[level-membership-for-emergency-medicine-category]
Section 10 Genitourinary
10.1 Acute kidney injury
Introduction
The first recognition of illness caused by a sudden decline in renal function (‘ischuria renalis’) was by William Heberden in 1802.1 In 1888 Delafield described a form of ‘acute Bright’s disease’ that was caused by toxins, various infectious diseases and extensive injuries, and where there was degeneration or death of tubule cells.2 Impaired renal function in injured soldiers was described in World War I (‘war nephritis’) and in World War II.3,4 The term ‘acute renal failure’ (ARF) was first used in 1951.5
The basic process in ARF is a rapid (hours to days) reduction in the glomerular filtration rate (GFR) due to renal hypoperfusion (prerenal causes), damage to glomeruli, tubules, interstitium or blood vessels (renal causes), or obstruction to urine flow (postrenal causes). The GFR is inversely related to the serum creatinine (SCr) concentration. The diagnosis of ARF is made when there is an increase in the SCr concentration, with or without a decrease in the urine output. A simple definition of ARF is an acute and sustained (lasting for 48 h or more) increase in the SCr of 44 μmol/L if the baseline is less than 221 μmol/L, or an increase in the SCr of more than 20% if the baseline is more than 221 μmol/L.6 A more comprehensive definition (the RIFLE system) is used to classify persons with acute impairment of renal function7 (Table 10.1.1).
| Stage | Serum creatinine (SCr) concentration | Urine output |
|---|---|---|
| RISK | Increase of 1.5 times the baseline | <0.5 mL/kg/h for 6 h |
| INJURY | Increase of 2.0 times the baseline | <0.5 mL/kg/h for 12 h |
| FAILURE | Increase of 3.0 times the baseline or SCr is 355 μmol/L or more when there has been an acute rise of greater than 44 μmol/L for 24 h or anuria for 12 h | <0.3 mL/kg/h |
| LOSS | Persistent acute renal failure; complete loss of kidney function for longer than 4 weeks | |
| END-STAGE RENAL DISEASE | End-stage renal disease for longer than 3 months |
Aetiology and pathogenesis
Prerenal acute kidney injury
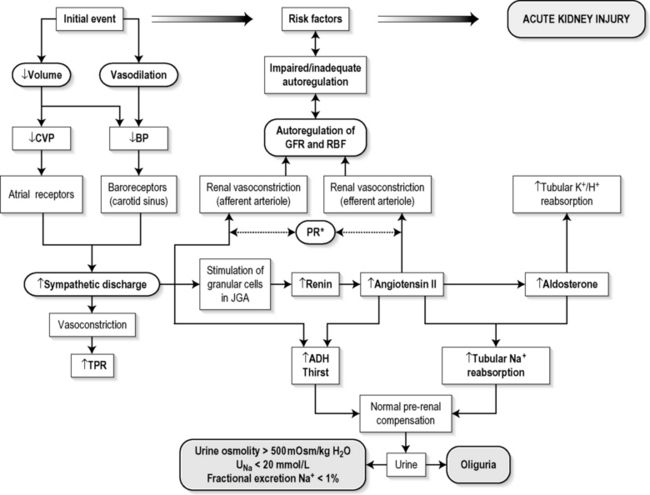
Reductions in renal blood flow (RBF) and GFR occur in the setting(s) of hypovolaemia, hypotension (cardiogenic shock, anaphylaxis, sepsis), oedematous states with a reduced ‘effective’ circulating volume (cardiac failure, hepatic cirrhosis, nephrotic syndrome) or renal hypoperfusion (renal artery stenosis, hepatorenal syndrome). Drugs that interfere with autoregulation (e.g. prostaglandin inhibitors, angiotensin converting enzyme (ACE) inhibitors or angiotensin II receptor antagonists) also reduce glomerular perfusion. The physiological responses to volume depletion and hypotension, and the link to prerenal AKI, are shown in Figure 10.1.1.
Renal (parenchymal) acute kidney injury
Acute tubular necrosis (ATN) is the most common pathological process that causes ARF. While the terminology suggests that the main cause is tubular damage, the actual pathophysiology is more complex: impaired autoregulation and marked intrarenal vasoconstriction (the main mechanism for the greatly reduced GFR), tubular damage (with cytoskeleton breakdown), increased tubuloglomerular feedback, endothelial cell injury, fibrin deposition in the microcirculation, release of cytokines, activation of inflammation and activation of the immune system.8,9
ATN is classified as ischaemic ATN or cytotoxic ATN (due to damage by toxins); both processes are present in some patients. In ischaemic ATN there is a continuum between prerenal azotaemia, the RISK and INJURY stages of AKI, and the development of ATN. ATN caused by administration of intravenous or intra-arterial contrast agents is due to renal vasoconstriction, reduced injury.10 The main mechanisms of AKI or ATN caused by drugs are vasoconstriction, altered intraglomerular haemodynamics, tubular cell toxicity, interstitial nephritis, crystal deposition, thrombotic microangiopathy and osmotic nephrosis.11
Important causes of cytotoxic ATN are listed in Table 10.1.2. Non-steroidal anti-inflammatory drugs (NSAIDs), ACE inhibitors and angiotensin receptor blockers (ARBs) often cause a gradual and asymptomatic decrease in the GFR, but can cause AKI (including ATN). NSAIDs do not impair renal function in a healthy person, but can reduce the GFR in elderly persons with atherosclerotic cardiovascular disease, in persons with chronic renal failure, when chronic prerenal hypoperfusion is present (e.g. cardiac failure, cirrhosis), or in persons using diuretics and calcium channel blockers.12
| Exogenous agents |
| Radiocontrast |
| Non-steroidal anti-inflammatory drugs |
| Antibiotics: aminoglycosides, amphotericin B |
| Antiviral drugs: aciclovir, foscarnet |
| Immunosuppressive drugs: ciclosporin |
| Organic solvents: ethylene glycol |
| Poisons: snake venom, paraquat, paracetamol |
| Chemotherapeutic drugs: cisplatin |
| Herbal remedies |
| Heavy metals |
| Endogenous agents |
| Haem pigments: haemoglobin, myoglobin |
| Uric acid |
| Myeloma proteins Correct intravascular volume depletion |
| Maintain perfusion pressure |
| Choice of resuscitation fluid |
| Diuresis in rhabdomyolysis |
| Avoid nephrotoxins |
| Use derived GFR or creatinine clearance when calculating drug doses |
Once ATN is established there is a persistent and marked reduction in RBF and in GFR that lasts for 1 to 2 weeks. During this time the patient is usually oliguric, and cannot excrete concentrated urine. Renal autoregulation is impaired, and renal perfusion depends directly on the systemic blood pressure. A fall in systemic blood pressure during the ATN phase causes more renal damage. Recovery from ATN is associated with increased renal blood flow (reperfusion), an increase in GFR and (often) a large volume urine output because the concentrating ability of the regenerating nephrons is impaired.
Abnormalities of renal interstitial structure and function are present in ATN. However, AKI and ATN can be caused by a primary abnormality of the interstitial tissues: acute tubulointerstitial nephritis (ATIN). The damage in ATIN is due to immunological mechanisms, the most important involving cell-mediated immunity. ATIN is usually due to a drug reaction, but can also be caused by infections (e.g. infection with hantavirus, a RNA virus that causes haemorrhagic fever with renal syndrome.13 Drugs that cause ATIN include antibiotics (β-lactam antibiotics, sulphonamides, fluoroquinolones), NSAIDs, cyclooxygenase-2 inhibitors, proton pump inhibitors, diuretics, phenytoin, carbamazepine and allopurinol.
Postrenal (obstructive) acute kidney injury
Obstructive uropathy refers to the functional or structural processes in the urinary tract that impede the normal flow of urine, and obstructive nephropathy is the renal damage caused by the obstruction. Hydronephrosis is a dilatation of the ureter(s); it can occur in the absence of obstruction, and some persons with obstruction do not have a dilated urinary collecting system.14–16
Epidemiology
The annual incidence of ARF in European communities is between 209 and 620 cases per million per year, with an incidence of severe acute renal failure (SCr greater than 500 μmol/L) of 172 cases per million per year.17–20 About 1% of patients in the USA have ARF on admission to hospital, and ARF develops in 5–7% of all hospitalized patients.21–23 The frequency of ARF in hospitalized patients is about 19 per 1000 admissions.24
Studies of the pathogenesis of community acquired ARF have produced conflicting results. In one study the major processes were identified as prerenal in 70% of cases, renal in 11% of cases and postrenal in 17% of cases.21 Other studies found a lower incidence of prerenal factors (present in 21–48% of cases) and a higher incidence of renal factors (present in 34–56% of cases, most commonly due to ATN).19,25 Acute on chronic renal failure was present in 13% of persons in one study.19 The basic processes in hospital acquired ARF are prerenal in 35–40% of cases, renal in 55–60% of cases and post-renal in 2–5% of cases.6
Using the RIFLE criteria the community incidence of AKI is 1811 per million of population, and AKI occurs in 18% of hospitalized patients (9% had changes in SCr concentration and urine output consistent with RISK, 5% had renal INJURY and 4% developed FAILURE).26,27
There are geographical differences in the causes of ATN. In Africa, India, Asia and Latin America ATN is usually caused by infections (e.g. diarrhoeal illnesses, malaria, leptospirosis), ingestion of plants or medicinal herbs, envenomation, intravascular haemolysis due to glucose-6-phosphate dehydrogenase deficiency or poisoning.28 The incidence of ATN due to crushing injuries is increased in earthquake-prone areas.
Prevention
The processes involved in the prevention of AKI are shown in Table 10.1.3.
