[level-membership-for-endocrinology-diabetes-and-metabolism-category]CHAPTER 38
Clinical biochemistry of the cardiovascular system
Clodagh M. Loughrey; Ian S. Young
CHAPTER OUTLINE
CARDIAC MUSCLE STRUCTURE AND BIOCHEMISTRY
ARTERIAL STRUCTURE AND FUNCTION
Biomarkers of acute myocardial damage
Cardiovascular risk assessment
Potentially modifiable risk factors
Laboratory assessment of hypertension
INTRODUCTION
The cardiovascular or circulatory system consists of the heart and blood vessels (arteries, arterioles, capillaries, venules and veins). When it functions normally, effective circulation of blood maintains perfusion of the tissues, so that substrates for cellular metabolism are provided and excretory products removed. Among other vital functions, it also allows hormones to be transported from their organs of origin to their target tissues, defends against infection through facilitation of the movement of white cells and cytokines, and promotes haemostasis through delivery of platelets and clotting factors to traumatized tissue.
Circulation of blood is maintained by the pumping action of the heart, a muscular organ consisting of two atria and two ventricles. The atria receive blood (the left atrium from the lungs and the right atrium from the rest of the body) and pass it to the ventricles. The right ventricle pumps deoxygenated venous blood to the lungs for oxygenation while the left ventricle supplies oxygenated blood to the rest of the body (including itself) via the aorta, arteries, arterioles and capillaries.
Cardiovascular disease
Cardiovascular disease (CVD) collectively comprises disease of the heart and of blood vessels (almost invariably arteries) supplying any organ. The organs most commonly affected by arterial disease are the heart (coronary artery disease), the brain (cerebrovascular disease) and the limbs (peripheral arterial disease). Renovascular disease is an important cause of chronic kidney disease and of hypertension.
Cardiovascular disease is the leading cause of death in England and Wales, currently responsible for one in three deaths. For every death due to CVD, there are at least two major non-fatal CVD events. It is also the leading cause of death globally: the World Health Organization (WHO) estimated that 17.3 million people died from CVD in 2008, representing 30% of all deaths. Of these, an estimated 7.3 million deaths were due to coronary heart disease and 6.2 million were due to stroke (cerebrovascular disease). Although approximately 80% of CVD is due to modifiable factors and is potentially preventable, CVD deaths continue to rise, largely because preventative measures are insufficient or ineffective. Low- and middle-income countries are disproportionately affected, with less than 20% of global deaths from CVD occurring in high-income countries (Fig. 38.1). Mortality due to all main communicable diseases, including HIV/AIDS, tuberculosis and malaria, is expected to decline globally between now and 2030. By 2030, it is anticipated that CVD alone will be responsible for more deaths in low-income countries than infectious diseases, maternal and perinatal conditions, and nutritional disorders combined, driven in particular by the increased global prevalence of obesity. Worldwide, it is estimated that in 2030, non-communicable diseases will account for more than three-quarters of all deaths, and almost 23.6 million people will die from CVD. Thus, CVD is today the largest single contributor to global mortality and is set to continue to dominate mortality trends on the world stage well into the future.
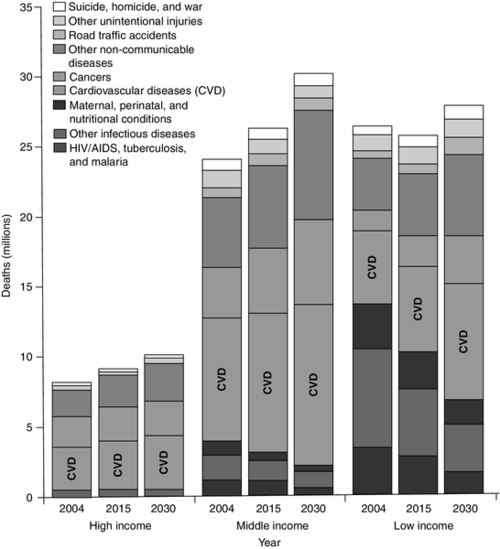
FIGURE 38.1 Projected deaths by cause in 2004, 2015 and 2030, in low-, middle- and high-income countries. Countries are categorized by the World Bank according to 2004 gross national income per person: low income < US$826; middle income US$826–$10 065; high income > US$10 066. Adapted from Figure 2 in Beaglehole R, Bonita R 2008 Global health: a scorecard. Lancet 372: 1988–1996, with permission.
Atherosclerosis is by far the most common cause of cardiovascular disease in developed countries: thus the term ‘cardiovascular disease’ is generally used and perceived to mean ‘atherosclerotic disease’, the term ‘coronary artery disease’ or ‘coronary heart disease’ to mean atherosclerosis of the coronary arteries, and ‘cerebrovascular disease’ to mean atherosclerosis of the cerebral vasculature. Coronary artery atherosclerosis leads to diminished blood supply to, or ischaemia of, the myocardium. When this becomes critical the heart muscles die, leading to a ‘myocardial infarction’ (MI), often, but not invariably, accompanied by diagnostic changes on the electrocardiogram (ECG). Acute coronary syndrome (ACS) is a blanket term for patients presenting acutely with clinical features of myocardial ischaemia, and encompasses ST elevation MI (STEMI), non-ST elevation MI (NSTEMI) and unstable angina (UA) (Fig. 38.2). Death from ischaemic heart disease leading to failure of the heart to maintain circulation may occur acutely in the setting of a myocardial infarction (either due to a large area of infarction or to cardiac dysrhythmia, which interferes with effective ventricular contraction) or in the setting of chronic heart failure. Treatment of MI with thrombolytic drugs or percutaneous coronary intervention (PCI) with or without stenting can be life-saving but depends on prompt and accurate diagnosis.
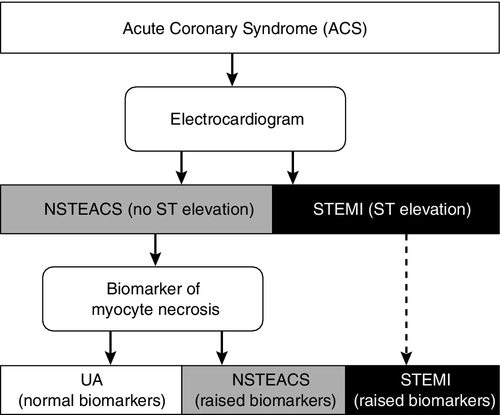
FIGURE 38.2 Categories of acute coronary syndromes, and the investigations used to discriminate between them. NSTEACS, non-ST elevation ACS; STEMI, ST elevation myocardial infarction; UA, unstable angina.
Role of the laboratory
The clinical biochemistry laboratory plays a critical role in the management of acute myocardial ischaemia, where delay in diagnosis and treatment directly increases morbidity and mortality. For many years, the diagnosis of acute MI relied on criteria that had been established by the World Health Organization (WHO) in 1979: rise in a biomarker of cardiac myocyte injury was one of the criteria but was not a prerequisite. The development of more specific and sensitive cardiac biomarkers has facilitated earlier and more accurate diagnosis, which permits more timely and appropriate intervention. In 2000, recognition of the fact that the newer cardiac biomarkers could identify myocardial micronecrosis, even when conventional diagnostic criteria excluded MI, led to a new joint European Society of Cardiology and American College of Cardiology (ESC/ACC) committee universal definition of ‘myocardial infarction’, in which biomarker measurement played a fundamental role. The definition was further revised in 2007 and again in 2012 by the Joint Task Force of the European Society of Cardiology, American College of Cardiology Foundation, the American Heart Association and the World Health Federation (ESC/ACCF/AHA/WHF). The current universal definition of acute MI has a rise and/or fall in cardiac biomarkers (preferably troponin) as the central feature of the definition (Box 38.1).
Identification of those at high risk of developing coronary atherosclerosis allows earlier intervention to prevent related clinical events, and there has also been sustained focus on developing biomarkers to aid prediction of cardiovascular risk. The laboratory also plays important roles in the diagnosis of heart failure and in the investigation of hypertension.
After sections describing the structure of heart muscle and arteries, and the pathogenesis of atherosclerosis, these roles will be explored in turn.
CARDIAC MUSCLE STRUCTURE AND BIOCHEMISTRY
Cardiac muscle is found only in the heart; it has much in common with skeletal muscle but is anatomically distinguishable from both skeletal muscle and smooth muscle. Like skeletal muscle, cardiac muscle consists of densely packed bundles of cylindrical muscle cells, multinucleated fibres (∼ 50 μm in diameter and up to several cm in length) which microscopically show characteristic cross-banding or striations. Unlike skeletal muscle cells, cardiac muscle cells are branched and interconnected; they do not have a nerve end plate but are myogenic, initiating contraction without neural control.
All striated muscle (i.e. both skeletal and cardiac, but not smooth muscle) fibres contain many myofibrils, cylindrical bundles composed of two types of contractile protein filaments: thick filaments of myosin (∼ 15 nm in diameter) and thin filaments of actin (∼ 7 nm in diameter). Muscle contraction involves the ATPase-dependent sliding of the thick myosin filaments across thin actin filaments. Each myofibril consists of a chain of contractile units called sarcomeres, each approximately 2.3 μm in length. Electron microscopy reveals that each sarcomere consists of several distinct regions consisting of dark bands alternating with light bands, giving rise to the striated appearance of skeletal and cardiac muscle. The light bands contain only thin actin filaments, whereas the dark bands contain thick myosin filaments overlapping with the actin filaments.
The cyclical interactions between actin and myosin that result in striated muscle contraction are regulated by intracellular calcium concentration, and are initiated by the release of calcium ions from the sarcoplasmic reticulum. The principal proteins which regulate these interactions, tropomyosin and troponin, are located on the actin filament (Fig. 38.3). Tropomyosin is an actin-binding protein which forms a continuous strand lying along the length of the actin filament. Troponin is a tadpole-shaped flexible complex of proteins that lies against the tropomyosin strand. It comprises three subunits:
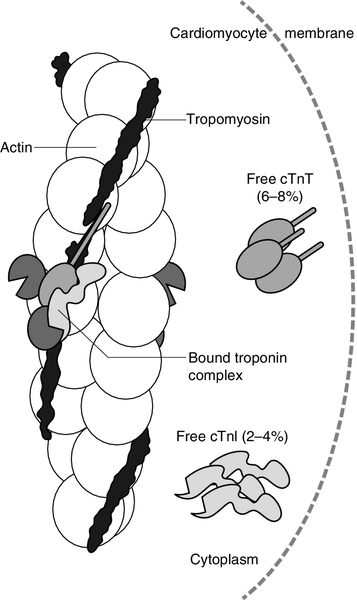
FIGURE 38.3 The thin filament of the cardiomyocyte, consisting of actin with the tropomyosin strand lying along its length and the troponin complex lying against the tropomyosin strand. The three proteins, which constitute the troponin complex (troponin T (TnT), troponin I (TnI) and troponin C (TnC)) are mostly bound to the myofibril, with small amounts of free TnT and TnI in the cytoplasm. From: Gaze D C, Collinson P O 2008 Multiple molecular forms of circulating cardiac troponin: analytical and clinical significance. Annals of Clinical Biochemistry 45(4): 349–355, with permission.
• troponin I (TnI, 22.5 kDa), holds T-TM complex in place by Inhibiting the activity of actin-myosin ATPase in the presence of low calcium, thus inhibiting contraction
• troponin C (TnC, 18 kDa), which senses and binds to Calcium and regulates contraction.
The structure of the ternary troponin complex implies that calcium binding to TnC removes the carboxy terminal of TnI from the actin filament. This alters the flexibility and mobility of the troponin complex, forcing tropomyosin away from the actin filaments and exposing the myosin binding site. Myosin cross-bridges then attach on to the actin filament resulting in muscle contraction.
Troponin C has only one form, which is distributed throughout all muscles, whereas TnT and TnI each have skeletal muscle and cardiac isoforms, the latter denoted by cTnT and cTnI respectively. Although most intracellular troponin is bound to the thin filament of the myofibril, a small amount of cTnT (6–8%) and cTnI (2–4%) exists free in the cytoplasm.
Effective ventricular pumping depends on effective contraction of the myocardium, the thick muscle that comprises most of the ventricular wall. Immediate energy for muscle contraction is supplied by the hydrolysis of ATP by actin-myosin ATPase, largely generated by the β-oxidation of fatty acids, with oxidative metabolism of ketone bodies and pyruvate generating a lesser amount of ATP. The ATP hydrolysis sites are on the cross-bridges formed between the interacting myosin and actin filaments, and the ATPase is highly active only when they interact in muscle contraction. Depleted ATP concentrations are maintained by the donation of phosphate groups to ADP by phosphocreatine in a rapid transphosphorylation reaction catalyzed by creatine kinase (see Chapter 33). The myocardium has a rich blood supply and normally extracts approximately 75% of the oxygen from blood passing through the left ventricle. This is facilitated by myoglobin, a cytoplasmic haem-containing protein, which is found in striated muscle and has a high affinity for oxygen. Cardiac muscle cells generate most ATP from the β-oxidation of fatty acids. They are highly dependent on aerobic respiration and do not function well when oxygen supply from coronary arteries is critical.
ARTERIAL STRUCTURE AND FUNCTION
The walls of medium-sized arteries, such as the coronary vessels consist of three cellular compartments separated by two layers of condensed elastic connective tissue (Fig. 38.4). The innermost tunica intima is separated from the media by a thin layer of elastic tissue called the internal elastic lamina. The media is composed of spirally arranged smooth muscle cells which maintain arterial tone and determine the luminal diameter. The tunica media is separated from the tunica adventitia by the external elastic lamina, which is a loose assembly of connective tissue and fibroblasts surrounding the artery. The intima is lined by a single-celled layer of endothelium, which separates the constituents of blood from the arterial wall. The endothelium appears to play a critical role in the regulation of vascular tone, as well as inhibition of leukocyte adhesion and platelet aggregation, through the release of mediators such as nitric oxide (NO) and prostacyclin. Endothelial cells also express tissue plasminogen activator (tPA) and plasminogen activation inhibitor-1 (PAI-1), thus controlling the relative balance between prothrombotic and fibrinolytic activity.
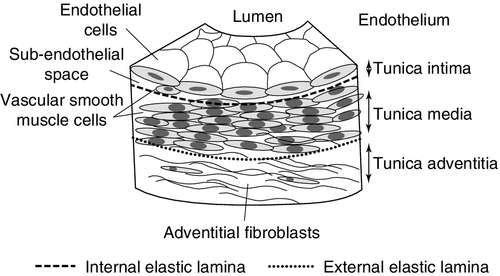
FIGURE 38.4 Structure of a normal medium-sized artery.
ATHEROSCLEROSIS
Atherosclerosis is a chronic, degenerative, inflammatory condition affecting medium-sized and large arteries. (Atherosclerosis does not normally affect veins, although, when they are used as arterial conduits, e.g. as coronary artery bypass grafts, atherosclerosis may develop very rapidly.) It involves the slowly progressive deposition of lipid and matrix proteins in the arterial wall, which eventually causes narrowing of the lumen. Clinical features of atherosclerosis are absent until an advanced stage (after several decades) when there is a critical reduction in blood supply to the affected tissues, causing ischaemia or infarction. The acute clinical events associated with atherosclerosis are in most cases due to lesions that are unstable and liable to rupture, leading to haemorrhage into the atherosclerotic plaque. The resultant thrombosis, the formation of an occluding clot within an artery, is the usual cause of myocardial infarction.
