[level-membership-for-internal-medicine-category]ANAEMIA
An Hb level of < 13 g/dL in men and < 12 g/dL in women is considered anaemic in common practice.
Ask about:
• the symptoms at presentation—patients may present with constitutional symptoms such as lethargy, malaise, exertional dyspnoea, fatigue or gastrointestinal blood loss in the form of haematemesis or melaena
• how long the patient has had anaemia—if they have had it since childhood, the likely diagnosis of a haemoglobinopathy should be considered
• easy bruising, uncontrolled blood loss, large joint arthropathy and excessive bleeding post-surgery—features suggesting a coagulation disorder/bleeding diathesis
• other symptoms that would help identify the aetiology, such as change in bowel habits or weight loss suggesting colonic cancer, abdominal discomfort, bloating, anorexia and weight loss of gastric carcinoma, abdominal pain suggesting peptic ulcer disease, night sweats, weight loss and fevers suggesting lymphoma.
Take a detailed medication history, looking for evidence of therapy with non-steroidal antiinflammatory agents, sulfur-containing medications (haemolysis) and myelosuppressive agents. Enquire about alcoholism and general nutrition. Check whether the patient has had any transfusions and whether there have been any adverse transfusion reactions or transmission of blood-borne infections such as hepatitis C and HIV. It is also useful to know the patient’s blood group.
Look for features of anaemia such as palmar crease pallor, conjunctival pallor, tachypnoea, tachycardia and evidence of high-output cardiac failure. Look for signs that suggest the likely aetiology (see box): characteristic copper-hue pigmentation (the combined effect of melanin deposition, icterus and pallor) of the thalassaemia patient with malar hyperplasia and often stunted growth, pigmented lesions in the mouth (Peutz-Jeghers syndrome), scleral icterus (haemolysis), epigastric tenderness (peptic ulcer disease), abdominal distension or mass lesions, splenomegaly, hepatomegaly, bony tenderness (malignancy), lymphadenopathy (lymphoma), impaired cognition, peripheral neuropathy and dorsal column signs (vitamin B
12 deficiency), arthropathy (anaemia of chronic disease or gastrointestinal blood loss secondary to NSAID use), slate-grey pigmentation, peripheral arteriovenous fistula, vascular access device or abdominal Tenckhoff catheter (renal anaemia).
Anaemia in the long case patient is often multifactorial. The diagnostic work-up should include a comprehensive battery of tests as guided by the clinical setting. Iron deficiency secondary to chronic blood loss, commonly from the gastrointestinal tract, is the most common cause of anaemia in this group of patients. Other differential diagnoses encountered commonly include anaemia of chronic disease, vitamin B12 and folate deficiency, myelodysplasia, aplastic anaemia, sideroblastic anaemia, myelosuppression, chronic renal failure, myelofibrosis and chronic alcoholism (due to a combination of bone marrow toxicity of alcohol, iron and folate deficiency and gastrointestinal bleeding).
Investigation of anaemia should be guided by the clinical picture. The more common conditions should be excluded first. Relevant investigations in the anaemic patient include:
Blood picture: Patients with anaemia of chronic disease usually have normocytic, normochromic anaemia. Those with iron deficiency, thallasaemia or sideroblastic anaemia have microcytic, hypochromic anaemia. Microcytic, hypochromic anaemia is also seen in myelodysplastic syndrome.
2. Coagulation profile—international normalised ratio (INR), activated partial thromboplastin time (APTT), bleeding time and clotting time, to exclude a bleeding diathesis
3. Haematinics—iron studies in the form of serum ferritin, serum iron and transferrin saturation, to exclude iron deficiency
4. Serum vitamin B12 and serum and red cell folate levels
5. Haemolytic screen—including blood film, serum haptoglobin level, serum lactate dehydrogenase (LDH) level, serum bilirubin level, the direct Coombs’ test, haemoglobin electrophoresis, Heinz body preparation and, if indicated, the cold agglutinin test
6. Erythrocyte sedimentation rate (ESR)—looking for evidence of chronic inflammation and paraproteinaemia
7. Electrolyte profile and the renal function indices—to exclude renal failure as a cause
8. Erythropoietin levels
9. Thyroid function tests—to exclude hypothyroidism, which can cause a macrocytic anaemia
10. Chest X-ray—to exclude chronic pulmonary disease and pulmonary malignancies
11. Serum protein electrophoresis and immunoelectrophoresis—to exclude any paraproteinaemia
12. If all non-invasive and minimally invasive tests do not help in the correct diagnosis, the next investigation is a bone marrow biopsy with both aspiration and trephine.
