Ultraviolet radiation carcinogenesis
James E. Cleaver, PhD  Susana Ortiz-Urda, MD, PhD, MBA
Susana Ortiz-Urda, MD, PhD, MBA  Radhika Gulhar, BA
Radhika Gulhar, BA  Sarah T. Arron, MD, PhD
Sarah T. Arron, MD, PhD  Lionel Brookes, 3rd PhD
Lionel Brookes, 3rd PhD  David L. Mitchell, PhD
David L. Mitchell, PhD
Overview
Skin carcinogenesis from solar ultraviolet (UV) exposure is initiated by UV photoproducts formed in the DNA of precursor cells that give rise to squamous cell carcinomas, basal cell carcinomas, and melanomas. These photoproducts consist of dimers between adjacent pyrimidines that are repaired by nucleotide excision repair (NER). Two branches of NER differ in mechanisms of damage recognition: in global genome repair (GGR), damage is recognized in nontranscribed DNA by the DDB2 and XPC proteins; in transcription-coupled repair (TCR), damage is recognized by arrest of the transcribing RNA polymerase at damaged sites. The T = C photoproduct is the predominant mutagenic product which, if unrepaired, gives rise to C to T or CC to TT mutations through replication by the low-fidelity polymerase Pol H. TCR reduces the mutation frequency in regions of transcription, in comparison to the rest of the genome subject to GGR. Deficiencies in GGR in the human disease xeroderma pigmentosum cause earlier onset and increased rates of skin cancer, which often contain UV-type mutations in genes that are rarely mutated in the general population. Deficiencies in TCR in the human disease Cockayne syndrome are associated with photosensitivity, developmental, and neurological disorders, but no cancer has been reported.
Epidemiology of skin cancer
Skin cancer frequency and age of onset
NMSCs (nonmelanoma skin cancers) are the most common cancers that occur in the United States each year,1, 2 comprising 30–40% of all cancers and have been increasing steadily for a century.3, 4 Skin cancer risks are associated with geographical location, skin type, various photosensitizing, enhancing, and protective applications and vitamin D.4–10 There is also a possibility of greater risk when the exposure is received during childhood and adolescence.5, 11 NMSC is, therefore, one of the few human malignancies for which there is clear evidence for the initiating agent. The relationship of melanoma to sun exposure and the possible action spectrum are less clear,12, 13 but may be related to acute burns rather than accumulated dose.5, 14
The importance of DNA as a target for ultraviolet radiation (UVR) is highlighted by the autosomal recessive disease XP (xeroderma pigmentosum). In this disease, a failure of DNA repair causes a large increase in NMSC and melanoma.12, 13 Median age for first diagnosis of NMSC in the general US population occurs after 60 years of age; in XP patients, carcinogenesis is accelerated and median age at first diagnosis is within the first decade (Figure 1).12, 13 Melanoma is also increased in XP patients, but the acceleration is less. Consequently, in XP patients, the onset of NMSC precedes melanoma, whereas in non-XP patients, melanomas generally occur earlier than NMSC.13
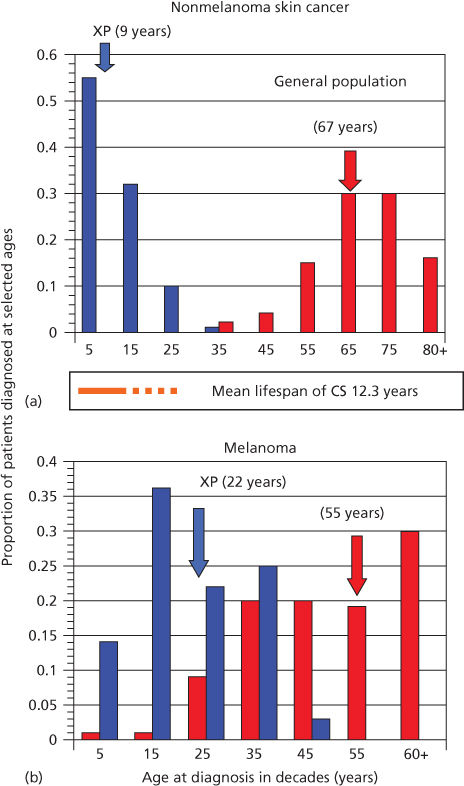
Figure 1 Diagnosis of first cancer by age and type in XP patients compared with the US general population. (a) Proportion of NMSC patients diagnosed at selected ages. (b) Proportion of melanoma patients diagnosed at selected ages. Individuals with both NMSC and melanoma were used in both panels. Ages shown are at the midpoint of each decade. XP: blue bars; general population: red bars. Colored arrows designate median ages of diagnosis for each population. The age span of Cockayne syndrome patients is also shown (orange): solid bar ends at mean age at death (12.25 years), with dashed line extending to maximum reported ages. XP and general population data redrawn from.27 Cockayne data drawn from.29
Sunlight spectrum and wavelengths responsible for skin cancer
UVR is divided into three wavelength ranges: UVA, UVB, and UVC. UVA (320–400 nm) is photocarcinogenic and involved in photoaging, but it is weakly absorbed in DNA and protein and may involve reactive oxygen species, which secondarily cause damage to DNA.15–18 Recent evidence, however, suggests that UVA may also induce DNA damage directly in human cells.19 UVB (290–320 nm) overlaps the upper end of the DNA and protein absorption spectra and is the range mainly responsible for skin cancer through direct photochemical damage to DNA. UVC (240–290 nm) is not present in ambient sunlight but is readily produced by low-pressure mercury sterilizing lamps (254 nm). UVC coincides with the peak of DNA absorption (260 nm) and is extensively used in experimental studies. Absorption of UVR by stratospheric ozone greatly attenuates these wavelengths, so that negligible radiation shorter than 300 nm reaches the earth’s surface. Hence, although UVA and UVB light constitute a minute portion of the emitted solar wavelengths (10−9), they are primarily responsible for the sun’s pathological effects.
