[level-membership-for-physical-medicine-and-rehabilitation-category]CHAPTER 132
Motor Neuron Disease
Nanette C. Joyce, DO, MAS; Gregory T. Carter, MD, MS
Definition
The term motor neuron disease encompasses a heterogeneous group of progressive neuromuscular disorders characterized by the selective loss of upper or lower motor neurons. However, motor neuron disease is also used interchangeably with amyotrophic lateral sclerosis (ALS), which can be confusing for the uninitiated. ALS is the most common adult motor neuron disease. For the diagnostic criteria for ALS to be met, both upper and lower motor neuron involvement is necessary [1,2]. In sporadic cases with only lower motor neuron dysfunction, the disease is called progressive muscular atrophy; if upper motor neuron dysfunction is singularly present, it is primary lateral sclerosis; and if dysfunction is localized to the bulbar region, the disease is called progressive bulbar palsy. Most patients initially diagnosed as having progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy eventually progress to meet diagnostic criteria for ALS [3]. Those who do not convert have a slower rate of disease progression. This chapter focuses on ALS as the management principles are similar for the entire class of motor neuron diseases.
The prevalence of ALS is about 6 to 8 per 100,000 people, with an annual incidence of approximately 2 cases per 100,000 people. Men are more commonly affected than women, with a ratio nearing 2:1. Most cases of ALS are sporadic, having unknown etiology. Only 5% to 10% of patients have familial ALS, which is most commonly transmitted in an autosomal dominant fashion. Approximately 30% to 40% of familial cases in the United States and Europe are caused by mutations in the C9orf72 gene; 20% worldwide are caused by mutations in the SOD1 (superoxide dismutase) gene; and rarer forms of familial ALS have been linked to mutations in the TARDBP, FUS, ANG, ALS2, SETX, and VAPB genes [4,5]. Other inherited adult motor neuron diseases are Kennedy disease (X-linked recessive) and adult spinal muscular atrophy (autosomal recessive), which are purely lower motor neuron disorders with greatly increased life span compared with ALS.
ALS causes rapid, progressive skeletal muscle weakness and atrophy, leading to premature death by respiratory failure. Weakness begins in a focal region, such as a single limb, the bulbar muscles, or the respiratory muscles, and spreads to affect other regions. Extraocular muscles and bowel and bladder sphincter function are often spared until late in the disease course. Mean age at onset is the mid-50s, but ALS may develop in adults of any age. Rare juvenile familial forms exist with onset before the age of 25 years. Mean survival, without tracheostomy, is 3 years from diagnosis but ranges from less than 1 year to more than 20 years. One explanation for this extreme variability is that ALS is multiple disorders without a single etiology but rather with multiple causes, sharing a common final step in the pathophysiologic pathway—motor neuron apoptosis. This is illustrated by the varying phenotypes associated with familial forms of ALS [3]. Theories about the pathogenesis of sporadic ALS have implicated RNA toxicity, glutamate excitotoxicity, oxidative stress, neuroinflammation, protein misfolding, glial cell activation, and mitochondrial dysfunction, to name a few [5].
Symptoms
Early symptoms of ALS can be subtle and include muscle twitching and cramping, weakness, and loss of coordination. Patients with a predominantly upper motor neuron syndrome often present with muscle stiffness, weakness, loss of dexterity, and loss of voluntary motor control from spasticity that may affect vocal quality or limb function. Patients with a predominantly lower motor neuron syndrome may present with weakness and muscle atrophy, fasciculations, muscle cramps, and flaccid dysarthria. Bulbar symptoms include dysarthria, dysphagia, sialorrhea (drooling), and pseudobulbar affect—laughing or crying in exaggeration of or incongruent with mood. Symptoms are initially painless and asymmetric across limbs. As the disease relentlessly progresses, weakness and atrophy spread to affect all skeletal muscles, causing significant impairment and disability. If patients do not succumb first to respiratory failure, they will ultimately transition from independent function to total dependence.
Respiratory failure is the presenting symptom in a rare few. Constitutional symptoms of weight loss and generalized fatigue are common. Cognitive symptoms including behavioral or executive dysfunction have been reported to occur in 33% to 51% of patients [6]. Most have milder symptoms; approximately 5% to 14% meet clinical criteria for a diagnosis of frontotemporal dementia [6].
Physical Examination
Physical examination of a patient with suspected motor neuron disease should be aimed at establishing the certainty of diagnosis. ALS is diagnosed clinically, and the patient with suspected motor neuron disease requires a thorough neurologic examination, assessing each of the four major body regions (bulbar, cervical, thoracic, and lumbar) for signs of upper and lower motor neuron involvement. The “gold standard” for the diagnosis of upper motor neuron disease is establishment of the presence of pathologic reflexes—a brisk jaw jerk, Hoffmann sign, abdominal skin reflex, and Babinski sign. Increased muscle stretch reflex responses as demonstrated by increased spread and amplitude or clonus are considered pathologic. Reflexes that would be graded normal but are elicited from atrophied and weak muscles should also be considered pathologic. Evidence of lower motor neuron disease includes muscle weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Patients with ALS may be hyperreflexic and hyporeflexic, depending on the stage at which they are in the disease process and whether they have a predominance of upper or lower motor neuron phenotype. For example, hyperreflexia occurs in the patient with upper motor neuron dysfunction, but this sign can be overcome and silenced by concomitant lower motor neuron loss causing muscle atrophy and hyporeflexia. Because ALS is an asymmetric and spreading process, the upper motor neuron signs may be more predominant than the lower motor neuron signs, or vice versa, within any single limb or between limbs and body regions. These examination findings will change over time as the disease progresses. The tongue should be examined for fasciculations, atrophy, strength, and range of motion. The patient’s mental status, nonmotor cranial nerve function, sensory examination, and cerebellar examination findings are usually normal.