Table 10.1.3 Use derived GFR or creatinine clearance when calculating drug doses
| Correct intravascular volume depletion |
| Maintain perfusion pressure |
| Choice of resuscitation fluid |
| Diuresis in rhabdomyolysis |
| Avoid nephrotoxins |
Maintaining intravascular volume and renal perfusion
Acute haemorrhage causes prerenal azotaemia, but the incidence of ARF is low (1.5% in major gastrointestinal bleeding29) if the hypovolaemia is treated promptly and there are no other risk factors. In severe trauma the incidence of ARF needing dialysis is about 0.1%.30 Severe progressive ARF after cardiopulmonary resuscitation is rare, and pre-existing and post resuscitation haemodynamics are more important risk factors for AKI than the degree of hypoperfusion during resuscitation.31
Choice of resuscitation fluid
Hypovolaemia is initially treated with crystalloid rather than colloid, with colloid being added in specific clinical settings (e.g. burns, anaphylaxis) or if there is no initial improvement in vital signs or urine output following crystalloid administration (e.g. in sepsis). The liberal use of crystalloid and colloid in high-output sepsis with AKI may increase the incidence of ARF or need for dialysis.32
Use of derived GFR in drug administration
Formulas are available to calculate the estimated creatinine clearance or estimated GFR from the measured SCr concentration.33,34
Rhabdomyolysis
Most studies on the prevention of ATN after rhabdomyolysis have been in persons with crush injury after earthquakes, where the incidence of AKI is about 50%. In this situation fluid resuscitation should, if possible, begin before the crush is relieved. These patients may require massive amounts of fluid because of fluid sequestration in the injured muscles. The goal of intravenous fluid treatment is to produce a urine output of 200–300 mL/h while myoglobinuria (discoloured urine) persists. There is no evidence to support this rate of fluid replacement in persons who have rhabdomyolysis and AKI without crush injury, although a urine output of 100 mL/h would be reasonable while the urine is discoloured. The intravenous administration of mannitol and sodium bicarbonate to produce an alkaline diuresis as a means of preventing ATN in severe rhabdomyolysis has not been shown to be effective.35
Clinical features
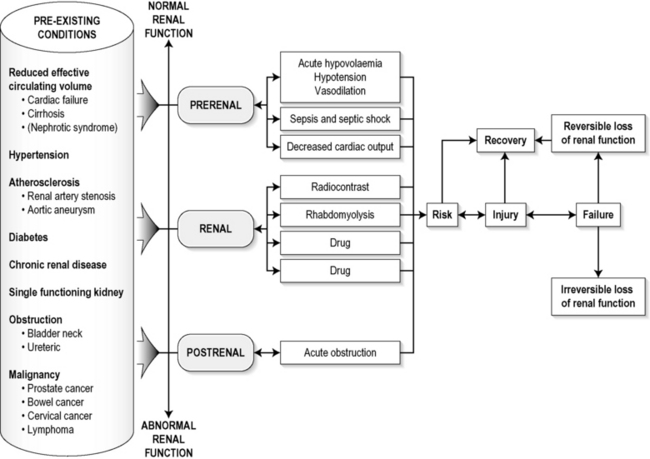
The diagnosis of AKI should be considered when there is a decrease in urine output, an elevated SCr concentration or increases in SCr concentration. The clinical features depend on the pre-existing conditions that increase the risk of developing AKI, the initiating factor(s), and the effects of AKI (Fig. 10.1.2). The history should include a detailed drug history, enquiry about recent invasive vascular or radiological procedures, and any family history of renal disease. This is followed by clinical examination and evaluation of investigations. A number of key issues then need to be resolved (Table 10.1.4).
| Assess the intravascular volume |
| Look for renovascular disease |
| Look for symptoms or signs of obstruction to urine flow |
| Systematic search for presence of infection or sepsis |
| Evaluate for pre-existing renal disease or chronic renal failure |
| Obtain a detailed history of medication or drug use |
| Consider possibility of glomerulonephritis |
Evaluation of prerenal (intravascular volume) status
Imprecise or lazy terminology such as ‘dry’ or ‘dehydrated’ should be avoided. ‘Dehydration’ refers to situations where more water than electrolyte(s) has been lost, shrinking body cells and increasing the serum sodium concentration and osmolality.36 In other words, ‘dehydration’ means water depletion. Hypovolaemia is a decrease in the intravascular volume due to loss of blood (haemorrhage, trauma) or loss of sodium and water (e.g. vomiting, diarrhoea, sequestration of fluid in the bowel, etc.).
The ‘typical’ features of intravascular volume depletion (tachycardia or hypotension or both in the supine position, or postural hypotension) are not as consistent or reliable as implied by textbook descriptions. About one-third of persons with hypovolaemia due to trauma have bradycardia rather than tachycardia.37,38 The presence of (supine) tachycardia has low sensitivity as a diagnostic feature of increasing acute blood loss in healthy persons.39 An increase in the pulse rate of 30 beats per minute or more between the supine value and the standing values is a highly sensitive and highly specific sign of hypovolaemia after phlebotomy of large volumes (600–1100 mL) of blood, but the sensitivity is much less after phlebotomy of smaller volumes.39 The inability to stand long enough for vital signs to be measured because of severe dizziness is a sensitive and specific feature of acute large blood loss.39 The persistence of tachycardia after intravenous administration of fluids in clinical conditions causing hypovolaemia suggests that hypovolaemia is still present, but tachycardia due to other causes (pain, fever) will persist after correction of hypovolaemia.
A systolic blood pressure of 95 mmHg or less in the supine position has high specificity but low sensitivity after acute blood loss.39 Postural hypotension is present in 10% of normovolaemic person younger than 65 years, and in up to 30% of normovolaemic person older than 65 years. Postural hypotension in persons who can stand without developing severe dizziness is of no diagnostic value after blood loss due to acute phlebotomy.39
The textbook descriptions of the signs of saline depletion in adults (dry mucous membranes, shrivelled tongue, sunken eyes, decreased skin turgor, weakness, confusion) are neither specific nor sensitive compared to laboratory tests for hypovolaemia. The presence of a dry axilla argues somewhat for the presence of saline depletion; the absence of tongue furrows and the presence of moist mucous membranes argue against the presence of saline depletion.39
Evaluation of the renovascular state
Atheromatous disease of the renal arteries is common in persons older than 50 years with widespread atherosclerosis. There is a 7% prevalence of renal artery stenosis in persons older than 65 years, and one-third of elderly persons with heart failure have renovascular disease.40,41 Persons with stenosis or occlusion of one or both renal arteries can develop an elevation in SCr concentration after starting treatment with ACE or ARB drugs, or develop acute on chronic renal failure.
Acute kidney injury and acute renal failure
The urine output usually decreases, and the patient may be oliguric (urine output less than 400 mL per day) or anuric (urine output less than 100 mL per day). Persons with AKI and oliguria have more severe kidney impairment than those without oliguria. Only a few conditions cause complete anuria: total obstruction, vascular lesions, severe ATN or rapidly progressive glomerulonephritis. The clinical features caused by ARF are shown in Table 10.1.5.
| 1. Anorexia, fatigue, confusion, drowsiness, nausea and vomiting, and pruritus |
| 2. Signs of salt and water retention in the intravascular and interstitial spaces: an elevated jugular venous pressure, peripheral oedema, pulmonary congestion, acute pulmonary oedema |
| 3. Abnormal plasma electrolyte concentrations, particularly hyperkalaemia |
| 4. Metabolic acidosis |
| 5. Anaemia |
| 6. Uraemic syndrome: ileus, asterixis, psychosis, myoclonus, seizures, pericardial disease (pericarditis, pericardial effusion, tamponade). |
Differential diagnosis
The diagnosis of AKI requires synthesis of data from the patient’s history, physical examination, laboratory studies and urine output. The category of AKI (RISK, INJURY or FAILURE) may be difficult to determine in the emergency department (ED) if the baseline SCr is unknown. The reversibility of the AKI may be inferred if there is a marked increase in urine output after correction of prerenal problems, but a reduction in SCr (due to an increase in GFR) may not be seen for 12–24 h.
Criteria for diagnosis
Serum biochemistry
The SCr concentration is the best available guide to the GFR. Acute reductions in GFR produce an increase in the SCr concentration. The changes in SCr concentration lag behind the change in GFR, and can be affected by the dilution effect of intravenous fluid. Correct interpretation of the SCr concentration extends beyond just knowing the normal values (Fig. 10.1.3). Creatinine is a metabolic product of creatine and phosphocreatine, which are found almost exclusively in skeletal muscle. The SCr concentration is affected by the muscle mass, meat intake, GFR, tubular secretion (which can vary in the same individual and increases as the GFR decreases) and breakdown of creatinine in the bowel (which increases in chronic renal failure). The GFR decreases by 1% per year after 40 years of age, yet the SCr concentration remains unchanged because the decrease in muscle mass with age reduces the production of creatinine. The GFR (corrected for body surface area) is 10% greater in males than females, but men have a higher muscle mass per kilogram of body weight. The SCr concentration in men is thus greater than in women.