Sunlight-induced photoproducts in DNA
The absorption spectrum of DNA correlates well with photoproduct formation, lethality, and mutation.20–24 The absorbed energy produces molecular changes, which involve single bases, interactions between adjacent and nonadjacent bases, and between DNA and proteins. The relative proportion of DNA photoproducts varies across the UV (ultraviolet) spectrum.
Dimerizations between adjacent pyrimidines are the most prevalent DNA photoproducts. Two major photoproducts are the cyclobutane pyrimidine dimer (CPD) and, at about 25% the frequency, the [6-4] pyrimidine dimer ([6-4]PD). [6-4]PDs cause a 47° helical bend in DNA, compared with 7° for CPDs. The [6-4]PD can further undergo a UVB-dependent conversion to its valence photoisomer, the Dewar pyrimidinone.25
The distribution of photoproducts in chromatin depends on base sequence, secondary DNA structure, and DNA–protein interactions.24, 26, 27 Because cytosine more efficiently absorbs longer wavelengths of UVR than thymine, CPDs containing this base are formed more readily after UVB irradiation.28 Cytosine CPDs and [6-4]PDs, which are preferentially induced at thymine–cytosine dipyrimidines, may play a major role in UVB (solar) mutagenesis.29 Cytosine methylation increases the formation of CPDs by UVB, but not UVC, by 1.7-fold.30 Methylation at PyrCG sequences in the p53 gene enhances formation of CPDs and [6-4]PDs at sites that are hotspots for mutations.31, 32 Other less common lesions include purine–purine and purine–pyrimidine photoadducts, photohydrates, and photooxidations.33 The total yield of these photoproducts is only 3–4% of the yield of CPDs, but a minor biological role as premutagenic lesions at specific sites cannot be excluded.
Genetic factors in skin carcinogenesis
Recognition of UV photoproducts in DNA
The repair of UV photoproducts in DNA [nucleotide excision repair (NER)] involves sequential steps of photoproduct recognition, assembly of the excision complex, displacement of the excised fragment, and polymerization of the replacement patch.34 The importance of NER arose from the discovery that cells from patients suffering from XP were deficient in NER.35, 36 Two major pathways of NER are known (Figure 2): transcription-coupled repair (TCR) and global genome repair (GGR), which differ in their mechanisms of damage recognition.34, 37–39 TCR removes damage more rapidly from the transcribed strands of transcriptionally active genes, whereas GGR acts more slowly on nontranscribed regions.40, 41 The half-life of [6-4]PDs is 2–6 h, whereas for CPDs, it is 12–24 h, and much longer in rodent cells.42, 43
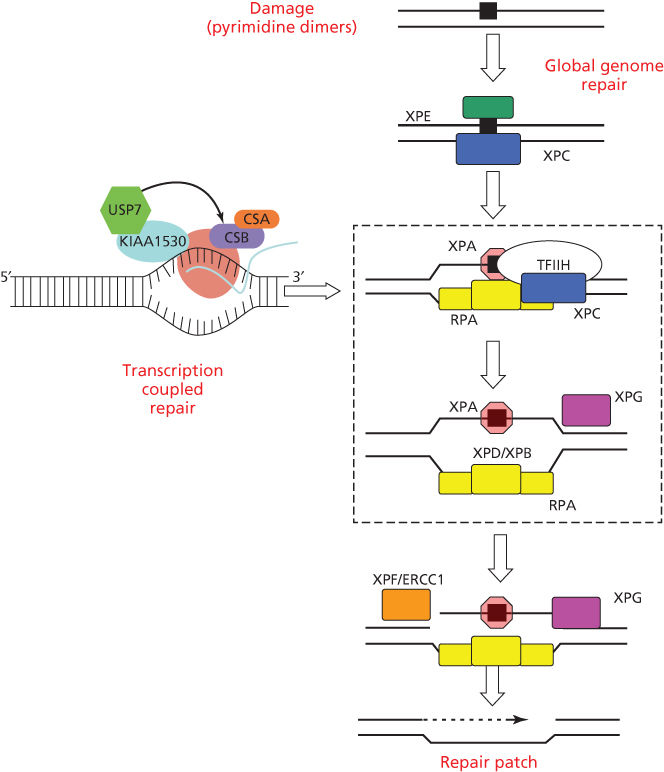
Figure 2 Biochemical steps for nucleotide excision repair of CPDs and [6-4]PDs by global genome repair or transcription-coupled repair. Initial recognition of damage by GGR involves the DDB2 and XPC gene products; initial recognition by TCR involves RNA polymerase II arrest and disengagement of the transcription complex from damage by CSA, CSB, and UVSSA. Subsequent steps involve XPA and RPA that bind to photoproducts and download the helicases XPB and XPD for local unwinding. Excision occurs when UV-specific endonucleases (XPF/ERCC1 and XPG) make incisions on the 5′ and 3′ sides. Excision and subsequent polymerization releases a 29 base oligonucleotide containing the CPD and activates the cryptic XPG endonuclease for the final cut.
