CHAPTER 33
Muscle disease
CHAPTER OUTLINE
FUNCTIONAL ANATOMY AND PHYSIOLOGY OF MUSCLE
DISEASES OF MUSCLE AND THEIR INVESTIGATION
BIOCHEMICAL INVESTIGATION OF MUSCLE DISEASE
Plasma creatine kinase activity
Other enzymes measurable in plasma
INVESTIGATION OF MUSCLE DISEASE
Non-metabolic, genetically determined myopathies
INTRODUCTION
Diseases affecting striated muscle are important causes of morbidity and mortality. They are common in clinical practice, and patients may be seen by a variety of clinical specialists, including neurologists, rheumatologists, orthopaedic surgeons and paediatricians. Investigating suspected muscle disease requires a combination of clinical and laboratory skills, including biochemical, genetic and pathological investigations, each assuming a different importance depending on the nature of the disorder. In some, biochemical studies play a minor role whereas in others, particularly the metabolic myopathies, biochemical investigations are crucial.
FUNCTIONAL ANATOMY AND PHYSIOLOGY OF MUSCLE
Skeletal muscle accounts for approximately 40% of total body weight and between 30% and 40% of total body oxygen consumption, even at rest. It is, therefore, an extremely important tissue in metabolic terms. Muscle is composed of multinucleated fibres that contain the contractile apparatus upon which movement depends. Although similar in structure, muscle fibres vary and three main types have been defined using metabolic and functional criteria (Fig. 33.1 and Table 33.1). Most skeletal muscles contain all three fibre types, although the proportions vary considerably depending on the function of the particular muscle.

FIGURE 33.1 Cross-section of normal muscle showing the histochemical reaction for ATPase activity. The three fibre types can be easily identified: type I (dark), type IIa and type IIb. (Photograph courtesy of Dr M A Johnson.)

The main function of muscle is to generate force in a controlled manner. This force, in the form of contraction (shortening), is produced in muscle fibres by the interaction of actin and myosin (Fig. 33.2), a process that is highly energy dependent. The energy required for muscle contraction comes from the hydrolysis of ATP, and maintenance of ATP concentration is critical. Any interference with ATP generation will inevitably impair the ability of muscle to produce force. ATP concentration is maintained by one of two mechanisms: ATP can be regenerated either from the energy storage molecule phosphocreatine or from ADP, or produced directly during glycolysis and mitochondrial oxidation. Regeneration is rapid, while the second process takes longer.
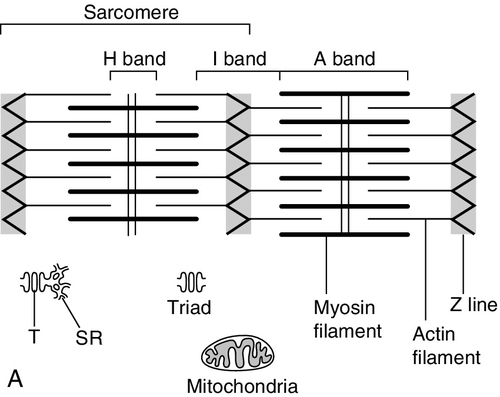

FIGURE 33.2 The structure of skeletal muscle. (A) Diagrammatic representation of the structural components within a single muscle fibre. Thin filaments (actin) are anchored to the Z line. Thick filaments are composed of multiple myosin molecules, each of which has a hinged end that can interact with the thin filament. The T tubule is a continuation of the sarcolemmal membrane, which interacts with the sarcoplasmic reticulum at specific sites (triad). The signal for contraction is transmitted along the sarcolemma to the T tubule, which in turn causes release of calcium from the sarcoplasmic reticulum (SR). Calcium release stimulates contraction. (B) Electron micrograph of normal muscle. This shows the bundles of myofilaments with Z lines (z), thin (I band) and thick (A band) filaments. Triad (Tr) and mitochondria (m) are identified. (Photograph courtesy of Dr M J Cullen. Magnification × 30 000.)
Phosphocreatine is present in large quantities in muscle (the other major site is brain) and acts as a reservoir of high-energy phosphate groups that it can donate to ADP in the following transphosphorylation reaction, catalysed by the enzyme creatine kinase:

The concentration of ATP does not fall significantly until nearly all of the phosphocreatine has been converted to creatine. A second transphosphorylation reaction, catalysed by adenylate kinase, seems to have a minor role in ATP production. The AMP formed is broken down further by AMP deaminase.


ATP can also be generated directly by glycolysis and the oxidative catabolism of carbohydrate and lipid fuels. These processes are slow compared with transphosphorylation, but nevertheless are essential for ATP generation. The breakdown of carbohydrate by glycolysis (Fig. 33.3) has a vital role in skeletal muscle since it permits ATP production under anaerobic conditions. When oxidative metabolism of pyruvate is impaired, for example during ischaemia, which includes high intensity activity, or in the presence of a defect of the respiratory chain, increasing amounts of lactate may be produced.
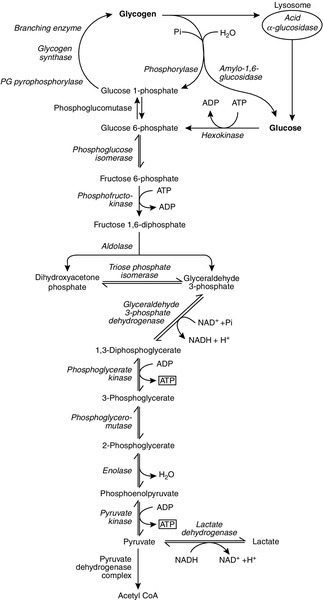
FIGURE 33.3 Glycogen breakdown and glycolysis. Under anaerobic conditions, when further oxidation of acetyl-CoA is impaired, pyruvate is metabolized to lactate. This will also occur in defects of pyruvate dehydrogenase complex (PDC) and of the respiratory chain, and is due to the failure to reoxidize intramitochondrial NADH, which in turn inhibits PDC. NADH formed during glycolysis (and other substrate oxidation) is reoxidized by complex I of the mitochondrial respiratory chain (see Fig. 33.7).
