[level-membership-for-emergency-medicine-category]
Section 29 Toxicology
29.1 Approach to the poisoned patient
Introduction
Deliberate self-poisoning is one of the commonest reasons for general hospital admission in the UK1 and accounts for 1–5% of all public hospital admissions in Australia.2,3 The bulk of the medical management of cases presenting to hospital is carried out in the emergency department (ED), and the emergency physician is expected to be expert in the field. Although the management must vary considerably according to the nature and severity of the poisoning, some general principles apply.
Above all, it must be remembered that the acute overdose presentation is only a discrete time-limited event in the course of the underlying condition, which is usually psychiatric or social in origin.
Pathophysiology and clinical features
The most frequent life-threatening respiratory complication of poisoning is ventilatory failure, which is usually a consequence of CNS depression. Less commonly, it is secondary to ventilatory muscle paralysis. The frequency and depth of respirations are reduced. Respiratory failure may also be caused by direct pulmonary toxicity, or complications such as pulmonary aspiration or non-cardiogenic pulmonary oedema (Table 29.1.1).
Cardiovascular manifestations of poisoning include tachycardia, bradycardia, hypertension, hypotension, conduction defects and arrhythmias (Table 29.1.2). Bradycardia is relatively rarely observed and is associated with a number of potentially life-threatening ingestions. Tachycardia is commonly observed and is usually benign. It may be due to intrinsic sympathomimetic or anticholinergic effects of a drug, or a reflex response to hypotension or hypoxia. Hypotension is also commonly observed and may be due to a number of different causes (Table 29.1.2). Hypertension is unusual. Severe hypertension is usually associated with illicit drug use and is important because it may produce complications such as intracerebral haemorrhage.
Table 29.1.2 Cardiovascular effects of poisoning
CNS manifestations of poisoning include decreased level of consciousness, agitation or delirium, seizures and disordered temperature regulation. A decreased level of consciousness is a common presentation of poisoning and is associated with many drugs, some of which are listed in Table 29.1.1. Although usually a direct drug effect, CNS depression is occasionally secondary to hypoglycaemia, hypoxia or hypotension. Common causes of agitation or delirium following overdose are listed in Table 29.1.3. Toxic seizures are potentially life-threatening, and important causes are listed in Table 29.1.4.
Table 29.1.3 Toxic causes of agitation or delirium
| Alcohol |
| Anticholinergic syndrome |
| Antidepressants |
| Atypical antipsychotic agents |
| Benzodiazepines and other sedative-hypnotics |
| Cannabis |
| Hallucinogenic agents |
| Serotonin syndrome |
| Sympathomimetic syndrome |
| Theophylline |
| Withdrawal syndromes |
Table 29.1.4 Toxic causes of seizures
| Amphetamines |
| Bupropion |
| Carbamazepine |
| Chloroquine |
| Cocaine |
| Isoniazid |
| Mefanamic acid |
| Theophylline |
| Tramadol |
| Tricyclic antidepressants |
| Venlafaxine |
Hypothermia is usually a complication of environmental exposure secondary to a decreased level of consciousness or altered behaviour. Hyperthermia is a direct toxic effect and causes are listed in Table 29.1.5. Severe hyperthermia is rapidly lethal if not corrected.
Table 29.1.5 Toxic causes of hyperthermia
| Amphetamines |
| Anticholinergics |
| Cocaine |
| MAO inhibitors |
| Salicylates |
| Serotonin syndrome |
Pulmonary aspiration frequently complicates a period of decreased level of consciousness or a seizure. It is a leading cause of in-hospital morbidity and mortality following overdose. This complication is characterized by rapid onset of dyspnoea, cough, fever, wheeze and cyanosis.
Assessment
Risk assessment
A risk assessment should be made as soon as possible in the management of the poisoned patient. Only resuscitation is a greater priority (see Table 29.1.6). Risk assessment is a distinct quantitative cognitive step through which the clinician attempts to predict the likely clinical course and potential complications for the individual patient at that particular presentation.4 An accurate risk assessment allows informed decision-making in regard to all subsequent management steps including duration and intensity of supportive care and monitoring, screening and specialized testing, decontamination, enhanced elimination, antidotes and disposition. Factors that are taken into account when formulating this risk assessment include: the agent(s), the dose, the time since ingestion, the clinical features present and patient factors (Table 29.1.6). Specialized testing may refine risk assessment. Access to specialized poisons information in the form of a poisons information centre or in-house databases is often necessary to formulate an accurate risk assessment.
Reproduced from Toxicology Handbook. Murray L, Daly F, Little M, Cadogan M. Elsevier, Sydney 2007.
Physical examination
The focused physical examination of the poisoned patient aims to:
Several toxic autonomic syndromes, or ‘toxidromes’, have been described in relation to poisoning. The principal ones are listed in Table 29.1.7. Identification of these syndromes may narrow the differential diagnosis in cases of unknown poisoning.5
| Toxidrome | Features | Common causes |
|---|---|---|
| Anticholinergic | Agitated delirium | Antihistamines |
| Tachycardia | Benztropine | |
| Hyperthermia | Carbamazepine | |
| Dilated pupils | Phenothiazines | |
| Dry flushed skin | Plant poisonings | |
| Urinary retention | Tricyclic antidepressants | |
| Ileus | ||
| Mixed cholinergic | Brady- or tachycardia | Organophosphates |
| Hypo- or hypertension | Carbamates | |
| Miosis or mydriasis | ||
| Sweating | ||
| Increased bronchial secretion | ||
| Gastrointestinal hyperactivity | ||
| Muscle weakness | ||
| Fasciculations | ||
| Mixed α- and β-adrenergic | Hypertension | Amphetamines |
| Tachycardia | Cocaine | |
| Mydriasis | ||
| Agitation | ||
| β-Adrenergic | Hypotension | Caffeine |
| Tachycardia | Salbutamol | |
| Hypokalaemia | Theophylline | |
| Hyperglycaemia | ||
| Serotonin | Altered mental status | Amphetamines |
| Autonomic dysfunction | Antihistamines | |
| Fever | Monoamine oxidase inhibitors | |
| Hypertension | NSSRIs | |
| Sweating | SSRIs | |
| Tachycardia | Tricyclic antidepressants (Usually combined overdose) | |
| Motor dysfunction | ||
| Hyperreflexia | ||
| Hypertonia (esp. lower limbs) | ||
| Myoclonus |
NSSRI, non-selective serotonin re-uptake inhibitor; SSRI, selective serotonin re-uptake inhibitor.
Poisons information
Information on the clinical course and toxic doses of specific pharmaceutical and non-pharmaceutical poisons is available on a 24 h basis throughout Australia by telephoning 131126. The poison information centres are staffed by pharmacists and are also able to refer cases to clinical toxicologists for consultation.
Treatment
The management of poisoning should be approached in a systematic way. Following initial resuscitation, further treatment is informed by the risk assessment (Table 29.1.6).
Resuscitation, supportive care and monitoring
Supportive care is the key element in the management of poisoning. The vast majority of poisonings result in temporary dysfunction of one or more of the body systems. If appropriate support of the system in question is instituted in a timely fashion and continued until the toxic substance is metabolized or excreted, a good outcome can be anticipated. In severe poisonings supportive care may be very aggressive, and possible interventions are listed in Table 29.1.8.
Table 29.1.8 Supportive care measures for the poisoned patient
| Airway | Endotracheal intubation |
| Breathing | Supplemental oxygen |
| Ventilation | |
| Circulation | Intravenous fluids |
| Inotropes | |
| Antihypertensives | |
| Antiarrhythmics | |
| Defibrillation/cardioversion | |
| Cardiac pacing | |
| Cardiopulmonary bypass | |
| Metabolic | Hypertonic dextrose |
| Hypertonic saline | |
| Insulin/dextrose | |
| Calcium salts | |
| Sodium bicarbonate | |
| Agitation/delirium | Benzodiazepines |
| Butyrophenones | |
| Seizures | Benzodiazepines |
| Barbiturates | |
| Body temperature | External rewarming |
| External cooling | |
| Impaired renal function | Rehydration |
| Haemodialysis |
Toxic hypertension rarely requires specific therapy. Most cases are mild and simple observation is sufficient. Agitation or delirium is a feature of many intoxications associated with hypertension, and adequate sedation with benzodiazepines usually lowers the blood pressure. Severe toxic hypertension is most likely in toxicity from cocaine or amphetamine-type drugs, and treatment may be indicated to avoid complications such as cardiac failure or intracerebral haemorrhage. The drug of choice in this situation is sodium nitroprusside by intravenous infusion. The extremely short duration of action of this vasodilator allows accurate control of hypertension during the toxic phase, and avoids the development of hypotension once toxicity begins to wear off.
Decontamination
Gastric emptying can be attempted by the administration of an emetic, most commonly syrup of ipecac, or by gastric lavage. In volunteer studies both of these techniques removed highly variable amounts of marker substances from the stomach even if performed immediately after ingestion, and the effect diminished rapidly with time to the point of being negligible after one hour.6,7 Clinical outcome trials have failed to demonstrate improved outcome as a result of routine gastric emptying in addition to administration of activated charcoal, except, perhaps, in patients presenting unconscious within 1 h of ingestion.8–10
The principal adsorbent available to clinicians is activated charcoal (AC), which effectively binds most pharmaceuticals and chemicals, and is currently the decontamination method of choice for most poisonings. Materials that do not bind well to charcoal are listed in Table 29.1.9.
Table 29.1.9 Materials that do not bind well to activated charcoal
| Alcohols |
Volunteer studies demonstrate that the effect of AC diminishes rapidly with time and that the greatest benefit occurs if it is administered within 1 h. There is as yet no evidence that AC improves clinical outcome.11
There is no evidence to suggest that the addition of a cathartic such as sorbitol to AC improves clinical outcome.12,13
Apart from rarely employed endoscopic and surgical techniques, whole-bowel irrigation (WBI) is the most aggressive form of gastrointestinal decontamination. Polyethylene glycol solution (Golytely™) is administered via a nasogastric tube at a rate of 2 L/h until a clear rectal effluent is produced. This usually takes about 6 h and requires one-to-one nursing. In volunteer studies, this technique reduced the absorption of slow-release pharmaceuticals and so may be of benefit in life-threatening overdoses of these agents.13,14 Again, clinical benefit has not yet been conclusively demonstrated.15 The use of WBI has also been reported in the management of potentially toxic ingestions of iron, lead and packets of illicit drugs. Whole-bowel irrigation is contraindicated if there is evidence of ileus or bowel obstruction, and in patients who have an unprotected airway or haemodynamic compromise.16
Enhanced elimination
A number of techniques are available to enhance the elimination of toxins from the body. Their use is rarely indicated, as only a very few drugs capable of causing severe poisoning have pharmacokinetic parameters that render them amenable to these techniques (Table 29.1.10).
| Technique | Suitable toxin |
|---|---|
| Repeat-dose activated charcoal | Carbamazepine |
| Dapsone | |
| Phenobarbitone | |
| Phenytoin | |
| Salicylate | |
| Theophylline | |
| Urinary alkalinization | Phenobarbitone |
| Salicylate | |
| Haemodialysis | Ethylene glycol |
| Lithium | |
| Methanol | |
| Salicylate | |
| Theophylline | |
| Haemoperfusion | Theophylline |
Repeat-dose AC (25–50 g every 3–4 h) may enhance drug elimination by interrupting the enterohepatic circulation or by ‘gastrointestinal dialysis’. Gastrointestinal dialysis is the movement of a toxin across the gastrointestinal wall from the circulation into the gut down a concentration gradient that is maintained by charcoal binding. For this technique to be effective, a drug must undergo considerable enterohepatic circulation or, in the case of ‘gastrointestinal dialysis’, have a small volume of distribution, small molecular weight, low protein binding, slow endogenous elimination and bind to charcoal.16,17 The advantages of this technique are that it is non-invasive and simple to carry out.
Alkalinization of the urine enhances urinary excretion of drugs that are filtered at the glomerulus and are unable to be reabsorbed across the tubular epithelium when in an ionized form at alkaline pH. For elimination to be effectively enhanced by this method, the drug must be predominantly eliminated by the kidneys in the unchanged form, have a low pKa, be distributed mainly to the extracellular fluid compartment and be minimally protein bound.18
Antidotes
Very few drugs have effective antidotes. Occasionally, however, timely use of an antidote may be life saving or substantially reduce morbidity, time in hospital or resource requirements. Antidotes that may be indicated in the ED setting are listed in Table 29.1.11. However, it must be remembered that antidotes are also drugs, and are frequently associated with adverse effects of their own. An antidote should only be used where a specific indication exists, and then only at the correct dose, by the correct route, and with appropriate monitoring. Because many antidotes are so infrequently used, obtaining sufficient supplies when the need arises can be difficult. Every ED must review its stocking of antidotes and have a plan for obtaining further supplies should the need arise.
| Poisoning | Antidote |
|---|---|
| Atropine | Physostigmine |
| Benzodiazepines | Flumazenil |
| Cyanide | Dicobalt edetate, hydroxocobalamin |
| Digoxin | Digoxin-specific Fab fragments |
| Insulin | Dextrose |
| Iron | Desferoxamine |
| Isoniazid | Pyridoxine |
| Methaemoglobinaemia | Methylene blue |
| Methanol and ethylene glycol | Ethanol, fomepizole |
| Organophosphates and carbamates | Atropine, oximes |
| Opioids | Naloxone |
| Paracetamol | N-acetyl cysteine |
| Sulphonylureas | Dextrose, octreotide |
| Tricyclic antidepressants | Sodium bicarbonate |
| Warfarin, brodifacoum | Vitamin K |
Clinical investigation
Measurement of serum drug concentrations is only useful if this provides important diagnostic or prognostic information, or assists in planning management. Some drug levels that may be useful are listed in Table 29.1.12. For most cases, drug overdose management is guided by clinical findings and not by drug levels. Some drugs commonly taken in overdose for which serum concentrations are of no value in planning management are listed in Table 29.1.13.
Table 29.1.12 Drug levels that may be helpful in the management of selected cases of overdose
| Carbamazepine |
| Digoxin |
| Dilantin |
| Lithium |
| Iron |
| Paracetamol |
| Phenobarbitone |
| Salicylate |
| Theophylline |
| Valproate |
Table 29.1.13 Drug levels that are not helpful in the management of overdose
| CNS drugs | Cardiovascular drugs |
|---|---|
| Antidepressants | ACE inhibitors |
| Benzodiazepines | Beta-blockers |
| Benztropine | Calcium channel blockers |
| Cocaine | Clonidine |
| Newer antipsychotics | |
| Opiates | |
| Phenothiazines |
Disposition
Both the medical and the psychiatric disposition of the overdose patient must be considered. A good risk assessment is essential to determining timely and safe disposition.
1 Hawton K, Fagg J. Trends in deliberate self-poisoning and self-injury in Oxford. British Medical Journal. 1992;304:1409-1411.
2 McGrath J. A survey of deliberate self-poisoning. Medical Journal of Australia. 1989;150:317-322.
3 Pond SM. Prescription for poisoning. Medical Journal of Australia. 1995;162:174-175.
4 Murray L, Daly F, Little M, Cadogan M, editors. Toxicology handbook. Sydney: Elsevier, 2007.
5 Kulig K. Initial management of ingestion of toxic substances. New England Journal of Medicine. 1992;326:1677.
6 Krenzelok EP, McGuigan M, Lheureux P. Position statement: ipecac syrup. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):699-709.
7 Vale JA. Position statement: gastric lavage. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):711-719.
8 Kulig K, Bar-Or D, Kantrill SV, et al. Management of acutely poisoned patients without gastric emptying. Annals of Emergency Medicine. 1990;14:562-567.
9 Merigian KS, Woodard M, Hedges JR, et al. Prospective evaluation of gastric emptying in the self-poisoned patient. American Journal of Emergency Medicine. 1990;8:479-483.
10 Pond SM, Lewis-Driver DJ, Williams G, et al. Gastric emptying in acute overdose: a prospective randomised controlled trial. Medical Journal of Australia. 1995;163:345-349.
11 Chyka PA, Seger D. Position statement: single-dose activated charcoal. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):721-741.
12 Barceloux D, McGuigan M, Hartigan-Go K. Position statement: cathartics. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):743-752.