GGR involves initial binding by the DDB1/DDB2(XPE) complex,44, 45 followed by recruitment of XPC/HR23B/centrin via ubiquitylation by the E3 ligase activity of DDB1/DDB2.46–49 The XPC protein binds to the undamaged strand opposite a PD, inserting a peptide chain within the helix to displace the dimer to an extra-helical position.50, 51
TCR is initiated by the arrest of RNA polymerase II at a damaged base, after which specific TCR factors CSA, CSB, UVSSA, and XABP, a binding partner of XPA, facilitate removal or degradation of arrested RNA polymerase II to permit access of NER proteins to damaged sites.52, 53 TCR is closely involved with the chromatin remodeling function of CSB that is a member of the SWI2/SNF2 chromatin remodeling protein family.54, 55 RNA polymerase II elongation is associated with cycles of ubiquitylation and deubiquitylation of histone H2B (H2Bub); arrest by UV damage tips the balance toward deubiquitylation and acetylation of histone H3.56 Transcription is restored along with H2Bub after excision of the arresting photoproducts.56
The mechanism of nucleotide excision repair
Subsequent to damage recognition, the open helix is stabilized by XPA/RPA, which binds with a higher affinity for the [6-4]PDs than CPDs and interacts with the unwinding activity of the 10 component transcription factor TFIIH, and the 3′–5′ (XPB) and 5′–3′ (XPD) nucleases.57, 58 The opened helix is cleaved first by the ERCC1/XPF nuclease presenting a 3′OH terminus for chain elongation by the DNA Pol D, proliferating cell nuclear antigen, and single-strand binding protein.59, 60 XPG is a cryptic nuclease that cuts 3′ to the lesion after the ERCC1/XPF nuclease has cut 5′ to the CPD and polymerization.60 A 27–29nt oligonucleotide containing the photoproduct is subsequently released and the patch is sealed by ligase I.61
Mutagenicity of UV photoproducts and low-fidelity DNA polymerases
Most photoproducts act as blocks to the replicative class B polymerases, Pol A, D, and E, but can be bypassed by damage-specific class Y DNA polymerases.62–64 The class Y polymerases, Pol H, I, and K, have low fidelity due to expanded active sites, which allow the polymerases to read-through noninformative sequence information.65 Pol H has the greatest capacity to replicate a large variety of DNA lesions,66 and preferentially inserts adenine in the nascent strand opposite the lesion (called the “A rule”).67 Hence, Pol H replicates a thymine-containing CPD faithfully.68–71 Pol I preferentially inserts guanines and is capable of replicating C-containing photoproducts.72, 73 Pol H or I, therefore, can insert bases opposite dipyrimidine photoproducts, but the 3′ complementary base can be mismatched by an erroneous insertion or the distortion caused by the photoproduct. Pol K or Pol Z can complete the replicative bypass by extension from the mismatched 3′ terminus.74–76 The absence of Pol H results in increased mutagenesis in the XPV group,77, 78 but the absence of Pol Z has the converse effect of reducing mutation rates.79
The replication by-pass mechanism has two important implications for UV mutagenicity. First, mutations will most often occur where cytosine is a component of the photoproduct as insertion of adenine opposite thymine is a correct and nonmutagenic event. Hence, CPDs that form between two thymine bases are not mutagenic. Second, the larger distortion of [6-4]PDs is more likely to be lethal than mutagenic.
Diseases of DNA repair
Xeroderma pigmentosum
XP is a rare autosomal recessive disease that occurs at a frequency of about 1 : 250,000 in the United States.12 Affected patients (homozygotes) have sun sensitivity resulting in progressive degenerative changes of sun-exposed portions of the skin and eyes, often leading to neoplasia. Some XP patients also have progressive neurologic degeneration. Obligate heterozygotes (parents) are asymptomatic. The median age of onset of symptoms is 1–2 years of age and the median age of first diagnosis of NMSC is 9 years of age (Figure 1).12, 13 The skin rapidly takes on the appearance of that seen in individuals with many years of sun exposure. Pigmentation is patchy, and skin shows atrophy and telangiectasia with development of NMSC and melanoma. The frequency of NMSC is about 2000 times that seen in the general population <20 years of age, with an approximate 30-year reduction in lifespan. Some patients develop myelodysplasia and leukemia in later life.
Among patients who are deficient in NER, there are eight complementation groups that correspond to components of NER, XPA through G and the exceedingly rare ERCC1.80 An additional group, the XP variant, has mutations in the low-fidelity polymerase Pol H (Table 1). UV-damaged plasmids passaged through XP cells show increased mutagenesis due to defective repair of CPDs and nondimer photoproducts.81 Carcinogenesis from UV damage in XP patients arises, therefore, from the loss of either NER capacity or Pol H; both lead to an increase in the amount of persistent genetic damage (mutations, gene rearrangements, deletions, and genomic instability).
Table 1 Genes involved in repair of UV damage in humans.d
| Group (gene) | Chromosome location | Central nervous system and developmental disorders | Relative DNA repair (%)a |
| Xeroderma pigmentosum | |||
| A | 9q34.1 | Yes | 2–5 |
| Bb | 2q21 | Yes | 3–7 |
| C | 3q25 | No | 5–20 |
| Db | 19q13.2 | Yes | 25–50 |
| E | 11p11-12 | No | 50 |
| F | 16q13.1 | No | 18 |
| G | 13q32.3 | Yes | <2 |
| V (pol H) | 6p21 | No | 100 |
| ERCC1c | 19q13.32 | Yes | 15 |
| Cockayne syndrome | |||
| A (ERCC8) | 5q12.1 | Yes | 100a |
| B (ERCC6) | 10q11.23 | Yes | 100a |
| UVSSA | 4p16.3 | No | 100a |
a Measurement of relative DNA repair represent mainly global genome repair. Cockayne syndrome is defective in transcription-coupled repair and has normal levels of global genome repair.
b Patients also exhibit symptoms commonly associated with Cockayne syndrome: dwarfism, cutaneous features, and mental retardation. Group B is designated ERCC3 and Group D is designated ERCC2, F is designated ERCC4, and G is designated ERCC5. Some patients also have symptoms of trichothiodystrophy.
c ERCC1 is rare and causes a neonatal fatal disorder, cerebro-oculo-facio-skeletal syndrome.
d Further details can be found in the web sites http://www.photobiology.info/ and http://www.cgal.icnet.uk/DNA_Repair_Genes.html.