Management depends on severity (level of Hb) and the patient’s cardiopulmonary status. Initial steps include identification and correction of reversible, causative factors and replacement of blood with packed cell transfusions and/or the supplementation of haematinics as indicated.
As a general rule, blood transfusion is indicated if Hb is < 7 g/dL. If Hb remains > 10 g/dL, transfusion should be considered only if there is a compelling clinical indication such as precipitation of coronary ischaemia or heart failure.
Gastrointestinal bleeding warrants endoscopy (colonoscopy or gastroscopy) to identify the exact location of the bleeding and for possible therapy or biopsy of suspicious lesions. If gastroscopy and colonoscopy fail to identify the source of bleeding, capsule endoscopy may be warranted, to look for a small intestinal focus.
Patients with chronic renal failure need iron supplementation and at times may need IV iron infusion. Patients with kidney disease may benefit from therapy with erythropoietin or darbapoietin.
It is recommended that patients with chronic kidney disease be maintained at an Hb level of 11 g/dL or more.
Patients with anaemia of chronic disease improve when the underlying disease is well treated. Haematinic replacement and therapy with erythropoietin or darbapoietin is also indicated in this group.
Management of anaemia due to cytotoxic chemotherapy in patients with non-myeloid malignancies is similar to the management of anaemia of chronic disease.
Haematological malignancies are commonly encountered in the long case. History taking should enquire into the events surrounding the diagnosis, such as presenting symptoms of fatigue, lethargy, weight loss, fever, night sweats, bone pain, how the diagnosis was made, the various investigations that have been carried out, including bone marrow biopsy and/or excision biopsy of lymph nodes, and the treatment given so far, including blood transfusions, chemotherapy and bone marrow or stem cell transplantation. Ask about the side effects of chemotherapy and any episodes of febrile neutropenia, life-threatening sepsis etc. Ask how the patient is coping with the disease, what knowledge they have about the prognosis of the condition, what expectations they have for the future, and the level of social support available.
Following is a discussion of the salient points relevant to the commonly encountered haematological malignancies. The candidate is expected to have an up-to-date knowledge of each disease and to be able to handle wisely the complicated clinical issues associated with the management of these conditions.
A 56-year-old man presents with significant lethargy, severe back pain, loss of sensation in the lower extremities and dizziness. He also complains of bleeding gums and frequent blurring of vision. Physical examination reveals mucosal and conjunctival pallor and bony tenderness of the lower back. There are signs of peripheral neuropathy. His blood tests reveal anaemia, renal impairment and hypercalcaemia.
Multiple myeloma is a common malignant condition encountered in the long case setting, because patients with multiple myeloma can have many secondary medical complications. Multiple myeloma is due to disordered proliferation of a clone of plasma cells secreting monoclonal immunoglobulins. The clinical presentation of multiple myeloma is variable.
Ask about pain, bony fractures, lethargy, fatigue, dyspnoea, mucosal bleeding, fevers and rigors. Enquire about loss of sensation, limb weakness, visual problems and dizziness.
Some common presentations are intractable bone pain, pathological fracture, symptomatic anaemia, pancytopenia, sepsis, renal failure or acute symptomatic hypercalcaemia. Some may present with the hyperviscosity syndrome, with associated neuropathy and coagulopathy (oral and nasal bleeding, blurred vision, headache and vertigo) or amyloidosis. Hyperviscosity syndrome is more common in Waldenström’s macroglobulinaemia, where the paraprotein is IgM. The most common presenting symptom, however, is bone pain, particularly in the vertebral column. Multiple myeloma should be suspected in any patient over the age of 40 years presenting with bone pain, pathological fracture, osteoporosis, lethargy, anaemia and recurrent infections. Incidental discovery of proteinuria, hypercalcaemia, acute renal failure, high ESR or rouleaux formation should alert the clinician to look for multiple myeloma.
Look for evidence of anaemia and sepsis. Do not forget to look at the temperature chart. Check the mucosa for bleeding. Perform a thorough neurological examination. Look for bony fractures and assess for bone tenderness.
For the diagnosis of multiple myeloma there should be one major criterion together with one minor criterion or three minor criteria present—these should be clinically manifest.
