Chapter 6 Vascular Pharmacology
Vascular Smooth Muscle Activation
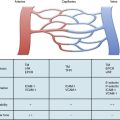
The actions of many drugs discussed in this chapter affect vascular smooth muscle cell (VSMC) contraction, and a basic understanding of contractile regulation is essential to understanding their mechanism of action. Contraction of vascular smooth muscle involves a sliding filament mechanism similar to that observed in other smooth muscle or in skeletal muscle. This topic has been reviewed in depth previously,1 is covered in detail in Chapter 3, and is therefore only briefly discussed here. The classical paradigm, depicted in Figure 6-1, is that increases in intracellular calcium lead to formation of a calcium-calmodulin complex. Calcium–CaM then binds and activates myosin light chain kinase (MLCK), which then phosphorylates myosin light chain (LC-20). Phosphorylation of LC-20 increases myosin adenosine triphosphatase (ATPase) activity, which leads to cross-bridge cycling and contraction. Myosin light chain phosphatase negatively regulates this process by dephosphorylating LC-20. Myosin light chain phosphatase is in turn inhibited by the small G-protein Rho and Rho kinase, which phosphorylates a subunit of myosin light chain phosphatase known as the myosin-binding subunit (MBS), leading to inhibition of phosphatase activity and favoring contraction. Myosin phosphatase is also inhibited by a 17-kDa protein known as CPI-17 (protein kinase C [PKC]–potentiated inhibitory protein of 17 kDa) that in turn is activated by PKC. Thus, activation of PKC can indirectly reduce myosin phosphatase activity, increase myosin phosphorylation, and promote vasoconstriction.
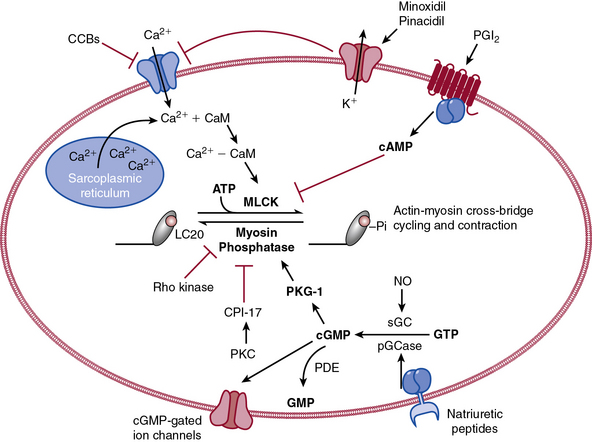
An important counterregulatory pathway in this scheme is the nitric oxide (NO) pathway. Nitric oxide acts on soluble guanylyl cyclase (sGC), which catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). In turn, cGMP acts as the only substrate for type 1 protein kinase G (PKG), which phosphorylates MBS, increasing its phosphatase activity and promoting vasodilation. Protein kinase G also phosphorylates and inhibits Rho, further reducing the propensity for vasoconstriction and promoting vasodilation. These pathways are targets of myriad vasoactive drugs that will be considered in greater depth in this chapter and are depicted in Figure 6-1.
It is now apparent that many pharmacological agents not only modulate vascular tone but also vascular growth, remodeling, inflammation, thrombosis, and vascular repair. As examples, many components of the contractile pathway discussed earlier exist in endothelial cells (ECs), including actin, myosin light chain, MLCK, Rho, and Rho kinase. These regulate endothelial shape, migration, cell-cell contact, and permeability. Myosin light chain kinase activation controls EC calcium entry, NO production, and release of endothelium-derived hyperpolarizing factor (EDHF).2 The Rho/Rho kinase pathway works in concert with other GTPases to modulate endothelial production of NO and reactive oxygen species (ROS) and gene expression.3 These aspects of vascular control have been the subject of substantial recent research, and new drugs have been developed to affect these targets. In addition, these pathways seem to be affected in an off-target fashion by several existing pharmacological agents.
Pharmacokinetics and Pharmacodynamics
The nature of a drug response helps classify the drug as a full or partial agonist, antagonist, or an inverse agonist (Fig. 6-2A) and may provide insight into the mechanism of drug action. For receptor conformation–specific drugs, pure antagonists stabilize the active and inactive conformations equally and have no net effect on basal activity. Inverse agonists preferentially stabilize the receptor’s inactive form, and agonists stabilize the active conformation.
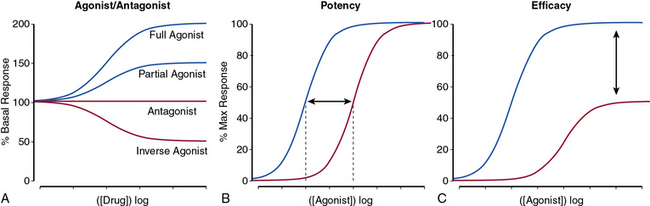
The potency of a drug refers to the molar concentration necessary to achieve a desired response (e.g., 50% maximal stimulation or inhibition; Fig. 6-2B), whereas efficacy reflects the drug’s maximal response relative to other agents (Fig. 6-2C). Clinical differences in drug potency may be overcome by increasing the dosage, whereas differences in drug efficacy cannot.
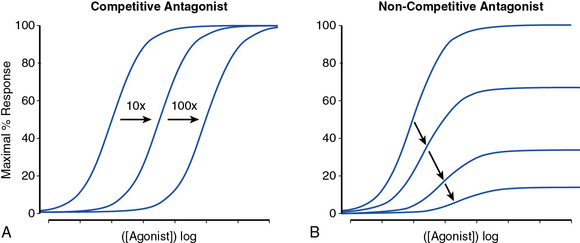
Receptor antagonists can be assessed by the response to a known stimulus in the presence of increasing antagonist concentration (Fig. 6-3). Antagonists that reversibly bind to the receptor can be overcome with increasing concentration of agonist (Fig. 6-3A). Antagonists that irreversibly bind their target impair the maximal response with increasing concentration (Fig. 6-3B). A number of drugs act in an allosteric manner by binding to a site on the receptor that is distinct from the native ligand, inducing a conformational change. Allosteric modulators can either increase or decrease agonist response by binding to a site distinct from the agonist binding site. An allosteric antagonist dose-response curve appears similar to that of a noncompetitive antagonist. Allosteric potentiators shift the agonist curve to the left (see Fig. 6-3A), while competitive antagonists shift the curve to the right.
Drugs That Affect Nitric Oxide/Guanylyl Cyclase/cGMP–Dependent Protein Kinase Pathway
The NO pathway plays a major role in modulating vascular reactivity; however, NO represents only one step in a complex pathway that can be affected by a variety of signaling molecules. This pathway is illustrated in the right portion of Figure 6-1, and involves the guanylyl cyclase enzymes, cGMP, and the binding targets of cGMP, which include the cGMP-dependent PKGs, ion channels regulated by cGMP, and phosphodiesterases (PDEs). The guanylyl cyclase/cGMP pathway is affected by a variety of agents, including NO and NO donors (the nitrovasodilators); other agents that activate guanylyl cyclase; agents that modulate degradation of cGMP; and agents that directly activate PKG.
Endogenously, NO is produced by the nitric oxide synthase (NOS) enzymes, and serves myriad signaling roles depending on the cell and tissue in which it is produced.4 Experimental studies have shown that NO produced by the endothelium not only mediates vasodilation, but also inhibits expression of adhesion molecules, reduces platelet adhesion, inhibits vascular smooth muscle growth and hypertrophy, and prevents vascular remodeling.
Nitrovasodilators
The nitrovasodilators produce their biological effects either by releasing NO or closely related molecules that are converted to NO in cells. The most commonly employed nitrovasodilators are the organic nitrates and sodium nitroprusside. It is useful to begin a discussion of these agents by comparing sodium nitroprusside and nitroglycerin, which are illustrated in Figure 6-4. As apparent, the oxidation state of the nitrogen that is ultimately released as NO differs in these molecules, and this basic structural property provides insight into their pharmacological profiles. Sodium nitroprusside requires a one-electron reduction to release NO, and this is readily accomplished nonenzymatically by a variety of reductants in the circulation, the interstitial space, and the cell. Thus, when infused intravenously, nitroprusside begins to release NO throughout the circulation and potently dilates all vessels. Moreover, given the short half-life of NO, the vasodilation caused by nitroprusside is short-lived once its infusion is discontinued.
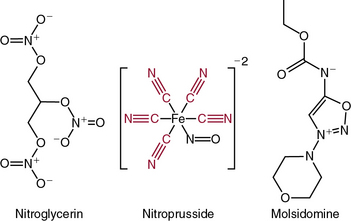
As is apparent from its structure, nitroprusside possesses five cyanide groups in each molecule (highlighted in red in Fig. 6-4), and prior studies have shown that each of these is reduced prior to the release of NO. The cyanide radicals react with hemoglobin (Hb) to form methemoglobin and are converted to thiocyanate in the liver. When these metabolic pathways are depleted, cyanide toxicity occurs, characterized by central nervous system (CNS) dysfunction, metabolic acidosis with a base deficit, and elevated plasma lactic acid concentrations.5 Fortunately, cyanide toxicity is infrequent during brief administration of sodium nitroprusside but occurs more commonly when infusion rates exceed 2 μg/kg/min and when the drug is infused for prolonged periods. In addition, the risk of cyanide toxicity is increased in patients with renal or hepatic failure, so sodium nitroprusside should be avoided in patients with these conditions. Owing to its capacity to rapidly release NO, sodium nitroprusside produces potent systemic vasodilation and is effective as an antihypertensive. It is still used for treatment of severe hypertension and, in some cases, for afterload reduction in patients with severe heart failure; however, newer agents with less potential toxicity are now more commonly used.
In contrast to sodium nitroprusside, nitroglycerin and other organic nitrates require a 3-electron reduction to yield NO. In the last several years, it has become clear that this is in large part accomplished by the action of the mitochondrial enzyme aldehyde reductase-2 (ADH2).6 Mice lacking this enzyme are resistant to the actions of nitroglycerin. Notably, about 40% of East Asians have a dominant negative mutation of ADH2 that causes intolerance to ethanol and markedly impaired vasodilator responses to nitroglycerin.7
As mentioned earlier, organic nitrates preferentially dilate larger arteries and veins while having less effect on arterioles, particularly at lower doses.8 This response profile is likely beneficial in alleviating angina because potent arteriolar dilators are prone to cause coronary steal and paradoxically worsen myocardial ischemia. Moreover, venous dilatation reduces left ventricular (LV) filling, alleviates pulmonary congestion, and can improve subendocardial perfusion in ischemic regions of the myocardium.
Experimental studies have shown that NO inhibits platelet adhesion, expression of adhesion molecules, and vascular smooth muscle proliferation and migration. Thus, one might expect that NO donors such as nitroglycerin would reduce atherosclerosis progression and potentially reduce major cardiovascular events in patients with coronary artery disease (CAD). Despite extensive use for alleviation of myocardial ischemia for almost a century and a half, no clinical trials have shown that these drugs reduce ischemic cardiovascular events. The GISSI-3 and ISIS-4 trials examined the effect of nitrates following myocardial infarction (MI) but failed to show a significant improvement in outcome.9,10 These trials only observed patients for 5 weeks to 6 months following MI, and therefore did not determine whether long-term nitrates might have a beneficial effect on outcome in patients with ischemic heart disease. Given the many putative beneficial effects of NO on vascular function, longer-term treatment might impart a beneficial effect on atherosclerosis, inflammation, vascular remodeling, or plaque stability. Indeed, a recent analysis of the GRACE registry, which includes patients admitted for ACS, showed that chronic nitrate users were much more likely to present with non–ST-segment elevation MI NSTEMI) than non-nitrate users.11 These data must be interpreted with caution because the use of nitrates was not randomized, and conclusions were derived from a retrospective analysis.
