CHAPTER 21
Disorders of puberty and sex development
S. Faisal Ahmed; Jane D. McNeilly
CHAPTER OUTLINE
Endocrinology of normal puberty
Physical signs of normal puberty
Terminology of disorders of sex development
General principles of management
General examination of a newborn with suspected DSD
Evaluation of the external genitalia
Evaluation of the internal anatomy
Investigating the newborn with DSD
Investigating the adolescent with DSD
Steroid measurement and its interpretation
The human chorionic gonadotrophin (hCG) stimulation test
The role of the clinical geneticist
Classification of disorders of sex development
Gonadotrophin dependent puberty (central causes)
INTRODUCTION
The clinician is most commonly faced with the investigation and management of abnormal sexual development at two age periods: birth and puberty. Abnormal sexual development at birth is the result of a disorder of sex development (DSD) presenting with genitalia that are either atypical for sex or ambiguous. Disorders of sex development can also present at adolescence as abnormalities of pubertal development. However, in most cases, abnormal pubertal development is due to an alteration in the regulation of puberty in an individual without a DSD. To understand these disorders there is a need to understand the normal processes of sex and pubertal development.
NORMAL SEX DEVELOPMENT
Male or female sex development is programmed at fertilization, depending on whether the zygote is heterogametic (46XY) or homogametic (46XX). A scheme summarizing the embryology and genetic control of sex development in the male and female fetus is shown in Figure 21.1. The indifferent, or primitive, gonad develops from a thickened ridge of coelomic epithelium that arises from the intermediate mesoderm. Cell precursors from this region give rise to the kidneys, adrenals and gonads. The adrenogenital primordium comprises a single population of steroidogenic factor-1 (SF-1) immunoreactive cells. The gonadal ridge becomes populated with primordial germ cells that have migrated from the wall of the yolk sac.
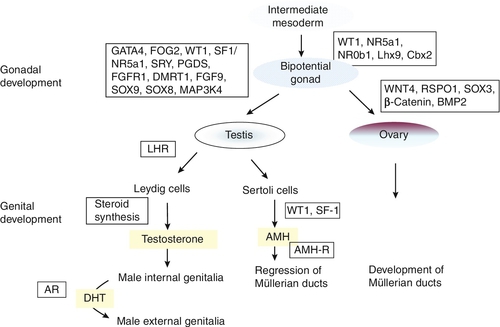
FIGURE 21.1 Embryology and genetic control of fetal sex development. LHR, LH receptor; AR, androgen receptor; DHT, dihydrotestosterone; AMH-R, anti-Müllerian hormone-receptor.
The first histological expression of fetal testicular development is the appearance of seminiferous or sex cords at between six and seven weeks of gestation. Surrounding the sex cords is the interstitial region, which contains the precursors of Leydig cells and also the peritubular myoid cells. Primitive interstitial cells differentiate later into Leydig cells, an event that coincides with the onset of testosterone synthesis by the fetal testis. Steroidogenesis is placental human chorionic gonadotrophin (hCG)-dependent and results in the production of fetal testosterone concentrations within the normal adult male range. The pivotal role for the Y chromosome in male sex differentiation has been recognized for more than 40 years, based on the phenotype of individuals with sex chromosome aneuploidies such as XXY and XO. The study of sex-reversed XX males and XY females led to the identification of the SRY gene (sex-determining region Y) on the short arm of the Y chromosome. That this is the prime testis-determining gene is illustrated by the introduction of the Sry homologue into a transgenic XX mouse and a resulting male phenotype. The majority of XX males are positive for the SRY gene, as a result of terminal exchange between the X and Y chromosomes during paternal meiosis. In the case of XY females, mutations have been identified that disrupt the function of the SRY protein, which acts as a transcription factor. However, these are found in only 15–20% of patients, indicating there are other genes also involved in testis determination. SRY acts to up-regulate the transcription of the related gene, Sox9 in the Sertoli cell lineage. High concentrations of SOX9 protein are maintained in these cells through a positive interaction with the secreted molecules fibroblast growth factor 9 (FGF9) and prostaglandin D2 (PGD2). These molecules are important in securing two other necessary steps in testis development: the recruitment of other somatic cells to differentiate into the Sertoli cell lineage and, in the case of FGF9, spread of the initial central testis-determining signal to the anterior and posterior poles of the gonad. Not only does SOX9 promote testis development, but it also antagonizes the Wnt signalling pathway and FOXL2, both of which are crucial for ovarian development and which can, themselves, antagonize SOX9 action.
With the committed development of the gonad as a testis, two trophic factors produced by the testis control subsequent differentiation of internal and external genitalia in the male. Anti-Müllerian hormone (AMH) is produced by the Sertoli cells and signals through two trans-membrane receptors expressed in the mesenchyme that gives rise to the Müllerian ducts. Anti-Müllerian hormone acts ipsilaterally to cause regression of the Müllerian ducts, the anlage (rudimentary precursor) of the uterus, fallopian tubes and upper two-thirds of the vagina. Circulating concentrations of AMH in males are high in early infancy and fall gradually throughout childhood, until they become undetectable after puberty; its measurement is useful for detecting testicular tissue, especially in early childhood. Testosterone is the other key trophic factor produced by the early developing testis. There is indirect evidence, based on luteinizing hormone (LH) receptor mouse knockout models and human LH β-chain mutations, that production is initially autonomous, but, thereafter, is under the control of hCG and then fetal pituitary gonadotrophin secretion. High local concentrations of testosterone stabilize Wolffian duct development in the male to differentiate into the vas deferens, epididymis and seminal vesicles. The external genitalia in both sexes develop from a common anlage, comprising the urogenital sinus, genital tubercle and swellings and urethral folds. In the male, androgens, again, play the key role in differentiation of these primordial structures, to develop as the penis, scrotum and opening of the urethra on the glans. Dihydrotestosterone (DHT) is necessary to provide an amplification of the androgenic effect. The evidence for DHT dependency is illustrated by the predominantly female external genital phenotype in 5α-reductase deficiency.
In the absence of the testis-determining factors, testosterone and AMH, and in the presence of the trophic proteins necessary for ovarian development, the gonad associated with the 46XX karyotype develops as an ovary and the Müllerian ducts differentiate normally into female internal genital ducts. The external genital anlage, in the absence of androgens, remains underdeveloped with respect to growth of the genital tubercle and the absence of midline fusion of the labioscrotal folds and swellings. The development of the lower part of the vagina from canalization of the vaginal plate is incompletely understood, but the process is probably influenced by oestrogens. Virilization of the female external genitalia from an extraneous source of androgens leaves the development of the female internal genitalia intact.
NORMAL PUBERTAL DEVELOPMENT
Endocrinology of normal puberty
The precise trigger that initiates the onset of puberty in the human remains an enigma. There is activation of pituitary gonadotrophin secretion both in early fetal life and, again, after birth for several months. During this latter period, plasma concentrations of gonadotrophins and sex steroids may reach values normally observed at puberty. Throughout later infancy and childhood, gonadotrophin concentrations remain low, although evidence of pulsatile LH secretion using more sensitive immunoassays can be detected in some children. Consequently, puberty represents the reactivation of gonadotrophin secretion that has been restrained during childhood.
The initial endocrine event at puberty is an increase in nocturnal pulsatile LH secretion in response to gonadotrophin releasing hormone (GnRH) released into the pituitary portal system. The neurons that release GnRH migrate to the hypothalamus, from the medial olfactory placode, during fetal development. A failure of such migration is considered to be the cause of some inherited forms of hypogonadotrophic hypogonadism that are associated with anosmia (Kallman syndrome). A GnRH pulse generator controls the onset of puberty by causing a progressive increase in amplitude and frequency of LH pulses, which occurs about one year before the onset of physical signs of puberty. The neuroendocrine mechanisms that cause activation of the GnRH generator at the appropriate age are extremely complex, but appear to involve excitatory amino acids, catecholamines, neuropeptide Y, leptin and acetylcholine, as well as external influences such as nutritional intake. Furthermore, an additional factor discovered as a result of studies in patients with hypogonadotrophic hypogonadism, is the kisspeptin/GPR54 system, which appears to be a gatekeeper of reproductive development. GPR54 is a G-protein-coupled receptor whose endogenous ligand is a 54-amino acid peptide, cleaved from the parent 145-amino acid peptide encoded by the KISS-1 gene. GPR54-deficient mice are infertile and have low circulating gonadotrophin concentrations. Mutations in the GPR54 gene cause hypogonadotrophic hypogonadism in humans. It appears that GPR54 is one of the key factors in the control of puberty. Administration of kisspeptin stimulates LH and follicle stimulating hormone (FSH) secretion, which can be blocked by GnRH antagonists, indicating that the kisspeptin/GPR54 system effect on the hypothalamic–pituitary–gonadal axis is mediated via GnRH release.
Increased gonadotrophin secretion leads to a gradual increase in the secretion of gonadal steroids, principally testosterone in males and oestradiol in females. Random, daytime sex steroid measurements are seldom predictive of pubertal events in an individual, although early morning testosterone concentration as a reflection of nocturnal secretion is more useful. Inhibin B, a marker of Sertoli cell function, increases early in puberty and reaches adult concentrations by mid-puberty. Spermatogenesis starts between 11 and 15 years of age and sperm can be detected in early morning urine specimens by 13 years of age.
The attainment of reproductive capacity in the female is dependent on cyclical gonadotrophin secretion to ensure ovulation. Follicle stimulating hormone-induced oestrogen secretion, by developing ovarian follicles, leads to a positive feedback on LH secretion and a midcycle surge that induces rupture of the mature follicle. After ovulation, the luteinized follicle secretes progesterone while oestradiol concentrations fall. Inhibin B secretion predominates during the follicular phase, while inhibin A secretion is dominant during the luteal phase. All these events are coordinated by pulsatile release of GnRH secretion and modulation of gonadotrophin and sex steroid production, by both negative and positive feedback loops. It is not surprising that there is generally a 1–2-year interval after menarche before the majority of girls are ovulating regularly. Ultrasonography can be used to assess the development of reproductive function in girls. Enlargement of the uterus and endometrial thickening are evident after birth because of the effect of maternally derived oestrogens. Multiple ovarian follicular cysts are often seen on ultrasound at this time. The effect of increasing secretion of oestradiol during puberty can be observed by appropriate morphological changes in the appearance of the uterus. The prepubertal uterus starts to increase in size from seven years of age onwards.
