[level-membership-for-endocrinology-diabetes-and-metabolism-category]
CHAPTER 1. What is diabetes?
David Matthews
Classification 2
Incidence and prevalence 6
Aetiology of diabetes 7
Type 1 diabetes 8
Type 2 diabetes 10
Insulin and its actions 13
Diabetic ketoacidosis 14
Hyperosmolar non-ketotic coma 15
The clinical differences between type 1 and type 2 diabetes 16
Conclusion 18
References 18
Diabetes mellitus is a chronic condition affecting around 171 million people worldwide; the World Health Organization (WHO) projects that this number will have more than doubled by 2030 (WHO 2005). The management of people with diabetes therefore poses a formidable challenge to the healthcare team, and to those in primary care in particular. Diabetes mellitus is a chronic condition characterised by hyperglycaemia due to deficiency or diminished effectiveness of insulin. This results in a disorder of carbohydrate metabolism; fat, protein and mineral metabolism can also be affected. Its importance as a disease is due to the irreversible tissue damage that results mainly from poor metabolic control. This irreversible tissue damage results in about 3.2 million deaths per annum from diabetes and in healthcare costs that range from 2.5 to 15% of overall budgets (WHO 2005). Diabetic retinopathy is the most common cause of blindness in persons aged less than 60 years in the UK and has an incidence of 50–65 per 100 000 of the diabetic population in Europe (Scottish Intercollegiate Guidelines Network (SIGN) 2001). One-third of people on programmes for the management of end-stage renal failure have diabetes. Having diabetes makes a person 2–3 times more likely to have a major vascular event such as myocardial infarction or stroke. Up to 15% of people with diabetes have a foot ulcer at some stage (Lancet 2005), with recurrence rates being greater than 50% after 3 years (Boulton et al 2005). Coupled with this, around 50% of people having amputations for non-traumatic reasons have diabetes. The resultant increased morbidity and mortality from diabetes is thus plain to see and explains why diabetes is a major consumer of healthcare resources world-wide.
Various classifications of diabetes were updated by the WHO in 1999 (Table 1.1). Type 2 diabetes is the most common, representing over 80% of all persons with diabetes; type 1 comprises most of the remaining 20%. Maturity onset diabetes of the young (MODY) and secondary diabetes are relatively uncommon and account for less than 5% of people. However, it is important to identify those individuals with secondary diabetes so that treatment can be directed to the underlying cause. Diabetes in association with other genetic syndromes is also very rare. In these instances, diabetes is another burden to be borne by these people.
| Primary | |
| Type 1 | |
| Type 2 | |
| MODY, maturity onset diabetes of the young | |
| Secondary to other disorders | |
| 1. Pancreatic disease |
Chronic pancreatitis
Haemochromatosis
Pancreatectomy
Carcinoma of pancreas
Cystic fibrosis
|
| 2. Endocrine disease |
Acromegaly
Cushing’s syndrome
Phaeochromocytoma
Gestational
|
| 3. Drugs |
Thiazide diuretics
Corticosteroids
|
| Associated with genetic syndromes | |
|
Friedreich’s ataxia
Muscular dystrophies
Down’s syndrome
Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD)
|
|
TYPE 1 DIABETES MELLITUS
The hallmark of type 1 diabetes is a dependence on exogenously injected insulin to prevent ketosis and maintain life. Without injected insulin, people with type 1 diabetes die. This was the situation before the discovery of insulin in 1921. Some died very quickly over a matter of days. Others struggled along miserably for 3 or 4 years by eating almost starvation-type diets (Bliss 1983). In type 1 diabetes, therefore, there is an absolute deficiency of insulin.
Joseph’s case is a fairly representative example of a person newly diagnosed with type 1 diabetes presenting in diabetic ketoacidotic coma (diabetic ketoacidosis is explained later in the chapter). Before the discovery of insulin, such a presentation was lethal. Joseph will be dependent on self-injected insulin for the rest of his life. Withdrawal of insulin would rapidly cause further ketoacidosis and he must be advised never to stop his insulin injections. Joseph and his family will require further support and education to give him the necessary skills for self-management (see Chapters 3, 5, 6, 7 and 11).
TYPE 2 DIABETES MELLITUS
Type 2 diabetes is the most common form of diabetes world wide and there is no requirement for insulin to prevent ketosis and preserve life. Many people with type 2 diabetes can be managed by dietary means alone (see Chapters 4 and 6). Sometimes, oral hypoglycaemic drugs are required in addition to diet. Most people are over the age of 30 years when diagnosed. The main associated feature is obesity and in such people the body tissues are relatively resistant to the effects of insulin, thus causing an elevation of blood glucose. By reducing body weight many people can make carbohydrate tolerance almost normal. The management of the person with type 2 diabetes is further expanded in Chapter 4.
There is a subgroup of people, however, who are not obese and who have a relative deficiency of insulin. These people cannot secrete enough insulin to cope with the carbohydrate load they consume. The pathological process in the cells of the islets of Langerhans in the pancreas is quite different from type 1 diabetes in that these individuals do not have autoimmune damage to the pancreas. However, they might eventually require insulin therapy, although they will not be classified as having true insulin dependence.
This is an inherited form of diabetes usually arising in a person’s teenage years or in his or her early twenties and, although rare, affects about 1–2% of those with diabetes. It is passed down from one family member to another and each child has a 50% chance of inheriting the affected gene and a very high risk of developing diabetes (Shepherd 2003a).
People with MODY are usually managed with diet and/or sulphonylureas, regardless of age, for many years and ultimately will require insulin therapy. Insulin would also be required in special circumstances, such as pregnancy. Most people who have this form of diabetes have a multigenerational family history, with autosomal dominant inheritance and are usually not obese.
The molecular consequences are now well defined, with mutations in at least six genes. This means that diagnostic and predictive testing is now possible in 50–80% of families with MODY, which has implications for professional management (Shepherd 2003b). People with MODY have a mutation in the glucokinase gene, which results in mildly elevated fasting hyperglycaemia (> 5.5 mmol/L) (Barrow et al 2005). MODY 1 is due to a mutation in hepatocyte nuclear factor (HNF) 4α; MODY 2 is due to a mutation in the glycolytic enzyme glucokinase. Similarly, MODY 3 relates to HNF 1α, MODY 4 to insulin promoter factor 1, MODY 5 to HNF 1β and MODY 6 to neurogenic differentiation factor 1 (Fajans et al 2001). Genetic testing can define the subtype of diabetes as this has implications for different treatment options (Shepherd 2003a).