Theories of early atherogenesis
Athere is the Greek word for ‘gruel’, and describes the appearance of the contents of the advanced plaque. Understanding of the cellular events that lead to coronary atherosclerosis has grown substantially over the last 2–3 decades due to: (1) application of cell-specific monoclonal antibodies for immunocytochemical analysis of human lesions; (2) the cloning and characterization of the genes of several cytokines, growth factors and cell surface receptors that may be involved in atherogenesis and that have allowed studies of gene expression, and (3) the use of experimental models including the development of gene knock-out animals.
The response-to-injury hypothesis
Ross first proposed his response-to-injury hypothesis in the early 1970s, focusing on the role of platelets as a possible source of growth factors, and their interaction with the damaged artery wall. With the subsequent realization that the endothelial layer remains intact until the stage of a very advanced ulcerated plaque, the theory was refined to emphasize the importance of more subtle forms of injury to the endothelium, without gross anatomical defects. It was proposed that a number of different agents could contribute to endothelial injury, including smoking, hypertension, hyperlipidaemia and viral infection (Box 38.2). This view has been supported by studies using measures of endothelial dysfunction.
The lipid oxidation hypothesis
The lipid oxidation hypothesis of Steinberg and colleagues provided a further mechanism of endothelial injury, as well as an explanation for the formation of macrophage-derived foam cells that are characteristic of the early lesions of atherosclerosis (Fig. 38.5).
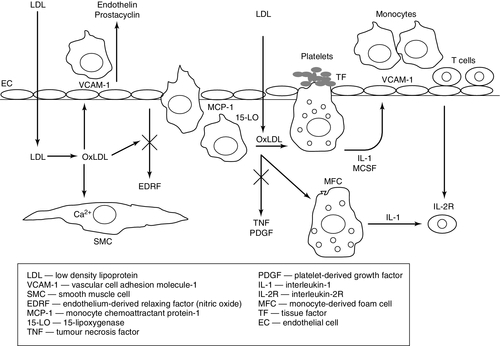
FIGURE 38.5 Steps in the lipid oxidation hypothesis of atherogenesis proposed by Steinberg and colleagues. Adapted from Steinberg, D, Parthasarathy S, Carew T E, Khoo J C, Witzum J L 1989 Beyond cholesterol. Modification of low density lipoprotein that increases its atherogenicity. N Engl J Med 1989; 320: 915–924.
Monocytes adhere to the endothelium and accumulate within the subendothelial space at an early stage of the process. Together with smooth muscle cells in the arterial wall, they take up cholesterol in an unregulated manner and are converted to lipid-laden foam cells within the arterial wall, giving rise to the first macroscopically evident lesion, the fatty streak. However, such excessive cellular uptake of cholesterol cannot occur via the low density lipoprotein (LDL) receptor pathway because of the tight regulation of LDL receptor expression by intracellular cholesterol. Goldstein and Brown proposed the existence of the ‘scavenger receptor’, which allows the unregulated uptake of cholesterol in the form of modified LDL. Several scavenger receptors have now been identified by molecular cloning, the most important of which appears to be CD36 (also called scavenger receptor B). Uptake by these receptors requires chemical modification of the LDL particle by enzymatic, non-oxidative alteration; oxidation, which accelerates the accumulation of cholesterol; glycosylation, or glycoxidation. The oxidation process modifies a lysine amino acid on the apolipoprotein B. Oxidation of LDL can occur in any of the cells within the artery, including the endothelial cells, macrophages, smooth muscle cells and T lymphocytes.
The oxidation of LDL results in the formation of isoprostanes which are chemically stable, free radical-catalysed products of arachidonic acid that are structural isomers of conventional prostaglandins. They reflect lipid peroxidation and are markers of oxidant stress in hypercholesterolemia and atherosclerosis. Levels of isoprostanes are increased in atherosclerotic lesions and localize to foam cells and the extracellular matrix.
Oxidized LDL particles promote atherosclerosis via one or more of the following effects:
• chemoattractant for monocytes
• promotion of inflammatory and immune changes via cytokine release from macrophages and antibody production
• unregulated uptake via the scavenger pathway leading to foam cell formation (foam cells can rupture, releasing oxidized LDL, intracellular enzymes, and oxygen free radicals that can further damage the vessel wall)
• induction of apoptosis of vascular smooth muscle and human endothelial cells, which suggests a mechanism for the response to injury hypothesis of atherosclerosis
• disruption of the endothelial cell surface which impairs endothelial function, reducing the release of nitric oxide (NO), which is a major mediator of endothelium-dependent vasodilation; damage to the endothelium also promotes platelet adherence and the release of cytokines that stimulate smooth muscle proliferation
• causes an increase in platelet aggregation and thromboxane release, which contributes to vasoconstriction and intravascular thrombus.
There is persuasive evidence supporting the notion that LDL oxidation occurs in vivo. Lines of argument include: (a) epitopes of oxidized LDL have been demonstrated in atherosclerotic lesions; (b) LDL isolated from lesions has similar properties to oxidized LDL, cross-reacts with antisera raised against malondialdehyde- and 4-hydroxynonenal-modified LDL, and is recognized by the scavenger receptor; (c) LDL modification also appears to lead to the expression of neo-antigens that elicit an autoimmune response – autoantibodies to oxidized LDL have been found in human plasma, and within human atherosclerotic lesions.
Inhibiting LDL oxidation in vivo by treatment with antioxidants such as vitamin E inhibits experimentally induced atherogenesis; vitamin E can also reduce the uptake of oxidized LDL in vivo by reducing expression of the CD36 receptor. Severity of atherosclerosis in several different animal models (rabbit, monkey, hamster, mouse) can be significantly ameliorated by treatment with a variety of antioxidant compounds. Large clinical trials of antioxidants (vitamin E or β-carotene) in the general population have, however, been singularly disappointing, with no effect on cardiovascular outcomes demonstrated by meta-analyses. Rather than the lipid oxidation theory not being relevant to human atherogenesis, this finding may be due to the wrong antioxidant being chosen, the wrong dose, the wrong subjects or the wrong stage of atherogenesis being targeted: foam cell formation is the earliest stage of the atheromatous plaque, and cardiovascular outcomes are noted with advanced plaque formation, generally several decades later.
The fibrofatty lesion
The mature plaque is characterized by a fibrous cap, composed of smooth muscle cells and extracellular matrix, overlying a pool of lipid, cholesterol crystals and inflammatory cells. The conversion of the fatty streak (Fig. 38.6A) into a fibrofatty plaque (Fig. 38.6B) necessitates the recruitment and proliferation of vascular smooth muscle cells. This process is driven by the synergistic interplay of several growth factors, such as platelet-derived growth factor, insulin-like growth factor and basic fibroblast growth factor. These factors are smooth muscle cell mitogens and/or chemotactic factors and are likely to be important contributors to the process.
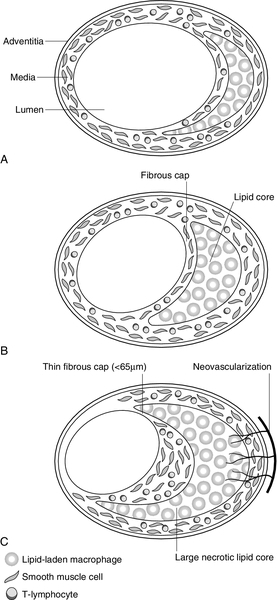
FIGURE 38.6 Features of an atherosclerotic plaque. (A) Fatty streak. Subendothelial collection of lipid-laden macrophages. Plaque grows in an outward direction so that the lumen is conserved. (B) Mature plaque/fibrofatty plaque, characterized by a fibrous cap consisting of smooth muscle cells and extracellular matrix, e.g. collagen. (C) Unstable plaque, characterized by more inflammatory cells and fewer muscle cells, particularly in the shoulder region. Large necrotic lipid core, thin fibrous cap and proliferation of adventitial vasa vasorum.
The complicated plaque/plaque rupture
Unstable plaques, prone to fissuring and rupture, are characterized by a large lipid pool, thin fibrous cap, fewer smooth muscle cells and larger numbers of inflammatory cells (Fig. 38.6C). Activated macrophages within the plaque are a rich source of matrix metalloproteinases (MMPs). These are a family of proteases which, together with other proteases such as cathepsins and elastases, play a key role in tissue remodelling. Under normal physiological circumstances, there is a balance between MMPs, which degrade cardiac extracellular matrix, and tissue inhibitors of metalloproteinases (TIMPs). However, increased MMP activation has been implicated in a wide variety of cardiovascular pathologies, including atherosclerosis. This has the potential to cause localized regions of de-endothelialization that may destabilize the plaque and lead to focal thrombosis and plaque rupture. The shoulder region of the plaque appears to be particularly vulnerable. In most cases of fatal myocardial infarction, at least one major coronary artery is narrowed by more than 75%, and this is usually associated with plaque fissuring and thrombosis.
ACUTE MYOCARDIAL DAMAGE
Chest pain is responsible for a large proportion of attendances at Accident and Emergency Departments; the spectrum of differential diagnoses is broad and, in addition to ACS, includes muscular pain, which is of little consequence, as well as other potentially fatal diagnoses such as pulmonary embolism. These diagnoses can often be distinguished clinically at the bedside on the basis of the history, examination and ECG findings.
Myocardial ischaemia due to obstructive but stable coronary artery atherosclerosis classically causes pain, discomfort or a feeling of tightness or heaviness in the central chest, left arm or jaw, usually precipitated by exertion and relieved by rest or anti-anginal drugs (e.g. sublingual glyceryl trinitrate). Stable angina results from ischaemia which is of insufficient severity to cause myocardial necrosis; transient ischaemic changes may be apparent on the ECG. Acute coronary syndrome results from erosion or rupture of atherosclerotic plaques in the coronary arteries, with release of the prothrombotic contents of the plaque and superimposed platelet aggregation and thrombosis. The pain is more severe, lasts longer than that of stable angina and does not improve with rest. Ischaemic changes on the ECG may be seen. Complete occlusion of a coronary vessel results in MI with classical ST elevation, i.e. STEMI.
However, history and ECG are not always helpful in differentiating causes of chest pain. A significant proportion of ACS patients do not describe classical pain, and many patients with non-cardiac pain describe features which may suggest cardiac pain. Typically ischaemic ECG abnormalities may not be present in ACS. Most patients with ACS have a partial or transient coronary artery occlusion causing myocardial ischaemia without persistent ST elevation, i.e. unstable angina (UA) or NSTEMI. The term ‘non-ST elevation ACS’ (NSTEACS) encompasses both UA and NSTEMI; it reflects the fact that both conditions are considered to be on the same spectrum of myocardial injury, sharing a common pathogenesis but differing in severity and prognosis. The principal feature distinguishing NSTEMI from UA is biochemical evidence of myocyte necrosis.
The importance of accurately and rapidly distinguishing ACS from non-cardiac chest pain is that early treatment of ACS significantly influences mortality and morbidity. Early exclusion of ACS will also help reduce costs of unnecessary hospital admission and investigations, as well as reducing the patient burden.
The relative frequency of the diagnoses in patients admitted with an ACS is approximately: STEMI 30–33%, NSTEMI 25% and UA 38–42%. In STEMI patients, immediate thrombolysis (either with drugs or PCI) is indicated as soon as possible after diagnostic ECG is performed, without waiting for confirmatory elevated serum biomarkers. Patients with NSTEACS benefit from potent antithrombotic treatment (e.g. low molecular weight heparin/platelet inhibition). The TACTICS-TIMI 18 trial (Fig. 38.7) has indicated that the NSTEMI subgroup may benefit from early coronary angiography and revascularization, whereas those with UA do not, although other studies have not confirmed this benefit and more work needs to be done.
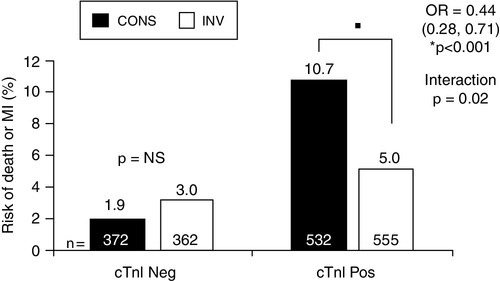
FIGURE 38.7 TACTICS-TIMI 18 trial, indicating improved outcome when patients with NSTEAC and elevated troponin (cTnI Pos) were treated with early invasive intervention (INV) compared to conservative management. The same benefit was not observed in NSTEAC patients without a raised troponin (cTnI Neg). Data from Morrow D A et al. 2007 National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines, Clinical Characteristics and Utilization of Biochemical Markers in Acute Coronary Syndromes. Clinical Chemistry 53: 552–574, with permission.
Nonetheless, it is clear that rapid and accurate diagnosis of ACS is key to improved outcomes. In both STEMI and NSTEMI, damaged myocytes release structural proteins into the circulation, which can be measured and used as biomarkers of acute myocardial injury. The precise definition of myocardial infarction, developed by the 2007 ECC/ACC/AHA/WHF Global Task Force, bases its first criterion on elevations of cardiac biomarkers (see Box 38.1). The current definition gives a preference for use of cardiac troponin, although there is an acknowledgement that the definition is likely to evolve with further scientific advances.
Biomarkers of acute myocardial damage
Within seconds of the onset of myocardial ischaemia, aerobic metabolism ceases within the myocyte, anaerobic glycolysis is initiated and potassium begins to leak out of the cell. This is followed within minutes by leakage of metabolites, a fall in pH and increase in intracellular calcium. Within hours, with sustained depletion of ATP, myocyte necrosis results in irreversible ultrastructural changes. These changes include disruption of the sarcolemma and leakage of macromolecules such as cTnT and TnI, CK-MB and myoglobin.
Since the development of thrombolytic therapies for acute myocardial injury, which must be administered early to be effective, it has become critical to be able to detect myocyte necrosis promptly and accurately. Throughout the 1980s and 1990s, the gold standard for diagnosis of MI was the cardiac-specific isoform of creatine kinase (CK-MB). However, it became apparent that serum CK-MB was not elevated in all cases of myocardial injury: biopsy of myocardium taken during coronary artery bypass surgery for unstable angina showed areas of myocardial micronecrosis, which had not been accompanied by a CK-MB rise. Thus, there has been a drive to develop more specific and sensitive biomarkers of cardiac necrosis, and there is a range of markers currently available, with varying tissue specificity, sensitivity and pattern of release into the circulation (Fig. 38.8).
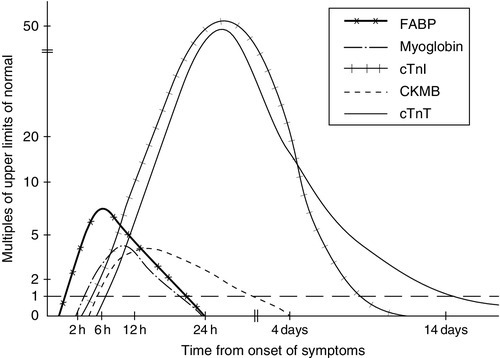
FIGURE 38.8 Pattern of release of some biomarkers into the circulation after acute myocardial infarction.
The pattern of release of biomarkers is influenced by:
• molecular mass: smaller molecules can enter the circulation directly via the microvascular endothelium but are also cleared through the glomerulus
• cytosolic enzymes: increased intracellular calcium activates cytosolic enzymes, including caspase, which promotes dissociation of structurally bound proteins such as cTnT and cTnI.
