of the evening injection of intermediate insulin to 2200 hours
Treatment of insulin-induced hypoglycaemia
Grade 3–4 hypoglycaemia
• Buccal glucose gel. Proprietary thick glucose gels (e.g. Glucogel, formerly known as Hypostop) or honey (with variable efficacy) can be smeared on the buccal mucosa.
• Glucagon. Parenteral glucagon (1 mg = 1 unit) can be given by intravenous, subcutaneous or, most reliably and conveniently, intramuscular injection. Glucagon is a hormone that rapidly counters the metabolic effects of insulin in the liver, causing glycogenolysis and release of glucose into the blood. Glucagon can raise the glucose by 3–6 mmol/L within minutes of administration.
Glucagon comes in a glucagon emergency rescue kit, which includes tiny vials containing 1 mg, the standard adult dose. The glucagon in the vial is a lyophilized pellet: it must be reconstituted with 1 ml sterile water, which is included in the ‘kit’. Relatives or friends can learn how to reconstitute and administer glucagon, and this should bolster confidence when dealing with a patient prone to recurrent, severe hypoglycaemia. If glucagon is ineffective within 10–15 min, intravenous dextrose should be administered.
• Intravenous glucose. Administer 25 mL 50% dextrose (or 100 mL 20% dextrose) into a large vein. If a person is unconscious, has had seizures, or has an altered mental status and cannot receive oral glucose gel or tablets, emergency personnel should establish a peripheral or central intravenous line and administer a solution containing dextrose and saline. Dextrose 25% and 50% are heavily necrotic owing to their hyperosmolarity, and should be given only through a patent intravenous line – any infiltration can cause tissue necrosis. Thrombophlebitis is also a well-recognized complication of intravenous delivery.
Behavioural problems specific to type 1 diabetes
Idiopathic ‘brittle diabetes’
Considerable difficulties in glycaemic control can occur as a result of a number of problems including delayed gastric emptying (gastroparesis) (see p. 18), drug interactions (e.g. β-blockers, steroids) and problems with insulin absorption.