SECONDARY DIABETES
This type of diabetes is secondary to other disease processes that cause either pancreatic damage or production of hormones that antagonise the action of insulin (Box 1.1).
Box 1.1
Hormones that are antagonistic to the action of insulin
▪ Growth hormone
▪ Cortisol
▪ Glucagon
▪ Adrenaline
▪ Placental steroids
▪ Noradrenaline
Pancreatic disease
The pancreas can be damaged as a result of frequent bouts of pancreatitis, which is often precipitated by alcohol abuse or the presence of gallstones in the common bile duct.
The presence of carcinoma within the pancreas can also cause destruction of normal insulin-secreting cells resulting in the development of diabetes. Pancreatic disorders causing diabetes usually require treatment with insulin.
Valentina is a 73-year-old woman who presents with a 9-kg weight loss associated with nausea, polydipsia, polyuria and vulvitis. Her random blood glucose is 19.7 mmol/L and she has 2% glycosuria. Valentina is diagnosed with diabetes and is treated with dietary measures and glipizide 2.5 mg to relieve her symptoms quickly. She returns to the GP surgery 3 weeks later. Her blood glucose has fallen to 9.1 mmol/L, she has no glycosuria, but her weight has fallen by a further 2 kg.
In Valentina’s case, it is unusual to be nauseated with primary diabetes and weight loss should stop when the energy-losing glycosuria is abolished. Further investigation was thus necessary and she was found to have a carcinoma of the tail of her pancreas. Carcinoma of the pancreas can present as an apparent acute onset of diabetes and it is therefore important to think of secondary causes at the time of diagnosis, especially in elderly individuals with marked weight loss.
Endocrine disease
The secondary endocrine causes of diabetes involve excess endogenous production of hormones that are insulin antagonists (see Box 1.1):
▪ In acromegaly, growth hormone is secreted in excess by the pituitary gland.
▪ In Cushing’s syndrome, the adrenal cortex makes excess cortisol due either to a primary tumour within the adrenal gland or to excess adrenocorticotrophic hormone (ACTH) production.
▪ In phaeochromocytoma there is excess secretion of adrenaline or noradrenaline by a tumour of the adrenal medulla or sympathetic plexus.
Hypertension, which is seen in acromegaly, Cushing’s syndrome and phaeochromocytoma, commonly accompanies type 2 diabetes. When faced with an obese, hypertensive person with an elevated blood glucose it is important to consider the possibility of an underlying endocrine disorder before accepting a diagnosis of type 2 diabetes
The exceedingly rare glucagonoma results in diabetes because glucagon also antagonises insulin.
Gestational diabetes occurs in about 4% of women with normal pregnancies and is due to the insulin-antagonising effects of placental steroids and human placental lactogen. Gestational diabetes has been defined as carbohydrate intolerance of variable severity with onset during pregnancy, although there is no consensus about its diagnosis, treatment or management (SIGN 2001). Some women with gestational diabetes might need insulin treatment towards the end of pregnancy. Postnatally, the diabetes goes into remission but these women will enter the ‘at risk’ category for developing type 2 diabetes in later years (see Chapter 2).
Drug therapy
The thiazide diuretics, which are commonly used to treat essential hypertension, have a blood-glucose-raising effect by their inhibitory action on insulin secretion. Other drugs, such as corticosteroids (e.g. prednisolone), also make tissues relatively resistant to the effects of insulin and thus unmask diabetes.
GENETIC SYNDROMES
The genetic syndromes in Table 1.1 are rare but can be associated with diabetes mellitus, although the mechanisms are unknown. The syndrome comprising diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) is a well-recognised entity that can present with varying clinical features of the syndrome. Acanthosis nigricans, a rare skin condition sometimes associated with hirsutism and polycystic ovaries, is associated with a very insulin-resistant type of diabetes.
INCIDENCE AND PREVALENCE
The incidence of a disease is the number of new cases arising each year. The prevalence is the number of people with a disease in the population at any one time. The incidence of diabetes varies from country to country and area to area within countries (Hjelm et al 2003, Yach et al 2006). In the UK, the prevalence of diabetes is now around 3% and rising.
TYPE 1 DIABETES MELLITUS
The incidence varies between population groups and in the UK the cumulative risk of developing type 1 diabetes in persons aged under 20 is now 0.3–0.4%, with the peak between 10 and 14 years. This is increasing in all age groups being most marked in the 0–4 years.
TYPE 2 DIABETES MELLITUS
Accurate figures of the incidence of type 2 diabetes are difficult to establish, despite this being the more frequent type, because of inconsistent diagnostic criteria. The diagnosis of diabetes is addressed in Chapter 2.
The true prevalence is also difficult to determine accurately because of the relatively large population of people with type 2 diabetes who remain undiagnosed. There is, however, gathering evidence of a worldwide epidemic (Hjelm et al 2003). The prevalence of type 2 diabetes varies from country to country and population to population. A main feature is the low prevalence among Europeans who remain within Europe; there is a high prevalence among the Pima Indians, urban New Guineans and the Nauru (Diamond 2003, Knowler et al 1981, WHO 2005, Zimmet et al 1990). The population groups with the projected greatest increase are in Asia and Africa (Hjelm et al 2003).
Type 2 diabetes becomes more common with increasing age, with over 50% of people attending diabetes services in the UK aged over 60 years. There are now a growing number of younger people acquiring type 2 diabetes in the Western world, although it is still rare (Ehtisham et al 2004). The prevalence within the UK is only 2 in every million children. These young people tend to present later than young people with type 1 diabetes, are overweight, are usually female and a greater proportion are of ethnic minority origins. It is thought that this is due to more sedentary lifestyles and consuming more processed foods that are high in fat content.
AETIOLOGY OF DIABETES
TYPE 1 DIABETES
There is good evidence that type 1 diabetes is a T-cell-mediated autoimmune disorder and that affected persons are born with the tendency to destroy their own insulin-producing beta cells in the islets of Langerhans. There is evidence of familial clustering but there must also be some environmental trigger, as twin studies have shown that in only around 50% of identical twin pairs does the other twin develop diabetes (Devendra et al 2004).
Modern science is continually expanding our knowledge while exploring the basic genetic defect, the nature of the environmental agent and the immunopathological processes. It would appear that there is interplay between genetic susceptibility and environmental factors in the pathogenesis of type 1 diabetes (Devendra et al 2004).
