[level-membership-for-emergency-medicine-category]
Section 11 Endocrine
11.1 Diabetes mellitus and hypoglycaemia: an overview
DIABETES MELLITUS
Classification system and diagnostic criteria
The classification system and diagnostic criteria for diabetes were re-examined in 1996 by the American Diabetes Association and the World Health Organization.1 The classification of type I and type II diabetes mellitus was retained, although the recommended criterion for the diagnosis of diabetes has become a fasting plasma glucose of 7 mmol/L or greater, or a random plasma glucose of over 11 mmol/L associated with polyuria, polydipsia and weight loss. The oral glucose tolerance test is no longer routinely recommended.
Emergency presentations of a high blood sugar
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic non-ketotic state (HHNS) are both life-threatening acute complications of diabetes mellitus. Although important differences do exist, the pathophysiology and treatment are similar. DKA is usually seen in type I diabetes and HHNS in patients with type II, but both complications can occur in type I and type II diabetes. See Chapter 11.2 for the diagnosis and management of DKA and HHNS.
General management of diabetes mellitus
Aims of long-term blood sugar control
The aim of excellent long-term blood sugar control is an HbA1c (glycated haemoglobin) level of less than 7.5% without frequent disabling hypoglycaemia for the prevention of microvascular disease, and 6.5% in those at increased risk of arterial disease.2 This should be represented by a pre-prandial blood glucose level of 4.0–7.0 mmol/L, and a post-prandial blood glucose level of less than 9.0 mmol/L.
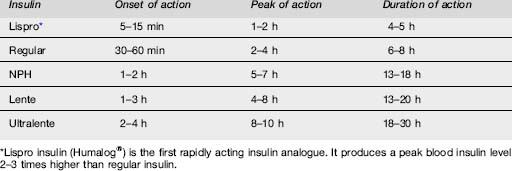
Insulins
Antidiabetic drugs
Two newer types of oral agents are available for the treatment of diabetes. The alpha-glucosidase inhibitor acarbose acts on the gastrointestinal tract to interfere with carbohydrate digestion. The other group is the thiazolidinediones, such as pioglitazone and rosiglitazone. The thiazolidinediones act primarily by reducing insulin resistance, thereby enhancing the effect of circulating insulin. However, roziglitazone increases the risk of myocardial infarction and cardiovascular deaths, and thus should be avoided in ischaemic heart disease.3 In addition, all thiazolidinediones must be avoided in people with moderate or severe heart failure.
DIABETIC HYPOGLYCAEMIA
Hypoglycaemia most commonly occurs in type I diabetes. The critical plasma level at which hypoglycaemia manifests varies between different individuals, but symptoms are likely below a plasma glucose of 3.5 mmol/L. Common precipitants include exercise, a late meal, inadequate carbohydrate intake, ethanol ingestion and errors of insulin dosage. Hypoglycaemia may also occur in the non-diabetic patient, precipitated by a variety of conditions (see Table 11.1.2).4
| Diabetic patients |
Management of hypoglycaemic coma
1 Alberti KGMM, DeFronzo RA, Keen H, et al. International textbook of diabetes mellitus. Chichester: John Wiley & Sons Ltd, 1992;31-98.
2 NICE Clinical Guideline 15. Type 1 diabetes: diagnosis and management of type 1 diabetes in children, young people and adults, July 2004. Available http://www.nice.org.uk/nicemedia/pdf/CG015NICEguideline.pdf (accessed Dec 2007)
3 Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007;356:2457-2471.
4 eMedicine. Hypoglycaemia, Dec 2007. Available http://www.emedicine.com/emerg/topic272.htm (accessed)
11.2 Diabetic ketoacidosis and hyperosmolar, hyperglycaemic non-ketotic state









Introduction
Diabetic ketoacidosis (DKA) is potentially fatal and overall is a common presentation to the emergency department (ED) for insulin-dependent diabetics. Shortfalls in the quality of care for patients with DKA have been highlighted. Hyperglycaemic, hyperosmolar non-ketotic state (HHNS) is less common than DKA, but has a worse prognosis, with an increased mortality and greater morbidity, often related to underlying chronic medical disorders.