13 Kirshenbaum LA, Mathew SC, Sitar DS, et al. Whole-bowel irrigation versus activated charcoal in sorbitol for the ingestion of modified-release pharmaceuticals. Clinical Pharmacology Therapy. 1989;46:264-271.
14 Smith SW, Ling LJ, Halstenson CE. Whole-bowel irrigation as a treatment for acute lithium overdose. Annals of Emergency Medicine. 1991;20:536-539.
15 Tenenbein M. Position statement: whole bowel irrigation. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):753-756.
16 Pond SM. Role of repeated oral doses of activated charcoal in clinical toxicology. Medical Toxicology. 1986;1:3-11.
17 Chyka PA. Multiple-dose activated charcoal and enhancement of systemic drug clearance: summary of studies in animals and human volunteers. Clinical Toxicology. 1995;33:399-405.
18 Winchester JF. Active methods for detoxification. In Haddad LM, Shannon MW, Winchester JF, editors: Clinical management of poisoning and drug overdose, 3rd edn, Philadelphia: WB Saunders, 1998.
19 Whyte IM, Dawson AH, Buckley NA, et al. A model for the management of self-poisoning. Medical Journal of Australia. 1997;167:142-146.
20 Lee V, Kerr JF, Braitberg G, et al. Impact of a toxicology service on a metropolitan teaching hospital. Emergency Medicine. 2001;13:37-42.
29.2 Cardiovascular drugs
Pharmacokinetics
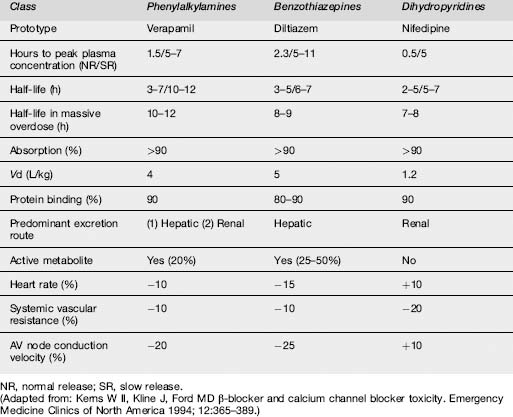
Standard CCB preparations are rapidly absorbed from the gastrointestinal tract, with onset of action occurring within 30 min.1 Pharmacokinetic parameters are shown in Table 29.2.1. Verapamil and diltiazem undergo significant first-pass hepatic clearance. Verapamil is metabolized to norverapamil, which possesses 15–20% of verapamil’s pharmacological activity and is renally excreted. Diltiazem is metabolized to deacetyldiltiazem, which has half the potency of the parent compound and undergoes biliary excretion. The elimination half-lives of all CCBs may be prolonged following massive overdose. Amlodipine has a longer plasma half-life (30–50 h) than other CCBs.
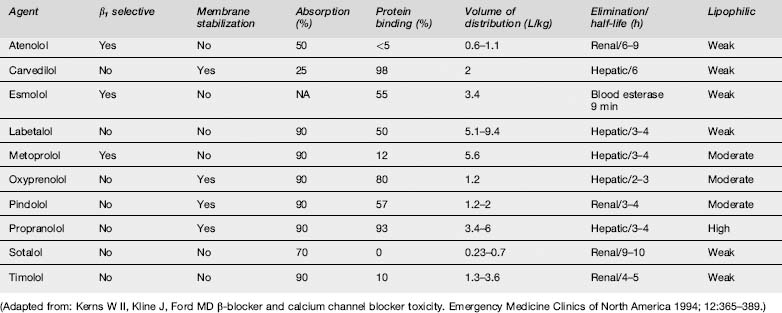
Absorption of β-blockers is rapid, with peak clinical effects occurring within 1–4 h. Pharmacokinetic parameters of the principal β-blockers are detailed in Table 29.2.2. Agents with high lipid solubility, such as propranolol, penetrate the blood–brain barrier better than the water-soluble agents, and hence cause greater central nervous system (CNS) toxicity.
Pathophysiology
CCBs antagonize the entry of extracellular calcium into cardiac and smooth muscle, but not skeletal muscle. Upon entry into cells, calcium participates in mechanical, electrical and biochemical reactions. It is involved in excitation–contraction of cardiac and smooth muscles, as well as phase 0 depolarization in the sinus and atrioventricular (AV) nodes by calcium influx through channels.2 CCBs affect myocardial contractility and slow conduction through the sinus and AV nodes. Contraction of smooth muscle is mediated by calcium influx, which is inhibited by CCBs. This results in vasodilatation and secondary reflex tachycardia from an increase in sympathetic activity.
The different classes of CCB have somewhat different pharmacological and toxic effects, as a consequence of their different binding characteristics to the dihydropyridine (DHP) receptors. Verapamil, a phenylalkylamine, produces more profound cardiac conduction defects and equal reductions in systemic vascular resistance when compared with other CCBs on a mg/kg basis.1 Verapamil is more likely to produce symptomatic decreases in blood pressure, heart rate and cardiac output than diltiazem, a benzothiazepine. The DHPs, which include amlodipine, felodipine, lercanidipine and nifedipine preferentially bind to vascular smooth muscle and predominantly decrease systemic and coronary vascular resistance. With the exception of felodipine, they also produce a reflex tachycardia by the unloading of baroreceptors.
The different β-blockers have slightly differing pharmacological properties, including selectivity for β adrenoreceptors, intrinsic sympathomimetic activity and membrane-stabilizing activity. The relative affinity for β adrenoreceptors may influence expression of toxicity. Atenolol, esmolol and metoprolol are β1-selective agents, and therapeutic use of these drugs is less likely to produce the peripheral vasoconstriction, bronchospasm and disturbances in glucose homoeostasis that result from β2 inhibition. However, pharmacological specificity decreases with increasing dose.3 Several β-blockers have partial agonist activity such that, although they block the β receptor to catecholamines, they also weakly stimulate the receptor. This partial agonist activity may have a protective effect in overdose.
Clinical features
Calcium channel blockers
The severity of toxicity is determined by a number of factors, including the amount and characteristics of the drug ingested, the underlying health of the patient, co-ingestants and delay until treatment. The majority of serious cases result from the ingestion of verapamil or diltiazem, the most toxic of the CCBs. Elderly patients and those with congestive cardiac failure are more prone to develop CCB poisoning. The principal clinical features are shown in Table 29.2.3. Ingestion of toxic amounts of standard preparations typically produces symptoms within 2 h, although maximal toxicity may not occur for up to 6–8 h. The slow-release preparations can produce significant toxicity, with onset of symptoms more than 6 h post ingestion. The major threats to life are myocardial depression and hypotension. Nifedipine produces tachycardia with normal blood pressure during the first 30 min, followed later by hypotension and bradycardia in large ingestions (>10 mg/kg). With verapamil and diltiazem poisoning, nausea, vomiting, hyperglycaemia and metabolic acidosis can develop. All CCBs can cause symptoms of cerebral hypoperfusion, such as syncope, lethargy, lightheadedness, dizziness, altered mental status, seizures and coma.
| Central nervous system |
β-blockers
In one large series of patients with β-blocker overdose, 30–40% of patients remained asymptomatic and only 20% developed severe toxicity.4 Toxicity is more likely to develop after ingestion of propranolol, in patients with pre-existing cardiac disease or where there is co-ingestion of other drugs with effects on the cardiovascular system, especially CCBs and cyclic antidepressants.4,5 If β-blocker toxicity is to develop, it is usually observed within 6 h of ingestion.5,6
Treatment
Oral-activated charcoal should be administered as soon as practicable to all patients presenting within 2–4 h of ingestion, and to all those presenting after ingestion of slow-release preparations. More aggressive decontamination, with whole-bowel irrigation, is indicated following overdose with slow-release CCBs.7
A number of drugs play a role in the management of significant CCB or β-blocker poisoning, although none is a completely effective antidote. Suggested doses are shown in Table 29.2.4.
Table 29.2.4 Useful drugs in the management of CCB and β-blocker toxicity
| CCBs | β-Blockers | |
|---|---|---|
| Calcium | 0.5–1 g (5–10 mLs) calcium chloride or 1–2 g (10–20 mLs) calcium gluconate i.v. over 5–10 minutes. Repeat every 10–15 minutes as required. Further administration guided by serum calcium concentrations. | |
| Catecholamines | Adrenaline (epinephrine) infusion started at 1 μg/kg/min and titrate to maintain organ perfusion. | Isoprenaline or adrenaline (epinephrine) infusion titrated to maintain organ perfusion. |
| Glucagon | A bolus dose of 5–10 mg followed by an infusion of 1–5 mg/h. | A bolus dose of 5–10 mg followed by an infusion of 1–5 mg/h. |
| Hyperinsulinaemia euglycaemia | Actrapid 1 U/kg i.v. bolus followed by 0.5–1 U/kg/hr infusion. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia. | Actrapid 1 U/kg i.v. bolus followed by 0.5–1 U/kg/hr infusion. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia. |
Calcium, an inotropic agent, is the initial drug of choice for CCB toxicity. Administration must be closely monitored, with ionized calcium measured 30 min after commencing the infusion, and then second-hourly.8 Catecholamines are useful in attempting to restore adequate tissue perfusion.
Glucagon, a polypeptide hormone of pancreatic origin, enhances myocardial performance by increasing intracellular cAMP concentrations. This increase in cAMP triggers the release of cAMP-dependent protein kinase, which activates the calcium channels, causing an increase in heart rate and myocardial contractility. It works independently from that of the β-adrenoreceptor stimulation of the heart. Use of glucagon is supported only by case reports and some animal studies. There are no clinical trials supporting its efficacy in either calcium channel or beta-blocker poisoning and its role in management of these poisoning is questioned.9 It is frequently difficult to source adequate stocks of glucagon for use as an inotropic agent.
Hyperinsulinaemic euglycaemia therapy (HIET) is increasingly advocated as therapy for hypotension unresponsive to fluids, calcium salts and inotropes. This therapy is supported by animal work10,11 and promising initial human case reports12 but again clinical trials are lacking. Insulin administration switches cardiac cell metabolism from fatty acids to carbohydrates. It restores calcium fluxes and improves myocardial contractility. The recommended initial dose of actrapid is 1 U/kg i.v. followed by an infusion of 0.5–1 U/kg/h. This should be accompanied by an initial bolus dose of 50 mL 50% dextrose followed by an infusion to maintain euglycaemia.13
Severe propranolol toxicity is usually due to sodium channel blockade and treatment as for tricyclic antidepressant poisoning, including intubation, ventilation and sodium bicarbonate, is appropriate.
There are no clinically effective methods of enhancing the elimination of CCBs or β-blockers.
Clinical features
Two distinct clinical presentations of digoxin toxicity are observed: acute and chronic. Both are characterized by cardiac arrhythmias, and virtually all types of arrhythmia have been reported in the context of digoxin toxicity.14
Clinical investigation
In acute poisoning the serum potassium rises as Na-K ATPase function is progressively impaired. Hyperkalaemia denotes significant acute digoxin toxicity. Prior to the availability of a specific antidote for digoxin poisoning, a serum potassium concentration >5.5 mEq/L was associated with a high probability of lethal outcome.16 Hyperkalaemia is not usually observed in chronic digoxin toxicity. In fact, these patients are frequently hypokalaemic and hypomagnesaemic secondary to chronic diuretic use. Both these electrolyte disorders are important as they exacerbate digoxin toxicity.
Treatment
The best outcome is associated with early recognition of digoxin toxicity.
For chronic toxicity with minimal symptoms, management may involve no more than observation, cessation of digoxin administration, correction of hypokalaemia and hypomagnesaemia and appropriate management of any factors that contributed to the development of toxicity. However, presence of any cardiovascular system effects, particularly in elderly patients, is an indication for the administration of Fab fragments of digoxin-specific antibodies. The potential lethality of chronic digoxin poisoning is often underestimated with the result that digoxin antibody fragments are inappropriately withheld.16 From a purely economic view, the reduction in length of stay as a result of treatment with digoxin-specific antibodies outweighs the expense of the therapy.17
The specific antidote to digoxin poisoning is Fab fragments of digoxin-specific antibodies, which should be administered as soon as possible in any potentially life-threatening digoxin intoxication. Commonly accepted indications for the administration of Fab fragments are listed in Table 29.2.5.
Table 29.2.5 Indications for administration of Fab fragments of digoxin-specific antibodies following acute overdose
| Hyperkalaemia (K > 5.0 mmol/L) associated with digoxin toxicity |
| History of ingestion of more than 10 mg of digoxin |
| Haemodynamically unstable cardiac arrhythmia |
| Cardiac arrest from digoxin toxicity |
| Serum digoxin concentration greater than 15 nmol/L |
Fab fragments of digoxin-specific antibodies
The extraordinary clinical efficacy of digoxin-specific fragments has been well documented in a multi-centre study.17 The same study also demonstrated the safety of the product, with the only adverse reactions reported being hypokalaemia (4% incidence) and worsening of congestive cardiac failure (3%).
Pharmacokinetics
Clonidine is well absorbed with a bioavailability of almost 100%. The peak concentration in plasma and effect is observed within 1–3 h. The elimination half-life is 6–24 h with a mean half-life of 12 h. Half of the administered dose is excreted unchanged by the kidney.19
Pathophysiology
Clonidine activates central α2 receptors. This results in a reduction in CNS sympathetic outflow at the vasomotor centre in the medulla oblongata. Clonidine is thought to reduce blood pressure through a reduction in cardiac output as well as its weak peripheral alpha adrenergic antagonist properties. Clonidine also stimulates parasympathetic outflow and this may contribute to the slowing of heart rate as a consequence of increased vagal tone. Paradoxically, clonidine overdose can result in an initial hypertension from its partial α1 adrenergic agonist effect. It is suggested that clonidine’s inhibition of sympathetic outflow is mediated through endogenous opiate release.
Clinical features
Clonidine can cause transient hypertension from initial vasoconstriction with parenteral administration followed by hypotension. In addition to bradycardia and conduction defects, it can cause a central chlorpromazine-like effect with sedation. Other CNS symptoms include coma, seizure, miosis, reduced respiration and hypothermia.20 The median onset of symptoms following clonidine ingestion is 30 min and patients are usually symptomatic on arrival at the ED.20 Symptoms usually resolve by 24 h.21
Investigations
The ECG is essential in evaluating and monitoring for bradycardia and conduction defects.
Disposition
1 Robertson RM, Robertson D. Drugs used for the treatment of myocardial ischaemia. In: Gilman AG, Hardman JG, Limbird LE, et al, editors. Goodman and Gilman’s: The pharmacological basis of therapeutics. 9th edn. New York: Pergamon Press; 1996:770.
2 Antman EM, Stone PH, Muller JE, et al. Calcium channel blocking agents in the treatment of cardiovascular diseases: Part E Basic and clinical electrophysiological effects. Annals of Internal Medicine. 1980;93:875-885.
3 Lewis RV, McDevitt DG. Adverse reactions and interactions with beta-adrenoreceptor blocking drugs. Medical Toxicology. 1986;1:343-361.
4 Taboulet P, Cariou A, Berdeaux A, et al. Pathophysiology and management of self-poisoning with beta-blockers. Journal of Toxicology and Clinical Toxicology. 1993;31:531-551.
5 Reith DM, Dawson AH, Epid D, et al. Relative toxicity of beta blockers in overdose. Journal of Toxicology and Clinical Toxicology. 1996;34:273-278.
6 Love J, Howell JM, Litovitz TL, et al. Acute beta blocker overdose: factors associated with the development of cardiovascular morbidity. Journal of Toxicology and Clinical Toxicology. 2000;38:275-281.
7 Buckley N, Dawson AH, Howarth D, et al. Slow release verapamil poisoning. Use of polyethylene glycol whole bowel lavage and high dose calcium. Medical Journal of Australia. 1993;158:202.
8 Pertoldi F, D’Orlando L, Mercante WP. Electromechanical dissociation 48 hours after atenolol overdose: usefulness of calcium chloride. Annals of Emergency Medicine. 1998;31:777-781.
9 Bailey B. Glucagon in beta-blocker and calcium channel blocker overdoses: a systematic review. Journal of Toxicology Clinical Toxicology. 2003;41:595-602.