Genetic predisposition
The risk of developing type 1 diabetes is mainly conferred by the inheritance of genes relating to the major histocompatibility complex (MHC) on chromosome 6. Gene mapping studies have shown that another important gene conferring susceptibility to diabetes is on chromosome 11, near the genes for insulin and insulin-like growth factor. In total, 20 independent chromosomal regions are associated with the genetic predisposition to type 1 diabetes (Atkinson & Eisenbarth 2001, Devendra et al 2004). Children are three times more likely to develop type 1 diabetes if their father has diabetes rather than their mother (Gale & Gillespie 2001).
The main susceptibility genotypes for type 1 diabetes are HLA-DR3 and DR4 with the DQ2 and DQ8 the high-risk alleles (Gillespie et al 2004). The mode of action of these susceptibility genes is unclear. Human genome wide screens looking for areas that associate with the risk of type 1 diabetes have shown that the area upstream of the insulin gene on chromosome 11 is most likely to confer risk.
ENVIRONMENTAL FACTORS
Evidence for environmental factors having a role in aetiology of diabetes is emerging. Devendra et al (2004) summarise 16 studies that have investigated the impact of environmental factors on the development of type 1 diabetes. The overall conclusion appears to be that environmental factors alone do not cause type 1 diabetes. It is thought that the interaction between genetic disposition and environmental factors act as a trigger for an autoimmune process that results in type 1 diabetes (Bach 2005, Devendra et al 2004, Gillespie et al 2004).
The low concordance rate in monozygotic twins, the changes in the incidence of the disease in migrants from low-incidence to high-incidence regions and the increasing incidence in stable genetic populations do, however, suggest something. Cows’ milk protein and Coxsackie virus B infection have been the most plausible postulates.
TYPE 1 DIABETES
PATHOGENESIS OF TYPE 1 DIABETES
Insulitis
Examination of the pancreases of people with long-standing type 1 diabetes has shown that there is destruction of the insulin-secreting beta cells while other cells are preserved. Even when pancreases of people newly diagnosed with diabetes are examined, most are deficient in beta cells. In other words, the destruction is highly selective to the beta cells.
In people newly diagnosed with diabetes there are large numbers of inflammatory cells including CD4 and CD8 T lymphocytes, B lymphocytes and others surrounding the beta cell (Atkinson & MacLaren 1994). This is referred to as insulitis. This inflammatory process is commonly associated with the presence of cytoplasmic antibodies in the peripheral blood. These circulating antibodies are more likely to be a marker of the insulitis rather than the cause. In people with type 1 diabetes, about 80% of the beta cells in the pancreas require to be destroyed by this autoimmune pathological process before the person presents with the more common symptoms of polyuria and polydipsia.
Circulating autoantibodies
Much has been learned about the natural history of type 1 diabetes by studying siblings of people with diabetes over a long period of time. A number of autoantibodies are specifically associated with type 1 diabetes (Kaufman 2003). Islet cell antibodies (ICA) are the most sensitive autoantibody marker of risk of future type 1 diabetes. Antibodies to other autoantigens, such as glutamic acid decarboxylase (GAD), IA-2 and IA-3 (phogrin), have also been found, although the role of these antibodies is unclear. Insulin autoantibodies (IAA) are directed against the insulin molecule but are indistinguishable from antibodies produced in response to exogenously injected insulin. IAA disappear with increasing ages of onset of type 1 diabetes so, if found, predict an early age of onset. GAD and ICA antibodies are more useful for predicting later-onset autoimmune diabetes. It is unlikely that these circulating antibodies contribute to the beta cell destruction as this is a predominantly T-cell-mediated phenomenon. The interaction between humoral and T-cell-mediated immunity in this situation remains unclear (Turner et al 1997).
Interestingly, in the UK Prospective Diabetes Study (UKPDS), ICA and GAD antibodies were measured in some 3672 people with typical type 2 diabetes, with 12% having at least one antibody (Turner et al 1997). This seemed to correlate with younger age, lower body mass index (BMI) and reduced beta cell function. This has led to the term latent autoimmune diabetes in adults (LADA) or slow onset type 1 diabetes (Davis et al 2005).
PREVENTION OF TYPE 1 DIABETES
Studies of the prevention of type 1 diabetes using a number of agents started late in the twentieth century but have proved uniformly disappointing (Devendra et al 2004, Diabetes Prevention Trial – Type 1 Diabetes Study Group 2002, European Nicotinamide Diabetes Intervention Trial (ENDIT) 2004). There is, however, some hope in preventing type 1 diabetes through preliminary work looking at interfering with the autoimmune processes by expanding specific cells or by blocking antibodies (von Boehmer 2004). This work is still at the early experimental stage.
There is no evidence that type 2 diabetes is an autoimmune process. Twin studies have shown almost 90% concordance for type 2 diabetes. It is more common in certain racial groups, such as Pima Indians, and is much more likely to be a predominantly inherited condition. The precise genetic mechanisms have yet to be determined and, although it has a strong genetic component, only a small number of genes have been identified (Stumvoll et al 2005).
Many people with type 2 diabetes also have associated obesity. Despite the tendency to obesity being partly hereditary, its development is mainly related to environmental factors, such as the enormous availability of food in developed nations and the cultural factors that make food such an important factor in physical and psychological well-being. It has been shown that individuals from fairly primitive societies, where the incidence of diabetes is low, who then move to societies where food is too readily available, often progress to develop diabetes (Diamond 2003). Recent studies might provide a link between obesity and type 2 diabetes through the identification of key neural pathways that control glucose production from the liver (Seeley & Tschop 2006).
Insulin resistance is also associated with diabetes and is the best predictor of its development in the children of people with type 2 diabetes. The mechanism whereby this occurs is, as yet, unknown, although it is associated with a reduction in mitochondrial function in muscle and an increase in lipid content (Petersen et al 2004, Taylor 2004).
Past epidemiological research has shown that people who develop type 2 diabetes in later life were small babies at birth, suggesting that some intrauterine factor(s) might be important in determining susceptibility (Hales et al 1991, Hattersley & Tooke 1999).
Current research is challenging the main causes of diabetes and beginning to question if it is a disease of the central nervous system. This might especially be the case with type 2 diabetes (Elmquist & Marcus 2003, Obici et al 2003). In some ways, this serves to demonstrate the ongoing research into the cause of diabetes and the interplay between glucose regulation, food intake and the endocrine system.