While small amounts of ATP can be generated by glycolysis in the cytosol, significantly greater amounts are produced by the oxidative breakdown of metabolic fuels (pyruvate, ketone bodies, fatty acids) that occurs within mitochondria. Long chain fatty acids, either from intracellular lipid stores or imported from the bloodstream, are first activated to their acyl-CoA esters before being transported into the mitochondrial matrix by the concerted action of carnitine palmitoyltransferase I, carnitine/acylcarnitine translocase and carnitine palmitoyltransferase II (Fig. 33.4). Short and medium chain fatty acids enter the mitochondria as the free acids and are activated to their acyl-CoA esters in the mitochondrial matrix. Inside the mitochondria, fatty acyl-CoA esters undergo β-oxidation, a series of four reactions that results in the production of acetyl-CoA and a chain-shortened fatty acid (Fig. 33.5); there are two or three enzymes with overlapping substrate specificities for each of these steps. Reducing equivalents generated by the process of β-oxidation are transferred to the respiratory chain. The acyl-CoA dehydrogenases transfer reducing equivalents to electron transfer flavoprotein (ETF) and thereafter to ETF dehydrogenase, which directly reduces ubiquinone of the respiratory chain (see Fig. 33.7). The 3-hydroxyacyl-CoA dehydrogenases reduce NAD+ to give NADH, which transfers its reducing equivalents to complex I of the respiratory chain. Acetyl-CoA generated either from carbohydrate or fatty acid oxidation is metabolized further by the tricarboxylic acid cycle (Fig. 33.6). The oxidation of fatty acids and glucose, as well as the subsequent metabolism of acetyl-CoA, generates more reduced cofactors (NADH and FADH2) that are re-oxidized by the respiratory chain, and the energy released by this process is conserved as ATP (Fig. 33.7).
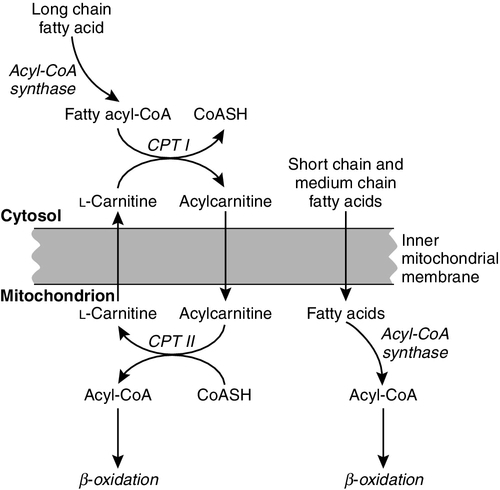
FIGURE 33.4 Transport of fatty acids across the inner mitochondrial membrane. Short and medium chain fatty acids do not require a specialized transport mechanism to cross the membrane. Long chain fatty acids are first acylated in the cytosol. Carnitine palmitoyltransferase (CPT) I on the outer side of the inner membrane converts the fatty acyl-CoA to an acylcarnitine ester and this is transported across the membrane linked to the export of carnitine. Inside the matrix, CPT II converts the acylcarnitine back to the fatty acyl-CoA, which is broken down by β-oxidation.
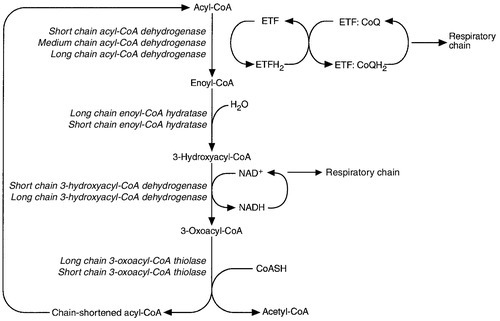
FIGURE 33.5 Mitochondrial β-oxidation of saturated fatty acids. ETF, electron transfer flavoprotein; CoQ, coenzyme Q.
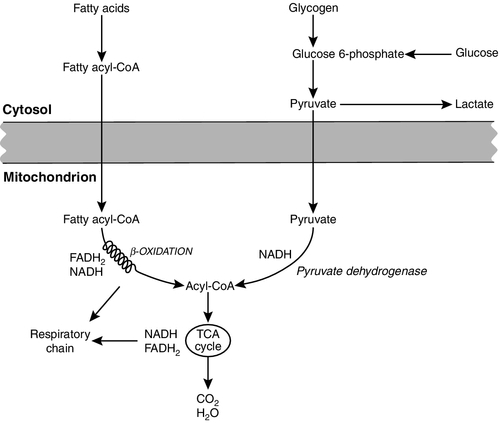
FIGURE 33.6 The production and further metabolism of acetyl-CoA.
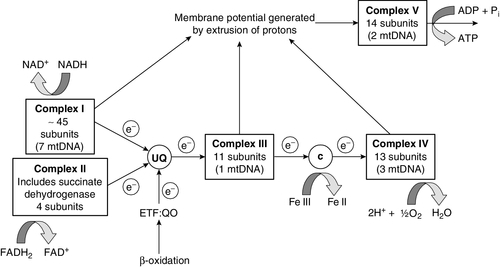
FIGURE 33.7 Components of the mitochondrial respiratory chain. NADH is reoxidized by complex I, while FADH2 donates electrons via complex II and ETF dehydrogenase (ETF:QO) donates electrons directly to uniquinone (UQ). Electron transport generates sufficient energy at three sites (complexes I, III and IV) to pump protons out of the matrix, producing an electrochemical gradient. This gradient is discharged by ATP synthase (complex V) and the energy released used to drive the phosphorylation of ADP to ATP. c, Cytochrome c; e−, electron.
The balance of muscle metabolism depends on the state of activity, diet and the influence of various hormones (particularly insulin, thyroxine, glucocorticoids). At rest, muscle predominantly oxidizes fatty acids to generate the energy for ATP synthesis. During exercise, the proportion of energy derived from carbohydrate or lipid depends on the degree and duration of this exercise and on the degree of physical fitness. High-intensity exercise at close to maximum oxygen uptake relies almost exclusively on carbohydrate metabolism, and glycogen depletion coincides with exhaustion. During moderate-intensity exercise for prolonged periods, there is a switch from carbohydrate to lipid metabolism.