(Adapted from Grogan T M 2001 Plasma cell neoplasms. In: Jaffe E S, Harris N L, Stein H et al (eds) World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon, p 142)
(Adapted from Grogan T M 2001 Plasma cell neoplasms. In: Jaffe E S, Harris N L, Stein H et al (eds) World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon, p 142)
(From Greipp P R, San Miguel J, Durie B J et al 2005 International staging system for multiple myeloma. Journal of Clinical Oncology 23(15):3412–3420)
(From Greipp P R, San Miguel J, Durie B J et al 2005 International staging system for multiple myeloma. Journal of Clinical Oncology 23(15):3412–3420)
Emergent management of acute complications:
General management strategy:
If the disease relapses or is progressive:
A 45-year-old man with massive hepatosplenomegaly was diagnosed with chronic myeloid leukaemia when he saw his GP to discuss his recent significant weight loss. He has a young family and he remains the sole breadwinner.
Chronic myelogenous leukaemia (CML) is a clonal myeloid stem cell disorder characterised by three discrete phases:
Without therapeutic intervention, the disease eventually and inevitably progresses to blast phase.
Individuals of all age groups can be affected by this condition, but it is particularly common among those over the age of 55 years.
Ask about the diagnosis and the initial symptoms. Ask about fatigue, weight loss, spontaneous bleeding and abdominal fullness. Patients present with the symptoms of fatigue, anorexia and weight loss. However, about 40% of patients are diagnosed incidentally.
Examine for abdominal organomegaly. On presentation the patient may have massive splenomegaly or hepatosplenomegaly.
A 63-year-old female presents with left-sided hemiparesis and visual impairment. She also complains of excessive fatigue preceding the neurological event, together with recent onset of back pain. She has a background history of Hodgkin’s lymphoma, treated curatively 5 years ago. Examination reveals, in addition to the neurological deficit, retinal ischaemia, evidence of anaemia, mucosal bleeding and gum hypertrophy. Blood tests reveal leucocytosis with myeloblasts.
The incidence of this condition increases with age, peaking at around age 65 years.
Look for evidence of anaemia, mucosal bleeding, bone tenderness, lymphadenopathy and organomegaly. Lymphadenopathy, hepatomegaly and splenomegaly are less common manifestations. Increased leucocyte count can lead to leucostasis, causing visual impairment due to retinal ischaemia, stroke, bleeding diathesis and acute respiratory distress syndrome. Collectively this is called ‘hyperleucocytosis syndrome’.
Some patients may present with hyperuricaemia, manifesting as gouty arthritis and ureteric colic, or with symptomatic hypercalcaemia. Check the temperature chart for fevers.
Management options differ depending on the patient’s age, performance status and cytogenetic risk category. Patients of good performance state and aged < 70 years are either recruited into a novel chemotherapy trial or treated with induction chemotherapy (e.g. ICE—idarubicin, cytarabine, etoposide or FLAG—fludarabine, cytarabine, combinations) followed by consolidation therapy.
This is followed by further therapy depending on the patient’s cytogenetic risk group (see box):
Patients with acute promyelocytic leukaemia (APML) should be investigated for evidence of disseminated intravascular coagulation.
Based on the cytogenetic analysis, patients are divided into good risk, standard risk and poor risk categories:
There are an evolving number of molecular variants of AML which, hopefully, will further stratify these cytogenetic subtypes.
These patients can be treated with all-transretinoic acid (ATRA) and idarubicin. Maintenance therapy consists of a combination of ATRA, methotrexate and 6-mercaptopurine.
At relapse, treatment is with arsenic trioxide (As2O3) and ATRA.
Acute lymphoblastic leukaemia (ALL) is less common, and in the adult patient the prognosis is relatively guarded. The prognosis depends on the cytogenetic analysis. Poor prognostic cytogenetic markers include t (4:11) and t (9:22). Patients are enrolled in a therapeutic clinical trial if eligible. The therapeutic regimen usually consists of multiple courses of high-dose prednisolone, combination chemotherapy and central nervous system prophylaxis (intrathecal methotrexate and cranial irradiation).
Consider allograft transplantation after first remission if the patient is less than 60 years old.