The onset of increased production of dehydroepiandrosterone (DHEA) and its sulphate (DHEAS) by the zona reticularis of the adrenals, between six and eight years of age, defines the phenomenon of adrenarche. Concentrations of DHEAS thereafter increase throughout puberty into early adult life in micromolar amounts and then start to decline gradually in older age (adrenopause). There is no concomitant increase in adrenal glucocorticoid and mineralocorticoid secretion, so ACTH is not the primary trophic factor controlling DHEA production. Intra-adrenal modulation of steroidogenesis, by post-translational regulation of the 17-hydroxylase enzyme (by phosphorylation on serine/threonine residues, and electron transfer by the P450-oxidoreductase enzyme), is the key factor in the production of C19 steroids such as DHEA. There is also a relative underexpression of the enzyme 3β-hydroxysteroid dehydrogenase enzyme in the zona reticularis that contributes to a preponderance of Δ5 steroids. Extra-adrenal factors postulated to play a role in adrenarche include prolactin, oestrogens, growth factors and cytokines. There is an association between leptin concentration and the timing of adrenarche in obese children.
Physical signs of normal puberty
Puberty is the transitional period between childhood and adulthood that spans adolescence and leads to the acquisition of reproductive capacity. The time span for the physical changes to take place is generally 4–5 years, but individual variations can result in a time interval of 2–6 years. This is referred to as the tempo of puberty. The age of onset of puberty also varies considerably, with epidemiological evidence suggesting that puberty may now be starting earlier.
The first sign of puberty in girls is breast development, starting, on average, at 11 years with an age range of 8–13 years (see Fig. 21.2A). This is termed ‘thelarche’ and starts as a small mound of tissue beneath the nipple manifest as a breast ‘bud’, which is usually distinguishable, on palpation, by its firmness compared with the softer and more diffuse texture of subcutaneous fat. Further development of the nipple, areola and underlying breast tissue takes place over the ensuing four years or so (Tanner stages B2–B5). Epidemiological studies conducted in the USA indicate that 25% of girls may have already started thelarche by eight years of age. However, it is unclear whether misinterpretation of adipose tissue as breast development may have skewed the data.
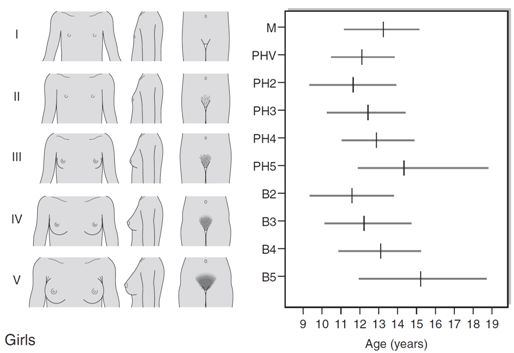
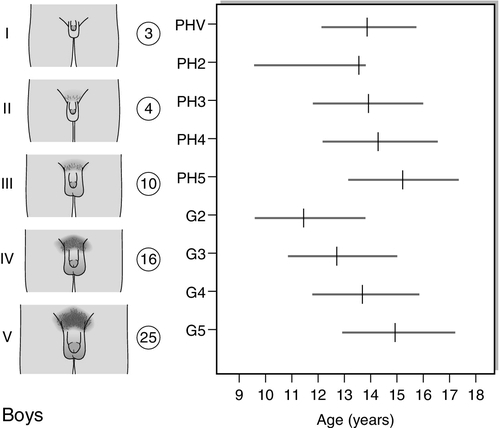
FIGURE 21.2 Tanner staging of physical development, based on external primary and secondary sexual characteristics in children, adolescence and adults. M, menarche; Circuled figures, testicular volume (mL); PHV, peak height velocity; PH, B, G, see text.
Coincident with breast development is the onset of growth of pubic hair which may precede thelarche in 10% of girls. Hair growth usually starts on the labia before spreading over the mons pubis and is quantified as Tanner stages PH2–5. Sometimes there may be further growth on the upper medial aspects of the thighs and along the linea alba (PH6). Axillary hair starts to appear at about 12.5 years of age and takes a further 18 months to reach adult distribution.
Menarche, the onset of menses, is a relatively late event in the pubertal process and occurs at around 12.5–13 years of age. This coincides with breast stage B4. A trend in the lowering of the age of menarche has occurred in the past, but the reduction has only been by a few months during the past 30 years. There are racial differences as well as geographical variations. The age of menarche tends to be slightly higher in Northern than Southern Europe.
Increased growth velocity is a measurable, indirect sign of puberty. In girls, peak height velocity (defined as the maximum rate of growth achieved during puberty, which is usually ~ 10 cm/year) is achieved relatively early in puberty (during breast stages B2–3) and before the onset of menarche. By the time of menarche, the majority of adult height has been achieved. Changes in body composition also occur, particularly with respect to fat distribution.
The first sign of puberty in boys is an increase in testis size (see Fig. 21.2B). Testicular size can be assessed clinically by the use of a series of standard ovoids (the Prader orchidometer). Testicular volume remains between 2–3 mL throughout infancy and childhood, an increase to 4 mL heralds the onset of puberty that occurs, on average, at 11.5 years with an age range of 9–14 years. Progressive enlargement of the testes takes place over three years with testicular volumes reaching up to 25 mL in adult life. Leydig cells constitute only a small part of testicular volume, the majority being the result of an increase in Sertoli cell and seminiferous tubule number and size.
Pubic hair growth, which may start as a few scrotal hairs, follows closely on testicular development and is quantified as Tanner stages PH2–5. Spread of hair up the abdominal wall is characteristic in many men and is classified as stage PH6. Growth of the penis, first in length and then in breadth, occurs concomitantly and is rated G1–G5. Growth of axillary and facial hair, are both later events in puberty, ranging in age from 14 to 16 years. ‘Breaking’ of the voice is due to enlargement of the larynx and elongation of the vocal cords and does not occur until stages G3–G4.
Peak height velocity in boys corresponds with stages G4 and PH4 and testicular volumes of 12–15 mL. This occurs, on average, at a chronological age of 14 years, as opposed to 12 years in girls. The adult male is, on average, taller than the adult female by 13 cm. This is partly due to the fact that boys start the growth spurt later and are, therefore, taller at its onset. In addition, the magnitude of growth achieved during the growth spurt is greater in boys (28 cm in males and 20 cm in females). Body composition alters in favour of increased muscle bulk and relatively less subcutaneous fat.
DISORDERS OF SEX DEVELOPMENT
Terminology of disorders of sex development
It is important to define terms used in relation to the investigation and management of disorders of sex development that present at birth or in childhood. Sex assignment (often used interchangeably with gender assignment), is the sexing of an infant at birth as being male or female. This is straightforward in the vast majority; indeed, sex assignment is increasingly an activity that occurs before birth. Gender identity refers to how individuals perceive themselves as being male or female, while gender role describes characteristics that are sexually dimorphic within a normal population. In childhood, for example, male-typical behaviour is attributed to toy preferences being vehicles and soldiers as opposed to playing with dolls, generally preferred by girls. This rather simplistic illustration of sexual dimorphism has some physiological underpinning from observations of girls’ play behaviour following exposure to androgens in utero. Thus, gender role is more male-typical in girls with congenital adrenal hyperplasia. There is less convincing evidence that prenatal hormones affect gender identity. Sexual orientation refers to the subject of erotic arousal, which may be heterosexual, homosexual or bisexual. Gender dysphoria or gender identity disorder are terms that describe gender dissatisfaction. The phenomenon appears to be more common in individuals with disorders of sex development than in the general population, but it is difficult to identify predisposing factors related to the specific disorder.
The use of terminology that is clear and easy to use and understand by all health professionals, patients and their families is fundamental to the understanding, investigation and management of affected newborns and children. In addition, terminology should respect the individual and avoid terms that might cause offence. The term ‘intersex’ has had variable connotations even among professionals; some employed it as a term that covered all affected newborns while at the other end of the spectrum, some believed that the term should only apply to those where there is complete mismatch between chromosomal and anatomic sex. The consensus reached in Chicago in 2005 on the management of these patients, stressed the importance of terminology and recommended a substitution for the term ‘intersex’ for ‘disorder of sex development (DSD)’, which is defined as any congenital condition in which development of chromosomal, gonadal or anatomic sex is atypical. It also recommended the abandonment of terms such as ‘pseudohermaphroditism’ and ‘true hermaphroditism’. While this new nomenclature (Table 21.1) is easier to use and understand, it will nevertheless evolve over time as our understanding of long-term outcome and molecular aetiology improves. Given that genital anomalies may occur as commonly as 1 in 300 births and may not always be associated with a functional abnormality, some have advocated the use of ‘differences’ in preference to the term ‘disorder’. The strength of the abbreviation ‘DSD’ is that it can be used to cover both differences and disorders of sex development. However, the likelihood of this difference existing as a disorder will depend on the functional implications of the condition, which may be heavily influenced by the social and cultural framework within which the child exists.
TABLE 21.1
Old and new terminologies for abnormal sex development
| Previous | Proposed |
| Intersex | Disorders of sex development (DSD) |
| Male pseudohermaphrodite. Undervirilization of an XY male, undermasculinization of an XY male | 46XY DSD |
| Female pseudohermaphrodite. Overvirilization of an XX female, masculinization of an XX female | 46XX DSD |
| True hermaphrodite | Ovotesticular DSD |
| XX male or XX sex reversal | 46XX testicular DSD |
| XY sex reversal | 46XY complete gonadal dysgenesis |
General principles of management
The management of a child with an abnormality of genital development can often be difficult, particularly in patients for whom the sex of rearing is uncertain. The initial contact with the parents of a child with a DSD is important as first impressions from these encounters often persist. A key point to emphasize is that a child with a DSD has the potential to become a well-adjusted, functional member of society. Establishing a dialogue and building rapport with the affected child and the parents, evaluating the child and then developing a logical, as well as pragmatic, plan for investigations are central to the initial approach and ongoing management. It is paramount that any child or adolescent with a suspected DSD is assessed by an expert with adequate knowledge about the range of variation in the physical appearance of genitalia. If there is any doubt, the patient should be discussed with the regional team. For most patients, particularly in the case of the newborn, the paediatric endocrinologist within the regional DSD team acts as the first point of contact. The underlying pathophysiology of DSD and the strengths and weaknesses of the tests that can be performed, should be discussed with the parents and child, as appropriate, and tests undertaken in a timely fashion. With babies in whom there is true genital ambiguity, it should be explained to the parents that the best course of action may not initially be clear, but the healthcare team will work with the family to reach the best possible set of decisions in the circumstances. Finally, in the field of rare conditions, it is imperative that the clinician shares the experience with others through national and international clinical and research collaboration.