DISEASES OF MUSCLE AND THEIR INVESTIGATION
There are a large number of different disorders of muscle, and while our classification includes the main categories (Box 33.1), more comprehensive lists are available (see Karpati et al. in Further reading, below). A detailed description of the clinical features associated with the different types of muscle disease is outside the scope of this chapter, but is discussed in several texts on muscle disease. The clinical features depend upon the age of the patient and the type of disease. For instance, a child with Duchenne muscular dystrophy will experience difficulty rising from sitting or lying and may have frequent falls. Such problems will prompt the parents to seek advice. In adults, the main forms of presentation are weakness, fatigue and pain. Less commonly, muscle wasting, swelling or twitching of the muscle or a skin rash may be a first symptom. In the genetically determined disorders, the weakness is usually gradually progressive and often follows a characteristic pattern. In other myopathies, there may be associated stigmata, for instance joint disease or skin rash suggesting a connective tissue disorder; anxiety, sweating and weight loss suggesting hyperthyroidism, or features compatible with high alcohol intake. The muscle pain described by patients with muscle disease may be important in suggesting whether there may be a metabolic cause. For instance, both defects of carbohydrate metabolism and fatty acid oxidation will cause muscle pain associated with exercise. The pain associated with defects of carbohydrate metabolism occurs during high intensity exercise when glycolysis generates most of the energy required for muscle contraction, whereas defects of fatty acid oxidation cause muscle pain after prolonged exercise at a time when fatty acids are the predominant metabolic fuels.
The clinician must evaluate the clinical features and decide which investigations are appropriate. In many patients with suspected muscle disease, this will involve a combination of biochemical, molecular genetic, neurophysiological and morphological investigations. While many biochemical and genetic studies are performed on blood samples, morphological study and biochemical analyses such as measurement of muscle enzyme activity, require tissue. Muscle biopsy is a relatively simple procedure and there are two main methods: an open biopsy, in which relatively large amounts (0.5–3 g) of muscle can be removed, and a needle biopsy, in which smaller amounts (50–200 mg) are obtained. For most biochemical and histochemical studies, small amounts are sufficient. Morphological changes alone may be sufficient to suggest a diagnosis, for example of Duchenne muscular dystrophy. The diagnosis of metabolic myopathies has been greatly improved by the development of cytochemical techniques that can show, for example, the abnormal storage of glycogen or lipid, or demonstrate the presence or absence of specific enzyme activities in situ. A further development in this area is the use of specific antisera to enable the precise localization (and therefore the presence or absence) of proteins at the cellular level. This technique of immunocytochemistry provides valuable additional information in the investigation of muscle disease.
BIOCHEMICAL INVESTIGATION OF MUSCLE DISEASE
‘Routine’ biochemical studies
These include the measurement of plasma sodium, potassium, chloride, urea, bicarbonate, glucose, calcium and phosphate, together with simple tests of endocrine function. While not all these tests are necessary for each patient with muscle disease, disturbances of each parameter can result in muscle disease, as shown by the following examples. Severe hypokalaemia associated, for example, with diuretic use or liquorice ingestion, can result in muscle weakness. Renal failure may lead to muscle weakness for several reasons, including electrolyte disturbance and altered calcium metabolism. It must also be remembered that acute muscle necrosis from any cause (e.g. malignant hyperpyrexia, drugs, injury or metabolic myopathy) may itself cause acute kidney injury owing to the tubulotoxic effect of myoglobin. Muscle symptoms are common in endocrine disturbances: hypothyroidism, for example, may be associated with proximal weakness, often with discomfort in the affected muscles.
Plasma creatine kinase activity
The measurement of plasma enzyme activity is important in the diagnosis of muscle disease, and while the activities of several enzymes may be elevated, creatine kinase (CK) is the most sensitive indicator of muscle damage. Skeletal muscle has the highest CK content of any tissue, more than three times as much as heart or brain, and consequently nearly all CK activity in normal plasma is derived from skeletal muscle. In addition, CK activity is more frequently abnormal than other enzymes in neuromuscular disease and the range of abnormal values is greater.
Human tissues contain three forms of creatine kinase, comprising dimers of the muscle and brain-type subunits, M and B. The combinations are CK-MM, CK-MB and CK-BB. Skeletal muscle contains mostly CK-MM with only a small amount of CK-MB, ranging from 0.2% to 15% of total enzyme activity (mean value of 5–6%). The proportion of CK-MB is higher in type I muscle fibres. Regenerating muscle fibres revert to an embryonic enzyme pattern and have about 40–50% CK-MB. Brain contains only CK-BB while heart muscle contains about 40% CK-MB, the rest being CK-MM. In normal adult plasma, CK activity is almost entirely due to the CK-MM isoform. Muscle damage will increase total CK activity and a proportion will be of the CK-MB type, but this is usually < 6%. High plasma CK activity associated with a CK-MB fraction > 6% may be associated with myocardial damage; however, there are several other situations in which the plasma CK-MB activity may be high:
• in patients with chronic neuromuscular diseases, there tends to be a higher CK-MB activity owing to the increased percentage of regenerating fibres
• in children younger than early teenage, the percentage of CK-MB in plasma is higher than in adults, in the range of 14–26%. This could complicate the investigation of heart disease in this age group.
The activity of CK in plasma will vary even in healthy subjects and, as noted above, may be significantly elevated following exercise. While CK activity may be raised immediately after exercise, it reaches its peak after 1–2 days. Generally, the more severe the exercise, the higher the total activity and the more delayed the peak. Thus, a 24-fold increase in CK activity has been reported after a 53 mile walk and transient CK values of 10 000–20 000 U/L may be seen after intense training in young men, in whom subsequent investigation fails to detect any abnormality. The effect on CK activity is more pronounced in untrained subjects compared with trained, but generally does not result in greater than a four-fold increase, unless the exercise performed is severe, and returns to normal with resting.
Conditions other than exercise may cause significant increases in plasma creatine kinase activity (Box 33.2). The importance of obtaining a good clinical history and measuring CK activity under basal conditions is, therefore, of paramount importance if unnecessary invasive investigations are to be avoided. Creatine kinase activity in plasma is raised whenever there is necrosis or regeneration of muscle and will, therefore, be elevated in most myopathies. Very high values are found in Duchenne muscular dystrophy and in conditions where severe muscle necrosis occurs, such as acute polymyositis and rhabdomyolysis associated with malignant hyperpyrexia or the metabolic myopathies. In myopathies where muscle destruction is not a major feature, for instance some endocrine myopathies, plasma CK activity may be normal or only mildly elevated. It must be remembered that plasma CK activity may also be elevated in severe neurogenic conditions that include active denervation, and thus it is not diagnostic of muscle conditions alone. Plasma CK activity may be used to follow the progress of myopathic disorders and their response to treatment. For example, the effective treatment of inflammatory muscle disease with steroids and/or other immunosuppressive treatments may be monitored by the fall in CK activity as well as by the improvement in muscle function.