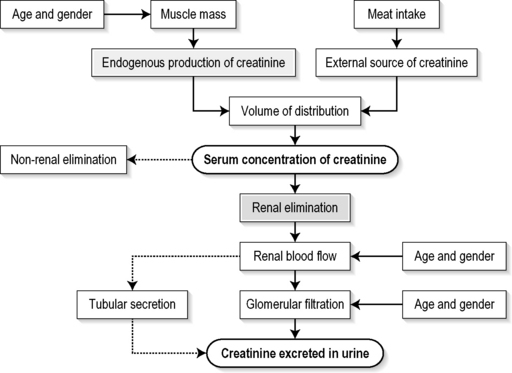
The creatinine clearance (CCr) or GFR are estimated indirectly using formulae (Cockcroft–Gault formula or the Modification of Diet in Renal Disease (MDRD) Study Equation) based on the SCr concentration33,34 (Fig. 10.1.4). These equations assume a steady-state SCr concentration, and are inaccurate if the GFR is changing rapidly. They will also be less accurate in amputees, very small or very large persons, or persons with muscle-wasting diseases.

Hyperkalaemia is a common complication, with the serum K+ usually rising by 0.5 mmol/L/day in ARF. The serum Ca2+ concentration may be normal or reduced in ARF. Both hypocalcaemia and hypercalcaemia may occur at different stages of ARF in rhabdomyolysis. Rhabdomyolysis is characterized by a very high blood CK concentration. Abnormal liver function tests invariably accompany hepatorenal syndrome and hepatic cirrhosis.
Urine tests
The measurement of the concentration of electrolytes in the urine, and the calculation of their fractional excretion, is of intellectual interest in understanding the pathophysiological responses of the nephron to different types of AKI. The calculations are cumbersome, the results are inconsistent, and the information obtained does not alter the patient’s immediate treatment.43
Imaging
A normal ultrasound examination can occur in the very early stages of obstruction, or if ureteric obstruction is due to retroperitoneal fibrosis or to infiltration by tumour.14–16 Hydronephrosis not due to obstruction occurs in pregnancy, vesicoureteric reflux or in diabetes insipidus.
Treatment
Monitoring and maintaining urine output
Urinary Catheter
Accurate measurement of urine output requires insertion of a urinary catheter, but this is not needed in the less severe forms of AKI if there is frequent spontaneous voiding. A catheter is required initially in persons with oliguria or (apparent) anuria, shock or obstruction to bladder outflow.
Electrolyte abnormalities
Potassium
Hyperkalaemia is due to an imbalance between potassium intake and renal potassium excretion, or follows redistribution of potassium from the intracellular to the extracellular space. Hyperkalaemia (often with metabolic acidosis) is a frequent finding in chronic obstruction.44,45 Hyperkalaemia in AKI can be asymptomatic, produce electrocardiogram (ECG) changes or cause potentially fatal changes in cardiac rhythm.
The initial ECG changes in hyperkalaemia are shortening of the PR and QT interval, followed by peaked T waves that are most prominent in leads II, III and V2 through V446,47 (Fig. 10.1.5). Marked ST-T segment elevation (pseudomyocardial infarction pattern) may occur.48 Bradycardia with sinoatrial (SA) block or atrioventricular block (including complete heart block) can develop and progress to periods of cardiac standstill or asystole. More commonly the PR interval is prolonged and the QRS complex is widened, with the QRS complex having a left or right bundle branch block configuration (Fig. 10.1.6). At high serum [K+] (8–9 mmol/L) the sinoatrial (SA) node may stimulate the ventricles without ECG evidence of atrial activity (sinoventricular rhythm).49 When the serum [K+] is 10 mmol/L or greater SA conduction no longer occurs and junctional rhythms are seen. The QRS complex width continues to increase and eventually the QRS complexes and the T wave blend, producing a sine wave ECG. At this stage ventricular fibrillation or asystole are imminent.
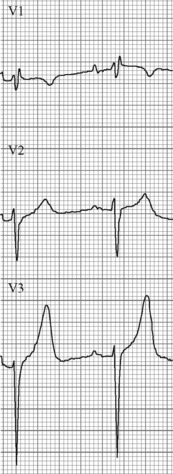
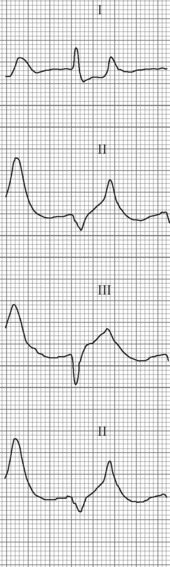
The higher the serum [K+] concentration the more likely is the occurrence of ECG changes or life-threatening arrhythmias or both. However, nearly half of persons with a serum [K+] greater than 6.8 mmol/L do not have ECG changes of hyperkalaemia.50,51 Physicians predict the presence of hyperkalaemia solely on the basis of ECG changes with a sensitivity of less than 45%.52
Drugs such as oral potassium tablets, ACE inhibitors and aldosterone antagonists should be ceased in AKI. Hyperkalaemia is treated when the serum [K+] is greater than 6.5 mmol/L (even if there are no ECG changes) or when there are ECG changes of hyperkalaemia. The emergency treatment of hyperkalaemia is covered in Chapter 12.2 Electrolyte disturbances.
Sodium
The sodium concentration in AKI may be normal, low (when water excess is present) or high (when water depletion is present). Hyponatraemia in ARF is usually asymptomatic and is treated by water restriction. Patients with ARF and symptomatic hyponatraemia should be treated with haemofiltration or dialysis. Hypernatraemia is treated by slow intravenous infusion of hypotonic saline or 5% dextrose.
Fluid overload
Some persons with severe hypertension and chronic renal impairment who have recurrent episodes of acute pulmonary oedema (‘flash’ pulmonary oedema) have significant renovascular disease. Renal revascularization in these patients can reduce or abolish the episodes of pulmonary oedema.53
Management of ATN
Reduction of damage/accelerating recovery
Despite much experimental laboratory work and numerous clinical trials, no therapeutic intervention has hastened the recovery of renal function in established ATN. Therapeutic trials of dopamine, atrial natriuretic peptide and various growth factors have been ineffective. The use of high-dose loop diuretics to convert oliguric ATN to non-oliguric ATN was based on the observation that patients with non-oliguric ATN had a lower mortality and better renal recovery rates than those with oliguric ATN. The use of high-dose loop diuretics does not affect the duration of ATN, the need for dialysis or the outcome.54
Supportive treatment
This includes monitoring fluid input and fluid output, measuring serum electrolyte values frequently, preventing sepsis by reducing the number of intravenous lines and removing urinary catheters, culturing periodically and using antibiotics when indicated clinically. The fluid intake is restricted to insensible water loss (about 500 mL per day in the absence of fever) plus all measured fluid losses (urine output, gastrointestinal losses, chest tube drainage). Nephrotoxic agents should be avoided and the dosage of renally excreted drugs reduced. Because the increase in SCr lags behind the decrease in GFR, drug doses should be calculated based on a GFR of less than 10 mL/min per 1.73 m2 rather than on the SCr value.
Renal replacement treatment
Renal replacement treatment (RRT) is required in most patients with oliguric ARF, and one-third of patients with nonoliguric ARF. The indications for RRT are summarized in Table 10.1.6.
Table 10.1.6 Indications for renal replacement treatment in acute kidney injury*
| Oliguria (urine output <200 mL/12 h) or anuria (urine output 0–50 mL/12 h) |
| Serum urea concentration >35 mmol/L |
| Serum creatinine concentration >400 μmol/L |
| Serum potassium concentration >6.5 mmol/L or rapidly rising |
| Serum sodium concentration <100 mmol/L or >160 mmol/L |
| Pulmonary oedema not responding to diuretics |
| Severe (uncompensated) metabolic acidosis with pH < 7.1 |
| Uraemic syndrome (asterixis, psychosis, myoclonus, seizures, pericarditis)Overdose with a toxin that is dialyzable |
* Presence of two or more indications in a patient means that renal replacement will be needed.
Prognosis
The prognosis of ARF is largely dependent on the underlying cause and the presence of comorbidities. Mortality varies from about 40% in those with no comorbidity to more than 80% in those who have three or more failed organ systems.
1 Eknoyan G. Emergence of the concept of acute renal failure. American Journal of Nephrology. 2002;22:225-230.
2 Delafield F. Acute Bright’s disease. Med Rec. 1888;33:151-155.
3 Davies F, Weldon R.A. contribution to the study of ‘war nephritis’. Lancet. 1917;II:118-120.
4 Bywaters EG, Beall D. Crush injuries with impairment of renal function. British Medical Journal. 1941;1:427-432.
5 Oliver J, Mac DM, Tracy A. The pathogenesis of acute renal failure associated with traumatic and toxic injury: renal ischemia, nephrotoxic damage and the ischemic episode. Journal of Clinical Investigation. 1951;30:1307-1439.
6 Singri N, Ahya SN, Levin ML. Acute renal failure. Journal of the American Medical Association. 2003;289:747-751.
7 Bellomo R, Ronco C, Kellum JA, et al. Acute renal failuredefinition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Critical Care. 2004;8:R204-R212.
8 Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Annals of Internal Medicine. 2002;137:744-752.
9 Gill N, Nally JV, Fatica RA. Renal failure secondary to acute tubular necrosis: epidemiology, diagnosis, and management. Chest. 2005;128:2847-2863.
10 Tumlin J, Stacul F, Adam A, et al. on behalf of the CIN Consensus Working Panel. Pathophysiology of contrast-induced nephropathy. American Journal of Cardiology. 2006;98(suppl):14K-20K.
11 Schetz M, Dasta J, Goldstein S, et al. Drug-induced acute kidney injury. Current Opinion in Critical Care. 2005;11(6):555-565.
12 Huerta C, Castellsague J, Varas-Lorenzo C, et al. Nonsteroidal anti-inflammatory drugs and the risk of ARF in the general population. American Journal of Kidney Diseases. 2005;45(3):531-539.