Epidemiology
An annual incidence of DKA of approximately 1:170 patients with type I diabetes has been reported.1
Clinical investigations in DKA
Urinalysis
This diagnosis is highly likely in an unwell patient in the presence of glycosuria and ketonuria.
Criteria for diagnosis
Treatment of DKA
The treatment of DKA is not complicated, but requires careful monitoring of the patient both clinically and biochemically. Ideally all observations and results should be recorded on a purpose-designed record sheet, such as an integrated care pathway that includes guidance and data recording.2
Fluids
Give patients who are not shocked 0.9% normal saline at a rate of 500 mL/h for 4 h, then 250 mL/h for the next 4 h.3 A suggested fluid regime is shown in Table 11.2.1.
Table 11.2.1 Replacement fluid regime in patients with DKA who are not haemodynamically compromised or in shock
| Litre | Timing (hours from starting treatment) |
|---|---|
| First at 500 mL/h | 0–2 |
| Second at 500 mL/h | 2–4 |
| Third at 250 mL/h | 4–8 |
| Fourth at 100 mL/h | 8–18 |
| Fifth at 100 mL/h | 18–28 |
| Sixth at 100 mL/h | 28–38 |
| Seventh at 100 mL/h | 38–48 |
Insulin
An intravenous insulin infusion should be started within 60 min of the patient’s arrival in the ED.
Insulin infusion regime
When the serum [glucose] is less than 15 mmol/L, halve the insulin infusion rate and then adjust it to maintain the serum [glucose] between 9 and 14 mmol/L.2
Potassium replacement
Hyperkalaemia and then hypokalaemia are the most common life-threatening electrolyte problems seen in DKA. Therefore the serum [K] should be monitored closely, and treatment planned to treat either condition rapidly, particularly the risk of hypokalaemia. The typical total body deficit of potassium in DKA is 3–5 mmol/kg.1
Usually adding 20 mmol/h is sufficient, but this may need to be altered in the light of serum [K] measurements, with the aim of maintaining serum [K] in the range 4–5 mmol/L. Use an intravenous fluid pump or driver to avoid unregulated or inadvertently rapid potassium infusion.
Treatment of HHNS
Important management differences
Miscellaneous issues
1 American Diabetes Association. Clinical practice recommendations. Diabetes Care. 2004;27(Suppl 1):S94-102.
2 McGeoch SC, Hutcheon SD, Vaughan SM, et al. Practical Diabetes International. 2007;24(5):257-261.
3 Adrogue HJ, Barrero J, Eknoyan G. Salutary effects of modest fluid replacement in the treatment of adults with diabetic ketoacidosis. Use in patients without extreme volume deficit. Journal of the American Medical Association. 1989;262(15):2108-2113.
4 Wallace TM, Matthews TR. Recent advances in the monitoring and management of diabetic ketoacidosis. Quarterly Journal of Medicine. 2004;97:773-780.
11.3 Thyroid and adrenal emergencies
THYROTOXICOSIS
Aetiology, genetics, pathogenesis and pathology
There are a number of pathological causes of thyrotoxicosis (see Table 11.3.1). Graves’ disease is an autoimmune condition related to a combination of genetic and environmental factors, including iodine intake, stress and smoking. The thyrotoxicosis of Graves’ disease is caused by autoantibodies, which stimulate the thyroid resulting in excess thyroid hormone production.
| Primary hyperthyroidism | Graves disease |
| Toxic multinodular goitre | |
| Toxic adenoma | |
| Thyroiditis | de Quervains |
| Postpartum | |
| Radiation | |
| Central hyperthyroidism | Pituitary adenoma |
| Ectopic thyroid tissue | |
| Metastatic thyroid tissue | |
| Drug induced | Lithium |
| Iodine (including radiographic contrast) | |
| Amiodarone | |
| Excess thyroid hormone ingestion (factitious thyrotoxicosis) |
Epidemiology
Graves’ disease accounts for at least 80% of cases of thyrotoxicosis.1 The prevalence increases in areas with high iodine intake. Graves’ disease has a strong female predominance, affecting up to 2% of all women.1, 2 Thyrotoxicosis due to Graves’ disease usually occurs in the second to fourth decades of life, whereas the prevalence of a toxic nodular goitre increases with age.