Occasionally, a significantly elevated CK is found unexpectedly. Many of these patients will be found to have muscle disease when formally examined, but there are also individuals with elevated CK who show no signs of neuromuscular disease, so-called ‘idiopathic hyperCKaemia’. Some of these prove to have ‘macro CK’ – an entity consisting of CK bound to an immunoglobulin (usually IgG) – which is cleared more slowly from the plasma than the unbound enzyme. Endocrine dysfunction, such as hypothyroidism, can elevate CK without obvious muscular dysfunction and it is important to remember that individuals with malignant hyperthermia can have elevated CK with no other features of neuromuscular disease.
Statin induced elevation of creatine kinase
Drug-induced elevation of CK occurs and, indeed, has become more common since the introduction of the HMG-CoA reductase inhibitors (statins). This class of drug frequently causes myalgia, often with no rise in plasma CK concentration, but only rarely causes muscle disease, although necrotising myopathy and rhabdomyolysis have been reported. Several risk factors that predispose to muscle disease in those taking statins are known, including genetic variants in drug transport molecules and hepatic drug metabolism. Factors that are associated with higher plasma concentrations of statins such as increasing age, female sex, liver or kidney disease, untreated hypothyroidism and drugs that inhibit the cytochrome P450 system, increase the risk of myalgia and rhabdomyolysis. Interfering drugs that are of particular concern include cyclosporin, fibrates, calcium channel blockers, protease inhibitors and warfarin; grapefruit juice also has a similar effect. Pravastatin is not metabolised by the cytochrome P450 system so may be a safer choice than other statins if combination treatment with these drugs is required. Low plasma concentrations of 25-hydroxycholecalciferol are associated with myalgia and may exacerbate statin associated muscle pain. There is some evidence that patients given vitamin D replacement before starting a statin are at lower risk of developing symptoms.
There is a strong association between statin induced myalgia and a common variant of the gene SLCO1B1, which codes for a transporter that mediates the hepatic uptake of statins apart from fluvastatin. In one study, > 60% of cases could be attributable to the variant, which impairs statin uptake. Cyclosporin is thought to inhibit the transporter and can therefore increase plasma concentrations of most statins, even of those that are not metabolised by the cytochrome P450 system.
The underlying mechanism of statin toxicity is still debated, but theories include alterations in myocyte membrane cholesterol content, depletion of isoprenoids that control myofibre apoptosis and depletion of coenzyme Q10 (ubiquinone). Hydrophilic statins such as pravastatin and rosuvastatin may be less myotoxic than other more lipophilic statins such as atorvastatin, simvastatin and lovastatin, possibly because of decreased penetration into muscle cells.
In most patients with statin-induced myalgia, the CK does not rise dramatically, if at all, and a minor rise may simply require monitoring and not cessation of treatment. Strategies for improving the tolerability of statin treatment include changing to a hydrophilic statin, reducing the frequency of doses to once or twice weekly, or supplementation with vitamin D and/or coenzyme Q10. However, much of the evidence of benefit of dosage variations and supplements has been anecdotal and there are few data on the cardiovascular effects of such strategies.
Persisting elevation of CK following withdrawal of statin treatment should be investigated, as there is more likely to be an underlying muscle disorder. The use of statins in patients with known muscle disease should be avoided if possible, although in individuals where there is a strong case for treatment, monitoring CK activity to detect progressive change is an alternative.
Other enzymes measurable in plasma
The activities of aldolase, lactate dehydrogenase and pyruvate kinase are also raised in destructive myopathies, but measurement of these enzymes provides no additional information to that given by CK measurement. Similarly, carbonic anhydrase III and a muscle-specific enolase, two enzymes that are specific to skeletal muscle, are also elevated in muscle disease, but the patterns of abnormality are similar to that of CK. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are also raised in the presence of muscle destruction and rise in parallel with CK. Measurement of plasma γ-glutamyl transferase activity may help to determine the tissue origin of high aminotransferases, as it is normal in muscle disease but usually raised with liver disease.
Myoglobinuria
In the presence of muscle damage, the muscle pigment myoglobin leaks into blood and urine. While myoglobin is a sensitive indicator of muscle destruction, it is neither significantly more sensitive nor more specific than CK. Myoglobin gives a positive reaction on urine dipstick testing for blood, which can be misinterpreted as being due to haematuria. In conditions in which there is severe muscle necrosis, rhabdomyolysis may occur. Causes include mechanical trauma, myotoxic agents (animal poisons, numerous drugs and alcohol), extremes of ambient temperature, infections and inherited muscle diseases such as malignant hyperpyrexia, some types of muscular dystrophy and defects of mitochondrial energy, carbohydrate and lipid metabolism. Myoglobin may be deposited in nephrons and cause acute kidney injury. The risk of injury appears to be associated with acute elevations of myoglobin.
INVESTIGATION OF MUSCLE DISEASE
Non-metabolic, genetically determined myopathies
The investigation of muscular dystrophies and other non-metabolic myopathies is largely dependent on morphological and/or genetic analyses. For example, the X-linked allelic disorders Duchenne and Becker muscular dystrophies may be diagnosed by identifying the defect at the genetic level; a deletion of the dystrophin gene is found in around 65% of Duchenne boys. The same is true for facioscapulohumeral muscular dystrophy, where a deletion of telomeric DNA on the long arm of chromosome 4 is seen in > 95% of cases. In the limb girdle group of muscular dystrophies, the same phenotype is produced by defects in very different proteins (and their corresponding genes) such that muscle biopsy and immunocytochemical studies are usually required.
In membrane disorders, such as the myotonic dystrophies and congenital myotonias, genetic analysis is paramount. In contrast, the periodic paralyses, which are also caused by ion channel dysfunction, are associated with electrolyte changes that are important to define. In hypokalaemic periodic paralysis, the patient may experience episodes of weakness that last from hours to days and which can be provoked by carbohydrate ingestion. Serum potassium measured at the onset of weakness is low, but may normalize quickly despite persisting symptoms. In hyperkalaemic periodic paralysis, the potassium concentration is elevated and there is also a form in which potassium appears not to change. In certain populations, particularly in Asia, the combination of hypokalaemic paralysis and hyperthyroidism is found. Treatment of the underlying thyroid dysfunction cures the potassium disturbance and the muscle disease.