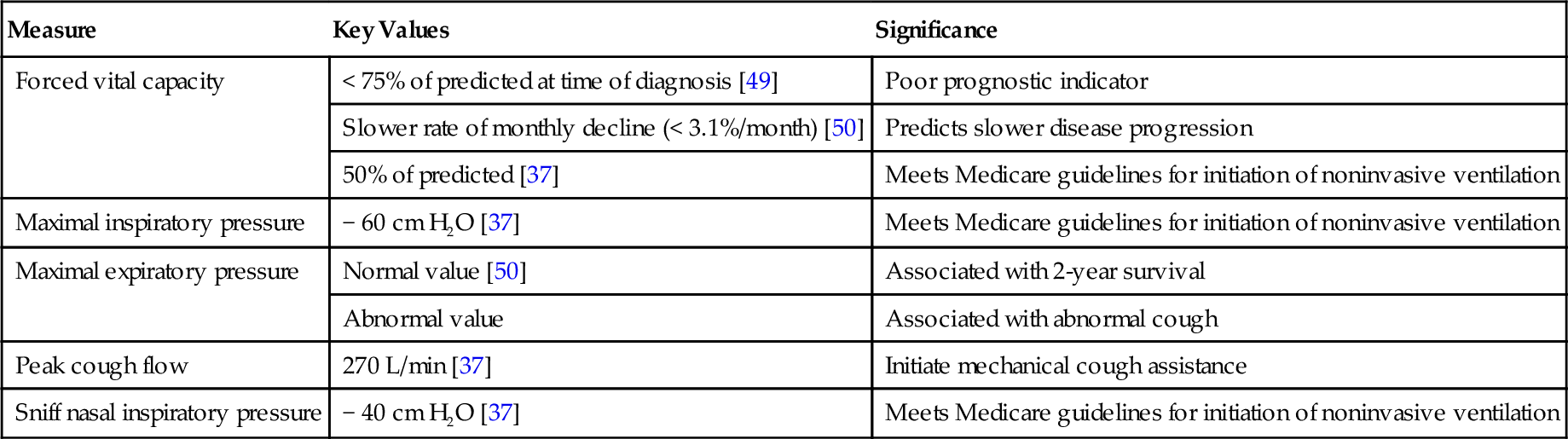
In patients with an established diagnosis, the physical examination documents disease progression and includes the musculoskeletal and cardiorespiratory systems in addition to the neurologic evaluation. The musculoskeletal examination focuses on assessment of range of motion and evaluation of painful joints or soft tissue structures. Because progressive respiratory failure is a ubiquitous manifestation of ALS, follow-up appointments should be scheduled regularly (i.e., every 3 months) and the cardiorespiratory system assessed at each visit. Forced vital capacity (FVC) and maximal inspiratory, maximal expiratory, and peak cough pressures can be measured with a spirometer in the office setting and should be considered part of the regular cardiorespiratory follow-up evaluation, providing relevant information for clinical decision-making and prognosis (Table 132.1).
Table 132.1
Pulmonary Function Testing in Amyotrophic Lateral Sclerosis
| Measure | Key Values | Significance |
| Forced vital capacity | < 75% of predicted at time of diagnosis [49] | Poor prognostic indicator |
| Slower rate of monthly decline (< 3.1%/month) [50] | Predicts slower disease progression | |
| 50% of predicted [37] | Meets Medicare guidelines for initiation of noninvasive ventilation | |
| Maximal inspiratory pressure | − 60 cm H2O [37] | Meets Medicare guidelines for initiation of noninvasive ventilation |
| Maximal expiratory pressure | Normal value [50] | Associated with 2-year survival |
| Abnormal value | Associated with abnormal cough | |
| Peak cough flow | 270 L/min [37] | Initiate mechanical cough assistance |
| Sniff nasal inspiratory pressure | − 40 cm H2O [37] | Meets Medicare guidelines for initiation of noninvasive ventilation |
Functional Limitations
The pattern of progressive functional limitation is directly related to the patient’s motor neuron disease phenotype. Bulbar-onset ALS initially affects the patient’s ability to speak and to swallow and typically spreads to involve the muscles of the upper extremities before the lower extremities [7]. These patients have a difficult time maintaining their weight both because of dysphagia and because of the loss of upper limb strength that impairs their ability to feed themselves. Eventually, bulbar-predominant patients become anarthric with accompanying severe dysphagia that limits their ability to control their secretions and to swallow their own saliva.
The functional limitations that develop in patients with spinal-onset ALS are the direct or indirect result of muscle weakness and atrophy. In lumbar spine onset, gait is abnormal early in the disease secondary to footdrop or hip flexion weakness. As the disease progresses, the patient’s mobility worsens. Eventually, even the most basic activities of daily living become impossible to perform. Patients often transition quickly from independence to total dependence.
Reactive depression, generalized fatigue, and musculoskeletal pain may further limit function.