In addition to their use as antianginal agents, the long-acting nitrates are now often employed for treatment of congestive heart failure (CHF), commonly in combination with hydralazine. Unlike the case for treatment of CAD, prospective randomized trials have shown that long-acting nitrates improve survival, reduce hospitalizations, and enhance quality of life in patients with CHF, particularly among African Americans.12 Precise mechanisms underlying the beneficial effects are unclear; however, long- acting nitrates appear to synergize with hydralazine as afterload- and preload-reducing agents. These agents might also improve renal hemodynamics and promote diuresis and, via release of NO, have beneficial effects on vascular and cardiac remodeling.
A major limitation to prolonged use of organic nitrates is development of tolerance. Within about 12 hours of administration, the hemodynamic effects of organic nitrates begin to abate, in part due to extravascular adaptations such as volume redistribution and neurohormonal activation. After several days of continuous nitrate therapy, the direct vascular actions of nitrates are lost, even when vessels are removed from the animal or human. The mechanisms of nitrate tolerance, and in particular this latter form of true vascular tolerance, remain uncertain but have been attributed to formation of ROS, nitrosation and oxidation of guanylyl cyclase, and changes in activity of ADH2.13 A number of strategies have been proposed to prevent nitrate tolerance, but the only approach accepted clinically is to allow a drug “holiday”; that is, to withdraw the nitrate for about 12 hours daily. The commonly employed isosorbide mononitrate preparations accomplish this by increasing blood levels of the drug for about 12 hours during waking hours, after which blood levels fall to near-undetectable levels. Experimental studies have shown that hydralazine prevents nitrate tolerance by reducing oxidative stress,13 which might explain the benefit of hydralazine when added to long-acting nitrates in the treatment of heart failure. The long-acting nitrate pentaerythritol tetranitrate seems not to cause tolerance in experimental animals, but this has not been proven in clinical studies.
Intravenous nitroglycerin has occasionally been used for treatment of hypertensive emergencies. This condition is often associated with a contracted blood volume. Owing to nitroglycerin’s propensity to produce venular dilation rather than arteriolar dilation, it has the potential to reduce cardiac output in this setting and may produce untoward effects in patients with compromised coronary, renal, or cerebral perfusion.14 Low-dose nitroglycerin might be useful in combination with other agents in treating a hypertensive emergency, particularly in patients with acute pulmonary edema, but other agents are available and likely more effective.
In addition to its reaction with sGC, NO can react with other heme proteins and radicals. Higher oxides of NO can also react with thiols, leading to formation of nitrosothiols.15 An important example of these reactions is the reversible NO reaction with cytochrome C, which modulates mitochondrial respiration and superoxide production.16 It is uncertain as to how important these reactions are in the overall response to nitrovasodilators.
Related to the chemistry mentioned earlier are reactions of inorganic nitrate (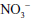 ) and nitrite (
) and nitrite (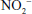 ). Although these are oxidation products of endogenously produced NO, they are also derived from dietary sources such as green leafy vegetables. Nitrate is rapidly converted to nitrite by bacteria in the oral cavity and gastrointestinal tract. Nitrite, in turn, can be reduced by various heme proteins, including deoxyhemoglobin, to NO. Studies have shown that the reaction of nitrite with deoxyhemoglobin promotes NO formation and vasodilation in regions of the circulation where oxygen tension is low, thereby improving oxygenation of hypoxic tissues.17 Thus, once considered an inactive metabolite of NO, nitrite likely has physiological significance and might have therapeutic utility.18
). Although these are oxidation products of endogenously produced NO, they are also derived from dietary sources such as green leafy vegetables. Nitrate is rapidly converted to nitrite by bacteria in the oral cavity and gastrointestinal tract. Nitrite, in turn, can be reduced by various heme proteins, including deoxyhemoglobin, to NO. Studies have shown that the reaction of nitrite with deoxyhemoglobin promotes NO formation and vasodilation in regions of the circulation where oxygen tension is low, thereby improving oxygenation of hypoxic tissues.17 Thus, once considered an inactive metabolite of NO, nitrite likely has physiological significance and might have therapeutic utility.18
Molsidomine (see Fig. 6-4) has also been used as an NO donor for treatment of angina, but it is not commonly employed clinically. The liver metabolizes molsidomine to release SIN-1, which in turn decomposes to NO and superoxide in equimolar amounts. These species can react rapidly with one another to yield the strong oxidant peroxynitrite. Because of this chemistry, SIN-1 oxidizes lipoproteins, damages DNA, and depletes antioxidants. This capacity to generate peroxynitrite has dampened enthusiasm for clinical use of molsidomine and related drugs, but SIN-1 is commonly used to produce peroxynitrite in experimental settings.
Unique Modulators of Soluble Guanylyl Cyclase
Compounds have been developed that activate sGC in an NO-independent fashion.19 Some of these, such as the pyrazolopyridine BAY 41-2272 and YC-1, interact with the heme group independent of NO, or can markedly enhance NO-stimulated enzyme activity. Others, such as BAY 58-2667 and HMR-1766, activate sGC in a heme-independent fashion and can stimulate cGMP formation even when the heme group is oxidized. Because these agents do not depend on endogenous production of NO, they have potential advantages over PDE inhibitors (see later discussion) in diseases where NO production is impaired. They also potentially bypass the problem of tolerance observed with various NO donors. These agents produce vasodilation, lower blood pressure, inhibit platelet aggregation, and have been shown to have therapeutic benefit in experimental models of systemic hypertension, pulmonary hypertension (PH), and heart failure.19 Like NO, they inhibit neointima formation following balloon injury in rats and therefore might be effective in treatment or prevention of restenosis and atherosclerosis. They also hold promise for treatment of erectile dysfunction (ED), liver fibrosis, and renal disease. Currently, clinical trials are underway to examine the efficacy of some of these agents in the treatment of heart failure and PH.
Natriuretic Peptides
Natriuretic peptides, including atrial (ANP), brain (BNP), and C-type (CNP) natriuretic peptides, are 17-amino-acid ring structures with an internal disulfide bond and are secreted as prohormones. Atrial natriuretic peptide and BNP are predominantly produced by atrial and ventricular myocytes; CNP is produced by vascular endothelial cells, the brain, and other peripheral tissues.20 Urodilatin, a related peptide processed from the ANP prohormone, is released from distal tubular cells of the kidney.21 The A and B natriuretic peptide receptors are homodimers that are widely distributed, particularly in the cardiovascular system and kidney.21 The cytoplasmic tails of these contain a guanylyl cyclase domain that is activated by binding with natriuretic peptides.20 There also exists a C-type natriuretic receptor that has a short cytoplasmic tail without a guanylyl cyclase domain and seems predominantly involved in clearing natriuretic peptides from the circulation.
As mentioned, ANP and BNP are produced predominantly in atrial myocytes. In the setting of a variety of conditions (e.g., heart failure, cardiac inflammation, fibrosis, hypoxia), BNP is expressed in large amounts by ventricular myocytes, leading to an elevation of circulating BNP. Thus, BNP and pro-BNP are commonly used as biomarkers for detection of various cardiac pathologies, and in particular for diagnosis and management of volume overload states.22
Activation of the A- and B-type natriuretic receptors leads to vasodilation and a variable diuretic and natriuretic response, depending on volume status. For this reason, a synthetic form of BNP known as nesiritide has been marketed and employed for treatment of decompensated heart failure. Like the nitrovasodilators, nesiritide infusion lowers pulmonary capillary wedge pressure (PCWP), right atrial pressure, and systemic vascular resistance, and improves symptoms of dyspnea.23 This agent also lowers circulating catecholamines, aldosterone, and angiotensin-(Ang) II levels, and aids diuresis. One study suggested that nesiritide was more effective that intravenous nitroglycerin treatment of patients with severe heart failure.23 An early meta-analyses suggested that nesiritide therapy was associated with an increase in mortality within 30 days of treatment, for uncertain reasons24; however, more recent meta-analysis of six randomized clinical trials showed no change in outcome at 10, 30, or 180 days following administration of this agent.25 A randomized trial of more than 7000 subjects has shown that treatment with nesiritide acutely improves patients with class IV heart failure, without worsening long-term outcome.26 This positive study is tempered by a very recent large study of 7143 patients with acute heart failure that showed no benefit of nesiritide in reducing symptoms or improving outcome at 30 days.27
Phosphodiesterase Inhibitors
As reflected in Figure 6-1, cGMP is rapidly inactivated to GMP by cellular PDEs. There are 11 PDE isoenzymes with varying specificities for the different cyclic nucleotides. Phosphodiesterases 5, 6, and 9 are highly selective for cGMP, while PDEs 3 and 10 are preferentially activated by cyclic adenosine monophosphate (cAMP). Phosphodiesterases 1, 2, and 11 have dual substrate specificity.28 In the cardiovascular system, the predominant PDEs are PDE1, 2, and 5. The PDEs are subject to substantial posttranslational regulation. As examples, PDE1 is calcium/CaM-dependent, cGMP stimulates PDE2 inactivation of cAMP, and binding of cAMP to PDE3 is inhibited by cGMP.
Several naturally occurring PDE inhibitors, such as caffeine, theophylline, and theobromines, are present in coffee, chocolates, and tea, and have been used since antiquity as stimulants.29 These are among the most widely distributed drugs in the world. Like cAMP and cGMP, the PDE inhibitors commonly contain a purine structure with linked pyrimidine and imidazole rings. These agents occupy the cAMP or cGMP PDE binding sites and inhibit respective PDE isoenzymes with varying degrees of selectivity.29 The immediate cardiovascular effects of nonselective PDE inhibition include vasodilation due to accumulation of cGMP and cAMP, increases in cardiac contractility due to accumulation of cAMP, and improvement in diastolic relaxation (lusitrophy) mediated by cAMP and cGMP.
In the past 30 years, a variety of PDE5 inhibitors, including sildenafil, tadalafil, and vardenafil, have been developed and are now used clinically (Table 6-1). Experimental studies have shown that the vasodilator effect of PDE5 inhibitors is almost exclusively dependent on endogenous NO release, and is prevented by inhibition of NOS and in conditions in which endogenous NO production is impaired.28 These agents also affect cardiac function. The PDE5 inhibitors acutely reduce cardiac contractility and precondition cardiac myocytes to reduce necrosis and apoptosis caused by experimental ischemia.30,31 Chronic PDE5 inhibition with sildenafil prevents experimental cardiac hypertrophy caused by transaortic constriction.32
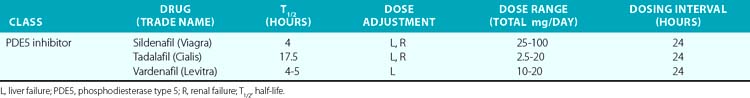
The PDE5 inhibitors were developed as antihypertensive agents, but because of their potent effect on the corpus cavernosa, they were initially approved and have become widely employed for treatment of erectile dysfunction. These agents are also potent dilators of the pulmonary circulation. Sildenafil and tadalafil been approved by the U.S. Food and Drug Administration (FDA) for treatment of pulmonary arterial hypertension (PAH). This disorder, defined by the hemodynamic parameters of a mean pulmonary artery pressure (PAP) above 25 mmHg and a PCWP 15 mmHg or lower, occurs as a primary condition and in the setting of a variety of diseases that affect the pulmonary circulation.33 (Also see Chapters 56 and 57.) A single dose of sildenafil was found to reduce PAP in patients with both primary and secondary PH and to augment the effect of inhaled NO in these subjects.34 Clinical studies have shown that chronic administration of PDE5 inhibitors reduces PAP and right ventricular (RV) mass, and improves exercise tolerance and functional status in patients with PAH.35 The recent SUPER-2 clinical trial showed that sildenafil improved 3-year survival in patients with PAH compared to historical controls.36 For these reasons, PDE5 inhibitors are now considered a mainstay of therapy for PAH. They have also been used with some success in neonates with persistent pulmonary hypertension of the newborn (PPHN).37
Phosphodiesterase type 5 inhibition has beneficial effects on hemodynamics and cardiac function in heart failure. In various experimental models of heart failure, PDE inhibitors prevent and reverse cardiac hypertrophy, reduce remodeling, and decrease myocardial fibrosis.38,39 In a recent placebo-controlled clinical trial of patients with severe heart failure, sildenafil treatment for 1 year improved ejection fraction, improved parameters of diastolic function, and reduced left atrial size while improving functional capacity and clinical status.40 This study was not designed to determine whether sildenafil improves survival; larger studies are needed with longer-term follow-up to discern whether PDE5 inhibition provides survival benefit. Nevertheless, these orally available agents, which avoid the problem of tolerance encountered with the nitrovasodilators, have substantial promise in treating ventricular dysfunction.