Malignant hyperpyrexia is a rare syndrome, in which there is a rapid rise of body temperature (up to 1 °C every 5 min), lactic acidosis, muscle rigidity, hyperkalaemia, very high plasma CK activity and myoglobinuria. The reaction may be triggered by several inhalational anaesthetics (e.g. halothane) and succinylcholine. The disorder has been linked to various different genetic defects including mutations in the ryanodine receptor gene. (Defects in this gene also give rise to the muscle disease, central core disease.) Plasma CK activity can also be increased in patients at risk. Diagnosis is dependent on in vitro testing of muscle strips for an abnormal contracture response to halothane, caffeine or a combination of both agents.
Metabolic, genetically determined myopathies
Whereas the biochemical investigations so far described are either general indicators of muscle damage or directed at systemic conditions in which muscle involvement occurs, the following section deals with specific investigations of genetically determined metabolic muscle disease. It is in this area that biochemical studies are of paramount importance. The three main areas to be described are disorders of carbohydrate metabolism, defects of the respiratory chain and defects of fatty acid oxidation. The discussion of each will be divided into sections dealing with the investigations performed in blood and/or urine, muscle tissue analyses (e.g. histocytochemical studies, when appropriate) and specific biochemical investigations.
Disorders of carbohydrate metabolism
Disorders of carbohydrate metabolism can be divided into two main groups:
• disorders resulting from lysosomal storage of glycogen (α-acid maltase deficiency) or from the deposition of abnormal polysaccharide (branching enzyme deficiency). For a detailed discussion of the clinical and biochemical features, the reader is referred to the review by Chen (2001) (see Further reading, below).
Dynamic/functional tests
In diseases related to failure to metabolize glucose, the symptoms are related to the lack of energy (ATP) under conditions where muscle relies on glycolysis to meet this demand. The main clinical manifestations are cramp-like muscle pain and, occasionally, the appearance of contractures. The inability to derive energy from glycolysis is easily demonstrated by making the patient perform muscular work and measuring the rise of blood lactate. This is usually done under ischaemic conditions, but is equally effective without ischaemia (see Appendix 33.1).
Histocytochemistry
Histocytochemical methods are used to study both the amount of glycogen and several of the enzymes of glycogen mobilization and glycolysis. Glycogen is usually demonstrated by some variant of the periodic acid Schiff (PAS) technique. Myophosphorylase, phosphofructokinase, myoadenylate deaminase and lactate dehydrogenase activities are all detectable cytochemically, and absence of enzyme activity is diagnostic in clinical terms. The finding of glycogen storage with grossly elevated membrane-bound acid phosphatase activity is virtually diagnostic of acid α-glucosidase deficiency.
Biochemical investigations
The enzymes of glycogenolysis and glycolysis are all highly active, and homogenates prepared from needle-biopsy specimens are usually adequate for analysis. The measurement of these enzymes has been reviewed elsewhere (see Further reading, below) and only a brief summary is given here.
Acid α-glucosidase (acid maltase) is measured by monitoring glucose release from maltose or glycogen at acid pH or by using 4-methylumbelliferyl α-glucoside as substrate and measuring the fluorescence of 4-methylumbelliferone released. The process has been simplified by the availability of a sensitive immune capture technique and activity can now be measured in dried blood spots saved on filter paper. The simplest technique for the assay of amylo-1, 6-glucosidase activity is based on the fact that the hydrolytic activity of this enzyme is partly reversible; activity may be followed by measuring the incorporation of [U-14C] glucose into glycogen in the presence of a muscle homogenate. Branching enzyme α-1:α-1,4-glucan:1,4-glucan-6-glucosyltransferase activity depends upon the stimulation of the rate of glycogen synthesis from glucose 1-phosphate catalysed by phosphorylase a (non-physiologically): activity is followed by the release of inorganic phosphate. The reaction catalysed by phosphorylase a is reversible, although, in vivo, the concentration of glucose 1-phosphate is at least two orders of magnitude too low to allow glycogen synthesis. The enzyme can therefore be measured in either direction. Phosphorylase kinase is measured by the rate of ATP-dependent conversion of phosphorylase a to phosphorylase b. Phosphofructokinase activity is measured in muscle homogenates by coupled enzyme assay in which the fructose 1,6-bisphosphate formed is converted to dihydroxyacetone phosphate, and this is converted to glycerol 3-phosphate with the oxidation of NADH. Phosphoglycerate kinase and phosphoglycerate mutase are measured in muscle homogenates by coupled enzyme assays. Lactate dehydrogenase is measured by a standard spectrophotometric assay.
Defects of the respiratory chain
These disorders were first identified in muscle, but are now known to produce a wide range of clinical disorders often involving the central nervous system (see Further reading, below). The respiratory chain is composed of five complexes (Fig. 33.7) and defects have been identified in each of them as well as in many of the proteins required to assemble and maintain this pathway and the mitochondrial genome that encodes some of the components.
Dynamic/functional tests in blood
Elevated plasma lactate, often associated with high concentrations of pyruvate and alanine, is a common, but not universal feature of respiratory chain dysfunction. In some patients, resting lactate is normal, but increases to very high concentrations during aerobic exercise. Aerobic exercise tests can, however, be difficult to perform in children and patients with significant physical disability. Examination of the CSF lactate is of value in patients who have predominantly CNS symptoms since the lactate concentration may be inappropriately high in this compartment, but normal or near normal in blood. In patients with the Kearns–Sayre syndrome, elevated total protein concentrations are found in CSF, and this is one of the diagnostic criteria of the disorder. Recently, a fibroblast growth factor, FGF21, was found to be elevated in plasma in mitochondrial disease and appears to be a better biomarker for mitochondrial muscle disease than metabolites such as lactate.
Histocytochemistry
The activity of several mitochondrial enzymes can be demonstrated cytochemically. Cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) are the most specific for the respiratory chain, but NADH oxidation and ATPase activity may also give non-specific clues to the presence of a disorder. The morphological hallmark of respiratory chain disease is the ragged red fibre originally described using the Gomori trichrome stain. The finding of COX positive and deficient muscle fibres (a mosaic) is highly suggestive of a defect in mitochondrial DNA (mtDNA), and this can easily be shown using COX and SDH stains in combination (Fig. 33.8). In this case, histochemical analysis is sufficient and the next step will be genetic analysis of mtDNA. Detailed biochemical analysis is still necessary, however, particularly for the investigation of complexes I and III. Newer techniques that combine gel electrophoresis, ‘in gel’ activity measurement and immunoblotting are also available and will certainly play a greater role in the diagnostic process in the future.