Due to the chronicity and the relative stability of the patient in the early stages, it is more likely for a candidate to encounter a patient with chronic lympohocytic leukaemia (CLL) in the examination setting. Patients usually present with lymphadenopathy, anaemia, infections and hepatosplenomegaly. Sometimes the diagnosis is made on incidental observation of an elevated WBC count. Disease stage and severity is classified according to the Rai or the Binet classification. The Binet classification is easy to remember, and goes as follows:
Staging has therapeutic as well as prognostic implications. Stages A and B disease warrant only watchful waiting in the older patient.
Therapy is indicated in advanced disease. Other indications for therapeutic intervention include bulky disease, B symptoms, autoimmune phenomena (autoimmune haemolytic anaemia), rapid doubling time (< 6 months), young age (< 40 years) and poor risk features (see box).
It is ideal to enrol the patient in a therapeutic clinical trial if suitable.
Those aged over 70 or with poor prognostic signs may be treated with fludarabine or chlorambucil together with rituximab.
A 35-year-old male presents to the emergency department with fevers, rigors and a productive cough. Examination reveals bronchial breath sounds in the right lower zone on his chest. Sputum mug contains rusty purulent sputum. Chest X-ray shows evidence of consolidation in the right lower zone. In addition, the chest X-ray displays a mass lesion in the mediastinum.
Ask about the initial diagnosis. Note the patient’s age. Hodgkin’s disease has a bimodal age-related prevalence with an initial peak in the third to fourth decade and a second peak in the sixth decade. The most common presentation is with singular or localised lymphadenopathy without any other symptoms. Some get diagnosed upon the incidental finding of a mediastinal mass on chest X-ray. However, upon enquiry, the patient may describe retrosternal chest discomfort, dyspnoea or cough. Ask about fevers, night sweats and recent weight loss. Some patients may complain of intractable pruritis and rash. Alcohol-induced pain is another late feature of this condition. Ask about any previous history of malignancies and family history of Hodgkin’s disease.
Identify the distribution of lymphadenopathy and establish its classic rubbery consistency. Examination of the neck region should be very thorough. Listen to the chest for evidence of tracheal or bronchial obstruction due to mediastinal mass. Examine the abdomen for splenomegaly and, less commonly, hepatomegaly. Study the temperature chart to establish the pattern of the fever, which classically is intermittent and often nocturnal (Pel-Ebstein fever). Look for a mediastinal mass in the chest X-ray.
Other investigations of prognostic significance:
The Ann Arbor staging classification of Hodgkin’s disease is as follows:
The disease is classified into early-stage disease (stage I, IB, IIA, non-bulky disease) and advanced-stage disease (bulky disease, stage IIB, III or IV) from a therapeutic perspective. Response to treatment is monitored with CT imaging and PET scanning.
Early-stage Hodgkin’s lymphoma
Advanced-stage Hodgkin’s lymphoma
Each adverse factor scores 1 point:
(From Hasenclever D, Diehl V 1998 A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. New England Journal of Medicine 339(21):1506–1514)
(From Hasenclever D, Diehl V 1998 A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. New England Journal of Medicine 339(21):1506–1514)
Patients with gastrointestinal lymphoma may have a history of coeliac disease. Ask about B symptoms of fevers, night sweats and weight loss.
Perform a thorough lymph node assessment for lymphadenopathy. Check for abdominal organomegaly. Don’t forget to look at the temperature chart.
CT scan, PET scan and bone marrow biopsy are used in the staging process.
The histological subtype of the disease is important in deciding on the management option. Disease is often categorised into aggressive disease or indolent disease, based on the histology.
The stage of the disease and the prognostic score, calculated according to the systems described in the boxes (for aggressive disease and indolent disease respectively), are important factors that influence the therapeutic plan. The scoring system also includes the patient’s age and the functional status, which have a major bearing on the management strategies.
Aggressive lymphomas include diffuse large B-cell lymphoma, mantle cell lymphoma, T-cell lymphoma, Burkitt’s lymphoma and lymphoblastic lymphoma. Indolent lymphomas include follicular lymphoma.