Prostaglandins and Thromboxane Agonists and Antagonists
Release of lipids from the cell membrane upon receptor binding or mechanical stimulation is a major signaling event in mammalian cells. One major class of lipid metabolites is the prostanoids, which include the prostaglandins (PGs) and thromboxane. The pathway leading to formation of these lipids is illustrated in Figure 6-5. They are formed from arachidonic acid, released from membrane phospholipids via the action of phospholipase A2. The initial step in prostanoid synthesis is conversion of arachidonic acid to the endoperoxide prostaglandin H2 (PGH2) by COX enzymes. Prostaglandin H2 is in turn a substrate for several enzymes including various PG synthases and thromboxane synthases (see Fig. 6-5), which leads to formation of multiple PG metabolites including PGE2, prostacyclin (PGI2), PGF2α, PGD2, and thromboxane A2 TxA2. Each of these has several G protein–linked receptors that are widely distributed and modulate myriad physiological and pathophysiological responses that include inflammation, vasomotor tone, hemostasis, renal function, and blood pressure.41,42 Vascular response to the various prostanoids depends on the category of the heterotrimeric G-protein receptor to which it binds. Vasodilator prostanoids, including PGI2 and PGD2, activate Gs, which leads to an increase in intracellular cAMP. The contractile prostanoids, including TxA2 and PGF2α, activate Gq, which leads to increased intracellular calcium. There are both Gs and Gq receptors for PGE2, which can therefore both vasodilate and vasoconstrict.
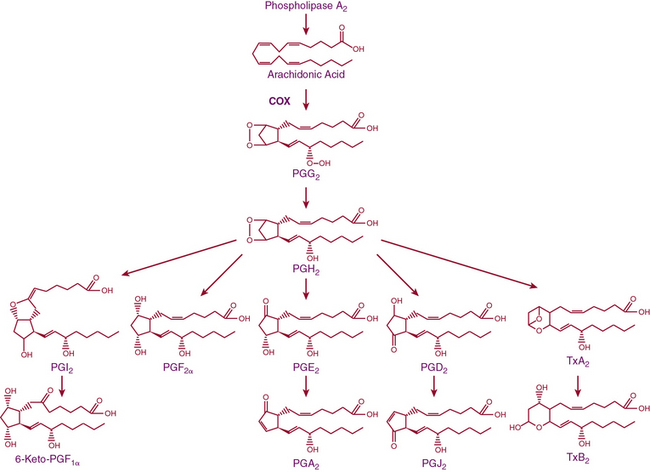
Figure 6-5 Arachidonic acid metabolic pathway.
COX, cyclooxygenase; PG, prostaglandin; PGI2, prostacyclin; Tx, thromboxane.
There are two isoforms of the COX enzymes: COX-1 and COX-2. Cyclooxygenase-1 is constitutively expressed and exerts housekeeping functions in many cells, including vascular cells. Cyclooxygenase-2 is generally considered an inducible enzyme, and its levels increase in the settings of inflammation, in particular when inflammatory cells enter the affected tissue.42 Cyclooxygenase-2 is also constitutively expressed in some cells, including ECs. The preferred substrate of COX-1 is arachidonic acid, but COX-2 can also produce unique antiinflammatory products from the endogenous cannabinoid 2-arachidonyl glycerol.43 Both COX-1 and COX-2 are activated by shear stress in the endothelium.44 The downstream products of COX are highly dependent on the cell type. In healthy blood vessels, the predominant arachidonic acid metabolite is PGI2, whereas platelets predominantly produce TxA2. In a variety of common cardiovascular diseases, however, vascular production of prostanoids can be shifted toward proinflammatory, procoagulant, and vasoconstrictor prostanoids.44 As an example, Ang-II stimulates COX-2 expression and production of PGE2 in VSMCs, and this response contributes to VSMC proliferation and migration in response to this hormone.45 In several experimental models of hypertension, obesity, and aging, the endothelium begins to produce prostanoid-contracting factors including PGH2, TxA2, and ROS generated as byproducts of COX activity.46
Cyclooxygenase Inhibitors
Aspirin has been studied extensively since the 1950s as a means of reducing cardiovascular events.47 Numerous large clinical trials performed in the 1980s supported the concept that aspirin decreases the occurrence of MI and stroke. A recent large meta-analysis showed that aspirin was effective in both primary and secondary prevention of total coronary events, ischemic stroke, and serious vascular events, with the greatest benefit observed in the case of secondary prevention.48 Another recent meta-analysis of nine trials that included 90,000 patients showed that aspirin is effective for primary prevention of nonfatal MI and total cardiovascular events, but not for stroke, cardiovascular mortality, or all- cause mortality.49 Of interest, several recent meta-analyses have suggested that aspirin might not be useful for primary prevention of events in the diabetic population.50,51
The beneficial effects of aspirin are generally considered a consequence of its antiplatelet effects and reduction of thrombosis. However, aspirin reduces levels of C-reactive protein (CRP) in patients with recent unstable coronary syndromes,52 and in experimental models of atherosclerosis, reduces atheroma burden, decreases inflammation, and improves endothelial function,53,54 suggesting that it might also have direct vascular effects.
Although aspirin has proven effective in reducing cardiovascular events, there are no clinical trials showing that other COX inhibitors convey similar cardiovascular benefit, and paradoxically, there is substantial evidence that these agents are harmful. The most striking example is that of the COX-2 inhibitor rofecoxib, which was withdrawn from the market because of increased thrombotic events55; however, other COX inhibitors might also increase cardiovascular risk, depending upon the relative COX-2–to–COX-1 selectivity.56,57 The precise mechanisms underlying this increased risk remain undefined, and it is unclear why aspirin, which inhibits the same enzyme, albeit via different mechanisms, is beneficial. These differences might relate to inhibition of vascular PGI2 and perhaps renal COX, which in turn could promote sodium retention and blood pressure elevation and worsen cardiovascular outcome. As previously mentioned, the downstream products and their receptors are myriad, so the in vivo actions of these agents are complex and difficult to predict. Nevertheless, NSAIDs other than aspirin should be used sparingly in patients with known cardiovascular diseases and currently have no role in preventing cardiovascular events.
Prostacyclin Analogs as Therapeutic Agents
Given its potent vasodilator effects, there is enormous interest in therapeutic use of PGI2 and its analogs. Several preparations have been developed. The most commonly employed are epoprostenol, a freeze-dried synthetic preparation of PGI2, and the PGI2 analogs iloprost, treprostinil, and beraprost. These agents have become a mainstay of treatment for PAH. Epoprostenol was initially approved for treatment of PAH following a 12-week trial in 81 patients prospectively randomized to either epoprostenol or conventional therapy.58 Among those treated with epoprostenol, there was improvement in exercise capacity and a decline in PAP. This was in contrast to those receiving conventional therapy, in whom walk time decreased and PAP increased. Patients treated with epoprostenol had greater symptomatic improvement, and most strikingly in this small study, eight patients died, all in the conventional therapy group. A second study showed that epoprostenol improved exercise duration and lowered PAP in patients with PH associated with scleroderma.59 Interestingly, these subjects showed a trend toward improvement of digital ulcers, suggesting that systemic vasodilation caused by this drug might also be beneficial. Subsequent long-term follow-up in large registries have confirmed a beneficial effect of continuous intravenous epoprostenol in PAH.
A downside of epoprostenol therapy is that it requires chronic central line placement, which is accompanied by risk of infection that might be related in part to prostanoid-mediated immunosuppression. The drug also often requires up-titration to overcome tachyphylaxis and is expensive.60 Owing to its short half-life, there is rebound PH that develops shortly after discontinuing the drug, which can have serious consequences. Common side effects include headaches, occasional cases of thyrotoxicosis, nausea, jaw pain, thrombocytopenia (in up to 34% of patients), flushing, skin rash, anorexia, arthralgias, and myalgias.
There is also lack of consensus on how to use these agents, which agents to use, and what dosing regimen is optimal.48 As discussed elsewhere in this chapter, these agents are often used in conjunction with PDE5 inhibitors and endothelin-1 (ET-1) receptor antagonists, again without uniformity across various centers.
Sympathetic and Parasympathetic Nervous Systems
Abrupt changes in blood pressure are buffered by the sympathetic and parasympathetic nervous system (Fig. 6-6). The baroreflex response helps integrate blood pressure detection and CNS response, and impairment of this response produces profound orthostatic intolerance and inability to maintain upright posture.61 Increased blood pressure stimulates baroreceptors located in the carotid sinus and aortic arch, which transmit their signals to the nucleus tractus solitarius in the CNS. The transmitted signal inhibits sympathetic outflow from the rostral ventrolateral medulla (RVLM). Sympathetic efferent preganglionic axons extend to the sympathetic ganglion, where acetylcholine serves as the principal neurotransmitter to postganglionic nicotinic receptors. Postganglionic sympathetic fibers extend to effector organs such as the heart and vasculature and release norepinephrine (NE) to produce vasoconstriction and increased contractility. The adrenal medulla is innervated directly by preganglionic sympathetic neurons and releases both NE and epinephrine into the circulation. At the same time, the reflex activates parasympathetic system and reduces heart rate via innervation of the cardiac conduction system. Therefore, the net effect of an abrupt increase in blood pressure is inhibition of the sympathetic system and activation of the parasympathetic nervous system.