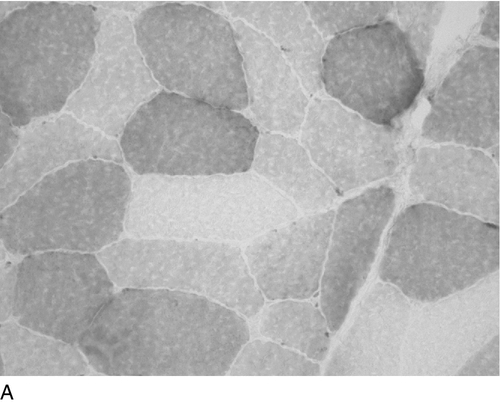
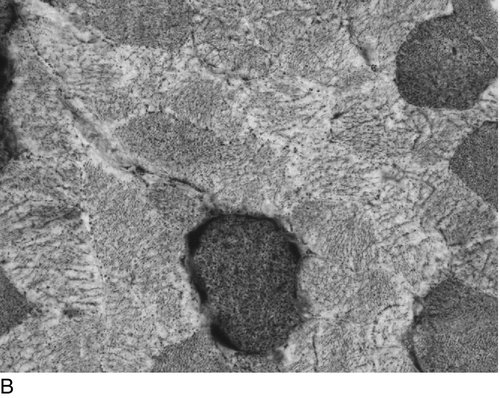
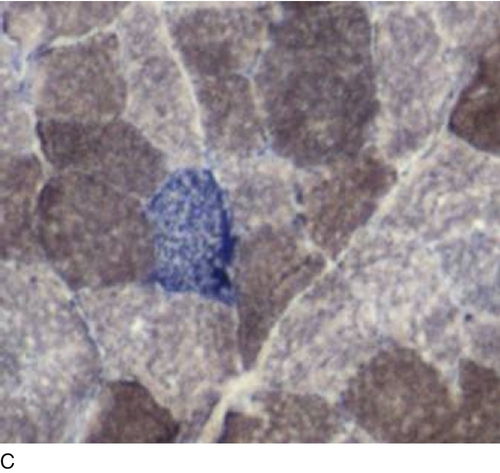
FIGURE 33.8 Histochemical demonstration of: (A) cytochrome c oxidase (complex IV) and (B) succinate dehydrogenase (complex II) activity. The reactions specifically identify respiratory chain complexes and the combination (C) permits the easy detection of muscle fibres lacking cytochrome c oxidase activity (light grey fibres). (Photographs courtesy of Dr B Krossnes.)
Biochemical investigations
Measurement of mitochondrial oxidations
The complete oxidation of metabolic fuels with concomitant synthesis of ATP can be studied in various ways, including overall flux from substrate to water, flux through various segments and the activities of individual components of the respiratory chain. These will be discussed below. In many instances, it is necessary to isolate mitochondria from tissue (usually muscle). This is a relatively straightforward, but critical, step, since the outcome of all further work will depend on the quality of the mitochondrial fraction prepared.
Flux through the whole respiratory chain can be studied either by measuring oxygen consumption or the production of ATP. Polarography measures the rate of change in oxygen concentration in solution, and since oxygen is the final electron acceptor of the respiratory chain, this provides direct measurement of activity. The working volume of the electrode determines how much mitochondrial protein is required and since human muscle biopsies are small, the smaller the volume the better. Several parameters may be assessed using polarography. First, the quality or state of the mitochondria may be inferred from the respiratory control ratio (RCR). This is the ratio of oxygen consumption in the presence of ADP to that when all ADP has been converted to ATP. The RCR in fact reflects the structural integrity of the mitochondria, since any damage to the inner membrane will dissipate the electrochemical gradient and thus the ability to synthesize ATP. The RCR in human skeletal muscle mitochondrial fractions is usually between 2 and 4. Flux through the respiratory chain is often expressed in terms of oxygen consumed per milligram of protein in the presence of ADP. It is also possible to measure ATP formed during respiration, either directly or by linking to a luminescent marker.
Flux through segments of the respiratory chain can be measured spectrophotometrically using artificial electron acceptors whose light absorption changes on reduction/oxidation. A commonly used acceptor is potassium ferricyanide. NADH produced by the oxidation of pyruvate, oxoglutarate or glutamate, yields electrons that enter the respiratory chain via complex I. Ferricyanide accepts electrons from cytochrome c with the result that it is possible to assess flux through the segment containing complex I, ubiquinone and complex III (see Fig. 33.7). Alternatively, using succinate as a substrate, electrons enter via succinate dehydrogenase that is part of the respiratory chain complex II. The ferricyanide-linked assay will then measure flux from complex II to cytochrome c.
Measurement of activity of individual respiratory chain complexes
The activity of these complexes is best measured in mitochondrial fractions. Where small amounts of tissue are available, partial purification is used.
Complex I activity is measured by determining rotenone-sensitive NADH-ubiquinone oxidoreductase activity. Low molecular weight, water-soluble ubiquinone or a ubiquinone analogue can be used as an electron acceptor. This assay requires that the mitochondria are made permeable to enable the NADH to reach its binding site on the inner aspect of the inner mitochondrial membrane. NADH oxidation is measured by the decrease in absorbance at 340 nm before and after the addition of rotenone. The use of rotenone, a specific complex I inhibitor, is essential since there are other enzymes capable of oxidizing NADH.
Complex II activity is measured by following the reduction of ubiquinone by succinate either directly or by linking the reaction to an artificial electron acceptor such as dichlorophenol-indophenol. The succinate dehydrogenase component of complex II can be further studied by using phenazine ethosulphate rather than ubiquinone as the intermediate electron acceptor.
Complex III activity is measured by following the reduction of cytochrome c by ubiquinol. Ubiquinol is synthesized from ubiquinone by reduction with dithionite followed by organic extraction. Further studies of part of complex III can be made by measuring the transhydrogenase reaction, which gives information on the b cytochromes.
Complex IV activity is measured by following the oxidation of reduced cytochrome c. Reduced cytochrome c is prepared by addition of ascorbate, which is removed by gel filtration. Polarographic measurement of complex IV activity is also widely used. In this reaction, ascorbate and tetramethyl-p-phenylenediamine dihydrochloride (TMPD) are used as artificial electron donors to cytochrome c.