General examination of a newborn with suspected DSD
The physical examination should determine whether there are any dysmorphic features and the general health of the baby. Affected infants, particularly those who have XY DSD, are more likely to be small for gestational age and may display other developmental abnormalities. In addition, the affected infant should be examined for midline defects which may point towards an abnormality of the hypothalamo-pituitary axis. The state of hydration and blood pressure should be assessed, as adrenal steroid biosynthetic defects can be associated with a variable extent of salt loss, masculinization and hypertension. In congenital adrenal hyperplasia (CAH), cardiovascular collapse with salt loss and hyperkalaemia does not usually occur until the second week of life (with salt loss usually evident from day four) and so will not be apparent at birth in the well neonate, but should be anticipated if the diagnosis is suspected. Jaundice (both conjugated and unconjugated) may be observed in babies with hypopituitarism or cortisol deficiency. Urine should be checked for protein as a screen for any associated renal anomaly (e.g. Denys–Drash or Frasier syndromes) and a pre-feed blood glucose concentration should be measured to exclude hypoglycaemia (suggestive of hypopituitarism, or occasionally CAH, e.g. 3β-hydroxysteroid dehydrogenase deficiency). Renal tract anomalies, such as ureteropelvic junction obstruction, vesicoureteric reflux, pelvic or horse-shoe kidney, crossed renal ectopia and renal agenesis, are reported to be more common in children with DSD.
Evaluation of the external genitalia
If the appearance of the external genitalia is sufficiently ambiguous to render sex assignment impossible, or the phenotype is not consistent with prenatal genetic tests, then investigations are clearly required. However, the ability to evaluate external genitalia fully may depend on the expertise of the observer and, before presentation to a specialist, the label of ambiguous genitalia has often already been assigned to newborns where the most appropriate sex of rearing was not clear to those present at the child’s birth. The birth prevalence of genital anomalies may be as high as 1 in 300 births but the birth prevalence of complex anomalies that may lead to true genital ambiguity on expert examination may be as low as 1 in 5000 births.
Apart from those whose genitalia are truly ambiguous, infants can often be divided into those who overall seem to have largely male or female genitalia but with some unusual features. However, it is very important to bear in mind that a 46XX newborn infant with congenital adrenal hyperplasia can either present as a girl with clitoromegaly or as a boy with bilateral undescended testes. When evaluating these infants, the clinical features of the external genitalia that require examination include the presence of gonads in the labioscrotal folds, the fusion of the labioscrotal folds, the size of the phallus and the site of the urinary meatus on the phallus, although the real site of the urinary meatus may, sometimes, only become clear on surgical exploration. These external features can be individually scored to provide an aggregate score, the external masculinization score (EMS) (see Fig. 21.3A), or they can be graded according to their overall description as classically described by Prader staging (see Fig. 21.3B).
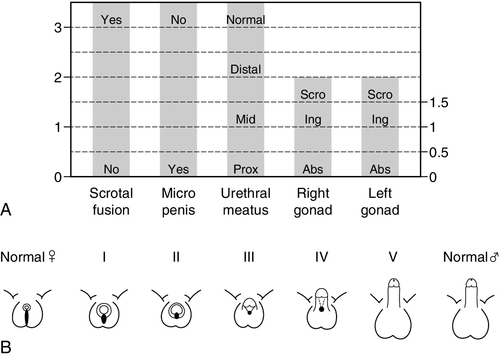
FIGURE 21.3 (A) External Masculinization Score. Each individual feature of the genitalia (phallus size, labioscrotal fusion, site of the gonads and location of urethral meatus) can be individually scored to provide a score out of 12. Microphallus refers to a phallus smaller than normal for age. Scro, scrotal; Ing, inguinal; Abs, abdominal or absent on examination. (B) Differential virilization of the external genitalia using the staging system of Prader from normal female (left) to normal male (right).
Infants with suspected DSD who require further clinical evaluation and need to be considered for investigation by a specialist, should include those with isolated perineal hypospadias, isolated micropenis, isolated clitoromegaly, any form of familial hypospadias and those who have a combination of genital anomalies with an EMS of less than 11. This will avoid unnecessary detailed investigations of boys with isolated glandular or mid-shaft hypospadias and boys with unilateral inguinal testis. The coexistence of a systemic metabolic disorder, associated malformations or dysmorphic features would lower the threshold for investigation as would a family history of consanguinity, stillbirths, multiple miscarriages, fertility problems, genital abnormalities, hernias, delayed puberty, genital surgery, unexplained deaths and the need for steroid replacement. In addition, maternal health and drug exposure during pregnancy, and the pregnancy history itself, may hold key information.
Evaluation of the internal anatomy
Examination and assessment by a paediatric surgeon with experience of DSD is critically important for defining the internal and the external anatomy in the affected patient. Combining examination with endoscopic visualization and radiological assessment can provide information on the location and state of the gonads, the urogenital sinus and Müllerian structures. Ultrasonography is the first-line imaging modality but the reliability is child and operator dependent. In the neonate the uterus, ovaries and adrenals should be identifiable. In the adolescent, it is sometimes difficult to confirm the presence of a pre-pubertal uterus by ultrasonography and there may be a place for repeat imaging after a six-month course of oestrogen. Magnetic resonance imaging (MRI) may be reserved for patients where ultrasonography has failed to delineate the relationship of the Müllerian structures and where there are abnormalities of the urinary tract. In adolescents, MRI can delineate structural anomalies such as hydrometrocolpos or hydronephrosis and identify secretory tumours. Nowadays, the ‘genitogram’ has been superseded by endoscopic examination of the genital tract (genitoscopy), which provides a more detailed and thorough assessment. Laparoscopy is a very effective method of visualizing the internal sex organs and facilitates direct inspection, biopsy or excision of intra-abdominal gonads. In 46XY DSD, laparoscopy is clearly indicated in all children with impalpable testes as the gonads need to be identified and brought down to the scrotum if possible.
Investigating the newborn with DSD
In all infants with ambiguous genitalia and/or bilateral impalpable gonads, a first tier of investigations should be undertaken to define the sex chromosomes, delineate the internal genitalia by pelvic ultrasound and to exclude life-threatening congenital adrenal hyperplasia (CAH) – the commonest cause of ambiguous genitalia of the newborn. This first tier should, therefore, also include plasma glucose, 17OH-progesterone (17OHP), and measurement of sodium and potassium. 17OH-Progesterone measurement is usually unreliable before the age of 36 h and in the salt losing form of CAH, electrolytes usually do not become abnormal before day four of life. The results of FISH (fluorescence in situ hybridization) analysis using Y- and X-specific probes should be available within one working day and the 17OHP results should be available within a maximum of two working days in all specialist DSD centres. In situations where the level of suspicion of CAH is very high and the infant needs immediate steroid replacement therapy, samples should be collected and stored before starting therapy. These should be of a sufficient volume to assess 17OHP, testosterone, androstenedione and, possibly, renin, in that order of priority. At least one spot or 24-h urine sample for a urine steroid profile should be collected before starting therapy. The results of these initial investigations will often dictate the second tier of investigations.
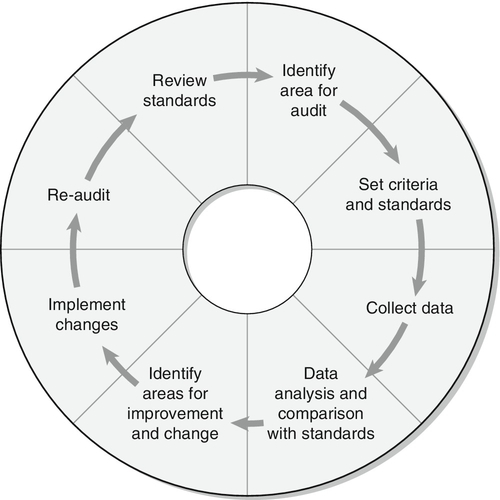
In an infant with impalpable gonads, a karyotype of 46XX, a significantly elevated plasma 17OHP concentration and the presence of a uterus, CAH due to 21-hydroxylase deficiency is the most likely diagnosis. A urine steroid profile can confirm this diagnosis and identify other rarer forms of CAH, which may also be associated with a raised 17OHP concentration in the newborn. In infants with sex chromosomes other than 46XX, a second tier of investigations is necessary to determine the presence of testes and the adequacy of androgen production and action. These tests include measurement of AMH, an hCG stimulation test, further imaging and laparoscopy. Confirmation of a specific diagnosis will often require biochemical identification of a defect in the androgen biosynthesis pathway and detailed genetic analysis.
Investigating the adolescent with DSD
Adolescents typically present with a suspected DSD in three ways – as a girl with primary amenorrhea (with or without breast development), as a girl who virilizes at puberty, or, as a boy with pubertal delay (see Fig. 21.4). The potential psychological impact of a thorough physical examination and medical photography on an adolescent should be carefully considered and, in some circumstances may be appropriate only under an anaesthetic. In girls with primary amenorrhoea, investigations should be considered at the age of 14 years if there is no pubertal development and at 16 years if other aspects of puberty, particularly breast development, have progressed normally. History should include a family history and an assessment of coexisting chronic disease, exercise and weight changes. Physical examination should include measurement of blood pressure, height and weight and assessment of secondary sexual characteristics including clitoral enlargement. Vaginal examination to assess vaginal length is rarely indicated if imaging is informative and, if carried out, should be clearly explained and performed by a gynaecologist. An initial investigation screen should comprise measurements of LH, FSH, prolactin, thyroid stimulating hormone (TSH), free thyroxine (FT4), sex hormone binding globulin (SHBG), androstenedione, oestradiol and testosterone, and transabdominal pelvic ultrasound, performed by a sonographer with experience of adolescent appearances. Raised gonadotrophins, or an absent uterus in the presence of normal breast development, are indications for karyotyping.
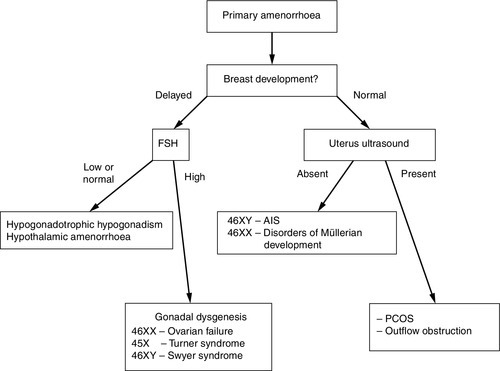
FIGURE 21.4 Approach to investigating adolescent girls with primary amenorrhoea. AIS, androgen insensitivity syndrome; PCOS, polycystic ovary syndrome.