PATHOGENESIS OF TYPE 2 DIABETES
There are three basic metabolic abnormalities in type 2 diabetes:
▪ insulin resistance
▪ impaired insulin secretion
▪ increased hepatic glucose production.
The sequence of events is shown in Fig. 1.1. The process from genetic disposition to the diagnosis of type 2 diabetes is progressive and will continue despite treatment.
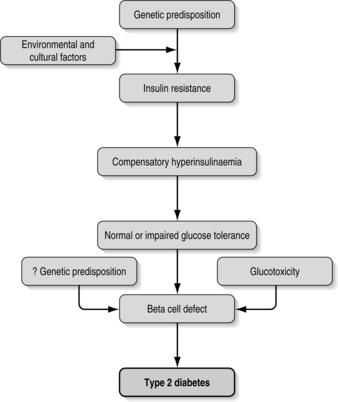 |
| Fig. 1.1Sequence of events leading to type 2 diabetes. |
The first step in developing type 2 diabetes is probably a defect in the action of insulin, resulting in impaired glucose uptake by the cells. To compensate for this, and to maintain normal blood glucose levels, there is a resulting increase in beta-cell insulin secretion with consequent hyperinsulinaemia (Fig. 1.1). This continues for a number of years, with insulin-secreting beta cells working very hard to maintain normal blood glucose (Kahn 1998, Taylor 2004).
As these overworked beta cells fail to keep pace with the insulin production required to maintain euglycaemia, impaired carbohydrate tolerance develops. Overt diabetes then follows, signalling that the beta cells are failing.
There is some evidence that this ineffectiveness of insulin action is inherited. The insulin receptor on the cell surface binds circulating insulin (Fig. 1.2). This receptor is made up of two extracellular alpha subunits. These bind insulin and are connected, through the cell membrane, to the intracellular milieu via two beta subunits. Binding of insulin to the alpha subunits results in the beta subunits triggering a cascade of metabolic events that involves the enzyme tyrosine kinase and culminates in the entry of glucose into the cell and subsequent metabolic processing. The entry of glucose is mediated by GLUT4, a cell membrane glucose transporter. Whereas abnormalities of this transporter might be expected to lead to type 2 diabetes, none has yet been identified.
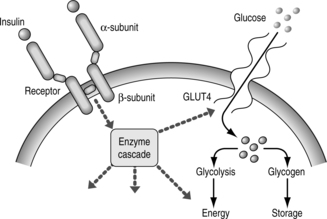 |
| Fig. 1.2The insulin receptor and its actions. |
There is no doubt that the state of insulin resistance is fairly dynamic and that certain physiological states influence this. Weight gain is known to increase insulin resistance and regular exercise to decrease it. This explains why losing weight and exercise prescription are important measures in the treatment of type 2 diabetes (see Chapter 4). Ongoing research is attempting to understand the link between obesity-related chronic inflammation and insulin resistance (de Luca & Olefsky 2006).
Impaired insulin secretion
This is the definitive event in the transition from insulin resistance to the diabetic state. As the beta cells fail and insulin levels fall, blood glucose rises and the person progresses from an insulin-resistant state to developing symptomatic diabetes. Whether the decline in beta-cell function is genetically preprogrammed or, whether it is acquired, remains unclear.
In early type 2 diabetes, basal insulin levels are normal or even increased but appear inappropriately low for the raised blood glucose level. The immediate postprandial insulin response is impaired and, as blood glucose rises, this in turn leads to further impairment. It appears as if higher glucose levels blunt subsequent insulin secretion – this is termed glucotoxicity. This glucotoxicity probably explains why so many people with acute symptoms of hyperglycaemia can respond fairly dramatically to the immediate removal or reduction of simple sugars in their diet. The rapid fall in blood glucose induced by dietary measures restores insulin secretion, which in turn helps to reduce the blood glucose still further.
Increased hepatic glucose output
In the pathogenesis of type 2 diabetes, there is reduced muscle and adipose cell glucose uptake as well as excessive glucose output from the liver. The liver can generate glucose from fat and protein (gluconeogenesis) and this process is under the influence of insulin. The mechanisms involved are not well understood but reduced insulin in portal venous blood is important. The overall result is enhanced gluconeogenesis and hence hyperglycaemia.
INSULIN AND ITS ACTIONS
The hormone insulin is a 51-amino-acid peptide secreted by the beta cells of the islets of Langerhans. The insulin gene is situated on the short arm of chromosome 11 and controls the formation of a large insulin precursor molecule called preproinsulin. In the cytoplasm of the beta cell, preproinsulin is cleaved by enzymes to form proinsulin, which in turn is cleaved by a peptidase enzyme to form insulin and C-peptide. The insulin molecule (Fig. 1.3) consists of an A-chain (21 amino acids) and a B-chain (30 amino acids). Three disulphide bridges link the structure.
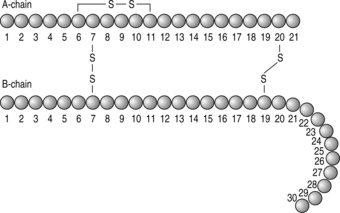 |
| Fig. 1.3The structure of insulin |
In people with diabetes the measurement of C-peptide (connecting-peptide) in plasma or urine allows the endogenous insulin-secretory status of the beta cells to be assessed. This is sometimes measured in people who are on insulin therapy to determine how much insulin their own pancreas is still producing.
The release of insulin from the beta cell can be stimulated by many factors, including glucose, amino acids and sulphonylureas. Glucose enters the beta cell and is probably metabolised there in order to stimulate insulin release.
Other agents can increase insulin release. These include the paracrine hormone, glucagon, which is released from the alpha cells, and another hormone, gastric inhibitory polypeptide (GIP), which stimulates insulin release. GIP is released when carbohydrate is eaten, which probably explains why ingested glucose is a much more potent stimulus of insulin release than intravenous glucose. This also accounts for the use of the oral glucose tolerance test, as opposed to the intravenous tolerance test, for diagnostic purposes in type 2 diabetes (see Chapter 2).
Glucose is normally absorbed from the small intestine and enters the portal vein. Most of the glucose then passes into the systemic circulation. The rise in blood glucose and GIP after a meal stimulates insulin release. Insulin then promotes glucose uptake from the circulation into fat and muscle cells and enhances the production of glycogen in liver and muscle. It increases amino acid uptake and protein synthesis in the tissues as well as preventing protein breakdown (Fig. 1.4). The increased insulin also inhibits both the breakdown of protein to amino acids and triglycerides to free fatty acids and glycerol.