Complex V is rarely measured in patients with suspected mitochondrial disease, but several methods to measure oligomycin-sensitive ATPase activity have been used for mitochondria from other species.
Molecular biology techniques
Mitochondria are unique in that they contain their own DNA (mtDNA). This small genome encodes 13 polypeptides (seven that go into complex I, one in complex III, three into complex IV and two into complex V) and 24 RNA species that participate in intramitochondrial protein synthesis. Disorders caused by mtDNA mutations are highly variable, but there are some classic syndromes that are easily recognizable. For example, Kearns–Sayre syndrome and chronic progressive external ophthalmoplegia that can be caused by gene rearrangements (most often deletions); Leber hereditary optic neuropathy is due to a point mutation in complex I protein-coding genes, and myoclonus, epilepsy with ragged red fibres (MERRF) and myopathy, encephalopathy, lactic acidosis and stroke (MELAS) are due to point mutations in different mitochondrial genes. Since the mitochondrial genome is inherited exclusively from the mother, disorders in which maternal inheritance occurs gave the first clue to the possibility of mtDNA mutations.
Since the first mtDNA mutations were described in 1988, more than 150 different mutations have been found including both rearrangements (deletions or duplications) and point mutations. In the last few years, mutations in nuclear encoded genes affecting mitochondria have also been identified and this group is expanding. As stated earlier, the finding of a mosaic of COX positive and deficient fibres is highly suggestive of a defect involving mtDNA. This can, however, either be a primary mtDNA mutation, as, for example, in the syndrome of MELAS, or a secondary mtDNA defect caused by a nuclear genetic defect in a protein that has a role in mtDNA maintenance.
Defects of fatty acid oxidation
Muscle symptoms associated with a defect of fatty acid oxidation can occur alone or as part of a systemic abnormality. Symptoms may arise acutely or more chronically and affected individuals are at risk of developing cardiomyopathy, hepatic dysfunction, sudden infant death and hypoglycaemic coma.
Dynamic/functional tests
Intermediary metabolites and metabolic fuels in blood
If a systemic defect is present, the inter-relationship between metabolic fuels is abnormal. In healthy subjects during stress or fasting, lipolysis is stimulated and free fatty acids are released into the circulation. Following uptake by tissues, fatty acids are converted to their acyl-CoA esters and oxidized by β-oxidation to generate acetyl-CoA. Acetyl-CoA may be further oxidized by the citrate cycle or converted to ketone bodies in the liver. Circulating ketone bodies are an important source of energy during stress or fasting and are readily oxidized by extrahepatic tissues, in particular muscle and brain. When fatty acid oxidation is impaired, the blood concentrations of free fatty acids increase, but there is no concomitant increase in the concentration of ketone bodies. Energy must, therefore, be derived from carbohydrate metabolism as long as glycogen stores last. Once these are depleted, hypoglycaemia can develop and may lead to permanent tissue damage or death. It is clear, therefore, that fasting is potentially dangerous for patients with fatty acid oxidation defects and should be avoided.
Measurement of plasma, tissue and urine carnitine concentrations
Patients with defects of fatty acid oxidation frequently have abnormalities of carnitine metabolism. Partial β-oxidation will continue to generate chain-shortened acyl-CoA ester intermediates that combine with carnitine in a reaction catalysed by carnitine acyltransferases. These carnitine esters are transported out of the mitochondrial matrix, resulting in a higher percentage of carnitine in the acylated form in blood and urine from patients compared with healthy subjects. In addition, in the presence of a defect in β-oxidation, there will be abnormal excretion of specific acylcarnitines, e.g. octanoylcarnitine in medium chain acyl-CoA dehydrogenase deficiency (MCAD). Secondary carnitine deficiency can occur in these individuals, although the mechanism remains uncertain, as total urinary excretion of acylcarnitines is not increased. Carnitine concentrations, free and acylated, are usually measured using tandem mass spectrometry. Since carnitine metabolism is perturbed by most defects of fatty acid oxidation, these measurements rarely help in making a specific diagnosis.
Mass spectroscopic measurement of specific acylcarnitines is a screening technique that is now widely used. This can be performed on dried blood spots (e.g. on a Guthrie card) and allows the detection of specific intermediates that are identified using tandem mass spectrometry. The metabolism of fatty acids proceeds by a chain shortening series of reactions that produce acetyl-CoA. In the presence of a block in this process, chain-shortened fatty acids will accumulate and these are esterified with carnitine, forming acylcarnitines that can be detected in blood. Figure 33.9 shows typical profiles for three different defects that can be identified using this technique in dried blood samples. Owing to the frequency of MCAD in certain populations and its association with sudden infant death, this technique is increasingly being used for screening of neonates.
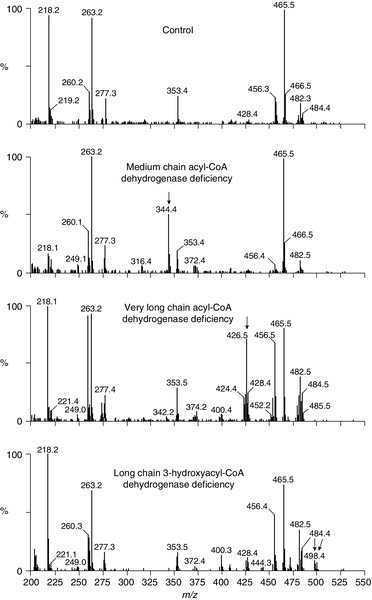
FIGURE 33.9 Analysis of acylcarnitine intermediates using mass spectrometry. Acylcarnitine profile in dried blood spot from a healthy subject (upper panel) and patients with defects of fatty acid oxidation. The numbers above the peaks are m/z values that correspond to the molecular ions of butylated acylcarnitine species: free carnitine (m/z 218), free carnitine internal standard (m/z 221), C2-carnitine (m/z 260), C2-carnitine internal standard (m/z 263), C3-carnitine (m/z 274), C3-carnitine internal standard (m/z 277), C8-carnitine (m/z 344), C8-carnitine internal standard (m/z 353), C12-carnitine (m/z 400), C14:1-carnitine (m/z 426), C14-carnitine (m/z 428), C16-carnitine (m/z 456), C16-carnitine internal standard (m/z 465), C18:1-carnitine (m/z 482), C18-carnitine (m/z 484), 3-OH-C18:1-carnitine (m/z 498), 3-OH-C18-carnitine (m/z 500). Major diagnostic analytes are shown by arrows. For example, in medium chain acyl-CoA deficiency, medium chain (C8)-carnitine esters accumulate (m/z 344). In very long chain acyl-CoA dehydrogenase deficiency, C14 and longer chain fatty acids accumulate, while in long chain 3-hydroxyacyl-CoA dehydrogenase deficiency (bottom panel), long chain hydroxy forms of the carnitine esters accumulate.
