CASE 1
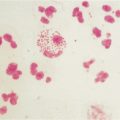
Mark is a 7-month-old boy who was born at term (40 weeks) weighing 4 kg, physically normal and apparently healthy. Although Mark was well the first couple of months of life, the last 3 months have shattered the illusion that Mark is healthy and normal. In the last few months, Mark has been plagued with fungal (diaper rash, oral candidiasis), viral (upper respiratory tract infections), and bacterial (otitis media) infections, all of which resolved with appropriate pharmacologic intervention. Mark has received routine childhood immunization, which his mother hoped would reduce the number of infections. Not surprising, Mark has not thrived and is well below the 50% percentile for weight. Mark was referred to a pediatrician who noted that Mark had (once again) a diaper rash and candidiasis (Fig. 1-1) and an upper respiratory tract infection. Despite these infections, his tonsils and lymph nodes were barely detectable. The pediatrician ordered a number of tests that included a complete blood cell count with differential, serum immunoglobulin (IgG), and a chest radiograph. The blood cell count indicated a low leukocyte count, profound lymphopenia, and very low serum IgG value.
QUESTIONS FOR GROUP DISCUSSION
RECOMMENDED APPROACH
Implications/Analysis of Laboratory Investigation
Although the chest radiograph did indicate that Mark has pneumonia, there is no mention of an anomaly regarding the thymic shadow, suggesting the organ of differentiation for T cells is intact (see Case 2). Laboratory investigations should focus on both T cell and B cell numbers and function.
THERAPY
Bone Marrow Transplant
SCID is often referred to as the “bubble boy” disease, in reference to David Vetter, a young boy who lived for 12 years in a plastic, sterile bubble but succumbed to infection after a bone marrow transplant from his sister who carried a latent Epstein-Barr virus (human herpesvirus 4) infection.
ETIOLOGY: SEVERE COMBINED IMMUNODEFICIENCY
Deficiency in CD132 (Common γ Chain)
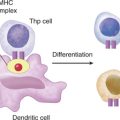
CD132, a signaling molecule in several cytokine receptor complexes, is encoded on the X chromosome and so this immunodeficiency is referred to as XSCID. Receptor complexes, in which CD132 is a component, bind the following interleukins: IL-2, IL-4, IL-7, IL-9, and IL-15 (Fig. 1-2). IL-7 plays an important role in the development of lymphoid progenitors during hematopoiesis; however, knockout mice for IL-7 are grossly deficient in T cells and only moderately deficient in B cells, suggesting that IL-7 is required for T cell maturation in the thymus.
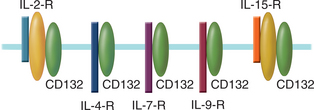
Mutations, ranging from nonsense mutations to deletion of entire exons, in the gene encoding CD132 lead to SCID because multiple cytokine systems are simultaneously affected. One of the diagnostic procedures for identifying these individuals is genomic sequencing of CD132. Because CD132 is encoded on the X chromosome, males are predominantly affected. Mark’s family history indicated that both males and females had presented with this disorder; therefore, it is unlikely that Mark has XSCID. Furthermore, the white blood cell differential indicated a gross deficiency of both B cells and T cells.
Deficiency in JAK 3
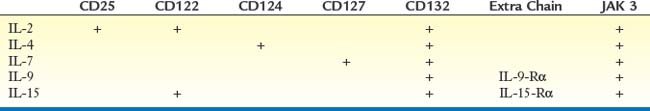
CD132 is a transmembrane protein whose cytoplasmic domain interacts with JAK 3 (Janus tyrosine kinase 3) after cytokine binding (Table 1-1). JAK 3 activates, by phosphorylation, the latent transcription factor, STAT 5 (signal transducer and activator of transcription), which then translocates to the nucleus. Stable T cell lines derived from patients with a deficiency in JAK 3 do not proliferate in response to IL-2, a T cell growth factor that signals via a receptor complex that includes CD132. An in vitro reconstitution study, in which a retroviral vector encoding JAK 3 was introduced into these cell lines, restored JAK 3 expression and proliferation of T cells in response to IL-2. Not surprisingly, JAK 3–deficient patients have a clinical phenotype virtually identical to that of XSCID, even though JAK 3 is encoded on chromosome 19 and not on the X chromosome.
Defect in Recombinase Activation Gene
Theoretically, in any given individual, it is possible to generate 1012 different T cell (or B cell) clones each bearing antigen specific receptors that consist of a unique variable and a constant region. This uniqueness is made possible because the variable regions are made up of sections of DNA referred to as variable (V), diversity (D), and joining (J) segments. Multiple versions of each segment are present in the germline genes (see Case 2). In the construction of a gene encoding any variable region, the gene segments are recombined in a different sequence from that present in germline DNA. This process, termed somatic recombination, requires the activation of recombinases, nucleoprotein products of the RAG1 and RAG2 genes (recombination-activating genes).