Cockayne syndrome
CS (Cockayne syndrome) is an autosomal recessive disease characterized by photosensitivity, cachectic dwarfism, retinal abnormalities, microcephaly, deafness, neural defects, and retardation of growth and development after birth.82, 83 A major symptom is cerebellar degeneration and Purkinje cell loss causing difficulties in walking and balance.84 Solar carcinogenesis is not seen in patients with CS, setting this disease apart from XP, even though their lifespan encompasses ages at which skin cancers are seen in XP (Figure 1).82 Mutations in either of two genes, CSA and CSB, are associated with CS.83, 85 Similar symptoms occur in XP groups B, D, and G.86 The CS proteins facilitate removal of RNA polymerase II from damaged sites permitting access for the NER machinery (Figure 2). An additional gene product UVSSA also contributes to TCR, but mutations in the UVSSA gene only give rise to mild photosensitivity.87, 88
CSA and CSB proteins also contribute to the redox balance of cells by interaction with oxidative phosphorylation in the mitochondria and mitophagy.89 Conceivably, their mitochondrial role may be more important in the developmental and neurological pathology of CS than their nuclear TCR function.90
Trichothiodystrophy
TTD (trichothiodystrophy) is a rare autosomal recessive disorder characterized by sulfur-deficient brittle hair that splits longitudinally into small fibers and ichthyosis. The levels of cysteine/cystine in hair proteins are 15–50% of those in normal individuals. Patients show physical and mental retardation of varying severity and often have an unusual facial appearance, with protruding ears and a receding chin. Mental abilities range from low normal to severe retardation.91 Several categories of the disease can be recognized on the basis of UV sensitivity and DNA repair deficiences.92, 93 The most severe cases with repair deficiencies have mutations in XPB and XPD.92 Several other TTD genes are known that are not involved in DNA repair.
Mutations in XPD that give rise to XP symptoms correspond to mis-sense mutations in the DNA helicase motifs, whereas those that give rise to TTD are mis-sense mutations in the RNA helicase motifs and the C-terminal end of the protein.94 Some TTD cases do not have mutations in XPB or XPD and are due to mutations in a small 8-kDa component of TFIIH that appears to regulate the overall level of the transcription factor.95
Carcinogenesis
Mutations and skin cancer types
Loeb was the first to recognize the high frequency of mutations in cancer cells and invoked the concept of a mutator phenotype.96 In triple-negative breast cancer, for example, the mutation frequency is 13.3 times that of the normal rate of 0.6 mutations per cell division.97 NMSC and melanoma have among the highest mutation frequencies, most of which are C to T or CC to TT transitions resulting from sun exposure.98, 99 The frequency of mutations in normal and cancer genomes is reduced in transcribed regions, compared with the rest of the genome, and shows the strand bias of TCR.97, 98, 100 Patients with reduced TCR, that is, CS patients, should therefore have higher frequencies of mutations in transcribed regions of the genome and hence more cancers. But they do not, unlike Cs-a or Cs-b mice that do develop skin cancer from carcinogen exposure.101 Mutant cells in CS must, therefore, be prevented or delayed from progressing to cancers.
Nonmelanoma skin cancer
SCCs and BCCs both have UV-type mutations in genes that drive development of these tumors. Actinic keratoses, precursors to SCCs, share many mutational spectrum and global gene expression profiles.102–104 The initiating molecular changes in SCC are UV-type mutations in p53 that result in expanding clones in the sun-exposed areas of the skin that are initially confined within the proliferating units.105 In normal skin, 50% of SCCs have p53 mutations; in XP patients, the frequency is 90%.106–110 Loss of p53 function causes genomic instability during DNA replication when subjected to further UV irradiation.111–113 Many of these are likely passenger mutations that do not confer a growth advantage; mutated p53 being the more potent driver of SCC.
SCC mutations have been identified in Notch, KNSTRN, a kinetochore gene, EGF, RAS, NFkb, JNK2, and MMP9.114–118 Activating mutations in H-RAS and N-RAS oncogenes at codon 61 have been associated with solar UV exposure even though they involve transversions at a TT site.119–122
BCCs and BCNS involve the Sonic Hedgehog pathway in which a transmembrane receptor, Patched (PTC), and the membrane protein, Smoothened (SMO), regulate signal transduction by the extracellular protein Hedgehog (HH) that binds to PTC.123 In the general population, most UV-type BCC mutations are in the PTC gene or less frequently SMO, and rarely HH.109, 124–126 In XP patients, however, a significant number of mutations occur in SMO and HH.127, 128
Melanoma
Melanomas involve a series of mutations, deletions, and amplifications (copy number changes) along the MAP kinase pathway, a phosphorylation cascade that regulates cell proliferations and differentiation.129–135 (Figure 3). A common mutation occurs in BRAF (V600E), but this sequence change is not a UV type.137, 136 In contrast, 10 of 11 mutations in BRAF from XP melanomas were UV type and different from V600E.136 Mutations of UV type have been identified in the promoter region of hTERT resulting in gene activation.138, 139 In addition to point mutations, melanomas have a significant frequency of translocations involving protein kinases.140 Nevi and melanomas in XP patients are generally lentiginous and have a high incidence (53–61%) of UV-type inactivating mutations in PTEN.136 These frequencies are much higher than those in BRAF, NRAS, and KIT, unlike the general population where PTEN mutations are lower.
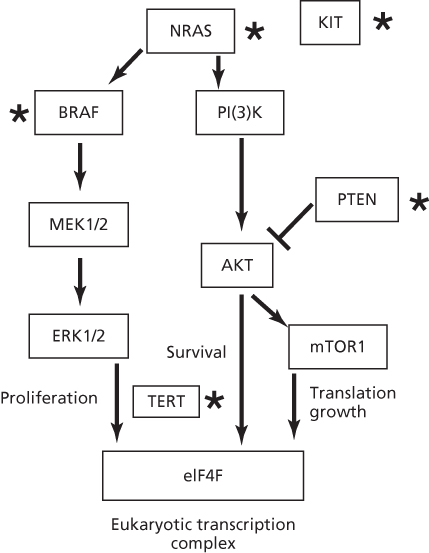
Figure 3 Pathways involved in melanoma. Proteins mutated by UV light in XP patients or the general population are designated by a star. Mutations in TERT are UV type but have yet to be reported in XP patients. Note the common BRAF melanoma mutation V600E is not induced by UV as it does not occur at a dipyrimidine site, but other mutations are formed by UV light in BRAF.136 The most common mutated gene in XP patients is PTEN, in contrast to the MAPK pathway in the general population.136
Acknowledgments
This work was supported by the E.A. Dickson Emeritus Professorship of the University of California San Francisco (JEC) and the Simon Memorial Fund of the UCSF Academic Senate (JEC).