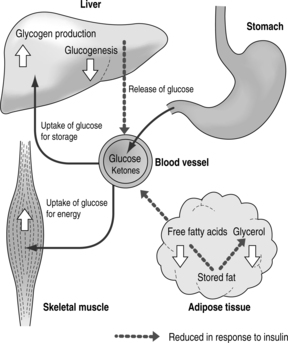 |
| Fig. 1.4The actions of insulin |
When insulin has done its work, driving the higher blood glucose out of the circulation into the tissues after a meal, its production wanes and the blood glucose levels are then maintained by the finely tuned liver production of glucose.
In the fasting state, insulin levels fall. The fall in insulin levels allows the breakdown of fat and proteins to occur. The fatty acids provide the main fuel for muscles, leaving glucose free to fuel the brain. The low continuing insulin concentrations are enough to limit ketone body formation and the normal buffering capacity of the blood prevents acidosis.
DIABETIC KETOACIDOSIS
In the pathological state of insulin deficiency there is an increased liver output of glucose from two sources. Glycogen is metabolised to glucose and glucose is also formed (gluconeogenesis) from a combination of the breakdown products of fatty acids and amino acids (see Fig. 1.4). In addition, the lack of insulin results in less glucose being taken up by the peripheral tissues such as muscle. Blood glucose thus rises, overcoming the renal threshold for glucose with resulting glycosuria. The resulting osmotic diuresis is responsible for the symptoms of polyuria, nocturia and polydipsia in uncontrolled diabetes.
In addition, fat and protein breakdown is quite unrestrained, with consequent weight loss. Fatty acid levels rise very high, which in turn causes the liver to increase the production of ketone bodies. Ketone levels eventually overwhelm the acid-buffering capacity of the blood and diabetic ketoacidosis (DKA) develops.
The combination of uncontrolled diabetes and acidosis with resultant circulatory collapse and clouded consciousness makes this a medical emergency that only intravenous fluids and insulin can reverse. Death can still occur from diabetic ketoacidosis, although this is now rare.
HYPEROSMOLAR NON-KETOTIC COMA
In a person presenting with hyperosmolar non-ketotic coma (HONK) there is a relative, rather than complete, lack of insulin. This differentiates it from diabetic ketoacidosis. Here, there is insufficient insulin production to prevent a rise in blood glucose yet there is just enough to prevent ketone body production. Hence ketonuria is not a feature.
Mabel, a 75-year-old woman, lives alone. Her carer, who visits once per week, found her lying on the floor. Mabel was semiconscious and unable to give an account of what had happened to her. The previous week, her carer had found her rather tired and lethargic when she had called and, on reflection, states that Mabel might have been more thirsty than normal. Mabel is immediately admitted to hospital, where she is found to be mildly hypothermic and clinically dehydrated. Her feet are mottled in colour and her peripheral pulses are poor. Initial biochemistry confirms a dehydrated state with a urea 22.5 mmol/L and creatinine 200 micromol/L. Her blood glucose is found to be 52 mmol/L but there is no evidence of any ketonuria.
HONK usually occurs in people with type 2 diabetes and especially in people who are as yet undiagnosed, as in Mabel’s situation, above. The elderly, who are at most risk of HONK, can become slowly unwell over a prolonged period as they fail to recognise the significance of their symptoms. This can result in the blood glucose rising to very high levels (often > 50 mmol/L), which causes a rise in plasma osmolality and results in severe dehydration. This is a medical emergency. Treatment is by slow intravenous fluids and insulin to gradually correct the blood glucose level and avoid the added complication of cerebral oedema. When restored to good health, the person is managed as an individual with type 2 diabetes. Long-term injected insulin is not usually required, suggesting that glucotoxicity might have played a role in the beta-cell failure.
A high mortality rate is associated with HONK, due to the fact that people can be comatose before they are discovered by relatives or friends. The dehydration reduces the circulation to the periphery and increases blood viscosity. Thrombosis of the arteries to the lower limbs and/or myocardial infarction is common and often contributes to the high mortality.
THE COUNTER-REGULATORY HORMONES
In both types of metabolic decompensation, DKA and HONK, it is important to recognise the actions of hormones that oppose the action of insulin (see Box 1.1).
Glucagon, adrenalin and noradrenaline modulate beta-cell insulin secretion. By increasing blood glucose levels they protect against hypoglycaemia and ensure fuel supplies to vital organs during stress. However, in the stressful situations of DKA or HONK, their actions are inappropriate and result in raising blood glucose to even greater levels.
THE CLINICAL DIFFERENCES BETWEEN TYPE 1 AND TYPE 2 DIABETES
AGE OF ONSET
People with newly diagnosed type 1 diabetes are usually children or young adults, although people with type 1 diabetes can be diagnosed at any age. Type 2 diabetes tends to present in people over the age of 30 years. However, increasing numbers of individuals are being identified at a younger age as the prevalence of obesity in younger people increases and as the public and healthcare professionals become more aware of type 2 diabetes.
GENDER
Type 1 diabetes is slightly more common in males, there being less sex bias than in type 2 diabetes.
PAST MEDICAL HISTORY
People with type 1 diabetes have usually been very healthy before the development of diabetes. It is very common, however, to find a history of hypertension and/or vascular disease in individuals who are diagnosed with type 2 diabetes.
Sometimes, people presenting with type 1 diabetes have a family history of diabetes, but this is more common in individuals developing type 2 diabetes.
PRESENTATION
People with type 1 diabetes usually present with classic symptoms of polydipsia, polyuria, weight loss and tiredness. They might present with diabetic ketoacidosis.
People with type 2 diabetes can also present with classic symptoms but never develop ketoacidosis. They more often present with symptoms associated with mild elevation of blood and urinary glucose, namely balanitis or vulvitis, blurred vision or leg cramps. Type 2 diabetes is more commonly diagnosed by the finding of glycosuria or hyperglycaemia in the course of assessment of other medical conditions or during routine screening tests (see Chapter 2). It is not uncommon for people with type 2 diabetes to present with an established microvascular complication of diabetes, such as background retinopathy during routine eye screening, or with an infected neuropathic foot ulcer.