Prognosis in non-Hodgkin’s lymphoma is described according to the International Prognostic Index (IPI). The disease is graded according to a scoring system, as follows:
(After Shipp M A, Harrington D P, Anderson J R et al 1993 A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. New England Journal of Medicine 329(14):987–994)
(After Shipp M A, Harrington D P, Anderson J R et al 1993 A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. New England Journal of Medicine 329(14):987–994)
(After Solal-Céligny P, Roy P, Colombat P et al 2004 Follicular Lymphoma International Prognostic Index. Blood 104:1258–1265)
(After Solal-Céligny P, Roy P, Colombat P et al 2004 Follicular Lymphoma International Prognostic Index. Blood 104:1258–1265)
This disease has a higher therapeutic response. Aggressive therapy is indicated if the patient’s cardiopulmonary performance status is adequate. Standard therapy is combination chemotherapy with cyclophosphamide, doxorubicin hydrochloride, vincristine and prednisolone (R-CHOP) and radiotherapy. Treatment intensification is considered for younger patients with poor prognostic indicators.
Consider intrathecal methotrexate if there is a high risk of central nervous system relapse. Features that suggest this possibility include high or high/intermediate IPI score, testicular or sinus involvement or stage IV disease.
Relapse of the disease is treated with second-line chemotherapy (fludarabine) followed by autologous stem cell transplants for those responsive to chemotherapy. If this strategy fails, further therapy with hypomethylating agents (epigenetic strategies), new monoclonal antibodies and non-ablative allograft transplantation should be considered.
Though this was considered a ‘low-grade’ lymphoma according to the working formulation, it is treated where feasible as an ‘aggressive lymphoma’ due to its poor prognosis.
There is no standard therapy for aggressive T-cell lymphomas. Generally CHOP with (or without) upfront autografting is considered.
These are managed according to the protocols used for the management of ALL (see p 121).
Indolent lymphomas such as follicular lymphoma are managed with watchful waiting, and active therapy when required (chemotherapy or radiation to localised disease)—similar to the indications for treatment in CLL (see p 121). Generally these diseases are considered incurable, though novel strategies (e.g. rituximab in combination) are starting to show survival benefits.
Myelodysplasic syndrome should be considered a possible diagnosis in the elderly patient who has a combination of macrocytic anaemia, neutropenia and thrombocytopenia. The condition could be idiopathic, or secondary to previous exposure to cytotoxic chemotherapy or radiotherapy. Diagnosis is made by bone marrow biopsy.
A 45-year-old female presents with progressive abdominal enlargement and bilateral pain in the upper abdominal quadrant. She denies smoking, alcohol excess or recent overseas travel, and her medication history is negative for potentially hepatotoxic agents. She denies ingestion of any herbal medications. On examination she is jaundiced. She has ascites, caput medusae and hepatosplenomegaly. She is particularly tender in the left upper quadrant with guarding. Liver function tests are grossly abnormal and ultrasonography reveals paucity of venous architecture. Full blood count shows erythrocytosis with low plasma erythropoietin levels. Hepatitis and HIV screen comes negative.
Polycythaemia vera is a myeloid stem cell disorder characterised by trilineage (erythrocytes, granulocytes and platelets) hyperplasia, high blood counts and splenomegaly. Polycythaemia rubra vera is characterised by predominant erythrocyte hyperplasia. Prior to the diagnosis of polycythaemia vera in any patient with erythrocytosis it is essential to exclude spurious polycythaemia and secondary polycythaemia.
Polycythaemia vera is common among middle-aged and older patients. Often it is an incidental diagnosis in a patient with a high haemoglobin level in the full blood count done for some other reason. Ask about constitutional symptoms of lethargy, malaise and weight loss. Check for neurological symptoms, such as headache, vertigo, tinnitus and visual disturbances. Thrombotic stroke is a possible complication. Some complain of pruritus, of which the precise aetiology is not clear. Ask about diaphoresis, which is due to hyperviscosity associated with erythrocytosis. Patients who present with erythema, warmness and pain in the distal extremities may be suffering from the condition called erythromelalgia, an association of polycythaemia vera. Enquire about bleeding in the form of epistaxis or gastrointestinal haemorrhage, which is a common presentation of this disease. Ask about acute joint pain due to gout.
Broad management objectives include: 1) prevent complications of raised haematocrit, such as thrombosis and hyperviscosity; 2) control organomegaly; and 3) manage the symptoms.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]ANAEMIA
An Hb level of < 13 g/dL in men and < 12 g/dL in women is considered anaemic in common practice.