Summary
Skin tumors in man account for about 30% of all new cancers reported annually.141, 1 Epidemiological and laboratory studies provide evidence for a direct causal role of sunlight exposure in the induction of most forms of skin cancer,142, 5 and the high rate of skin carcinogenesis is a direct result of the high dose rate from the UV light component. Nonmelanoma skin cancers (NMSCs, basal cell, and squamous cell carcinomas) and melanomas are found on sun-exposed parts of the body (e.g., the face and trunk in men, face and legs in women), and their incidence is correlated with cumulative or acute sunlight exposure. Tumor incidence and mortality increases with occupational exposure, such as in ranchers fishermen and even flight crew members.6, 143–145 There is also an increased risk of melanoma for people using tanning parlors,145, 146 PUVA or UVA therapy,147 or sildenafil (Viagra).148 The use of sunscreens can also contribute to melanoma if their use results in persons staying in the sun for increased times.149 Melanomas are additionally found on protected sites, so other factors contribute to the incidence as well as sun exposure.12, 150, 151
Human skin can be classified into types I–IV, ranging from individuals who always burn and never tan, to those who tan but never burn; skin cancer susceptibility varies accordingly.152 The most dramatic examples of variations in human susceptibility to sunburn and skin cancer are the human genetic disorders: XP, CS, TTD, basal cell nevus syndrome (BCNS), the porphyrias, albinism, and phenylketonuria.12 XP, CS, and TTD involve genes with major roles in repair of UV damage to DNA. Other disorders associated with acquired sun sensitivity include polymorphous light eruption, actinic reticuloid and prurigo, solar urticaria, lupus erythematosus, and Darier’s disease as well as medication and immunological status. Sunlight exposure can have an immunosuppressive effect leading to loss of antigen-presenting Langerhans cells and the appearance of dyskeratotic keratinocytes (apoptotic sunburn cells) in the upper epidermis.The erythemal response is also associated with vasodilation caused by a release of prostaglandins.153 Immunosuppression in organ transplant and HIV patients also increases skin cancer incidence.154
“Mad Dogs and Englishmen Go Out in the Mid-Day Sun”—Noel Coward.
References
- 1 Scotto J, Fraumeni JF Jr. Skin (other than melanoma). In: Schottenfeld D, Fraumeni JR Jr, eds. Cancer Epidemiology and Prevention. Philadelphia, PA: W. B. Saunders; 1982:996–1011.
- 2 Serrano H, Scotto J, Shornick G, et al. Incidence of nonmelanoma skin cancer in New Hampshire and Vermont. J Am Acad Dermatol. 1991;24:574–579.
- 3 Boring CC, Squire TS, Tong T. Cancer statistics 1993. Cancer J Clin. 1993;43:7–26.
- 4 Gallagher RP, Ma D, McLean DI, et al. Trends in basal cell carcinoma, squamous carcinoma and melanoma of the skin from 1973 through 1987. J Am Acad Dermatol. 1990;23:413–421.
- 5 Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8–18.
- 6 Jablonski NG, Chaplin G. The evolution of human skin coloration. J Hum Evol. 2000;39:57–106.
- 7 Davies RE, Forbes PD, Urbach F. Effects of chemicals on photobiologic reactions of the skin. Basic Life Sci. 1990;53:127–135.
- 8 Robinson JK, Rademaker AW. Relative importance of prior basal cell carcinomas, containing sun exposure and circulating T lymphocytes on the development of basal cell carcinoma. J Invest Dermatol. 1992;99:227–231.
- 9 Marks R, Staples M, Giles GG. Trends in non-melanomic skin cancer treated in Australia: The second national survey. Int J Cancer. 1993;53:585–590.
- 10 Thompson SC. Reduction of solar keratoses by regular sunscreen use. N Engl J Med. 1993;3291:1147–1151.
- 11 Marks R, Jolley D, Lectas S, Foley P. The role of childhood exposure to sunlight in the development of solar keratoses and non-melanocytic skin cancer. Med J Aust. 1990;152:62–65.
- 12 Kraemer KH, Lee MM, Andrews AD, Lambert WC. The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol. 1994;130:1018–1021.
- 13 Bradford PT, Goldstein AM, Tamura DT, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–176.
- 14 Armstrong BK. Epidemiology of malignant melanoma: intermittent or total accumulated exposure to the sun? J Dermatol Surg Oncol. 1988;14:835–849.
- 15 Tyrrell RM, Keyse SM. New trends in photobiology: the interaction of UVA radiation with cultured cells. J Photochem Photobiol B. 1990;4:349–361.
- 16 Smith PJ, Paterson MC. Abnormal responses to mid-ultraviolet light of cultured fibroblasts from patients with disorders featuring sunlight sensitivity. Cancer Res. 1981;41:511–518.
- 17 Elkind MM, Han A, Chiang-Liu C-M. “Sunlight”- induced mammalian cell killing: a comparative study of ultraviolet and near-ultraviolet inactivation. Photochem Photobiol. 1978;27:709–715.
- 18 Tyrrell RM, Pidoux M. Endogenous glutathione protects human skin fibroblasts against the cytotoxic action of UVB, UVA and near-visible radiations. Photochem Photobiol. 1986;44:561–564.
- 19 Courdavault S, Baudouin C, Charveron M, et al. Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells. Mutat Res. 2004;556(1–2):135–142.
- 20 Jones CA, Huberman E, Cunningham ML, Peak MJ. Mutagenesis and cytotoxicity in human epithelial cells by far- and near-ultraviolet radiations: action spectra. Radiat Res. 1987;110:244–254.
- 21 Niggli HJ, Cerutti PA. Cyclobutane-type pyrimidine photodimer formation and excision in human skin fibroblasts after irradiation with 313-nm ultraviolet light. Biochemistry. 1983;22:1390–1395.
- 22 Tyrrell RM, Pidoux M. Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet, and violet regions of sunlight on epidermal keratinocytes. Cancer Res. 1987;47:1825–1829.
- 23 Pfeifer GP, Drouin R, Riggs AD, Holmquist GP. In vivo mapping of a DNA adduct at nucleotide resolution: detection of pyrimidine [6-4] pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc Natl Acad Sci U S A. 1991;88:1374–1378.