The ideal biomarker of myocardial injury has a detectable increase in the circulation early, is specific for myocyte necrosis and is not elevated in non-cardiac conditions. None of the currently available cardiac biomarkers meet these criteria, but awareness of their limitations considerably improves clinical utility. The current marker of choice is cTn, with little to choose between cTnI and cTnT.
Troponins
Over the past 20 years, cardiac troponin (cTn) measurement has revolutionized diagnosis of patients with suspected acute coronary syndromes. Monoclonal antibody-based assays specific for the cardiac isoforms of TnT and TnI have been developed that reliably identify myocardial micronecrosis even when CK-MB is not elevated. Recognition of their superior clinical value led in 2000 to the new universal definition of MI based on the elevation of cardiac troponins, updated in 2007 and 2012 to include evidence-based clinical and analytical guidance on use of biomarkers in management of ACS. Cardiac troponins now play a pivotal role in the diagnosis of AMI as well as an important role in the risk stratification of ACS. The current definition of AMI requires detection of a rise and/or fall of cTn (ideally), with at least one measurement > 99th centile of a reference population, together with clinical or ECG evidence of ischaemia.
Although most usually found in the myofibril, a small proportion of total cTn also exists in a cytosolic pool (approximately 6% of cTnT and 3% of cTnI) (see Fig. 38.3). The biphasic release of cTnT from damaged heart muscle probably reflects this distribution: the initial release into the circulation following membrane damage during severe ischaemia becomes detectable at 3 h with a peak at 14 h. This is followed by slow dissociation from and degradation of myofilaments, leading to continuous release over 3–5 days. Elevations persist for up to ten days, permitting late diagnosis of MI. There is some debate over whether cTn is released before cell death occurs, i.e. at a reversible stage of injury. In support of this is the finding of transient minor elevations in circulating cTn in some triathletes following extreme exercise. However, in practical terms this is not a critical issue, since, in the majority of situations apart from extreme exercise, increased cTn is associated with a poorer outcome regardless of whether the injury is perceived to be reversible. As well as being the basis for diagnosis of MI, cTn is also helpful for risk assessment and management of suspected ACS patients. Patients previously classified as UA on the basis of normal CK-MB, may be reclassified as NSTEMI, with increased serum concentrations of troponins associated with higher risk of recurrent cardiac events.
In some cases, cardiac TnT antibodies also cross-react 0.5–2.0% with skeletal muscle TnT, thus skeletal muscle may be a source of raised plasma levels. TnI is thought not to be expressed at all in skeletal muscle and may therefore be more specific for myocyte necrosis. However, in the majority of clinical situations, the specificity of cTnT is comparable to cTnI.
There is only one commercially available cTnT assay, but a range of immunoassays for cTnI, which exhibit different analytical sensitivities and specificities. Potential causative factors include a lack of standardization, the occurrence of post-translational modifications of both cTnI and cTnT, and variation in cross-reactivities of the antibody to the various detectable forms of cTnI resulting from its degradation.
The ESC/ACC consensus document on the definition of myocardial infarction recommends that individual laboratories use a cut-off value equal to the 99th percentile of the reference population. At present, cTnT and cTnI are normally undetectable in healthy subjects using conventional assays; therefore, the 99th centile is very low and most assays do not have good precision at this low concentration. This limits the capacity to detect change with serial samples but does not significantly increase the number of false positive results. Thus, in an appropriate clinical scenario, any elevation of cardiac troponin T or I concentration above the 99th centile for a reference population indicates myocardial infarction. The absolute cut-off varies with the assay used, although for cTnT (for which assay there is currently only one manufacturer), this value is often taken to be 0.1 μg/L. It is important that clinicians are familiar with the 99th centile value of the assay used in their own laboratory and are aware that decision limits will vary with different assays. A regularly updated table showing the characteristics of commercially available cTn assays is maintained by the International Federation of Clinical Chemistry and Laboratory Medicine and can be found at: http://www.ifcc.org/ifcc-scientific-division/documents-of-the-sd/.
As data have accumulated, it has become evident that even small elevations of troponin are associated with poorer outcomes. In patients with ACS whose troponin concentration is less than the 99th centile, there is still an association between troponin value and clinical outcome. It is thus also useful to define the lower limit of detection. This may be taken to be the lowest concentration at which a between-batch CV of 10% is achievable, and for cTnT this is around 0.03 μg/L. However, most cTn assays in current routine clinical use have poor precision below the 99th centile. The recognition that the assay has clinical utility in this range has led to a continued drive to develop tests which reliably detect lower concentrations, resulting in several generations of troponin assays.
Although terminology for naming generations of cTn assays has been inconsistent, contemporary assays are generally referred to as ‘sensitive’ assays. The next generation of assays should be able to measure cardiac troponin with a total CV of < 10% at concentrations significantly lower than the 99th centile of the normal reference population. Thus they are likely to be termed ‘high sensitivity’ cTn (hs-cTn) assays. They should reliably measure troponin in most normal individuals and be able to detect changes in serial measurements in the range below the 99th centile, potentially resulting in earlier diagnosis and better management of ACS.
High-sensitivity troponins
The newest generation of Tn assays have a limit of detection that is of the order of 10–100-fold lower than the ‘sensitive’ assays currently available. By definition, these hs-cTn assays measure cTn in the majority of normal healthy subjects with acceptable precision. They have the potential to increase the promptness and accuracy of diagnosis of ACS. There are a number of different hs-cTn assays in various stages of development; in the case of hs-cTn, a value > 14 ng/L is considered abnormal. Separate decision limits are required for different hs-cTnI assays in the absence of standardization.
Because small differences can be very significant when dealing with such low values, hs-cTn assays are more subject to analytical factors than sensitive assays. Even mild haemolysis reduces hs-cTn values, and sample type (heparin vs EDTA vs serum) can also influence the result. There is greater potential for increase originating from skeletal muscle with hs-cTn compared with the sensitive assay. Some assays show gender-based differences in the 99th centile value.
Since hs-cTn assays can detect cTn in individuals without apparent myocardial damage, it is possible to establish a reference interval for healthy subjects. Values above the 99th centile (a considerably reduced diagnostic threshold compared to 99th centile of the sensitive assays) are associated with a poorer prognosis, and emphasis on detection of a rise and/or fall is likely to be important. Early studies indicate that hs-cTn is likely to facilitate the early diagnosis of AMI as it is presently defined. However, the trade-off for increased diagnostic sensitivity is a reduction in diagnostic specificity. The proportion of patients falsely diagnosed with an MI increases if the diagnosis is based on hs-cTn, particularly if clinical symptoms or ECG changes are uncertain. Even within the reference interval, it is apparent that the higher the value, the greater the risk to the individual, and hs-cTn thus appears to be a better marker for stratifying long-term risk in ACS compared with sensitive assays.
There remain many unanswered questions relating to the use of hs-cTn assays in ACS, including what degree of change can be called an increase, exactly what an increase means, what are its prognostic implications and whether it can be used to influence management to improve outcomes. Until these questions can be answered, the role of hs-cTn in the acute setting will continue to evolve, and must be interpreted in conjunction with the clinical picture. There may be particular problems if the assay is used unselectively in patients with low likelihood of ACS.
Utilization of hs-cTn measurements may be more useful for risk stratifying patients in settings other than acute coronary syndromes including renal insufficiency, heart failure, cardiac amyloid and the elderly.
Other causes of elevated cTn
cTnT and TnI are sensitive and specific markers of myocardial injury when used as recommended by the current universal definition of myocardial infarction, with a cut-off point of the 99th centile. However, lowering the cut-off point may lead to interpretation of an elevated cTn as due to ischaemia, when the elevation is due to myocardial damage due to other causes. To avoid misdiagnosis, it is important to recognize the wide spectrum of disease states other than ACS that can cause raised cTn. These include disorders that would be in the differential diagnosis of MI such as pulmonary embolism, heart failure and myocarditis. The differential diagnosis is given in Box 38.3. Although increased cTn that is not due to ACS may be perceived to be a confounder of clinical interpretation, careful consideration of the source of the elevation will help the clinician determine its clinical significance. There have been occasional descriptions of falsely elevated cTn as a result of analytical interference, but this is a rare occurrence and most frequently elevated cTn in the absence of ischaemia reflects myocardial damage from another cause. Abnormal results may also occur as a result of microparticle interference in anticoagulated samples.
Creatine kinase-MB (CK-MB)
Creatine kinase (CK) is present in large amounts in both skeletal muscle as well as heart muscle, and is also found in brain. Its lack of specificity limits its effectiveness as a cardiac marker, false positives in trauma or post-surgery patients being a particular problem. The enzyme is formed from two dimers, M and B (each with a molecular weight of 40 kDa), and thus three different isoenzymes are possible: CK-MM, CK-BB and CK-MB. The MB isoenzyme is predominantly found in the heart, comprising about 40% of the CK activity in cardiac muscle, and 2% or less of the activity in most skeletal muscle groups and other tissues.
CK-MB isoenzyme is quickly and easily measured and is widely available; it was the gold standard cardiac biomarker until the discovery of the more specific and sensitive cardiac troponin. Measurement of MB isoenzyme can be undertaken by either measurement of the catalytic activity of the MB fraction after antibody inactivation of MM (‘activity’ measurement) or by direct measurement of CK-MB by immunological detection using antibodies (‘mass’ measurement). Most current assays measure CK-MB mass, which is more sensitive than activity. Mass assays also largely avoid detection of macro-CKs (CK linked to IgG and dimers of mitochondrial CK), which can confound diagnosis using assays of CK activity. The presence of macro-CK should be considered when CK-MB is a very high percentage (> 20%) of total CK.
Even though CK-MB is largely confined to the heart, there are measurable amounts in skeletal muscle, accounting for up to 20% in some muscle groups, although the overall figure is about 1%. Thus, coexisting skeletal muscle injury (including exercise and rhabdomyolysis) can reduce the specificity of CK-MB. Following MI, plasma CK-MB achieves a peak slightly before total CK and also returns to normal more quickly. CK-MB rises at about 3–4 h after myocardial necrosis, peaks in 10–24 h, and returns to normal within 72 h. Although potentially helpful in the diagnosis of MI at an early stage, it is less useful for the confirmation of diagnosis of late-presenting MI, since the concentration of enzyme generally returns to within the reference limit by three days. The same property makes CK-MB useful for detection of reinfarction, as cTn does not normalize so quickly; however, further increase of cTn from a high baseline also indicates re-infarction.
CK-MB is outperformed in specificity by contemporary cTn assays, and the newer more sensitive cTn assays are likely to take over any remaining clinical utility for CK-MB.
Myoglobin
Myoglobin is a relatively small (17.8 kDa) haem-containing protein found in abundance in the cytoplasm of striated muscle cells, where its main function is oxygen transport. Myoglobin accounts for about 2% of total muscle protein. Its high tissue:plasma ratio, in combination with its small size, means that it rapidly increases in the circulation after myocyte necrosis. Indeed, it is the earliest indicator routinely available for the detection of both skeletal and cardiac muscle damage, appearing in the circulation within 1–3 h of myocardial necrosis, peaking at 6–9 h, before returning to normal by 24–36 h. Its small molecular size also results in a relatively short time frame for its detection as it is also cleared rapidly by the kidneys.
Myoglobin has long been used as an indicator of muscle damage and has received much attention as a cardiac biomarker because of the rapidity of its release. However, its generalized distribution in all striated muscle limits its specificity as a marker of myocardial damage and its usefulness has declined with the increasing availability of cardiac-specific biomarkers. Its relatively high negative predictive value implies that it may be most useful in the early ruling out of acute MI.
Myoglobin can be measured by radioimmunoassay in serum. However, biomarkers of ACS require a short analytical turnaround time if measurements are to be clinically useful. More rapid measurement is provided by latex agglutination immunoassay or fluoroimmunoassay. Clinical utility of point-of-care measurement of myoglobin in combination with cTn with or without CK-MB mass in excluding ACS is currently being evaluated.
Heart-type fatty acid binding protein (H-FABP)
These are also relatively small (15 kDa) proteins found in large amounts in organs associated with significant fatty acid metabolism, which includes the liver and intestine as well as the heart. There are nine distinct types. The H-FABP isoform is present at high concentration within cardiac myocytes, although it is also found at lower concentration in other tissues such as skeletal muscle, distant tubules of the kidneys, the brain, lactating mammary glands and placenta. Under aerobic conditions, long-chain free fatty acid (FFA) metabolism represents the major source of ATP generated in the myocardium. Free fatty acids are poorly soluble in the aqueous phase and circulate in plasma mainly bound to albumin. In the myocyte cytoplasm, long-chain FFAs are reversibly bound to H-FABP, which is thought to facilitate their transport to the mitochondrial outer membrane where the process of beta-oxidation is initiated.
The potential of H-FABP as a biochemical marker of myocardial injury has been recognized since it was demonstrated to be released from injured myocardium in 1988. Several studies reported its clinical usefulness as an early marker of acute MI (using the World Health Organization criteria as was standard clinical practice at that time). In general it has been shown to perform at least as well or better than myoglobin: this is probably due to its higher relative concentration in cardiac muscle, myoglobin being found in similar amounts in cardiac and skeletal muscle. After acute MI a rise is detectable in plasma as early as 1.5 h after symptom onset, a peak concentration is reached after 4–6 h and, due to rapid renal clearance, the concentration returns to baseline within 20 h.
The normal basal concentration of H-FABP detectable in plasma is likely to be due to continuous release from damaged skeletal muscle. Although its small size makes it useful as an early marker, it also means that it is normally cleared rapidly by the kidney. In renal impairment the plasma level of H-FABP is markedly raised, rendering interpretation difficult. With normal renal function, sustained high concentrations might be indicative of re-infarction, which may be missed by markers such as cTn, which returns to baseline more slowly.
Modern H-FABP assays rely on monoclonal antibodies that have no cross-reactivity (unlike the earlier assays). Point-of-care testing has recently become available. As a standalone test, current H-FABP assays have not been yet shown to have the requisite specificity or sensitivity to safely diagnose early MI. Further work needs to be done to examine its potential role, which may be as part of a panel with a marker which has a complementary timeframe.
Other
Copeptin, a peptide of 39 amino acids, is the C-terminal part of pro-arginine vasopressin (AVP) and is released together with AVP during processing of the precursor peptide. It is thus a surrogate marker for vasopressin, which is significantly involved in the regulation of the endogenous stress response and is present at elevated concentrations in MI, heart failure and different states of shock. Copeptin concentrations are elevated 0–4 h after the onset of symptoms of acute MI, and may be of value in combination with troponin.
Other potential biomarkers of acute myocardial injury that have been evaluated but where insufficient evidence exists to support routine use include ischaemia-modified albumin (IMA), glycogen phosphorylase isoenzyme (GP-BB) and soluble CD40 ligand, among others.
Tests for other causes of chest pain
Measurement of D-dimer may be of some use in the diagnosis of pulmonary embolism. D-dimer is a degradation product released into the circulation when fibrin (involved in thrombus formation) undergoes endogenous fibrinolysis. A low plasma concentration in the setting of a low clinical risk of pulmonary embolism excludes the diagnosis of pulmonary embolism. However, if the clinical risk is high then the patient needs further investigation (e.g. ventilation-perfusion scanning, CT pulmonary angiography) even if the D-dimer concentration is low. Elevated concentrations of D-dimer occur non-specifically in other conditions, for example MI, pneumonia and other forms of sepsis.