Clinical features
The signs and symptoms of hyperthyroidism are secondary to the effects of excess thyroid hormone in the circulation. The severity of the signs and symptoms is related to the duration of the illness, the magnitude of the hormone excess and the age of the patient. These symptoms and signs are summarized in Table 11.3.2, which illustrates the wide spectrum of possible clinical features.
| Nervousness, irritability |
| Heat intolerance and increased sweating |
| Tremor |
| Weight loss and alterations in appetite |
| Palpitations and tachycardia, in particular atrial fibrillation |
| Widened pulse pressure |
| Exertional intolerance and dyspnoea |
| Frequent bowel movements |
| Fatigue and muscle weakness |
| Thyroid enlargement (depending on cause) |
| Pretibial myxoedema (with Graves’ disease) |
| Menstrual disturbance and impaired fertility |
| Mental disturbances |
| Sleep disturbances |
| Changes in vision, photophobia, eye irritation, diplopia, lid lag or exophthalmos |
| Dependent lower extremity oedema |
| Sudden paralysis, with or without hypokalaemia. |
A comprehensive history and physical examination should be performed, with particular attention to weight, blood pressure, pulse rate and rhythm, looking specifically for cardiac failure, palpation and auscultation of the thyroid to determine thyroid size, nodularity and vascularity, neuromuscular examination, and an eye examination for evidence of exophthalmos or ophthalmoplegia.
Clinical investigation and criteria for diagnosis1–3
Other laboratory and isotope tests may include:
Treatment
Give carbimazole 10–45 mg daily bd or tds, or propylthiouracil 200–600 mg daily bd or tds initially, using the larger doses for more severe cases.4 It is preferable to discuss initiation of these agents with the physician who will be managing the patient after their discharge from the ED. Ninety per cent of patients will be controlled within weeks with these drugs.5 Treatment for 12–18 months will result in a long-term remission in 40–60% of patients with Graves’ disease.2
Thyroid storm
Clinical features



Treatment
β-blockers
β-blockade is the most important factor in decreasing morbidity and mortality. Many of the peripheral manifestations of hyperthyroidism, in particular the cardiovascular effects, are reduced by the use of propranolol. Propranolol inhibits the peripheral conversion of T4 to T3 as well as antagonizing the effects of thyroid hormones and the hypersensitivity to catecholamines.
Thyroid-blocking drugs
Iodine in large doses inhibits the synthesis and release of thyroid hormones, and may be given either orally as Lugol’s iodine, 30–60 drops daily in divided doses, or intravenously as sodium iodide 1 g 12-hourly. Lithium carbonate may be used in patients allergic to iodine or added when there is difficulty with control.4
Epidemiology
Iodine deficiency is the most common cause worldwide, whereas in areas of iodine sufficiency, autoimmune disease and hypothyoidism secondary to treatment of hyperthyroid disease are most common. The prevalence of hyperthyroidism in adults is of the order of 1.4% in women and <0.1% in men.6 Congenital hypothyroidism is rare, occurring in about 1:4000 births.
Clinical features
The symptoms of hypothyroidism are related to the duration and severity of hypothyroidism, the rapidity with which hypothyroidism occurs and the psychological characteristics of the patient. These are summarized in Table 11.3.3.
| Dry skin and cold intolerance |
| Coarse facial features |
| Enlarged tongue |
| Coarse brittle hair or loss of hair, loss of outer third of eyebrows |
| Periorbital oedema |
| Fatigue |
| Constipation |
| Weight gain/obesity |
| Memory and mental impairment, decreased concentration |
| Depression, personality changes |
| Yellow skin |
| Swelling of ankles |
| Irregular or heavy menses and infertility |
| Hoarseness |
| Myalgias |
| Goitre |
| Hyperlipidaemia |
| Delayed relaxation phase of tendon reflexes, ataxia |
| Sinus bradycardia (atrioventricular block, rare) |
| Cardiac failure, pericardial effusion (rare) |
| Hypothermia (uncommon) |
Clinical investigation and criteria for diagnosis
Laboratory evaluation
Perform a TSH assay as the primary test to establish the diagnosis of hypothyroidism. The reference range is 0.4–5.0 mIU/L depending on the method. Additional tests may include free thyroxine assay and thyroid autoantibodies. A combination of an elevated TSH and low free thyroxine is diagnostic.3,4,6 A patient may be hypothyroid with a TSH greater than twice the reference interval, but with a free thyroxine within the normal range. Subnormal thyroxine with a normal TSH can occur in secondary hypothyroidism.