Diagnostic Studies
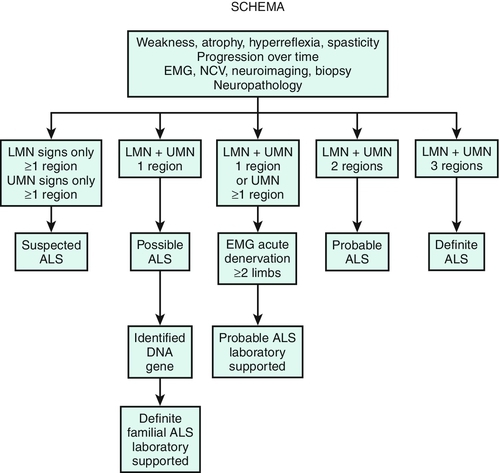
The diagnosis of ALS is based on the combined clinical and electrodiagnostic examinations. Neuroimaging and clinical laboratory studies are used to exclude other conditions that mimic ALS. All patients thought to have a motor neuron disease should undergo electrodiagnostic testing. The Awaji-shima revised El Escorial criteria (Table 132.2) are used to establish the certainty level of the diagnosis of ALS [1,2]. These criteria were developed as a tool for clinical trial enrollment but are commonly used in the clinic. The revised criteria classify the certainty level of diagnosis into one of three categories: definite, probable, and possible, which no longer includes the categories of Suspected ALS or Probable ALS Laboratory Supported that were part of the El Escorial criteria (Fig. 132.1). In addition to both upper and lower motor neuron findings, a diagnosis of ALS requires evidence of progressive spread of signs or symptoms within a single body region or from one of the four body regions to another. Certainty level of diagnosis depends on how many regions reveal upper motor neuron and lower motor neuron disease [1]. Electrophysiologic findings of denervation, including positive sharp waves, fibrillation potentials, and fasciculation potentials, are used to confirm lower motor neuron dysfunction in clinically affected regions and to detect subclinical lower motor neuron dysfunction, thereby extending the clinical examination. Signs of denervation observed during electromyography are now considered equivalent to lower motor neuron symptoms on clinical examination [1].
Table 132.2
Awaji-shima Revised El Escorial Criteria: Clinical Certainty Levels for the Diagnosis of Amyotrophic Lateral Sclerosis [1]
| Clinical Certainty | Clinical or Electrophysiologic Evidence |
| Clinically definite | UMN and LMN findings in at least three body regions |
| Clinically probable | UMN and LMN findings in at least two body regions with UMN findings rostral to LMN findings |
| Clinically possible | UMN and LMN findings in one body region, or UMN findings in at least two body regions without LMN findings, or LMN findings that are rostral to UMN findings |

LMN, lower motor neuron; UMN, upper motor neuron. Four body regions: bulbar, cervical, thoracic, lumbar.
Imaging studies are used to exclude possibilities other than motor neuron disease from the differential diagnosis. Magnetic resonance imaging is the primary imaging modality in the evaluation of patients with suspected ALS. Almost all patients should have magnetic resonance imaging of the cervical spine to exclude cord compression, syrinx, or other spinal cord disease. The location of symptoms will dictate whether other regions of the spinal cord should be imaged. In those presenting with bulbar symptoms, brain magnetic resonance imaging should be performed to exclude stroke, tumor, syringobulbia, and other pathologic processes.
In most neuromuscular clinics, a routine panel of laboratory tests is performed for all patients thought to have ALS. A suggested set of such tests is provided in Table 132.3. The rationale behind the performance of this extensive battery of tests is to assess the general health of the patient and to exclude treatable conditions. The differential diagnosis, developed after the history and physical examination, may suggest that more specialized testing be performed. Table 132.4 suggests additional tests that may be warranted when the presentation is atypical with a progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy phenotype. When there is a family history of motor neuron disease, genetic testing may be considered.
Table 132.4
Specialized Laboratory Testing
| Phenotype | Test | Diagnosis Excluded |
| Progressive muscular atrophy | DNA test: CAG repeat on X chromosome | Kennedy disease |
| DNA test: SMN gene mutation | Spinal muscular atrophy | |
| Hexosaminidase A | Hexosaminidase A deficiency (heterozygous Tay-Sachs disease) | |
| Voltage-gated calcium channel antibody test | Lambert-Eaton myasthenic syndrome | |
| Cerebrospinal fluid examination | Polyradiculopathy, infectious or neoplastic | |
| GM1 antibody panel | Multifocal motor neuropathy | |
| Primary lateral sclerosis | Very long chain fatty acids | Adrenoleukodystrophy |
| Human T-lymphotropic virus 1 (HTLV-1) antibodies | HTLV-1 myelopathy (tropical spastic paraparesis) | |
| Parathyroid hormone | Hyperparathyroid myelopathy | |
| Cerebrospinal fluid examination | Multiple sclerosis | |
| Progressive bulbar palsy | Acetylcholine receptor antibodies | Myasthenia gravis |
| DNA test: CAG repeat on X chromosome | Kennedy disease | |
| Cerebrospinal fluid examination | Multiple sclerosis |
From Krivickas LS. Motor neuron disease. In Frontera WR, Silver JK, Rizzo TD Jr, eds. Essentials of Physical Medicine and Rehabilitation, 2nd ed. Philadelphia, WB Saunders, 2008.
Treatment
Initial
Pharmacologic
There is no cure for ALS; however, ongoing research is focused on identifying disease mechanisms and finding drugs to slow disease progression. Riluzole (Rilutek) is the only medication approved by the Food and Drug Administration (FDA) specifically for treatment of ALS. It is an antiglutamate agent that was approved by the FDA for treatment of ALS in 1995 after two clinical trials showed that the drug slowed disease progression [8,9]. A Cochrane review reporting a meta-analysis of riluzole clinical trials showed a survival benefit of approximately 2 months [10]. Unfortunately, no functional benefit was derived as strength declined at a rate similar to that in those taking riluzole and placebo. The recommended dose of riluzole is 50 mg twice daily. The most common adverse side effects are fatigue, nausea, and elevation of hepatic enzymes. Riluzole is contraindicated in those with hepatic enzyme activity of more than five times the upper limit of normal.