Ask about:
• the symptoms at presentation—patients may present with constitutional symptoms such as lethargy, malaise, exertional dyspnoea, fatigue or gastrointestinal blood loss in the form of haematemesis or melaena
• how long the patient has had anaemia—if they have had it since childhood, the likely diagnosis of a haemoglobinopathy should be considered
• easy bruising, uncontrolled blood loss, large joint arthropathy and excessive bleeding post-surgery—features suggesting a coagulation disorder/bleeding diathesis
• other symptoms that would help identify the aetiology, such as change in bowel habits or weight loss suggesting colonic cancer, abdominal discomfort, bloating, anorexia and weight loss of gastric carcinoma, abdominal pain suggesting peptic ulcer disease, night sweats, weight loss and fevers suggesting lymphoma.
Take a detailed medication history, looking for evidence of therapy with non-steroidal antiinflammatory agents, sulfur-containing medications (haemolysis) and myelosuppressive agents. Enquire about alcoholism and general nutrition. Check whether the patient has had any transfusions and whether there have been any adverse transfusion reactions or transmission of blood-borne infections such as hepatitis C and HIV. It is also useful to know the patient’s blood group.
Look for features of anaemia such as palmar crease pallor, conjunctival pallor, tachypnoea, tachycardia and evidence of high-output cardiac failure. Look for signs that suggest the likely aetiology (see box): characteristic copper-hue pigmentation (the combined effect of melanin deposition, icterus and pallor) of the thalassaemia patient with malar hyperplasia and often stunted growth, pigmented lesions in the mouth (Peutz-Jeghers syndrome), scleral icterus (haemolysis), epigastric tenderness (peptic ulcer disease), abdominal distension or mass lesions, splenomegaly, hepatomegaly, bony tenderness (malignancy), lymphadenopathy (lymphoma), impaired cognition, peripheral neuropathy and dorsal column signs (vitamin B
12 deficiency), arthropathy (anaemia of chronic disease or gastrointestinal blood loss secondary to NSAID use), slate-grey pigmentation, peripheral arteriovenous fistula, vascular access device or abdominal Tenckhoff catheter (renal anaemia).
Anaemia in the long case patient is often multifactorial. The diagnostic work-up should include a comprehensive battery of tests as guided by the clinical setting. Iron deficiency secondary to chronic blood loss, commonly from the gastrointestinal tract, is the most common cause of anaemia in this group of patients. Other differential diagnoses encountered commonly include anaemia of chronic disease, vitamin B12 and folate deficiency, myelodysplasia, aplastic anaemia, sideroblastic anaemia, myelosuppression, chronic renal failure, myelofibrosis and chronic alcoholism (due to a combination of bone marrow toxicity of alcohol, iron and folate deficiency and gastrointestinal bleeding).
Investigation of anaemia should be guided by the clinical picture. The more common conditions should be excluded first. Relevant investigations in the anaemic patient include:
Blood picture: Patients with anaemia of chronic disease usually have normocytic, normochromic anaemia. Those with iron deficiency, thallasaemia or sideroblastic anaemia have microcytic, hypochromic anaemia. Microcytic, hypochromic anaemia is also seen in myelodysplastic syndrome.
2. Coagulation profile—international normalised ratio (INR), activated partial thromboplastin time (APTT), bleeding time and clotting time, to exclude a bleeding diathesis
3. Haematinics—iron studies in the form of serum ferritin, serum iron and transferrin saturation, to exclude iron deficiency
4. Serum vitamin B12 and serum and red cell folate levels
5. Haemolytic screen—including blood film, serum haptoglobin level, serum lactate dehydrogenase (LDH) level, serum bilirubin level, the direct Coombs’ test, haemoglobin electrophoresis, Heinz body preparation and, if indicated, the cold agglutinin test
6. Erythrocyte sedimentation rate (ESR)—looking for evidence of chronic inflammation and paraproteinaemia
7. Electrolyte profile and the renal function indices—to exclude renal failure as a cause
8. Erythropoietin levels
9. Thyroid function tests—to exclude hypothyroidism, which can cause a macrocytic anaemia
10. Chest X-ray—to exclude chronic pulmonary disease and pulmonary malignancies
11. Serum protein electrophoresis and immunoelectrophoresis—to exclude any paraproteinaemia
12. If all non-invasive and minimally invasive tests do not help in the correct diagnosis, the next investigation is a bone marrow biopsy with both aspiration and trephine.
Management depends on severity (level of Hb) and the patient’s cardiopulmonary status. Initial steps include identification and correction of reversible, causative factors and replacement of blood with packed cell transfusions and/or the supplementation of haematinics as indicated.