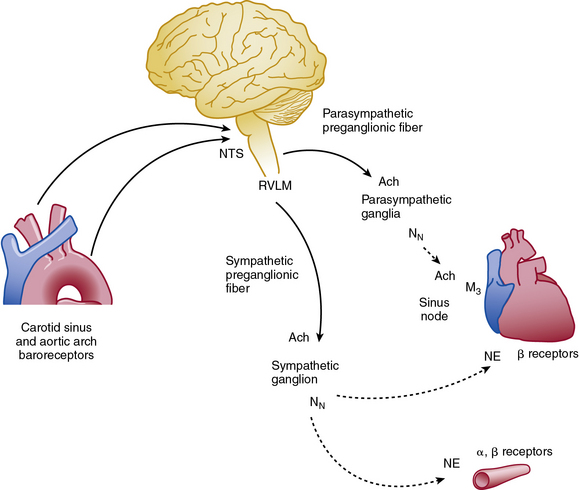
Vascular Parasympathetic System
Postganglionic parasympathetic fibers release acetylcholine, which stimulates muscarinic and nicotinic receptors. Most blood vessels lack parasympathetic innervation, although some notable exceptions exist (e.g., coronary arteries), and the physiological role of endogenous acetylcholine in vasodilation is uncertain.62 The vasculature does contain muscarinic receptors and responds to exogenously administered acetylcholine or mimetics (e.g., methacholine). Exogenous acetylcholine dilates blood vessels by its actions on the vascular endothelium, but it produces vasoconstriction if the endothelial layer is injured or removed. This discovery demonstrated the importance of the endothelium as an active participant in vascular reactivity and eventually led to the discovery of endothelium-derived relaxing factors (e.g., NO, PGI2.63 Patients with cardiovascular disease exhibit an impaired vasodilatory response to acetylcholine (endothelial dysfunction) but often have a normal response to direct vasodilators such as nitroprusside. Impaired vascular reactivity in both the coronary and forearm vasculature predicts future cardiovascular events,64,65 and the endothelium-dependent response may be improved with drug therapy, exercise, or risk factor modification (e.g., smoking cessation).66–68
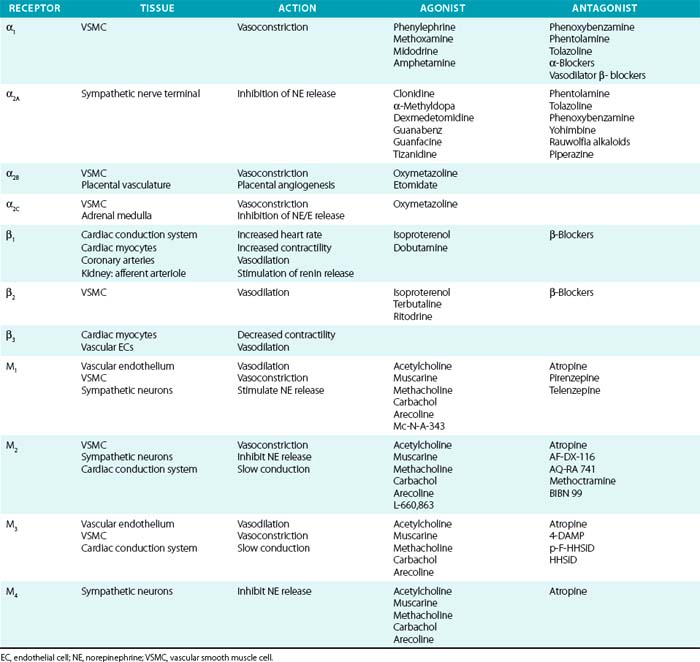
Acetylcholine receptors (AchRs) are classified by their ability to respond to either muscarine (M1-M5) or nicotine (nAchR). Muscarinic receptors are classic G protein–coupled receptors (GPCRs), coupled to Gi, which inhibits cAMP production. Nicotinic AchRs are ligand-gated voltage channels. Vascular M1, M2, and M3 receptors have been described and produce vasodilation via endothelial, or vasoconstriction via VSMC, receptors69 (Table 6-2). Acetylcholine is a nonselective agonist; there are no clinically available subtype-selective agents, although a number of investigational drugs exist. Methacholine is frequently used in clinical research because of its longer half-life and stability. Atropine is a nonselective muscarinic antagonist used mainly to increase heart rate by its effects on cardiac M2 and M3 receptors. Muscarinic receptors are also located on postsynaptic sympathetic nerve terminals and inhibit NE release. Peripheral neuronal nicotinic AchRs (NN) transmit sympathetic impulses in autonomic ganglia and adrenal medulla to stimulate NE and epinephrine release. Trimethaphan inhibits NN and was one of the earliest antihypertensive agents available, although it is no longer used, owing to resulting severe autonomic impairment and intolerable side effects.
Adrenergic Receptors and Agonist Selectivity
Sympathetic postganglionic neurons richly innervate the vasculature and release NE, whereas the adrenal medulla secretes epinephrine in addition to NE. These catecholamines activate adrenergic receptors, which are classic seven-transmembrane receptors coupled to G proteins. They are further classified as either α (α1 and α2) or β receptors (β1, β2, and β3). α-Receptor subtypes have also been identified (α1A, α1B, α1D, α2A, α2B, and α2C), although no subtype-specific antagonists are available. Their physiological effects have been determined in part by the study of receptor knockout models.70,71 In general, α1 is coupled to Gq (stimulation of phospholipase C/D/A2), α2 to Gi (inhibition of adenylate cyclase), and β-receptors to Gs (stimulation of adenylate cyclase).
Distribution of tissue adrenergic receptors is a major determinant of the agonist response because they are relatively nonselective for epinephrine and NE (see Table 6-2). Vascular smooth muscle cells (venous, arterial, and arteriolar) are richly innervated by sympathetic nerve terminals and possess adrenergic receptors (α1, α2, and β2). These receptors can have opposing actions within the vasculature, as demonstrated by α-mediated vasoconstriction and β2-mediated vasodilation, and the vascular response is determined by the relative activation of α1, α2, and β2 receptors. Vascular α1 receptors produce vasoconstriction, whereas presynaptic α2 receptors suppress NE release. Cardiovascular β1 receptors are expressed primarily within the cardiac conduction system and cardiomyocytes, rather than in the vascular bed. However, vascular β1 receptors mediate vasodilation within coronary arteries and stimulate renin secretion in the renal juxtaglomerular apparatus.72 The β3 receptor is primarily expressed on adipocytes, where it stimulates lipolysis; β3 receptors may counteract adrenergic stimulation via β1 receptors in cardiac myocytes and contribute to control of vasodilation by vascular ECs.
Pharmacological Interruption of Catecholamine Metabolism
Catecholamine metabolism is an important target of therapeutic drugs and other chemical agents. Catecholamines are produced locally within the sympathetic neurons by metabolism of tyrosine (Fig. 6-7) to dopamine. Dopamine is concentrated into vesicles via vesicular monoamine transporters. Once in the vesicles, dopamine is converted into NE. Norepinephrine is then secreted and activates adrenergic receptors, provides positive or negative feedback, or is taken back up into the cell via NE transporter (NET). Norepinephrine transporters and similar transporters also transport other neurotransmitters such as epinephrine, dopamine, and serotonin. Norepinephrine is metabolized via monoamine oxidases (MAO-A and MAO-B) after reuptake into the cell, or by catechol-O-methyltransferase (COMT) after diffusion into the circulation.
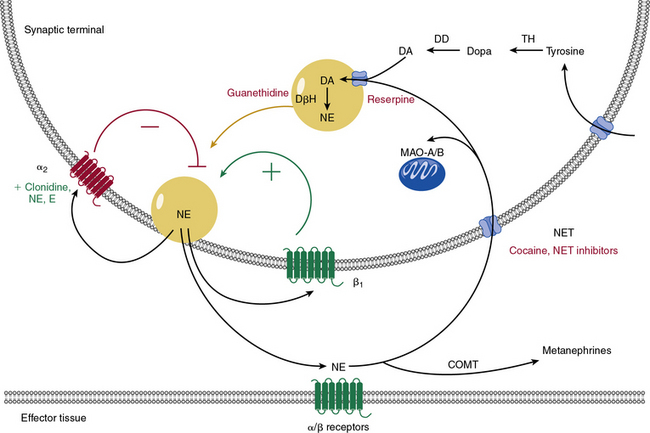
Many weight-loss supplements, decongestant preparations, and herbal supplements act as α1-agonists (direct sympathomimetics) or stimulate release of catecholamines (indirect sympathomimetics).73–75 Whereas epinephrine and NE are rapidly metabolized via COMT, many synthetic sympathomimetic drugs are resistant to this effect, and are therefore effective when ingested by mouth. Ephedra (or ma huang) is a sympathomimetic herbal extract used for asthma treatment, weight loss, and enhanced athletic performance. It can cause severe hypertension, cardiovascular events, and even death in young, apparently healthy individuals. Caffeine coadministration likely exacerbates ephedra-related complications.76 Performance athletes or enthusiastic weight lifters may also take sympathomimetic supplements, which comprise many of the medications banned by the World Anti-Doping Agency.74
Adrenergic Agonists and Antagonists
Vascular α- and β-receptor agonists and antagonists are listed in Table 6-2, and their vascular actions can generally be inferred from the respective receptor actions. Physiological effects of endogenous and synthetic catecholamines are complex because they activate multiple receptors, exhibit dose-dependent responses, and activate compensatory reflexes.
Epinephrine is primarily secreted by the adrenal medulla, where it constitutes roughly 80% of total catecholamine content. Depending on the dose and route of administration, epinephrine may produce divergent vascular responses (Table 6-3). Acute intravenous administration produces marked vasoconstriction, tachycardia, and elevated blood pressure. Continuous infusion or subcutaneous administration of epinephrine increases heart rate and cardiac contractility, systolic blood pressure, and mean arterial blood pressure. Diastolic blood pressure is affected to a lesser extent, resulting in a marked increase in pulse pressure. At lower doses, epinephrine reduces vascular resistance secondary to β2-receptor stimulation and vasodilation, which may reduce blood pressure. Epinephrine is commonly used to treat anaphylactic reactions, bronchoconstriction, and refractory bradycardia and hypotension. Epinephrine is less often used than NE for treatment of septic shock because of tachycardia and concerns for worsened splanchnic ischemia compared to other agents.
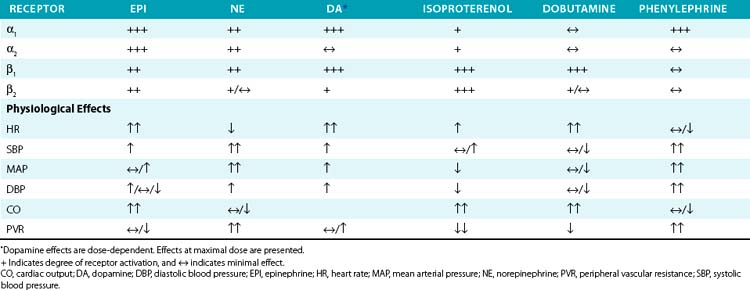
Norepinephrine produces vasoconstriction with lesser direct cardiac effects and β2 activity than epinephrine, which increases both blood pressure and peripheral vascular resistance. Heart rate decreases due to the baroreflex response. Norepinephrine is useful for treating hypotension refractory to fluid resuscitation (e.g., septic shock). Although there is debate regarding the optimal vasopressor in septic shock, NE has proven as effective as comparable agents, possibly with fewer complications.77–81 Norepinephrine appears to produce less splanchnic vasoconstriction and intestinal ischemia than epinephrine or phenylephrine.