Offering patients pharmacologic treatment of their disease and access to clinical trials has psychological benefits that may outweigh the actual slowing of disease progression currently able to be achieved. The following is an abbreviated list of compounds and biologics that are currently in clinical trial for ALS: arimoclomol, creatine combined with tamoxifen, edaravone, neural stem cells, and mesenchymal stem cells. Negative ALS clinical trials have tested many therapeutics and supplements, including multiple growth factors, dexpramipexole, creatine, lithium, ceftriaxone, glutamate antagonists, anti-inflammatory agents (celecoxib), calcium channel blockers, and amino acids [11]. Whereas many ALS patients take multiple supplements, no double-blind, placebo-controlled trials have proved their efficacy. In the future, a cocktail approach to slowing of disease progression may be the ideal treatment strategy [12].
Symptomatic
A number of drugs are useful for treatment of spasticity, sialorrhea, pseudobulbar affect, depression, and anxiety (Table 132.5).

Spasticity requires treatment only if it interferes with function. Nonpharmacologic management involves teaching patients stretching exercises and positioning techniques that decrease muscle tone. Baclofen (Lioresal) is the most effective pharmacologic agent, followed by tizanidine (Zanaflex). Diazepam (Valium) should be avoided because it may suppress respiration, and dantrolene (Dantrium) is not recommended because it causes excessive muscle weakness. In general, pharmacologic management of spasticity is less successful in ALS than in multiple sclerosis or spinal cord injury because the lower motor neuron component of ALS makes patients extremely susceptible to the development of excessive weakness.
Patients with bulbar dysfunction experience sialorrhea because they have difficulty swallowing and managing the oral secretions they normally produce. A variety of anticholinergic drugs may be used to dry the mouth. Tricyclic antidepressants are often tried first but may not be tolerated because of adverse side effects (excessive dry mouth, somnolence, urinary retention). One benefit of the tricyclic antidepressants is that they may treat other ALS-related symptoms, such as pseudobulbar affect, insomnia, and pain. Tricyclic antidepressants are contraindicated in patients with cardiac arrhythmia or conduction disorder. When tricyclic antidepressants are not tolerated, a scopolamine patch (Transderm Scōp) or glycopyrrolate (Robinul) may be helpful. If these treatments are inadequate, patients may benefit from salivary gland botulinum toxin injection [13–15].
Pseudobulbar affect, sometimes called emotional incontinence, may become distressing for both the patient and the family. Amitriptyline (Elavil) or fluvoxamine (Luvox) may help blunt the intensity of these inappropriate or exaggerated reactions. A combination preparation of dextromethorphan and quinidine (Nuedexta) was found to be effective in a large randomized clinical trial [16] and was granted FDA approval in late 2010 for the treatment of pseudobulbar affect. Nuedexta is contraindicated in patients with heart failure, long QT syndrome, and conduction block.
Reactive depression and anxiety are both normal responses to a diagnosis of ALS [17]. Patients and their families may benefit from individual counseling and participation in ALS support groups. Anxiety may be treated with benzodiazepines as long as the patient does not have significant reduction of vital capacity. Undetected hypoventilation may produce or contribute to preexisting feelings of anxiety. Depression should be treated pharmacologically because not treating it may have a significant negative impact on the quality of life remaining [18,19]. Selective serotonin reuptake inhibitors (SSRIs) are good first choices because of their minimal side effects. Tricyclic antidepressants may be preferred if they are also needed to treat other symptoms, such as sialorrhea or pseudobulbar affect. Caution should be taken in prescribing both an SSRI and a tricyclic antidepressant as SSRIs will increase the serum level of the tricyclic antidepressant and increase the potential for serious drug-related events.
Rehabilitation
Exercise
Skeletal muscle weakness is the primary impairment in ALS and causes the majority of the clinical problems. In the early stages of ALS, patients often inquire about the role of exercise in preventing or forestalling the development of weakness. There remains scarce evidence on which to base recommendations for exercise in ALS. Little research has been published since the 2008 Cochrane review of Dal Bello–Haas et al [20]. However, a search of www.clinicaltrials.gov identified two ongoing clinical trials, one studying the effects of resistance training in patients with ALS and the other studying aerobic exercise training [21]. Rehabilitation strategies later in the disease focus on augmenting function by compensating for muscle weakness to maintain independence.
Three forms of exercise training are most relevant to patients with ALS: flexibility, strengthening, and aerobic exercise. Flexibility training helps prevent the development of painful contractures, is considered first-line treatment of spasticity, and aborts painful muscle spasms. A home stretching program, designed by a knowledgeable therapist, is appropriate for the ALS patient throughout the course of the disease. The stretching routine initially may be created for independent performance but necessarily will be transitioned to caregiver-assisted exercises when the patient is no longer capable of performing them alone.
Physicians caring for ALS patients have traditionally been reluctant to recommend strengthening exercises. The reasons for this appear to be twofold; epidemiologic studies have inconsistently suggested a link between high-intensity physical exercise and disease onset, and reports of overuse weakness accelerating disease progression have appeared in the literature [22,23]. This philosophy promotes the development of disuse weakness, compounding the disability produced by ALS itself. The literature supporting the development of overwork weakness in neuromuscular patients is anecdotal and has not been demonstrated in controlled prospective studies. A single randomized trial of strength training in ALS has been published [24]. It demonstrated a slowing of decline in physical function as measured by the ALS Functional Rating Scale out to 6 months after exercise initiation compared with the control group. No adverse effects were observed. Studies of patients with more slowly progressive neuromuscular diseases and postpoliomyelitis syndrome suggest that only muscles mildly affected by the disease process can be strengthened by a moderate-intensity resistance program [25–27]. Interested ALS patients should be encouraged to begin or to continue a resistance program to maximize the strength of unaffected or mildly affected muscles with the goal of prolonging function. Until further studies have been completed examining the effects of high-intensity strength training in ALS, resistance exercise can safely be accomplished by instructing the patient to use a weight that can be lifted 20 times in submaximal sets of 10 to 15 repetitions. This ensures that the training is performed at a low to moderate level of resistance. Common sense would suggest that if an exercise regimen consistently produces immediate or severe delayed muscle soreness or fatigue lasting longer than a half-hour after exercise, it is too strenuous and is impairing function. If the patient chooses to exercise, the level of postexercise fatigue and recovery time should be ascertained during the regular follow-up appointments. Recommendations for modification of exercise intensity and duration should be provided on the basis of the patient’s response.