HEART FAILURE
Heart failure (HF) is a failure of the heart to fill with (diastolic HF) or to eject (systolic HF) blood, or both. The term ‘congestive heart failure’ implies congestion in the lungs due to back pressure caused by failure of the left ventricle to pump blood around the body. By far, the commonest cause is ischaemic heart disease, but it may also result from any structural or functional cardiac pathology, including valvular heart disease, hypertension or viral cardiomyopathy. Dyspnoea is the predominant symptom, although chronic condition heart failure may be relatively asymptomatic in the early stages. It may also present as a medical emergency (acute life-threatening pulmonary oedema). There is a broad spectrum of severity between these extremes and symptomatic heart failure is a relatively common cause of attendance to emergency departments.
Heart failure has an overall prevalence of approximately 2%, although this is markedly higher in the elderly, affecting up to 15% of those aged over 85 years. It is likely to increase in prevalence with ‘ageing of the population’. It is more common in men than women at all ages. The condition is progressive, and patients with heart failure have a shortened life expectancy and impaired quality of life. In the UK, over 40% of patients do not survive 18 months from the time of diagnosis.
Both mortality and morbidity can be improved with appropriate treatment (e.g. ACE inhibitors and certain β-blockers), which relies on accurate diagnosis. Clinical uncertainty is known to contribute to poorer outcome in patients presenting with dyspnea. However, diagnostic certainty is not always straightforward. Two-dimensional and Doppler echocardiography are widely used and in addition to demonstrating left or right systolic or diastolic ventricular impairment may also provide information on aetiology. However, in many healthcare systems, access to such tests may be limited and there has therefore been considerable interest in biochemical tests for screening, diagnosis, prognosis and monitoring of treatment.
Natriuretic peptides
There has been much interest in the natriuretic peptides (NPs) that are secreted by the myocardium, as potential markers for HF. B-type natriuretic peptide (BNP) was originally identified in the brain but is now known to be released primarily by the heart, particularly the ventricles. It is synthesized as a prohormone, the C-terminal of which is cleaved on release from the myocardium to produce BNP (a 32-amino acid biologically active peptide) and an N-terminal fragment of the prohormone called NT-proBNP (76-amino acid and biologically inert). Both can be measured in plasma, and in normal subjects, the plasma concentrations of BNP and NT-proBNP are similar (approximately 10 pmol/L). However in both systolic and diastolic heart failure, plasma concentrations of both rise, plasma NT-proBNP proportionately more than BNP, with NT-proBNP concentrations approximately four-fold higher than BNP concentrations.
Critical values
Measurement of either BNP or NT-proBNP helps to discriminate between HF and other causes of dyspnoea. A normal value of either (BNP < 100 ng/L [29 pmol/L] or NT-proBNP < 400 ng/L [47 pmol/L]) makes decompensated heart failure highly unlikely (‘rule-out’ values) and suggests a respiratory or other cause of dyspnoea. High plasma concentrations (BNP > 400 ng/L or NT-proBNP > 2000 ng/L) strongly support a diagnosis of abnormal ventricular function (‘rule-in values’). Intermediate values (BNP 100–400 ng/L [29–116 pmol/L] or NT-proBNP 400–2000 ng/L [47–236 pmol/L]) should prompt a search for a non-cardiac cause of dyspnoea: for example, COPD. Approximately 75% of patients in this grey zone will have HF, which is relatively mild and has a good prognosis.
Non-HF factors influencing NPs
Measurements vary with the assay used and with age, gender, and body mass index: normal values tend to increase with age and to be higher in women than men and, for BNP, lower in obese individuals. Plasma concentrations of both tend to be less elevated in heart failure with preserved ejection fraction (EF) than in heart failure with low EF. A raised concentration may occur in other conditions such as hypoxia (e.g. pulmonary embolism, chronic obstructive pulmonary disease), myocardial ischaemia, tachycardia, atrial fibrillation, left ventricular hypertrophy, right ventricular overload, renal impairment (particularly NT-proBNP), liver cirrhosis, diabetes and sepsis. It is not necessary to adjust rule-out values for age or gender, but it has been suggested that obese (BMI > 30 kg/m2) individuals should have their BNP doubled to use the cut-off points above. To date, no correction has been suggested for NT-proBNP in obesity. Plasma concentrations of both, but particularly NT-proBNP, should be interpreted in conjunction with an estimate of renal function.
Clinical utility
When raised concentrations are due to heart failure they are a useful prognostic indicator. Trials with either of these diagnostic biomarkers suggest that their use may reduce both the length of hospital stay and the overall cost of treatment. Both markers provide prognostic information in patients with acute and chronic HF, and plasma BNP also has prognostic value in patients with asymptomatic or minimally symptomatic LV dysfunction. Guidance from the UK National Institute for Health and Care Excellence (NICE) (2010) suggests measurement of BNP or NT-proBNP as first line investigation in patients clinically suspected of having HF, where there has not been a previous MI (Fig. 38.9). It also suggests that patients with high concentrations (BNP > 400 ng/L or NT-proBNP > 2000 ng/L) should be referred within two weeks for echocardiography and specialist advice, because of the implications for poor prognosis. A raised BNP or NT-proBNP is also an independent prognostic indicator of mortality in patients at high risk of CHD and in those with established CHD. The value of serial measurements to guide management of acute HF has not yet been conclusively demonstrated, possibly due to the considerable intra-individual biological and analytical variability.
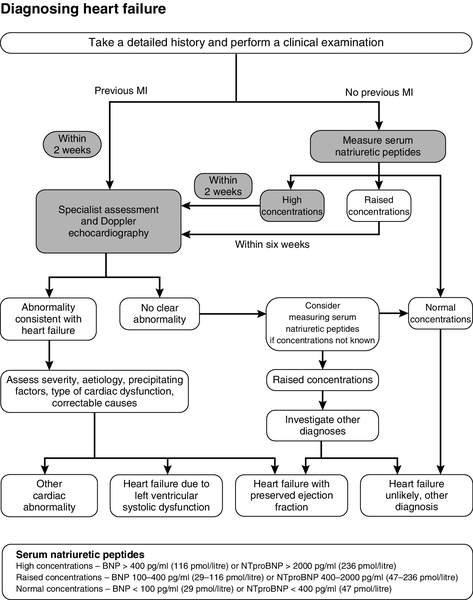
FIGURE 38.9 Diagnosis of heart failure algorithm. From NICE 2010 Clinical Guideline 108 Chronic Heart Failure: management of chronic heart failure in adults in primary and secondary care, with permission.
Work is also currently underway to assess diagnostic and prognostic benefits of mid-regional pro-atrial natriuretic peptide (MR-proANP) in HF.
CARDIOVASCULAR RISK FACTORS
Cardiovascular risk assessment
Assessment of cardiovascular risk is important to allow targeting of preventative measures to those who will achieve the greatest benefit. The most widely used cardiovascular risk prediction equation was developed from data collected in the Framingham Heart Study which was initiated in 1948 in the town of Framingham, near Boston, USA. It set out to identify the factors contributing to coronary disease using a cohort design in patients initially without coronary disease. The Framingham Risk Score has proved remarkably useful in diverse populations considering that it was derived from data on a predominantly middle-class, middle-aged, Caucasian population, and therefore underestimates overall lifetime risk and risk in the elderly. It is reasonably accurate at predicting coronary events, with an area under the receiver operating characteristic (ROC) curve of 0.71–0.76 in males and 0.76–0.81 in females (Fig. 38.10).
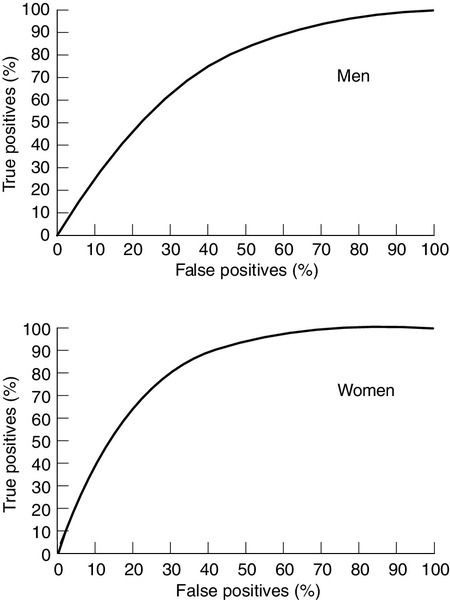
FIGURE 38.10 Receiver operating characteristic (ROC) curves for the Framingham algorithm in men (upper graph) and women (lower graph) using National Health and Nutritional Examination Survey (NHANES) follow-up data. Adapted from Liao Y, McGee D L, Cooper R S, Sutkowski M B 1999 How generalizable are coronary risk prevention models? Comparison of Framingham and two national cohorts. American Heart Journal 137: 837–845, with permission.
The Framingham risk score continues to be used in the UK, although it somewhat overestimates cardiovascular risk in most subjects. In recent years, it has been surpassed in terms of accuracy by the QRISK and QRISK2 scores, which are derived from the UK general practice database and it is likely that QRISK2 or its future iterations will become the generally used cardiovascular risk prediction equation in the UK. QRISK2 employs the same core set of risk factors as Framingham but assigns different weightings to some and incorporates additional risk factors (notably postcode as a marker of social class), which impact significantly on cardiovascular risk in the UK. Alternative risk scores have been developed for use in other populations, notably EUROSCORE, which is widely used across Europe and the Reynolds risk score, which incorporates CRP as an additional risk factor. The major risk factors for cardiovascular disease are similar across all populations, although their relative importance may differ.
Unmodifiable risk factors
Age
Although post-mortem studies have demonstrated that fatty streaks and more advanced lesions are present in the second decade of life, these do not usually become clinically evident until the fifth or sixth decades. Experimental models of atherosclerosis suggest that some of these early lesions are, to some extent, reversible. Atherosclerosis becomes increasingly prevalent after the age of 20 years. This may be partly associated with the increase in plasma cholesterol concentration and other coronary risk factors with age. The extent of coronary and aortic atherosclerosis increases with age (Fig. 38.11) and risk factor burden (Fig. 38.12). Although the absolute risk of CHD rises with age, the relative risk with increasing cholesterol is steeper in the younger age groups.
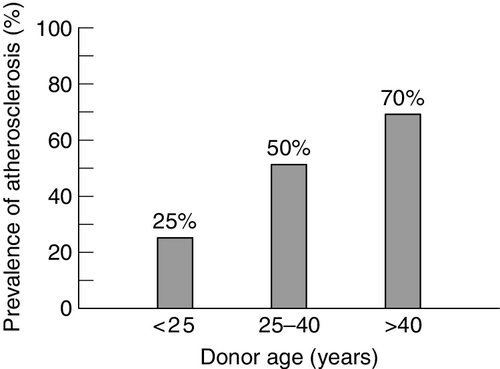
FIGURE 38.11 Prevalence of atherosclerosis identified by intravascular ultrasound in the coronary arteries of heart donors by age. From: Tuzcu E M et al. 2001 High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults. Circulation 103: 2705–2710, with permission.
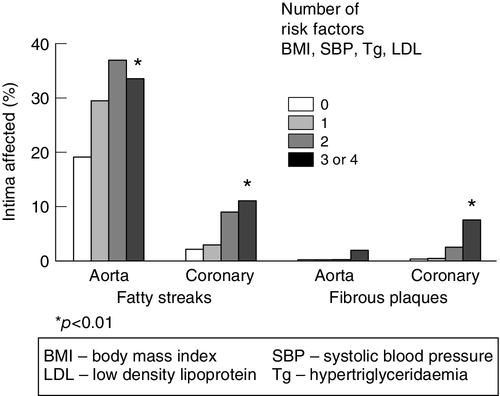
FIGURE 38.12 Effects of cumulative risk factors on coronary and aortic lesion development in children and young adults. Data from Berenson G S et al. 1998 Association between cardiovascular multiple risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. New England Journal of Medicine 338: 1650–1656, with permission.
Gender
The risk of CHD in premenopausal women is approximately 30% of that in men at any particular age, irrespective of smoking status and the presence of hypertension. In women, a greater proportion of cholesterol is present as high density lipoprotein (HDL) cholesterol and this may be protective. Furthermore, it is possible that oestrogens have a direct protective effect on the vasculature. After the menopause, plasma LDL-cholesterol concentrations rise, HDL-cholesterol (HDL-C) falls, there is an increase in visceral adiposity and the risk of CHD rises considerably.
Race
The incidence of CHD and stroke varies with race. In the USA, black people have a higher risk of CHD than white people, while Hispanic groups have lower rates than either group. In the UK, men and women of South Asian descent have a particularly high incidence of CHD (approximately 50% higher rates of premature CHD death compared with the Caucasian population). The difference is widening, as rates do not appear to be falling as fast among this subgroup as for the UK as a whole. This may relate, in part, to a higher prevalence of diabetes in this group. Other factors contributing to this excess mortality may be socioeconomic status, a proatherogenic diet, lack of exercise, enhanced inflammatory status and high plasma concentrations of homocysteine and Lp(a). QRISK2 performs significantly better than the Framingham risk equation for estimating cardiovascular risk in South Asian subjects providing ethnicity is appropriately coded.
Family history
A family history of premature CVD is an important risk factor. In some cases this may be attributable to a single gene effect such as is observed in familial hypercholesterolaemia (see below), although, in most cases, the basis of the underlying genetic predisposition remains unknown and is likely to be polygenic. Family history is a particularly important contributor to risk in men in the lowest quintiles for calculated CHD risk. In women, age- adjusted risk is increased almost three-fold if either parent has CHD before 60 years of age.
Genetic factors
There are a number of inherited traits that are associated with increased CHD susceptibility. Familial hypercholesterolaemia (incidence ∼ 1 in 500 in the UK) and familial combined hyperlipidaemia (1 in 100) account for a large proportion of cases. More common genetic variants have a more subtle effect on coronary risk. For example, the apolipoprotein E gene polymorphism is associated with a modest effect on serum cholesterol concentrations, and compared with individuals with the E-3/E-3 genotype, carriers of the E-4 allele have a 42% higher risk of CHD, whereas the apo E-2 allele appears not to confer significantly increased risk. The apolipoprotein (a) (apo(a)) gene polymorphism, due to tandem repeat within the kringle 4 region, affects the concentration and molecular weight of plasma Lp(a), and high plasma concentrations of Lp(a) are found more commonly among patients with coronary disease.
Low birth weight
Barker and colleagues have demonstrated that a low birth weight is associated with an increased risk of CVD and type 2 diabetes mellitus. This may reflect a permanent metabolic change resulting from undernutrition during critical stages of early development, which may become manifest as impaired endothelial function later in life. Low birth weight and high body mass in adulthood appear to interact to predict CHD.