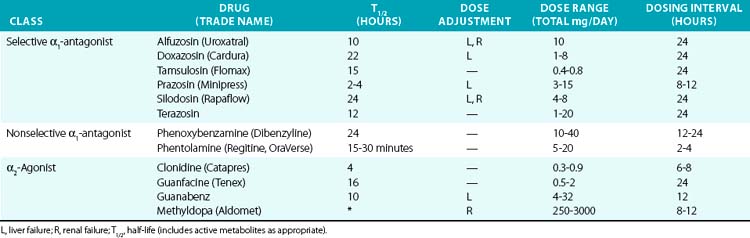
α1-Antagonists
Most clinically available α-antagonists are α1-selective and produce vascular relaxation, vasodilation, and reduction in blood pressure (Table 6-4). These agents are most commonly used for treatment of urinary retention in prostatic hypertrophy because of their inhibitory actions on the prostatic urethra smooth muscle. They are therefore useful for hypertension treatment in patients with concomitant chronic urinary retention. Side effects are nasal congestion, fatigue, and those in common with other vasodilators (peripheral edema, reflex tachycardia, and postural hypotension). The major dose-limiting side effects are postural hypotension and fluid retention. α-Blockers have also been linked to the rare occurrence of “intraoperative floppy iris syndrome,” which may result in permanent vision loss after eye surgery. α-Blockers are not generally recommended as hypertension monotherapy, owing to side effects and increased occurrence of cardiovascular events, compared to the thiazide diuretic chlorthalidone in the Antihypertensive and Lipid-Lowering Treatement to Prevent Heart Attack Trial (ALLHAT) trial.82
α2-Agonists
Activation of the α2 receptor within the CNS provides negative feedback inhibition of sympathetic activity and NE release. Clonidine and other α2-agonists (see Table 6-4) suppress sympathetic and increase parasympathetic activity by actions within the CNS. Evidence for the central effect is obtained from in vivo studies demonstrating no effect of clonidine after spinal cord transection. Clonidine can also produce vasoconstriction via activation of peripheral α2B receptors, although this usually only occurs after intravenous administration or accidental overdose.83 However, this effect may be evident after oral clonidine administration in some patients with autonomic dysfunction.84 Methyldopa is metabolized similarly to NE and acts as a false transmitter and α2-agonist. Methyldopa is commonly used in pregnancy for its history of safety, and also remains an effective alternative in resistant hypertension. Other α2-agonists, such as tizanidine and dexmedetomidine, are used for their sedative effects but may affect blood pressure regulation as a side effect. Etomidate is a sedative with pressor effects that appear to be mediated via α2B.
β-Adrenergic Antagonists
Historically, β-adrenergic antagonists (β-blockers) have been classified by receptor subtype specificity and intrinsic sympathomimetic activity (ISA). In addition, some β-blockers also inhibit α1 receptors, producing a vasodilatory effect. Intrinsic sympathomimetic activity reflects the drug’s ability to activate receptors when administered in the absence of any endogenous sympathetic activity (e.g., sympathetic denervation). This effect likely reflects the relative stabilization of the inactive/active receptor conformations as discussed in the pharmacodynamics section. Drugs with ISA tend to produce less bradycardia and may directly reduce vascular resistance, although evidence that this translates into hard clinical outcomes remains debatable. Analyses suggest that β-blockers with ISA do not reduce cardiovascular mortality and may actually worsen outcomes.85
Drugs with β-blocking ability are summarized in Table 6-5. Propranolol was the first clinically available β-blocker, and is nonselective. Second-generation agents offer increased β1 selectivity. Recently, vasodilatory β-blockers entered the market and produce additional blood pressure lowering effects via α1 blockade and possibly via β3 activation.86 β-Blockers are commonly used to treat hypertension, acute MI, heart failure, angina, and supraventricular arrhythmias. In acute MI, atenolol reduces in-hospital mortality by 15%, although benefit with prolonged therapy is less well established. Perioperative β-adrenergic blockade also reduces in-hospital mortality in patients with high cardiovascular risk.85
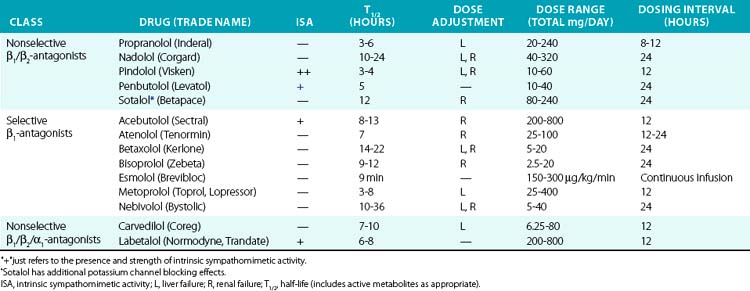
In the past, β-blockers were withheld in patients with systolic heart failure, owing to concerns of worsening contractile function and intolerance. However, the observation that the sympathetic nervous system is activated in severe heart failure and predicted mortality supported the concept of sympathetic blockade in CHF.87 Randomized clinical trials have definitively demonstrated that metoprolol, bisoprolol, and carvedilol improve systolic function and reduce mortality in CHF. These agents should be introduced gradually and titrated upwards as tolerated in patients with severe CHF.
Dopamine and Dopaminergic Agonists
Dopamine is endogenously produced in both peripheral and central neuronal cells and in the adrenal gland via the action of dopa decarboxylase on dopa (see Fig. 6-7). Dopamine acts on one of 5 G protein–linked receptors, termed D1 through D5, which are further classified into two major groups termed D1 and D2. The D1 class of dopamine receptors, D1 and D5, are Gαs-linked receptors that activate adenylyl cyclase; the D2 class receptors are linked to Gαi/o and inhibit adenylyl cyclase. Perturbations of dopamine signaling in the CNS have been linked with a variety of disorders, including Parkinson’s disease, Huntington’s disease, Tourette’s syndrome, schizophrenia, and major depression. In addition to CNS receptors, dopamine receptors are widely present in peripheral tissues including the kidney, gastrointestinal tract, heart, adrenal glands, and vasculature. Dopamine receptor signaling has recently been reviewed in depth.88
The predominant clinical use of dopamine has been for circulatory support in critically ill patients in settings such as shock or the postoperative period. The clinical response to dopamine is complex and depends on the dose. At low doses (1-4 μg/kg/min), often referred to as “renal doses,” dopamine acts on D1-like receptors and β-adrenergic receptors to promote renal arterial vasodilation and improve renal blood flow. As the dose is increased, dopamine begins to exert greater effects at β- and α-adrenergic receptors, and the α-adrenergic effects begin to predominate at doses exceeding 10 μg/kg/min. There is also substantial variability in these responses, such that the precise effect of dopamine in an individual patient is difficult to predict. The potential increase renal blood flow, due to D1-like receptor activation, has not proven to have significant clinical benefit. Recent clinical trials have shown no benefit of dopamine over NE infusion in patients with septic shock, with substantially more cardiac arrhythmias and sinus tachycardia caused by dopamine.89,90
Despite these potentially beneficial effects of fenoldopam, its clinical use in severe hypertension remains limited, largely because several other drugs are quite effective. In prior clinical trials, fenoldopam showed no benefit over sodium nitroprusside in lowering blood pressure,91 and it is considerably more expensive.
Based on its ability to enhance renal perfusion and sodium excretion, fenoldopam has been used as a renal protectant in critically ill patients. A recent meta-analysis of 16 randomized trials involving 1290 patients indicated that fenoldopam reduced the need for renal replacement therapy, in-hospital mortality, and length of stay in the intensive care unit in postoperative or critically ill patients.92 Similar results were obtained from a meta-analysis of patients undergoing cardiovascular surgery.93 Such analyses can be flawed by publication bias, and prospective trials are needed to establish a benefit of fenoldopam in this setting.
There was initial enthusiasm for use of fenoldopam to prevent contrast-induced nephropathy. However, a rigorous randomized prospective trial showed no benefit of this agent in preventing changes in renal function in patients undergoing angiography procedures,94 and its use in this setting is no longer recommended. Dopexamine, which is a combined D1-like and β2-adrenergic agonist, has been studied in a variety of settings involving critically ill patients, but it has not proven beneficial in randomized prospective trials.95,96
Vascular Potassium and Calcium Channels
Direct vasodilators reduce blood pressure by acting on vascular smooth muscle and ultimately impair myosin light chain phosphorylation and contraction (see Fig. 6-1). Minoxidil, for example, activates KATP channels, which hyperpolarizes the cell and prevents calcium entry and contraction.97,98 Channel blocking agents are presented in Table 6-6.
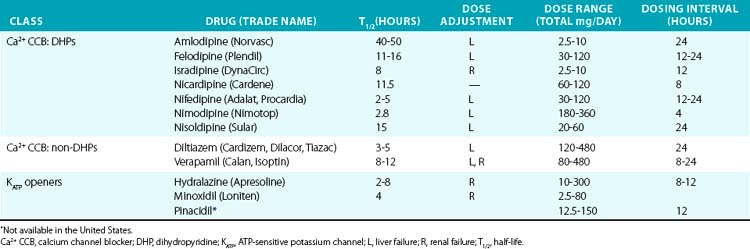
Dihydropyridines produce relatively selective vascular effects in vivo and do not significantly slow cardiac conduction. In some patients, vasodilation may produce reflex tachycardia and vasodilatory edema. This may cause tachycardia and rarely precipitate angina, especially if given acutely. The rapid hypotensive effect of immediate-release nifedipine, particularly when given sublingually, can actually increase cardiovascular events and should be avoided by using only slow-release formulations.99 In contrast, long-acting DHPs have a good safety profile and reduce hypertensive complications.97 Because multiple other agents have proven effectiveness in CHF, and CCBs may worsen cardiac function, they should not be used in this class of patients. Vasodilatory edema during treatment with CCBs is typically refractory to diuretic treatment, but the incidence is reduced by concomitant treatment with an angiotensin-converting enzyme inhibitor (ACEI) or angiotensin receptor blocker (ARB).
Because of their frequent side effects, minoxidil and hydralazine are direct vasodilators typically reserved for refractory hypertension.100 Minoxidil acts on the sulfonylurea receptor-2 (SUR2) component of the KATP channel in VSMCs, and in turn increases K+ flux, hyperpolarizes the cell, and produces vasodilation. Although sulfonylurea drugs (e.g., glibenclamide, glyburide, glipizide) stimulate insulin secretion via opposite effects on SUR1, evidence that they cause vasoconstriction via SUR2 in vivo is lacking. Hydralazine is a direct vasodilator, although the exact mechanism of action is poorly understood. Minoxidil is more effective than hydralazine and can be effective in patients who have not responded to hydralazine. They must be administered with a rate-controlling agent and diuretic to prevent reflex tachycardia and fluid retention, which limit their antihypertensive effectiveness. Minoxidil may worsen LV hypertrophy despite adequate hypertension control, in part because of these compensatory responses. During long-term use, excessive hair growth also occurs and is particularly worrisome to female patients. Hydralazine may also produce a lupus-like syndrome. Both drugs may also produce pericardial or pleural effusions. Reflex sympathetic activation may precipitate cardiac ischemia in some patients. Use of these agents in the setting of acute aortic dissection should be avoided because of reflex sympathetic activation. Hydralazine has been approved for treatment of heart failure in African Americans in combination with a nitrate, as discussed earlier.12
Renin-Angiotensin-Aldosterone System
Regulation of the Renin-Angiotensin-Aldosterone System
The renin-angiotensin-aldosterone system (RAAS) is highly coordinated to maintain blood volume and blood pressure, and is of major importance during sodium and/or fluid depletion (Fig. 6-8). The RAAS is stimulated under pathological conditions that cause reduced renal perfusion, such as heart failure, aortic coarctation, or renal artery stenosis. In addition, this system is inappropriately activated in obesity and diabetes.