Aerobic exercise helps maintain cardiorespiratory fitness. A study of moderate aerobic activity in patients with ALS demonstrated a short-term positive effect on disability up to 3 months after exercise initiation compared with controls [28]. Multiple studies in transgenic mouse models of ALS have shown that aerobic exercise prolongs survival [29–31]. Given the lack of any apparent contraindication, aerobic exercise training is recommended for patients with ALS as long as it can be performed safely without a risk of falling or injury. In addition to the physical benefits, strengthening and aerobic exercise may have a beneficial effect on mood, psychological well-being, bone health, appetite, and sleep. Further studies are needed to assess the impact of regular exercise and varying exercise intensities on the patient with ALS.
Assistive Technology for Mobility
As ALS progresses, the rehabilitation focus shifts from exercise to maintenance of independent mobility and function for as long as possible. Interventions include assistive devices such as canes, walkers, braces, hand splints, wheelchairs, and scooters; home equipment such as dressing aids, adapted utensils, grab bars, raised toilet seats, shower benches, and lifts; home modifications (ramps, wide doorways); and automobile adaptations, such as hand controls, environmental control units, and voice-activated software. These rehabilitation interventions are best provided by a multidisciplinary team that includes a physiatrist, physical therapist, occupational therapist, and orthotist.
Pain Management
Patients frequently have musculoskeletal pain syndromes, such as adhesive capsulitis (frozen shoulder), low back pain, and neck pain due to muscle weakness and inability to change positions. Measures to prevent adhesive capsulitis include range of motion exercises and adequate support for a hypotonic arm rather than allowing it to dangle at the patient’s side. Low back pain can be triggered by an ill-fitting seating system. Preventive measures include a lumbar support for the wheelchair, a good cushion on a solid seat, the encouragement of frequent weight shifts, and a reclining back and tilt-in-space wheelchair. Neck pain associated with head drop is one of the most difficult musculoskeletal pain issues to remedy. A variety of cervical collars may be tried, but a head support on the wheelchair or a reclining lounge chair may be more comfortable than a collar. Nonpharmacologic measures such as massage and transcutaneous electrical nerve stimulation can also be used for pain control. Acetaminophen alone or combined with nonsteroidal anti-inflammatory drugs, lidocaine patches, or, if necessary, opioid medications can be used to alleviate musculoskeletal pain. The major concerns with opiate use are respiratory depression and constipation. The respiratory depression of opiates may be acceptable in the late and terminal stages of the disease when morphine is needed to relieve air hunger.
Dysphagia
Adequate swallowing function is necessary to maintain the nutritional status of the patient with ALS (unless a feeding tube is in place). If nutritional status is not properly maintained, patients tend to use muscle as fuel and thus lose muscle mass and strength earlier than they would otherwise [32]. Swallowing dysfunction may also precipitate aspiration pneumonia or respiratory failure. Early signs and symptoms of dysphagia are drooling, a wet voice, coughing during or after drinking thin liquids, nasal regurgitation, and requiring an excessive amount of time to complete meals. Patients should be referred to a speech pathologist when the first signs of dysphagia develop. Those with mild swallowing difficulties can be taught compensatory techniques to reduce the risk of aspiration and choking [33]. Recommendations may be given concerning modification of food consistencies. Feeding tube placement is indicated with the development of aspiration pneumonia, with loss of more than 10% of body weight, and when an excessive amount of time is required to eat such that quality of life is impaired.
Dysarthria
Early or mild dysarthria may be managed by having a speech therapist teach patients adaptive strategies, such as overarticulation and slowing of the speaking rate to avoid vocal fatigue. Speech therapy with the intent of improving speech quality has not been shown to be effective and is not indicated for ALS patients. In patients with hypernasal speech caused by palatal weakness and primarily lower motor neuron dysfunction, a palatal lift or augmentation prosthesis may improve speech clarity [34]. As dysarthria worsens, patients require alternative forms of communication. Low-technology interventions include letter and word boards for written communication in those with good hand function. Higher technology solutions include iPad (Apple, Inc., Cupertino, Calif) voice applications and more traditional augmentative communication devices with a voice synthesizer (i.e., DynaVox, Pittsburgh, Pa; Tobii Technology, Stockholm, Sweden). As long as one muscle, somewhere in the body, can be voluntarily activated (including the extraocular muscles), the patient should be able to operate a communication device. High-technology solutions to communication problems are not suitable for all patients. The most enduring systems are flexible so that the method of access can be modified as weakness progresses.
Pulmonary Rehabilitation (see also Chapter 150)
Respiratory failure is the primary cause of death in ALS. In the absence of underlying intrinsic pulmonary disease, the respiratory failure in ALS is purely mechanical. Because of muscle weakness, the lungs do not inflate fully on inspiration. Most patients with ALS remain asymptomatic until the FVC is less than 50% of predicted. Pulmonary function tests, particularly the FVC, should be monitored every few months, depending on rate of disease progression. Nocturnal hypoventilation is typically the earliest manifestation of respiratory insufficiency; symptoms include poor sleep with frequent awakening, nightmares, early morning headaches, and excessive daytime fatigue and sleepiness [35]. Another early sign of respiratory muscle weakness is a weak cough and difficulty in clearing secretions.