The appearance of clitoromegaly and hirsutism at puberty, in the presence of primary amenorrhoea, is a classical presentation of two 46XY DSDs: 17β-hydroxysteroid dehydrogenase type 3 deficiency and 5α-reductase type 2 deficiency. It is less typical of partial androgen insensitivity syndrome (PAIS), which is usually associated with ambiguous genitalia at birth. Müllerian structures will not be detectable in any of these conditions. In partial gonadal dysgenesis and ovotesticular DSD, mild clitoromegaly that may have been present at birth but may have been overlooked, becomes a more prominent feature at adolescence. The differential diagnosis would also include CAH and androgen-secreting tumours of the ovary or adrenal gland, but in these conditions Müllerian structures are present. Investigations include measurement of LH, FSH, DHEAS, SHBG, androstenedione, testosterone, dihydrotestosterone (DHT) and 17OHP. A 24 h urine collection for urinary steroid profiling will confirm 5α-reductase type 2 deficiency, CAH or adrenocortical tumour. A pelvic ultrasound will assess the presence of a uterus and determine the need for a karyotype.
Although the commonest cause of delayed puberty is constitutional delay, all boys with delayed puberty who are over the age of 14 years should be assessed. Overweight boys need careful examination so that a buried penis is not mistaken for micropenis. Rarely, PAIS, a disorder of testosterone biosynthesis or mild forms of testicular dysgenesis can present in this age group; often there is a history of hypospadias repair or orchidopexy. Investigations include a bone age and measurements of LH, FSH, testosterone and prolactin. For those with raised gonadotrophins, karyotype should be performed to exclude disorders such as Klinefelter syndrome (47XXY and variants) or 45XO/46XY mosaicism.
Steroid measurement and its interpretation
Steroid hormone analysis is a vital component of the biochemical evaluation of the child with DSD but the method of analysis can have a significant impact on the result. Analysis is most often performed by non-extraction (direct) immunoassays on automated platforms and these are subject to concerns of analytical specificity and variability between manufacturers. Liquid chromatography linked with tandem mass spectrometry (LC-MS/MS) allows multiple analyte analysis from a single sample whilst maintaining analytical specificity. Thus, in patients with DSD steroid measurement by either LC-MS/MS or immunoassay after organic solvent extraction is preferred. As these methods tend to be labour intensive, close communication between the clinical and laboratory personnel within the DSD team is vital to ensure timely availability of results.
Urinary steroid profile (USP) analysis by gas chromatography mass spectrometry (GC-MS) provides qualitative and quantitative data on excretion of steroid metabolites (see Fig. 21.5). It is ideal for detecting altered steroid metabolites, especially in patients with CAH, where the activity of a combination of steroidogenic enzymes can produce unusual metabolites which can cross-react in traditional direct serum assays. The diagnosis of rarer forms of CAH such as P450 oxidoreductase deficiency (ORD) is best established using urinary GC-MS analysis as it allows for concurrent determination of all adrenal-derived steroid metabolites. As concentrations of gonadotrophins, androgens and precursors fluctuate markedly over the first few months of life it may be appropriate to consider an early neonatal collection as well as further samples at a later stage. A urine sample can be frozen and stored for many years and may help with a review of the diagnosis at a later stage. Urinary steroid profiling is not appropriate for suspected cases of 5α-reductase type 2 deficiency until after three months of age as diagnostic ratios of 5β to 5α reduced metabolites are not detectable before then. Infants, particularly boys, normally have significant changes in steroid and other endocrine hormone concentrations during the first 100 days of life. In boys, serum testosterone and DHT may initially be high at birth but decline to < 1 nmol/L and undetectable, respectively by around day 30. Concentrations then rise to peak at day 70 before declining to normal prepubertal values. These normal variations may influence the interpretation of sex steroid and gonadotrophin measurements as well as the results of an hCG stimulation test (see p. 422). Furthermore, the actual value for the hormone concentration will vary depending on the assay methodology; it is therefore essential that age and method-related reference ranges are available to facilitate appropriate interpretation of results.
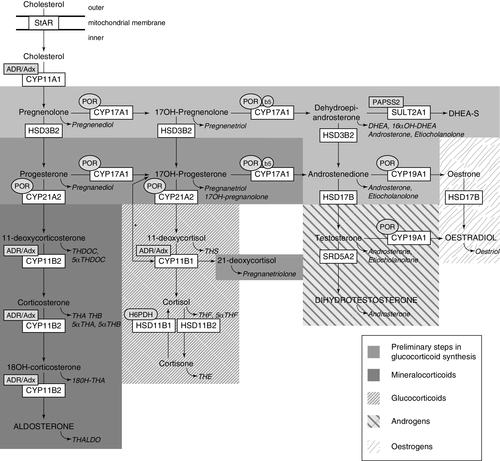
FIGURE 21.5 Synthesis and metabolism of hormonal steroids. This figure illustrates the formation of the major hormone classes from cholesterol. Steroid names in conventional script are steroid hormones and precursors; those in italics are urinary metabolites of the aforementioned. The major transformative enzymes are in rectangular boxes, the cofactor (‘facilitator’) enzymes in ovals. Mitochondrial CYP type I enzymes requiring electron transfer via adrenodoxin reductase (ADR) and adrenodoxin (Adx) CYP11A1, CYP11B1, CYP11B2, are marked with a labelled box ADR/Adx. Microsomal CYP type II enzymes receive electrons from P450 oxidoreductase (POR), CYP17A1, CYP21A2, CYP19A1, are marked by circled POR. The 17,20-lyase reaction catalyzed by CYP17A1 requires in addition to POR also cytochrome b5 indicated by a circled b5. Similarly, hexose-6-phosphate dehydrogenase (H6PDH) is the cofactor-generating enzyme for 11β-HSD1. The asterisk (*) indicates the 11-hydroxylation of 17OHP to 21-deoxycortisol in 21-hydroxylase deficiency. The conversion of androstenedione to testosterone is catalysed by HSD17B3 in the gonad and AKR1C3 (HSD17B5) in the adrenal. StAR, steroidogenic acute regulatory protein; CYP11A1, P450 side-chain cleavage enzyme; HSD3B2, 3β-hydroxysteroid dehydrogenase type 2; CYP17A1, 17α-hydroxylase; CYP21A2, 21-hydroxylase; CYP11B1, 11β-hydroxylase; CYP11B2, aldosterone synthase; HSD17B, 17β-hydroxysteroid dehydrogenase; CYP19A1, P450 aromatase; SRD5A2, 5α-reductase type 2; SULT2A1, sulfotransferase 2A1; PAPPS2, 3′-phosphoadenosine 5′-phosphosulfate synthase 2; TH, tetrahydro. With permission: Krone et al. J Steroid Biochem Mol Biol 2010; 121:496–504.
Anti-Müllerian hormone
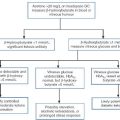
Anti-Müllerian hormone, also known as Müllerian inhibiting substance, is strongly expressed in Sertoli cells from the time of testicular differentiation to puberty and to a much lesser degree in granulosa cells from birth to menopause. Measurement of AMH concentration in the adult female is used to assess ovarian reserve. Published information on circulating AMH concentrations have to be interpreted with caution owing to differences in primary antibody, standard calibration and units used for measurement in individual immunoassays. In boys, AMH is detectable at birth at much higher circulating concentrations than in girls and these concentrations rise during infancy before gradually declining at puberty. Therefore, it may be helpful to repeat AMH measurement later in infancy if a newborn boy is found to have a low concentration. As summarized in Table 21.2, measurement of AMH is a powerful tool to assess Sertoli cell activity in children with suspected DSD and may also have diagnostic utility in conditions associated with androgen deficiency or insensitivity.
TABLE 21.2
Interpretation of serum Anti-Müllerian hormone concentrations in DSD
| Serum AMH | Testicular tissue | Interpretation |
| Undetectable | Absent | 46XX,CAH Complete gonadal dysgenesis PMDS due to AMH gene defect |
| Within female age-related reference range | Usually absent | 46XX, CAH Dysgenetic testes or ovotestes |
| Below male/above female age-related reference range | Present | Dysgenetic testes Ovotestes |
| Within male age-related reference range | Usually normal | Non-specific XY DSD Hypogonadotrophic hypogonadism PMDS due to AMH-R defect 46XX testicular DSD Ovotestes |
| Above male age-related reference range | Present | AIS especially complete androgen insensitivity syndrome 5α-reductase deficiency Testosterone biosynthetic defect Leydig cell hypoplasia |
PMDS, Persistent Müllerian duct syndrome; AIS, androgen insensitivity syndrome; CAH, congenital adrenal hyperplasia.
Insulin-like factor 3
Insulin-like factor 3 (INSL3) is one of the hormones secreted by the Leydig cells in the testis, and plays a pivotal role in testicular descent during fetal development. In males, circulating INSL3 concentrations display a characteristic pattern with a transient perinatal increase, low concentrations during childhood, increasing during puberty with highest concentrations found in adulthood when it is expressed constitutively. Insulin-like factor 3 is increasingly being recognized as a more sensitive marker of current Leydig cell differentiation and function than testosterone and, thus, has potential clinical value when used as an adjunct to standard investigations for DSD and may avoid reliance on hCG stimulation tests to assess testicular function adequately. In females, INSL3 is produced by theca cells and the corpus luteum, therefore concentrations are undetectable until puberty, when active ovarian cycling occurs. Circulating INSL3 concentrations are significantly lower in females than males. Lack of age-appropriate reference values, robust, sensitive commercial immunoassays and larger-scale studies are major barriers to the use of INSL3 in routine practice.
Inhibins
Inhibins, produced by granulosa and theca cells of the ovary and Sertoli cells of the testes, play an important role in the negative feedback control of pituitary gonadotrophin secretion. The two major isoforms present in circulation are inhibin A and B. In human males, inhibin B, a marker of Sertoli cell function, is the only biologically active form present. After an initial rise in concentration shortly after birth, inhibin B remains low until the onset of puberty, when increasing concentrations denote Sertoli cell maturation and accurately reflect spermatogenesis. In females, both inhibin A and B are produced. Inhibin B concentrations show a biphasic pattern with a peak at around three months, a pre-pubertal quiescent phase and a gradual rise several years prior to the onset of puberty, suggesting pre-pubertal increase in follicular activity. Inhibin A becomes detectable only in the latter stages of puberty. During the menstrual cycle, inhibin B predominates during the follicular phase reflecting recruitment of pre-antral follicles, whereas inhibin A is the major isoform produced during the luteal phase, being secreted by dominant follicles and the corpus luteum.