- 24 Pfeifer GP, Drouin R, Riggs AD, Holmquist GP. Binding of transcription factors creates hotspots for UV photoproducts. Mol Cell Biol. 1992;12:1798–1804.
- 25 Taylor JS, Cohrs MP. DNA, light. and Dewar pyrimidinones: the structure and significance of TpT3. J Am Chem Soc. 1987;109:2834–2835.
- 26 Mitchell DL, Jen J, Cleaver JE. Relative induction of cyclobutane dimers and cytosine photohydrates in DNA irradiated in vitro and in vivo with ultraviolet C and ultraviolet B light. Photochem Photobiol. 1991;54:741–746.
- 27 Mitchell DL, Jen J, Cleaver JE. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res. 1992;20:225–229.
- 28 Ellison MJ, Childs JD. Pyrimidine CPDs induced in Escherichia coli DNA by ultraviolet radiation present in sunlight. Photochem Photobiol. 1981;34:465–469.
- 29 Mitchell DL, Cleaver JE. Photochemical alterations of cytosine account for most biological effects after ultraviolet irradiation. Trends Photochem Photobiol. 1990;1:107–119.
- 30 Rochette PJ, Lacoste S, Therrien JP, et al. Influence of cytosine methylation on ultraviolet-induced cyclobutane pyrimidine dimer formation in genomic DNA. Mutat Res. 2009;665:7–13.
- 31 Tommasi S, Denissenko MF, Pfeifer GP. Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res. 1997;57:4727–4730.
- 32 Mitchell DL. Effects of cytosine methylation on pyrimidine dimer formation in DNA. Photochem Photobiol. 2000;71:162–165.
- 33 Cadet J, Vigney P. The photochemistry of nucleic acids. In: Morrison H, ed. Bioorganic Photochemistry: Photochemistry and the Nucleic Acids. New York: John Wiley and Sons, Inc.; 1990:1–273.
- 34 Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374.
- 35 Cleaver JE. Defective repair replication in xeroderma pigmentosum. Nature. 1968;218:652–656.
- 36 Cleaver JE. Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective. Proc Natl Acad Sci U S A. 1969;63:428–435.
- 37 Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756–768.
- 38 Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85.
- 39 Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–481.
- 40 Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266:1957–1958.
- 41 Ford JM, Hanawalt PC. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem. 1997;272:28073–28080.
- 42 Cleaver JE. DNA damage and repair in normal, xeroderma pigmentosum, and XP revertant cells analyzed by gel electrophoresis: excision of cyclobutane dimers from the whole genome is not necessary for cell survival. Carcinogenesis. 1989;10:1691–1696.
- 43 Freeman SE. Variations in excision repair of UVB-induced pyrimidine CPDs in DNA of human skin in situ. J Invest Dermatol. 1988;90:814–817.
- 44 Hwang BJ, Toering S, Francke U, Chu G. p48 activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol Cell Biol. 1998;18:4391–4399.
- 45 Robu M, Brind’Amour J, Kandan-Kulangara F, et al. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. 2013;110:1658–1663.
- 46 Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003;276:46909–46910.
- 47 Shivji MK, Eker AP, Wood RD. DNA repair defect in xeroderma pigmentosum group C and complementing factor from HeLa cells. J Biol Chem. 1994;269:22749–22757.
- 48 Masutani C, Sugasawa K, Yanagisawa J, et al. Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J. 1994;13:1831–1843.
- 49 Groisman R, Polanowska J, Kuraoka I, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367.
- 50 Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575.
- 51 Maillard O, Solyom S, Naegeli H. An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol. 2007;5:e79.
- 52 Lindsey-Boltz LA, Sancar A. RNA poymerase: the most specific damage recognition protein in cellular responses to DNA damage. Proc Natl Acad Sci U S A. 2007;104:13213–13214.
- 53 Schwertman P, Lagarou A, Dekkers DHW, et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44:598–602.
- 54 Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482.
- 55 Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618.
- 56 Mao P, Meas R, Dorgan KM, Smerdon MJ. UV damage-induced RNA polymerase II stalling stimulates H2B deubiquitylation. Proc Natl Acad Sci U S A. 2014;111:12811–12816.
- 57 Sugasawa K, Ng JMY, Masutani C, et al. Xeroderma pigmentosum group C protein complex is the initiator of global nucleotide excision repair. Mol Cell. 1998;2:223–232.
- 58 Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81:39–44.
- 59 Sancar A. Mechanisms of DNA excision repair. Science. 1994;266:1954–1956.
- 60 Fagbemi AF, Orelli B, Scharer OD. Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair. 2011;10:722–729.
- 61 Huang JC, Sancar A. Determination of minimum substrate size for human excinuclease. J Biol Chem. 1994;269:19034–19044.
- 62 Cleaver JE. Stopping DNA replication in its tracks. Science. 1999;285:212–213.
- 63 Burgers PM, Koonin EV, Bruford E, et al. Eukaryotic DNA polymerases: proposal for a revised nomenclature. J Biol Chem. 2001;276:43487–43490.
- 64 Ohmori H, Friedberg EC, Fuchs RPP, et al. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8.
- 65 Trincao J, Johnson RE, Escalante CR, et al. Structure of the catalytic core of S. cerevisiae DNA polymerase h: implications for translesion synthesis. Mol Cell. 2001;8:417–426.
- 66 Johnson RE, Washington MT, Prakash S, Prakash L. Fidelity of human DNA polymerase h. J Biol Chem. 2000;275:7447–7450.
- 67 Tessman I. In: Bukhari A, Ljungquist E, eds. Abstracts of the Bacteriophage Meeting. New York: Cold Spring Harbor Laboratory Press; 1976:87.
- 68 Johnson RE, Prakash S, Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase eta. Science. 1999;283:1001–1004.
- 69 McCulloch SD, Kokoska RJ, Masutani C, et al. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100.
- 70 Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science. 1999;264:263–265.
- 71 Masutani C, Kusumoto R, Yamada A, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase h. Nature. 1999;399:700–704.