10 Kline JA, Leonova E, Raymond RM. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Critical Care Medicine. 1995;23:1251-1263.
11 Holger JS, Engerbretsen KM, Fritzlar SJ, et al. Insulin versus vasopressin and epinephrine to treat b-blocker toxicity. Clinical Toxicology. 2007;45:396-401.
12 Yuan I, Kerns WP, Tomaszewski CA, et al. Insulin glucose as adjunctive therapy for severe calcium channel antagonist poisoning. Journal of Toxicology Clinical Toxicology. 1999;37:463-474.
13 Megarbane B, Karyo S, Baud FJ. The role of insulin and glucose (hyperinsulinaemia/euglycaemia) therapy in acute calcium and beta-blocker poisoning. Toxicology Reviews. 2004;23(4):214-222.
14 Moorman JR, Pritchett ELC. The arrhythmias of digitalis intoxication. Archives of Internal Medicine. 1985;145:1289-1292.
15 Bismuth C, Gaultier M, Conso F, et al. Hyperkalemia in acute digitalis poisoning: prognostic significance and therapeutic implications. Clinical Toxicology. 1973;6:153-162.
16 Marik PE, Fromm L. A case series of hospitalised patients with elevated digoxin levels. American Journal of Medicine. 1998;105(2):110-115.
17 DiDomenico RJ, Walton SM, Sanoski CA, et al. Analysis of the use of digoxin immune Fab for the treatment of non-life-threatening digoxin toxicity. Journal of Cardiovascular Pharmacology & Therapeutics. 2000;5(2):77-85.
18 Antman EM, Wenger FL, Butler VP, et al. Treatment of 150 cases of life threatening digitalis intoxication with digoxin specific Fab antibody fragments: final report of multicenter study. Circulation. 1990;81:1744-1752.
19 Seger D. Clonidine Toxicity Revisited. Clinical Toxicology. 2002;40(2):145-155.
20 Nichols MH, King WD, James LP. Clonidine poisoning in Jefferson County, Alabama. Annals of Emergency Medicine. 1997;29:511-517.
21 Erickson SJ, Duncan A. Clonidine poisoning – an emerging problem: Epidemiology, clinical features, management and preventative strategies. Journal of Paediatrics and Child Health. 1998;34(3):280-282.
22 McVey FK, Corke CF. Extracorporeal circulation in the management of massive propranolol overdose. Anaesthesia. 1991;46:744-746.
29.3 Central nervous system drugs
Benzodiazepines
Pharmacology
Gamma-aminobutyric acid (GABA) is an inhibitory neurotransmitter found predominantly in the basal ganglia, the hippocampus, hypothalamus, cerebellum and the dorsal horn of the spinal cord.1 GABA interacts with two receptors, GABA-A and GABA-B, resulting in the influx of chloride through a ligand-gated ion channel. The former receptor is the predominant site of benzodiazepine action. By binding to the GABA-A receptor complex at a specific site, benzodiazepines enhance the binding of GABA at GABA-A, which in turn opens more chloride channels, and hence produces their sedative, hypnotic, anxiolytic and anticonvulsant effects. GABA-B is mainly involved in feedback mechanisms and the control of muscle tone. GABA receptor subunits have been identified, and the sensitivity and specificity of individual benzodiazepines is determined by their interaction with these subunits. Tolerance develops to most of the effects of benzodiazepines, and may be associated with downregulation of GABA receptors.
Clinical features
Overdose
Death as a result of pure benzodiazepine overdose is very uncommon.2 When death is reported, it is usually in the setting of a mixed overdose including other CNS depressants such as alcohol, antidepressants, phenothiazines and narcotics. The most common manifestation of overdose is drowsiness, which may progress to stupor depending on patient characteristics, co-ingestants and dose. Coma is uncommon. Other features characteristic of benzodiazepine overdose are respiratory depression, hypothermia and hypotension, though these are not usually life-threatening.3 The duration of effect varies from 6 to 36 h, depending on the drug.
Respiratory insufficiency in the setting of benzodiazepine overdose may be due to an increase in upper airway resistance and work of breathing rather than central apnoea.4
The duration of benzodiazepine effect varies between 6 and 36 h depending on the agents(s) involved. The effects on the CNS are exacerbated if there is co-ingestion of other CNS depressants such as alcohol, antidepressants, phenothiazines or narcotics. A fatal outcome is more likely in this setting. Acute alcohol ingestion tends to delay benzodiazepine metabolism whereas chronic alcohol ingestion induces metabolic pathways and may increase clearance rates of these drugs.5
Treatment
The role of flumazenil, a specific benzodiazepine receptor antagonist, in the management of overdose is limited. Recovery from benzodiazepine overdose is usually uncomplicated with simple supportive care and the use of flumazenil may precipitate acute withdrawal syndrome in benzodiazepine-dependent patients and seizures in those with co-ingestants which lower the seizure threshold.6 Flumazenil does have a role as a diagnostic agent to confirm benzodiazepine overdose, in the reversal of post-operative benzodiazepine-induced sedation and those patients with a pure benzodiazepine overdose, who would otherwise require intubation.
Non-benzodiazepine sedative-hypnotics
Zopiclone
Zopiclone also acts on the GABA-A receptor complex, but at a separate site to that of the benzodiazepines and barbiturates. Furthermore, unlike the benzodiazepines where the binding to GABA-A receptors is modulated by the presence of GABA itself, the binding of zopiclone is not. It is also suggested that zopiclone binds to the same receptor site as benzodiazepines, but the resulting conformational change is different.
Clinical features
Chloral hydrate
As well as CNS and respiratory depression, overdose with chloral hydrate may produce severe gastrointestinal irritation, with haematemesis, gastric ulceration and oesophageal stricture formation. More importantly, chloral hydrate overdose characteristically produces cardiac rhythm disturbances, including simple ventricular ectopics, ventricular tachycardia, torsades de pointes and ventricular fibrillation. Fatalities continue to be reported.7
Zopiclone
Overdose with zopiclone produces CNS depression and potentially respiratory depression. First-degree heart block8 and fatalities are reported.9,10
Zolpidem
As with the other sedative-hypnotics, the primary clinical effects observed following zolpidem overdose are CNS and respiratory depression. Pure ingestions of <400 mg in adults tend to produce sedation and amnesia only.11 Fatalities have been ascribed to zolpidem overdose on the basis of postmortem toxicological analyses.12
Treatment
As with the benzodiazepines, the most important aspect of management for patients presenting following an overdose of the other sedative-hypnotic drugs is good supportive care. Careful attention must be paid to maintaining an adequate airway, ventilation and blood pressure. Hypotension usually responds to intravenous fluid administration, but may require administration of inotropes. Activated charcoal can be considered if patients present within 1–2 h of ingestion. Specific medications may be of use with certain types of overdose. Flumazenil has been shown to reverse the effect of zopiclone, and its use in pure zopiclone overdoses with respiratory depression that might otherwise require intubation should be considered.13–15 In the setting of chloral hydrate overdose, ventricular arrhythmias may be resistant to usual therapies but will often respond to the use of β-blockers, such as intravenous propranolol.16,17 Hypoxia and electrolyte disturbances should also be corrected.
Antipsychotic Drugs
Pharmacology
This large group of drugs can be classified as typical or atypical (Table 29.3.1), according to structure (Table 29.3.2) or according to neuroreceptor-binding affinity. The latter may offer the most reliable prediction of the risk of toxicity.18
| Typical | Atypical |
|---|---|
| Chlorpromazine | Mesoridazine |
| Fluphenazine | Thioridazine |
| Perphenazine | Clozapine |
| Prochlorperazine | Olanzapine |
| Trifluoperazine | Risperidone |
| Haloperidol | Remoxipride |
| Thiothixene | Loxapine |
| Molindone | Quetiapine |
Table 29.3.2 Structural classification of the antipsychotics
| Structural class | Generic name |
|---|---|
| Phenothiazines | |
| Aliphatic | Chlorpromazine |
| Triflupromazine | |
| Promethazine | |
| Piperazine | Fluphenazine |
| Perphenazine | |
| Prochlorperazine | |
| Trifluoperazine | |
| Piperidine | Mesoridazine |
| Thioridazine | |
| Butyrophenone | Haloperidol |
| Thioxanthene | Droperidol |
| Chlorprothixene | |
| Thiothixene | |
| Dihydroindolone | Molindone |
| Dibenzoxazepine | Loxapine |
| Clozapine | |
| Olanzapine | |
| Diphenylbutylpiperidine | Pimozide |
| Benzisoxazole | Risperidone |
| Benzamides | Sulpiride |
| Remoxipride |
Atypical drugs are defined as such on clinical and pharmacological grounds. Clinically, they produce fewer extrapyramidal side effects and tardive dyskinesias. For this reasons the newer atypical antipsychotic agents have largely replaced the traditional agents as first-line treatment of schizophrenia. Pharmacologically, they may be regarded as atypical for a variety of reasons including low D2-dopamine receptor potency, low D2-receptor occupancy in the mesolimbic and nigrostriatal areas and high affinities for M1-muscarinic, D1– and D4-dopamine and 5-HT1A- and 5-HT2A-serotonin receptors.19 This produces three broad functional groups of atypical antipsychotics: the D2-, D3-receptor antagonists such as amisulpiride, the D2, α1, 5-HT2A-receptor antagonists such as risperidone, and the broad-spectrum multiple receptor antagonists such as clozapine, quetiapine and olanzapine.19
Clinical features
Adverse effects
Extrapyramidal movement disorders
Up to 90% of patients receiving antipsychotic medication will experience some extrapyramidal side effects, and these often result in the cessation of treatment.20 Of the four recognized extrapyramidal syndromes, acute dystonia, parkinsonism and akathisia are reversible and tend to occur early in a course of treatment. Tardive dyskinesia is irreversible but occurs after months to years of treatment. Clozapine, olanzapine and quetiapine are not associated with extrapyramidal syndromes.19
The pathophysiology of acute dystonic reactions is not fully understood, but involves disruption of the dopaminergic-cholinergic-GABA balance in the basal ganglia. Reactions are idiosyncratic and equally frequent following a single therapeutic ingestion or an overdose. Risk factors for developing an acute dystonic reaction following antipsychotic medication are the use of antipsychotic drugs with a high D2-dopaminergic, low M1-muscarinic and low 5-HT2A-serotonergic receptor binding affinity; young and male patients; the use of depot preparations and the recent use of alcohol.21,22 Reactions may present in varied forms and may be spasmodic or sustained, but are always involuntary. The muscles of the face, trunk and neck are commonly involved, but other sites may also be affected. About half of all cases occur within 48 h of dosing.20 The overall incidence of acute dystonic reactions varies considerably: rates of 3.5% have been reported for chlorpromazine, and 16% for haloperidol.21
Akathisia is dose-related, can occur at any age, and tends to appear some days after beginning treatment. It is thought to be due to D2-dopaminergic blockade in the mesocortical pathways.23 Drug-induced parkinsonism is more common in the elderly and tends to be seen with high-potency agents that block the postsynaptic D2-dopaminergic receptors in the nigrostriatal area. Tardive dyskinesia appears after months or years of antipsychotic treatment. It is seen with all antipsychotics except clozapine, and has a prevalence of between 27% and 35% in patients on long-term therapy.24 It is thought to be the result of an increased number and sensitivity of dopaminergic receptors in the nigrostriatal area of the brain, a response to long-term blockade.
Seizures
Antipsychotic drugs lower the seizure threshold.25 They also produce EEG changes that vary depending on the agent.26 Organic brain disease, epilepsy, drug-associated seizures and polypharmacy are risk factors for the development of seizures. They are more likely with chlorpromazine, clozapine and loxapine.
Cardiovascular
Postural hypotension and ECG changes can occur with therapeutic dosing of antipsychotics. Postural hypotension is multifactorial, with α1-adrenergic blockade, central vasomotor reflex depression and direct myocardial depression all playing a part. ECG changes can be diverse, with QRS and QT prolongation, a right axis shift, ST segment depression and T-wave inversion/flattening. QT prolongation is less evident with the newer atypical antipsychotics.27 Torsades des pointes is reported following high therapeutic dosing with haloperidol, thioridazine and mesoridazine.
Neuroleptic malignant syndrome
This idiosyncratic adverse reaction to antipsychotic medication therapy is rare. It occurs early in the treatment course or after changes in dose. Neuroleptic malignant syndrome (NMS) has been reported with all typical antipsychotics but is particularly associated with higher potency drugs such as haloperidol and fluphenazine. In the atypical group, NMS has been reported with clozapine, olanzapine and risperidone.28–30 Pooled data studies suggest the incidence is somewhere between 0.07% and 0.2%, although some have described incidences of up to 12.2%.
Typically, patients are male (male : female ratio 2 : 1), with symptoms developing over 1–3 days. Risk factors associated with the development of NMS include the use of high-potency agents and depot preparations, organic brain disease, past history of NMS, dehydration and interactions with other drugs such as lithium and anticholinergics.31 The characteristic clinical features are a temperature ≥38 °C, muscle rigidity, altered consciousness and autonomic dysfunction. Other features that may be seen are an elevated creatine kinase, leukocytosis, elevated hepatic transaminases, renal failure and metabolic acidosis. There is no specific test to confirm or exclude the diagnosis, which is reliant upon clinical and historical data. Alternative diagnoses must be excluded, especially infection (including meningitis and encephalitis). Other differential diagnoses include heat stroke, thyrotoxicosis, intracranial haemorrhage, phaeochromocytoma, tetanus, serotonin syndrome, drug overdose (MAOI, sympathomimetics and lithium), substance/alcohol withdrawal and malignant hyperthermia. The mortality rate has been reduced from 30% to 5–11% mainly as a result of improved intensive supportive care.32 Death is usually secondary to respiratory or cardiovascular failure; however, renal failure secondary to myoglobinuria, arrhythmias, pulmonary embolism and disseminated intravascular coagulation are also reported.
Other
Clozapine is associated with idiosyncratic agranulocytosis. The incidence is between 0.6% and 2.0% and usually occurs within the first 18 weeks of therapy. The mortality rate of clozapine-induced agranulocytosis, once established, is up to 85%.18 Other phenothiazines have also been associated with agranulocytosis, but with a much lower incidence.
Overdose
Most patients with serious poisoning display manifestations of cardiovascular and/or CNS toxicity. Isolated antipsychotic overdose is rarely fatal. Peak toxicity is usually seen from 2 to 6 h following ingestion but may be delayed especially after thioridazine overdose.33 Delayed onset of life-threatening cardiotoxicity is also reported following amisulpride overdose.34 CNS effects vary greatly, depending on individual susceptibility, dose ingested and the presence of co-ingestants. Lethargy is common to most patients, with effects potentially progressing to confusion, ataxia, coma and seizures. Ingestion of more than 300 mg of olanzapine or 3 g of quetiapine is likely to cause CNS depression significant enough to require intubation. Seizures are more often seen following overdose with loxapine or clozapine, and are usually generalized.35 Paradoxically, agitation may also be observed, especially in the setting of mixed overdose, and following overdose with clozapine, olanzapine or thioridazine.
Life-threatening cardiotoxicity is unusual, except in the setting of piperidine phenothiazine overdoses, e.g. thioridazine, which is associated with QRS widening, QT prolongation, ventricular tachycardia and torsade des pointes.36,37 Torsades des pointes is also reported in large haloperidol overdoses, either following deliberate self-poisoning or in the setting of excessive intravenous therapy in critically ill patients.38 More common cardiovascular effects are hypotension (initially postural) and tachycardia. Uncommon effects are hypertension and bradycardia. ECG abnormalities are not unusual in significant overdoses and can range from simple ventricular ectopics to conduction abnormalities, QRS widening, ventricular tachycardia and torsades des pointes. QT prolongation has been reported in the setting of thioridazine overdose and more recently quetiapine and amisulpride ingestions.34,39–41 The prolonged corrected QT observed in a number of reported quetiapine overdoses may be the result of the underlying sinus tachycardia observed, rather than an indicator of significant cardiotoxicity.39,40 Amisulpride overdose can result in severe cardiotoxicity, characterized by intraventricular conduction abnormalities, QT prolongation and torsades des pointes.34
The diagnosis of antipsychotic drug overdose is based on a history of ingestion and the presence of symptoms and signs in keeping with the expected findings, as outlined above. Qualitative serum and urine drug screening can be used to detect the presence of many of the antipsychotic drugs, but a high false–negative rate makes these screens notoriously unreliable. Quantitative levels may also be performed, but the results do not correlate with clinical findings and do affect management. The differential diagnosis of antipsychotic drug overdose includes meningitis and other CNS infections, stroke and head injury, as well as other drug toxicities, including tricyclic antidepressants, sedatives, alcohols, anticholinergic drugs and anticonvulsants. Many other agents have also been reported as causing acute dystonic reactions, including tricyclic antidepressants, antihistamines and anticonvulsants.