Original radioimmunoassays for inhibins were not able to distinguish between non-biologically active free alpha inhibin and biologically active inhibin A or B. Therefore, it was not until the development of sensitive and specific immunoassays for inhibin A and B that their potential diagnostic utility in various fields of reproductive endocrinology was truly recognized. When used in combination with standard baseline investigations of reproductive function, inhibin B has considerable potential in aiding diagnoses of disorders of pubertal development, (delay and precocity) and premature ovarian and testicular failure.
The human chorionic gonadotrophin (hCG) stimulation test
Stimulation with hCG allows the identification of functioning testicular tissue, as well as biosynthetic defects in testosterone synthesis (see Fig. 21.6). However, it is an invasive test which should only be performed as a second-line investigation after discussion with the paediatric endocrinologist in the regional DSD team. Most protocols for hCG stimulation in the UK use intramuscular hCG 1000–1500 units on three consecutive days. This can be followed by further hCG stimulation with 1500 units on two days a week for the following two weeks. In young infants and older children, three days of hCG stimulation may be sufficient. In the very young infant with an intrinsically active gonadal axis, an hCG stimulation test may not be necessary if serial blood samples show raised serum testosterone concentrations. A testosterone response to hCG may be labelled as normal if absolute testosterone concentrations reach a level that is above the upper limit of the normal prepubertal range, or rise by more than twice the baseline value. As a minimum, other androgens that should be assessed include dihydrotestosterone (DHT) and androstenedione. For these two metabolites, the post-hCG, day four sample is more important than the pre-hCG sample. Following prolonged hCG stimulation there is no additional benefit of analysing a sample for these two metabolites on day 22. However, a day 22 sample collected for testosterone measurement can be stored and used to measure DHT or androstenedione if analysis was not achieved on day four. In the presence of a poor testosterone response following hCG stimulation, assessment of adrenal function by a standard short tetracosactide stimulation test should be considered. There is currently insufficient evidence to recommend that every child with XY DSD should have a tetracosactide stimulation test, but clinicians should be aware of the clear association between some forms of DSD and primary adrenal insufficiency. They should consider thorough assessment of adrenal function in children with diagnoses where an association has already been described and in children with any clinical suspicion of adrenal insufficiency, especially those with low steroid precursors on USP.
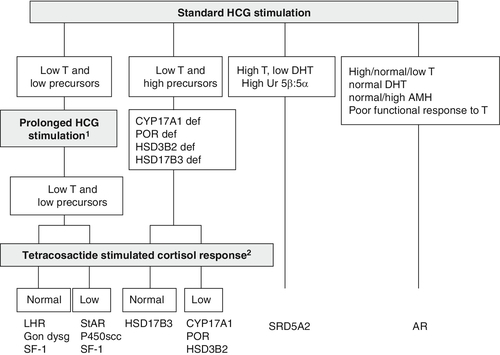
FIGURE 21.6 Interpretation of the results of the human chorionic gonadotrophin (hCG) stimulation test when investigating XY DSD and pointers for consideration of prolonged hCG and adrenocorticotropin hormone stimulation. 1Prolonged hCG stimulation test should be considered in those cases where there is a poor testosterone (T) response to a standard hCG stimulation test. 2A tetracosactide stimulation test should be considered in those patients who show a poor testosterone response to hCG stimulation. 46XY children with lipoid congenital adrenal hyperplasia due to a steroidogenic acute regulatory defect or P450scc deficiency due to a CYP11A1 defect will have female genitalia and present in a salt-losing state in the first days or weeks of life before a tetracosactide test is performed. Gon dysg, gonadal dysgenesis; for other abbreviations, see text and Table 21.5.
The role of the clinical geneticist
Establishing a specific molecular diagnosis is helpful in the clinical management of patients and in offering accurate genetic counselling for the family. However, the number of diagnostic gene tests that are available in clinically accredited DNA laboratories in the UK, or internationally, is limited and testing is costly. As developments in DNA and chromosomal analysis accelerate biomedical research, many techniques such as multiplex ligation-dependent probe amplification (MLPA) and comparative genomic hybridization (CGH) have the potential to become routine in clinical practice. Next generation DNA sequencing platforms will allow whole genome sequencing for rare diseases to become a reality at a realistic price. The clinical geneticist at the specialist DSD centre is correctly placed to judge which technique is appropriate and cost-effective for each clinical situation. DNA can be stored indefinitely with consent, so when further diagnostic testing opportunities arise, aliquots of DNA can be accessed, avoiding repeated venepuncture. If a molecular diagnosis is reached in a research laboratory, then confirmation by a clinically accredited laboratory should be sought where possible, before disclosing results to the family. Close involvement of the clinical genetics service can ensure that the multidisciplinary team covers all aspects of genetic counselling, including provision of information to the family, the mode of inheritance of the disorder and the choices or options available for dealing with this risk. Established links with the clinical genetics service are also essential when considering any prenatal investigations or interventions.
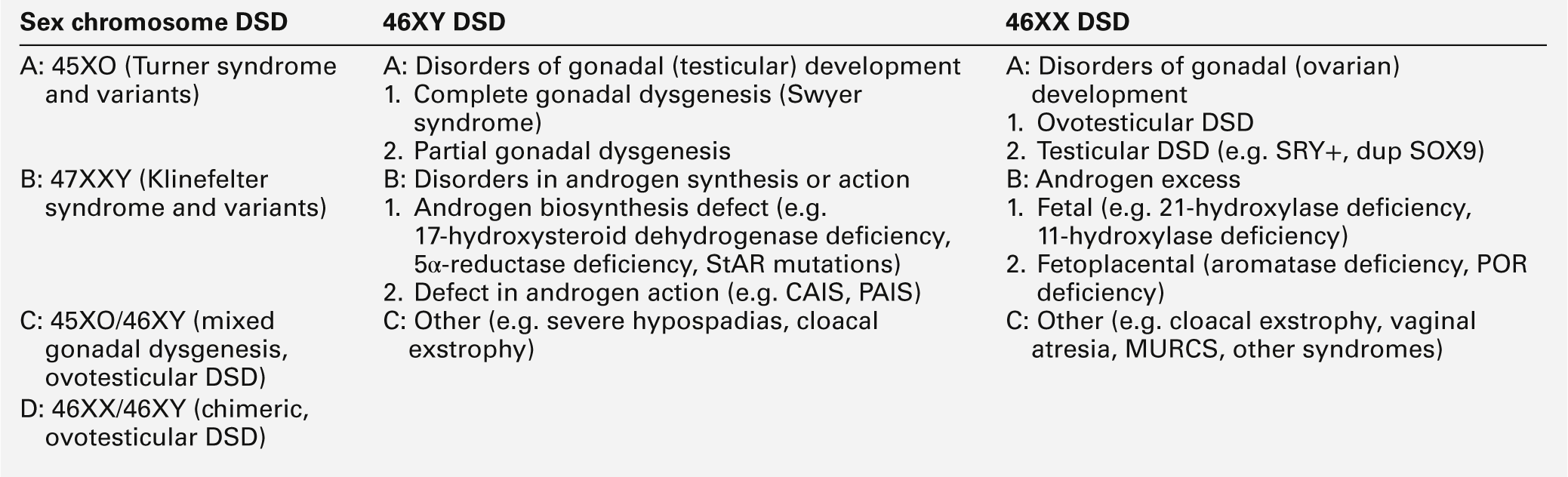
Classification of disorders of sex development
XX DSD
46XX DSD can be classified into disorders of ovarian development, conditions with androgen excess and other syndromes, which are often associated with other developmental abnormalities (Table 21.3).
Disorders of androgen excess
Congenital adrenal hyperplasia is the commonest cause of 46XX DSD with ambiguous genitalia in the neonatal period or early infancy; it is characterized by androgen excess, a variable alteration in glucocorticoid and mineralocorticoid function and a specific profile of steroid hormones. A urinary steroid profile can identify the enzyme defects, including deficiency of 21α-hydroxylase (90–95% of cases), 11β-hydroxylase (4–8% of cases), 3β-hydroxysteroid dehydrogenase type 2 (rare) and P450 oxidoreductase (unknown prevalence). In its most severe form, 21α-hydroxylase deficiency presents as an acute salt-wasting crisis during the second or third week of life. Early biochemical features are hyperkalaemic acidosis, and increased urinary sodium excretion before plasma sodium starts to fall. P450 oxidoreductase deficiency (ORD) manifests biochemically as apparent combined CYP17A1 and CYP21A2 deficiency, sometimes also resembling CYP19A1 (aromatase) deficiency. Unlike other forms of CAH, ORD is characterized by increased androgen concentrations only during the prenatal and early neonatal period, after which sex hormone deficiency develops rapidly. Further details of these enzyme defects are outlined in Table 21.4.