Potentially modifiable risk factors
Smoking
The seminal studies of Doll and colleagues were the first to identify the dangers of cigarette smoking with regards to CHD risk and death: approximately 20% of cardiovascular deaths can be attributed to smoking. The injurious agents in cigarette smoke probably include tar, carbon monoxide and free radicals. The assessment of the risk of cigarette smoking is confounded by a number of other variables including social class, ethnicity and gender, but it remains important even when allowance is made for them. The relative risk of CHD in smokers is particularly high in young adults, especially women. Smoking rates differ regionally and, to some degree, contribute to the geographical, racial and socioeconomic differences in CHD rates. Furthermore, smoking also has affects on other risk factors such as plasma lipid concentrations, particularly HDL-C, clotting factors and fasting glucose concentrations. Cross-cultural studies indicate that smoking by itself is insufficient to cause CHD, but increases susceptibility to other risk factors. Passive smoking is also associated with an increased risk of coronary disease (∼15–50%). The prevalence of cigarette smoking in the UK has fallen over the past three decades, as has overall tobacco consumption. Smoking cessation is associated with a substantial fall in coronary risk. The benefit of smoking cessation is particularly marked in patients who have already suffered an MI and is associated with a substantial reduction (36%) in mortality in patients with established CHD.
Lipids and lipoproteins
Numerous studies have demonstrated a positive association between CHD and plasma total cholesterol concentration. The association is continuous, exponential and shows no threshold, even at very low concentrations. However, the distributions of plasma total cholesterol concentrations among patients with and without CHD overlap to a considerable degree. Plasma apolipoprotein B (apo B) concentrations appear to be more discriminating: however, they are of less practical utility, as few clinical studies have used apo B concentrations as a basis for treatment (see also Chapter 37).
An inverse relationship between plasma HDL-C concentrations and CHD risk has also been demonstrated in various studies. HDL-C therefore forms an important component of risk prediction equations. However, the results of recent clinical trials using drugs to modify HDL-C levels have generally been disappointing, so HDL-C is not currently a therapeutic target. Just as apoB may be more discriminating than total or LDL-C, so there is some evidence that apoA1 may be more discriminating than HDL-C.
Over recent years, the importance of triglycerides as a CHD risk factor has received increasing attention. There is a very strong inverse relationship between plasma HDL-C (particularly the HDL2 fraction) and triglycerides, and, consequently, it has been difficult to demonstrate an independent relationship. However, recent data do support a role for triglycerides as an independent risk factor. Concentrations of plasma triglycerides above approximately 1.7 mmol/L are associated with the formation of the more atherogenic, small, dense LDL. However, triglycerides themselves also have proatherogenic effects by promoting a procoagulant state, being associated with enhanced factor VII activity.
Most primary (genetic) dyslipidaemias predispose to premature CHD, although hyperchylomicronaemia is associated with acute pancreatitis rather than CHD. However, while in the general population approximately 50% of the variability in plasma cholesterol is genetically determined, these monogenic disorders account for only a small proportion of it, and the basis of the greater part remains unknown.
Thrombogenesis, rheology and clotting factors
Thrombosis is the usual terminal event in coronary atherosclerosis. Several studies have revealed the importance of plasma clotting factor concentrations as risk factors for CVD. Fibrinogen forms the substrate for thrombin and represents the final step in the coagulation cascade. It is essential for platelet aggregation, modulates endothelial function and promotes smooth muscle proliferation. A recent systematic review has shown that plasma fibrinogen concentration is moderately strongly associated with CHD, stroke and vascular mortality in middle-aged adults. Fibrinogen concentrations are also related to risk of ischaemic stroke and CHD events in patients who have had a previous stroke. The relationship between plasma fibrinogen and coronary risk may underlie the positive association between plasma viscosity and CHD. However, it is unclear whether these associations are direct, or related to other risk factors such as smoking, so lowering fibrinogen may not itself reduce risk.
Endothelial injury causes the release of tissue factor that, in turn, activates the intrinsic clotting cascade. Platelet activation and aggregation are crucial processes in atherothrombogenesis, and platelet reactivity has been reported to be elevated in subjects with unstable angina and diabetes. The clinical benefit of antiplatelet drugs such as aspirin in patients at coronary risk is now well established.
There is a balance between the formation of clot and its inhibition and dissolution by factors such as proteins C and S and plasmin. The effectiveness of the fibrinolytic system depends on the balance between tissue plasminogen activator (tPA) and inhibitors of plasminogen activation including PAI-1. Tissue plasminogen activator converts plasminogen to plasmin, which acts on fibrin, causing clot dissolution. This process is inhibited by PAI-1, high concentrations of which are associated with a high risk of reinfarction. Evidence from the Framingham study indicates that plasma PAI-1 concentrations rise with increasing systolic and diastolic blood pressure. Atherosclerotic lesions from diabetic subjects have been shown to contain high concentrations of PAI-1, and plasma PAI-1 is strongly associated with several CHD risk factors including body mass index (BMI), lipids and alcohol intake, and these effects appear to be cumulative.
Apolipoprotein(a) is a glycoprotein that has structural homologies with plasminogen. It is attached to apo B by a disulphide bond and, in some individuals, comprises the major cholesterol rich lipoprotein. High concentrations of Lp(a) have been shown to be associated with an increased risk of CHD, particularly when associated with raised concentrations of LDL-C or homocysteine. The structural similarities between apo(a) and plasminogen have led to the proposition that Lp(a) inhibits plasmin activity, leading to a prothrombotic state. Plasma concentrations of Lp(a) are largely genetically determined, but can be modified, to a limited degree, by dietary fatty acids, oestrogen, the lipid-lowering agent, nicotinic acid and alcohol.
Hypertension
Coronary heart disease risk increases both with increasing systolic and diastolic blood pressure (BP). Blood pressure, like plasma cholesterol, is a continuous variable and there is no clear cut-off value, but hypertension doubles the risk of CHD at any given concentration of cholesterol. Because intra-subject blood pressure measurements can vary so much over time, the diagnosis of hypertension relies on the measurement of blood pressure on several occasions. Blood pressure increases with age and its prevalence varies with ethnicity. Hypertension is also associated with obesity and dyslipidaemia, often as part of the metabolic syndrome.
Obesity
Obesity is an independent risk factor for CHD and is rising in prevalence throughout the developed and developing world (see Chapter 11). Body mass is positively related to fasting triglyceride concentrations, plasma cholesterol and blood pressure, and inversely related to HDL-C. The distribution of body fat appears to be particularly important. It is central or visceral obesity, measured as waist circumference, that is most strongly related to insulin resistance and CHD risk. Waist circumference is a significantly better index of insulin resistance than either waist/hip ratio or BMI. A cut-off value for waist circumference of < 100 cm excludes insulin resistance in both sexes with optimal sensitivity and specificity. It has been proposed that hyperinsulinaemia stimulates 11β-hydroxysteroid dehydrogenase in omental adipose tissue, generating cortisol and promoting a cushingoid fat distribution. Adipose tissue is now recognized to be a source of a number of inflammatory cytokines (interleukin-6, tumour necrosis factor α), growth factors (heparin binding epidermal growth factor) and hormone-like substances (leptin, adiponectin, resistin).
Weight loss is associated with an improvement in a number of coronary risk factors including LDL, HDL, triglycerides, systolic BP and fasting blood glucose. Rapid weight gain in childhood (between 2 and 11 years) appears to predict coronary disease in adulthood.
Impaired glucose tolerance and diabetes
Both diabetes mellitus and impaired glucose tolerance are important risk factors for CVD (Fig. 38.13) and epidemiological data have led to the notion that diabetes confers a similar risk of a cardiovascular event to a prior myocardial infarction (Fig. 38.14). In patients with diabetes, a high haemoglobin A1c (HbA1c) (>10mmol/mol) is associated with an increased risk of CHD; relative risk increases by 1.2-fold for each percentage point increase of HbA1c. However, the value of tight glucose control in preventing macrovascular disease is less clear than its benefits in the prevention of microvascular complications. This may be in part because the atherogenic lipid profile typically associated with type 2 diabetes tends to persist even with excellent glycaemic control.
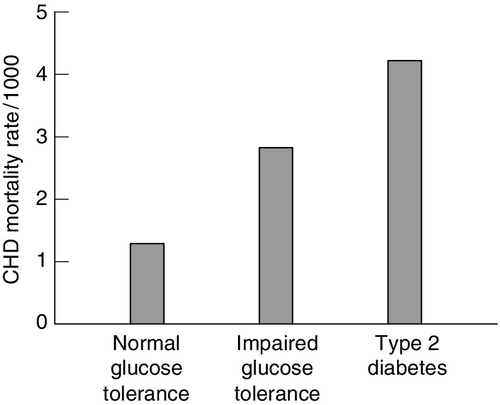
FIGURE 38.13 The effects of glucose tolerance on CHD mortality rates. From: Hsueh W A, Law R E 1998 Cardiovascular risk continuum: implication of insulin resistance and diabetes. American Journal of Medicine 105: 4 s–14 s, with permission.
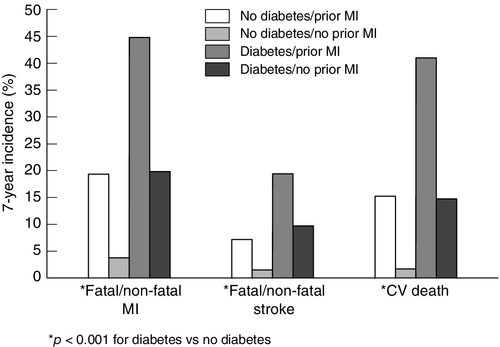
FIGURE 38.14 Diabetes mellitus confers a similar risk for a cardiovascular event as a prior myocardial infarction (MI). Data from Haffner S M et al. 1998 Mortality from coronary heart disease in subjects with type 2 diabetes mellitus and in non diabetic subjects without prior myocardial infarction. New England Journal of Medicine 339: 229–234, with permission.
Although the absolute CHD mortality is higher for diabetic men than for diabetic women, the gender-related protection in women is lost if they have diabetes.
Metabolic syndrome
The clustering of several coronary risk factors (high triglycerides, low HDL, obesity, hyperuricaemia, hyperinsulinaemia and hypertension) has been known for several decades, and led Reaven and others to propose the existence of a syndrome with a common underlying metabolic defect. The genetic basis for this, if one exists, has yet to be identified. This is further complicated by the many synonyms and definitions in common use. These include definitions developed by the World Health Organization (WHO), International Diabetes Federation (IDF), American Diabetic Association, American Association of Clinical Endocrinologists (AACE), American Heart Association and the National Cholesterol Education Programme Adult Treatment Panel III (NCEP-ATPIII) (Table 38.1). Insulin resistance and hyperinsulinaemia appear to be characteristic features of the syndrome. The latter has also been shown to be associated with coronary events (Fig. 38.15). No matter how defined, there is a high prevalence of metabolic syndrome in westernized populations, with particularly high rates in some ethnic groups. Recent definitions have included ethnic-specific criteria. The prevalence of metabolic syndrome is approximately 22–39%, dependent on the definition. At any particular level of coronary risk, patients with metabolic syndrome appear to have higher event rates than predicted. Furthermore, the metabolic syndrome also predicts the future development of diabetes mellitus, and is associated with a raised concentration of inflammatory markers, such as C-reactive protein (CRP).
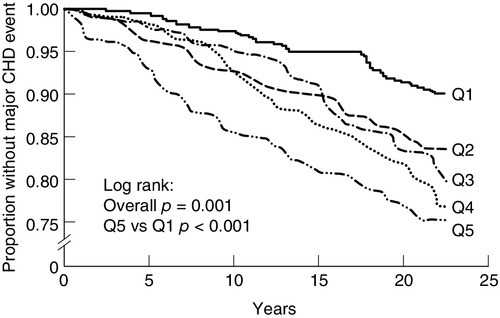
FIGURE 38.15 High plasma insulin concentrations predict increased CHD events in non-diabetic subjects (Q1-Q5 are quintiles of insulin concentration: subjects with the highest concentrations are in Q1, those with the lowest in Q5). From: Pyöräala M et al. 1998. Hyperinsulinaemia predicts coronary heart disease risk in healthy middle-aged men. Circulation 98: 398–404, with permission.
Physical activity
A high level of physical activity protects against CHD and probably does so by a number of different mechanisms. For example, it is associated with a reduction in blood pressure and improved lipid profile. Recent data indicate that approximately 30 min/day of moderate exercise is required to have a significant impact on coronary risk. Exercise-based rehabilitation for patients with CHD is effective in reducing total mortality and lipid concentrations. High levels of physical activity also decrease the risk of stroke in a dose-dependent manner. Reduction in fitness predicts cardiovascular death in middle-aged men.
Psychological factors
Data from the mid-1970s indicated that a type A personality, characterized by high achievement and drive, was associated with increased risk of CHD. However, more recent data do not support these findings. Stress adversely affects blood pressure, sleeping patterns and plasma lipid concentrations and there is clearly a plausible interaction between stress and personality. Some studies have demonstrated an increased risk of CHD in patients with depressive illness, though the basis of the association is unclear.
Inflammation and infection
Atherosclerosis bears many hallmarks of a chronic inflammatory disease, and at every stage of its evolution is characterized by macrophage and T lymphocyte infiltration. The possible stimuli for this inflammatory process include oxidized LDL, homocysteine, free radicals generated from cigarette smoking and infectious microorganisms. If the original insult were not adequately neutralized, the inflammation might persist, causing the local and systemic release of growth factors and cytokines. These can cause intimal thickening by stimulating smooth muscle cell migration, proliferation and extracellular matrix elaboration. The release of IL-1β and IL-6 from activated leukocytes may also lead to an induction of hepatic CRP synthesis.
Over the past few years, there has been an increasing interest in the use of inflammatory markers to estimate the risks of acute events in patients with established coronary disease. In part, the predictive value of these markers may be related to their ability to identify patients with vulnerable plaques, which are rich in activated leukocytes. The risk associated with a high plasma CRP concentration is stronger than that associated with raised von Willebrand factor or erythrocyte sedimentation rate (ESR) but is estimated to be weaker than an increased cholesterol or positive smoking habit. It has also been reported that plasma concentrations of CRP are positively associated with subclinical carotid and femoral atherosclerosis, and a number of established coronary risk factors in healthy, middle-aged men. Among these factors were smoking habit, indices of adiposity, blood pressure, triglycerides and HDL. Of these, smoking habit showed the strongest association with plasma CRP. C-reactive protein is deposited in lipid rich regions of human coronary arteries, and its accumulation may precede monocyte recruitment. It is chemotactic for monocytes, and may also be involved in the activation of complement. A relationship between plasma concentrations of CRP and basal endothelial cell nitric oxide (NO) synthesis has also been reported, suggesting that CRP may indeed be involved early in the pathogenesis of atherosclerosis.
It remains unclear whether CRP is a risk marker or risk factor for atherosclerotic cardiovascular disease. Mendelian randomization studies suggest that genetic variation associated with increased CRP levels is not associated with increased cardiovascular risk, suggesting that CRP is a risk marker. However, it is now recognized that a range of conditions associated with chronic inflammation and elevated CRP, such as rheumatoid arthritis and psoriasis, are associated with a significant increased risk of atherosclerotic vascular disease.
Relative importance of coronary risk factors
Although many risk factors for CHD have been described, and others no doubt will become recognized in the future, the contribution of many of them is relatively minor, certainly in comparison with diabetes, cigarette smoking, hypertension and hypercholesterolaemia. It is to these risk factors that the greatest effort should be directed in seeking to ameliorate an individual’s risk of CHD.
Dietary factors
Diet is known to modulate several established coronary risk factors, and dietary change is an important element of coronary risk management as outlined by various dietary guidelines. These are targeted at optimizing the lipid profile, attaining ideal body weight and reducing blood pressure.