Measurement of dicarboxylic acids and acylglycines in urine
When mitochondrial β-oxidation is impaired, dicarboxylic acids, e.g. adipic, suberic and sebacic, are generated by the partial oxidation of fatty acids in the endoplasmic reticulum (ω-oxidation) and by β-oxidation in peroxisomes. These dicarboxylic acids are excreted in the urine, and the pattern of the organic aciduria, detected by gas chromatography, may be helpful in the diagnosis of a defect of fatty acid oxidation. Acylglycines are formed in the liver by the combination of acyl-CoA esters and glycine, a reaction catalysed by glycine-N-acylase. Glycine conjugates are excreted in the urine and can be detected by gas chromatography. An isotope dilution analysis of urinary acylglycines has been introduced, which seems to be a very sensitive test for defects of medium chain acyl-CoA dehydrogenase deficiency.
Specific biochemical investigation
Measurement of flux through β-oxidation
Measurement of flux is an important investigation in defects of fatty acid oxidation. There are, however, many problems in accurately determining the flux in muscle preparations for diagnostic purposes. One of the most frequently used methods measures 14CO2 formed by the oxidation of 14C-labelled fatty acids. This method is inaccurate since 14CO2 is only released by the further oxidation of acetyl-CoA by the citrate cycle, which is incomplete and not necessarily proportional to the flux through β-oxidation. Measurement of 14C-labelled acid-soluble products is a much better indicator of flux, but this method is not suitable for 14C-labelled medium and short chain fatty acids. An alternative approach is to determine 3H2O formed during the oxidation of 3H-labelled fatty acids. Unfortunately, this technique is also limited because of the relatively few 3H-labelled fatty acids available. Acylcarnitine profiles can be studied in fibroblasts using the same technique as for dried blood spots.
Measurement of carnitine transport and enzyme activity
Primary carnitine deficiency, due to a defect of carnitine transport, can be investigated by measuring the uptake of labelled carnitine into fibroblasts. Carnitine palmitoyltransferase activity is measured radiochemically. A number of different spectrophotometric and fluorometric techniques have been used to measure acyl-CoA dehydrogenase activities and the methods of choice are a dye reduction assay and the fluorometric ETF-reduction assay. Electron transfer flavoprotein activity can be measured by a dye reduction assay or by a catalytic assay using a physiological electron donor, reduced medium chain acyl-CoA dehydrogenase and the physiological electron acceptor ETF dehydrogenase. Electron transfer flavoprotein dehydrogenase activity can be measured by two different methods. One measures the NADH-ETF reductase activity anaerobically following the reduction of the ETFox flavin to the semiquinone. The other measures the co-proportionation of oxidized- and two-electron reduced to one-electron reduced ETF semiquinone by following the disappearance of the ETFox flavin fluorescence. Enoyl-CoA hydratase activity is measured by following the hydration of the double bond of 2-enoyl-CoA esters. 3-Hydroxyacyl-CoA dehydrogenase activity is measured in the reverse direction following the oxidation of NADH or in the forward direction by linking the reaction with 3-oxoacyl-CoA thiolase. 3-Oxoacyl-CoA thiolase activity is measured by following the decrease in absorbance resulting from the disappearance of a Mg2 +-enolate complex using a long chain 3-oxoacyl-CoA ester and acetoacetyl-CoA as substrates.
CONCLUSION
The investigation of muscle disease requires a combination of clinical investigation, morphological, biochemical and molecular genetic studies. Clinicians must recognize the characteristic patterns of weakness and important associated clinical features and then decide which investigations are appropriate. Biochemical studies will play an important part in the investigation, particularly in those patients with suspected metabolic myopathies. Many of the routine biochemical studies will be available at all hospitals, but the specialized tests to investigate metabolic myopathies are best performed in the few centres with a research interest in muscle disease.
ACKNOWLEDGEMENTS
I am grateful to my colleagues Mike Cullen, Margaret Johnson, Mori Pourfarzam and Bård Krossnes for giving me their illustrations and their experience and to Petter Sanaker for reading through the manuscript.
Further reading
APPENDIX 33.1 THE FOREARM EXERCISE TEST
This consists of measuring lactate production following 1 min of exercise, which can be performed under ischaemic conditions or without. The use of ischaemia in patients with glycogen breakdown defects such as McArdle disease can be associated with rhabdomyolysis and should be avoided. The test is otherwise the same. If used, ischaemia is induced in the forearm by the use of a sphygmomanometer cuff placed around the upper arm and inflated to just above systolic pressure (note: a reliable cuff is essential since gradual loss of pressure will allow the entry of fresh blood).
The subject is asked to squeeze a (spare) sphygmomanometer bulb once every second for 1 min and then rest with the cuff still inflated for 1 min to allow lactate to leak out of the forearm muscles. The cuff is then released. Blood samples are taken prior to inflating the cuff and immediately following release, and at intervals (e.g. 2, 5, 7 and 12 min) after exercise. The pre-exercise blood sample must be withdrawn without stasis so a small, indwelling cannula (e.g. 21-G butterfly) is placed in an antecubital vein. Should stasis be required to place this, 10 min should be left before taking the pre-exercise sample. Samples taken into anticoagulants (e.g. fluoride oxalate) must be analysed within 30 min. Alternatively, samples can be mixed with ice-cold perchloric acid.
The normal response is for lactate to rise in the first sample after ischaemic exercise to between three and five times pre-exercise concentrations and then gradually decline. Healthy subjects perform this test easily, but patients with disorders of glycolysis often have to stop the exercise owing to pain and/or cramp. In disorders of glycogen mobilization or glycolysis, little or no rise in venous lactate will be seen. Since myoadenylate deaminase deficiency gives a similar clinical picture to myophosphorylase deficiency, ammonia concentration should be measured simultaneously with lactate. In myoadenylate deficiency, lactate rises normally, but there is no (normal) rise in ammonia. This provides, moreover, an internal control for the investigation in most instances, since failure of lactate to rise can also be due to poor effort during ischaemic exercise.