- 72 Tissier A, Frank EG, McDonald JP, et al. Misinsertion and bypass of thymine-thymine dimers by human DNA polymerase iota. EMBO J. 2000;19:5259–5266.
- 73 Tissier A, McDonald JP, Frank EG, Woodgate R. Pol iota, a remarkable error-prone human DNA polymerase. Genes Dev. 2000;14:1642–1650.
- 74 Woodgate R. Evolution of the two-step model for UV-mutagenesis. Mutat Res. 2001;485:83–92.
- 75 Washington MT, Johnson RE, Prakash L, Prakash S. Human DINB1-encoded DNA polymerase k is a promiscuous extender of mispaired promer termini. Proc Natl Acad Sci U S A. 2002;99:1910–1914.
- 76 Johnson RE, Washington MT, Haracska L, et al. Eukaryotic polymerases i and z act sequentially to bypass DNA lesions. Nature. 2000;406:1015–1019.
- 77 Wang YC, Maher VM, McCormick JJ. Xeroderma pigmentosum variant cells are less likely than normal cells to incorporate dAMP opposite photoproducts during replication of UV-irradiated plasmids. Proc Natl Acad Sci U S A. 1991;88:7810–7814.
- 78 Wang YC, Maher VM, Mitchell DL, McCormick JJ. Evidence from mutation spectra that the UV hypermutability of xeroderma pigmentosum variant cells reflects abnormal error-prone replication on a template containing photoproducts. Mol Cell Biol. 1993;13:4276–4283.
- 79 Gibbs PE, McGregor WG, Maher VM, et al. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc Natl Acad Sci U S A. 1998;95:6876–6880.
- 80 Jaspers NG, Raams A, Silengo MC, et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80:457–466.
- 81 Parris CH, Kraemer KH. Ultraviolet-light induced mutations in Cockayne syndrome cells are primarily caused by cyclobutane dimer photoproducts while repair of other photoproducts is normal. Proc Natl Acad Sci U S A. 1993;90:7260–7264.
- 82 Nance MA, Berry SA. Cockayne syndrome:review of 140 cases. Am J Med Genet. 1992;42:68–84.
- 83 Cleaver JE, Kraemer KH. Xeroderma pigmentosum and Cockayne Syndrome. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease, 6th ed. Vol. III. New York: McGraw-Hill; 1995:4393–4419.
- 84 Laposa RR, Feeney L, Crowley E, et al. p53 suppression overwhelms DNA polymerase h deficiency in determining the cellular UV DNA damage response. DNA Repair. 2007;6:1794–1804.
- 85 Lehmann AR. Three complementation groups in Cockayne syndrome. Mutat Res. 1982;106:347–356.
- 86 Weeda G, van Ham R, Vermeulen W, et al. A presumed helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne’s syndrome. Cell. 1990;62:777–791.
- 87 Nakazawa Y, Sasaki K, Mitsutake N, et al. KIAA1530/UVSSA is responsible for UV-sensitive syndrome that facilitates damage-dependent processing of stalled RNA polymerase IIo in TC-NER. Nat Genet. 2012;44:586–592.
- 88 Zhang X, Horibata K, Saijo M, et al. Mutations in KIAA1530/UVSSA cause UV-sensitive syndrome destabilizing ERCC6 in transcription-coupled DNA repair. Nat Genet. 2012;44:593–597.
- 89 Scheibye-Knudsen M, Croteau DL, Bohr VA. Mitochondrial deficiency in Cockayne syndrome. Mech Aging Dev. 2013;134:275–283.
- 90 Cleaver JE, Brennan-Minnella AM, Swanson RA, et al. Mitochondrial reactive oxygen species are scavenged by Cockayne Syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci U S A. 2014;111:13487–13492.
- 91 Lehmann AR, Arlett CF, Broughton BC, et al. Trichothiodystrophy, a human DNA repair disorder with heterogeneity in the cellular response to ultraviolet light. Cancer Res. 1988;48:6090–6096.
- 92 Broughton BC, Lehmann AR, Harcourt SA, et al. Relationship between pyrimidine dimers, 6-4 photoproducts, repair synthesis and cell survival: studies using cells from patients with trichothiodystrophy. Mutat Res. 1990;235:33–40.
- 93 Weber CA, Salazar EP, Stewart SA, Thompson LH. ERCC2: cDNA cloning and molecular characterization of a human nucleotide excision repair gene with high homology to yeast RAD3. EMBO J. 1990;9:1437–1447.
- 94 Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol. 2001;44:891–920.
- 95 Giglia-Mari G, Coin F, Ranish JA, et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet. 2004;36:714–719.
- 96 Loeb L. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079.
- 97 Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160.
- 98 Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218.
- 99 Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196.
- 100 Behjati S, Huch M, van Boxtel R, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425.
- 101 Berg RJ, Rebel H, van der Horst GT, et al. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res. 2000;60:2858–2863.
- 102 Fuchs A, Marmur E. The kinetics of skin cancer:progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33:1099–1101.
- 103 Ratushny V, Gober MD, Hick R, et al. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Invest. 2014;122:464–472.
- 104 Lambert SR, Mladkova N, Gulati A, et al. Key differences identified between actinic keratosis and cutaneous squamous cell carcinoma by transcriptome profiling. Br J Cancer. 2014;110:520–529.
- 105 Zhang W, Remenyik E, Zelterman D, et al. Escaping the stem cell compartment:sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc Natl Acad Sci U S A. 2001;98:13948–13953.
- 106 Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88:10124–10128.
- 107 Ziegler A, Jonason AS, Leffell DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776.
- 108 Nataraj AJ, Trent JC, Ananthaswamy HN. p53 gene mutations and photocarcinogenesis. Photochem Photobiol. 1995;62:165–177.
- 109 Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma cancer. Adv Exp Med Biol. 2008;624:162–178.
- 110 Giglia-Maria G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21:217–228.
- 111 Limoli CL, Giedzinski E, Bonner WM, Cleaver JE. UV-induced replication arrest in the xeroderma pigmentosum variant leads to double strand breaks, g-H2Ax formation, and Mre11 relocalization. Proc Natl Acad Sci U S A. 2002;99:233–238.