The management of respiratory failure in ALS involves prevention of infection and provision of noninvasive or invasive mechanical ventilatory assistance. All patients with ALS should receive a yearly influenza vaccination and a pneumococcal vaccination with boosters every 5 years. Patients with an inadequate cough, as determined by a peak cough flow measurement of 270 L/min or less, can be helped by manually assisted coughing or an in-exsufflator device that mechanically augments the cough [35]. Providing patients with supplemental oxygen should typically be avoided as it may suppress respiratory drive, exacerbate alveolar hypoventilation, and ultimately lead to carbon dioxide retention and respiratory arrest [36]. Supplemental oxygen is recommended only for patients with a concomitant pulmonary disease or as a comfort measure for those who decline assisted ventilation. Noninvasive positive pressure ventilation (NIPPV) is considered standard of care for patients with ALS. The American Academy of Neurology’s practice parameter on ALS recommends that NIPPV be introduced when the FVC falls to 50% of predicted or earlier if the patient is symptomatic [37]. However, others have suggested that earlier introduction of NIPPV may further improve survival and quality of life [38,39]. With good patient acceptance, NIPPV can extend the life of an ALS patient well beyond that of available disease-modifying medications.
NIPPV can be delivered through a variety of oral or nasal mask interfaces with use of bilevel positive airway pressure machines or portable volume-cycled ventilators. Bilevel positive airway pressure is currently the most commonly used form of NIPPV. Gaining popularity are newer technologies that offer volume-assured pressure support and provide automatic inspiratory and expiratory pressure adjustments to meet the tidal volume or alveolar ventilation needs of patients with progressive restrictive lung disease. Initially, NIPPV is used at night. As FVC continues to decline, ventilator use can be extended into the day and eventually become continuous. Although studies have confirmed that use of NIPPV prolongs survival and may slow the decline of FVC [40,41], it is a temporary measure. Patients wishing to prolong their life to the fullest extent possible may consider invasive mechanical ventilation.
Discussion concerning respiratory failure should be initiated soon after the diagnosis of ALS is confirmed so that patients and their families can learn about their choices and, ideally, make a decision about invasive ventilator use in a noncrisis situation. These discussions should include both the benefits and limitations of invasive ventilation. An increasing number of patients are choosing tracheotomy and mechanical ventilation. In a recently published study, 31.3% of patients attending an ALS clinic chose mechanical ventilation with a median increase in survival of 16 months compared with nontracheostomized patients [42].
Before initiation of mechanical ventilation, patients should outline their wishes about withdrawal of treatment as part of an advance directive and inform their family and physicians; continued muscle atrophy and weakness may lead to a “locked-in” state in which patient communication is no longer possible. In one retrospective study, the most common reason for withdrawal of mechanical ventilation by ALS patients was a “loss of meaning in life.” [43]
Procedures
Management of Sialorrhea
Botulinum toxin injection of the salivary glands is the preferred method for management of sialorrhea in patients who do not respond to pharmacologic therapy [13–15]. Botulinum toxins A and B have been found to be equally effective. The safety of injection is increased with ultrasound guidance. Another treatment option for the patient refractive to more conservative treatments is radiotherapy of the parotid glands [44].
Gastrostomy Tube
A gastrostomy tube is recommended in ALS patients when they show signs of nutritional deficiency (i.e., lose 10% of their weight). The American Academy of Neurology’s practice parameter on ALS states that the morbidity and mortality of percutaneous endoscopic gastrostomy tube placement increase when the FVC falls below 50% of predicted [37] and recommends placement before that time. Some studies have shown that percutaneous endoscopic gastrostomy tubes can be safely placed with bilevel positive airway pressure assistance in patients with lower FVCs [45,46]. In addition, radiologically inserted gastrostomy tube placement appears to be safer, better tolerated in patients with ALS, and preferable in patients with low FVCs; less sedation is required, incidence of aspiration is lower, tube placement is more often successful, and acute respiratory decompensation observed with percutaneous endoscopic gastrostomy is avoided [47,48].
Tracheostomy
Every effort should be made to have a tracheotomy performed on a planned basis after a patient has chosen to use invasive ventilation. However, this is often not the case, and the procedure is commonly performed during a crisis situation. Patients should be given early access to unbiased literature describing the procedure and reviewing care and cost considerations to enable an informed decision. Once it is placed, a cuffless tracheostomy tube or a tube with a deflated cuff is preferred.
Potential Disease Complications
Disease complications include progressive weakness, joint contractures, musculoskeletal pain syndromes, osteoporosis, fractures due to falls, dysphagia, dehydration, impaired nutrition, aspiration, dysarthria, depression, progressive respiratory failure, pneumonia, pressure wounds, deep venous thrombosis, and death.
Potential Treatment Complications
The potential treatment complications include drug reactions (e.g., to riluzole, tricyclic antidepressants) and misplacement, malfunction, or infection of the percutaneous endoscopic gastrostomy tube or radiologically inserted gastrostomy tube.
Complications of long-term ventilation by tracheostomy are tracheomalacia, pneumonia, loss of all skeletal muscle function including extraocular movements causing a locked-in state, and dementia.