As a general rule, blood transfusion is indicated if Hb is < 7 g/dL. If Hb remains > 10 g/dL, transfusion should be considered only if there is a compelling clinical indication such as precipitation of coronary ischaemia or heart failure.
Gastrointestinal bleeding warrants endoscopy (colonoscopy or gastroscopy) to identify the exact location of the bleeding and for possible therapy or biopsy of suspicious lesions. If gastroscopy and colonoscopy fail to identify the source of bleeding, capsule endoscopy may be warranted, to look for a small intestinal focus.
Patients with chronic renal failure need iron supplementation and at times may need IV iron infusion. Patients with kidney disease may benefit from therapy with erythropoietin or darbapoietin.
It is recommended that patients with chronic kidney disease be maintained at an Hb level of 11 g/dL or more.
Patients with anaemia of chronic disease improve when the underlying disease is well treated. Haematinic replacement and therapy with erythropoietin or darbapoietin is also indicated in this group.
Management of anaemia due to cytotoxic chemotherapy in patients with non-myeloid malignancies is similar to the management of anaemia of chronic disease.
Haematological malignancies are commonly encountered in the long case. History taking should enquire into the events surrounding the diagnosis, such as presenting symptoms of fatigue, lethargy, weight loss, fever, night sweats, bone pain, how the diagnosis was made, the various investigations that have been carried out, including bone marrow biopsy and/or excision biopsy of lymph nodes, and the treatment given so far, including blood transfusions, chemotherapy and bone marrow or stem cell transplantation. Ask about the side effects of chemotherapy and any episodes of febrile neutropenia, life-threatening sepsis etc. Ask how the patient is coping with the disease, what knowledge they have about the prognosis of the condition, what expectations they have for the future, and the level of social support available.
Following is a discussion of the salient points relevant to the commonly encountered haematological malignancies. The candidate is expected to have an up-to-date knowledge of each disease and to be able to handle wisely the complicated clinical issues associated with the management of these conditions.
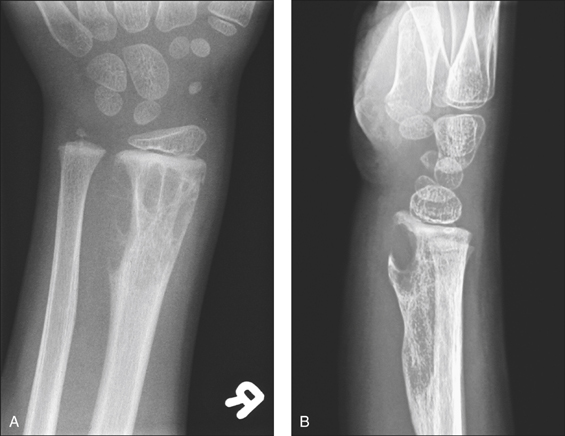
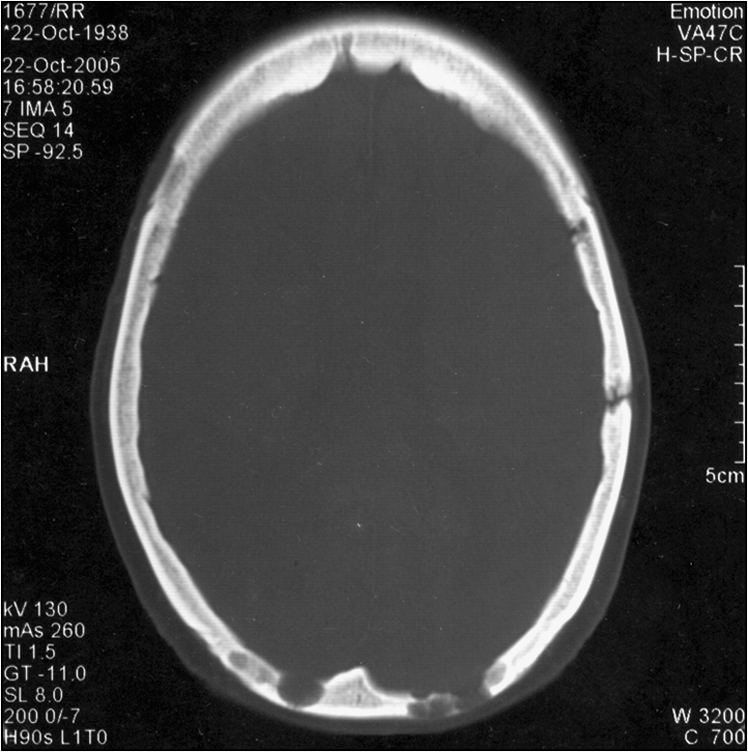
A 56-year-old man presents with significant lethargy, severe back pain, loss of sensation in the lower extremities and dizziness. He also complains of bleeding gums and frequent blurring of vision. Physical examination reveals mucosal and conjunctival pallor and bony tenderness of the lower back. There are signs of peripheral neuropathy. His blood tests reveal anaemia, renal impairment and hypercalcaemia.