Salt
In experimental models, an increased dietary intake of sodium chloride raises blood pressure. This appears to be confirmed by clinical studies such as the INTERSALT study. Sodium excretion has been found to be related to median systolic and diastolic BP. A 5.9 g difference in intake of salt was associated with a 3–6 mmHg difference in blood pressure. A reduction of dietary salt causes a significant reduction in blood pressure, although sensitivity to salt intake varies between individuals. Up to 50% of hypertensive subjects are sensitive to salt loading, an effect that varies with ethnicity. The intake of other minerals also appears to modify blood pressure; these include potassium, calcium and magnesium, the intakes of which are inversely related to blood pressure.
Simple sugars
Dietary simple sugars (sucrose, lactose, glucose and fructose) comprise approximately 25% of total energy intake. An association between sucrose intake and CHD risk was first proposed in the 1960s, but this has not been supported by recent studies. Carbohydrates differ in their effects on blood sugar and insulin concentrations. Low glycaemic index (GI) foods have less effect on postprandial blood glucose and insulin. The Nurses’ Health study has shown that a high glycaemic load is associated with an increased risk of CHD. This may be related to adverse effects on HDL and triglycerides, and increased risk of diabetes. However, low-GI diets appear to have only a modest impact on CHD risk. Low-GI diets also appear to have little effect on lipid concentrations or HbA1c, although most studies to date have been short term. Diets rich in protein or unsaturated fat have been found to reduce blood pressure more than diets high in carbohydrate in obese subjects with mild hypertension.
Ethanol
Several population studies have reported that the relationship between ethanol consumption and CHD risk is J-shaped, with a nadir for risk at approximately three units (30 mL) of ethanol per day. However, confounding factors make it difficult to interpret the relative benefits of moderate drinking. People who never drink alcohol appear to have a higher prevalence of several other cardiovascular risk factors. Although systolic blood pressure rises with increasing alcohol intake (> 3 units/day is associated with a significant increase in systolic blood pressure and other adverse events), alcohol intake is positively associated with HDL-cholesterol, particularly the HDL2 subfraction. Plasma triglycerides also increase with alcohol consumption, but this is associated with an increase in VLDL and the large, buoyant LDL subfraction, rather than small, dense LDL. The relative benefits of various alcoholic beverages have been debated, and related to their antioxidant content. However, the form of alcohol consumed is likely to be confounded by lifestyle, diet and cultural factors. For example, the prevalence of wine drinking varies with age, race, smoking, ethnicity and educational background.
Fish and fish oils
A relationship between fish consumption and protection against CVD was first mooted in the 1960s because of the low rates of coronary disease observed in the Inuit people. Subsequent epidemiological studies have shown that consuming more than 35 g of fish per day is associated with a substantial reduction in CHD risk. This effect is dose dependent, each 20 g increase in fish intake being associated with a 7% lower risk for CHD mortality. The Nurses’ Health Study population revealed similar findings with up to 34% lower CHD mortality when fish was eaten more than five times per week.
It has been proposed that the protection conferred by increasing fish consumption may relate to the original risk status of the population concerned, and the kind of fish consumed.
Soy protein
Soy is an edible protein component of soya bean. A metaanalysis of 38 clinical studies has shown that consumption of soy protein can lower plasma LDL-cholesterol by as much as 13% and triglycerides by 10%, but only in subjects with higher starting concentrations. Several mechanisms for this have been proposed but both soy proteins and isoflavones appear to be required. It is noteworthy that the per capita consumption in Japan (where the incidence of CHD is low) is as much as 55 g/day, whereas in the USA (where it is much higher), it is < 5 g/day. An intake of >25 g/day is required to significantly improve the lipid profile.
Fatty acids
Changing the fatty acid composition of the diet can have a substantial effect on plasma cholesterol concentrations and on coronary risk. Dietary fatty acids can have a profound effect on the inflammatory process and consequently may also affect plaque composition and stability. A high intake of α-linolenic acid is associated with reduced risk of fatal CHD, but in men, an increased risk of prostate cancer has also been reported.
Plant sterols
Sterols are alcohol derivatives of cyclopentanoperhydrophenanthrene, and are essential constituents of all cell membranes. Cholesterol is a mammalian sterol, containing 27 carbon atoms, whereas sitosterol, campesterol and stigmasterol are plant sterols with 28 and 29 carbon atoms, due to the presence of an additional methyl or ethyl side chain, respectively. Dietary cholesterol has a modest effect on plasma cholesterol concentrations, and an intake of < 300 mg/day is recommended for the general population (< 200 mg/day in patients with CVD). Plant sterols are normally poorly absorbed by the human intestine and inhibit cholesterol absorption. Five g/day of sitostanol ester reduces plasma cholesterol concentration by approximately 10–20%.
Fibre
Dietary fibre intake is inversely related to CHD risk. A 10 g/day increase in fibre has been shown to be associated with a 14% decrease in coronary events and a 27% decrease in coronary deaths. Intake of whole grain wheat is associated with a 26% reduction in coronary disease, which may be related to a reduction in LDL and triglycerides. High dietary fibre intake is also associated with a reduced prevalence of diabetes, hypertension and obesity. It has been proposed that the fibre content of soy may account for some of its cholesterol-lowering properties. However, the effect of fibre on cholesterol is a modest one: there is approximately a 0.045 mmol/L fall in plasma cholesterol per gram of dietary fibre. The mechanisms of action are thought to include: bile salt binding, in much the same way that resins work; altered gut motility; an effect on satiety, and increased insulin sensitivity. Overall, high dietary fibre intake is associated with a substantially lower relative risk of fatal and non-fatal CVD. It should also be noted that soy contains phyto-oestrogens, and whole grain wheat is rich in selenium, so that the beneficial effects of these foods may not be entirely attributable to their fibre content.
Fruit and vegetables, tea and coffee
Higher rates of coronary disease have been found in regions with low fruit and vegetable consumption, and a diet rich in fruit and vegetables is associated with a 5% reduction in stroke risk per portion of fruit and vegetables per day. Vegetarians have been reported to have a lower rate of CHD, although these data are confounded by other associated patterns of lifestyle, including a lower fat and tobacco consumption and higher socioeconomic class. The Dietary Approaches to Stop Hypertension (DASH) trial found that a diet high in fruit and vegetables and low in fat reduced systolic blood pressure by 5.5 and diastolic blood pressure by 3 mmHg. Fruit and vegetables are also a rich source of antioxidants, such as vitamins C and E, carotene and polyphenols.
Green and black teas contain a complex mixture of polyphenolic compounds that include tannins. This spectrum of components and their relative content depends on the conditions of brewing and their bioavailability depends on whether the tea is taken with milk. However, drinking tea more than three times per day is associated with reduced stroke and CHD. Drinking boiled coffee is associated with an increase in plasma cholesterol. Plasma homocysteine concentrations are also increased in subjects with a high coffee consumption. However, there is little evidence that coffee intake is related to overall cardiovascular risk.
Dietary pattern
While individual dietary constituents may ameliorate cardiovascular risk as outlined above, increasing emphasis is placed on having a healthy dietary pattern rather than individual dietary components. Healthy diet is one which, first, maintains BMI in the normal range and minimizes the risk of overweight or obesity. A number of scores of been developed to identify a healthy or prudent dietary pattern. However, one dietary pattern that has considerable evidence to support benefits for cardiovascular health and that is currently recommended in NICE cardiovascular disease prevention guidelines is the Mediterranean diet. The Lyon Heart Study examined the effects of a Mediterranean diet (30% of calories from fat, of which 8% were saturated) compared with a prudent Western diet (34% of calories from fat of which 12% was saturated fat) on coronary events in patients with established coronary disease. The Mediterranean diet contained more bread, fruit, root and green vegetables, poultry and fish, and less red meat; dairy products were replaced with a high α-linolenic acid margarine. After a 46-month follow-up period, there was a 50–70% lower risk of cardiovascular events in the Mediterranean diet group.
HYPERTENSION
Hypertension, or raised blood pressure, is one of the single most common and preventable causes of premature morbidity and mortality. It is a major risk factor for myocardial infarction; heart failure; left ventricular hypertrophy; stroke (both ischaemic and haemorrhagic); chronic kidney disease; peripheral vascular disease; cognitive decline, and premature death. It can also (rarely) present as malignant hypertension, an acute, life-threatening emergency (see below).
The laboratory is involved in:
• assessment of end-organ damage in all hypertensive patients
• monitoring for complications of some commonly used antihypertensive drugs
• investigation for causes of secondary hypertension, particularly screening for renal disease (all) and investigation of endocrine causes (a selected sub-group).
Definition
Blood pressure is measured as diastolic and systolic pressures, measured in millimetres of mercury (mmHg). Systolic pressure represents the peak blood pressure during ventricular contraction (systole); diastolic pressure represents the pressure during ventricular relaxation (diastole).
The blood pressure of individuals within a population is continuously distributed in a Gaussian (normal) manner. Although it would be possible to define a reference interval or range that comprised 95% of an apparently healthy population, this range (as with that for plasma cholesterol concentration) would include significant numbers of people whose blood pressure would cause increased risk of the long-term complications. There is no cut-off value above which hypertension definitively exists; thus any definition of hypertension is to some extent arbitrary.
Epidemiological studies demonstrate increased risk of adverse effects above blood pressures of 115/70 mmHg in all age groups. The relationship is continuous and progressive: for every 2 mmHg rise in blood pressure, the risk of death due to IHD increases by 7% and the risk of death from stroke by 10%. However, this observed association does not prove causality, which would require randomized trials demonstrating risk reduction with blood pressure reduction. Thus, the level of blood pressure determining the presence of hypertension is defined as the level of blood pressure above which treatment has been shown to reduce the development or progression of disease. This is currently generally accepted by most bodies internationally to be 140/90 mmHg.
Blood pressure measurement outside a clinic environment has been shown to correlate more strongly with hypertension-related morbidity and mortality than that measured in the clinic setting, and the latest (2011) NICE guidance recommends that home ambulatory blood pressure monitoring be used to confirm hypertension detected in the clinic.
Hypertension is usually asymptomatic, and diagnosed incidentally, either as part of routine assessment in primary care, as part of a general medical examination when a patient presents with an unrelated condition or as a result of one of its complications (e.g. angina due to coronary atherosclerosis, breathlessness due to cardiac failure). Presenting features in malignant hypertension (see below) include headache, visual disturbances and fits.
Cause
In at least 90% of individuals with sustained hypertension, no primary cause is identifiable, and the condition is termed ‘primary hypertension’ (previously ‘essential hypertension’). Up to 10% have ‘secondary hypertension’, for which it is possible to identify a specific cause, such as renovascular disease, phaeochromocytoma or hyperaldosteronism. The clinical picture can help target investigation appropriately (Table 38.2).
TABLE 38.2
Clinical pointers to secondary hypertension
| Clinical feature | Condition |
| Drug-resistant hypertension Acute hypertension with previously stable values Diagnosis before age 30 in non-obese Severe hypertension with evidence of end-organ damage |
Any |
| Raised creatinine/reduced eGFR | Renal impairment |
| Acute rise in serum creatinine (> 30%) with ACE inhibitor or angiotensin II receptor blockade Moderate to severe hypertension with known vascular disease, asymmetry on renal ultrasound, flash pulmonary oedema Abdominal bruit (low sensitivity) |
Renovascular hypertension |
| Hypokalaemia (mild or easily-induced) | Primary aldosteronism (serum potassium more often normal) |
| Mild hypernatraemia | Primary aldosteronism |
| Cushingoid facies, truncal obesity, proximal myopathy, bruising | Cushing syndrome |
| Adrenal ‘incidentaloma’ | Primary aldosteronism Cushing disease |
| Oral contraceptives Glucocorticoids Ciclosporin Erythropoietin Illicit drug use (esp. cocaine, ecstasy) Excessive alcohol consumption |
Drug-induced |
| Paroxysmal hypertension, esp. with headaches, sweating, palpitations | Phaeochromocytoma (rarely asymptomatic) |
| Obesity (particularly men) Heavy snoring Daytime drowsiness / headache |
Sleep apnoea syndrome |
| Delayed or reduced femoral pulse Aortic ‘machinery’ murmur |
Coarctation of aorta |
| Symptoms of hypothyroidism Raised TSH |
Primary hypothyroidism |
| Hypercalcaemia | Primary hyperparathyroidism |
Primary hypertension
Primary hypertension is very common in Western populations and its prevalence is strongly influenced by age as well as lifestyle factors. Blood pressure tends to rise with age (to a greater extent in men than in women), although, over the age of 70 years, diastolic blood pressure may decline. It is estimated that at least 25% of the UK adult population have hypertension, as well as more than 50% of those aged more than 60 years. Systolic pressure is more commonly elevated in patients in older patients, while raised diastolic pressure is a more prevalent feature of hypertension in those aged less than 50 years.
The aetiology of primary hypertension is poorly understood but is likely to be complex and multifactorial, including genetic, intrauterine and lifestyle factors. Increased sympathetic neural activity and increased angiotensin II activity have both been implicated, although no abnormalities have consistently been identified.
Blood pressure tends to exhibit a familial concordance, to an extent that cannot be explained by a shared environment: having one or more hypertensive parent approximately doubles an individual’s risk of hypertension. An influence of intrauterine environment is indicated by the fact that babies born at term with lower birth weights tend to have higher blood pressure as adults, possibly as a result of reduced nephron mass.
Important lifestyle factors include obesity, high sodium intake and a high alcohol intake (though individuals who consume moderate alcohol tend to have slightly lower blood pressures than abstainers). A meta-analysis of 18 studies has recently indicated an association between hypertension and vitamin D deficiency.
The ‘metabolic syndrome’ comprises hyperinsulinaemia, glucose intolerance, central obesity and dyslipidaemia (all secondary to insulin resistance) and hypertension; insulin resistance and hypertension also frequently co-exist in patients with polycystic ovary syndrome, but in neither case is the mechanism linking insulin resistance and hypertension fully understood. Dyslipidaemia also shows an association with higher blood pressure, which is independent of body mass. Blood pressure also varies between populations, tending to be higher in black Africans than in white Europids.
The prevalence of primary hypertension will continue to rise with increasing longevity, particularly with the concurrently rising prevalence of obesity. Lifestyle modification is an important aspect of the management of hypertension.
Secondary hypertension
Kidney disease
Hypertension is an important complication of both acute and chronic kidney disease, most commonly glomerular or vascular. It is detected by blood testing (elevated serum creatinine and urea) and/or dipstick urinalysis which may show proteinuria or haematuria. Disease is usually bilateral. Both activation of the renin–angiotensin–aldosterone axis and retention of sodium and water may contribute, the latter becoming more important with deteriorating renal function, and peripheral oedema may be observed. Less frequently, unilateral renovascular disease is responsible.
Endocrine disease
Phaeochromocytoma, hyperaldosteronism and Cushing syndrome are all well-recognized (though individually relatively uncommon) causes of hypertension. Patients with thyrotoxicosis and hypothyroid patients may both have hypertension. There is also an association between primary hyperparathyroidism and hypertension. Hypertension is present in some 40% of patients with acromegaly, although some of this is likely to be essential hypertension. In addition, there are several rare, inherited endocrine conditions whose manifestations include hypertension: these are summarized in Table 38.3.