WEIGHT
People with type 1 diabetes have commonly lost weight at diagnosis, but not always. Many people with type 2 diabetes have coexistent obesity at diagnosis and it is not uncommon for some to claim recent great success in losing weight. This is, of course, due to the amount of energy lost in persisting glycosuria.
URINALYSIS
People who present with the classic symptoms of type 1 diabetes of polyuria and polydipsia will have glycosuria; ketonuria will be evident if presentation is with ketoacidosis. Most people with newly diagnosed type 1 diabetes will have significant ketonuria. Some people presenting with type 2 diabetes can have small or moderate amounts of ketonuria. If this occurs, it is usually because the person has reduced his/her calorie intake for whatever reason.
Sometimes, despite these clinical differences, it is difficult to tell whether people have type 1 or type 2 diabetes. This might not matter too much in the sense that initial treatments might not differ. However, it could be crucial in some individuals to know if they are likely to require insulin treatment because this might have serious implications in relation to employment, insurance and driving (see Chapter 11).
REFERENCES
MA Atkinson, GS Eisenbarth, Type 1 diabetes: new perspectives on disease pathogenesis and treatment, Lancet 358 (2001) 221–229.
MA Atkinson, NK MacLaren, The pathogenesis of insulin dependent diabetes, New England Journal of Medicine 331 (1994) 1428–1436.
JF Bach, A toll-like trigger for autoimmune disease, Nature Medicine 11 (2) (2005) 120–121.
BA Barrow, S Ellard, MH Shepherd, et al., Patient recruitment strategy for family studies investigating novel genetic causes of maturity-onset diabetes of the young, European Diabetes Nursing 2 (1) (2005) 24–29.
M Bliss, The discovery of insulin. (1983) Paul Harris Publishing, Edinburgh .
AJM Boulton, L Vileikyte, G Ragnarson-Tennvall, J Apelqvist, The global burden of diabetic foot disease, Lancet 366 (2005) 1719–1724.
TME Davis, AD Wright, ZM Mehta, et al., Islet autoantibodies in clinically diagnosed type 2 diabetes: prevalence and relationship with metabolic control (UKPDS 70), Diabetologia 48 (2005) 695–702.
C De Luca, JM Olefsky, Stressed out about obesity and insulin resistance, Nature Medicine 12 (1) (2006) 41–42.
D Devendra, E Liu, GS Eisenbarth, Type 1 diabetes: recent developments, British Medical Journal 328 (2004) 750–754.
Diabetes Prevention Trial — Type 1 Diabetes Study Group, Effects of insulin in relatives of patients with type 1 diabetes mellitus, New England Journal of Medicine 346 (22) (2002) 1685–1691.
J Diamond, The double puzzle of diabetes, Nature 423 (2003) 599–602.
S Ehtisham, AT Hattersley, DB Dunger, TG Barrett, First UK survey of paediatric type 2 diabetes and MODY, Archives of Disease in Childhood 89 (2004) 526–529.
JK Elmquist, JN Marcus, Rethinking the central causes of diabetes, Nature Medicine 9 (6) (2003) 645–647.
European Nicotinamide Diabetes Intervention Trial (ENDIT), A randomised controlled trial of intervention before the onset of type 1 diabetes, Lancet 363 (2004) 925–931.
SS Fajans, GI Bell, KS Polonsky, Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young, New England Journal of Medicine 345 (2001) 971–980.
EA Gale, KM Gillespie, Diabetes and gender, Diabetologia 44 (2001) 3–15.
KM Gillespie, SC Bain, AH Barnett, et al., The rising incidence of childhood type 1 diabetes and reduced contribution of high-risk HLA haplotypes, Lancet 364 (2004) 1699–1700.
C Hales, DJP Barker, PMS Clark, et al., Fetal and infant growth and impaired glucose tolerance at age 64 years, British Medical Journal 303 (1991) 1019–1022 .
AT Hattersley, JE Tooke, The fetal insulin hypothesis: an alternative explanation of the association of low birth weight with diabetes and vascular disease, Lancet 353 (1999) 1789–1792.
K Hjelm, E Mufunda, G Nambozi, J Kemp, Preparing nurses to face the pandemic of diabetes mellitus: a literature review, Journal of Advanced Nursing 41 (5) (2003) 424–434.
BB Kahn, Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance, Cell 92 (1998) 593–596.
DL Kaufman, Murder mysteries in type 1 diabetes, Nature Medicine 9 (2) (2003) 161–162.
WC Knowler, DJ Pettit, PH Bennett, PI Savage, Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes, American Journal of Epidemiology 113 (1981) 144–156.
Lancet, Putting feet first in diabetes [editorial], Lancet 366 (2005) 1674.
S Obici, Z Feng, A Arduini, et al., Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production, Nature Medicine 9 (2003) 756–761.
KF Petersen, S Dufour, D Befroy, et al., Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes, New England Journal of Medicine 350 (2004) 664–671.
Scottish Intercollegiate Guidelines Network (SIGN), SIGN 55: management of diabetes. (2001) SIGN, Edinburgh .
RJ Seeley, M Tschop, How diabetes went to our heads, Nature Medicine 12 (1) (2006) 47–49.
M Shepherd, ‘I’m amazed I’ve been able to come off injections’: patients’ perceptions of genetic testing in diabetes, Practical Diabetes International 20 (9) (2003) 338–343.
M Shepherd, Genetic testing in maturity onset diabetes of the young (MODY) — practical guidelines for professionals, Practical Diabetes International 20 (3) (2003) 108–110.
M Stumvoll, BJ Goldstein, TW van Haeften, Type 2 diabetes: principles of pathogenesis and therapy, Lancet 365 (2005) 1333–1346.
R Taylor, Causation of type 2 diabetes — the Gordian knot unravels, New England Journal of Medicine 350 (7) (2004) 639–641.
R Turner, I Stratton, S Manley, et al., UKPDS 2 Autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes, Lancet 350 (1997) 1288–1293.
H Von Boehmer, Type 1 diabetes: focus on prevention, Nature Medicine 10 (8) (2004) 783–784.
World Health Organization (WHO) 1999 Online. Available: www.who.int/mediacentre/factsheets/fs138/en/index.html
World Health Organization (WHO) 2005 Online. Available: www.who.int/dietphysicalactivity/publications/facts/diabetes/en/
D Yach, D Stuckler, KD Brownell, Epidemiological and economic consequences of the global epidemic of obesity and diabetes, Nature Medicine 12 (1) (2006) 62–66.