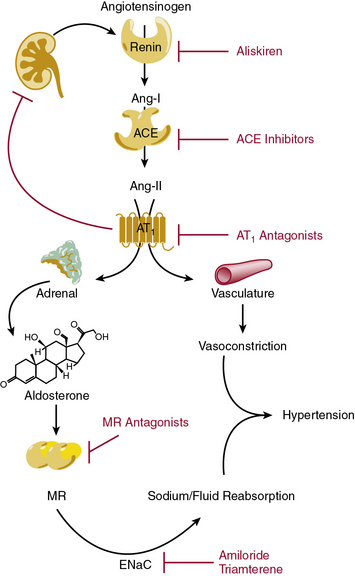
Renin secretion by renal juxtaglomerular cells, the rate-limiting step in the RAAS cascade, is stimulated by reduced sodium chloride delivery to the macula densa, reduced renal perfusion pressure, and sympathetic stimulation.101 Upon release into the circulation, renin cleaves circulating angiotensinogen to angiotensin (Ang-I). Although Ang-I is inactive, it is rapidly converted into Ang-II by angiotensin-converting enzyme (ACE), which is abundantly expressed within the pulmonary vasculature and to a lesser extent in the peripheral circulation. In addition to the endothelial membrane-bound form, ACE also circulates in a soluble form. Angiotensin-II is a potent vasoconstrictor, acting directly on the Ang-II type 1 receptors (AT1) on VSMCs. Within the kidney, Ang-II acts upon the renal afferent and efferent arteriole, to a greater extent on the efferent arteriole. During periods of volume depletion, this efferent selectivity serves to preserve glomerular filtration by increasing intraglomerular pressure. Angiotensin-II also stimulates aldosterone secretion from the adrenal gland. Aldosterone reinforces the vasoconstrictor effect of Angiotensin-II by increasing renal sodium reabsorption and expanding intravascular volume via the mineralocorticoid receptor (MR) in principal cells of the kidney and activation of the epithelial sodium channel (ENaC). Angiotensin-converting enzyme is the principal metabolizing enzyme for a number of other vasoactive peptides, notably bradykinin, which may confer some of the beneficial antihypertensive and antithrombotic effects observed during ACE inhibition.
Receptors and Novel Mediators in RAAS Signaling
The AT1 and AT2 receptors are the principal Ang-II receptors in humans and are widely expressed, including in areas important for blood pressure regulation (vascular smooth muscle, kidney, adrenal cortex, brain). AT1 is a classic seven-transmembrane domain GPCR that signals via Gαq and phospholipase C, as well as other G protein–independent pathways.102,103 Upon Ang-II binding, angiotensin receptor–associated protein (ATRAP) facilitates AT1 internalization and desensitization.104 AT1 mediates the classic Ang-II effects including vasoconstriction, adrenal aldosterone secretion, and renal proximal tubule sodium reabsorption. In addition, Ang-II participates in a negative feedback loop in the kidney to inhibit renin secretion via AT1. In mice, two AT1 receptors have been identified, AT1a and AT1b, with AT1a responsible for most of the pressor and mitogenic effects, although a single AT1 receptor is present in humans.
In general, the actions of the AT2 receptor tend to oppose those of the AT1 receptor, although some effects are inconsistent with this generalization.105,106 AT2 stimulation produces vasodilation, in part via an increase in bradykinin and receptor heterodimerization with the bradykinin receptor. AT2 has an antinatriuretic effect within renal tubules. However, AT2 and AT1 similarly suppress renin secretion. Although investigational agonists and antagonists for the AT2 receptor are available, these agents are not available clinically. Therefore, the clinical implication of the AT2 receptor remains unproven. Angiotensin-II decreases during ACE inhibition but increases during AT1 antagonism. However, the AT2 receptor remains available for Ang-II activation during chronic AT1 antagonism and may promote beneficial effects. This rationale has led some to argue the benefit of AT1 antagonism over ACE inhibition.
Aldosterone and other corticosteroids activate the MR within principal cells in the cortical collecting duct. Angiotensin-II, aldosterone, and MR activation induce multiple proteins that coordinate to increase renal sodium and water reabsorption.107–109 The MR is a classic nuclear receptor localized to the cytosol in its inactive form, which dimerizes and translocates to the nucleus and activates nuclear transcription when activated. Although aldosterone appears to be the critical physiological stimulus, cortisol, corticosterone, and other steroids have a similar affinity for the MR. However, within epithelial target tissues, 11-β-hydroxysteroid dehydrogenase type 2 (11βHSD2) inactivates these hormones and prevents inappropriate MR activation. Either inhibition of this enzyme by licorice or genetic deficiency produces unregulated MR activation and hypertension with metabolic alkalosis and hypokalemia. The MR is also expressed within vascular smooth muscle and ECs, where it may contribute to vascular injury via activation of NADPH oxidase, generation of ROS, and inflammation.
Greater complexity of the RAAS has emerged with the discovery of novel angiotensin peptides and receptors. Angiotensin-(1-7) is formed by cleavage of Ang-I by neprilysin or prolyl-endopeptidase or from cleavage of Ang-II by ACE2.110 Angiotensin-(1-7) acts via the Mas receptor, a G protein–coupled cell-surface receptor generally opposing AT1 effects.111 Angiotensin-(1-7) and ACE2 confer protection against Ang-II-mediated cardiovascular injury, and ACE2-deficient mice have accentuated Ang-II-induced injury.112 Angiotensin-II is also metabolized in vivo by aminopeptidase A to Ang-III and Ang-IV, which may have important physiological effects within the CNS.103
Additional interest has focused on the (pro)-renin receptor (PRR), which binds either renin or prorenin.113 The PRR can exist as a full-length transmembrane protein, a soluble circulating form, or a truncated protein (transmembrane/cytoplasmic portion). The full-length transmembrane PRR can bind and activate prorenin by inducing a conformational change that exposes the catalytic site. In addition, (pro)-renin activates PRR and cellular signaling events (e.g., mitogen-activated protein kinase [MAPK] pathways) independent of renin activity.114 Prorenin circulates in marked excess of active renin, and the prorenin/renin ratio is further increased in diabetes, raising the possibility that (pro)renin-PRR signaling or PRR-induced activation of prorenin and local angiotensin production could contribute to cardiovascular injury.
Drugs That Inhibit the Renin-Angiotensin-Aldosterone System
The first ACE inhibitor was serendipitously discovered as a bradykinin-potentiating factor isolated from venom of the pit viper Bothrops jararaca. Subsequent studies demonstrated its activity against ACE, suggesting that this enzyme played a key role in regulating both the RAAS and the kallikrein-kinin systems. Isolation of the responsible peptide sequences led to development of captopril, one of the earliest examples of structure-based drug design.115 Captopril’s success in treatment of cardiovascular disease was critical to the development of other drugs that block the RAAS (Table 6-7). Drugs are now clinically available to block the RAAS cascade at nearly every level (see Fig. 6-8).
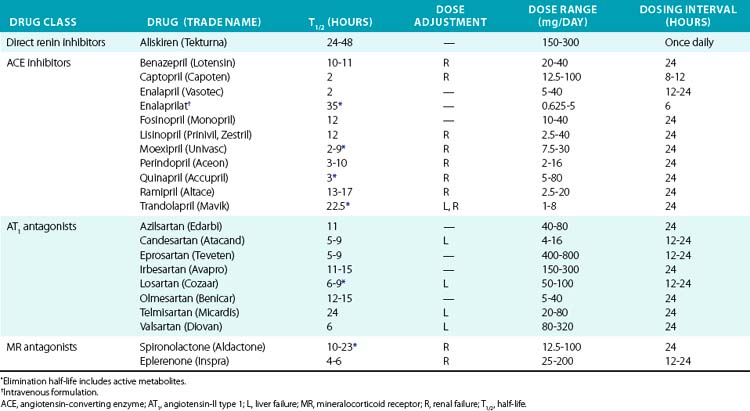
Direct renin inhibitors are the most recent class of RAAS blocking agents. Although renin is the rate-limiting enzyme in the RAAS pathway and a logical drug target, development of clinical renin inhibitors was hindered by poor potency, stability, and oral bioavailability.116 Development of aliskiren overcame these issues, and other agents are in clinical studies. Aliskiren selectively inhibits renin activity and dose-dependently reduces Ang-I and Ang-II production and blood pressure. Renin secretion markedly increases during aliskiren therapy, and attention to the assay method is needed if plasma renin concentration is measured.117 Plasma renin activity (assessed by in vitro Ang-I generation) remains inhibited, and thus compensatory renin secretion does not appear to overcome the effect of aliskiren or increase blood pressure.118 Aliskiren is well tolerated and has a low rate of side effects, which are principally gastrointestinal. Aliskiren effectively reduces blood pressure when used in alone or in combination with diuretic therapy, ACE inhibitors, or ARBs.116,119–121 Addition of aliskiren to maximal-dose losartan reduced proteinuria compared to placebo in a population with diabetic proteinuira.122 Aliskiren provided similar LV mass reduction compared to losartan in a group of overweight subjects with hypertension but provided no additional benefit in combination.123 Further studies are needed to investigate hard cardiovascular endpoints.
Angiotensin-II can also be generated by enzymes other than ACE (e.g., chymase, cathepsin G), providing a rationale for combination therapy with ARBs and ACE inhibitors. Angiotensin-II type 1 receptor antagonists (ARBs) also provide an alternative treatment option for patients who are intolerant of ACE inhibitors. Early studies were done with saralasin, an intravenous peptide Ang-II analog, which demonstrated effectiveness of ARBs and led to the development of orally available agents.124 Since then, multiple agents have been developed and approved for hypertension treatment and prevention of cardiovascular complications (see Table 6-7). Angiotensin receptor blockers are remarkably well tolerated and may even reduce the incidence of some complaints such as headache. All of the ARBs are reliably absorbed, highly protein bound, and selective for the AT1 receptor. Telmisartan and the losartan metabolite EXP3174 can also activate peroxime proliferator-activated receptor gamma (PPARγ), which may explain improvement in insulin sensitivity.125,126 Losartan has a short half-life, but has an active metabolite (EXP3174) with a long half-life. Elimination is primarily hepatic for most ARBs, but dose adjustment is usually needed only in severe liver impairment.
RAAS blockade with any of these drugs (ACE inhibitors, ARBs, or renin inhibitors) is contraindicated during pregnancy because of the risk of congenital renal and other malformations and should be used with extreme caution in women of childbearing potential.127 These agents also carry a risk of hyperkalemia and worsening renal insufficiency.128 RAAS blockade should not be used in the setting of bilateral renal artery stenosis because of the risk of worsening renal failure. Angiotensin-converting enzyme inhibitors rarely cause potentially fatal angioedema, which is more frequent in African Americans. Angiotensin-converting enzyme inhibitors commonly cause a cough, which may be bothersome enough to require cessation, but is a separate pathogenesis than angioedema.129
Spironolactone and eplerenone are MR antagonists whose main effect is mediated by antagonizing MR activity in the distal kidney. These agents also produce systemic vascular effects such as reducing inflammation, improving vascular endothelial function, and promoting fibrinolysis. Mineralocorticoid receptor antagonists reduce mortality in patients with chronic heart failure and after acute MI, and are very effective in the treatment of drug-resistant hypertension.130–133 Spironolactone possesses progesterone-like activity and can cause gynecomastia and/or breast tenderness in 5% to 10% of patients, which resolves upon cessation. Eplerenone does not cause gynecomastia because it is MR specific and therefore provides an alternative for those who are spironolactone intolerant. As opposed to other diuretics, MR antagonists do not require filtration into the urinary space to achieve their effect, but renal insufficiency carries an increased risk of hyperkalemia, and they should be used with caution if at all in this setting. Eplerenone carries additional risk of drug interactions due to moderate CYP3A4 inhibition.
Endothelin Receptor Antagonists
Endothelin-1 is a vasoactive peptide initially described in 1988, and among the most potent vasoconstrictor substances known.134,135 Endothelin-1 is converted by endothelin-converting enzyme from a precursor protein, big ET-1, and is secreted from the cell. Although multiple isoforms exist, ET-1 produces most of the important cardiovascular effects. Endothelin-1 is secreted abluminally (e.g., by ECs toward VSMC) and produces responses that are highly tissue dependent. Although ET-1 is found in the circulation, local paracrine and autocrine actions are more important than endocrine effects. Endothelin-1 acts via ET-1 type A (ETA) and B (ETB) receptors that are widely distributed throughout the body. In vascular smooth muscle, ETA and ETB produce vasoconstriction, whereas endothelial ETB mediates NO-dependent vasodilation. ETB also contributes to clearance of ET-1 by internalization and cellular degradation.