13 Settergren B, Ahlm C, Alexeyev O, et al. Pathogenetic and clinical aspects of the renal involvement in hemorrhagic fever with renal syndrome. Renal Failure. 1997;19(1):1-14.
14 James B, Naidich JB, Rackson ME, et al. Nondilated obstructive uropathy: percutaneous nephrostomy performed to reverse renal failure. Radiology. 1986;160:653-657.
15 Charasse C, Camus C, Darnault P, et al. Acute nondilated anuric obstructive nephropathy on echography: difficult diagnosis in the intensive care unit. Intensive Care Medicine. 1991;17:387-391.
16 Leong W, Sells H, Moretti KL. Anuric, obstructive uropathy in the absence of obvious radiological evidence of obstruction. Australian and New Zealand Journal of Surgery. 2004;74(7):611-613.
17 Khan IH, Catto GR, Edward N, et al. Acute renal failure: factors influencing nephrology referral and outcome. QJM. 1997;90(12):781-785.
18 Stevens PE, Tamimi NA, Al-Hasani MK, et al. Non-specialist management of acute renal failure. QJM. 2001;94:533-540.
19 Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicentre, community based study. Madrid Acute Renal Failure Study Group. Kidney International. 1996;50(3):811-818.
20 Feest TG, Round A, Hamad S. Incidence of severe acute renal failure in adults: results of a community based study. British Medical Journal. 1993;306:481-483.
21 Kaufman J, Dhakal M, Patel B, et al. Community acquired acute renal failure. American Journal of Kidney Disorder. 1991;17(12):191-198.
22 Shusterman N, Strom BL, Murray TG, et al. Risk factors and outcomes of hospital acquired ARF: clinical epidemiologic study. American Journal of Medicine. 1987;83:65-71.
23 Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. American Journal of Kidney Diseases. 2002;39(5):930-936.
24 Liangos O, Wald R, O’Bell JW, et al. Epidemiology and outcomes of acute renal failure in hospitalized patients: a national survey. Clinical Journal of American Society of Nephrology. 2006;1(1):43-51.
25 Sesso R, Roque A, Vicioso B, et al. Prognosis of ARF in hospitalized elderly patients. American Journal of Kidney Diseases. 2004;44(3):410-419.
26 Ali T, Khan I, Simpson W, et al. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. Journal of American Society of Nephrology. 2007;18:1292-1298.
27 Uchino S, Bellomo R, Goldsmith D, et al. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Critical Care Medicine. 2006;34(7):1913-1917.
28 Uchino S. The epidemiology of acute renal failure in the world. Current Opinion in Critical Care. 2006;12:538-543.
29 Anderson RJ, Schrier RW. Acute renal failure. In: Schrier RW, Gottschalk CW, editors. Diseases of the kidney. 6th edn. Boston: Little Brown; 1997:1069-1113. Chapter 41
30 Morris JA, Mucha P, Ross SE, et al. Acute post traumatic renal failure: a multicentre perspective. Journal of Trauma. 1991;31(12):1584-1590.
31 Domanovits H, Schillinger M, Mullner M, et al. Acute renal failure after successful cardiopulmonary resuscitation. Intensive Care Medicine. 2001;27(7):1194-1199.
32 Bagshaw SM, Bellomo R. Fluid resuscitation and the septic kidney. Current Opinion in Critical Care. 2006;12:527-530.
33 Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31-41.
34 Stevens LA, Coresh J, Greene T, et al. Assessing kidney function-measured and estimated glomerular filtration rate. New England Journal of Medicine. 2006;354:2473-2483.
35 Brown CV, Rhee P, Chan L, et al. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? Journal of Trauma. 2004;56(6):1191-1196.
36 Mange K, Matsuura D, Cizman B, et al. Language guiding therapy: the case of dehydration versus volume depletion. Annals of Internal Medicine. 1997;127:848-853.
37 Demetriades D, Chan LS, Bhasin P, et al. Relative bradycardia in patients with traumatic hypotension. Journal of Trauma. 1998;45(3):534-539.
38 Victorino GP, Battistella FD, Wisner DH. Does tachycardia correlate with hypotension after trauma? Journal of American College of Surgeons. 2003;196:679-684.
39 McGee S. Evidence based physical diagnosis, 2nd edn, Saunders Elsevier: St Louis; 2007:94-96. 153–173
40 MacDowall P, Kalra PA, O’Donoghue DJ, et al. Risk of morbidity from renovascular disease in elderly patients with congestive cardiac failure. Lancet. 1998;352:13-16.
41 Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population based study. Journal of Vascular Surgery. 2002;36(3):43-51.
42 Schrier RW, Wang W. Acute renal failure and sepsis. New England Journal of Medicine. 2004;351:159-169.
43 Bagshaw SM, Langenberg C, Bellomo R. Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. American Journal of Kidney Diseases. 2006;48:695-705.
44 Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructed uropathy. New England Journal of Medicine. 1981;304(7):373-380.
45 Pelleya R, Oster JR, Perez GO. Hyporeninemic hypoaldosteronism, sodium wasting and mineralocorticoid-resistant hyperkalemia in two patients with obstructive uropathy. American Journal of Nephrology. 1983;13(4):223-227.
46 Dittrich KL, Walls RM. Hyperkalemia: ECG manifestations and clinical considerations. Journal of Emergency Medicine. 1986;4(6):449-455.
47 Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Annals of Emergency Medicine. 1994;24:305-311.
48 Simon BC. Pseudomyocardial infarction and hyperkalemia: a case report and subject review. Journal of Emergency Medicine. 1988;6(6):511-515.
49 Cohen HC, Gozo EGJr, Pick A. The nature and type of arrhythmias in acute experimental hyperkalemia in the intact dog. American Heart Journal. 1971;82(6):777-785.
50 Acker CG, Johnson JP, Palevsky PM, et al. Hyperkalemia in hospitalized patients: causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Archives of Internal Medicine. 1998;158(8):917-924.
51 Martinez-Vea A, Bardaji A, Garcia C, et al. Severe hyperkalemia with minimal electrocardiographic manifestations: a report of seven cases. Journal of Electrocardiology. 1999;32(1):45-49.
52 Wrenn KD, Slovis CM, Slovis BS. The ability of physicians to predict hyperkalemia from the ECG. Annals of Emergency Medicine. 1991;20(11):1229-1232.
53 Bloch MJ, Trost DW, Pickering TG, et al. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. American Journal of Hypertension. 1999;12(1):1-7.
54 Ho KM, Sheridan DJ. Meta-analysis of frusemide to prevent or treat acute renal failure. British Medical Journal. 2006;333:420-425.
10.2 The acute scrotum
Aetiology
Torsion is due to a powerful contraction of the cremaster muscles in an abnormally attached testis. A normal testis is anchored posterolaterally to the scrotal sac and is, therefore, fixed in place. The main abnormality found in patients with torsion is an enlarged tunica vaginalis, which surrounds the whole of the testes and epididymis, preventing the testis from creating any attachment to the scrotal wall. The testis, therefore, floats freely like a clapper inside a bell. The contraction of the cremaster causes the testes and adnexa to be rotated, thereby twisting the cord.1
Pathology
The extent and rapidity of the damage depends on the degree of torsion, that is the number of turns:
Clinical presentation
There is a sudden onset of severe scrotal or abdominal pain. There are no irritative voiding symptoms. Between 29 and 50% of patients have had previous episodes of acute scrotal pain.3,4
The patient looks pale and may vomit. The testis is tender and riding high in the scrotum. Other signs are loss of cremasteric reflex, scrotal oedema testicular swelling and retraction. One study in which all children had mandatory exploration found these clinical signs had sensitivities of 60–91% and specificities of 27–68%.5
Systemic signs such as fever are classically absent. Urinalysis is normal.
Intermittent torsion of the testis
This is a syndrome of recurrent acute scrotal pain, usually lasting less than 2 h, which resolves spontaneously. Creagh and McDermott4 describe a series of 27 patients who underwent elective orchidopexy for these symptoms. Three patients developed acute torsion while on the waiting list; of those coming to operation, one had an atrophic testis and four had evidence of torsion of the appendages of the testis. One patient subsequently had torsion after surgery because absorbable sutures were used.4
Differential diagnosis of acute testicular pain
Differential diagnoses to consider in acute testicular pain are listed in Table 10.2.1.