Myxoedema coma
The mortality for myxoedema coma remains up to 50% despite aggressive treatment.
Clinical features
Treatment
Treatment should commence on clinical suspicion.
Administration of thyroid hormones
Tri-iodothyronine
There is no consensus as to whether T3 or T4 replacement is preferable.7,8 Intravenous T3 may give a faster clinical response in myxoedema coma, as it is the active form of the hormone. Give T3 as an initial i.v. bolus of 25–50 μg followed by 10–20 μg 8-hourly to a maximum of 60 μg per day. Alternatively, commence an infusion with a lower total dose of 20 μg per day, as large initial doses appear unnecessary for recovery and may in fact be harmful. Oral or nasogastric replacement of T3 is not recommended in the initial phase of management because of unreliable gastrointestinal absorption.
Thyroxine
The use of T4 is supported as the gradual delivery of T3 through the peripheral conversion of T4 is better tolerated and as the onset of action is more predictable. Give a 400–500 μg i.v. bolus (300 μg/m2), followed by 50 μg i.v. daily until oral therapy is tolerated. Combined approaches are now also described.
HYPOADRENAL STATES
Aetiology, genetics, pathogenesis and pathology
The adrenals normally respond within minutes by elevating corticosteroid levels in response to any physiological or pathological stress. When glucocorticoid insufficiency is present such stressors may result in hypotension, shock and ultimately death if left untreated.
Secondary adrenal insufficiency
Secondary adrenal insufficiency is due to failure of adequate adrenocorticotrophic hormone (ACTH) from the pituitay gland (Table 11.3.4). Hyponatraemia still occurs in secondary adrenal insufficiency, but is due to cortisol deficiency.9,10
| Primary | Addison’s disease |
| Surgical removal | |
| Infectious (TB, viral, fungal) | |
| Haemorrhage, including Waterhouse–Friedrichsen syndrome | |
| Congenital | |
| Secondary | Exogenous steroid suppression (single most common cause of hypoadrenalism overall) |
| Endogenous steroid (tumour) | |
| Pituitary failure |
Causes of primary or secondary adrenal insufficiency
The cause of 80% of primary adrenal insufficiency is autoimmune (Addison’s disease). Other causes include primary or secondary malignancy, infection such as tuberculosis, adrenal infarction or haemorrhage (Waterhouse-Friedrichsen syndrome) seen in meningococcaemia or severe sepsis, and drugs. Primary adrenal insufficiency also occurs in up to 20% of patients with AIDS. Up to 60% of patients with sepsis have a low baseline cortisol level, although fewer meet criteria of insufficiency on suppression testing.11,12
Clinical features
Hypercalcaemia and/or hyperkalaemia may be the first sign of adrenal insufficiency in the critically ill patient.10 The other features of adrenal insufficiency may be masked by coexisting illness, but the possibility of adrenal insufficiency should always be considered in such cases.
Clinical investigation
Treatment
Corticosteroid replacement
Do not delay treatment awaiting confirmatory results if acute adrenal insufficiency is suspected.
General supportive measures
General supportive measures include treatment of hypoglycaemia and other electrolyte replacement abnormalities, although most will be corrected with saline rehydration alone. Mineralocorticoid replacement is usually not necessary in the acute crisis if salt and water replacement is adequate.
Response to severe illness
The normal response to severe illness should see cortisol levels rising to at least 500 nmol/L. States of ‘relative adrenal insufficiency’ are described where glucocorticoid administration diminishes or even eliminates the requirements for vasopressor agents, even though measured cortisol levels are normal or close to normal.11 There is no concensus on what constitutes ‘normal’ cortisol levels in severe illness.
Up to 60% of patients in sepsis may have some degree of adrenal insufficiency depending upon the threshold cortisol level used.12 Moreover, it appears that it is the delta cortisol rather than the basal cortisol level that is associated with clinical outcome.13 Repeat adrenal function testing is indicated in patients with severe illness who remain unstable or who fail to improve with aggressive supportive therapy.14
The use of hydrocortisone has been recommended in septic shock after a 250 μg Synacthen® stimulation test.15–17 This should continue for a week if adrenal insufficiency is confirmed.
