[level-membership-for-physical-medicine-and-rehabilitation-category]CHAPTER 135
Myopathies
Definition
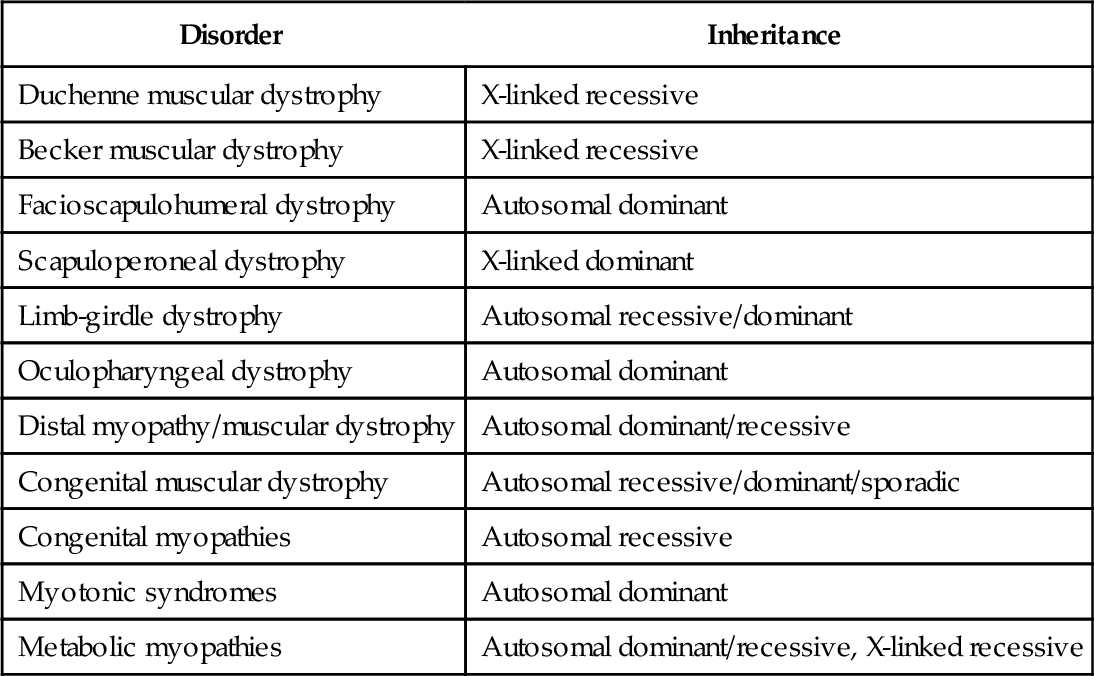
Myopathy is the common name for diseases derived from the muscle. Myopathies have different causes and different courses, that is, they may have acute, subacute, or chronic presentations (Table 135.1). Myopathies affect proximal or distal muscle groups; some of them also affect heart muscle, leading to cardiomyopathy [1]. Many myopathies are inherited disorders (Table 135.2); in an increasing number of disorders, the genetic background and the abnormal or missing muscle protein have been revealed during the last decade. An updated gene table is published online [2]. There is also a gene table updated for 2012 and published in Neuromuscular Disorders [3]. Muscle diseases are rare, with a prevalence of approximately 50 per 100,000. Myopathy is more common in females than in males, although Duchenne muscular dystrophy is found only in males because of an X-linked inheritance.
Muscular Dystrophies
Muscular dystrophies are inherited disorders of muscle due to abnormal structural muscle proteins, for example, dystrophin in Duchenne muscular dystrophy and sarcoglycans, calpain, and dysferlin in different limb-girdle dystrophies [2,3]. They are characterized by progressive course and early onset.
Congenital Myopathies
Congenital myopathies form a clinically heterogeneous group characterized by findings on muscle biopsy. They are slowly progressive or nonprogressive and usually are manifested in the neonatal period.
Metabolic Myopathies Including Mitochondrial Myopathies
Metabolic myopathies form a clinically heterogeneous group of muscle disorders resulting from inherited defects in intracellular energy production. They may be manifested as cramps and myoglobinuria. Patients with cramps and myoglobinuria often have disorders in the glycogen or lipid metabolism pathways. They may be asymptomatic at rest, but symptoms develop after exercise. Mitochondrial myopathies may be a part of a neurologic syndrome often involving the central nervous system.