Table 10.2.1 Differential diagnosis of acute testicular pain
| Epididymo-orchitis |
| Strangulated hernia |
| Haematocoele |
| Hydrocoele |
| Testicular tumour |
| Henoch Schonlein purpura in children |
| Idiopathic scrotal oedema. |
Traps in the clinical diagnosis
There are many potential pitfalls in the clinical diagnosis of the acute scrotum:
Clinical findings remain misleading and none can reliably exclude the diagnosis of torsion.7
Investigations
Surgical exploration of the scrotum
This is the investigation of choice where the diagnosis of torsion is likely, and maximizes the chance of saving the testis. Delaying the diagnosis is ‘castration by neglect’.3,7 Surgical exploration requires only a skin incision and has no major complications.5,7
Role of investigations in suspected testicular torsion
When the diagnosis of torsion remains probable then the only investigation is surgical exploration of the scrotum, and any investigations that delay theatre are unnecessary. If torsion is unlikely clinically, but needs to be excluded, then CDI can be used, provided it is available on an urgent basis.1,3,7 It is important that there is early communication and discussion with the responsible surgeon so no unavoidable delays are instituted between the suspicion of torsion and any surgical intervention.1–3
Treatment
Manual untwisting
This manoeuvre is not universally recommended and should be done only as a temporizing measure or when surgical exploration cannot be performed. The spermatic cord is infiltrated with local anaesthetic and the testis is untwisted. Untwisting is done by turning the left testis anticlockwise (outward) and the right one clockwise, like opening the pages of a book.8
Surgery
The scrotum is opened and the testis is delivered, untwisted and inspected for return of colour and bleeding. An obviously infarcted testis is removed at the initial surgery. A viable testis is sutured into place on the scrotal wall. The tunica should be inverted and also sutured to the scrotal wall. It is vital that the normal side is also explored and fixed to the scrotal wall, as the abnormality is bilateral in most cases. Retorsion following orchidopexy has occurred when absorbable sutures have been used.1,3–6
Prognosis
Testicular salvage (return of circulation at surgery) does not mean no injury to the testicle. Long-term follow-up of salvaged testes shows that 75% have a reduction in volume. Abnormalities are also seen in sperm volume, motility and morphology. These abnormalities are not seen in patients who have had an infarcted testis removed at the initial operation. This suggests some antispermatogenesis effect caused by the damaged testicle.1,3,4
Torsion of a testicular appendage
These are embryological remnants with no function. They are small (<5 mm) pedunculated structures that may twist on their pedicle. If the appendage can be isolated in the scrotum a small blue lump may be isolated: ‘the blue dot sign’. These do not need surgery and can be treated with analgesia. Late presentations may have scrotal or testicular swelling, in which case they should be treated as torsion until proved otherwise.1
Aetiology
A variety of organisms may be responsible for EDO (Table 10.2.2).
| Bacterial: Neisseria gonorrhoeae, Escherichia coli, Pseudomonas aeruginosa, coliforms, Klebsiella, Mycobacterium tuberculosis |
| Chlamydial: C. trachomatis |
| Viral: mumps |
| Drugs: amiodarone epididymitis |
| Fungal: cryptococcal |
| Parasitic: filariasis (usually chronic). |
EDO is usually thought to be an ascending infection from the urethra or prostate, but it can be part of a generalized systemic disease. The infection spreads from epididymis to testicle, and eventually they may become one large inflammatory mass. Isolated orchitis is rare and usually due to viral causes, which are spread via the bloodstream.9–11
Investigations
Urethral swabs
Urethral discharge may not be seen if the patient has just voided, so a urethral swab and smear should be examined for white blood cells (WBC). If there are more than five WBC per high-powered field, then urethritis is likely. The presence of intracellular diplococci confirms the diagnosis of gonorrhoea; their absence suggests chlamydia.9
Differential diagnosis
In the acute non-traumatic setting the most important differential diagnosis is torsion of the testicle.9 If the clinical features, urethral swabs or mid-stream urine do not differentiate, then ultrasound or isotope scans may help. In young men, if these are not available and there is no evidence of UTI or urethritis, then surgical exploration may be necessary. Ultrasound can help differentiate other causes of the acute scrotum.
Blunt traumatic injury to the testicle
The mobility of the testicle, cremaster muscle contraction and the tough capsule usually protect the testicle from injury. However, a direct blow that drives the testicle against the symphysis pubis may result in contusion or rupture of the testicle. Typical mechanisms are a direct kick to the groin, or handlebar and straddle injuries.12,13
The most serious is testicular rupture, where the tunica is split, allowing blood and seminiferous tubules to extrude into the tunica vaginalis. This occurs in up to 50% of blunt trauma. Complete disruption of the testis may occur.12–14
Indications for exploratory surgery include:
It should be noted that 10–15% of testicular tumours present after an episode of trauma, and so any abnormalities on ultrasound examination should be followed to resolution if surgery is not performed.15
Early surgical exploration with evacuation of blood clots in the tunica vaginalis and repair of testicular rupture, if present, results in a shortened hospital stay, a greatly reduced period of disability and a faster return to normal activity compared to patients managed conservatively. Conservative management is complicated by secondary infection of the haematocoele, frank acute necrosis of the testis and delayed atrophy due to pressure effects of haematoma. The orchidectomy rate for early exploration is only 9%, compared to 45% for those managed non-operatively.13
Controversies and future directions
1 Lutzker LG, Zuckier LS. Testicular scanning and other applications of radionuclide imaging of the genital tract. Seminars in Nuclear Medicine. 1990;20(2):159-188.
2 Herbener TE. Ultrasound in the assessment of the acute scrotum. Journal of Clinical Ultrasound. 1996;24:405-421.
3 Cass AS. Torsion of the testis. Postgraduate Medicine. 1990;87:69-74.
4 Creagh TA, McDermott TE, McLean PA, et al. Intermittent torsion of the testis. British Medical Journal. 1988;297:525-526.
5 Van Glabeke E, Khairouni A, Larroquet M, et al. Acute scrotal pain in children: results of 543 surgical explorations. Pediatric Surgery International. 1999;15:353-357.
6 Rajfer J. Testicular torsion. Walsh PC, Retik AB, Darracott VE, Wein AJ, editors. Campbell’s urology, 7th edn, Vol. 2. London: WB Saunders, 1997;2184-2186.
7 Murphy FL, Fletcher L, Pease P. Early Scrotal exploration in all cases is the investigation and intervention of choice in the acute paediatric scrotum. Pediatric Surgery. 2006;22(5):413.
8 Schneider RE. Testicular torsion. In Tintinalli JE, Ruiz E, Krome RL, editors: Emergency medicine: a comprehensive study guide, 4th edn, New York: McGraw-Hill, 1996.
9 Berger R. Epididymitis. Walsh PC, Retik AB, Darracott VE, Wein AJ, editors. Campbell’s urology, 7th edn, Vol. 1. London: WB Saunders, 1997;670-673.
10 Tintanalli JE, Ruiz E, Krome RL. Epididymitis. In Tintinalli JE, Krome RL, editors: Emergency medicine: a comprehensive study guide, 4th edn, New York: McGraw-Hill, 1996.
11 Therapeutic Guidelines. Antibiotics, 10th edn. Melbourne: Therapeutic Guidelines Limited; 2006. March
12 Bertini JE, Corriere JN. The etiology and management of genital injuries. Journal of Trauma. 1990;28:1278-1281.
13 Cass AS. Testicular trauma. Journal of Urology. 1983;129:299-300.
14 Kukadia AN, Ercole CJ, Gleich P, et al. Testicular trauma: potential impact on reproductive function. Journal of Urology. 1996;156:1643-1646.
15 Cass AS, Luxenberg M. Testicular injuries. Urology. 1991;38:528-530.
10.3 Renal colic
Introduction
Nephrolithiasis is a common disorder affecting 2–5% of the population at some point in their lives.1 It occurs most frequently between the ages of 20 and 50 years, with a male:female ratio of approximately 3:1. About 50% of patients have a single episode but the remaining 50% have recurrent episodes within 5 years.2
Predisposing factors for stone formation include prolonged immobilization, strong family history of nephrolitiasis, hyperparathyroidism or peptic ulcer disease (hyperexcretion of calcium), small bowel disease, such as Crohn’s disease or ulcerative colitis (hyperoxaluria), and gout (hyperuricaemia). Myeloproliferative disorders, malignancy, glycogen storage disorders, renal tubular acidosis and the use of certain medications (calcium supplements, acetazolamide, vitamins C and D, and antacids) may also be conducive to nephrolithiasis.3
Pathophysiology of pain
Acute obstruction of the upper urinary tract from a calculus results in increased pressure in the renal pelvis, which, in turn, induces the synthesis and secretion of renal prostaglandins, in particular PGE2, which promotes a diuresis by causing dilatation of the afferent arteriole, further elevating the renal pelvic pressure.4,5 The acute obstruction and renal capsular tension are believed to be the cause of the constant ache in the costovertebral angle.
In experiments with isolated ureteric smooth muscle, prostaglandins have also been shown to increase phasic and tonic contractile activity,6 resulting in ureteric spasm and severe, colicky pain.
Presentation
The pain of renal colic has been described as the worst pain a person can endure. The classic textbook description is of severe, intermittent, flank pain of abrupt onset originating from the area of the costovertebral angle and radiating anteriorly to the lower abdominal and inguinal regions. Testicular or labial pain may be present and may suggest the location of the stone as a low ureteric position. Urinary frequency or urgency often develops as the stone nears the bladder, and nausea and vomiting frequently accompany the pain. One-third of patients complain of gross haematuria.7
Urinalysis usually shows red blood cells, although the absence of red cells in the urine in the setting of colicky flank loin to groin pain does not rule out nephrolithiasis, and between 10 and 30% of patients with documented nephrolithiasis do not have haematuria.8 Nitrites, leukocytes or micro-organisms in the urine suggest either the complication of infection or a diagnosis of acute pyelonephritis. Urine culture is indicated to rule out infection with urea-splitting organisms such as Klebsiella and Proteus spp. Electrolyte studies may demonstrate obstruction or suggest an underlying metabolic abnormality such as hypercalcaemia, hyperuricaemia or hypokalaemia. A slightly elevated white blood cell count may occur with renal colic, but a count greater than 15 000/mm3 suggests active infection.
Many conditions may have a similar presentation to renal colic, and examination and investigations should be directed towards confirming the diagnosis of nephrolithiasis and excluding the other conditions in the differential diagnosis (Table 10.3.1).
| • Renal carcinoma producing blood clots temporarily occluding the ureter |
| • Ectopic pregnancy |
| • Ovarian torsion |
| • Abdominal aortic aneurysm |
| • Acute intestinal obstruction |
| • Pyelonephritis |
| • Appendicitis |
| • Diverticulitis |
| • Narcotic seekers and Munchausen’s syndrome |
Radiological examination
A variety of imaging modalities is used to evaluate renal colic. Their pros and cons are listed in Table 10.3.2.