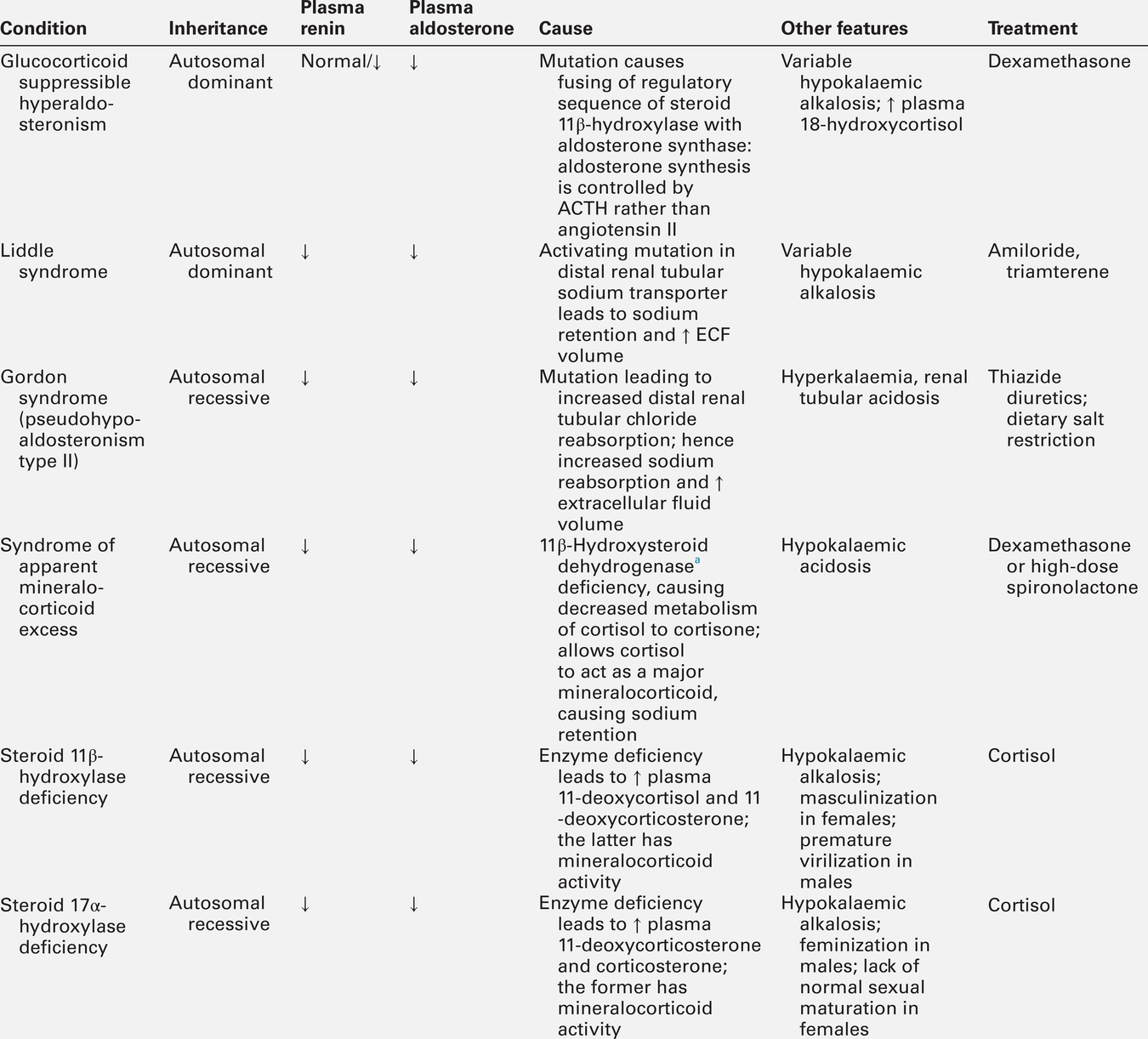
Laboratory assessment of hypertension
All patients should have formal cardiovascular risk prediction using one of the risk prediction equations discussed above. Electrolytes and creatinine (with estimation of glomerular filtration rate) should be measured as baseline before treatment is started with drugs which are potentially nephrotoxic or associated with deranged electrolytes. Renal damage can also both cause hypertension and be affected by target organ damage, and all newly diagnosed hypertensive patients should also be tested for haematuria using a reagent strip at the clinic, and for microalbuminuria with a urinary albumin:creatinine ratio.
Investigation for secondary causes
Comprehensive investigation of every patient with newly diagnosed hypertension is not an efficient use of healthcare resources, since approximately 90% of patients will have primary hypertension and by definition no cause will be found. It is important to bear in mind and target patients with signs and symptoms suggesting a secondary cause of hypertension for specialist endocrine or radiological investigations. Specific clinical features that may suggest specific diagnoses are given in Table 38.2. General features which should prompt comprehensive investigation include:
• an acute rise in blood pressure in a patient with previously well-controlled measurements
• aged less than 40 years in a Caucasian patient without a family history of hypertension or other risk factor such as obesity
• prepubertal onset
• malignant or accelerated hypertension (e.g. severe hypertension plus signs of end-organ damage such as retinal hemorrhages or papilloedema, heart failure or acute kidney injury).
Renovascular hypertension
A proportion of the 10% of all hypertensive patients who have secondary hypertension present with chronic kidney disease which, if not clinically apparent, will be obvious on initial laboratory screening. Renovascular hypertension is the most common remediable cause of secondary hypertension. It may occur in the setting of a patient with other known vascular disease such as CHD or peripheral vascular disease. Particularly suggestive clinical clues include:
• severe hypertension in a patient with asymmetric kidneys or an unexplained atrophic kidney seen on renal ultrasound
• severe hypertension in a patient with recurrent acute pulmonary oedema, or severe heart failure with impaired renal function
• severe hypertension in conjunction with a systolic–diastolic abdominal bruit that lateralizes to one side.
Any of these features should trigger prompt discussion with the local nephrology service and imaging of the renal arteries.
Primary aldosteronism (hyperaldosteronism)
Primary aldosteronism (PA) is the commonest identifiable, specifically treatable and potentially curable form of hypertension. The triad of hypertension, unexplained hypokalemia, and an aldosterone-producing adenoma (APA) of the adrenal gland, was first described in 1955 by Jerome Conn, in a 34-year-old female who had a seven-year history of intermittent muscle weakness, muscle spasms and cramping of her hands. This clinical picture is not specific for Conn adenoma but is associated with any cause of mineralocorticoid excess. Metabolic alkalosis is also a common feature.
Primary aldosteronism comprises a group of disorders characterized by inappropriately high secretion of aldosterone, which is autonomous and is not suppressed by saline. A growing body of research has implicated aldosterone in the pathogenesis of vascular disease independently of blood pressure, possibly via promotion of systemic inflammation and oxidative stress, which contribute to insulin resistance and vascular abnormalities. The recognition that most patients with PA (> 75%) are actually normokalaemic has led to an appreciation that it is much more common than previously recognized. As many as 5–13% of patients with hypertension are currently thought to have PA.
Forms of PA
Around 30% have a unilateral APA, which is potentially resectable by laparoscopic adrenalectomy (hypokalaemia is more common in this group). Most of the remainder have bilateral idiopathic hyperaldosteronism (IHA), which responds best to aldosterone-antagonist drugs. A rare cause is glucocorticoid-suppressible aldosteronism (GRA). Very rarely, an adrenal carcinoma is responsible, and it is occasionally a component of multiple endocrine neoplasia (MEN) type 1. In general, hypertension and hypokalaemia are more severe in patients with APA than bilateral forms.
The diagnosis of PA should be considered in:
• hypertensive patients with spontaneous or diuretic-induced hypokalaemia (see Table 38.4 for differential diagnoses)
TABLE 38.4
Differential diagnosis of primary aldosteronism (PA)
| Hypertensive condition | Differentiating clinical features | Differentiating tests |
| Primary hypertension | None | Aldosterone/renin ratio normal off interferent drugs × 2–4 weeks. Normal ratio on drugs that can potentially cause false positives makes PA highly unlikely Hypokalaemia not a feature of primary HT (only infrequently seen in PA) |
| Diuretic-induced hypokalaemia in patient with primary hypertension | Drug history | Aldosterone/renin ratio normal after correction of hypokalaemia and diuretic withdrawal for at least six weeks |
| Secondary hypertension | Known vascular disease or risk factors; known renal artery stenosis | Aldosterone/renin ratio normal or low Imaging studies(to demonstrate renal artery stenosis or reninoma) |
| Ectopic ACTH syndrome | Features of underlying malignancy (usually bronchial small cell tumour; rarely thymus pancreas or bronchial carcinoid) Rarely Cushingoid features (may be present if slow-growing tumour) |
Aldosterone/renin ratio usually normal despite hypokalaemia and suppressed renin (aldosterone also low) Evidence of malignancy on imaging studies Cortisol and ACTH levels elevated and non-suppressible with high-dose dexamethasone |
| Liddle syndrome | Childhood presentation. Family history (autosomal dominant) | Aldosterone/renin ratio usually normal despite hypokalaemia and suppressed renin (aldosterone also low) |
| Syndrome of apparent mineralocorticoid excess | Childhood presentation of primary form. Family history (autosomal recessive) May be acquired due to excessive liquorice consumption |
Aldosterone/renin ratio usually normal despite hypokalaemia and suppressed renin (aldosterone also low) Raised urinary free cortisol/cortisone ratio |
| Hypertensive forms of congenital adrenal hyperplasia | Childhood presentation. History of either virilization (in 11β-hydroxylase deficiency) or feminization (in 17α-hydroxylase deficiency) Family history of congenital adrenal hyperplasia due to 11β-hydroxylase or 17α-hydroxylase deficiency (autosomal recessive) |
Aldosterone/renin ratio usually normal despite hypokalaemia and suppressed renin (aldosterone also low) 11β-hydroxylase deficiency: low plasma cortisol and corticosterone; raised basal or ACTH-stimulated levels of deoxycorticosterone and 11-deoxycortisol 17α-hydroxylase deficiency: low plasma 17α-hydroxyprogesterone, 11-deoxycortisol, and cortisol; increased gonadotrophins (LH and FSH) |
| Primary glucocorticoid resistance | Family history (although may be acquired) May be associated with androgenization |
Aldosterone/renin ratio normal despite hypokalaemia and suppressed renin (aldosterone also low) Raised ACTH and cortisol; resistance of cortisol to dexamethasone suppression in absence of clinical features of Cushing syndrome |
| Activating mutations of the mineralocorticoid receptor | Family history Pregnancy-induced worsening of HT and development of hypokalaemia |
Aldosterone/renin ratio usually normal despite hypokalaemia and suppressed renin (aldosterone also low) |
| Pseudohypoaldosteronism type II (Gordon syndrome) | Family history (autosomal dominant) Hyperkalaemia; mild normal anion gap metabolic acidosis Increased distal tubular Cl− reabsorption |
Low renin, normal aldosterone; often raised aldosterone/renin ratio |
• patients with moderate, severe or resistant hypertension (less common in mild hypertension)
• patients with adrenal incidentaloma and hypertension
• patients with the onset of hypertension under the age of 20
• patients with hypertension and a family history of young-onset hypertension or CVA at young age (< 40 years), who may have the rare, inherited glucocorticoid-remediable form of PA.
Biochemical investigation
Determination of the ratio of plasma aldosterone concentration to plasma renin activity is widely accepted as the optimal test for screening, after controlling for factors (including medicines) that may confound results. (See Appendix 1 for detailed protocol.)
Confounding factors
Posture
Assuming upright posture effects a rise in plasma aldosterone via increased renin, released from the juxtaglomerular apparatus in response to a slight fall in renal perfusion. All APA, and most with IHA, retain normal responsiveness to upright posture, and measurement in a sample taken when upright may be the most sensitive in detecting raised aldosterone. In practice, a mid-morning upright sample, taken after sitting for 5–15 min appears to be sensitive as well as pragmatic.
Time of day
In PA patients, suppression of renin makes aldosterone more strongly influenced by ACTH, which has a circadian rhythm, being highest in the morning, then falling rapidly. Thus, morning samples are most likely to show elevated aldosterone.
Drugs
See Box 38.4.
Relevant drugs should be withdrawn at least two weeks before testing, or for diuretics, at least four weeks. Where antihypertensive drugs cannot be safely withheld, verapamil slow-release, hydralazine and/or prazosin have lesser effects on the aldosterone–renin system.
Dietary sodium
Salt restriction stimulates renin production, which may lower the aldosterone:renin ratio, thus patients are advised to maintain a liberal salt intake before testing.
Plasma potassium
Hypokalaemia suppresses aldosterone secretion and may cause a false negative result, thus any hypokalaemia should be corrected as far as possible with oral potassium supplements for several days prior to testing.
Phase of menstrual cycle
Higher aldosterone concentrations seem to be measured in the luteal phase relative to the follicular phase, almost certainly mediated by sex hormones, and it may thus be preferable to screen premenopausal women early in the menstrual cycle. More research is required in this area before firm recommendations can be given.
Renal impairment/elderly patients
Renin levels are lower in renal impairment due to reduced nephron mass as well as sodium and water retention. Similarly elderly patients secrete less renin and false positive ratios may result in both circumstances.
Other conditions
Co-existing conditions, which may give rise to false negatives, include pregnancy, renal artery stenosis and malignant hypertension.
Differences between laboratories in assays used and units reported give rise to variability in cut-off values used by different groups; this makes standardization of diagnostic criteria difficult. Renin measurement can be in terms of its enzymatic activity (plasma renin activity, PRA) or its mass (direct active renin concentration, DAR). Automated immunometric assays for measuring DAR have been widely adopted as they are faster and more convenient than PRA. However, considerable work is required to validate these methods, and in the meantime, PRA remains the preferred measurement.
Because of inherent variability in both renin and aldosterone, measurements should be repeated at least once for confirmation. In patients with repeatedly elevated aldosterone/renin ratios, definitive confirmation or exclusion of diagnosis involves careful suppression testing with measurement of aldosterone response to fludrocortisone or to salt loading.
Genetic testing for the hybrid gene causing familial hyperaldosteronism type I (glucocorticoid-remediable aldosteronism (GRA)) allows subtype differentiation, which allows treatment to be tailored. However this is rare, and the majority of genetic tests will be negative.
Localization
The next step should be to differentiate unilateral APA amenable to surgery from bilateral forms of PA which respond to drugs. Computerized tomography (CT) and MRI scanning are both used. Adrenal CT on its own is unreliable, as it may miss small secreting APAs, and misdiagnose larger non-functional adrenal tumours (‘incidentalomas’) as APA, leading to unnecessary surgery. The most sensitive technique for the localization of an isolated adenoma is selective adrenal vein blood sampling with measurement of the aldosterone/cortisol ratio, although this is a technically demanding technique.
The investigation of Cushing syndrome is discussed in Chapter 18.
Phaeochromocytoma
Phaeochromocytoma and functional paraganglioma (PGL) are catecholamine-secreting tumours of the adrenal medulla and extra-adrenal sympathetic nervous tissue (e.g. sympathetic ganglia), respectively. Because of their similarity in presentation and management principles, the term phaeochromocytoma is often used to referred to both, and this convention is also used here. Approximately 10% of phaeochromocytomas are extra-adrenal.
A patient identified with phaeochromocytoma is usually symptomatic. The classical triad of paroxysmal headache, sweating and palpitations is due to excess tumoral secretion of noradrenaline, adrenalin, and dopamine, and is said to have high predictive value. Pallor, lethargy, tremor, nausea, flushing and orthostatic hypotension can also occur. Episodes last from minutes up to an hour and are self-limiting. Symptoms are characteristically made worse by β-blockade because their use permits unopposed α-adrenergic stimulation.