[/level-membership-for-physical-medicine-and-rehabilitation-category][not-level-membership-for-physical-medicine-and-rehabilitation-category]CHAPTER 132
Motor Neuron Disease
Nanette C. Joyce, DO, MAS; Gregory T. Carter, MD, MS
Definition
The term motor neuron disease encompasses a heterogeneous group of progressive neuromuscular disorders characterized by the selective loss of upper or lower motor neurons. However, motor neuron disease is also used interchangeably with amyotrophic lateral sclerosis (ALS), which can be confusing for the uninitiated. ALS is the most common adult motor neuron disease. For the diagnostic criteria for ALS to be met, both upper and lower motor neuron involvement is necessary [1,2]. In sporadic cases with only lower motor neuron dysfunction, the disease is called progressive muscular atrophy; if upper motor neuron dysfunction is singularly present, it is primary lateral sclerosis; and if dysfunction is localized to the bulbar region, the disease is called progressive bulbar palsy. Most patients initially diagnosed as having progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy eventually progress to meet diagnostic criteria for ALS [3]. Those who do not convert have a slower rate of disease progression. This chapter focuses on ALS as the management principles are similar for the entire class of motor neuron diseases.
The prevalence of ALS is about 6 to 8 per 100,000 people, with an annual incidence of approximately 2 cases per 100,000 people. Men are more commonly affected than women, with a ratio nearing 2:1. Most cases of ALS are sporadic, having unknown etiology. Only 5% to 10% of patients have familial ALS, which is most commonly transmitted in an autosomal dominant fashion. Approximately 30% to 40% of familial cases in the United States and Europe are caused by mutations in the C9orf72 gene; 20% worldwide are caused by mutations in the SOD1 (superoxide dismutase) gene; and rarer forms of familial ALS have been linked to mutations in the TARDBP, FUS, ANG, ALS2, SETX, and VAPB genes [4,5]. Other inherited adult motor neuron diseases are Kennedy disease (X-linked recessive) and adult spinal muscular atrophy (autosomal recessive), which are purely lower motor neuron disorders with greatly increased life span compared with ALS.
ALS causes rapid, progressive skeletal muscle weakness and atrophy, leading to premature death by respiratory failure. Weakness begins in a focal region, such as a single limb, the bulbar muscles, or the respiratory muscles, and spreads to affect other regions. Extraocular muscles and bowel and bladder sphincter function are often spared until late in the disease course. Mean age at onset is the mid-50s, but ALS may develop in adults of any age. Rare juvenile familial forms exist with onset before the age of 25 years. Mean survival, without tracheostomy, is 3 years from diagnosis but ranges from less than 1 year to more than 20 years. One explanation for this extreme variability is that ALS is multiple disorders without a single etiology but rather with multiple causes, sharing a common final step in the pathophysiologic pathway—motor neuron apoptosis. This is illustrated by the varying phenotypes associated with familial forms of ALS [3]. Theories about the pathogenesis of sporadic ALS have implicated RNA toxicity, glutamate excitotoxicity, oxidative stress, neuroinflammation, protein misfolding, glial cell activation, and mitochondrial dysfunction, to name a few [5].
Symptoms
Early symptoms of ALS can be subtle and include muscle twitching and cramping, weakness, and loss of coordination. Patients with a predominantly upper motor neuron syndrome often present with muscle stiffness, weakness, loss of dexterity, and loss of voluntary motor control from spasticity that may affect vocal quality or limb function. Patients with a predominantly lower motor neuron syndrome may present with weakness and muscle atrophy, fasciculations, muscle cramps, and flaccid dysarthria. Bulbar symptoms include dysarthria, dysphagia, sialorrhea (drooling), and pseudobulbar affect—laughing or crying in exaggeration of or incongruent with mood. Symptoms are initially painless and asymmetric across limbs. As the disease relentlessly progresses, weakness and atrophy spread to affect all skeletal muscles, causing significant impairment and disability. If patients do not succumb first to respiratory failure, they will ultimately transition from independent function to total dependence.
Respiratory failure is the presenting symptom in a rare few. Constitutional symptoms of weight loss and generalized fatigue are common. Cognitive symptoms including behavioral or executive dysfunction have been reported to occur in 33% to 51% of patients [6]. Most have milder symptoms; approximately 5% to 14% meet clinical criteria for a diagnosis of frontotemporal dementia [6].
Physical Examination
Physical examination of a patient with suspected motor neuron disease should be aimed at establishing the certainty of diagnosis. ALS is diagnosed clinically, and the patient with suspected motor neuron disease requires a thorough neurologic examination, assessing each of the four major body regions (bulbar, cervical, thoracic, and lumbar) for signs of upper and lower motor neuron involvement. The “gold standard” for the diagnosis of upper motor neuron disease is establishment of the presence of pathologic reflexes—a brisk jaw jerk, Hoffmann sign, abdominal skin reflex, and Babinski sign. Increased muscle stretch reflex responses as demonstrated by increased spread and amplitude or clonus are considered pathologic. Reflexes that would be graded normal but are elicited from atrophied and weak muscles should also be considered pathologic. Evidence of lower motor neuron disease includes muscle weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Patients with ALS may be hyperreflexic and hyporeflexic, depending on the stage at which they are in the disease process and whether they have a predominance of upper or lower motor neuron phenotype. For example, hyperreflexia occurs in the patient with upper motor neuron dysfunction, but this sign can be overcome and silenced by concomitant lower motor neuron loss causing muscle atrophy and hyporeflexia. Because ALS is an asymmetric and spreading process, the upper motor neuron signs may be more predominant than the lower motor neuron signs, or vice versa, within any single limb or between limbs and body regions. These examination findings will change over time as the disease progresses. The tongue should be examined for fasciculations, atrophy, strength, and range of motion. The patient’s mental status, nonmotor cranial nerve function, sensory examination, and cerebellar examination findings are usually normal.