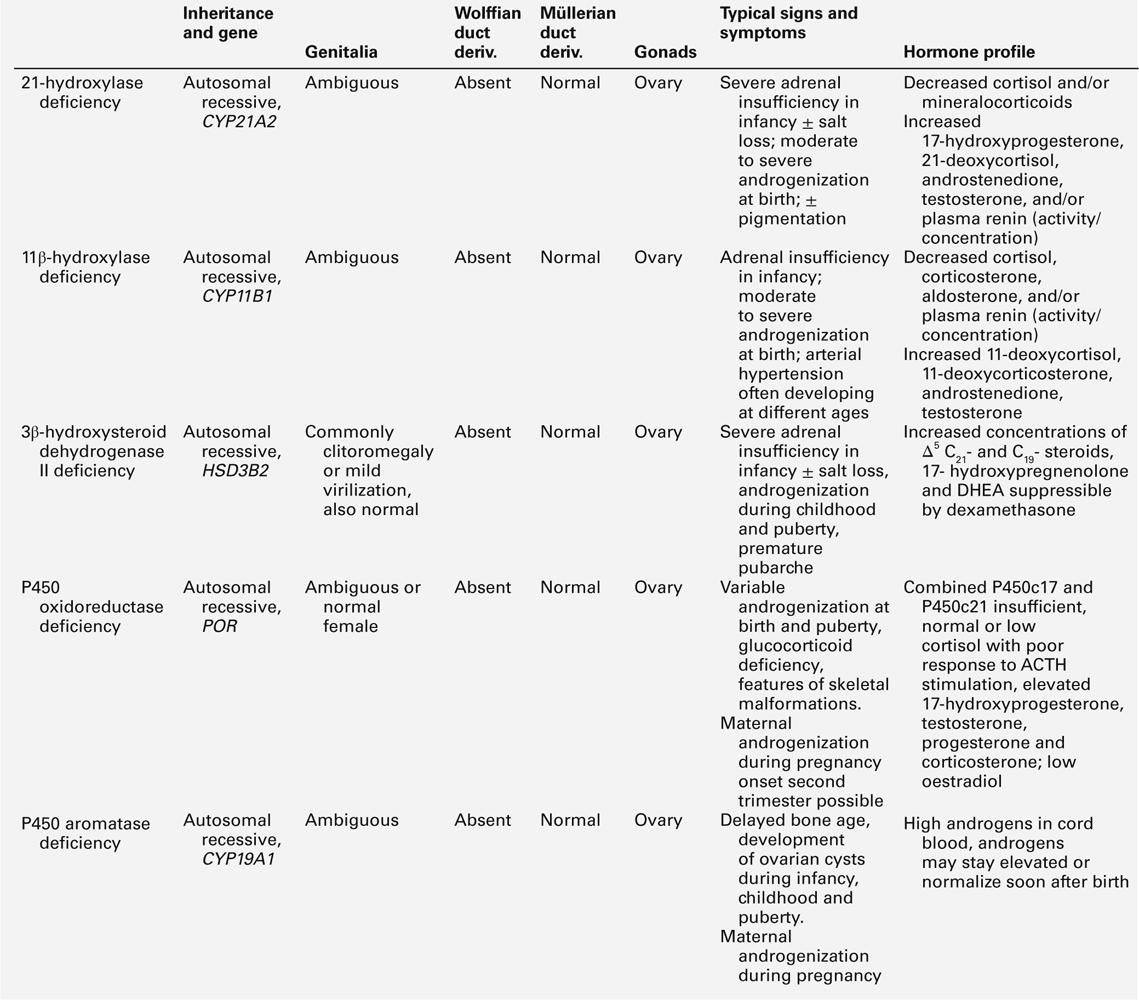
46XX DSD also includes disorders of gonadal development including 46XX ovotesticular DSD and 46XX testicular DSD. 46XX ovotesticular DSD refers to the presence of both ovarian tissue containing follicles and testicular tissue, previously termed ‘true hermaphroditism’. It commonly presents at birth with ambiguous genitalia and progressive virilization during puberty and the majority of such patients are raised as boys. The internal genitalia usually comprise a uterus and genital ducts, which have developed according to the nature of the ipsilateral gonad. The minority of infants raised as girls have breast development at puberty and may also menstruate. Fertility has been reported in some cases. In contrast, individuals with 46XX testicular DSD usually have a normal male phenotype and absent Müllerian structures and are often diagnosed after karyotype analysis during investigation for infertility. In 46XX testicular DSD, about 80–90% of patients will have Y chromosomal material including a translocated SRY gene, which is only rarely detected in 46XX ovotesticular DSD. In other patients with 46XX testicular DSD, duplications of the SOX9 gene and mutations of the RSOP1 gene have been described. In patients with the suspicion of 46XX ovotesticular DSD, there is a need to assess the functional potential of testicular and ovarian tissue by a combination of biochemical testing, imaging and surgical exploration.
Disorders of Müllerian development are another group of 46XX DSD and in these patients ovarian function is usually normal but often associated with cloacal anomalies and other characteristic malformations. Although most cases of Müllerian development disorders are not associated with androgen excess, the presence of the latter, particularly in the adolescent, should alert the clinician to a possible abnormality of the WNT4 gene.
XY DSD with low testosterone and low precursor concentrations
The differential diagnosis of 46XY DSD associated with low testosterone and precursor concentrations includes: defects occurring early in steroid synthesis (steroidogenic acute regulatory (StAR) protein, P450 side-chain cleavage (scc) enzyme/CYP11A1, sometimes Smith–Lemli–Optiz/DHCR7); LH receptor defects (LHCGR), and partial and complete forms of gonadal (testicular) dysgenesis (Table 21.5).
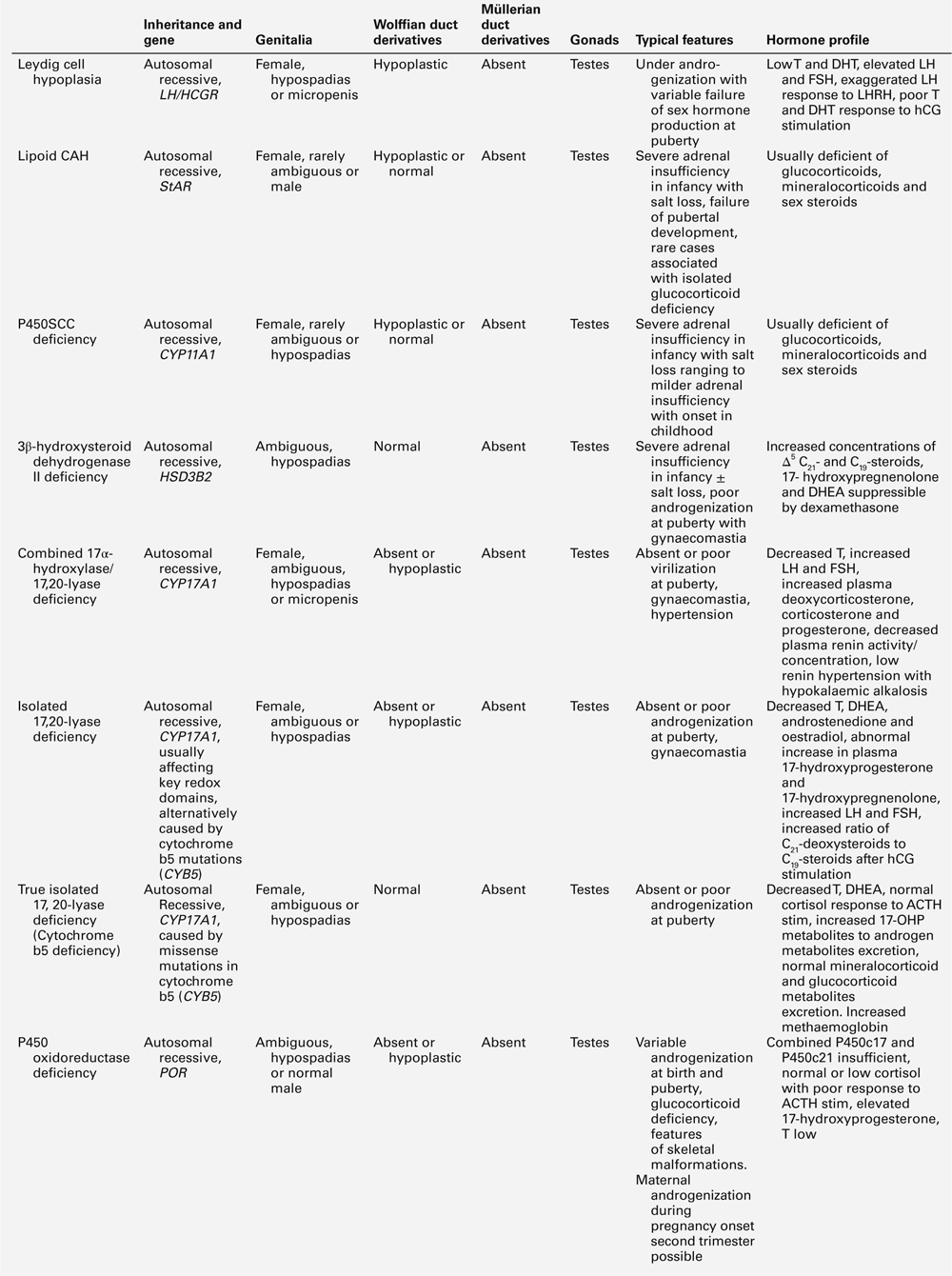

Of note, complete or partial combined 17α-hydroxylase/17,20-lyase deficiency (CYP17A1) may also present with ‘low testosterone and precursor’ concentrations if DHEAS and androstenedione are the only intermediates measured. The actual diagnosis can be reached by assessment of adrenal function by measuring ACTH, tetracosactide-stimulated cortisol, plasma renin activity (PRA), 11-deoxycorticosterone (DOC), aldosterone, Δ5-(pregnenolone, 17OHPreg) and Δ4-(progesterone, 17OHP) precursors or by urine steroid analysis. Isolated 17,20-lyase deficiency and ORD might also be diagnosed by this approach. Proximal blocks (StAR, P450scc) in the pathway affect steroidogenesis in the adrenal gland as well as the developing gonad.
LH receptor defects (‘Leydig cell hypoplasia’) typically result in elevated basal LH concentrations, hyper-responsive LH to GnRH stimulation, low testosterone and precursor concentrations and impaired androgen response to hCG stimulation. No Müllerian structures will be present and adrenal function is normal. A spectrum of phenotypes has been reported including ambiguous genitalia and micropenis. In some patients, basal LH may not be elevated when the hypothalamo–pituitary–gonadal axis is quiescent (six months to late childhood).
In complete gonadal dysgenesis (Swyer syndrome), children will usually have a female phenotype with intra-abdominal streak gonads. In some situations, ovotestes or even ovaries may be found. Müllerian structures are usually present owing to impaired AMH secretion in early fetal life. Androgens and their precursor concentrations will be low, LH elevated, depending on age, and a poor or absent testosterone response to hCG stimulation is seen. Anti-Müllerian hormone concentrations will be low or undetectable. Adrenal function is usually normal, unless the underlying defect is in steroidogenic factor-1 (SF-1) or related adrenal or gonadal factors.
Partial gonadal (testicular) dysgenesis can present with a spectrum of phenotypes ranging from clitoromegaly, to ambiguous genitalia or severe hypospadias. Müllerian structures may or may not be present and testes of variable size and architecture are present along the path of descent. The biochemical profile is similar to complete gonadal dysgenesis, but generally less severe. If mild degrees of clitoromegaly in infancy are overlooked, a 46XY child with partial gonadal dysgenesis may first present at puberty with progressive androgenization. Genetic analysis and associated features may be useful in defining the molecular aetiology of some forms of gonadal dysgenesis.
Frasier syndrome is a rare form of gonadal dysgenesis in which there is also chronic kidney disease and a risk of gonadoblastoma arising in the streak gonads. The syndrome is caused by mutation in the WT1 gene, located on chromosome 11p13, which encodes a zinc finger protein that functions as a transcription factor.