Inflammatory Myopathies
Inflammatory myopathies are characterized by inflammatory changes in the muscle and are associated with infections or an immunologic process. They are divided into polymyositis, dermatomyositis, and inclusion body myositis [4]. The course is acute or subacute and is almost always associated with an elevated serum creatine kinase (CK) level.
Drug-Induced and Endocrine Myopathies
Drug-induced myopathies are caused by different drugs, for example, colchicine, azidothymidine (AZT), chloroquine, hydroxychloroquine, and corticosteroids. Myopathy due to intake of statins, the cholesterol-lowering agents, has been reported [5]. Endocrine myopathies include both hyperthyroid and hypothyroid myopathies as well as myopathy due to hyperparathyroidism.
Myotonic Syndromes
A number of disorders are associated with clinical or electrical myotonia. The myotonic disorders are divided into myotonic dystrophies and “pure” myotonia [6]. The myotonias are inherited disorders due to alterations in ion channels in the muscle and seldom give rise to persistent muscle weakness. Myotonic dystrophy (MD 1) or Steinert disease exists in a congenital and an adult form. Individuals with myotonic dystrophy may not notice any problems until adolescence or early adult life [7]. The first symptom may be difficulty in releasing an object due to myotonia. Progressive weakness with onset in distal muscles follows. The genetic explanation to MD 1 is an expansion of a CTG repeat in chromosome 19 (DMPK gene). The gene encodes a protein kinase that occurs in different tissues, leading, for example, to cardiomyopathy. The clinical affection is correlated to the number of repeats, and the number of repeats increases between generations, leading to a clinical anticipation. There is also a more rare form with later onset, a slower course, and proximal muscle weakness (MD 2). The genetic background to MD 2 is a CCTG repeat in chromosome 3 (ZNF9 gene) [8].
Symptoms
Muscle weakness, most often affecting proximal muscles, is the cardinal symptom. Almost all myopathies affect the proximal muscles to the greatest extent. Another prominent symptom is muscle fatigue. The earliest symptoms are often related to weakness of the hip and proximal leg muscles; patients experience difficulty in rising from a chair and often require support of their arms. Compensation for leg extensor weakness by bracing of the legs with the hands and climbing with the hands on the legs when rising to a standing position is known as the Gower maneuver. Walking up and down stairs may be difficult because of quadriceps and hip extensor weakness, respectively. Weakness of proximal muscles in the upper extremities can be manifested as fatigue or inability to perform overhead tasks, such as hair brushing, brushing teeth, and lifting objects to elevated shelves.
In hereditary distal myopathies [9] and inclusion body myositis, the distal muscle weakness leads to footdrop and ankle instability as well as difficulty with manual tasks, such as turning doorknobs and opening jars.
Pain is not a common symptom in myopathies. However, inflammatory and metabolic myopathies are associated with pain. Exercise-induced muscle pain suggests a metabolic myopathy. The pain has an aching, dull, and crampy quality and is usually poorly localized. An exercise-induced weakness suggests a neuromuscular junction disorder. Symptoms of hypoventilation and cardiac failure should be considered.
Physical Examination
A general examination is performed to assess for signs of cardiac failure and rashes found in dermatomyositis. Muscle atrophy assessment and testing of muscle strength are central and should involve examination of proximal and distal muscles in all extremities as well as facial muscles and neck flexors and extensors. Hip girdle muscles are best isolated for strength testing while the patient is in the supine and prone positions. The abilities of walking, rising from a chair (or floor in pediatric patients), and stepping onto a low chair are often helpful in evaluating leg weakness. Examination of the shoulder may reveal winging of the scapula, a characteristic finding in facioscapulohumeral muscular dystrophy. Facial weakness and temporalis muscle wasting are also present in facioscapulohumeral muscular dystrophy and in myotonic dystrophy.
Range of motion at joints should be examined because contractures may have marked functional effects. Reflexes should be normal or decreased proportional to muscle weakness in myopathies. Abnormalities on sensory testing suggest involvement of sensory nerves (i.e., a neuromuscular disorder).