Bupropion
Pharmacology
Bupropion is a monocyclic antidepressant with structural similarities to amphetamine and diethylpropion. It is a selective inhibitor of the reuptake of catecholamines with minimal effect on the reuptake of serotonin. It also possesses mild anticholinergic activity.42 Bupropion is metabolized to hydroxy-bupropion, with plasma protein binding of 84% and 77%, respectively. The elimination half-life of bupropion is between 13 and 20 h, while that of hydroxy-bupropion is approximately 20 h. Bupropion is widely distributed with an apparent volume of distribution of approximately 2000 L.43
Clinical features
Adverse effects
The adverse effects of bupropion are relatively mild compared to other antidepressants. Mild hypertension has been noted, but has usually occurred in the already hypertensive.44 Postural hypotension has also been observed in sporadic patients.45 QRS or QT prolongation is not seen with bupropion at therapeutic doses.46 Neurological side effects occur more commonly with headache, insomnia, agitation and seizures being reported.47,48 Minor gastrointestinal irritation and priapism are also reported.
Overdose
The most significant clinical feature of overdose of the sustained release product is seizures and, importantly, the onset of the first seizure is commonly delayed until 6 to 8 h following ingestion. In a series of 59 overdoses, 19 of which involved sustained-release bupropion alone, the clinical effects noted were sinus tachycardia (83%), hypertension (56%) and seizures (37%). Seizures were dose-dependent, occurring in 30% of patients ingesting <4.5 g, in 50% of those ingesting 4.5–9 g and in 100% of cases involving >9 g (n = 2).49
Cardiotoxicity is rarely reported following overdose but may occur following massive ingestion (>9 g).50,51 It is suggested that QTc prolongation (>440 ms) observed following bupropion overdose is an overcorrection of the QTc due to the tachycardia, rather than a change indicative of cardiotoxicity.52
Fatalities are reported following bupropion overdose. In one case a 26-year-old male who ingested 23 g of bupropion became hypoxic following recurrent seizure activity and died despite resuscitation after a cardiac arrest.54
Treatment
The management of bupropion overdose is essentially supportive. Patients should be managed in an area equipped for cardiorespiratory monitoring and resuscitation. Close observation for at least 12 h and until the patient is asymptomatic is essential. Staff should be prepared to recognize and treat seizures. Intravenous benzodiazepines are the agent of choice. Prophylactic administration of titrated intravenous benzodiazepines to patients with agitation or tachycardia may be useful.
Tricyclic antidepressants
Tricyclic antidepressants (TCAs) have long been the leading cause of death from prescription drug overdose. However, they are increasingly being replaced in clinical practice by newer agents, which appear to be significantly safer in overdose. The TCAs currently available in Australia are listed in Table 29.3.3. Reported mortality rates for intentional TCA overdose range from 2% to 5%.54 The vast majority of successful TCA suicides do not reach hospital but die at home.55 The ingestion of 10 mg/kg or more of a TCA is potentially fatal, though there are differences in toxicity within the group. In Australia, doxepin is associated with the greatest lethality.56
Table 29.3.3 Tricyclic antidepressants available in Australia
| Amitriptyline |
| Clomipramine |
| Dothiepin |
| Doxepin |
| Imipramine |
| Nortriptyline |
| Trimipramine |
Pharmacology
TCAs have a distinct chemical structure, comprised of three aromatic rings. TCAs are non-selective agents that exhibit a large number of pharmacological effects. The therapeutic effect is most likely due to the inhibition of amine reuptake in the CNS, particularly serotonin and dopamine.57 This effect is not responsible for the toxicity of TCA overdose, but forms the basis of the role of TCAs in the development of serotonin syndrome.58
The major features of TCA overdose are related to the following pharmacological actions:
Clinical features
The clinical features of TCA overdose include anticholinergic, cardiovascular and CNS effects. The type and severity of clinical manifestations are dose-related (Table 29.3.4) Onset is usually rapid and, following large ingestions, rapid deterioration in clinical status within 1–2 h is characteristic.
Table 29.3.4 Tricyclic antidepressants: dose-related risk assessment
| Dose | Effect |
|---|---|
| <5 mg/kg | Minimal symptoms |
| 5–10 mg/kg | Drowsiness and mild anticholinergic effects Major toxicity not expected |
| >10 mg/kg | Potential for all major effects to occur within 2–4 h of ingestion |
| >30 mg/kg | Severe toxicity with pH-dependent cardiotoxicity and coma expected to last >24 h |
Adapted from Toxicology Handbook. Murray L, Daly F, Little M, Cadogan M (eds), Sydney: Elsevier; 2007.
The clinical features of central and peripheral anticholinergic toxicity are described elsewhere in this book. Anticholinergic delirium is most commonly observed following a modest TCA overdose or early in the course of more significant ingestions. Large overdoses usually lead to coma, which obscures any evidence of anticholinergic delirium. Seizures are characteristic of TCA overdose and usually occur early in the clinical course. Overall the rate is quoted to be 3–4%.59 Myoclonic jerking is also associated with TCA overdose.
Sinus tachycardia is commonly observed following TCA overdose and is usually due to the anticholinergic effects of the TCA, rather than sodium channel blockade. More serious cardiac arrhythmias can develop as a consequence of the effects on the fast sodium channels and cardiac depolarization and conduction. These include supraventricular tachycardia (with or without aberrancy), ventricular tachycardia, torsades des pointes (augmented by potassium channel blocking effects) and ventricular fibrillation. Junctional or idioventricular rhythms, second- or third-degree heart block or asystole can also occur.60 Hypotension is commonly observed and is due to both peripheral vasodilatation and impaired myocardial contractility.
Clinical investigation
Serum TCA concentrations correlate poorly with the clinical severity of TCA intoxication. The single most important investigation in assessing the patient following a TCA overdose is the 12-lead ECG. The degree of prolongation of the QRS interval is predictive of the risk of both ventricular arrhythmias and seizures.61 The positive and negative predictive values of ECG changes in TCA poisoning in one study were 66% and 100%, respectively.62 A QRS duration of >120 ms in the setting of a TCA overdose indicates cardiotoxicity. A terminal R wave >3 mm in lead aVR may be a more useful predictor of seizures or arrhythmias than QRS duration.63 A patient may exhibit significant CNS toxicity despite a normal ECG.
Treatment
Hypotension should initially be managed with i.v. fluids. If blood pressure fails to respond to infusions of crystalloid or colloid, then sodium bicarbonate should be tried even in the presence of a normal QRS. If there is still no response inotropes should be started. The ideal inotrope is one that will overcome α-adrenergic blockade and have little stimulatory effect on β receptors. For these reasons, noradrenaline (norepinephrine) is usually regarded as the inotrope of choice. Dopamine is best avoided as it stimulates β receptors (and may lead to a paradoxical decrease in blood pressure) and, as an indirectly acting sympathomimetic, it will become ineffective when neuronal stores of noradrenaline (norepinephrine) are depleted in the presence of a potent reuptake pump inhibitor such as TCAs.64
Seizures, delirium and hyperthermia should be controlled using standard techniques.
Flumazenil should be avoided in the setting of a TCA overdose because its action may precipitate refractory seizures and increase morbidity and mortality.65
Sodium bicarbonate
Sodium bicarbonate is regarded as a specific antidote in the management of TCA poisoning. It offers a hypertonic source of sodium, which competitively overcomes sodium channel blockade. It appears that the pH alteration also contributes to improved sodium channel function.66,67 However, manipulation of pH by hyperventilation is not reliably effective in reducing QRS duration.
Disposition
Patients with a history of TCA ingestion and who have received oral-activated charcoal but show no signs of toxicity after 6 h of observation are safe for medical discharge and ready for psychiatric evaluation.68 Those with significant cardiovascular or CNS toxicity should be admitted to an intensive care environment. Those with mild CNS manifestations only should be observed in hospital until these resolve.
Selective serotonin reuptake inhibitors (And Atypical Antidepressants)
The selective serotonin reuptake inhibitors (SSRIs) have now replaced the TCAs as the first-line drug therapy for depression, bringing with them the advantages of fewer adverse effects and relative safety in overdose. These drugs are also used in the treatment of obsessive-compulsive disorder, panic disorders and eating disorders including bulimia nervosa. Currently available SSRIs together with the atypical antidepressants that modulate serotonin neurotransmission are listed in Table 29.3.5.
Table 29.3.5 Seletive serotonin reuptake inhibitors and atypical antidepressants available in Australia
| Selective serotonin reuptake inhibition |
Pharmacology
SSRIs raise synaptic concentrations of serotonin by inhibiting serotonin uptake into presynaptic neurons. In addition, serotonin release from neurons, like other biogenic amines, is subject to autoregulation by presynaptic serotonin receptors that mediate negative feedback. SSRIs desensitize presynaptic serotonin autoreceptors, resulting in increased serotonin release. The rise in synaptic serotonin concentration and resultant stimulation of serotonin receptors (at least 14 different receptors discovered to date) is thought to explain SSRIs’ antidepressant activity.69
The atypical antidepressants have other effects apart from those on serotonin neurotransmission and these are listed in Table 29.3.5. The SSRIs and atypical antidepressants are generally rapidly and well absorbed after oral administration. Importantly, an extended release formulation of venlafaxine is widely prescribed. These drugs display diverse elimination patterns and have numerous active metabolites, which results in an extended therapeutic effect but also prolongs the time during which drug interactions and adverse effects can occur.
Clinical features
Adverse effects
The most common adverse effects attributed to the SSRIs are gastrointestinal symptoms, sexual dysfunction, headache, insomnia, jitteriness, dizziness and fatigue.70 Inappropriate antidiuretic hormone secretion is also reported, particularly in the elderly.71 The adverse effect most likely to result in presentation to the ED is the development of serotonin syndrome (see below) as a result of an interaction between two drugs that enhance serotonergic activity or where there has been an insufficient ‘wash-out’ period between ceasing one such drug and commencing another.
Overdose
Overdose of the SSRIs and atypical antidepressant generally follows a relatively benign course with the vast majority of patients remaining asymptomatic or experiencing minor self-limiting symptoms only. Venlafaxine with its significant noradrenaline reuptake inhibitor properties is an exception. It is associated with seizures and, in large doses (>4.5 g), cardiotoxicity. Citalopram is associated with QT prolongation although this is rarely of clinical significance.73,74 For most SSRIs the major concerns are the adverse interactions and serotonin syndrome.
Serotonin syndrome
Previously known as serotonin behavioural syndrome, this was first described in the late 1960s and early 1970s when rats, after being given a combination of a non-selective monoamine oxidase inhibitor and L-tryptophan, developed resting tremor, rigidity and abnormal limb, tail and head movements. Subsequent experiments showed that any drug capable of increasing synaptic levels of serotonin could induce a similar syndrome in larger animals. Human reports of recognized serotonin syndrome first appeared in the literature in the early 1980s.75,76
Clinically relevant drugs that can increase synaptic serotonin levels and have been implicated in the development of serotonin syndrome are listed in Table 29.3.6. The mechanisms by which these agents increase synaptic serotonin levels in the cortex, lower brain stem and spinal cord regions are variable and described elsewhere. The postsynaptic receptor subtype 5-HT1A appears to be mainly responsible.69
| Increased serotonin production |
Symptoms usually begin shortly after the commencement of a serotoninergic drug, or the administration of two different classes of drugs that increase serotonin levels synergistically, for example lithium and fluoxetine. In addition, potential drug interaction may arise when the appropriate ‘change-over’ period between drugs is not observed.77 A severe form of the syndrome may develop some hours following overdose with an SSRI or, more commonly, following overdose with multiple serotonergically active drugs.78–80
The diagnosis of serotonin syndrome is clinical and based upon the presence of the triad of alteration in behaviour-cognitive ability, autonomic function and neuromuscular activity. A grading system has been proposed.81 In its most benign form the patient experiences anxiety and apprehension, but altered sensorium with confusion occurs in 50% of reported cases.69 Seizures may occur.79 Abnormal neuromuscular activity, caused by increased brainstem and spinal-cord serotonin levels, manifests as increased rigidity (more in the lower than the upper limbs), hyperreflexia, involuntary jerks and resting extremity tremor. Hyperthermia, secondary to increased muscle activity is a common feature and may lead to confusion with NMS. Diaphoresis, diarrhoea and rigors are common. Cardiovascular instability may occur. Although most patients recover, fatalities are reported.79,80 There is no correlation with drug levels, and serotonin syndrome remains a clinical diagnosis.69 The differential diagnosis includes NMS, acute dystonia, hyperadrenergic states (e.g. cocaine toxicity), anticholinergic syndrome and malignant hyperthermia. Decision algorithms have been developed to help the clinician distinguish serotonin syndrome from other conditions.82
Treatment
Non-specific 5-HT1 and 5-HT2 antagonists such as propranolol, cyproheptadine, chlorpromazine and olanzapine have been tried.83–85 There are no controlled trials using these agents, but anecdotally cyproheptadine appears to be effective without the adverse effects of the other drugs.58 A dose of 4–8 mg 8-hourly is recommended.
Anticonvulsants
Anticonvulsants are frequently taken in deliberate self-poisoning. Toxicity resulting in ED attendance also results as a consequence of therapeutic administration. The benzodiazepines and phenobarbitone are discussed earlier in this chapter. This section discusses the traditional anticonvulsants, carbamazepine, phenytoin and sodium valproate – all of which have important toxic syndromes, and the newer anticonvulsants.
Carbamazepine
Clinical features
Onset of clinical features of carbamazepine toxicity may be delayed many hours following overdose due to delayed absorption.86,87 The clinical features are predominantly neurological and include CNS depression which may progress to coma, drowsiness, ataxia, nystagmus and dystonia. Paradoxical seizures are also reported in severe poisoning.88,89 Carbamazepine toxicity may also manifest as the anticholinergic syndrome, although the delirium may be masked by coma as the intoxication progresses.
Minor ECG changes may be observed in severe carbamazepine poisoning but significant cardiovascular effects are rare.90
Phenytoin
Clinical presentation
Chronic toxicity
With a relatively low therapeutic index and multiple drug interactions phenytoin toxicity occurs relatively frequently with therapeutic dosing. This is most likely to occur after injudicious dose adjustment.91,92 These patients usually present with neurological disturbance characterized by ataxia, dysarthria and nystagmus.
Overdose
Similar neurological disturbances are observed following acute overdose, although following large overdoses, a more severe neurological disturbance and even progressive CNS depression may develop. Paradoxical seizures may also occur.93
Oral phenytoin overdose is not associated with cardiovascular effects of clinical significance. Cardiotoxicity in the form of hypotension, dysrhythmias and death is only reported followed over-rapid administration or excessive doses of intravenous phenytoin.94 This cardiotoxicity is thought to be due to the propylene glycol diluent rather than the phenytoin per se.
Treatment
The management of phenytoin overdose is principally supportive. A single dose of activated charcoal should be administered in the patient who presents early. Cardiac monitoring is not required where phenytoin is the only agent ingested. Serial phenytoin levels may be useful to confirm the diagnosis and guide therapy. Therapeutic levels are 40–80 μmol/L (10–20 mg/L). Progressive toxicity albeit with much individual variation is seen as levels extending above that range (Table 29.3.7).
