[level-membership-for-internal-medicine-category]
(See also Chap. 148) In addition to receiving antibiotic prophylaxis, transplant recipients should be vaccinated against likely pathogens (Table 169-6). In the case of HSC transplant recipients, optimal responses cannot be achieved until after immune reconstitution, despite previous immunization of both donor and recipient. Recipients of an allogeneic HSC transplant must be reimmunized if they are to be protected against pathogens. The situation is less clear-cut in the case of autologous transplantation. T and B cells in the peripheral blood may reconstitute the immune response if they are transferred in adequate numbers. However, cancer patients (particularly those with Hodgkin’s disease, in whom vaccination has been extensively studied) who are undergoing chemotherapy do not respond normally to immunization, and titers of antibodies to infectious agents fall more rapidly than in healthy individuals. Therefore, even immunosuppressed patients who have not undergone HSC transplantation may need booster vaccine injections. If memory cells are specifically eliminated as part of a stem cell “cleanup” procedure, it will be necessary to reimmunize the recipient with a new primary series. Optimal times for immunizations of different transplant populations are being evaluated. Yearly immunization of household and other contacts (including health care personnel) against influenza benefits the patient by preventing local spread.
|
VACCINATION OF HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) AND SOLID ORGAN TRANSPLANT (SOT) RECIPIENTS |
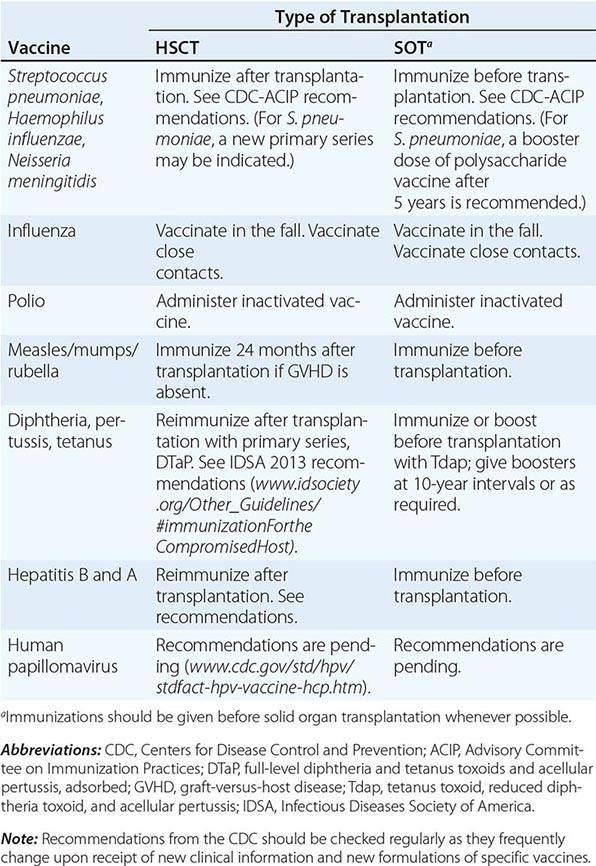
In the absence of compelling data as to optimal timing, it is reasonable to administer the pneumococcal and H. influenzae type b conjugate vaccines to both autologous and allogeneic HSC transplant recipients beginning 12 months after transplantation. A series that includes both the 13-valent pneumococcal conjugate vaccine (Prevnar) and the 23-valent pneumococcal polysaccharide vaccine (Pneumovax) is now recommended (according to CDC guidelines). The pneumococcal and H. influenzae type b vaccines are particularly important for patients who have undergone splenectomy. The Neisseria meningitidis polysaccharide conjugate vaccine (Menactra or Menveo) also is recommended. In addition, diphtheria, tetanus, acellular pertussis, and inactivated polio vaccines can all be given at these same intervals (12 months and, as required, 24 months after transplantation). Some authorities recommend a new primary series for tetanus/diphtheria/pertussis and inactivated poliovirus vaccines beginning 12 months after transplantation. Vaccination to prevent hepatitis B and hepatitis A (both killed vaccines) also seems advisable. A formal recommendation regarding immunization with the tetravalent HPV virus-like particle vaccine (Gardasil) after HSC transplantation has not been issued. However, HPV vaccination, which can prevent genital warts as well as specific cancers, is recommended through age 26 for healthy young adults who previously have not been vaccinated or have not received the full series. Live-virus measles/mumps/rubella (MMR) vaccine can be given to autologous HSC transplant recipients 24 months after transplantation and to most allogeneic HSC transplant recipients at the same point if they are not receiving maintenance therapy with immunosuppressive drugs and do not have ongoing GVHD. The risk of spread from a household contact is low for MMR vaccine. In parts of the world where live poliovirus vaccine is used, patients as well as contacts should be advised to receive only the killed vaccine. In the rare setting where both donor and recipient are