Functional Limitations
The most common functional limitations are related to the prominent symptom of proximal weakness, which can have a marked effect on transfers, ascending and descending stairs, and ambulation. In severe myopathies, patients may be restricted to wheelchair mobility. Proximal upper extremity weakness can interfere with activities of daily living, such as dressing, grooming, and cooking. Fatigue is common secondary to the increased effort required with weakened muscles. Cardiac failure and respiratory insufficiency requiring ventilation can result in difficulty with daily activities and increased fatigue. The dysphagia involved in some myopathies may make eating time-consuming and difficult.
Diagnostic Studies
The serum CK concentration is the most important test in myopathies. Patients with Duchenne muscular dystrophy may have very high CK values. Serum CK concentration is usually elevated in inflammatory myopathies and metabolic myopathies. It is often normal or nearly normal in congenital myopathies.
Exercise, especially if it is strenuous or performed in a sedentary individual, can cause marked CK elevation. Thus, patients should be advised to abstain from strenuous exercise for 5 days before serum CK testing. CK concentration may also be elevated in neuromuscular disorders, such as motor neuron disease.
Nerve conduction studies are important in cases of suspected myopathy. Sensory and motor nerve conduction studies are usually normal. However, distal compound muscle action potentials may be reduced. Exceptions include patients with inclusion body myositis (30% have a sensory or sensorimotor polyneuropathy) [4]. In cases with abnormal nerve conduction velocities, coexisting neuropathies such as diabetic polyneuropathy should be considered.
The electromyographic examination is helpful in the evaluation of myopathies. Primarily, the size of motor units is decreased by the dysfunction or loss of individual muscle fibers, leading to motor unit action potentials that characteristically have decreased duration, decreased amplitude, and increased phases in contrast with the neuropathic motor unit action potential findings. In addition, denervating potentials (fibrillations and positive sharp waves) occur in many myopathic disorders.
Muscle biopsy is often useful in the diagnosis of myopathy. The selection of muscle for biopsy is important because a muscle that is end stage is likely to show only fibrotic replacement of muscle tissue, and an unaffected muscle may be normal. In an acute myopathy, it is best to select a muscle for biopsy that is clinically weak; in a chronic myopathy, it is preferable to select a muscle that is only mildly weak. The muscle selected should not have been sampled by needle electromyography, which may result in temporary inflammation. The pathologist, to investigate possible causes, performs specific histochemical and immunohistochemical stains and occasionally electron microscopy on the specimen.
Magnetic resonance imaging may be an additional diagnostic tool in clinical practice. The pattern of muscle involvement is often specific for the different entities [10].
Genetic testing is nowadays routine in the evaluation of many of the muscular dystrophies and is also useful in the evaluation of other chronic myopathies. With similar phenotypes, such as limb-girdle muscular dystrophy, the only way to accurately diagnose the condition is by genetic testing. A helpful resource is the gene table published online [2].
Treatment
Initial
Of great importance is an initial detailed explanation to the patient and relatives. Patients are informed that they must not exert themselves to the point of exhaustion. Referral to the Muscular Dystrophy Association is helpful for education and support of the patient.
Steroids are often effective in the treatment of inflammatory myopathies (as are other immunosuppressants) and have been shown to slow progression in some muscular dystrophies. In a recent Cochrane report [11], corticosteroids were shown to improve muscle strength and function in Duchenne muscular dystrophy in the short term. Carnitine is used in lipid storage myopathies. Much work is now devoted to development of new therapeutic strategies, including gene [12] and stem cell [13] therapies. Otherwise, the only therapeutic option is symptomatic treatment (e.g., pain treatment with different analgesics).
Rehabilitation
Physical therapy and occupational therapy are often necessary for gait training and stretching. Assistive devices, such as canes, walkers, and wheelchairs, as indicated in a particular patient, can minimize disability. Assistive devices should be used, preferably after training with a physical therapist. Bracing may be helpful for footdrop.
Adaptive equipment may be prescribed to assist a patient to perform daily activities. Home adaptations, such as tub bars and entrance ramps, may be of great assistance to patients with proximal muscle weakness.
Exercise can help maintain joint range of motion. There has been debate about whether patients with muscle disorders benefit from exercise training. There is an increasing knowledge on the effect of exercise training in different muscle disorders [14,15]. High-resistance training at submaximal level seems to be beneficial in slowly progressive disorders in the short term. In rapidly progressive disorders, such as Duchenne muscular dystrophy, high-resistance training is questionable [14]. On the basis of this, moderate exercise, not to the point of exhaustion, is preferable. It is important to start exercise training early in the course of the disease so that there are still trainable muscle fibers left.