Table 29.3.7 Correlation between serum phenytoin concentration and clinical features
| Phenytoin concentration (μmol/L) | Clinical features |
|---|---|
| 80–120 | Horizontal nystagmus |
| 120–160 | Vertical nystagmus, ataxia, dysarthria |
| >160 | CNS depression, coma, seizures |
Valproic acid
Pharmacology
Valproic acid is a simple monocarboxylic acid, chemically unrelated to any other class of antiepileptic drug. Its mechanism of action is thought to involve but not be limited to a decrease in breakdown of GABA-A and increased conversion of GABA from glutamate.95
Oral absorption of valproic acid is rapid with peak levels usually occurring within 4 h but this may be delayed following overdose.96 The volume of distribution is very small at 0.13–0.23 L/kg and there is extensive plasma protein binding which may be saturated in overdose. The drug undergoes extensive hepatic metabolism and has active metabolites. The elimination half-life of 7–15 h may be prolonged in overdose.97
Clinical presentation
The clinical course following valproate overdose is dose-dependent. Ingestions of less than 200 mg/kg are usually asymptomatic or result in minor drowsiness only.98 For ingestions >200 mg/kg, coma may develop and, for ingestions >400 mg/kg, there is a risk of prolonged profound coma and metabolic disorders including hyperammonaemia, hypernatraemia, hypocalcaemia and bone-marrow depression.99 Death from cerebral oedema is reported.
L-carnitine
L-carnitine is an amino acid carrier molecule used to transport long-chain fatty acids across to mitochondria. It is synthetized chiefly in liver and kidney. It is available in oral and intravenous forms and appears to have an acceptable safety profile.100 It is postulated that L-carnitine could provide benefit in patients with concomitant hyperammonemia encephalopathy and/or hepatotoxicity as there is some evidence that it reduces ammonia concentrations in acute valproate overdose.101,102 While L-carnitine has been used in a number of case reports definitive evidence of efficacy is lacking.
Newer antiepileptic drugs
A number of new antiepileptic drugs with differing pharmacokinetic properties and mechanisms of action have been introduced into clinical practice over the last decade. These include oxcarbazepine, gabapentin, felbamate, vigabatrin, topiramate and tiagabine. The toxicity profiles of these drugs in overdose are not yet well established. Gabapentin, felbamate and lamotrigine are reported to cause only minor CNS effects in overdose.99–103 Vigabatrin overdose has resulted in severe agitation.104
1 Goodchild CS. GABA receptors and benzodiazepines. British Journal of Anaesthesia. 1993;71:127-133.
2 Greenblatt DJ, Allen MD, Noel BJ, et al. Acute overdosage with benzodiazepine derivatives. Clinical Pharmacology and Therapy. 1977;21:497-514.
3 Busto U, Kaplan HL, Sellers EM. Benzodiazepine-associated emergencies in Toronto. American Journal of Psychiatry. 1980;137:224-227.
4 Gueye PN, Lofaso F, Borron SW, et al. Mechanism of respiratory insufficiency in pure or mixed drug-induced coma involving benzodiazepines. Clinical Toxicology. 2002;40:35-47.
5 Tanaka E. Toxicological interactions between alcohol and benzodiazepines. Clinical Toxicology. 2002;40:69-75.
6 Haverkos GP, DiSalvo RP, Imhoff TE. Fatal seizures after flumazenil administration in a patient with mixed overdose. Annals of Pharmacotherapy. 1994;28:1347-1349.
7 Gaulier JM, Merle G, Lacassie E, et al. Fatal intoxications with chloral hydrate. Journal of Forensic Sciences. 2001;46:1507-1509.
8 Regouby Y, Delomez G, Tisserant A. First-degree heart block caused by voluntary zopiclone poisoning. Therapie. 1990;45:162.
9 Bramness JG, Arnestad M, Karinen R, et al. Fatal overdose of zopiclone in an elderly woman with bronchogenic coma. Journal of Forensic Sciences. 2001;46:1247-1249.
10 Boniface PF, Russel SG. Two cases of fatal zopiclone overdose. Journal of Analytical Toxicology. 1996;20:131-133.
11 Meram D, Descotes J. Acute poisoning with zolpidem. Revue Medicine Interne. 1989;10:466.
12 Gock SB, Wong SHY, Nuwayhid N, et al. Acute zolpidem overdose – report of two cases. Journal of Analytical Toxicology. 1999;23:559-562.
13 Ahmad Z, Herepath M, Ebden P. Diagnostic utility of flumazenil in coma with suspected poisoning. British Medical Journal. 1991;302:292.
14 Lheureux P, Debailleul G, De Witte O, et al. Zolpidem intoxication mimicking narcotic overdoes: response to flumazenil. Human and Experimental Toxicology. 1990;9:105-107.
15 Lheureux P. Continuous flumazenil for zolpidem toxicity – commentary. Clinical Toxicology. 1998;36:745-746.
16 Graham SR, Day RO, Lee R, et al. Overdose with chloral hydrate: a pharmacological and therapeutic review. Medical Journal of Australia. 1988;149:686-688.
17 Zahedi A, Grant MH, Wong DT. Successful treatment of chloral hydrate cardiac toxicity with propranolol. American Journal of Emergency Medicine. 1999;17:490-491.
18 Black JL, Richelson E, Richardson JW. Antipsychotic agents: a clinical update. Mayo Clinic Proceedings. 1985;60:777.
19 Burns MJ. The pharmacology and toxicology of atypical antipsychotic agents. Clinical Toxicology. 2001;39:1-14.
20 Casey DE, Keepers GA. Neuroleptic side effects: acute extrapyramidal syndromes and tardive dyskinesia. Casey DE, Christensen AV, editors. Psychopharmacology: current trends. 1988, 74-83. Springer-Verlag
21 Rupniak NJ, Jenner P, Marsden CD. Acute dystonia induced by neuroleptic drugs. Psychopharmacology. 1986;88:403.
22 Swett C. Drug induced dystonia. American Journal of Psychiatry. 1975;132:532.
23 Marsden CD, Jenner P. The pathophysiology of extrapyramidal side effects of neuroleptic drugs. Psychological Medicine. 1980;10:55.
24 Yassa R, Ananth J, Cordozo S, et al. Tardive dyskinesia in an outpatient population: prevalence and predisposing factors. Canadian Journal of Psychiatry. 1983;28:391.
25 Marks RC, Luchins DJ. Antipsychotic medications and seizures. Psychiatric Medicine. 1991;9:37.
26 Logothetis J. Spontaneous epileptic seizures and electroencephalographic changes in the course of phenothiazine therapy. Neurology. 1967;17:869.
27 Reilly JG, Ayis SA, Ferrier IN, et al. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048-1052.
28 Kariagianis JL, Phillips LC, Hogan KP, et al. Clozapine-associated neuroleptic malignant syndrome: two new cases and a review of the literature. Annals of Pharmacotherapy. 1999;3(5):623-630.
29 Levin GM, Lazowick AL, Powell HS. Neuroleptic malignant syndrome with risperidone. Journal of Clinical Psychopharmacology. 1996;16:192-193.
30 Burkhard PR, Vingerhoets FLG. Olanzapine-induced neuroleptic malignant syndrome (letter). Archives of General Psychiatry. 1999;56:101-102.
31 Nierenberg D, Disch M, Manheimer E, et al. Facilitating prompt diagnosis and treatment of the neuroleptic malignant syndrome. Clinical Pharmacology and Therapy. 1991;50:580.
32 Shalev A, Hermesh H, Munitz H. Mortality from neuroleptic malignant syndrome. Journal of Clinical Psychiatry. 1989;50:18.
33 Blaye IL, Donatini B, Hall M, et al. Acute overdosage with thioridazine: a review of the available clinical exposure. Veterinary and Human Toxicology. 1993;35:147-150.
34 Isbister GK, Murray L, John S, et al. Amisulpride deliberate self-poisoning causing severe cardiac toxicity including QT prolongation and torsades de pointes. Medical Journal of Australia. 2006;184:354-356.
35 Haag S, Spigset O, Edwardsson H, et al. Prolonged sedation and slowly decreasing clozapine serum concentrations after an overdose. Journal of Clinical Psychopharmacology. 1999;19:282-284.
36 Buckley NA, Whyte IM, Dawson AH. Cardiotoxicity is more common in thioridazine overdose than with other neuroleptics. Clinical Toxicology. 1995;33:199-204.
37 Schmidt W, Lang K. Life-threatening dysrhythmias in severe thioridazine poisoning treated with physostigmine and transient atrial pacing. Critical Care Medicine. 1997;25:1925-1930.
38 Sharma ND, Roman HS, Padhi D, et al. Torsades de pointes associated with intravenous haloperidol in critically ill patients. American Journal of Cardiology. 1998;81:238-240.
39 Ward DI. Two cases of amisulpride overdose: A cause for prolonged QT syndrome. Emergency Medical Association. 2005;17:274-276.
40 Balit CR, Isbister GK, Hackett PL, et al. Quetiapine Poisoning: A Case Series. Annals of Emergency Medicine. 2003;42:751-758.
41 Hunfeld NGM, Westerman EM, Boswijk DJ, et al. Quetiapine in overdosage. a clinical and pharmacokinetic analysis of 14 cases. Therapeutic Drug Monitoring. 2006;28:185-189.
42 Bryant SG, Guernsey BG, Ingrim NB. Review of bupropion. Clinical Pharmacology. 1983;2:525-537.
43 Lai AA, Schroeder DH. Clinical pharmacokinetics of bupropion: a review. Journal of Clinical Psychiatry. 1983;44:82-84.
44 Roose SP, Dalack GW, Glassman AH, et al. Cardiovascular effects of bupropion in depressed patients with heart disease. American Journal of Psychiatry. 1991;148:512-516.
45 Szuba MP, Leuchter AF. Falling backward in two elderly patients taking bupropion. Journal of Clinical Psychiatry. 1992;53:157-159.
46 Wenger TL, Stern WC. The cardiovascular profile of bupropion. Journal of Clinical Psychiatry. 1983;44:176-182.
47 Settle EC, Stahl SM, Batey SR, et al. Safety profile of sustained-release bupropion in depression: results of three clinical trials. Clinical Therapeutics. 1999;21:454-463.
48 Davidson J. Seizures and bupropion: a review. Journal of Clinical Psychiatry. 1989;50:256-261.
49 Balit CR, Lynch CN, Isbister GK. Bupropion poisoning: a case series. Medical Journal of Australia. 2003;178:61-63.
50 Druteika D, Zed PJ. Cardiotoxicity following bupropion overdose. Annals of Pharmacotherapy. 2002;36:1791-1795.
51 Biswas AK, Zabrocki LA, Mayes KL, et al. Cardiotoxicity associated with intentional ziprasidone and bupropion overdose. Journal of Toxicology, Clinical Toxicology. 2003;41:101-104.
52 Isbister GK, Balit CR. Bupropion overdose: QTc prolongation and its clinical significance. Annals of Pharmacotherapy. 2003;37:999-1002.
53 Harris CR, Gualtieri J, Stark G. Fatal bupropion overdose. Clinical Toxicology. 1997;35:321-324.
54 Mills KC. Tricyclic antidepressants. In: Tintinalli JE, Ruiz E, Krome RL, editors. Emergency medicine: a comprehensive study guide. 4th edn. Ohio: McGraw Hill; 1996:740.
55 Buckley NA, Dawson AH, Whyte IM, et al. Six years of self-poisoning in Newcastle: 19871992. Medical Journal of Australia. 1995;162:190-193.
56 Buckley NA, Dawson AH, Whyte IM, et al. Greater toxicity of dothiepin in overdose than of other tricyclic antidepressants. Lancet. 1994;343:159-162.
57 Curry SC. Neurotransmitter principles. In: Goldfrank LR, Weissman RS, Flomenbaum NE, Howard MA, Lewin NA, Hoffman RS, editors. Goldfrank’s toxicologic emergencies. 5th edn. Norwalk, Connecticut: Appleton and Lange; 1994:231-257.
58 Chan BC, Graudins A, Whyte IA, et al. Serotonin syndrome resulting from drug interactions. Medical Journal of Australia. 1998;169:523-525.
59 Wedin GP, Odra GM, Klein-Schwartz W, et al. Relative toxicity of cyclic antidepressants. Annals of Emergency Medicine. 1986;15:797.
60 Dzuikas LJ, Vohra J. Tricyclic antidepressant poisoning. Medical Journal of Australia. 1991;154:344-350.
61 Boehnert MT, Lovejoy FH. Value of the QRS duration versus the serum drug level in predicting seizures and ventricular arrhythmias after an acute overdose of tricyclic antidepressants. New England Journal of Medicine. 1985;313:474-479.
62 Niemann JT, Benson HA, Rothstein RJ, et al. Electrocardiographic criteria for tricyclic antidepressant cardiotoxicity. American Journal of Cardiology. 1986;57:1154-1159.
63 Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Annals of Emergency Medicine. 1995;26:195-201.
64 Buchamn AL, Dauer J, Giederan J. The use of vasoactive agents in the treatment of antidepressant overdose. Journal of Clinical Psychopharmacology. 1990;10:409-413.
65 Spivey WH. Flumazenil and seizures: Analysis of 33 cases. Clinical Therapy. 1992;14(2):292-305.
66 Nattel S, Mittleman M. Treatment of ventricular tachyarrhythmias from amitriptyline toxicity in dogs. Journal of Pharmacology and Experimental. Therapy. 1984;1231:430-435.
67 Nattel S, Keable H, Sasyniuk BI. Experimental amytriptyline intoxication: electrophysiologic manifestations and management. Journal of Cardiovascular Pharmacology. 1984;6:83-89.
68 Callaham M, Kassel D. Epidemiology of fatal tricyclic antidepressant ingestion: implications for management. Annals of Emergency Medicine. 1985;14:1-9.
69 Mills K. Serotonin toxicity: a comprehensive review for emergency medicine. Topics in Emergency Medicine. 1993;15:54-73.
70 Woodrum ST, Brown CS. Management of SSRI-induced sexual dysfunction. Annals of Pharmacotherapy. 1998;32:1209-1215.
71 Kirchner V, Silver LE, Kelly CA. Selective serotonin reuptake inhibitors and hyponatremia: review and proposed mechanisms in the elderly. Journal of Psychopharmacology. 1998;12:396-400.
72 Whyte IM, Dawson AH, Buckley NA. Relative toxicity of venlafaxine and selective serotonin reuptake inhibitors in overdose compared to tricyclic antidepressants. Quarterly Journal of Medicine. 2003;96:369-374.
73 Isbister GK, Bowe SJ, Dawson A, et al. Relative toxicity of selective serotonin re-uptake inhibitors (SSRIs) in overdose. Clinical Toxicology. 2004;42:277-285.
74 Isbister GK Fridberg LE, Duffull SB. Application of pharmacokinetic-pharmacodynamic modelling in the management of QT abnormalities after citalopram overdose. Intensive Care Medicine. 2006;32:1060-1065.
75 Insel TR, Roy BF, Cohen RM, et al. Possible development of the serotonin syndrome in man. American Journal of Psychiatry. 1982;139:954-955.
76 Sternbach H. The serotonin syndrome. American Journal of Psychiatry. 1991;148:705-714.
77 DeVane DL. Pharmacokinetics of the selective serotonin reuptake inhibitors. Journal of Clinical Psychiatry. 1992;53:13-19. (Supplement)
78 Kaminski CA, Robbins MS, Weibley RE. Sertraline intoxication in a child. Annals of Emergency Medicine. 1994;23:1371-1374.
79 Kline SS, Mauro LS, Scala-Barnett DM, et al. Serotonin syndrome versus neuroleptic malignant syndrome as a cause of death. Clinical Pharmacology. 1989;8:510-514.
80 Power BM, Pinder M, Hackett LP, et al. Fatal serotonin syndrome following a combined overdose of moclobemide, clomipramine and fluoxetine. Anaesthetics and Intensive Care. 1995;23:499-502.
81 Dunkley EJ, Isister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. Quarterly Journal of Medicine. 2003;96:635-642.
82 Boyer EW, Shannon M. The serotonin syndrome. New England Journal of Medicine. 2005;352:1112-1120.
83 Graudins A, Aaron CK. Delayed peak serum valproic acid in massive divalproex overdose treatment with charcoal hemoperfusion. Journal of Toxicology, Clinical Toxicology. 1996;34:335-341.
84 Guze BH, Baxter LR. The serotonin syndrome: case responsive to propranolol (letter). Journal of Clinical Psychopharmacology. 1986;6:119-120.