VZV naïve and the recipient is no longer receiving acyclovir or ganciclovir prophylaxis, the patient should be counseled to receive varicella-zoster immune globulin (VariZIG) up to 10 days after an exposure to a person with chickenpox or uncovered zoster; such patients should avoid close contact with persons recently vaccinated with Varivax. A formal recommendation regarding Varivax immunization of such patients is not currently available. Neither patients nor their household contacts should receive vaccinia vaccine unless they have been exposed to smallpox virus. Among patients who have active GVHD and/or are taking high maintenance doses of glucocorticoids, it may be prudent to avoid all live-virus vaccines.
In the case of SOT recipients, administration of all the usual vaccines and of the indicated booster doses should be completed before immunosuppression, if possible, to maximize responses. For patients taking immunosuppressive agents, the administration of pneumococcal vaccine should be repeated every 5 years. No data are available for the meningococcal vaccine, but it is probably reasonable to administer it along with the pneumococcal vaccine. H. influenzae conjugate vaccine is safe and should be efficacious in this population; therefore, its administration before transplantation is recommended. Booster doses of this vaccine are not recommended for adults. SOT recipients who continue to receive immunosuppressive drugs should not receive live-virus vaccines. A person in this group who is exposed to measles should be given measles immune globulin. Similarly, an immunocompromised patient who is seronegative for varicella and who comes into contact with a person who has chickenpox should be given varicella-zoster immune globulin as soon as possible (optimally within 96 h; up to 10 days after contact); if this is not possible, a 10- to 14-day course of acyclovir therapy should be started immediately. Upon the discontinuation of treatment, clinical disease may still occur in a small number of patients; thus vigilance is indicated. Rapid re-treatment with acyclovir should limit the symptoms of disease. Household contacts of transplant recipients can receive live attenuated VZV vaccine, but vaccinees should avoid direct contact with the patient if a rash develops. Virus-like particle vaccines have been licensed for the prevention of infection with several HPV serotypes most commonly implicated in cervical and anal carcinomas and in anogenital and laryngeal warts. These vaccines are not live; however, no information is yet available about their immunogenicity or efficacy in transplant recipients.
Immunocompromised patients who travel may benefit from some but not all vaccines (Chaps. 148 and 149). In general, these patients should receive any killed or inactivated vaccine preparation appropriate to the area they are visiting; this recommendation includes the vaccines for Japanese encephalitis, hepatitis A and B, poliomyelitis, meningococcal infection, and typhoid. The live typhoid vaccines are not recommended for use in most immunocompromised patients, but an inactivated or purified polysaccharide typhoid vaccine can be used. Live yellow fever vaccine should not be administered. On the other hand, primary immunization or boosting with the purified-protein hepatitis B vaccine is indicated. Inactivated hepatitis A vaccine should also be used in the appropriate setting (Chap. 148). A vaccine is now available that provides dual protection against hepatitis A and hepatitis B. If hepatitis A vaccine is not administered, travelers should consider receiving passive protection with immune globulin (the dose depending on the duration of travel in the high-risk area).