P Zimmet, G Dowse, C Finch, et al., The epidemiology and natural history of NIDDM-lessons to be learnt from the South Pacific, Diabetes/Metabolism Reviews 6 (1990) 91–124.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
CHAPTER 1. What is diabetes?
David Matthews
Classification 2
Incidence and prevalence 6
Aetiology of diabetes 7
Type 1 diabetes 8
Type 2 diabetes 10
Insulin and its actions 13
Diabetic ketoacidosis 14
Hyperosmolar non-ketotic coma 15
The clinical differences between type 1 and type 2 diabetes 16
Conclusion 18
References 18
Diabetes mellitus is a chronic condition affecting around 171 million people worldwide; the World Health Organization (WHO) projects that this number will have more than doubled by 2030 (WHO 2005). The management of people with diabetes therefore poses a formidable challenge to the healthcare team, and to those in primary care in particular. Diabetes mellitus is a chronic condition characterised by hyperglycaemia due to deficiency or diminished effectiveness of insulin. This results in a disorder of carbohydrate metabolism; fat, protein and mineral metabolism can also be affected. Its importance as a disease is due to the irreversible tissue damage that results mainly from poor metabolic control. This irreversible tissue damage results in about 3.2 million deaths per annum from diabetes and in healthcare costs that range from 2.5 to 15% of overall budgets (WHO 2005). Diabetic retinopathy is the most common cause of blindness in persons aged less than 60 years in the UK and has an incidence of 50–65 per 100 000 of the diabetic population in Europe (Scottish Intercollegiate Guidelines Network (SIGN) 2001). One-third of people on programmes for the management of end-stage renal failure have diabetes. Having diabetes makes a person 2–3 times more likely to have a major vascular event such as myocardial infarction or stroke. Up to 15% of people with diabetes have a foot ulcer at some stage (Lancet 2005), with recurrence rates being greater than 50% after 3 years (Boulton et al 2005). Coupled with this, around 50% of people having amputations for non-traumatic reasons have diabetes. The resultant increased morbidity and mortality from diabetes is thus plain to see and explains why diabetes is a major consumer of healthcare resources world-wide.
Various classifications of diabetes were updated by the WHO in 1999 (Table 1.1). Type 2 diabetes is the most common, representing over 80% of all persons with diabetes; type 1 comprises most of the remaining 20%. Maturity onset diabetes of the young (MODY) and secondary diabetes are relatively uncommon and account for less than 5% of people. However, it is important to identify those individuals with secondary diabetes so that treatment can be directed to the underlying cause. Diabetes in association with other genetic syndromes is also very rare. In these instances, diabetes is another burden to be borne by these people.
| Primary | |
| Type 1 | |
| Type 2 | |
| MODY, maturity onset diabetes of the young | |
| Secondary to other disorders | |
| 1. Pancreatic disease |
Chronic pancreatitis
Haemochromatosis
Pancreatectomy
Carcinoma of pancreas
Cystic fibrosis
|
| 2. Endocrine disease |
Acromegaly
Cushing’s syndrome
Phaeochromocytoma
Gestational
|
| 3. Drugs |
Thiazide diuretics
Corticosteroids
|
| Associated with genetic syndromes | |
|
Friedreich’s ataxia
Muscular dystrophies
Down’s syndrome
Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD)
|
|
TYPE 1 DIABETES MELLITUS
The hallmark of type 1 diabetes is a dependence on exogenously injected insulin to prevent ketosis and maintain life. Without injected insulin, people with type 1 diabetes die. This was the situation before the discovery of insulin in 1921. Some died very quickly over a matter of days. Others struggled along miserably for 3 or 4 years by eating almost starvation-type diets (Bliss 1983). In type 1 diabetes, therefore, there is an absolute deficiency of insulin.
Joseph’s case is a fairly representative example of a person newly diagnosed with type 1 diabetes presenting in diabetic ketoacidotic coma (diabetic ketoacidosis is explained later in the chapter). Before the discovery of insulin, such a presentation was lethal. Joseph will be dependent on self-injected insulin for the rest of his life. Withdrawal of insulin would rapidly cause further ketoacidosis and he must be advised never to stop his insulin injections. Joseph and his family will require further support and education to give him the necessary skills for self-management (see Chapters 3, 5, 6, 7 and 11).
TYPE 2 DIABETES MELLITUS
Type 2 diabetes is the most common form of diabetes world wide and there is no requirement for insulin to prevent ketosis and preserve life. Many people with type 2 diabetes can be managed by dietary means alone (see Chapters 4 and 6). Sometimes, oral hypoglycaemic drugs are required in addition to diet. Most people are over the age of 30 years when diagnosed. The main associated feature is obesity and in such people the body tissues are relatively resistant to the effects of insulin, thus causing an elevation of blood glucose. By reducing body weight many people can make carbohydrate tolerance almost normal. The management of the person with type 2 diabetes is further expanded in Chapter 4.
There is a subgroup of people, however, who are not obese and who have a relative deficiency of insulin. These people cannot secrete enough insulin to cope with the carbohydrate load they consume. The pathological process in the cells of the islets of Langerhans in the pancreas is quite different from type 1 diabetes in that these individuals do not have autoimmune damage to the pancreas. However, they might eventually require insulin therapy, although they will not be classified as having true insulin dependence.
This is an inherited form of diabetes usually arising in a person’s teenage years or in his or her early twenties and, although rare, affects about 1–2% of those with diabetes. It is passed down from one family member to another and each child has a 50% chance of inheriting the affected gene and a very high risk of developing diabetes (Shepherd 2003a).
People with MODY are usually managed with diet and/or sulphonylureas, regardless of age, for many years and ultimately will require insulin therapy. Insulin would also be required in special circumstances, such as pregnancy. Most people who have this form of diabetes have a multigenerational family history, with autosomal dominant inheritance and are usually not obese.
The molecular consequences are now well defined, with mutations in at least six genes. This means that diagnostic and predictive testing is now possible in 50–80% of families with MODY, which has implications for professional management (Shepherd 2003b). People with MODY have a mutation in the glucokinase gene, which results in mildly elevated fasting hyperglycaemia (> 5.5 mmol/L) (Barrow et al 2005). MODY 1 is due to a mutation in hepatocyte nuclear factor (HNF) 4α; MODY 2 is due to a mutation in the glycolytic enzyme glucokinase. Similarly, MODY 3 relates to HNF 1α, MODY 4 to insulin promoter factor 1, MODY 5 to HNF 1β and MODY 6 to neurogenic differentiation factor 1 (Fajans et al 2001). Genetic testing can define the subtype of diabetes as this has implications for different treatment options (Shepherd 2003a).