85 Geer SC, Baldessarini RJ. Motor effects of serotonin in the central nervous system. Life Sciences. 1980;27:1435-1451.
86 Sandyk R. L dopa induced ‘serotonin syndrome’ in a parkinsonian patient on bromocriptine (letter). Journal of Clinical Psychopharmacology. 1980;6:194-195.
87 Sullivan JB, Rumack BH, Peterson RG. Acute carbamazepine toxicity resulting from overdose. Neurology. 1981;31:621-624.
88 Sethna M, Solomon G, Cedarbaum J, et al. Successful treatment of massive carbamazepine overdose. Epilepsia. 1989;30:71-73.
89 Hojer J, Malmlund HO, Berg A. Clinical features in 28 consecutive cases of laboratory confirmed massive poisoning with carbamazepine alone. Clinical Toxicology. 1993;3:449-458.
90 Tibballs J. Acute toxic reaction to carbamazepine: clinical effects and serum concentrations. Journal of Pediatrics. 1992;121:295-299.
91 Apfelbaum JD, Caravati EM, Kerns WP, et al. Cardiovascular effects of carbamazepine toxicity. Annals of Emergency Medicine. 1995;25:631-635.
92 Morgan F. Fortnightly review: drug treatment of epilepsy. British Medical Journal. 1999;318:106-109.
93 Maloteaux EG. Pharmacological management of epilepsy. Mechanism of action, pharmacokinetic drug interactions, and new drug discovery possibilities. International Journal of Clinical Pharmacology and Therapeutics. 1988;36:181-184.
94 Peruca E, Gram L, Avanzi G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5-17.
95 Russell MA, Bousvaros G. Fatal results from diphenylhydantoin administered intravenously. Journal of the American Medical Association. 1968;20:2118-2119.
96 Curry SC, Mills KC, Graeme KA. Neurotransmitters. Goldfrank LR, editor. Goldfranks toxicologic emergencies, 7th edn. 2002, 133-165. New York: McGraw-Hill
97 Brubacher JR, Dahghani P, McKnight D. Delayed toxicity following ingestion of enteric-coated divalproex sodium (Epival). Journal of Emergency Medicine. 1999;17:463-467.
98 Garnier R, Boudignat O, Fournier PE. Valproate poisoning. Lancet. 1982;2:97.
99 Anderson GO, Ritland S. Life-threatening intoxication with sodium valproate. Clinical Toxicology. 1995;33:279-284.
100 LoVecchio F, Shriki J, Samaddar R. L-carnitine was safely administered in the setting of valproate toxicity. Am J Emerg Med. 2005.
101 Yehya N, Saldarini CT, Koski ME, et al. Valproate-induced hyperammonemic encephalopathy. [Case Reports. Letter] Journal of the American Academy of Child & Adolescent Psychiatry. 2004;43(8):926-927.
102 Perez A, McKay CA. Role of carnitine in valproic acid toxicity. Journal of Toxicology Clinical Toxicology, 2003;1901-2100.
103 Nagel TR, Schunk JE. Felbamate overdose: a case report and discussion of a new antiepileptic drug. Pediatric Emergency Care. 1995;11:369-371.
104 O’Donnel J, Bateman ND. Lamotrigine overdose in an adult. Clinical Toxicology. 2000;38:659-660.
29.4 Lithium
Clinical features
Chronic lithium toxicity
Chronic lithium toxicity may develop in association with prolonged excessive dosing or, more commonly, as a result of impaired lithium excretion due to intercurrent illness or a drug interaction. Lithium excretion is impaired in renal failure and congestive cardiac failure because of reduced filtration at the glomerulus and also in water or sodium depletion states because of increased reabsorption of sodium (and lithium) in the proximal tubule. A number of drugs including non-steroidal anti-inflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors (SSRIs), angiotesin converting enzyme (ACE) inhibitors, thiazide diuretics and topiramate may also impair lithium excretion.
The clinical features of chronic lithium toxicity are almost exclusively neurological and the following severity grading system is widely used:1
Lithium toxicity is generally not associated with significant cardiovascular effects although delayed onset of conduction disturbances is reported.2 Minor benign ECG changes are more commonly observed.3
Acute lithium overdose
Acute lithium overdose is much less likely to result in significant neurotoxicity than is chronic lithium toxicity.4 Neurotoxicity could theoretically slowly develop following acute overdose if renal clearance were sufficiently impaired so as to allow redistribution of sufficient lithium from the intravascular compartment to tissue compartments before it could be excreted. This situation may develop if there is pre-existing renal failure or if inadequate fluid resuscitation leads to dehydration, sodium depletion or renal impairment as a consequence of the fluid losses from gastrointestinal toxicity.
Clinical investigation
Therapeutic serum lithium concentrations are generally quoted as 0.6–1.2 mEq/L, although clinical evidence of lithium toxicity can be observed at concentrations within this range, particularly in the elderly.5 More commonly in cases of chronic intoxication, mild toxicity is observed at lithium concentrations of 1.5–2.5 mEq/L, severe toxicity at concentrations of 2.5 to 3.5 mEq/L, and life-threatening toxicity at concentrations >3.5 mEq/L. Following acute overdose, serum lithium concentrations do not correlate with clinical severity as they do not reflect CNS concentrations; however, when performed serially, they are useful in guiding management. Peak serum lithium concentrations >4.0 mEq/L are frequently observed following acute overdose in patients who do not go on to develop neurotoxicity.
Treatment
Chronic lithium toxicity
Appropriate supportive care measures should be instituted on arrival. Once the diagnosis of chronic lithium toxicity is confirmed, further care is oriented towards management of the precipitating medical condition and enhancing lithium excretion by optimizing renal function and correcting any water or sodium deficits with intravenous normal saline. Therapy with lithium carbonate and any drugs contributing to lithium toxicity should be immediately discontinued.6
Enhanced elimination of lithium by haemodialysis may be attempted in severe or worsening chronic lithium neurotoxicity. The aim of this intervention is to minimize the duration of neurological dysfunction and avoid permanent neurological sequelae. Lithium has physicochemical and pharmacokinetic properties that render it very suitable for enhancing elimination by haemodialysis: low molecular weight, high water solubility, small volume of distribution, no plasma protein binding and an endogenous renal clearance rate much lower than that achieved by haemodialysis.7 There is, however, no evidence that haemodialysis improves clinical outcome or survival rates.
The indications for haemodialysis are difficult to define. It should be considered in any patient with an elevated serum lithium concentration and severe or life-threatening neurotoxicity. It may be considered in the patient with less severe toxicity in whom adequate renal function and a falling lithium concentration are unable to be established with initial fluid resuscitation. Once instituted haemodialysis should be continued until the serum lithium is <1 mEq/L. Some rebound in serum lithium may be noted after intermittent haemodialysis is discontinued, which may be avoided if continuous arterio-venous (AV) or veno-venous (VV) haemodiafiltration is sustained for >16 h.8,9 The decision to dialyse can usually be made some 8–12 h after admission.7
Acute lithium overdose
In contrast to chronic toxicity, the vast majority of acute poisonings can be managed solely with good supportive care. Intravenous access should be established and infusion of normal saline commenced during the initial assessment. Administration should be sufficient to correct any sodium or water deficits arising as a result of the toxic gastroenteritis and to ensure a good urine output. Excessive administration of normal saline or attempts at forced diuresis do not further enhance lithium excretion.10 A serum lithium concentration, renal function and electrolytes should be performed as part of the initial assessment and repeated as necessary to guide further management. In particular, the serum lithium should be followed until falling and <2 mEq/L.
Activated charcoal does not bind lithium well and need not be administered unless there has been a significant co-ingestion. Sodium polystyrene sulfonate has been proposed as an effective alternative absorbent but is not widely used and repeated administration can cause hypokalaemia.11 On the basis of a single volunteer study, whole-bowel irrigation has been recommended for overdose of extended-release preparations12 but the gastrointestinal upset renders this intervention technically difficult in patients with large ingestions.
Disposition and prognosis
Patients with chronic lithium intoxication require admission for management of their fluid and electrolyte status, monitoring of renal function and serum lithium concentration and management of intercurrent illnesses. Ideally, admission should be to an institution with a capacity to perform haemodialysis where toxicity is moderate or severe. Following haemodialysis, neurological recovery may be delayed well beyond the removal of lithium and permanent neurological deficits are reported.13,14
1 Hansen HE, Amdisen A. Lithium intoxication. Quarterly Journal of Medicine. 1978;47:123-144.
2 Waring WS. Delayed cardiotoxicity in chronic lithium poisoning: discrepancy between serum lithium concentrations and clinical status. Basic and Clinical Pharmacology and Toxicology. 2007;100(5):353-355.
3 Tilkian AG, Schroeder JS, Kao JJ. Cardiovascular effects of lithium in man: a review of the literature. American Journal of Medicine. 1976;61:665-667.
4 Oakley PW, Whyte IM, Carter GL. Lithium toxicity: an iatrogenic problem in susceptible individuals. Australian & New Zealand Journal of Psychiatry. 2001;35:833-840.
5 Strayhorn JM, Nash JL. Severe neurotoxicity despite ‘therapeutic’ serum lithium levels. Diseases of the Nervous System. 1977;38:107-111.
6 Eyer F, Pfab R, Felgenhauer N, et al. Lithium poisoning: pharmacokinetics and clearance during different therapeutic measures. Journal of Clinical Psychopharmacology. 2006;26(3):325-330.
7 Jaeger A, Saunder P, Kopferschmidt J, et al. When should dialysis be performed in lithium poisoning? A kinetic study in 14 cases of lithium poisoning. Clinical Toxicology. 1993;31(3):429-447.
8 LeBlanc M, Raymond M, Bonnardeau A, et al. Lithium poisoning treated by high-performance continuous arteriovenous and venovenous hemodiafiltration. American Journal of Kidney Disease. 1996;27:365-372.
9 Waring WS. Management of lithium toxicity. Toxicology Reviews. 2006;25(4):221-230.
10 Amidsen A. Clinical features and management of lithium poisoning. Medical and Toxicological Adverse Drug Experiences. 1988;3:18-32.
11 Roberge RJ, Martin TG, Schneider S. Use of sodium polystyrene sulfonate in a lithium overdose. Annals of Emergency Medicine. 1993;22:1911-1915.
12 Smith S, Ling L, Halstenson C. Whole-bowel irrigation as a treatment for acute lithium overdose. Annals of Emergency Medicine. 1991;20:536-539.
13 Verdoux H, Bougeois M. A case of lithium neurotoxicity with irreversible cerebellar syndrome. Journal of Nervous and Mental Disorders. 1990;178:761.
14 Shou M. Long lasting neurological sequelae after lithium intoxication. Acta Psychiatrica Scandinavica. 1984;70:594.
29.5 Antihistamine and anticholinergic poisoning
Introduction
Anticholinergic toxicity is a common side effect of many pharmaceutical agents, natural remedies and plants, both in therapeutic dosing and in overdose (see Table 29.5.1). Symptoms and signs may range from mild manifestations of the syndrome (e.g. dry mouth and blurred vision) to severe anticholinergic delirium with agitation, hallucinations and aggressive behaviour.
| Pharmaceuticals |
Antihistamine agents are relatively easy to obtain and frequently used in overdose for attempted suicide and abused recreationally for their sedating and anticholinergic effects. The incidence of antihistamine poisoning and abuse in Australia is not well characterized. Other prescription drugs may also result in anticholinergic toxicity both in therapeutic dosing and in overdose. These may also be intentionally abused for their anticholinergic effects.1–4 Chinese and traditional herbal medicines may result in anticholinergic toxicity either directly from the herbal agent ingested or as a result of contamination with anticholinergic agents such as atropine or scopolamine.5–7 The intentional abuse of botanicals (e.g. Datura spp.) may also present with anticholinergic toxicity.8–10 In view of the easy availability of many of these pharmaceutical and herbal agents, the emergency physician should include a detailed drug history in the evaluation of any patient presenting with evidence of mental status change and anticholinergic symptoms and signs. In particular, polypharmacy and drug interactions between multiple agents with the potential for anticholinergic effects should be included in the differential diagnosis of elderly patients presenting with mental status changes.
Pharmacodynamics and pharmacokinetics
The H1-antagonists are a diverse group of agents that reversibly block the action of histamine at H1 receptors. High lipid solubility results in central nervous system (CNS) penetration and sedation. The first generation agents also block muscarinic, α-adrenergic and serotonergic receptors. Local anaesthetic effects due to sodium channel blockade may mimic the antiarrhythmic properties of class 1A antiarrhythmic agents.11 Diphenhydramine, dimenhydrinate and cyproheptadine, in particular, may prolong the cardiac muscle cell action potential duration by this mechanism.12,13
The second generation H1-antagonists (fexofenadine, loratadine) have much less CNS penetration and are more histamine receptor specific with little or no effect at other receptor subtypes.11
All the H1-antagonists are well absorbed orally with peak serum concentrations occurring within 2 to 4 h. Absorption may be delayed in overdose due to anticholinergic effects seen with the first-generation agents. Bioavailability is limited by significant first-pass metabolism. Some agents may be converted to active metabolites (e.g. hydroxyzine). Volume of distribution and protein binding are generally high. Elimination half-lives for the first generation agents are between 2 and 6 h.11 The second generation agents generally have longer half-lives (e.g. loratadine 8.3 h).11
Clinical features
The anticholinergic toxidrome is usually manifest by a combination of peripheral and central muscarinic, cholinergic receptor blockade. Peripheral effects may include sinus tachycardia, cutaneous vasodilatation and flushing, low-grade temperature, warm dry skin with an absence of axillary sweat, dry mucous membranes, gastrointestinal ileus and urinary retention. CNS effects include mydriasis with blurred vision due to the inhibition of visual accommodation, delirium, confusion, visual hallucinations, incoherent speech, agitation, combativeness, aggression and coma.14 Patients presenting with anticholinergic syndrome will often have an impaired perception of their environment. This may result in behaviour that could injure the patient. Anticholinergic symptoms and signs may be prominent with ingestion of first-generation antihistamines.15–17 Even therapeutic doses of some of the H1-antagonist agents may be sufficient to produce an anticholinergic delirium in susceptible individuals (especially the elderly and children). Topical use of these agents, particularly on broken skin surfaces, may also result in anticholinergic delirium.18,19
In patients who present to hospital several hours following poisoning with an anticholinergic agent, the peripheral features of the toxidrome may be absent.20–22 This may also occur in elderly people with mild-to-moderate anticholinergic delirium resulting from the side effects of therapeutic drug administration.22
Other manifestations of H1-antagonist toxicity may include CNS and cardiovascular effects, and rhabdomyolysis. Overdose of first-generation agents commonly produces drowsiness, sedation, confusion, agitation and ataxia. Large ingestions may result in coma.23,24 Seizures may also occur. Pheniramine, a commonly abused antihistamine in Australia, appears to be more proconvulsant than other agents following overdose, with a reported incidence of seizures of 30%.17 Seizures have been reported with other first-generation H1-antagonists, such as diphenhydramine, in doses as small as 150 mg in children.23,25 Fatal doses of diphenhydramine in adults range from 20 to 40 mg/kg.26 Doxylamine poisoning may result in non-traumatic rhabdomyolysis.27,28 Hypotension, due to α-receptor blockade, can occur following large ingestions of first-generation agents. Conduction defects are infrequent following poisoning with first-generation H1-antagonists. Diphenhydramine and dimenhydrinate poisoning can result in QRS-interval prolongation, broad-complex tachycardia and ventricular arrhythmias similar to that seen in cyclic antidepressant poisoning.12,13 This effect has not been reported with other first-generation H1-antagonists.