HYPERADRENAL STATES
Aetiology, pathogenesis and epidemiology
The incidence of Cushing’s syndrome ranges from 0.7 to 2.4 per million population per year, but the reported prevalence in obese patients with type II diabetes may be between 2 and 5%.18
Clinical investigation and criteria for diagnosis
Overnight dexamathasone suppression test
This is an outpatient screening test for Cushing’s syndrome:3
1 Cooper DS. Hyperthyroidism. Lancet. 2003;362(9382):459-468.
2 Pearce EN. Diagnosis and management of thyrotoxicosis. British Medical Journal. 2006;332:1369-1372.
3 Royal College of Pathologists of Australasia. Manual Version 4.0. Surrey Hills: Royal College of Pathologists of Australasia, 2004.
4 Endocrinology Expert Group. Endocrinology. Guidelines. Melbourne: Therepeutic Guidelines Limited, 2004.
5 Cooper DS. Antithyroid drugs. New England Journal of Medicine. 2005;352:905-917.
6 Lindsay RS, Toft AD. Hypothyroidism. Lancet. 1997;349:413-417.
7 Jordan RM. Myxedema coma. Medical Clinics of North America. 1995;79:185-194.
8 Vedig AE. Thyroid emergencies. Oh’s intensive care manual, 5th edn. Elsevier, Edinburgh, 2004.
9 Oelkers W. Adrenal insufficiency. New England Journal of Medicine. 1996;335:1206-1212.
10 Nair G, Simmons D. Adrenal insufficiency presenting as hypercalcemia. Hospital Physician. 2003:33-35.
11 de Herder W, van der Lely A. Addisonian crisis and relative adrenal failure. Reviews in Endocrine & Metabolic Disorders. 2003;4:143-147.
12 Marik P, Zaloga G. Adrenal insufficiency during septic shock. Critical Care Medicine. 2003;31:141-145.
13 Lipner Fredman D, Sprung C, et al. Adrenal function in sepsis: the retrospective Corticus cohort study. Critical Care Medicine. 2007;35:1012-1018.
14 Marik P. Adrenal-exhaustion syndrome in patients with liver disease. Intensive Care Medicine. 2006;134:275-280.
15 Cooper MS, Stewart PM. Current concepts: corticosteroid insufficiency in acutely ill patients. New England Journal of Medicine. 2003;348:727-734.
16 Annane D. Glucocorticoids in the treatment of severe sepsis and septic shock. Current Opinion in Critical Care. 2005;11:449-453.
17 Luce JM. Physicians should administer low-dose corticosteroid selectively to septic patients until an ongoing trial is completed. Annals of Internal Medicine. 2004;141:70.
18 Newall-Price J, Bertagna X, Grossman AB, et al. Cushing’s syndrome. Lancet. 2006;367:1605-1617.
Jameson L, Weetman AP. Disorders of the thyroid gland. In Kaspar DL, et al, editors: Harrison’s principles of internal medicine, 16th edn, New York: McGraw-Hill, 2005.
Maclean A, Dunn R. Endocrinology. The emergency medicine manual, 4th edn. Venom Publishing, Adelaide, 2006.
Vedig AE. Thyroid emergencies. Oh’s intensive care manual, 5th edn. Elsevier, Edinburgh, 2004.
Vedig AE. Adrenocortical insufficiency. Oh’s intensive care manual, 5th edn. Edinburgh, Elsevier, 2004.
Williams GH, Dluhy RG. Disorders of the adrenal cortex. In Kaspar DL, et al, editors: Harrison’s principles of internal medicine, 16th edn, New York: McGraw-Hill, 2005.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Section 11 Endocrine
11.1 Diabetes mellitus and hypoglycaemia: an overview
DIABETES MELLITUS
Classification system and diagnostic criteria
The classification system and diagnostic criteria for diabetes were re-examined in 1996 by the American Diabetes Association and the World Health Organization.1 The classification of type I and type II diabetes mellitus was retained, although the recommended criterion for the diagnosis of diabetes has become a fasting plasma glucose of 7 mmol/L or greater, or a random plasma glucose of over 11 mmol/L associated with polyuria, polydipsia and weight loss. The oral glucose tolerance test is no longer routinely recommended.