SECONDARY DIABETES
This type of diabetes is secondary to other disease processes that cause either pancreatic damage or production of hormones that antagonise the action of insulin (Box 1.1).
Box 1.1
Hormones that are antagonistic to the action of insulin
▪ Growth hormone
▪ Cortisol
▪ Glucagon
▪ Adrenaline
▪ Placental steroids
▪ Noradrenaline
Pancreatic disease
The pancreas can be damaged as a result of frequent bouts of pancreatitis, which is often precipitated by alcohol abuse or the presence of gallstones in the common bile duct.
The presence of carcinoma within the pancreas can also cause destruction of normal insulin-secreting cells resulting in the development of diabetes. Pancreatic disorders causing diabetes usually require treatment with insulin.
Valentina is a 73-year-old woman who presents with a 9-kg weight loss associated with nausea, polydipsia, polyuria and vulvitis. Her random blood glucose is 19.7 mmol/L and she has 2% glycosuria. Valentina is diagnosed with diabetes and is treated with dietary measures and glipizide 2.5 mg to relieve her symptoms quickly. She returns to the GP surgery 3 weeks later. Her blood glucose has fallen to 9.1 mmol/L, she has no glycosuria, but her weight has fallen by a further 2 kg.
In Valentina’s case, it is unusual to be nauseated with primary diabetes and weight loss should stop when the energy-losing glycosuria is abolished. Further investigation was thus necessary and she was found to have a carcinoma of the tail of her pancreas. Carcinoma of the pancreas can present as an apparent acute onset of diabetes and it is therefore important to think of secondary causes at the time of diagnosis, especially in elderly individuals with marked weight loss.
Endocrine disease
The secondary endocrine causes of diabetes involve excess endogenous production of hormones that are insulin antagonists (see Box 1.1):
▪ In acromegaly, growth hormone is secreted in excess by the pituitary gland.
▪ In Cushing’s syndrome, the adrenal cortex makes excess cortisol due either to a primary tumour within the adrenal gland or to excess adrenocorticotrophic hormone (ACTH) production.
▪ In phaeochromocytoma there is excess secretion of adrenaline or noradrenaline by a tumour of the adrenal medulla or sympathetic plexus.
Hypertension, which is seen in acromegaly, Cushing’s syndrome and phaeochromocytoma, commonly accompanies type 2 diabetes. When faced with an obese, hypertensive person with an elevated blood glucose it is important to consider the possibility of an underlying endocrine disorder before accepting a diagnosis of type 2 diabetes
The exceedingly rare glucagonoma results in diabetes because glucagon also antagonises insulin.
Gestational diabetes occurs in about 4% of women with normal pregnancies and is due to the insulin-antagonising effects of placental steroids and human placental lactogen. Gestational diabetes has been defined as carbohydrate intolerance of variable severity with onset during pregnancy, although there is no consensus about its diagnosis, treatment or management (SIGN 2001). Some women with gestational diabetes might need insulin treatment towards the end of pregnancy. Postnatally, the diabetes goes into remission but these women will enter the ‘at risk’ category for developing type 2 diabetes in later years (see Chapter 2).
Drug therapy
The thiazide diuretics, which are commonly used to treat essential hypertension, have a blood-glucose-raising effect by their inhibitory action on insulin secretion. Other drugs, such as corticosteroids (e.g. prednisolone), also make tissues relatively resistant to the effects of insulin and thus unmask diabetes.
GENETIC SYNDROMES
The genetic syndromes in Table 1.1 are rare but can be associated with diabetes mellitus, although the mechanisms are unknown. The syndrome comprising diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) is a well-recognised entity that can present with varying clinical features of the syndrome. Acanthosis nigricans, a rare skin condition sometimes associated with hirsutism and polycystic ovaries, is associated with a very insulin-resistant type of diabetes.
INCIDENCE AND PREVALENCE
The incidence of a disease is the number of new cases arising each year. The prevalence is the number of people with a disease in the population at any one time. The incidence of diabetes varies from country to country and area to area within countries (Hjelm et al 2003, Yach et al 2006). In the UK, the prevalence of diabetes is now around 3% and rising.
TYPE 1 DIABETES MELLITUS
The incidence varies between population groups and in the UK the cumulative risk of developing type 1 diabetes in persons aged under 20 is now 0.3–0.4%, with the peak between 10 and 14 years. This is increasing in all age groups being most marked in the 0–4 years.
TYPE 2 DIABETES MELLITUS
Accurate figures of the incidence of type 2 diabetes are difficult to establish, despite this being the more frequent type, because of inconsistent diagnostic criteria. The diagnosis of diabetes is addressed in Chapter 2.
The true prevalence is also difficult to determine accurately because of the relatively large population of people with type 2 diabetes who remain undiagnosed. There is, however, gathering evidence of a worldwide epidemic (Hjelm et al 2003). The prevalence of type 2 diabetes varies from country to country and population to population. A main feature is the low prevalence among Europeans who remain within Europe; there is a high prevalence among the Pima Indians, urban New Guineans and the Nauru (Diamond 2003, Knowler et al 1981, WHO 2005, Zimmet et al 1990). The population groups with the projected greatest increase are in Asia and Africa (Hjelm et al 2003).
Type 2 diabetes becomes more common with increasing age, with over 50% of people attending diabetes services in the UK aged over 60 years. There are now a growing number of younger people acquiring type 2 diabetes in the Western world, although it is still rare (Ehtisham et al 2004). The prevalence within the UK is only 2 in every million children. These young people tend to present later than young people with type 1 diabetes, are overweight, are usually female and a greater proportion are of ethnic minority origins. It is thought that this is due to more sedentary lifestyles and consuming more processed foods that are high in fat content.
AETIOLOGY OF DIABETES
TYPE 1 DIABETES
There is good evidence that type 1 diabetes is a T-cell-mediated autoimmune disorder and that affected persons are born with the tendency to destroy their own insulin-producing beta cells in the islets of Langerhans. There is evidence of familial clustering but there must also be some environmental trigger, as twin studies have shown that in only around 50% of identical twin pairs does the other twin develop diabetes (Devendra et al 2004).
Buy Membership for Endocrinology, Diabetes and Metabolism Category to continue reading. Learn more here
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