Overdose with H2-antagonists, such as cimetidine, usually results in little or no evidence of toxicity. Doses of up to 15 g have failed to produce clinical toxicity.29
Treatment
The reversible acetylcholinesterase inhibitor physostigmine has been used in the management and diagnosis of anticholinergic agitation and delirium.30–32 Physostigmine rapidly reverses the effects of anticholinergic delirium and may prevent the need for escalating doses of benzodiazepines to control agitation. Physostigmine may decrease the need for other interventions such as cerebral computerized tomography scanning and lumbar puncture in patients with suspected anticholinergic delirium.31
When using physostigmine, an initial test dose 0.5 mg i.v. is followed by 1.0–2.0 mg over the following 3–5 min in an adult. A partial response may necessitate further 0.5–1.0 mg boluses. Clinical effects may last from 30 to 120 min. Caution should be exercised in using physostigmine in patients with suspected acute cyclic antidepressant poisoning or ECG evidence of cardiac conduction delay because of the risk of precipitating cardiac asystole.33,34
Broad-complex tachycardia resulting from severe poisoning with diphenhydramine or dimenhydrinate should be treated with serum alkalinization with intravenous sodium bicarbonate boluses (0.5–1.0 mmol/kg) as for severe cyclic antidepressant poisoning.12,13 Symptomatic bradycardia and high degree atrioventricular block should be initially treated with atropine. Unresponsive cases may need cardiac pacing or inotropic support. Class-1a, -1c or -3 anti-arrhythmic agents should be avoided in cases of antihistamine-induced arrhythmias.
1 Acri AA, Henretig FM. Effects of risperidone in overdose. American Journal of Emergency Medicine. 1998;16:498-501.
2 Fisher RS, Cysyk B. A fatal overdose of carbamazepine: case report and review of literature. Journal of Toxicology Clinical Toxicology. 1988;26:477-486.
3 Graudins A, Peden G, Dowsett RP. Massive overdose with controlled-release carbamazepine resulting in delayed peak serum concentrations and life-threatening toxicity. Emergency Medicine (Fremantle). 2002;14:89-94.
4 Yang CC, Deng JF. Anticholinergic syndrome with severe rhabdomyolysis – an unusual feature of amantadine toxicity. Intensive Care Medicine. 1997;23:355-356.
5 Chan TY. Anticholinergic poisoning due to Chinese herbal medicines. Veterinary & Human Toxicology. 1995;37:156-157.
6 Chan JC, Chan TY, Chan KL, et al. Anticholinergic poisoning from Chinese herbal medicines. Australian & New Zealand Journal of Medicine. 1994;24:317-318.
7 Chan TY, Tang CH, Critchley JA. Poisoning due to an over-the-counter hypnotic, Sleep-Qik (hyoscine, cyproheptadine, valerian). Postgraduate Medical Journal. 1995;71:227-228.
8 Finlay P. Anticholinergic poisoning due to Datura candida. Tropical Doctor. 1998;28:183-184.
9 Hanna JP, Schmidley JW, Braselton WE. Datura delirium. Clinical Neuropharmacology. 1992;15:109-113.
10 Mahler DA. Anticholinergic poisoning from Jimson weed. Journal of the American College of Emergency Physicians. 1976;5:440-442.
11 Rimmer SJ, Church MK. The pharmacology and mechanism of action of histamine H1 antagonists. Clinical and Experimental Allergy. 1990;20:3.
12 Clark RF, Vance MV. Massive diphenhydramine poisoning resulting in a wide-complex tachycardia: successful treatment with sodium bicarbonate. Annals of Emergency Medicine. 1992;21:318-321.
13 Farrell M, Heinrichs M, Tilelli JA. Response of life threatening dimenhydrinate intoxication to sodium bicarbonate administration. Journal of Toxicology – Clinical Toxicology. 1991;29:527-535.
14 Feldman MD. The syndrome of anticholinergic intoxication. American Family Physician. 1986;34:113-116.
15 Jones IH, Stevenson J, Jordan A, et al. Pheniramine as an hallucinogen. Medical Journal of Australia. 1973;1:382-386.
16 Jones J, Dougherty J, Cannon L. Diphenhydramine-induced toxic psychosis. American Journal of Emergency Medicine. 1986;4:369-371.
17 Buckley NA, Whyte IM, Dawson AH, et al. Pheniramine-a much abused drug. Medical Journal of Australia. 1994;160:188-192.
18 Schipior PG. An unusual case of antihistamine intoxication. Journal of Pediatrics. 1967;71:589-591.
19 Reilly JFJr., Weisse ME. Topically induced diphenhydramine toxicity. Journal of Emergency Medicine. 1990;8:59-61.
20 Rupreht J, Dworacek B. Central anticholinergic syndrome in anesthetic practice. Acta Anaesthesiologica Belgica. 1976;27:45-60.
21 Koppel C, Hopfe T, Menzel J. Central anticholinergic syndrome after ofloxacin overdose and therapeutic doses of diphenhydramine and chlormezanone. Journal of Toxicology – Clinical Toxicology. 1990;28:249-253.
22 Feinberg M. The problems of anticholinergic adverse effects in older patients. Drugs Aging. 1993;3:335-348.
23 Koppel C, Ibe K, Tenczer J. Clinical symptomatology of diphenhydramine overdose: an evaluation of 136 cases in 1982 to 1985. Journal of Toxicology – Clinical Toxicology. 1987;25:53-70.
24 Koppel C, Tenczer J, Ibe K. Poisoning with over-the-counter doxylamine preparations: an evaluation of 109 cases. Human Toxicology. 1987;6:355-359.
26 Krenzelok EP, Anderson GM, Mirick M. Massive diphenhydramine overdose resulting in death. Annals of Emergency Medicine. 1982;11:212-213.
27 Mendoza FS, Atiba JO, Krensky AM, et al. Rhabdomyolysis complicating doxylamine overdose. Clinical Paediatrics. 1987;26:595-597.
28 Frankel D, Dolgin J, Murray BM. Non-traumatic rhabdomyolysis complicating antihistamine overdosage. Clinical Toxicology. 1990;31:493.
29 Krenzelok EP, Litovitz T, Lippold KP, et al. Cimetidine toxicity: an assessment of 881 cases. Annals of Emergency Medicine. 1987;16:1217-1221.
30 Beaver KM, Gavin TJ. Treatment of acute anticholinergic poisoning with physostigmine. American Journal of Emergency Medicine. 1998;16:505-507.
31 Burns MJ, Linden CH, Graudins A, et al. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Annals of Emergency Medicine. 2000;35:374-381.
32 Mendelson G. Pheniramine aminosalicylate overdosage. Reversal of delirium and choreiform movements with tacrine treatment. Archives of Neurology. 1977;34:313.
33 Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Annals of Emergency Medicine. 1980;9:588-590.
34 Suchard JR. Assessing physostigmine’s contraindication in cyclic antidepressant ingestions. Journal of Emergency Medicine. 2003;25:185-191.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Section 29 Toxicology
29.1 Approach to the poisoned patient
Introduction
Deliberate self-poisoning is one of the commonest reasons for general hospital admission in the UK1 and accounts for 1–5% of all public hospital admissions in Australia.2,3 The bulk of the medical management of cases presenting to hospital is carried out in the emergency department (ED), and the emergency physician is expected to be expert in the field. Although the management must vary considerably according to the nature and severity of the poisoning, some general principles apply.
Above all, it must be remembered that the acute overdose presentation is only a discrete time-limited event in the course of the underlying condition, which is usually psychiatric or social in origin.
Pathophysiology and clinical features
The most frequent life-threatening respiratory complication of poisoning is ventilatory failure, which is usually a consequence of CNS depression. Less commonly, it is secondary to ventilatory muscle paralysis. The frequency and depth of respirations are reduced. Respiratory failure may also be caused by direct pulmonary toxicity, or complications such as pulmonary aspiration or non-cardiogenic pulmonary oedema (Table 29.1.1).
Cardiovascular manifestations of poisoning include tachycardia, bradycardia, hypertension, hypotension, conduction defects and arrhythmias (Table 29.1.2). Bradycardia is relatively rarely observed and is associated with a number of potentially life-threatening ingestions. Tachycardia is commonly observed and is usually benign. It may be due to intrinsic sympathomimetic or anticholinergic effects of a drug, or a reflex response to hypotension or hypoxia. Hypotension is also commonly observed and may be due to a number of different causes (Table 29.1.2). Hypertension is unusual. Severe hypertension is usually associated with illicit drug use and is important because it may produce complications such as intracerebral haemorrhage.
Table 29.1.2 Cardiovascular effects of poisoning
CNS manifestations of poisoning include decreased level of consciousness, agitation or delirium, seizures and disordered temperature regulation. A decreased level of consciousness is a common presentation of poisoning and is associated with many drugs, some of which are listed in Table 29.1.1. Although usually a direct drug effect, CNS depression is occasionally secondary to hypoglycaemia, hypoxia or hypotension. Common causes of agitation or delirium following overdose are listed in Table 29.1.3. Toxic seizures are potentially life-threatening, and important causes are listed in Table 29.1.4.
Table 29.1.3 Toxic causes of agitation or delirium
| Alcohol |
| Anticholinergic syndrome |
| Antidepressants |
| Atypical antipsychotic agents |
| Benzodiazepines and other sedative-hypnotics |
| Cannabis |
| Hallucinogenic agents |
| Serotonin syndrome |
| Sympathomimetic syndrome |
| Theophylline |
| Withdrawal syndromes |
Table 29.1.4 Toxic causes of seizures
| Amphetamines |
| Bupropion |
| Carbamazepine |
| Chloroquine |
| Cocaine |
| Isoniazid |
| Mefanamic acid |
| Theophylline |
| Tramadol |
| Tricyclic antidepressants |
| Venlafaxine |
Hypothermia is usually a complication of environmental exposure secondary to a decreased level of consciousness or altered behaviour. Hyperthermia is a direct toxic effect and causes are listed in Table 29.1.5. Severe hyperthermia is rapidly lethal if not corrected.
Table 29.1.5 Toxic causes of hyperthermia
| Amphetamines |
| Anticholinergics |
| Cocaine |
| MAO inhibitors |
| Salicylates |
| Serotonin syndrome |
Pulmonary aspiration frequently complicates a period of decreased level of consciousness or a seizure. It is a leading cause of in-hospital morbidity and mortality following overdose. This complication is characterized by rapid onset of dyspnoea, cough, fever, wheeze and cyanosis.
Assessment
Risk assessment
A risk assessment should be made as soon as possible in the management of the poisoned patient. Only resuscitation is a greater priority (see Table 29.1.6). Risk assessment is a distinct quantitative cognitive step through which the clinician attempts to predict the likely clinical course and potential complications for the individual patient at that particular presentation.4 An accurate risk assessment allows informed decision-making in regard to all subsequent management steps including duration and intensity of supportive care and monitoring, screening and specialized testing, decontamination, enhanced elimination, antidotes and disposition. Factors that are taken into account when formulating this risk assessment include: the agent(s), the dose, the time since ingestion, the clinical features present and patient factors (Table 29.1.6). Specialized testing may refine risk assessment. Access to specialized poisons information in the form of a poisons information centre or in-house databases is often necessary to formulate an accurate risk assessment.
Reproduced from Toxicology Handbook. Murray L, Daly F, Little M, Cadogan M. Elsevier, Sydney 2007.
Physical examination
The focused physical examination of the poisoned patient aims to:
Several toxic autonomic syndromes, or ‘toxidromes’, have been described in relation to poisoning. The principal ones are listed in Table 29.1.7. Identification of these syndromes may narrow the differential diagnosis in cases of unknown poisoning.5
| Toxidrome | Features | Common causes |
|---|---|---|
| Anticholinergic | Agitated delirium | Antihistamines |
| Tachycardia | Benztropine | |
| Hyperthermia | Carbamazepine | |
| Dilated pupils | Phenothiazines | |
| Dry flushed skin | Plant poisonings | |
| Urinary retention | Tricyclic antidepressants | |
| Ileus | ||
| Mixed cholinergic | Brady- or tachycardia | Organophosphates |
| Hypo- or hypertension | Carbamates | |
| Miosis or mydriasis | ||
| Sweating | ||
| Increased bronchial secretion | ||
| Gastrointestinal hyperactivity | ||
| Muscle weakness | ||
| Fasciculations | ||
| Mixed α- and β-adrenergic | Hypertension | Amphetamines |
| Tachycardia | Cocaine | |
| Mydriasis | ||
| Agitation | ||
| β-Adrenergic | Hypotension | Caffeine |
| Tachycardia | Salbutamol | |
| Hypokalaemia | Theophylline | |
| Hyperglycaemia | ||
| Serotonin | Altered mental status | Amphetamines |
| Autonomic dysfunction | Antihistamines | |
| Fever | Monoamine oxidase inhibitors | |
| Hypertension | NSSRIs | |
| Sweating | SSRIs | |
| Tachycardia | Tricyclic antidepressants (Usually combined overdose) | |
| Motor dysfunction | ||
| Hyperreflexia | ||
| Hypertonia (esp. lower limbs) | ||
| Myoclonus |
NSSRI, non-selective serotonin re-uptake inhibitor; SSRI, selective serotonin re-uptake inhibitor.
Poisons information
Information on the clinical course and toxic doses of specific pharmaceutical and non-pharmaceutical poisons is available on a 24 h basis throughout Australia by telephoning 131126. The poison information centres are staffed by pharmacists and are also able to refer cases to clinical toxicologists for consultation.
Treatment
The management of poisoning should be approached in a systematic way. Following initial resuscitation, further treatment is informed by the risk assessment (Table 29.1.6).
Resuscitation, supportive care and monitoring
Supportive care is the key element in the management of poisoning. The vast majority of poisonings result in temporary dysfunction of one or more of the body systems. If appropriate support of the system in question is instituted in a timely fashion and continued until the toxic substance is metabolized or excreted, a good outcome can be anticipated. In severe poisonings supportive care may be very aggressive, and possible interventions are listed in Table 29.1.8.
Table 29.1.8 Supportive care measures for the poisoned patient
| Airway | Endotracheal intubation |
| Breathing | Supplemental oxygen |
| Ventilation | |
| Circulation | Intravenous fluids |
| Inotropes | |
| Antihypertensives | |
| Antiarrhythmics | |
| Defibrillation/cardioversion | |
| Cardiac pacing | |
| Cardiopulmonary bypass | |
| Metabolic | Hypertonic dextrose |
| Hypertonic saline | |
| Insulin/dextrose | |
| Calcium salts | |
| Sodium bicarbonate | |
| Agitation/delirium | Benzodiazepines |
| Butyrophenones | |
| Seizures | Benzodiazepines |
| Barbiturates | |
| Body temperature | External rewarming |
| External cooling | |
| Impaired renal function | Rehydration |
| Haemodialysis |
Toxic hypertension rarely requires specific therapy. Most cases are mild and simple observation is sufficient. Agitation or delirium is a feature of many intoxications associated with hypertension, and adequate sedation with benzodiazepines usually lowers the blood pressure. Severe toxic hypertension is most likely in toxicity from cocaine or amphetamine-type drugs, and treatment may be indicated to avoid complications such as cardiac failure or intracerebral haemorrhage. The drug of choice in this situation is sodium nitroprusside by intravenous infusion. The extremely short duration of action of this vasodilator allows accurate control of hypertension during the toxic phase, and avoids the development of hypotension once toxicity begins to wear off.
Decontamination
Gastric emptying can be attempted by the administration of an emetic, most commonly syrup of ipecac, or by gastric lavage. In volunteer studies both of these techniques removed highly variable amounts of marker substances from the stomach even if performed immediately after ingestion, and the effect diminished rapidly with time to the point of being negligible after one hour.6,7 Clinical outcome trials have failed to demonstrate improved outcome as a result of routine gastric emptying in addition to administration of activated charcoal, except, perhaps, in patients presenting unconscious within 1 h of ingestion.8–10
The principal adsorbent available to clinicians is activated charcoal (AC), which effectively binds most pharmaceuticals and chemicals, and is currently the decontamination method of choice for most poisonings. Materials that do not bind well to charcoal are listed in Table 29.1.9.