Vascular bed–specific differences exist, with the renal vasculature being exquisitely sensitive to the effects of ET-1. However, the predominant effect of ET-1 within the kidney is to increase natriuresis and free water excretion. Renal ET-1 is produced predominantly in the medulla, which contributes to long-term blood pressure control via ETB in the distal nephron.134 Knockout of ET-1 within the cortical collecting duct results in hypertension and inability to excrete a sodium load, which is improved by ENaC blockade with amiloride.
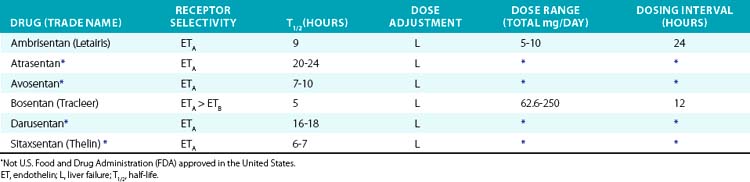
In the pulmonary vasculature, ETA acts as a potent vasoconstrictor and also promotes vascular smooth muscle hypertrophy and proliferation, making the endothelin system a logical pharmacological target. Endothelin receptor antagonists (ETRAs) achieved initial clinical success in patients with PAH. Bosentan was the first ETRA and blocks both ETA and ETB. Ambrisentan is ETA selective. Bosentan and ambrisentan are effective in PH and have demonstrated improved exercise capacity and hemodynamics.136,137 Improved long-term survival has been suggested by observational studies but not in randomized clinical trials.
The role of ETRAs in hypertension, diabetic nephropathy, and heart failure is still developing and has mainly been tested using experimental agents (Table 6-8). Clinical trials in hypertension have demonstrated that the ETA-selective antagonist darusentan is effective in patients with resistant hypertension.138 The ETA-selective antagonist avosentan reduced albuminuria by 40% to 50% in patients with diabetic nephropathy, but also significantly increased the occurrence of fluid overload and clinical heart failure. In heart failure, ETRAs appear to improve hemodynamic endpoints, but in longer-term trials do not improve clinical symptoms or mortality.139,140 Endothelin receptor antagonists commonly produce edema, headache, and a decrease in Hb. Hepatic toxicity (increase in serum transaminase) is the most serious adverse effect and requires close monitoring.
1 Kim H.R., Appel S., Vetterkind S., et al. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12(6A):2165–2180.
2 Watanabe H., Tran Q.K., Takeuchi K., et al. Myosin light-chain kinase regulates endothelial calcium entry and endothelium-dependent vasodilation. FASEB J. 2001;15(2):282–284.
3 Budzyn K., Sobey C.G. Vascular Rho kinases and their potential therapeutic applications. Curr Opin Drug Discov Devel. 2007;10(5):590–596.
4 Bryan N.S., Bian K., Murad F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci. 2009;14:1–18.
5 Friederich J.A., Butterworth J.F. Sodium nitroprusside: twenty years and counting. Anesth Analg. 1995;81(1):152–162.
6 Chen Z., Zhang J., Stamler J.S. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99(12):8306–8311.
7 Li Y., Zhang D., Jin W., et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116(2):506–511.
8 Chihara E., Manyari D.E., Isaac D.L., et al. Comparative effects of nitroglycerin on intestinal vascular capacitance and conductance. Can J Cardiol. 2002;18(2):165–174.
9 ISIS-4: a randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. Lancet. 1995;345(8951):669–685.
10 Six-month effects of early treatment with lisinopril and transdermal glyceryl trinitrate singly and together withdrawn six weeks after acute myocardial infarction: the GISSI-3 trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico. J Am Coll Cardiol. 1996;27(2):337–344.
11 Ambrosio G., Del P.M., Tritto I., et al. Chronic nitrate therapy is associated with different presentation and evolution of acute coronary syndromes: insights from 52,693 patients in the Global Registry of Acute Coronary Events. Eur Heart J. 2010;31(4):430–438.
12 Taylor A.L., Ziesche S., Yancy C., et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351(20):2049–2057.
13 Daiber A., Munzel T., Gori T. Organic nitrates and nitrate tolerance–state of the art and future developments. Adv Pharmacol. 2010;60:177–227.
14 Rhoney D., Peacock W.F. Intravenous therapy for hypertensive emergencies, part 2. Am J Health Syst Pharm. 2009;66(16):1448–1457.
15 Thomas D.D., Miranda K.M., Colton C.A., et al. Heme proteins and nitric oxide (NO): the neglected, eloquent chemistry in NO redox signaling and regulation. Antioxid Redox Signal. 2003;5(3):307–317.
16 Erusalimsky J.D., Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27(12):2524–2531.
17 Lundberg J.O., Weitzberg E., Gladwin M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167.
18 Dejam A., Hunter C.J., Tremonti C., et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116(16):1821–1831.
19 Evgenov O.V., Pacher P., Schmidt P.M., et al. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov. 2006;5(9):755–768.
20 Misono K.S., Philo J.S., Arakawa T., et al. Structure, signaling mechanism and regulation of the natriuretic peptide receptor guanylate cyclase. FEBS J. 2011;278(11):1818–1829.
21 Clerico A., Giannoni A., Vittorini S., et al. Thirty years of the heart as an endocrine organ: Physiological role and clinical utility of cardiac natriuretic hormones. Am J Physiol Heart Circ Physiol. 2011.
22 Palazzuoli A., Gallotta M., Quatrini I., et al. Natriuretic peptides (BNP and NT-proBNP): measurement and relevance in heart failure. Vasc Health Risk Manag. 2010;6:411–418.
23 Colucci W.S., Elkayam U., Horton D.P., et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343(4):246–253.
24 Sackner-Bernstein J.D., Kowalski M., Fox M., et al. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900–1905.
25 Abraham W.T., Trupp R.J., Jarjoura D. Nesiritide in acute decompensated heart failure: a pooled analysis of randomized controlled trials. Clin Cardiol. 2010;33(8):484–489.
26 Hernandez A.F., O’Connor C.M., Starling R.C., et al. Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial (ASCEND-HF). Circulation. 2010;122(21):2217.
27 O’Connor C.M., Starling R.C., Hernandez A.F., et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32–43.
28 Kass D.A., Takimoto E., Nagayama T., et al. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res. 2007;75(2):303–314.
29 Francis S.H., Sekhar K.R., Ke H., et al. Inhibition of cyclic nucleotide phosphodiesterases by methylxanthines and related compounds. Handb Exp Pharmacol. 200. 2011:93–133.
30 Das A., Xi L., Kukreja R.C. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005;280(13):12944–12955.
31 Borlaug B.A., Melenovsky V., Marhin T., et al. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005;112(17):2642–2649.
32 Takimoto E., Champion H.C., Li M., et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11(2):214–222.
33 Badesch D.B., Abman S.H., Simonneau G., et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131(6):1917–1928.
34 Michelakis E., Tymchak W., Lien D., et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105(20):2398–2403.
35 Benedict N., Seybert A., Mathier M.A. Evidence-based pharmacologic management of pulmonary arterial hypertension. Clin Ther. 2007;29(10):2134–2153.
36 Rubin L.J., Badesch D.B., Fleming T.R., et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: SUPER-2. Chest. 2011.
37 Cruz-Blanquel A., Espinosa-Oropeza A., Romo-Hernandez G., et al. Persistent pulmonary hypertension in the newborn: therapeutic effect of sildenafil. Proc West Pharmacol Soc. 2008;51:73–77.
38 Fisher P.W., Salloum F., Das A., et al. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111(13):1601–1610.
39 Nagayama T., Hsu S., Zhang M., et al. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009;53(2):207–215.
40 Guazzi M., Vicenzi M., Arena R., et al. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4(1):8–17.
41 Hao C.M., Breyer M.D. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357–377.
42 Tilley S.L., Coffman T.M., Koller B.H. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108(1):15–23.
43 Rockwell C.E., Raman P., Kaplan B.L., et al. A COX-2 metabolite of the endogenous cannabinoid, 2-arachidonyl glycerol, mediates suppression of IL-2 secretion in activated Jurkat T cells. Biochem Pharmacol. 2008;76(3):353–361.
44 Feletou M., Huang Y., Vanhoutte P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011.
45 Hu Z.W., Kerb R., Shi X.Y., et al. Angiotensin II increases expression of cyclooxygenase-2: implications for the function of vascular smooth muscle cells. J Pharmacol Exp Ther. 2002;303(2):563–573.
46 Tang E.H., Leung F.P., Huang Y., et al. Calcium and reactive oxygen species increase in endothelial cells in response to releasers of endothelium-derived contracting factor. Br J Pharmacol. 2007;151(1):15–23.
47 Bunimov N., Laneuville O. Cyclooxygenase inhibitors: instrumental drugs to understand cardiovascular homeostasis and arterial thrombosis. Cardiovasc Hematol Disord Drug Targets. 2008;8(4):268–277.
48 Baigent C., Blackwell L., Collins R., et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373(9678):1849–1860.
49 Bartolucci A.A., Tendera M., Howard G. Meta-analysis of multiple primary prevention trials of cardiovascular events using aspirin. Am J Cardiol. 2011;107(12):1796–1801.
50 Stavrakis S., Stoner J.A., Azar M., et al. Low-dose aspirin for primary prevention of cardiovascular events in patients with diabetes: a meta-analysis. Am J Med Sci. 2011;341(1):1–9.
51 Simpson S.H., Gamble J.M., Mereu L., et al. Effect of aspirin dose on mortality and cardiovascular events in people with diabetes: A meta-analysis. J Gen Intern Med. 2011.
52 Kronish I.M., Rieckmann N., Shimbo D., et al. Aspirin adherence, aspirin dosage, and C-reactive protein in the first 3 months after acute coronary syndrome. Am J Cardiol. 2010;106(8):1090–1094.
53 Yamamoto Y., Yamashita T., Kitagawa F., et al. The effect of the long term aspirin administration on the progress of atherosclerosis in apoE-/- LDLR-/- double knockout mouse. Thromb Res. 2010;125(3):246–252.
54 Cyrus T., Sung S., Zhao L., et al. Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2002;106(10):1282–1287.
55 Bresalier R.S., Sandler R.S., Quan H., et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102.
56 Mitchell J.A., Warner T.D. COX isoforms in the cardiovascular system: understanding the activities of nonsteroidal antiinflammatory drugs. Nat Rev Drug Discov. 2006;5(1):75–86.
57 Antman E.M., DeMets D., Loscalzo J. Cyclooxygenase inhibition and cardiovascular risk. Circulation. 2005;112(5):759–770.
58 Barst R.J., Rubin L.J., Long W.A., et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296–302.
59 Badesch D.B., Tapson V.F., McGoon M.D., et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132(6):425–434.
60 Chen Y.F., Jowett S., Barton P., et al. Clinical and cost-effectiveness of epoprostenol, iloprost, bosentan, sitaxentan and sildenafil for pulmonary arterial hypertension within their licensed indications: a systematic review and economic evaluation. Health Technol Assess. 2009;13(49):1–320.
61 Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med. 2008;358(6):615–624.
62 Sequeira I.M., Haberberger R.V., Kummer W. Atrial and ventricular rat coronary arteries are differently supplied by noradrenergic, cholinergic and nitrergic, but not sensory nerve fibres. Ann Anat. 2005;187(4):345–355.
63 Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355(19):2003–2011.