XY DSD with low testosterone and high steroid precursor concentrations
46XY DSD with low testosterone and increased precursor concentrations can be caused by several variants of CAH, namely 17α-hydroxylase (CYP17A1) deficiency, ORD and 3β-hydroxysteroid dehydrogenase type 2 (3βHSD2) deficiency, caused by inactivating mutations in the corresponding genes CYP17A1, POR and HSD3B2. In addition, 46XY DSD with low testosterone and increased precursor concentrations, can typically be found in individuals affected by 17β-hydroxysteroid dehydrogenase type 3 (17βHSD3) deficiency, caused by HSD17B3 mutations (see Table 21.5).
About 1% of patients with 46XY DSD have deficiency of CYP17A1. Characteristically, affected individuals present with female genitalia and low DHEA, androstenedione and testosterone concentrations. In ORD, sex steroid concentrations are characteristically low, sometimes low normal, while pregnenolone and progesterone and their metabolites accumulate, because of the combined block of CYP21A2 and CYP17A1 activities. There is often a relative preponderance of mineralocorticoid over glucocorticoid metabolites in affected patients but hypertension manifests only in adolescence, or later. Although baseline glucocorticoid secretion is usually sufficient, in the majority of patients, the stress response to ACTH is significantly impaired, requiring hydrocortisone cover at least in times of stress or permanent glucocorticoid replacement. 3β-hydroxysteroid dehydrogenase type 2 deficiency (also termed Δ4–Δ5 isomerase deficiency) invariably leads to glucocorticoid deficiency, as well as a variable degree of mineralocorticoid deficiency; its characteristic features are outlined in Table 21.5. 17β-hydroxysteroid dehydrogenase type 3 is responsible for the conversion of androstenedione to testosterone in the gonad and deficiency has no effect on adrenal steroidogenesis. Characteristically, androstenedione concentration is increased while that of testosterone is low, particularly after hCG stimulation. However, a low testosterone to androstenedione ratio may also occur in patients with gonadal dysgenesis and the reliability of a low ratio in identifying 17β-HSD3 deficiency is unclear. In urine, the typical finding is an increase in the androgen and androstenedione metabolites, androsterone and etiocholanolone, but it is unclear whether this applies across all age groups.
XY DSD with normal testosterone, normal precursor and low DHT concentrations
The 5α-reductase type 2 (SRD5A2) isoenzyme is highly expressed in androgen sensitive tissues and converts testosterone to the more potent androgen, dihydrotestosterone (DHT) required for the development of external male genitalia. At birth, the external appearance of the genitalia of a child with SRD5A2 deficiency can range from a completely female phenotype, through hypospadias of varying severity, to isolated micropenis. A positive family history is often present in this autosomal recessive condition. In serum, the testosterone:DHT ratio following hCG stimulation usually exceeds 30:1. In infants over 3–6 months, the defect should be easily identifiable on a urine sample which shows a decreased ratio of 5α:5β-reduced C21 and C19 steroids and the diagnosis can therefore be made in situations where a child has had early gonadectomy. Early diagnosis of this condition is important as the affected infant may need sex reassignment if initially raised as a girl. In the child raised as a boy, application of topical DHT cream may be a way of assessing the potential of the genitalia to virilize over the longer term.
XY DSD with normal testosterone, normal precursor and normal DHT concentrations
A defect in androgen signalling is most likely to be due to dysfunction of the androgen receptor (AR) and mutations resulting in a complete lack of function of the AR cause complete androgen insensitivity syndrome (CAIS). This presents in the newborn infant as a discordance between a female phenotype and a prenatal karyotype of 46XY, as inguinal swellings in an otherwise normal girl or during a postnatal check because of a positive family history. Complete androgen insensitivity syndrome usually presents in adolescence as primary amenorrhoea with normal breast development. The presence of pubic hair is often reported in CAIS and should not be used to exclude the diagnosis. Mutations which result in some residual AR function and varying degrees of androgenization cause partial androgen insensitivity syndrome (PAIS). Although children with PAIS typically have a normal testosterone and DHT response to hCG stimulation and a normal urinary steroid profile, some demonstrate a poor response to hCG stimulation. The serum AMH concentration is normal or may even be elevated. Luteinizing hormone concentrations are increased in the face of normal or elevated serum testosterone, reflecting a state of androgen resistance. A family history of X-linked inheritance is informative, although one-third of cases are the result of spontaneous new mutations.
A functional assessment of androgen sensitivity may include the clinical effect of a short course of testosterone applied to the phallus, or the effect of systemic testosterone following hCG stimulation. However, there is no consensus on the choice of androgen, dosage, method of administration, timing or duration of treatment or on the definition of a satisfactory response in the growth of the phallus. Androgen sensitivity can also be assessed by measuring the change in SHBG, an androgen-responsive protein which normally decreases in concentration following androgen exposure. This fall in SHBG is absent in CAIS, variable in PAIS and difficult to interpret in young infants who have highly variable circulating SHBG concentrations. Androgen receptor analysis may reveal a mutation in over 80% of patients with a CAIS phenotype and 30% of patients with a PAIS phenotype and androgen receptor binding studies are not necessary for routine diagnosis of AIS. A number of patients with XY DSD are loosely labelled as ‘PAIS’ when no conclusive biochemical or genetic abnormalities are identified in gonadal function, androgen synthesis or androgen action. The term PAIS should be reserved for those children who have XY DSD and a pathogenetic mutation in AR.
DISORDERS OF PUBERTY
Precocious puberty
Signs of puberty that occur before the ages of eight years in girls and nine years in boys are generally considered to indicate abnormally early puberty. The problem is more common in girls, in whom a cause is not often found. The earlier occurrence of puberty in the population has been attributed to the effects of xenoestrogenic chemicals in the environment and perhaps also to the increasing incidence of obesity. Adipocytes are a source of oestrogens through peripheral aromatization. Precocious puberty is increasingly seen in girls migrating from deprived environments for adoption in the developed world. Previous exposure to chemicals such as pesticides coupled with refeeding and catch-up growth in the new environment may be the trigger for early puberty. Box 21.1 lists some of the known causes of precocious puberty in both sexes. Structural lesions in the hypothalamic-pituitary region are increasingly recognized through sensitive imaging techniques. Severe head trauma may be followed quite abruptly by the onset of puberty. It is presumed that premature activation of the GnRH pulse generator has occurred. Similar mechanisms may operate in hydrocephalus, cerebral palsy, spina bifida and intracranial infections.
Gonadotrophin dependent puberty (central causes)
Although the cause of precocious puberty in a large majority of children remains unknown (idiopathic), some children may require a thorough endocrine evaluation of the pubertal axis as listed in Table 21.6. Important causes outlined below should be considered if clinically appropriate.
TABLE 21.6
Basic endocrine investigation of disorders of puberty
| Precocious puberty | Delayed puberty |
| Basal LH, FSH, testosterone or oestradiol | Basal LH, FSH, testosterone or oestradiol |
| Acute GnRH stimulation test | Acute GnRH stimulation test |
| Thyroid hormone profile (TSH & FT4) | Thyroid function tests (TSH & FT4) |
| Prolactin | Peripheral karyotype (females) |
| β-hCG | Possibly hCG stimulation test (males) |
| Skeletal age (bone age) | Skeletal age (bone age) |
Tumours in the region of the hypothalamus known to be associated with precocious puberty include hamartoma, astrocytoma, neurofibroma and, occasionally, craniopharyngioma. While the prevalence of such tumours causing precocious puberty is the same in boys and girls, a tumour is more likely to be the cause of precocious puberty in a boy. Hypothalamic-pituitary damage from irradiation to intracranial tumours causes pituitary deficiency of growth hormone and thyrotrophin releasing hormone (TRH) but, paradoxically, premature reactivation of the GnRH pulse generator and precocious puberty.
The McCune–Albright syndrome comprises the triad of café-au-lait skin pigmentation, fibrous dysplasia of bone (affecting particularly long bones and the base of the skull) and precocious puberty. In girls, the latter is typically gonadotrophin independent, as shown by a prepubertal LH and FSH response to acute GnRH stimulation. Autonomously functioning multiple ovarian cysts occur, as well as evidence of dysfunction in other endocrine glands such as the thyroid (thyrotoxicosis), adrenal (Cushing syndrome), pituitary (gigantism and hyperprolactinaemia) and parathyroid (hyperparathyroidism). The condition may present in early childhood and progress to gonadotrophin-dependent ovarian hyperfunction and the consequent problems of menses in a young child. The McCune–Albright syndrome is less common in boys and may manifest with asymmetrical enlargement of the testes as well as the development of secondary sexual characteristics. Somatic activating mutations of the G-protein α subunit (GSα) component of the guanine nucleotide binding protein, alpha stimulating (GNAS) complex that couples seven transmembrane receptors to the cAMP signal transduction pathway, cause the widespread distribution of tissue abnormalities in the syndrome. The predominant mutation is substitution of an arginine at codon 201 in the GSα gene. Heterozygous inactivating GSα mutations cause Albright hereditary osteodystrophy, a syndrome characterized by short stature, obesity, short metacarpals, subcutaneous calcification and behavioural problems.
Autonomously hyperfunctioning testes, independent of gonadotrophic stimulation, also cause precocious puberty in males, owing to Leydig cell hyperplasia. Familial forms are recognized that present with signs of virilization, but with testes inappropriately small for the advanced stage of puberty. The onset of precocious puberty often occurs in early childhood and may even occur in infancy. Testosterone concentrations are increased, while basal and GnRH-stimulated concentrations of gonadotrophins are suppressed. Histological examination of the testes shows Leydig cell hyperplasia and spermatogenesis. This form of familial, male-limited precocious puberty is autosomal dominant and is caused by an activating mutation in the G-protein-LH-coupled receptor. Somatic LH receptor mutations are also found in males with Leydig cell adenomas without a history of precocious puberty, but a Leydig cell adenoma rarely occurs in the precocious puberty phenotype. This is at variance with the phenotype associated with activating mutations in the TSH receptor where nodular thyroid hyperplasia is often an associated histological feature of this form of hyperthyroidism. Treatment for male-limited, familial precocious puberty comprises an anti-androgen agent, such as spironolactone, together with an aromatase enzyme inhibitor such as testolactone or anastrozole, but the long-term therapeutic benefits of this approach are unclear. Use of a GnRH analogue is not appropriate initially as gonadotrophin concentrations are already suppressed. However, the precocious puberty may become gonadotrophic dependent at a later stage, when a long-acting GnRH analogue can be added to the treatment regimen.