Table 29.1.9 Materials that do not bind well to activated charcoal
| Alcohols |
Volunteer studies demonstrate that the effect of AC diminishes rapidly with time and that the greatest benefit occurs if it is administered within 1 h. There is as yet no evidence that AC improves clinical outcome.11
There is no evidence to suggest that the addition of a cathartic such as sorbitol to AC improves clinical outcome.12,13
Apart from rarely employed endoscopic and surgical techniques, whole-bowel irrigation (WBI) is the most aggressive form of gastrointestinal decontamination. Polyethylene glycol solution (Golytely™) is administered via a nasogastric tube at a rate of 2 L/h until a clear rectal effluent is produced. This usually takes about 6 h and requires one-to-one nursing. In volunteer studies, this technique reduced the absorption of slow-release pharmaceuticals and so may be of benefit in life-threatening overdoses of these agents.13,14 Again, clinical benefit has not yet been conclusively demonstrated.15 The use of WBI has also been reported in the management of potentially toxic ingestions of iron, lead and packets of illicit drugs. Whole-bowel irrigation is contraindicated if there is evidence of ileus or bowel obstruction, and in patients who have an unprotected airway or haemodynamic compromise.16
Enhanced elimination
A number of techniques are available to enhance the elimination of toxins from the body. Their use is rarely indicated, as only a very few drugs capable of causing severe poisoning have pharmacokinetic parameters that render them amenable to these techniques (Table 29.1.10).
| Technique | Suitable toxin |
|---|---|
| Repeat-dose activated charcoal | Carbamazepine |
| Dapsone | |
| Phenobarbitone | |
| Phenytoin | |
| Salicylate | |
| Theophylline | |
| Urinary alkalinization | Phenobarbitone |
| Salicylate | |
| Haemodialysis | Ethylene glycol |
| Lithium | |
| Methanol | |
| Salicylate | |
| Theophylline | |
| Haemoperfusion | Theophylline |
Repeat-dose AC (25–50 g every 3–4 h) may enhance drug elimination by interrupting the enterohepatic circulation or by ‘gastrointestinal dialysis’. Gastrointestinal dialysis is the movement of a toxin across the gastrointestinal wall from the circulation into the gut down a concentration gradient that is maintained by charcoal binding. For this technique to be effective, a drug must undergo considerable enterohepatic circulation or, in the case of ‘gastrointestinal dialysis’, have a small volume of distribution, small molecular weight, low protein binding, slow endogenous elimination and bind to charcoal.16,17 The advantages of this technique are that it is non-invasive and simple to carry out.
Alkalinization of the urine enhances urinary excretion of drugs that are filtered at the glomerulus and are unable to be reabsorbed across the tubular epithelium when in an ionized form at alkaline pH. For elimination to be effectively enhanced by this method, the drug must be predominantly eliminated by the kidneys in the unchanged form, have a low pKa, be distributed mainly to the extracellular fluid compartment and be minimally protein bound.18
Antidotes
Very few drugs have effective antidotes. Occasionally, however, timely use of an antidote may be life saving or substantially reduce morbidity, time in hospital or resource requirements. Antidotes that may be indicated in the ED setting are listed in Table 29.1.11. However, it must be remembered that antidotes are also drugs, and are frequently associated with adverse effects of their own. An antidote should only be used where a specific indication exists, and then only at the correct dose, by the correct route, and with appropriate monitoring. Because many antidotes are so infrequently used, obtaining sufficient supplies when the need arises can be difficult. Every ED must review its stocking of antidotes and have a plan for obtaining further supplies should the need arise.
| Poisoning | Antidote |
|---|---|
| Atropine | Physostigmine |
| Benzodiazepines | Flumazenil |
| Cyanide | Dicobalt edetate, hydroxocobalamin |
| Digoxin | Digoxin-specific Fab fragments |
| Insulin | Dextrose |
| Iron | Desferoxamine |
| Isoniazid | Pyridoxine |
| Methaemoglobinaemia | Methylene blue |
| Methanol and ethylene glycol | Ethanol, fomepizole |
| Organophosphates and carbamates | Atropine, oximes |
| Opioids | Naloxone |
| Paracetamol | N-acetyl cysteine |
| Sulphonylureas | Dextrose, octreotide |
| Tricyclic antidepressants | Sodium bicarbonate |
| Warfarin, brodifacoum | Vitamin K |
Clinical investigation
Measurement of serum drug concentrations is only useful if this provides important diagnostic or prognostic information, or assists in planning management. Some drug levels that may be useful are listed in Table 29.1.12. For most cases, drug overdose management is guided by clinical findings and not by drug levels. Some drugs commonly taken in overdose for which serum concentrations are of no value in planning management are listed in Table 29.1.13.
Table 29.1.12 Drug levels that may be helpful in the management of selected cases of overdose
| Carbamazepine |
| Digoxin |
| Dilantin |
| Lithium |
| Iron |
| Paracetamol |
| Phenobarbitone |
| Salicylate |
| Theophylline |
| Valproate |
Table 29.1.13 Drug levels that are not helpful in the management of overdose
| CNS drugs | Cardiovascular drugs |
|---|---|
| Antidepressants | ACE inhibitors |
| Benzodiazepines | Beta-blockers |
| Benztropine | Calcium channel blockers |
| Cocaine | Clonidine |
| Newer antipsychotics | |
| Opiates | |
| Phenothiazines |
Disposition
Both the medical and the psychiatric disposition of the overdose patient must be considered. A good risk assessment is essential to determining timely and safe disposition.
1 Hawton K, Fagg J. Trends in deliberate self-poisoning and self-injury in Oxford. British Medical Journal. 1992;304:1409-1411.
2 McGrath J. A survey of deliberate self-poisoning. Medical Journal of Australia. 1989;150:317-322.
3 Pond SM. Prescription for poisoning. Medical Journal of Australia. 1995;162:174-175.
4 Murray L, Daly F, Little M, Cadogan M, editors. Toxicology handbook. Sydney: Elsevier, 2007.
5 Kulig K. Initial management of ingestion of toxic substances. New England Journal of Medicine. 1992;326:1677.
6 Krenzelok EP, McGuigan M, Lheureux P. Position statement: ipecac syrup. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):699-709.
7 Vale JA. Position statement: gastric lavage. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):711-719.
8 Kulig K, Bar-Or D, Kantrill SV, et al. Management of acutely poisoned patients without gastric emptying. Annals of Emergency Medicine. 1990;14:562-567.
9 Merigian KS, Woodard M, Hedges JR, et al. Prospective evaluation of gastric emptying in the self-poisoned patient. American Journal of Emergency Medicine. 1990;8:479-483.
10 Pond SM, Lewis-Driver DJ, Williams G, et al. Gastric emptying in acute overdose: a prospective randomised controlled trial. Medical Journal of Australia. 1995;163:345-349.
11 Chyka PA, Seger D. Position statement: single-dose activated charcoal. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):721-741.
12 Barceloux D, McGuigan M, Hartigan-Go K. Position statement: cathartics. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):743-752.
13 Kirshenbaum LA, Mathew SC, Sitar DS, et al. Whole-bowel irrigation versus activated charcoal in sorbitol for the ingestion of modified-release pharmaceuticals. Clinical Pharmacology Therapy. 1989;46:264-271.
14 Smith SW, Ling LJ, Halstenson CE. Whole-bowel irrigation as a treatment for acute lithium overdose. Annals of Emergency Medicine. 1991;20:536-539.
15 Tenenbein M. Position statement: whole bowel irrigation. American Academy of Clinical Toxicology; European Association of Poisons Centres and Clinical Toxicologists. Journal of Toxicology – Clinical Toxicology. 1997;35(7):753-756.
16 Pond SM. Role of repeated oral doses of activated charcoal in clinical toxicology. Medical Toxicology. 1986;1:3-11.
17 Chyka PA. Multiple-dose activated charcoal and enhancement of systemic drug clearance: summary of studies in animals and human volunteers. Clinical Toxicology. 1995;33:399-405.
18 Winchester JF. Active methods for detoxification. In Haddad LM, Shannon MW, Winchester JF, editors: Clinical management of poisoning and drug overdose, 3rd edn, Philadelphia: WB Saunders, 1998.
19 Whyte IM, Dawson AH, Buckley NA, et al. A model for the management of self-poisoning. Medical Journal of Australia. 1997;167:142-146.
20 Lee V, Kerr JF, Braitberg G, et al. Impact of a toxicology service on a metropolitan teaching hospital. Emergency Medicine. 2001;13:37-42.
29.2 Cardiovascular drugs
Pharmacokinetics
Standard CCB preparations are rapidly absorbed from the gastrointestinal tract, with onset of action occurring within 30 min.1 Pharmacokinetic parameters are shown in Table 29.2.1. Verapamil and diltiazem undergo significant first-pass hepatic clearance. Verapamil is metabolized to norverapamil, which possesses 15–20% of verapamil’s pharmacological activity and is renally excreted. Diltiazem is metabolized to deacetyldiltiazem, which has half the potency of the parent compound and undergoes biliary excretion. The elimination half-lives of all CCBs may be prolonged following massive overdose. Amlodipine has a longer plasma half-life (30–50 h) than other CCBs.
Absorption of β-blockers is rapid, with peak clinical effects occurring within 1–4 h. Pharmacokinetic parameters of the principal β-blockers are detailed in Table 29.2.2. Agents with high lipid solubility, such as propranolol, penetrate the blood–brain barrier better than the water-soluble agents, and hence cause greater central nervous system (CNS) toxicity.
Pathophysiology
CCBs antagonize the entry of extracellular calcium into cardiac and smooth muscle, but not skeletal muscle. Upon entry into cells, calcium participates in mechanical, electrical and biochemical reactions. It is involved in excitation–contraction of cardiac and smooth muscles, as well as phase 0 depolarization in the sinus and atrioventricular (AV) nodes by calcium influx through channels.2 CCBs affect myocardial contractility and slow conduction through the sinus and AV nodes. Contraction of smooth muscle is mediated by calcium influx, which is inhibited by CCBs. This results in vasodilatation and secondary reflex tachycardia from an increase in sympathetic activity.
The different classes of CCB have somewhat different pharmacological and toxic effects, as a consequence of their different binding characteristics to the dihydropyridine (DHP) receptors. Verapamil, a phenylalkylamine, produces more profound cardiac conduction defects and equal reductions in systemic vascular resistance when compared with other CCBs on a mg/kg basis.1 Verapamil is more likely to produce symptomatic decreases in blood pressure, heart rate and cardiac output than diltiazem, a benzothiazepine. The DHPs, which include amlodipine, felodipine, lercanidipine and nifedipine preferentially bind to vascular smooth muscle and predominantly decrease systemic and coronary vascular resistance. With the exception of felodipine, they also produce a reflex tachycardia by the unloading of baroreceptors.
The different β-blockers have slightly differing pharmacological properties, including selectivity for β adrenoreceptors, intrinsic sympathomimetic activity and membrane-stabilizing activity. The relative affinity for β adrenoreceptors may influence expression of toxicity. Atenolol, esmolol and metoprolol are β1-selective agents, and therapeutic use of these drugs is less likely to produce the peripheral vasoconstriction, bronchospasm and disturbances in glucose homoeostasis that result from β2 inhibition. However, pharmacological specificity decreases with increasing dose.3 Several β-blockers have partial agonist activity such that, although they block the β receptor to catecholamines, they also weakly stimulate the receptor. This partial agonist activity may have a protective effect in overdose.
Clinical features
Calcium channel blockers
The severity of toxicity is determined by a number of factors, including the amount and characteristics of the drug ingested, the underlying health of the patient, co-ingestants and delay until treatment. The majority of serious cases result from the ingestion of verapamil or diltiazem, the most toxic of the CCBs. Elderly patients and those with congestive cardiac failure are more prone to develop CCB poisoning. The principal clinical features are shown in Table 29.2.3. Ingestion of toxic amounts of standard preparations typically produces symptoms within 2 h, although maximal toxicity may not occur for up to 6–8 h. The slow-release preparations can produce significant toxicity, with onset of symptoms more than 6 h post ingestion. The major threats to life are myocardial depression and hypotension. Nifedipine produces tachycardia with normal blood pressure during the first 30 min, followed later by hypotension and bradycardia in large ingestions (>10 mg/kg). With verapamil and diltiazem poisoning, nausea, vomiting, hyperglycaemia and metabolic acidosis can develop. All CCBs can cause symptoms of cerebral hypoperfusion, such as syncope, lethargy, lightheadedness, dizziness, altered mental status, seizures and coma.
| Central nervous system |
β-blockers
In one large series of patients with β-blocker overdose, 30–40% of patients remained asymptomatic and only 20% developed severe toxicity.4 Toxicity is more likely to develop after ingestion of propranolol, in patients with pre-existing cardiac disease or where there is co-ingestion of other drugs with effects on the cardiovascular system, especially CCBs and cyclic antidepressants.4,5 If β-blocker toxicity is to develop, it is usually observed within 6 h of ingestion.5,6
Treatment
Oral-activated charcoal should be administered as soon as practicable to all patients presenting within 2–4 h of ingestion, and to all those presenting after ingestion of slow-release preparations. More aggressive decontamination, with whole-bowel irrigation, is indicated following overdose with slow-release CCBs.7
A number of drugs play a role in the management of significant CCB or β-blocker poisoning, although none is a completely effective antidote. Suggested doses are shown in Table 29.2.4.
Table 29.2.4 Useful drugs in the management of CCB and β-blocker toxicity
| CCBs | β-Blockers | |
|---|---|---|
| Calcium | 0.5–1 g (5–10 mLs) calcium chloride or 1–2 g (10–20 mLs) calcium gluconate i.v. over 5–10 minutes. Repeat every 10–15 minutes as required. Further administration guided by serum calcium concentrations. | |
| Catecholamines | Adrenaline (epinephrine) infusion started at 1 μg/kg/min and titrate to maintain organ perfusion. | Isoprenaline or adrenaline (epinephrine) infusion titrated to maintain organ perfusion. |
| Glucagon | A bolus dose of 5–10 mg followed by an infusion of 1–5 mg/h. | A bolus dose of 5–10 mg followed by an infusion of 1–5 mg/h. |
| Hyperinsulinaemia euglycaemia | Actrapid 1 U/kg i.v. bolus followed by 0.5–1 U/kg/hr infusion. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia. | Actrapid 1 U/kg i.v. bolus followed by 0.5–1 U/kg/hr infusion. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia. |
Calcium, an inotropic agent, is the initial drug of choice for CCB toxicity. Administration must be closely monitored, with ionized calcium measured 30 min after commencing the infusion, and then second-hourly.8 Catecholamines are useful in attempting to restore adequate tissue perfusion.
Glucagon, a polypeptide hormone of pancreatic origin, enhances myocardial performance by increasing intracellular cAMP concentrations. This increase in cAMP triggers the release of cAMP-dependent protein kinase, which activates the calcium channels, causing an increase in heart rate and myocardial contractility. It works independently from that of the β-adrenoreceptor stimulation of the heart. Use of glucagon is supported only by case reports and some animal studies. There are no clinical trials supporting its efficacy in either calcium channel or beta-blocker poisoning and its role in management of these poisoning is questioned.9 It is frequently difficult to source adequate stocks of glucagon for use as an inotropic agent.
Hyperinsulinaemic euglycaemia therapy (HIET) is increasingly advocated as therapy for hypotension unresponsive to fluids, calcium salts and inotropes. This therapy is supported by animal work10,11 and promising initial human case reports12 but again clinical trials are lacking. Insulin administration switches cardiac cell metabolism from fatty acids to carbohydrates. It restores calcium fluxes and improves myocardial contractility. The recommended initial dose of actrapid is 1 U/kg i.v. followed by an infusion of 0.5–1 U/kg/h. This should be accompanied by an initial bolus dose of 50 mL 50% dextrose followed by an infusion to maintain euglycaemia.13
Severe propranolol toxicity is usually due to sodium channel blockade and treatment as for tricyclic antidepressant poisoning, including intubation, ventilation and sodium bicarbonate, is appropriate.
There are no clinically effective methods of enhancing the elimination of CCBs or β-blockers.
Clinical features
Two distinct clinical presentations of digoxin toxicity are observed: acute and chronic. Both are characterized by cardiac arrhythmias, and virtually all types of arrhythmia have been reported in the context of digoxin toxicity.14
Clinical investigation
In acute poisoning the serum potassium rises as Na-K ATPase function is progressively impaired. Hyperkalaemia denotes significant acute digoxin toxicity. Prior to the availability of a specific antidote for digoxin poisoning, a serum potassium concentration >5.5 mEq/L was associated with a high probability of lethal outcome.16 Hyperkalaemia is not usually observed in chronic digoxin toxicity. In fact, these patients are frequently hypokalaemic and hypomagnesaemic secondary to chronic diuretic use. Both these electrolyte disorders are important as they exacerbate digoxin toxicity.