64 Schachinger V., Britten M.B., Zeiher A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899–1906.
65 Perticone F., Ceravolo R., Pujia A., et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104(2):191–196.
66 Pretorius M., Rosenbaum D.A., Lefebvre J., et al. Smoking impairs bradykinin-stimulated t-PA release. Hypertension. 2002;39(3):767–771.
67 Pretorius M., Rosenbaum D., Vaughan D.E., et al. Angiotensin-converting enzyme inhibition increases human vascular tissue-type plasminogen activator release through endogenous bradykinin. Circulation. 2003;107(4):579–585.
68 DeSouza C.A., Shapiro L.F., Clevenger C.M., et al. Regular aerobic exercise prevents and restores age-related declines in endothelium-dependent vasodilation in healthy men. Circulation. 2000;102(12):1351–1357.
69 van Zwieten P.A., Doods H.N. Muscarinic receptors and drugs in cardiovascular medicine. Cardiovasc Drugs Ther. 1995;9(1):159–167.
70 Brede M., Philipp M., Knaus A., et al. Alpha2-adrenergic receptor subtypes–novel functions uncovered in gene-targeted mouse models. Biol Cell. 2004;96(5):343–348.
71 Civantos C.B., Aleixandre de A.A. Alpha-adrenoceptor subtypes. Pharmacol Res. 2001;44(3):195–208.
72 Gao F., de Beer V.J., Hoekstra M., et al. Both beta1- and beta2-adrenoceptors contribute to feedforward coronary resistance vessel dilation during exercise. Am J Physiol Heart Circ Physiol. 2010;298(3):H921–H929.
73 Davis E., Loiacono R., Summers R.J. The rush to adrenaline: drugs in sport acting on the beta-adrenergic system. Br J Pharmacol. 2008;154(3):584–597.
74 Docherty J.R. Pharmacology of stimulants prohibited by the World Anti-Doping Agency (WADA). Br J Pharmacol. 2008;154(3):606–622.
75 Grossman E., Messerli F.H. Secondary hypertension: interfering substances. J Clin Hypertens (Greenwich). 2008;10(7):556–566.
76 Haller C.A., Benowitz N.L. Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med. 2000;343(25):1833–1838.
77 Morelli A., Ertmer C., Rehberg S., et al. Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care. 2008;12(6):R143.
78 De B.D., Creteur J., Silva E., et al. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med. 2003;31(6):1659–1667.
79 Nygren A., Thoren A., Ricksten S.E. Vasopressors and intestinal mucosal perfusion after cardiac surgery: norepinephrine vs. phenylephrine. Crit Care Med. 2006;34(3):722–729.
80 Levy B., Perez P., Perny J., et al. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med. 2011;39(3):450–455.
81 Russell J.A., Walley K.R., Singer J., et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358(9):877–887.
82 Furberg C.D., Wright J.T., Davis B.R. Major cardiovascular events in hypertensive patients randomized to doxazosin vs. chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA. 2000;283(15):1967–1975.
83 Frye C.B., Vance M.A. Hypertensive crisis and myocardial infarction following massive clonidine overdose. Ann Pharmacother. 2000;34(5):611–615.
84 Robertson D., Goldberg M.R., Hollister A.S., et al. Clonidine raises blood pressure in severe idiopathic orthostatic hypotension. Am J Med. 1983;74(2):193–200.
85 Cruickshank J.M. Are we misunderstanding beta-blockers. Int J Cardiol. 2007;120(1):10–27.
86 Dessy C., Balligand J.L. Beta3-adrenergic receptors in cardiac and vascular tissues emerging concepts and therapeutic perspectives. Adv Pharmacol. 2010;59:135–163.
87 Schrier R.W., Abraham W.T. Hormones and hemodynamics in heart failure. N Engl J Med. 1999;341(8):577–585.
88 Beaulieu J.M., Gainetdinov R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217.
89 De B.D., Biston P., Devriendt J., et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362(9):779–789.
90 Patel G.P., Grahe J.S., Sperry M., et al. Efficacy and safety of dopamine versus norepinephrine in the management of septic shock. Shock. 2010;33(4):375–380.
91 Devlin J.W., Seta M.L., Kanji S., et al. Fenoldopam versus nitroprusside for the treatment of hypertensive emergency. Ann Pharmacother. 2004;38(5):755–759.
92 Landoni G., Biondi-Zoccai G.G., Tumlin J.A., et al. Beneficial impact of fenoldopam in critically ill patients with or at risk for acute renal failure: a meta-analysis of randomized clinical trials. Am J Kidney Dis. 2007;49(1):56–68.
93 Landoni G., Biondi-Zoccai G.G., Marino G., et al. Fenoldopam reduces the need for renal replacement therapy and in-hospital death in cardiovascular surgery: a meta-analysis. J Cardiothorac Vasc Anesth. 2008;22(1):27–33.
94 Stone G.W., McCullough P.A., Tumlin J.A., et al. Fenoldopam mesylate for the prevention of contrast-induced nephropathy: a randomized controlled trial. JAMA. 2003;290(17):2284–2291.
95 Gopal S., Jayakumar D., Nelson P.N. Meta-analysis on the effect of dopexamine on in-hospital mortality. Anaesthesia. 2009;64(6):589–594.
96 Davies S.J., Yates D., Wilson R.J. Dopexamine has no additional benefit in high-risk patients receiving goal-directed fluid therapy undergoing major abdominal surgery. Anesth Analg. 2011;112(1):130–138.
97 Basile J. The role of existing and newer calcium channel blockers in the treatment of hypertension. J Clin Hypertens (Greenwich). 2004;6(11):621–629.
98 Abernethy D.R., Schwartz J.B. Calcium-antagonist drugs. N Engl J Med. 1999;341(19):1447–1457.
99 Grossman E., Messerli F.H., Grodzicki T., et al. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328–1331.
100 Sica D.A. Minoxidil: an underused vasodilator for resistant or severe hypertension. J Clin Hypertens (Greenwich). 2004;6(5):283–287.
101 Peti-Peterdi J., Harris R.C. Macula densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol. 2010;21(7):1093–1096.
102 Crackower M.A., Sarao R., Oudit G.Y., et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828.
103 Oliveira L., Costa-Neto C.M., Nakaie C.R., et al. The angiotensin II AT1 receptor structure-activity correlations in the light of rhodopsin structure. Physiol Rev. 2007;87(2):565–592.
104 Oppermann M., Gess B., Schweda F., et al. Atrap deficiency increases arterial blood pressure and plasma volume. J Am Soc Nephrol. 2010;21(3):468–477.
105 Carey R.M., Padia S.H. Angiotensin AT2 receptors: control of renal sodium excretion and blood pressure. Trends Endocrinol Metab. 2008;19(3):84–87.
106 Porrello E.R., Delbridge L.M., Thomas W.G. The angiotensin II type 2 (AT2) receptor: an enigmatic seven transmembrane receptor. Front Biosci. 2009;14:958–972.
107 Butterworth M.B., Edinger R.S., Frizzell R.A., et al. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol. 2009;296(1):F10–F24.
108 Hoorn E.J., Nelson J.H., McCormick J.A., et al. The WNK Kinase Network Regulating Sodium, Potassium, and Blood Pressure. J Am Soc Nephrol. 2011;22(4):605–614.
109 Vallon V., Schroth J., Lang F., et al. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol. 2009;297(3):F704–F712.
110 Ferrario C.M., Varagic J. The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol. 2010;298(6):F1297–F1305.
111 Alenina N., Xu P., Rentzsch B., et al. Genetically altered animal models for Mas and angiotensin-(1-7). Exp Physiol. 2008;93(5):528–537.
112 Zhong J., Basu R., Guo D., et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122(7):717–728. 18
113 Nguyen G. Renin, (pro)renin and receptor: an update. Clin Sci (Lond). 2011;120(5):169–178.
114 Feldt S., Batenburg W.W., Mazak I., et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51(3):682–688.
115 Cushman D.W., Ondetti M.A. History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension. 1991;17(4):589–592.
116 Jensen C., Herold P., Brunner H.R. Aliskiren: the first renin inhibitor for clinical treatment. Nat Rev Drug Discov. 2008;7(5):399–410.
117 Campbell D.J. Interpretation of plasma renin concentration in patients receiving aliskiren therapy. Hypertension. 2008;51(1):15–18.
118 Stanton A.V., Gradman A.H., Schmieder R.E., et al. Aliskiren monotherapy does not cause paradoxical blood pressure rises: meta-analysis of data from 8 clinical trials. Hypertension. 2010;55(1):54–60.
119 Oparil S., Yarows S.A., Patel S., et al. Efficacy and safety of combined use of aliskiren and valsartan in patients with hypertension: a randomised, double-blind trial. Lancet. 2007;370(9583):221–229.
120 Nussberger J., Wuerzner G., Jensen C., et al. Angiotensin II suppression in humans by the orally active renin inhibitor aliskiren (SPP100): comparison with enalapril. Hypertension. 2002;39(1):E1–E8.
121 Stanton A., Jensen C., Nussberger J., et al. Blood pressure lowering in essential hypertension with an oral renin inhibitor, aliskiren. Hypertension. 2003;42(6):1137–1143.
122 Parving H.H., Persson F., Lewis J.B., et al. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358(23):2433–2446.
123 Solomon S.D., Appelbaum E., Manning W.J., et al. Effect of the direct renin inhibitor aliskiren, the angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation. 2009;119(4):530–537.
124 Burnier M. Angiotensin II type 1 receptor blockers. Circulation. 2001;103(6):904–912.
125 Benson S.C., Pershadsingh H.A., Ho C.I., et al. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43(5):993–1002.
126 Kappert K., Tsuprykov O., Kaufmann J., et al. Chronic treatment with losartan results in sufficient serum levels of the metabolite EXP3179 for PPARgamma activation. Hypertension. 2009;54(4):738–743.
127 Cooper W.O., Hernandez-Diaz S., Arbogast P.G., et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354(23):2443–2451.
128 Palmer B.F. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med. 2004;351(6):585–592.
129 Sica D.A. Angiotensin-converting enzyme inhibitors side effects–physiologic and non-physiologic considerations. J Clin Hypertens (Greenwich). 2004;6(7):410–416.
130 Zannad F., McMurray J.J., Krum H., et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364(1):11–21.
131 Pitt B., Remme W., Zannad F., et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309–1321.
132 Pitt B., Zannad F., Remme W.J., et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341(10):709–717.
133 Vaclavik J., Sedlak R., Plachy M., et al. Addition of Spironolactone in Patients With Resistant Arterial Hypertension (ASPIRANT): a randomized, double-blind, placebo-controlled trial. Hypertension. 2011;57(6):1069–1075.
134 Kohan D.E., Rossi N.F., Inscho E.W., et al. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91(1):1–77.
135 Dhaun N., Goddard J., Kohan D.E., et al. Role of endothelin-1 in clinical hypertension: 20 years on. Hypertension. 2008;52(3):452–459.
136 Channick R.N., Simonneau G., Sitbon O., et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358(9288):1119–1123.
137 Anderson J.R., Nawarskas J.J. Pharmacotherapeutic management of pulmonary arterial hypertension. Cardiol Rev. 2010;18(3):148–162.
138 Weber M.A., Black H., Bakris G., et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9699):1423–1431.
139 Anand I., McMurray J., Cohn J.N., et al. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364(9431):347–354.
140 McMurray J.J., Teerlink J.R., Cotter G., et al. Effects of tezosentan on symptoms and clinical outcomes in patients with acute heart failure: the VERITAS randomized controlled trials. JAMA. 2007;298(17):2009–2019.