Table 10.3.2 Pros and cons of imaging modalities in renal colic
| Pros | Cons | |
|---|---|---|
| CT | High sensitivity (97%) | Exposes patient to radiation |
| High specificity (96%) | Higher cost | |
| Nearly all stones opaque | ||
| Can accurately measure stone size | ||
| Can detect obstruction | ||
| Can diagnose other causes of flank pain | ||
| Can avoid the use of contrast | ||
| Abdominal radiography (KUD) | Readily available | Low sensitivity |
| Fast | Exposes patient to radiation | |
| Intravenous urography | Provides information regarding size and location of stone | Potential for contrast reaction |
| Measure of renal function | Exposes patient to radiation | |
| More time-consuming than CT | ||
| Unable to exclude alternative diagnoses | ||
| MRI | Useful in pregnant patients | Not readily available |
| Does not use ionizing radiation | Time consuming | |
| Does not use contrast | Accuracy may be less than IVU | |
| Ultrasound | Non invasive | Lower sensitivity than IVU |
| No exposure to ionizing radiation | Size of stone cannot be accurately measured | |
| Modality of choice in pregnant patients | May not be available 24 h | |
| Requires skilled operator |
Most stones (90%) are radio-opaque and theoretically should be visible on plain X-ray; if seen, they are irregularly shaped densities on abdominal radiography (KUB). However, a KUB alone is not usually sufficient to make the diagnosis of nephrolithiasis as it has poor sensitivity of between 58 and 62%.9 Phlebitis in the pelvic veins and calcified mesenteric lymph nodes may add confusion, and many small stones may be obscured by the bony density of the sacrum. Thus, plain X-ray should only be used in conjunction with another imagingmodality such as ultrasound in the setting of renal colic.
Computerized tomography (CT), with or without contrast, is the first-line test in many centres, and has become the adopted gold standard with high sensitivity (97%) and specificity (96%) for ureterolithiasis.10 Nearly all stones are opaque on CT, and thus the size of the stone and its position can be accurately measured. Other positive findings include perinephric stranding, dilatation of the kidney (hydronephrosis) or ureter, and low density of the kidney, suggesting oedema. Non-contrast CT is equivalent to intravenous urography (IVU) in the diagnosis of obstruction and is more reliable in the detection of ureterolithiasis.11 It is also useful in the exclusion or confirmation of the other intra-abdominal differential diagnoses such as appendicitis, abdominal aortic aneurysm or diverticulitis. As no contrast is used there is not the risk of contrast reaction that is associated with IVU. It is more rapid than IVU and does not depend on the technical expertise required by other imaging modalities such as ultrasound, but it does subject the patient to a larger dose of radiation than IVU.
The intravenous pyelogram had been the standard investigation for the evaluation of renal colic until the widespread adoption of CT. It establishes the diagnosis of calculus disease in 96% of cases and determines the severity of obstruction.12 Classic findings of acute obstruction include a delay in the appearance of one kidney, a dilated ureter and a dilated renal pelvis.13 IVU is useful in estimating the size of the stone, in identifying extravasation of dye and in evaluating renal function. Its main disadvantage is the use of ionizing radiation, although less than in CT, and the administration of intravenous iodinated contrast media with its risk of contrast reaction. Compared with CT it is time-consuming and unable to offer alternative diagnoses.
Ultrasonography is a useful, safe and a non-invasive alternative when renal function is impaired or contrast media contraindicated. It can identify the stone, its location and demonstrate proximal obstruction such as hydroureter or a dilated pelvis, as well as the size and configuration of each kidney, but unfortunately not size of the stone. Ultrasound has significantly lower sensitivity than IVU and misses more than 30% of stones.14.
Magnetic resonance imaging (MRI) can easily depict a dilated ureter and demonstrate the level of obstruction without using ionizing radiation or contrast. The accuracy of MRI for stones may be lower than IVU as its special resolution is not high enough to detect small stones, but when used in combination with ultrasound it may have a role in the evaluation of loin pain, especially in the pregnant patient. MRI is, however, expensive, time-consuming and usually not readily available to most emergency departments (EDs).
Management
Intravenous narcotics provide rapid analgesia, are titratable to effect and relieve anxiety in most cases. However, prolonged use may cause dependence and tolerance. Side effects are common and include nausea, vomiting, drowsiness, constipation and with larger doses precipitate respiratory depression and hypotension. The data are very variable with regards to the effect of opioids on ureteric tone. Results indicate an increase in ureteric tone or no effect at all.15
Codeine, a less potent opioid than morphine, is effective for relieving mild to moderate pain associated with renal colic. Constipation is a significant side effect and limits its long-term use. One hundred milligrams of tramadol, an opioid-like agents but with fewer side effects when used for treating renal colic, has been shown to be as effective as pethidine 50 mg in one study,16 but more research is needed before adopting tramadol as an alternative to conventional opioids.
NSAIDs appear to be equally effective when compared with opioids.17 A double-blind study comparing diclofenac and an opioid demonstrated a better effect with diclofenac and fewer side effects, but slower onset of action.18 There are many NSAIDs available, differing in preparation and route of administration, the major differences between them being the incidence and nature of side effects, predominantly gastric irritation, ulceration and precipitation of renal failure. Ibuprofen has the fewest side effects and the lowest risk of gastrointestinal effects, but the weakest analgesic action. Naproxen and diclofenac provide stronger analgesia and a relatively low incidence of side effects. Oral diclofenac and oral/rectal indometacin have both been shown to be effective in reducing the number of new renal colic episodes as well as further admission to hospital, but have no effect on spontaneous stone passage rates.19,20 Thus, it has been suggested that one should give both a rapidly acting titratable opioid and a slower acting NSAID, which may result in earlier discharge from the ED.21 Intravenous preparations of NSAIDs have limited availability in Australian EDs and have been reported to have a faster onset of action but a higher incidence of side effects, and therefore if available should be used with caution.
Buscopan, an antimuscarinic agent used for treating smooth muscle spasm, has been shown to decrease ureteric activity to some degree in 80% of the subjects studied.22 However, one study comparing its use to a NSAID found that buscopan was less effective23 and was associated with significant side effects, including dry mouth, photophobia, urgency, retention and constipation, significantly limiting its use in renal colic.
Recently the use of alpha-blockers in renal colic has been reported, with a number of studies showing that patients treated with alpha-blockers as well as standard therapy achieve stone clearance more often and take less time to do so than controls.24
The size, shape and site of the stone at initial presentation are factors that determine whether a stone passes spontaneously or requires removal. Stones less than 5 mm in patients without associated infection or anatomic abnormality pass within 1 month in 90% of cases, stones 4–6 mm pass 50% of the time but only 5% of stones larger than 7 mm pass, and hence usually require elective surgical removal.8 The overall passage rate for ureteral stones is:
Indications for admission to hospital are listed in Table 10.3.3.
Table 10.3.3 Indications for hospital admission in renal colic
| • Presence of infection |
| • Deteriorating renal function |
| • Persistent pain requiring parenteral narcotics |
| • Stone greater than 5 mm in diameter |
| • Extravasation of dye (uncommon) |
Precautions
Renal colic, with its minimal findings on examination, is a commonly used presentation for those seeking narcotics or with Munchausen’s syndrome, and treating physicians should be aware of this. However, it is essential to give analgesia to those patients suffering from renal colic and it is probably preferable to give patients analgesia unnecessarily than cause unnecessary suffering. Features suggesting narcotic seeking are discussed in Chapter 21.5.
Conclusion
Renal colic is an acutely distressing medical condition that requires a careful evaluation of symptoms and signs to ensure timely analgesia, recognition of other causes of acute abdominal pain and avoidance of inappropriate narcotic usage.
1 Lingeman J. Calculous disease of the kidney and bladder. In: Harwood-Nuss A, editor. The clinical practice of emergency medicine. Philadelphia: JB Lippincott, 1991.
2 Trivedi BK. Nephrolithiasis. Postgraduate Medicine. 1996;100(6):3-78.
3 Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. New England Journal of Medicine. 1992;327(16):1141-1152.
4 Holmlund D. The pathophysiology of ureteric colic. Scandinavian Journal of Urology and Nephrology. 1983;75(suppl):25-27.
5 Nishikawa K, Morrisin A, Needleman P. Exaggerated prostaglandin biosynthesis and its influence on renal resistance in the isolated hydronephrotic rabbit kidney. Journal of Clinical Investigation. 1977;59:1143-1150.
6 Cole RS, Fry CH, Shuttleworth KED. The action of prostaglandins on isolated human ureteric smooth muscle. British Journal of Urology. 1988;61:19-26.
7 Smith DR. General urology. Los Altos, 9th edn. Lange Medical Publishers, California, 1978.
8 Teichman JM. Acute renal colic from ureteral calculus. New England Journal of Medicine. 2004;350:684.
9 Mutgi A, Willliams JW, Nettleman M, Renal colic. The utility of the plain abdominal roentgenogram. Archives of Internal Medicine. 1991;151:1589-1592.
10 Kenney PJ. CT evaluation of urinary lithiasis. The Radiological Clinics of North America. 2003;41:979-999.
11 Sourtzis S, Thibeau JF, Damry N, et al. Radiologic investigation of renal colic: unenhanced helical CT compared with excretory urography. American Journal of Roentgentology. 1999;172:1491-1494.