Emergency presentations of a high blood sugar
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycaemic non-ketotic state (HHNS) are both life-threatening acute complications of diabetes mellitus. Although important differences do exist, the pathophysiology and treatment are similar. DKA is usually seen in type I diabetes and HHNS in patients with type II, but both complications can occur in type I and type II diabetes. See Chapter 11.2 for the diagnosis and management of DKA and HHNS.
General management of diabetes mellitus
Aims of long-term blood sugar control
The aim of excellent long-term blood sugar control is an HbA1c (glycated haemoglobin) level of less than 7.5% without frequent disabling hypoglycaemia for the prevention of microvascular disease, and 6.5% in those at increased risk of arterial disease.2 This should be represented by a pre-prandial blood glucose level of 4.0–7.0 mmol/L, and a post-prandial blood glucose level of less than 9.0 mmol/L.
Insulins
Antidiabetic drugs
Two newer types of oral agents are available for the treatment of diabetes. The alpha-glucosidase inhibitor acarbose acts on the gastrointestinal tract to interfere with carbohydrate digestion. The other group is the thiazolidinediones, such as pioglitazone and rosiglitazone. The thiazolidinediones act primarily by reducing insulin resistance, thereby enhancing the effect of circulating insulin. However, roziglitazone increases the risk of myocardial infarction and cardiovascular deaths, and thus should be avoided in ischaemic heart disease.3 In addition, all thiazolidinediones must be avoided in people with moderate or severe heart failure.
DIABETIC HYPOGLYCAEMIA
Hypoglycaemia most commonly occurs in type I diabetes. The critical plasma level at which hypoglycaemia manifests varies between different individuals, but symptoms are likely below a plasma glucose of 3.5 mmol/L. Common precipitants include exercise, a late meal, inadequate carbohydrate intake, ethanol ingestion and errors of insulin dosage. Hypoglycaemia may also occur in the non-diabetic patient, precipitated by a variety of conditions (see Table 11.1.2).4
| Diabetic patients |
Management of hypoglycaemic coma
1 Alberti KGMM, DeFronzo RA, Keen H, et al. International textbook of diabetes mellitus. Chichester: John Wiley & Sons Ltd, 1992;31-98.
2 NICE Clinical Guideline 15. Type 1 diabetes: diagnosis and management of type 1 diabetes in children, young people and adults, July 2004. Available http://www.nice.org.uk/nicemedia/pdf/CG015NICEguideline.pdf (accessed Dec 2007)
3 Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007;356:2457-2471.
4 eMedicine. Hypoglycaemia, Dec 2007. Available http://www.emedicine.com/emerg/topic272.htm (accessed)
11.2 Diabetic ketoacidosis and hyperosmolar, hyperglycaemic non-ketotic state
Introduction
Diabetic ketoacidosis (DKA) is potentially fatal and overall is a common presentation to the emergency department (ED) for insulin-dependent diabetics. Shortfalls in the quality of care for patients with DKA have been highlighted. Hyperglycaemic, hyperosmolar non-ketotic state (HHNS) is less common than DKA, but has a worse prognosis, with an increased mortality and greater morbidity, often related to underlying chronic medical disorders.
Epidemiology
An annual incidence of DKA of approximately 1:170 patients with type I diabetes has been reported.1
Clinical investigations in DKA
Urinalysis
This diagnosis is highly likely in an unwell patient in the presence of glycosuria and ketonuria.
Criteria for diagnosis
Treatment of DKA
The treatment of DKA is not complicated, but requires careful monitoring of the patient both clinically and biochemically. Ideally all observations and results should be recorded on a purpose-designed record sheet, such as an integrated care pathway that includes guidance and data recording.2
Fluids
Give patients who are not shocked 0.9% normal saline at a rate of 500 mL/h for 4 h, then 250 mL/h for the next 4 h.3 A suggested fluid regime is shown in Table 11.2.1.
Table 11.2.1 Replacement fluid regime in patients with DKA who are not haemodynamically compromised or in shock
| Litre | Timing (hours from starting treatment) |
|---|---|
| First at 500 mL/h | 0–2 |
| Second at 500 mL/h | 2–4 |
| Third at 250 mL/h | 4–8 |
| Fourth at 100 mL/h | 8–18 |
| Fifth at 100 mL/h | 18–28 |
| Sixth at 100 mL/h | 28–38 |
| Seventh at 100 mL/h | 38–48 |