Variants of early puberty
The most frequent variant of early puberty is premature thelarche, or isolated early breast development. The usual age of onset is 1–3 years and occasionally breast development may have persisted from the neonatal period. The breast enlargement may be unilateral or bilateral and typically tends to wax and wane over time. No other signs of puberty occur and the rate of linear growth is normal for chronological age. Serum oestradiol and gonadotrophin concentrations remain prepubertal, although most immunoassays are insufficiently sensitive to detect fluctuations in oestradiol concentrations in prepubertal girls. It has been proposed that breast enlargement is the result of enhanced tissue responsiveness to a transient increase in circulating oestradiol. Increased concentrations of SHBG have been detected in some girls with premature thelarche, thereby resulting in an increase in the ratio of free oestradiol to testosterone concentrations. This may explain the breast development, even though total oestradiol concentration is not increased. The source of oestrogen is ovarian, and cysts of the ovary are sometimes detected on ultrasound. While population studies imply the possibility of exposure to chemicals in the environment acting as xenoestrogens, it is difficult to prove such exposure in individual patients. The natural history of premature thelarche is the onset of puberty and menarche at a normal age, as well as normal reproductive potential. A form of thelarche variant is also recognized that is associated with some increase in growth rate and skeletal maturation and a slightly exaggerated LH and FSH response to acute GnRH stimulation.
Isolated recurrent vaginal bleeding can sometimes occur in girls before puberty. It is important to exclude local lesions of the genital tract, self-harm by the child or child abuse. Generally, it is associated with increased FSH concentrations that produce sufficient oestradiol to stimulate the endometrium and induce a subsequent withdrawal bleed. Sometimes, ovarian cysts are found on ultrasound. This form of premature isolated menarche is generally self-limiting and has no adverse consequences for subsequent puberty and reproductive capacity.
Premature adrenarche is the term applied to the early development of pubic hair without any other signs of puberty. It is more common in girls, occurring usually between six and eight years of age. The cause is an increase in adrenal androgen production associated with elevated concentrations of DHEA and its sulphate, as well as androstenedione. It is partly explained by a change in steroidogenesis regulated by the orphan nuclear receptor, NGF1B. Premature adrenarche is generally followed by a normal progression through puberty. There is some evidence that girls with premature adrenarche have a higher risk of later developing polycystic ovarian syndrome.
Delayed puberty
The wide range for the age of onset of puberty in normal children has previously been emphasized. Puberty is regarded as delayed if physical signs have not started by 13 years in girls and 14 years in boys. Box 21.2 lists some of the causes of delayed puberty in both sexes. The list is not an exhaustive one but encompasses the majority of important causes. During the clinical assessment it is important to exclude a chronic disease that may manifest solely as slow growth and delayed puberty, examples include cystic fibrosis, inflammatory bowel disease and anorexia nervosa.
Delayed growth and puberty
Delayed puberty presents as a clinical problem far more often in boys than in girls. This may be an ascertainment bias, perhaps because of the greater social pressures of delayed puberty (and hence short stature) in a boy than a girl. Most delayed puberty just represents the extreme lower end of the physiological range for pubertal events. Since it is inevitably accompanied by short stature due to the delayed appearance of the pubertal growth spurt, this common cause is called constitutional delay in growth and development. There is often a family history of delayed puberty. The natural outcome is for puberty to develop spontaneously (according to skeletal maturation as assessed by bone age examination rather than chronological age) and lead to normal, albeit delayed, final height according to genetic potential. Sometimes, low-dose sex steroids are given to prime the pubertal growth spurt. Final height may be increased in boys with constitutional delay by treatment with aromatase inhibitors during adolescence, but there are concerns that such treatment may have undesired effects on bone health at this critical period of skeletal development.
Delayed puberty is a disturbance in the tempo of growth and pubertal maturation associated with a delay in activation of the hypothalamic GnRH pulse generator. Random and GnRH-stimulated plasma gonadotrophin concentrations are low and appropriate for physiological development, as are those of gonadal steroids. A similar endocrine profile is found in delayed puberty secondary to hypogonadotrophic hypogonadism in which spontaneous onset of puberty does not occur. It can be difficult to distinguish these two variants, even when short-term pulsatile GnRH therapy is used in the prepubertal state. Spontaneous onset of puberty is the ultimate decider. Familial clustering may suggest rare causes such as mutations affecting the GnRH receptor gene or the GPR54 gene. No mutation has yet been found involving the GnRH gene and delayed puberty. Short stature in delayed puberty is partly the result of an insufficient production of growth hormone (GH) reflected in decreased pulse amplitude. This, in turn, results in less generation of insulin-like growth factor 1, which may be relevant for gonadal function. Puberty is a complex interplay of sex hormones, GH and growth factors activated as a result of polygenic and environmental factors. While the discovery of GPR54 has provided a molecular explanation for a rare form of hypogonadotrophic hypogonadism, no GPR54 mutations have been reported in constitutional delayed puberty.
Organic causes affecting the hypothalamic-pituitary area result in hypogonadotrophic hypogonadism and delayed puberty. Numerous examples exist such as craniopharyngioma, optic glioma, germinoma, astrocytoma, head trauma (which may also lead to early puberty), effects of irradiation, infiltrative diseases and post-infectious lesions. An organic cause should be considered when puberty has started, but then becomes arrested in progress.
Hypogonadotrophic hypogonadism
Syndromes associated with gonadotrophin deficiency include Prader–Willi, Lawrence–Moon–Biedl and Kallman syndromes. Failure of migration of the GnRH neurons from the olfactory placode to the medial basal hypothalamus causes Kallman syndrome and explains the association with anosmia. An X-linked form of Kallman syndrome is caused by mutations in the KAL1 gene, which encodes for the anosmin-1 protein. An autosomal dominant form is caused by heterozygous mutations in the FGFR1 gene (fibroblast growth factor receptor-1). Inactivating mutations in the GPR54 gene cause isolated hypogonadotrophic hypogonadism.
Isolated gonadotrophin deficiency may occur in association with congenital adrenal hypoplasia. Affected individuals present in infancy with a salt-losing crisis and masquerade as if the diagnosis were congenital adrenal hyperplasia. However, adrenal steroid concentrations are low and unresponsive to ACTH stimulation. The diagnosis may be suspected before birth because of low maternal oestriol excretion resulting from decreased steroid substrates from the affected fetal adrenals. The disorder is X-linked; affected boys fail to enter puberty spontaneously because of an associated gonadotrophin deficiency. Adult males have azoospermia and a poor response to pulsatile GnRH treatment. Mutations in DAX-1, a gene on Xp21.3–21.2 that encodes an orphan nuclear hormone receptor, are described in patients with X-linked congenital adrenal hypoplasia and hypogonadotrophic hypogonadism. A variant of the syndrome may present for the first time in adulthood with mild adrenal insufficiency and partial hypogonadism. Another orphan nuclear receptor involved in both adrenal and hypothalamic development is steroidogenic factor-1 (SF-1). Rare mutations cause adrenal failure and generally XY sex reversal.
Primary hypogonadism
The other major groups of disorders that manifest with delayed puberty are those associated with primary hypogonadism. In these, gonadotrophin concentrations are elevated. The chromosome aneuploidies, Klinefelter syndrome (47XXY) and Turner syndrome (45XO and variants), are important causes of delayed puberty. Turner syndrome has an incidence of about 1 in 2000 live-born girls. It is estimated that 1–2% of conceptuses have the syndrome with only 1% of these not aborted spontaneously. Two constant clinical features of this syndrome are short stature and failure to enter puberty spontaneously because of premature ovarian failure. The former feature dictates that a peripheral karyotype should be performed on any short girl in whom there is no readily apparent cause for the growth failure.
The typical physical features of Turner syndrome may not always be present. They include a short, broad neck which may be webbed, a low hairline, low-set ears, high-arched palate, ptosis, hypoplastic nails, short metacarpals, cubitus valgus (increased carrying angle), pigmented naevi and cardiovascular anomalies. Affected girls are often small at birth and have peripheral lymphoedema. This usually is an alert to making the diagnosis. The results of a pilot study using FSH measurement in blood spots for newborn screening were not sufficiently specific. Pyrosequencing assays for quantitative genotyping of informative single nucleotide polymorphism (SNP) markers spanning the X chromosome in a newborn screening programme reliably detect complete or partial X deletions and mosaicisms. Growth failure is invariably present by 2–3 years of age. Ovarian failure is a progressive process starting in late gestation and appears to be an acceleration of the diminution in the number of primary oocytes that occurs between fetal and postnatal life in the normal female. Generally, streak gonads are the only remnants of ovaries, although 5–10% of Turner syndrome girls do have spontaneous puberty and menstruate. A small number are able to become pregnant, but this is associated with a high rate of miscarriage, stillbirths and congenital malformations. Management of Turner patients requires oestrogen replacement at the time of expected puberty and the early use of GH to enhance adult height. Numerous studies confirm that GH can significantly increase final height. Chromosomal mosaicism in the form of 45XO/46XY may show some features of the Turner phenotype, such as short stature, but the presentation may be at birth because of abnormal genitalia. Adult patients with Turner syndrome need regular surveillance of cardiovascular parameters, metabolic abnormalities such as insulin resistance, renal disorders and sensorineural hearing deficits. Turner syndrome represents one example of spontaneous premature ovarian failure. In addition, there are other causes of acquired as well as genetic causes of ovarian failure.
Klinefelter syndrome affects about 1 in 1000 males and is characterized by tall stature, small firm testes, gynaecomastia and infertility in adults. These men are also hypogonadal and, although they enter puberty normally, they undergo pubertal arrest and progress through puberty does not occur as expected. Serum testosterone concentrations increase appropriately according to the stages of puberty, while a characteristic elevation of gonadotrophin concentrations is displayed from the age of 13 years. Fertility may be achievable using testicular extraction for intracytoplasmic sperm injection. Other causes of primary hypogonadism in the male include testosterone biosynthetic defects and the effects of chemotherapy in conditions such as leukaemia. Anorchia (absence of both testes at birth) is relatively common and is sometimes referred to as the vanishing testis syndrome. Genital development in affected boys is otherwise normal, which implies that testicular function (including testosterone and AMH production) was also normal in early gestation. The cause of testicular regression is not known but is postulated to be the result of intrauterine testicular torsion. The presence or absence of testicular tissue after birth can be assessed by a prolonged hCG stimulation test to determine any response in serum testosterone. Serum AMH is also a useful marker that avoids the necessity of undertaking an hCG stimulation test. At the expected age of puberty, there is an increase in plasma gonadotrophins.
Further reading
Disorders of sex development
Ahmed SF, Hughes IA. The genetics of male undermasculinisation. Clin Endocrinol (Oxf). 2002;56:1–18.
Federman DD. The biology of human sex differences. N Engl J Med. 2006;354:1507–1514.
Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–788.
Puberty