CASE 10
Barry is 16 months of age and since his adoption at birth has been hospitalized on several occasions with multiple and severe gram-positive bacterial infections of the respiratory tract, skin, soft tissues, gastrointestinal tract, and even bones. On two occasions he has suffered from meningitis, and on at least one occasion he developed nearly overwhelming septicemia, with systemic spread of gram-negative bacteria. Laboratory analysis at the time of these various infections indicated that he had had infections with Mycobacterium avium, as well as with both gram-positive (Streptococcus pneumoniae, Staphylococcus aureus) and gram-negative (Haemophilus influenzae and Pseudomonas aeruginosa) organisms.
Interestingly, despite childhood immunization with H. influenzae, as well as infection with S. pneumoniae and H. influenzae, there was a dearth of specific antipolysaccharide antibodies, probably contributing to the susceptibility to encapsulated bacteria. IgM levels were elevated, with a slight diminution in IgG and IgA levels. Levels of T cells and B cells and macrophages/neutrophils were normal, as was the subset distribution of T cells and the surface expression of CD40L/CD154 (see Case 4). Analysis of phagocyte function (see Case 6) and proliferation of mitogen-stimulated T cells were at the low end of normal (see Case 2). NK cell levels were normal, with evidence for some decrease in killing function (˜threefold on a cell-for-cell basis compared with normal).
QUESTIONS FOR GROUP DISCUSSION
RECOMMENDED APPROACH
Implications/Analysis of Laboratory Investigation
B cell activation leading to isotype switching requires T cell–derived cytokines as well as cognate interaction with T cells. Therefore, a deficiency or defect in T cells could lead to a problem in B cell activation and isotype switching. However, we are told that T cell function is normal, as is the expression of CD40L whose defect on T cells leads to an immunodeficiency disorder characterized by an inability to isotype switch (see Case 4).
Additional Laboratory Tests
Detailed laboratory investigation led to a diagnosis of EDA-ID. Laboratory results consistent with a diagnosis of EDA-ID would include a defect in signaling via various cytokines, including interleukin [IL]-1, tumor necrosis factor [TNF], and IL-18. Because all of the receptors for these cytokines signal via NFκB (nuclear factor kappa B), mutational analysis of various proteins required for NFκB signaling would have shown a mutation in NEMO (NFκB essential modifier). NEMO is the noncatalytic component of IKK, a serine kinase complex that is required for activation of NFκB. NFκB is also a key signaling component in TLRs (see Case 7), NOD activation (see Case 9), and even for T cell and B cell receptor mediation activation.
ETIOLOGY: EDA-ID
Hypohidrotic (anhidrotic) ectodermal dysplasia (HED/EDA) was described first in Great Britain in about 1848. About 150 types of ectodermal dysplasias have been described, with the common feature being that the affected tissues are derived mainly from the ectoderm germ layer. Of the 150 ectodermal dysplasias, the hypohidrotic/anhidrotic form is the most common. Hypohidrotic derives from the word “hypohidrosis” and indicates a severe decrease in sweat production. The cause of HED/EDA is a mutation in ectodysplasmin, a protein that controls normal ectodermal growth/differentiation.
NFκB and IκB
Patients have been identified whose clinical presentation is consistent with HED/EDA but who do not have mutations in the gene encoding ectodysplasmin. Rather, these patients have a mutation in a noncatalytic component of IKK, the serine kinase complex essential for NFκB activation (Fig. 10-1). NFκB was described initially as a transcription factor required for transcription of the kappa light chain constant region in B cells, so patients with mutations in IKK would express immunoglobulins with lambda light chains. NFκB is now known to be a family of transcription factors composed of homo-heterodimers made up of the structurally related (and evolutionarily conserved) proteins, NFκB1 (p50) and NFκB2 (p52), RelA/p65, RelB, and c-Rel. These proteins are constitutively expressed and are sequestered in the cytosol where they remain inactive as a result of their association with inhibitor proteins (IκB) that block sequences required for NFκB localization to the nucleus (i.e., NFκB activation).
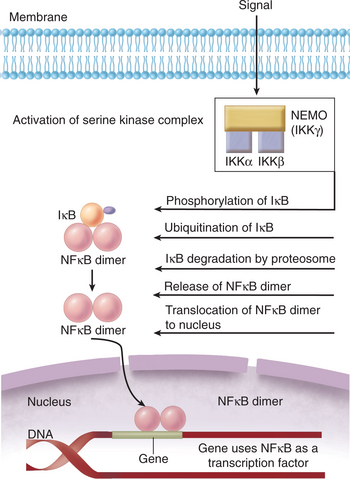
NEMO versus Ectodysplasmin Defects
A defect in NEMO is much more profound (than a defect in ectodysplasmin) because NFκB activation regulates expression of genes important for adaptive immunity, for adhesion and cell motility, for cell proliferation/apoptosis, and for cytokine and chemokine expression. In essence, activation of NFκB is required for the activation of numerous genes that play a role in immunity and inflammation. Therefore, it is thus not a surprise that interference with this pathway, and in NEMO, can have such profound defects. Patients with a mutation in the NEMO gene also present with hyper-IgM syndrome (see Case 4), an immunodeficiency disorder caused by a defect in CD40 or CD40L/CD154, because CD40 ligation itself also leads to activation of NFκB.
Severe NEMO Mutations
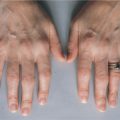
NEMO mutations can result in different clinical syndromes depending on the nature of the defect (Fig. 10-2). EDA-ID results from hypomorphic mutations in coding regions (reduced level of activity). Documented cases of patients with defects in the NEMO stop codon exhibit similar pathologic processes but also present with osteopetrosis and lymphedema (OL-EDA-ID). In contrast, deletions or mutations that abolish NEMO function lead to incontinentia pigmenti, an X-linked dominant disorder that is lethal in utero for males. Females that inherit one copy of the defective gene develop symptoms similar to those of EDA as well as hyperpigmentation of the skin and central nervous system defects.