In patients with an established diagnosis, the physical examination documents disease progression and includes the musculoskeletal and cardiorespiratory systems in addition to the neurologic evaluation. The musculoskeletal examination focuses on assessment of range of motion and evaluation of painful joints or soft tissue structures. Because progressive respiratory failure is a ubiquitous manifestation of ALS, follow-up appointments should be scheduled regularly (i.e., every 3 months) and the cardiorespiratory system assessed at each visit. Forced vital capacity (FVC) and maximal inspiratory, maximal expiratory, and peak cough pressures can be measured with a spirometer in the office setting and should be considered part of the regular cardiorespiratory follow-up evaluation, providing relevant information for clinical decision-making and prognosis (Table 132.1).
Table 132.1
Pulmonary Function Testing in Amyotrophic Lateral Sclerosis
| Measure | Key Values | Significance |
| Forced vital capacity | < 75% of predicted at time of diagnosis [49] | Poor prognostic indicator |
| Slower rate of monthly decline (< 3.1%/month) [50] | Predicts slower disease progression | |
| 50% of predicted [37] | Meets Medicare guidelines for initiation of noninvasive ventilation | |
| Maximal inspiratory pressure | − 60 cm H2O [37] | Meets Medicare guidelines for initiation of noninvasive ventilation |
| Maximal expiratory pressure | Normal value [50] | Associated with 2-year survival |
| Abnormal value | Associated with abnormal cough | |
| Peak cough flow | 270 L/min [37] | Initiate mechanical cough assistance |
| Sniff nasal inspiratory pressure | − 40 cm H2O [37] | Meets Medicare guidelines for initiation of noninvasive ventilation |
Functional Limitations
The pattern of progressive functional limitation is directly related to the patient’s motor neuron disease phenotype. Bulbar-onset ALS initially affects the patient’s ability to speak and to swallow and typically spreads to involve the muscles of the upper extremities before the lower extremities [7]. These patients have a difficult time maintaining their weight both because of dysphagia and because of the loss of upper limb strength that impairs their ability to feed themselves. Eventually, bulbar-predominant patients become anarthric with accompanying severe dysphagia that limits their ability to control their secretions and to swallow their own saliva.
The functional limitations that develop in patients with spinal-onset ALS are the direct or indirect result of muscle weakness and atrophy. In lumbar spine onset, gait is abnormal early in the disease secondary to footdrop or hip flexion weakness. As the disease progresses, the patient’s mobility worsens. Eventually, even the most basic activities of daily living become impossible to perform. Patients often transition quickly from independence to total dependence.
Reactive depression, generalized fatigue, and musculoskeletal pain may further limit function.
Diagnostic Studies
The diagnosis of ALS is based on the combined clinical and electrodiagnostic examinations. Neuroimaging and clinical laboratory studies are used to exclude other conditions that mimic ALS. All patients thought to have a motor neuron disease should undergo electrodiagnostic testing. The Awaji-shima revised El Escorial criteria (Table 132.2) are used to establish the certainty level of the diagnosis of ALS [1,2]. These criteria were developed as a tool for clinical trial enrollment but are commonly used in the clinic. The revised criteria classify the certainty level of diagnosis into one of three categories: definite, probable, and possible, which no longer includes the categories of Suspected ALS or Probable ALS Laboratory Supported that were part of the El Escorial criteria (Fig. 132.1). In addition to both upper and lower motor neuron findings, a diagnosis of ALS requires evidence of progressive spread of signs or symptoms within a single body region or from one of the four body regions to another. Certainty level of diagnosis depends on how many regions reveal upper motor neuron and lower motor neuron disease [1]. Electrophysiologic findings of denervation, including positive sharp waves, fibrillation potentials, and fasciculation potentials, are used to confirm lower motor neuron dysfunction in clinically affected regions and to detect subclinical lower motor neuron dysfunction, thereby extending the clinical examination. Signs of denervation observed during electromyography are now considered equivalent to lower motor neuron symptoms on clinical examination [1].
Table 132.2
Awaji-shima Revised El Escorial Criteria: Clinical Certainty Levels for the Diagnosis of Amyotrophic Lateral Sclerosis [1]
| Clinical Certainty | Clinical or Electrophysiologic Evidence |
| Clinically definite | UMN and LMN findings in at least three body regions |
| Clinically probable | UMN and LMN findings in at least two body regions with UMN findings rostral to LMN findings |
| Clinically possible | UMN and LMN findings in one body region, or UMN findings in at least two body regions without LMN findings, or LMN findings that are rostral to UMN findings |
LMN, lower motor neuron; UMN, upper motor neuron. Four body regions: bulbar, cervical, thoracic, lumbar.
Imaging studies are used to exclude possibilities other than motor neuron disease from the differential diagnosis. Magnetic resonance imaging is the primary imaging modality in the evaluation of patients with suspected ALS. Almost all patients should have magnetic resonance imaging of the cervical spine to exclude cord compression, syrinx, or other spinal cord disease. The location of symptoms will dictate whether other regions of the spinal cord should be imaged. In those presenting with bulbar symptoms, brain magnetic resonance imaging should be performed to exclude stroke, tumor, syringobulbia, and other pathologic processes.
In most neuromuscular clinics, a routine panel of laboratory tests is performed for all patients thought to have ALS. A suggested set of such tests is provided in Table 132.3. The rationale behind the performance of this extensive battery of tests is to assess the general health of the patient and to exclude treatable conditions. The differential diagnosis, developed after the history and physical examination, may suggest that more specialized testing be performed. Table 132.4 suggests additional tests that may be warranted when the presentation is atypical with a progressive muscular atrophy, primary lateral sclerosis, or progressive bulbar palsy phenotype. When there is a family history of motor neuron disease, genetic testing may be considered.
Table 132.4
Specialized Laboratory Testing
| Phenotype | Test | Diagnosis Excluded |
| Progressive muscular atrophy | DNA test: CAG repeat on X chromosome | Kennedy disease |
| DNA test: SMN gene mutation | Spinal muscular atrophy | |
| Hexosaminidase A |