Prevalence in the general population is uncertain, but it accounts for fewer than 0.2% of patients with hypertension. Its importance lies in its being malignant in about 10% of patients (more frequently in the context of multiple endocrine neoplasia, MEN, type 2) and being potentially curable. Malignant tumours biochemically and histologically resemble benign tumours, and can only be diagnosed for certain by the presence of local or distant spread which may occur many years after the primary tumour is resected.
Approximately 90% of phaeochromocytomas are sporadic: these are usually single adenomas. Approximately 10% are inherited: these are often bilateral and multiple. Inherited phaeochromocytomas may be isolated (inherited as an autosomal dominant trait) or be a component of multiple endocrine neoplasia (MEN 2A and 2B, in which they have a combined prevalence of 40%). They also occur as the sole manifestation of MEN 2A in approximately 25% of heterozygotes for this condition. (See Chapter 41 for further details of inherited phaeochromocytomas.)
Patients with phaeochromocytomas may come to medical attention because of their hypertension (paroxysmal in about one-third of patients, but more often sustained though variable) or because of the clinical features of increased secretion of catecholamines.
The diagnosis should be suspected in individuals with one or more of the following:
• paroxysmal self-limiting episodes of typical adrenergic symptoms (see above)
• resistant or labile hypertension (although 5–15% are normotensive at diagnosis)
• familial syndrome predisposing to catecholamine-secreting tumours (e.g. MEN2)
• family history of phaeochromocytoma
• incidentally discovered adrenal mass (25% of diagnoses)
• onset of hypertension at a young age (e.g. < 20 years)
• hypertension with new onset or atypical diabetes mellitus
• hypertensive response during anesthesia, surgery or angiography
• idiopathic dilated cardiomyopathy
• history of gastric stromal tumour or pulmonary chondromas (Carney triad).
Biochemical investigation
Diagnosis of phaemochromocytoma involves first, demonstrating excessive secretion of catecholamines, followed by radiological localization of the causative tumour(s). Consideration of the diagnosis and a low threshold for investigation are key initial factors in successful diagnosis. Because the symptoms are so non-specific, diagnosis is rarely confirmed biochemically in patients with suggestive symptoms. The investigation strategy in a particular patient will be dictated by the availability of biochemical tests in any centre. However, as it is so important not to miss the diagnosis, testing with a high diagnostic sensitivity is critical, particularly when screening asymptomatic patients with an inherited risk of phaeochromocytoma.
Traditional measurement of urinary catecholamines is gradually being superseded by HPLC measurement of fractionated metanephrines (metabolites of catecholamines), either in a 24 h urine collection or in plasma, both of which offer better diagnostic sensitivity and specificity (Table 38.5). The improved sensitivity is a result of continuous production of metanephrines by the tumour, which contains a high concentration of catechol-O-methyltransferase. Even when the tumour release of catecholamines by the tumour is sporadic, this enzyme ensures continued metabolism of catecholamines to metanephrines. The sensitivity of metanephrine measurement to detect phaeochromocytoma is 99% for plasma and 97% for urine, which compare favourably with 86% for urinary catecholamines, among symptomatic patients, those with a previous phaeochromocytoma or a family history, and those with an incidental adrenal mass. The diagnostic specificities for plasma and urinary metanephrines are 89% and 93%, respectively, compared with 88% for urinary catecholamines. False positive results are common owing to the presence of other medical conditions (such as obstructive sleep apnoea) and medications (Table 38.6) that can cause elevation of metanephrines. The relative infrequency of phaeochromocytoma in those tested also contributes to the high rate of false positives. In addition, physical and psychological stress (e.g. surgery, severe pain or anxiety) can potentially increase catecholamine secretion, leading to false positive results.
TABLE 38.5
Sensitivities and specificities of biochemical tests for phaeochromocytoma in 214 patients
| Sensitivity (%) | Specificity (%) | |
| P free metadrenalines | 99 | 89 |
| U fractionated metadrenalines (HPLC) | 97 | 64 |
| P catecholamines | 81 | 81 |
| U catecholamines | 85 | 86 |
| U total metadrenalines (spectrophotometry) | 74 | 93 |
| U vanillylmandelic acid (VMA) | 62 | 93 |
Data from Lenders et al. JAMA 2002; 287:1427–1434.
P, plasma;U, urine.
TABLE 38.6
Some drug interferents with measurement of catecholamines and metadrenalines
| Interferent | Effect |
| Tricyclic antidepressants | ↑ catecholamines and metadrenalines in U and P |
| Phenoxybenzamine | ↑ catecholamines and metadrenalines in U and P |
| Monoamine oxidase inhibitors | ↑ metadrenalines in U and P |
| Buspirone | ↑ metadrenalines in U |
| Calcium-channel blockers | ↑ catecholamines in U and P |
| Nicotine | ↑ catecholamines in U and P |
| Caffeine | ↑ catecholamines in U and P Analytical interference with HPLC assays |
| Methyldopa | Analytical interference with HPLC assays |
| Labetalol | Analytical interference with HPLC assays |
| Levodopa | Analytical interference with HPLC assays ↑ catecholamines and metadrenalines in U and P? |
U, urine; P, plasma.
Concentrations of metanephrines that are over four times normal are associated with a very high probability of phaeochromocytoma; however, intermediate raised levels will require further biochemical investigation, which may include repeat testing after withdrawal of implicated medication. By contrast with the, now infrequently used, measurement of urine vanillylmandelic acid, dietary restrictions are not needed when performing these investigations.
Localization
The purpose of localization is to guide surgical intervention after biochemical confirmation, as well as to look for possible metastatic disease. Direct anatomical imaging methods by computed (CT) tomography or magnetic resonance imaging (MRI) should be used as first-line to locate the primary tumours and is particularly helpful in the relatively large, sporadic, symptomatic tumours. Both techniques have high sensitivity but lower specificity, due to the high prevalence of the benign adrenal cortical adenoma (‘adrenal incidentaloma’). Meta-iodobenzylguanidine (MIBG) scintigraphy is particularly helpful for detecting smaller, presymptomatic tumours, extra-adrenal tumours, including metastases and recurrent tumours. However, false positive results may occur in patients with carcinoid tumours and, because the isotopes are excreted in the urine, false negatives can occur with tumours in the bladder or head of the pancreas (which is closely related to the right kidney). Overall, MIBG scanning has higher specificity than anatomical imaging. Newer functional imaging techniques include 18 F-fluorodopamine positron emission tomography (PET) and 111In-octreotide single photon emission computed tomography (SPECT) scanning.
Management
It is beyond the scope of this book to discuss the management of phaeochromocytoma in any detail. Even following apparently successful surgical resection, patients should be followed-up long term, both clinically (blood pressure measurements) and biochemically. Hypertension may persist and tumours may recur.
Malignant hypertension
Malignant hypertension is defined as markedly elevated blood pressure (diastolic BP often > 140 mmHg) with retinal haemorrhages, exudates or papilloedema with or without evidence of target organ damage, typically acute and progressive kidney injury with proteinuria and haematuria (‘malignant nephrosclerosis’). Its management is a medical emergency and should take precedence over investigations to determine whether there is an underlying cause. Neurological signs in malignant hypertension may be due to intracerebral or subarachnoid haemorrhage or lacunar infarct, or to hypertensive encephalopathy. The latter is associated with insidious onset and non-lateralizing symptoms, such as headache, vomiting, restlessness, confusion. It may be confirmed radiologically on the basis of MRI, which shows oedema of the white matter in the occipito-parietal regions, termed ‘reversible posterior leucoencephalopathy’. (The distinction is important as hypertensive encephalopathy mandates aggressive reduction of blood pressure, not usually indicated for haemorrhage or infarct.)
Hypertension in pregnancy
Hypertension developing in pregnancy constitutes a significant risk to the mother and the fetus. Pre-eclampsia is the development of new hypertension and proteinuria in the second half of pregnancy. It is a multisystem disorder and if untreated, the condition can progress to severe hypertension, acute kidney injury and seizures (eclampsia) and with high fetal and maternal morbidity and mortality. An increase in plasma urate concentration can be a sensitive indicator of deteriorating renal function in this condition. Low platelet count, microangiopathic haemolysis seen on blood film examination, rising creatinine and liver transaminases appear to be particularly predictive of adverse outcome. Pre-eclampsia may sometimes be a feature of the haemolysis, elevated liver enzymes and low platelet (HELLP) syndrome, which is discussed in Chapter 22.
Management of hypertension
Except in malignant hypertension, or in secondary causes of hypertension for which there are specific treatments, management should always begin with efforts to bring about appropriate lifestyle changes, for example attainment of ideal body weight, reduction in any excessive alcohol intake, reduction in dietary salt intake and attention to other cardiovascular risk factors.
The classes of drugs available include diuretics, calcium channel antagonists, α- and β-blockers, ACE inhibitors and angiotensin receptor antagonists, and centrally acting agents. Combinations of drugs are often required to achieve adequate control of blood pressure; if three or more are required in a young (< 40 years) patient with normal renal function, a secondary cause of hypertension should be considered. Compliance with treatment can pose a considerable challenge, particularly because hypertension is generally asymptomatic but also because drug treatment may cause significant adverse effects.
CONCLUSION
Biochemical investigations play a key role in the management of several distinct areas of cardiovascular disease.
Plasma concentrations of markers of myocyte necrosis form the basis of evidence-based criteria that influence decisions in the management of acute coronary syndrome. Increase in cardiac troponin concentrations is embedded in the universal definition of myocardial infarction, and new cardiac biomarkers, as well as panels of biomarkers with complementary timeframes, are currently being developed and evaluated.
The diagnostic and prognostic value of natriuretic peptides in heart failure is reflected in their inclusion as recommended first-line investigations, although they have limitations including lack of specificity.
Measurement of plasma lipids is well-established as a key component of the estimation of cardiovascular risk, and work is currently underway to examine the potential role of inflammatory markers in further stratifying this risk.
The biochemistry laboratory is involved in investigation of end-organ damage in hypertension, monitoring for side-effects of antihypertensive agents, and in the diagnosis of causes of secondary hypertension. A low threshold for investigation is necessary to avoid missing disorders such as phaeochromocytoma and primary aldosteronism, which are probably underdiagnosed.
Cardiovascular disorders are associated with high morbidity and mortality. They contribute significantly to the workload of modern routine clinical biochemistry laboratories in developed countries, and are likely to continue to do so for the foreseeable future.
Further reading
Atherogenesis
Myocardial injury
Heart failure
Cardiovascular risk factors and prevention
Hypertension
APPENDIX 1 PROTOCOL FOR INVESTIGATION OF ALDOSTERONISM: SCREENING AND CONFIRMATORY TESTS
Patient preparation
Ideally, all drugs that have an effect on the renin–aldosterone system should be discontinued for two weeks before samples are collected. Aldosterone antagonists (e.g. spironolactone) and oestrogens must be discontinued for at least six weeks. Where antihypertensive drugs cannot safely be withheld, verapamil (slow-release), hydralazine, doxazosin or prazosin can be used as these have minimal effect on the renin– aldosterone system. If essential to continue other drugs to control blood pressure, as a minimum, β-blockers and ACE inhibitors should be stopped for two weeks and calcium channel blockers withheld on the day of the test until after its completion. The potential effects of interfering drugs must be taken into account in the interpretation of the renin and aldosterone results if they have to be continued (see Box 38.4).
The patient must be encouraged to take a liberal amount of dietary sodium (typically 100–150 mmol/day). Administer potassium salts orally, or if necessary intravenously, to restore plasma potassium concentration to within the reference range or, if this is not attainable, to the maximum concentration possible. Discontinue this supplementation 24 h before blood samples are taken. The patient must be well hydrated at the time of the test.
Screening procedure
Collect blood mid-morning, after the patient has been up (sitting, standing or walking) for at least two hours and then seated for 5–15 min.
Draw blood into a tube containing anticoagulant (type required is assay dependent). The sample should be taken rapidly to the laboratory, but not on ice as cooling of the sample promotes conversion of inactive renin precursor to active renin. Care must be taken to avoid haemolysis, which will result in an artefactual increase in plasma potassium concentration.
Measure plasma aldosterone, renin and electrolytes.
Interpretation of results of screening test
A high aldosterone:renin ratio indicates probable primary aldosteronism (PA). The particular decision limits used are method dependent and therefore vary from laboratory to laboratory, although if the ratio of aldosterone (in pmol/L) to PRA (in pmol/mL/h) is > 2000, the patient almost certainly has PA. Patients with a positive screening test should proceed to a confirmatory test before further investigations are undertaken to determine the cause and location of the disease. If either aldosterone or renin results are abnormal but the ratio is not clearly diagnostic, the screening test should be repeated after a further period off-treatment from all potentially interfering drugs, if some had initially been continued. Other causes of abnormal aldosterone or renin results should be reviewed (see Table 38.4).
CONFIRMATORY TESTS
A number of different confirmatory tests have been described, and there is no definitive evidence as to which has the best diagnostic performance. Of the two tests most commonly used in the UK, the saline suppression test is more convenient and safer to perform than the fludrocortisone suppression test.
Saline suppression test
The test is only indicated when the screening test demonstrates a high aldosterone:renin ratio. The same patient preparation requirements as for the screening test apply. The saline suppression test is contraindicated in patients with severe uncontrolled hypertension, chronic kidney disease, heart failure, cardiac arrhythmia or severe hypokalemia.
Procedure
1. Start test between 08.00 and 09.30 h.
2. Place indwelling catheter in antecubital fossa for infusion of 0.9% saline.
3. Place indwelling catheter in opposite arm for blood sampling.
4. Position patient in recumbent position for 1 h prior to commencing the infusion, and throughout test. Blood pressure and heart rate must be monitored throughout the test.
5. Draw blood into a tube containing anticoagulant (type required is assay dependent), for aldosterone, renin and electrolytes – do not place on ice but send to the laboratory immediately.
6. Infuse 2 L of 0.9% saline over 4 h.
7. At completion of infusion, immediately (with patient still recumbent) draw another blood sample for measurement of aldosterone and renin – do not place on ice but send to the laboratory immediately.
Interpretation
Plasma aldosterone concentration of > 140 pmol/L at the end of the test confirms a diagnosis of PA.
Fludrocortisone suppression test
The test is only indicated when the screening test demonstrates a high aldosterone:renin ratio. The same patient preparation requirements as for the screening test apply. The fludrocortisone suppression test is contraindicated in the elderly and those with severe hypertension owing to the risks associated with sodium retention. This test should be performed on an in-patient basis.
Procedure
2. Administer fludrocortisone 0.1 mg 6-hourly for four days.
3. Administer oral slow-release sodium tablets 30 mmol 8-hourly with meals for four days.
4. Administer slow-release potassium tablets in sufficient quantity to maintain plasma potassium concentration close to 4.0 mmol/L. Monitor plasma potassium concentration at least twice daily.
5. Day 4 (mid-morning). Ensure that the patient is upright for at least 30 min and repeat blood sample as on day 1.
Interpretation
Upright plasma aldosterone > 170 pmol/L on day 4 confirms PA, provided renin is fully suppressed.
Reference
Funder JW, Carey RM, Fardella C et al. Case Detection, Diagnosis, and Treatment of Patients with Primary Aldosteronism: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008;93: 3266–3281.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]C
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