12 Harrison JH, et al, editors. Campbell’s urology, 4th edn, Vol. 1. Philadelphia: WB Saunders, 1987.
13 Samm BJ, Dmochowski RR. Urologic emergencies. Postgraduate Medicine. 1996;100(4):177-184.
14 Svedstorm E, Alanen A, Nurmi M. Radiologic diagnosis of renal colic: the role of plain films, excretory urography and sonography. European Journal of Radiology. 1990;11:180-183.
15 Lennon GM, Bourke J, Ryan PC, et al. Pharmacological options for the treatment of acute ureteric colic. British Journal of Urology. 1993;71:401-407.
16 Salehi M, Ghaserni H, Shiery H, et al. Intramuscular tramadol versus intramuscular pethidine for the treatment of acute renal colic. Journal of Endourology. 2003;17(suppl 1):A243.
17 Cordell WH, Larson TA, Lingerman JE, et al. Indomethacin suppositories versus intravenous titrated morphine for treatment of ureteric colic. Annals of Emergency Medicine. 1994;23:262-269.
18 Lundstam SO, Leissner KH, Wahlandar LA, et al. Prostaglandin synthetase inhibition of diclofenac in the treatment of renal colic: comparison with use of a narcotic analgesic. Lancet. 1982:1096-1097.
19 Laerum E, Omundsen OE, Gronseth JE, et al. Oral diclofenac in the prophylactic treatment of recurrent renal colic. European Journal of Urology. 1995;28:108-111.
20 Grenabo L, Holmlund D. Indomethacin as prophylaxis against recurrent ureteral colic. Scandinavian Journal of Urology and Nephrology. 1984;18:325-327.
21 Larkin GL, Peacock WF, Pearl SM, et al. Efficiency of ketorolac tromethamine verses meperidine in ED treatment of acute renal colic. American Journal of Emergency Medicine. 1999;17(1):6-10.
22 Ross JA, Edmond P, Kirkland IS. The action of drugs on the intact human ureter. In: Behaviour of the human ureter in health and disease. Churchill Livingstone; 1972:118-129. Chapter 9
23 Al-waili NS, Saloom KY. Intravenous tenoxicam to treat acute renal colic: comparison with buscopan. Journal of the Pakistan Medical Association. 1998;48(12):370-372.
24 De Sio M, Autorino R, Lorenzo GD, et al. Medical expulsive treatment of distal-ureteral stones using tamsulosin. Journal of Endourology. 2006;20(1):12-16.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Section 10 Genitourinary
10.1 Acute kidney injury
Introduction
The first recognition of illness caused by a sudden decline in renal function (‘ischuria renalis’) was by William Heberden in 1802.1 In 1888 Delafield described a form of ‘acute Bright’s disease’ that was caused by toxins, various infectious diseases and extensive injuries, and where there was degeneration or death of tubule cells.2 Impaired renal function in injured soldiers was described in World War I (‘war nephritis’) and in World War II.3,4 The term ‘acute renal failure’ (ARF) was first used in 1951.5
The basic process in ARF is a rapid (hours to days) reduction in the glomerular filtration rate (GFR) due to renal hypoperfusion (prerenal causes), damage to glomeruli, tubules, interstitium or blood vessels (renal causes), or obstruction to urine flow (postrenal causes). The GFR is inversely related to the serum creatinine (SCr) concentration. The diagnosis of ARF is made when there is an increase in the SCr concentration, with or without a decrease in the urine output. A simple definition of ARF is an acute and sustained (lasting for 48 h or more) increase in the SCr of 44 μmol/L if the baseline is less than 221 μmol/L, or an increase in the SCr of more than 20% if the baseline is more than 221 μmol/L.6 A more comprehensive definition (the RIFLE system) is used to classify persons with acute impairment of renal function7 (Table 10.1.1).
| Stage | Serum creatinine (SCr) concentration | Urine output |
|---|---|---|
| RISK | Increase of 1.5 times the baseline | <0.5 mL/kg/h for 6 h |
| INJURY | Increase of 2.0 times the baseline | <0.5 mL/kg/h for 12 h |
| FAILURE | Increase of 3.0 times the baseline or SCr is 355 μmol/L or more when there has been an acute rise of greater than 44 μmol/L for 24 h or anuria for 12 h | <0.3 mL/kg/h |
| LOSS | Persistent acute renal failure; complete loss of kidney function for longer than 4 weeks | |
| END-STAGE RENAL DISEASE | End-stage renal disease for longer than 3 months |
Aetiology and pathogenesis
Prerenal acute kidney injury
Reductions in renal blood flow (RBF) and GFR occur in the setting(s) of hypovolaemia, hypotension (cardiogenic shock, anaphylaxis, sepsis), oedematous states with a reduced ‘effective’ circulating volume (cardiac failure, hepatic cirrhosis, nephrotic syndrome) or renal hypoperfusion (renal artery stenosis, hepatorenal syndrome). Drugs that interfere with autoregulation (e.g. prostaglandin inhibitors, angiotensin converting enzyme (ACE) inhibitors or angiotensin II receptor antagonists) also reduce glomerular perfusion. The physiological responses to volume depletion and hypotension, and the link to prerenal AKI, are shown in Figure 10.1.1.
Renal (parenchymal) acute kidney injury
Acute tubular necrosis (ATN) is the most common pathological process that causes ARF. While the terminology suggests that the main cause is tubular damage, the actual pathophysiology is more complex: impaired autoregulation and marked intrarenal vasoconstriction (the main mechanism for the greatly reduced GFR), tubular damage (with cytoskeleton breakdown), increased tubuloglomerular feedback, endothelial cell injury, fibrin deposition in the microcirculation, release of cytokines, activation of inflammation and activation of the immune system.8,9
ATN is classified as ischaemic ATN or cytotoxic ATN (due to damage by toxins); both processes are present in some patients. In ischaemic ATN there is a continuum between prerenal azotaemia, the RISK and INJURY stages of AKI, and the development of ATN. ATN caused by administration of intravenous or intra-arterial contrast agents is due to renal vasoconstriction, reduced injury.10 The main mechanisms of AKI or ATN caused by drugs are vasoconstriction, altered intraglomerular haemodynamics, tubular cell toxicity, interstitial nephritis, crystal deposition, thrombotic microangiopathy and osmotic nephrosis.11
Important causes of cytotoxic ATN are listed in Table 10.1.2. Non-steroidal anti-inflammatory drugs (NSAIDs), ACE inhibitors and angiotensin receptor blockers (ARBs) often cause a gradual and asymptomatic decrease in the GFR, but can cause AKI (including ATN). NSAIDs do not impair renal function in a healthy person, but can reduce the GFR in elderly persons with atherosclerotic cardiovascular disease, in persons with chronic renal failure, when chronic prerenal hypoperfusion is present (e.g. cardiac failure, cirrhosis), or in persons using diuretics and calcium channel blockers.12
| Exogenous agents |
| Radiocontrast |
| Non-steroidal anti-inflammatory drugs |
| Antibiotics: aminoglycosides, amphotericin B |
| Antiviral drugs: aciclovir, foscarnet |
| Immunosuppressive drugs: ciclosporin |
| Organic solvents: ethylene glycol |
| Poisons: snake venom, paraquat, paracetamol |
| Chemotherapeutic drugs: cisplatin |
| Herbal remedies |
| Heavy metals |
| Endogenous agents |
| Haem pigments: haemoglobin, myoglobin |
| Uric acid |
| Myeloma proteins Correct intravascular volume depletion |
| Maintain perfusion pressure |
| Choice of resuscitation fluid |
| Diuresis in rhabdomyolysis |
| Avoid nephrotoxins |
| Use derived GFR or creatinine clearance when calculating drug doses |
Once ATN is established there is a persistent and marked reduction in RBF and in GFR that lasts for 1 to 2 weeks. During this time the patient is usually oliguric, and cannot excrete concentrated urine. Renal autoregulation is impaired, and renal perfusion depends directly on the systemic blood pressure. A fall in systemic blood pressure during the ATN phase causes more renal damage. Recovery from ATN is associated with increased renal blood flow (reperfusion), an increase in GFR and (often) a large volume urine output because the concentrating ability of the regenerating nephrons is impaired.
Abnormalities of renal interstitial structure and function are present in ATN. However, AKI and ATN can be caused by a primary abnormality of the interstitial tissues: acute tubulointerstitial nephritis (ATIN). The damage in ATIN is due to immunological mechanisms, the most important involving cell-mediated immunity. ATIN is usually due to a drug reaction, but can also be caused by infections (e.g. infection with hantavirus, a RNA virus that causes haemorrhagic fever with renal syndrome.13 Drugs that cause ATIN include antibiotics (β-lactam antibiotics, sulphonamides, fluoroquinolones), NSAIDs, cyclooxygenase-2 inhibitors, proton pump inhibitors, diuretics, phenytoin, carbamazepine and allopurinol.