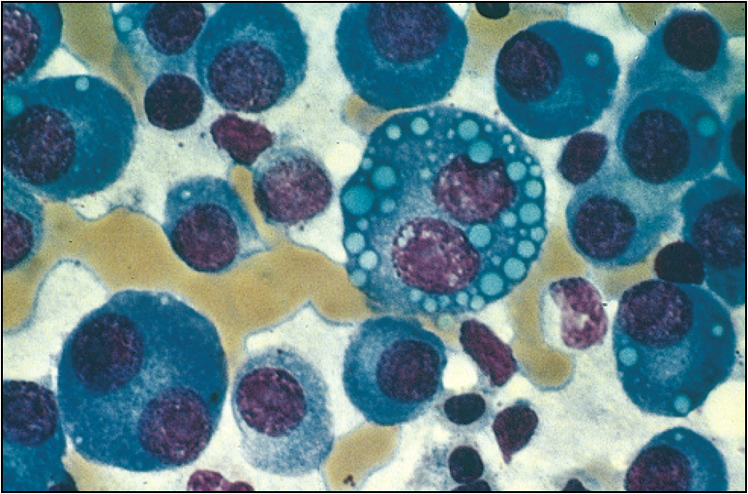
Multiple myeloma is a common malignant condition encountered in the long case setting, because patients with multiple myeloma can have many secondary medical complications. Multiple myeloma is due to disordered proliferation of a clone of plasma cells secreting monoclonal immunoglobulins. The clinical presentation of multiple myeloma is variable.
Ask about pain, bony fractures, lethargy, fatigue, dyspnoea, mucosal bleeding, fevers and rigors. Enquire about loss of sensation, limb weakness, visual problems and dizziness.
Some common presentations are intractable bone pain, pathological fracture, symptomatic anaemia, pancytopenia, sepsis, renal failure or acute symptomatic hypercalcaemia. Some may present with the hyperviscosity syndrome, with associated neuropathy and coagulopathy (oral and nasal bleeding, blurred vision, headache and vertigo) or amyloidosis. Hyperviscosity syndrome is more common in Waldenström’s macroglobulinaemia, where the paraprotein is IgM. The most common presenting symptom, however, is bone pain, particularly in the vertebral column. Multiple myeloma should be suspected in any patient over the age of 40 years presenting with bone pain, pathological fracture, osteoporosis, lethargy, anaemia and recurrent infections. Incidental discovery of proteinuria, hypercalcaemia, acute renal failure, high ESR or rouleaux formation should alert the clinician to look for multiple myeloma.
Look for evidence of anaemia and sepsis. Do not forget to look at the temperature chart. Check the mucosa for bleeding. Perform a thorough neurological examination. Look for bony fractures and assess for bone tenderness.
For the diagnosis of multiple myeloma there should be one major criterion together with one minor criterion or three minor criteria present—these should be clinically manifest.
(Adapted from Grogan T M 2001 Plasma cell neoplasms. In: Jaffe E S, Harris N L, Stein H et al (eds) World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon, p 142)
(Adapted from Grogan T M 2001 Plasma cell neoplasms. In: Jaffe E S, Harris N L, Stein H et al (eds) World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon, p 142)
(From Greipp P R, San Miguel J, Durie B J et al 2005 International staging system for multiple myeloma. Journal of Clinical Oncology 23(15):3412–3420)
(From Greipp P R, San Miguel J, Durie B J et al 2005 International staging system for multiple myeloma. Journal of Clinical Oncology 23(15):3412–3420)
Emergent management of acute complications:
General management strategy:
If the disease relapses or is progressive:
Mastering the Medical Long Case