The common opinion on exercise training in inflammatory myopathies has changed during recent years. Patients with inflammatory myopathies have been strongly advised to avoid exercise. However, results from training studies have shown that exercise training has beneficial effects on muscle function [16].
Procedures
Assisted ventilation, including negative pressure ventilation, noninvasive positive pressure ventilation, and invasive positive pressure ventilation (i.e., endotracheal tube or tracheostomy), may be indicated for patients with insufficient ventilation [17]. Feeding tubes may be necessary for patients with dysphagia due to severe bulbar myopathy.
Surgery
Patients with muscular dystrophies may require contracture release and spine stabilization surgeries.
Potential Disease Complications
Severe myopathies, including muscular dystrophies, can cause restrictive pulmonary disease due to chest wall muscle weakness and scoliosis. Assisted ventilation, including negative pressure ventilation, noninvasive positive pressure ventilation, and invasive positive pressure ventilation (by endotracheal tube or tracheostomy), may be indicated. Decreased mobility can result from proximal lower and upper extremity weakness. Contractures can be caused by disuse and fibrosis as well as by scoliosis due to paraspinal muscle weakness. Cardiac involvement is present in some forms of muscle disorders [1]. Gastrointestinal symptoms due to smooth muscle involvement are common in patients with muscular dystrophies.
Potential Treatment Complications
Immunosuppression may have associated side effects. Steroid use and decreased mobility may lead to osteoporosis and the subsequent risk of pathologic fractures, and analgesics have well-known side effects. Special attention should be paid to patients undergoing general anesthesia because of the risk of malignant hyperthermia, which is a potentially lethal condition appearing in different myopathies, including a specific disorder with a mutation in the skeletal muscle ryanodine receptor.
[/level-membership-for-physical-medicine-and-rehabilitation-category][not-level-membership-for-physical-medicine-and-rehabilitation-category]CHAPTER 135
Myopathies
Definition
Myopathy is the common name for diseases derived from the muscle. Myopathies have different causes and different courses, that is, they may have acute, subacute, or chronic presentations (Table 135.1). Myopathies affect proximal or distal muscle groups; some of them also affect heart muscle, leading to cardiomyopathy [1]. Many myopathies are inherited disorders (Table 135.2); in an increasing number of disorders, the genetic background and the abnormal or missing muscle protein have been revealed during the last decade. An updated gene table is published online [2]. There is also a gene table updated for 2012 and published in Neuromuscular Disorders [3]. Muscle diseases are rare, with a prevalence of approximately 50 per 100,000. Myopathy is more common in females than in males, although Duchenne muscular dystrophy is found only in males because of an X-linked inheritance.
Muscular Dystrophies
Muscular dystrophies are inherited disorders of muscle due to abnormal structural muscle proteins, for example, dystrophin in Duchenne muscular dystrophy and sarcoglycans, calpain, and dysferlin in different limb-girdle dystrophies [2,3]. They are characterized by progressive course and early onset.
Congenital Myopathies
Congenital myopathies form a clinically heterogeneous group characterized by findings on muscle biopsy. They are slowly progressive or nonprogressive and usually are manifested in the neonatal period.
Metabolic Myopathies Including Mitochondrial Myopathies
Metabolic myopathies form a clinically heterogeneous group of muscle disorders resulting from inherited defects in intracellular energy production. They may be manifested as cramps and myoglobinuria. Patients with cramps and myoglobinuria often have disorders in the glycogen or lipid metabolism pathways. They may be asymptomatic at rest, but symptoms develop after exercise. Mitochondrial myopathies may be a part of a neurologic syndrome often involving the central nervous system.
Inflammatory Myopathies
Inflammatory myopathies are characterized by inflammatory changes in the muscle and are associated with infections or an immunologic process. They are divided into polymyositis, dermatomyositis, and inclusion body myositis [4]. The course is acute or subacute and is almost always associated with an elevated serum creatine kinase (CK) level.
Drug-Induced and Endocrine Myopathies
[/not-level-membership-for-physical-medicine-and-rehabilitation-category]