Postrenal (obstructive) acute kidney injury
Obstructive uropathy refers to the functional or structural processes in the urinary tract that impede the normal flow of urine, and obstructive nephropathy is the renal damage caused by the obstruction. Hydronephrosis is a dilatation of the ureter(s); it can occur in the absence of obstruction, and some persons with obstruction do not have a dilated urinary collecting system.14–16
Epidemiology
The annual incidence of ARF in European communities is between 209 and 620 cases per million per year, with an incidence of severe acute renal failure (SCr greater than 500 μmol/L) of 172 cases per million per year.17–20 About 1% of patients in the USA have ARF on admission to hospital, and ARF develops in 5–7% of all hospitalized patients.21–23 The frequency of ARF in hospitalized patients is about 19 per 1000 admissions.24
Studies of the pathogenesis of community acquired ARF have produced conflicting results. In one study the major processes were identified as prerenal in 70% of cases, renal in 11% of cases and postrenal in 17% of cases.21 Other studies found a lower incidence of prerenal factors (present in 21–48% of cases) and a higher incidence of renal factors (present in 34–56% of cases, most commonly due to ATN).19,25 Acute on chronic renal failure was present in 13% of persons in one study.19 The basic processes in hospital acquired ARF are prerenal in 35–40% of cases, renal in 55–60% of cases and post-renal in 2–5% of cases.6
Using the RIFLE criteria the community incidence of AKI is 1811 per million of population, and AKI occurs in 18% of hospitalized patients (9% had changes in SCr concentration and urine output consistent with RISK, 5% had renal INJURY and 4% developed FAILURE).26,27
There are geographical differences in the causes of ATN. In Africa, India, Asia and Latin America ATN is usually caused by infections (e.g. diarrhoeal illnesses, malaria, leptospirosis), ingestion of plants or medicinal herbs, envenomation, intravascular haemolysis due to glucose-6-phosphate dehydrogenase deficiency or poisoning.28 The incidence of ATN due to crushing injuries is increased in earthquake-prone areas.
Prevention
The processes involved in the prevention of AKI are shown in Table 10.1.3.
Table 10.1.3 Use derived GFR or creatinine clearance when calculating drug doses
| Correct intravascular volume depletion |
| Maintain perfusion pressure |
| Choice of resuscitation fluid |
| Diuresis in rhabdomyolysis |
| Avoid nephrotoxins |
Maintaining intravascular volume and renal perfusion
Acute haemorrhage causes prerenal azotaemia, but the incidence of ARF is low (1.5% in major gastrointestinal bleeding29) if the hypovolaemia is treated promptly and there are no other risk factors. In severe trauma the incidence of ARF needing dialysis is about 0.1%.30 Severe progressive ARF after cardiopulmonary resuscitation is rare, and pre-existing and post resuscitation haemodynamics are more important risk factors for AKI than the degree of hypoperfusion during resuscitation.31
Choice of resuscitation fluid
Hypovolaemia is initially treated with crystalloid rather than colloid, with colloid being added in specific clinical settings (e.g. burns, anaphylaxis) or if there is no initial improvement in vital signs or urine output following crystalloid administration (e.g. in sepsis). The liberal use of crystalloid and colloid in high-output sepsis with AKI may increase the incidence of ARF or need for dialysis.32
Use of derived GFR in drug administration
Formulas are available to calculate the estimated creatinine clearance or estimated GFR from the measured SCr concentration.33,34
Rhabdomyolysis
Most studies on the prevention of ATN after rhabdomyolysis have been in persons with crush injury after earthquakes, where the incidence of AKI is about 50%. In this situation fluid resuscitation should, if possible, begin before the crush is relieved. These patients may require massive amounts of fluid because of fluid sequestration in the injured muscles. The goal of intravenous fluid treatment is to produce a urine output of 200–300 mL/h while myoglobinuria (discoloured urine) persists. There is no evidence to support this rate of fluid replacement in persons who have rhabdomyolysis and AKI without crush injury, although a urine output of 100 mL/h would be reasonable while the urine is discoloured. The intravenous administration of mannitol and sodium bicarbonate to produce an alkaline diuresis as a means of preventing ATN in severe rhabdomyolysis has not been shown to be effective.35
Clinical features
The diagnosis of AKI should be considered when there is a decrease in urine output, an elevated SCr concentration or increases in SCr concentration. The clinical features depend on the pre-existing conditions that increase the risk of developing AKI, the initiating factor(s), and the effects of AKI (Fig. 10.1.2). The history should include a detailed drug history, enquiry about recent invasive vascular or radiological procedures, and any family history of renal disease. This is followed by clinical examination and evaluation of investigations. A number of key issues then need to be resolved (Table 10.1.4).
| Assess the intravascular volume |
| Look for renovascular disease |
| Look for symptoms or signs of obstruction to urine flow |
| Systematic search for presence of infection or sepsis |
| Evaluate for pre-existing renal disease or chronic renal failure |
| Obtain a detailed history of medication or drug use |
| Consider possibility of glomerulonephritis |
Evaluation of prerenal (intravascular volume) status
Imprecise or lazy terminology such as ‘dry’ or ‘dehydrated’ should be avoided. ‘Dehydration’ refers to situations where more water than electrolyte(s) has been lost, shrinking body cells and increasing the serum sodium concentration and osmolality.36 In other words, ‘dehydration’ means water depletion. Hypovolaemia is a decrease in the intravascular volume due to loss of blood (haemorrhage, trauma) or loss of sodium and water (e.g. vomiting, diarrhoea, sequestration of fluid in the bowel, etc.).
The ‘typical’ features of intravascular volume depletion (tachycardia or hypotension or both in the supine position, or postural hypotension) are not as consistent or reliable as implied by textbook descriptions. About one-third of persons with hypovolaemia due to trauma have bradycardia rather than tachycardia.37,38 The presence of (supine) tachycardia has low sensitivity as a diagnostic feature of increasing acute blood loss in healthy persons.39 An increase in the pulse rate of 30 beats per minute or more between the supine value and the standing values is a highly sensitive and highly specific sign of hypovolaemia after phlebotomy of large volumes (600–1100 mL) of blood, but the sensitivity is much less after phlebotomy of smaller volumes.39 The inability to stand long enough for vital signs to be measured because of severe dizziness is a sensitive and specific feature of acute large blood loss.39 The persistence of tachycardia after intravenous administration of fluids in clinical conditions causing hypovolaemia suggests that hypovolaemia is still present, but tachycardia due to other causes (pain, fever) will persist after correction of hypovolaemia.
A systolic blood pressure of 95 mmHg or less in the supine position has high specificity but low sensitivity after acute blood loss.39 Postural hypotension is present in 10% of normovolaemic person younger than 65 years, and in up to 30% of normovolaemic person older than 65 years. Postural hypotension in persons who can stand without developing severe dizziness is of no diagnostic value after blood loss due to acute phlebotomy.39
The textbook descriptions of the signs of saline depletion in adults (dry mucous membranes, shrivelled tongue, sunken eyes, decreased skin turgor, weakness, confusion) are neither specific nor sensitive compared to laboratory tests for hypovolaemia. The presence of a dry axilla argues somewhat for the presence of saline depletion; the absence of tongue furrows and the presence of moist mucous membranes argue against the presence of saline depletion.39
Evaluation of the renovascular state
Atheromatous disease of the renal arteries is common in persons older than 50 years with widespread atherosclerosis. There is a 7% prevalence of renal artery stenosis in persons older than 65 years, and one-third of elderly persons with heart failure have renovascular disease.40,41 Persons with stenosis or occlusion of one or both renal arteries can develop an elevation in SCr concentration after starting treatment with ACE or ARB drugs, or develop acute on chronic renal failure.
Acute kidney injury and acute renal failure
The urine output usually decreases, and the patient may be oliguric (urine output less than 400 mL per day) or anuric (urine output less than 100 mL per day). Persons with AKI and oliguria have more severe kidney impairment than those without oliguria. Only a few conditions cause complete anuria: total obstruction, vascular lesions, severe ATN or rapidly progressive glomerulonephritis. The clinical features caused by ARF are shown in Table 10.1.5.
| 1. Anorexia, fatigue, confusion, drowsiness, nausea and vomiting, and pruritus |
| 2. Signs of salt and water retention in the intravascular and interstitial spaces: an elevated jugular venous pressure, peripheral oedema, pulmonary congestion, acute pulmonary oedema |
| 3. Abnormal plasma electrolyte concentrations, particularly hyperkalaemia |
| 4. Metabolic acidosis |
| 5. Anaemia |
| 6. Uraemic syndrome: ileus, asterixis, psychosis, myoclonus, seizures, pericardial disease (pericarditis, pericardial effusion, tamponade). |
Differential diagnosis
The diagnosis of AKI requires synthesis of data from the patient’s history, physical examination, laboratory studies and urine output. The category of AKI (RISK, INJURY or FAILURE) may be difficult to determine in the emergency department (ED) if the baseline SCr is unknown. The reversibility of the AKI may be inferred if there is a marked increase in urine output after correction of prerenal problems, but a reduction in SCr (due to an increase in GFR) may not be seen for 12–24 h.
Criteria for diagnosis
Serum biochemistry
The SCr concentration is the best available guide to the GFR. Acute reductions in GFR produce an increase in the SCr concentration. The changes in SCr concentration lag behind the change in GFR, and can be affected by the dilution effect of intravenous fluid. Correct interpretation of the SCr concentration extends beyond just knowing the normal values (Fig. 10.1.3). Creatinine is a metabolic product of creatine and phosphocreatine, which are found almost exclusively in skeletal muscle. The SCr concentration is affected by the muscle mass, meat intake, GFR, tubular secretion (which can vary in the same individual and increases as the GFR decreases) and breakdown of creatinine in the bowel (which increases in chronic renal failure). The GFR decreases by 1% per year after 40 years of age, yet the SCr concentration remains unchanged because the decrease in muscle mass with age reduces the production of creatinine. The GFR (corrected for body surface area) is 10% greater in males than females, but men have a higher muscle mass per kilogram of body weight. The SCr concentration in men is thus greater than in women.
The creatinine clearance (CCr) or GFR are estimated indirectly using formulae (Cockcroft–Gault formula or the Modification of Diet in Renal Disease (MDRD) Study Equation) based on the SCr concentration33,34 (Fig. 10.1.4). These equations assume a steady-state SCr concentration, and are inaccurate if the GFR is changing rapidly. They will also be less accurate in amputees, very small or very large persons, or persons with muscle-wasting diseases.