- 112 Bertrand P, Saintigny Y, Lopez BS. p53’s double life:transactivation-independent repression of homologous recombination. Trends Genet. 2004;202:235–243.
- 113 Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci U S A. 2007;104:1389–1394.
- 114 Wang NJ, Sanborn Z, Arnett KL, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:17761–17766.
- 115 Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. J Dermatol Sci. 1998;17:1–7.
- 116 Mock BA, Lowry DT, Rehman I, et al. Multigenic control of skin tumor susceptibility in SENCARA/Pt mice. Carcinogenesis. 1998;19:1109–1115.
- 117 Nagase H, Mao JH, de Kooning JP, et al. Epistatic interactions between skin tumor modifier loci in interspecific (spretus/musculus) backcross mice. Cancer Res. 2001;61:1305–1308.
- 118 Lee CS, Bhaduri A, Mah A, et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat Genet. 2014, in press.
- 119 Ananthaswamy HN, Price JE, Goldberg LH, Bales ES. Detection and identification of activated oncogenes in human skin cancers occurring on sun-exposed body sites. Cancer Res. 1988;48:3341–3346.
- 120 Keijzer W, Mulder MP, Langeveld JC, et al. Establishment and characterization of a melanoma cell line from a xeroderma pigmentosum patient: activation of N-ras at a potential pyrimidine CPD site. Cancer Res. 1989;49:1229–1235.
- 121 Suarez HG, Daya-Grosjean L, Schlaifer D, et al. Activated oncogenes in human skin tumors from a repair-deficient syndrome, xeroderma pigmentosum. Cancer Res. 1989;49:1223–1228.
- 122 Bredberg A, Kraemer KH, Seidman MM. Restricted ultraviolet mutational spectrum in a shuttle vector propagated in xeroderma pigmentosum cells. Proc Natl Acad Sci U S A. 1986;83:8273–8277.
- 123 Toftgard R. Hedgehog signalling in cancer. Cell Mol Life Sci. 2000;57:1720–1731.
- 124 Epstein EH. Basal cell carcinomas:attack of the hedgehog. Nat Rev Cancer. 2008;8:743–754.
- 125 Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671.
- 126 Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell. 1996;85:841–851.
- 127 Couve-Privat S, Bouadjar B, Avril MF, et al. Significantly high levels of ultraviolet-specific mutations in the smoothened gene in basal cell carcinomas from DNA repair-deficient xeroderma pigmentosum patients. Cancer Res. 2002;62:7185–7189.
- 128 Couvé-Privat S, Le Bret M, Traiffort E, et al. Functional analysis of novel sonic hedgehog gene mutations identified in basal cell carcinomas from xeroderma pigmentosum patients. Cancer Res. 2004;64:3559–3565.
- 129 Goldstein AM, Tucker MA. Genetic epidemiology of cutaneous melanoma: a global perspective. Archiv Dermatol. 2001;137:1493–1496.
- 130 Kamb A, Shattuck-Eidens D, Eeles R, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:23–26.
- 131 Nobori T, Miura K, Wu DJ, et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756.
- 132 Bastian B, LeBoit PE, Hamm H, et al. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genome hybridization. Cancer Res. 1998;58:2170–2175.
- 133 Bastian BC, Wesselmann U, Pinkel D, Leboit PE. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J Invest Dermatol. 1999;113:1065–1069.
- 134 Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increases of HRAS in Spitz nevi with distinctive histopathologic features. Am J Pathol. 2000;157:967–972.
- 135 Pollock PM, Trent JM. The genetics of cutaneous melanoma. Clin Lab Med. 2000;20:667–690.
- 136 Masaki T, Wang Y, DiGiovanna JJ, et al. High frequency of PTEN mutations in nevi and melanomas from xeroderma pigmentosum patients. Pigment Cell Melanoma Res. 2014;27:454–464.
- 137 Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954.
- 138 Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961.
- 139 Scott GA, Laughlin TS, Rothberg PG. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod Pathol. 2014;27:516–523.
- 140 Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
- 141 Scotto J, Fears TR, Fraumeni JF. Incidence of Nonmelanoma Skin Cancer in the United States. Bethesda, MD: U.S. Department of Health and Human Services, NIH Publication No. 83–2433; 1983.
- 142 Fitzpatrick TB, Sober AJ. Sunlight and skin cancer. N Engl J Med. 1985;313:818–820.
- 143 Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2014, in press.
- 144 Grossman L, Wei Q. DNA repair and epidemiology of basal cell carcinoma. Clin Chem. 1995;41(Part 2):1854–1863.
- 145 Wehner MR, Chren MM, Nameth D, et al. International prevalence of indoor tanning: a systematic review and meta-analysis. JAMA Dermatol. 2014;150:390–400.
- 146 Diffey BL. A quantitative estimate of melanoma mortality from ultraviolet A sunbed use in the U.K. Br J Dermatol. 2003;149:578–591.
- 147 Archier E, Devaux S, Castela E, et al. Carcinogenic risks of psoralen UV-A therapy and narrowband UV-B therapy in chronic plaque psoriasis: a systematic literature review. J Eur Acad Dermatol Venereol. 2012;(Suppl. 3):22–31.
- 148 Li WQ, Qureshi AA, Robinson KC, Han J. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964–970.
- 149 Jou PC, Tomecki KJ. Sunscreens in the United States: current status and future outlook. Adv Exp Med Biol. 2014;810:464–484.
- 150 De Fabo EC, Noonan FP, Fears T, Merlino G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 2004;64:6372–6376.
- 151 Noonan FP, Recio JA, Takayama H, et al. Neonatal sunburn and melanoma in mice. Nature. 2001;413:271–272.
- 152 Vitaliano PP, Urbach F. The relative importance of risk factors in nonmelanoma skin cancer. Arch Dermatol. 1980;116:454–456.
- 153 Kripke ML. Immunological effects of ultraviolet radiation. J Dermatol. 1991;18:429–433.
- 154 London NJ, Farmery SM, Will EJ, et al. Risk of neoplasia in renal tansplant patients. Lancet. 1995;346:403–406.