SECTION 4 |
APPROACH TO THERAPY FOR BACTERIAL DISEASES |
170 |
Treatment and Prophylaxis of Bacterial Infections |
Antimicrobial agents have had a major impact on human health. Together with vaccines, they have contributed to reduced mortality, extended lifespan, and enhanced quality of life. Among drugs used in human medicine, however, they are distinctive in that their use promotes the occurrence of drug resistance in the pathogens they are designed to treat as well as in other “bystander” organisms. Indeed, the history of antimicrobial development has been driven in large part by the medical need engendered by the emergence of resistance to each generation of agents. Thus, the careful and appropriate use of antimicrobial drugs is particularly important not only for optimizing efficacy and minimizing adverse effects but also for minimizing the risk of resistance and preserving the value of existing agents. Although this chapter focuses on antibacterial agents, the optimal use of all antimicrobials depends on an understanding of each drug’s mechanism of action, spectrum of activity, mechanisms of resistance, pharmacology, and adverse effect profile. This information is then applied in the context of the patient’s clinical presentation, underlying conditions, and epidemiology to define the site and likely nature of the infection or other condition and thus to choose the best therapy. Gathering of microbiologic information is important for refining therapeutic choices on the basis of documented pathogen and susceptibility data whenever possible; this information also makes it possible to choose more targeted therapy, thereby reducing the risk of selection of resistant bacteria. Durations of therapy are chosen according to the nature of the infection and the patient’s response to treatment and are informed by clinical studies when they are available, with the understanding that shorter courses are less likely than longer ones to promote the emergence of resistance. This chapter provides specific information that is necessary for making informed choices among antibacterial agents.
MECHANISMS OF ACTION AND RESISTANCE
The mechanisms of action of and resistance to antibacterial agents are discussed in detail in the text and are summarized for the most commonly used groups of agents in Table 170-1. A schematic of antibacterial targets is provided in Fig. 170-1.
|
MECHANISMS OF ACTION OF AND RESISTANCE TO ANTIBACTERIAL AGENTS |
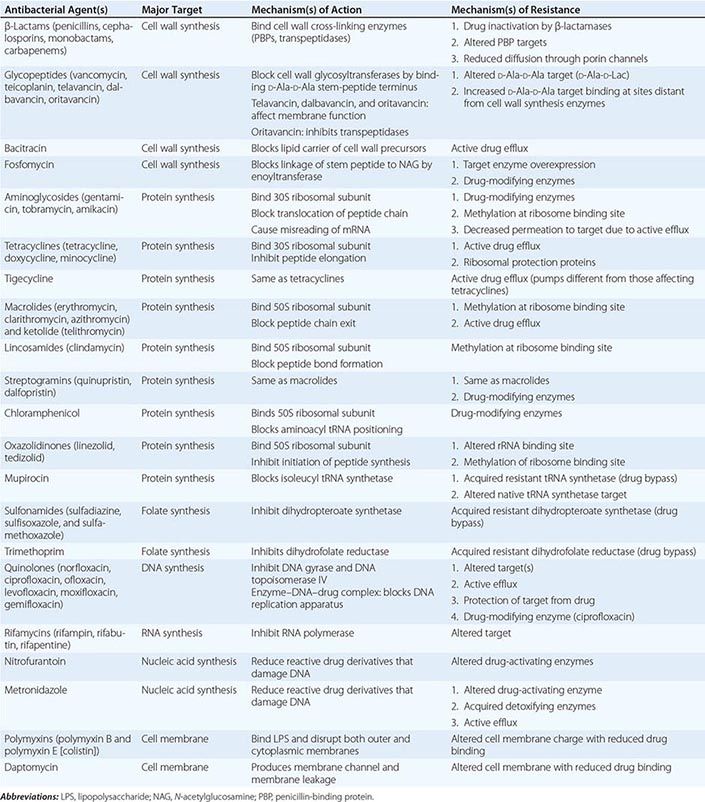
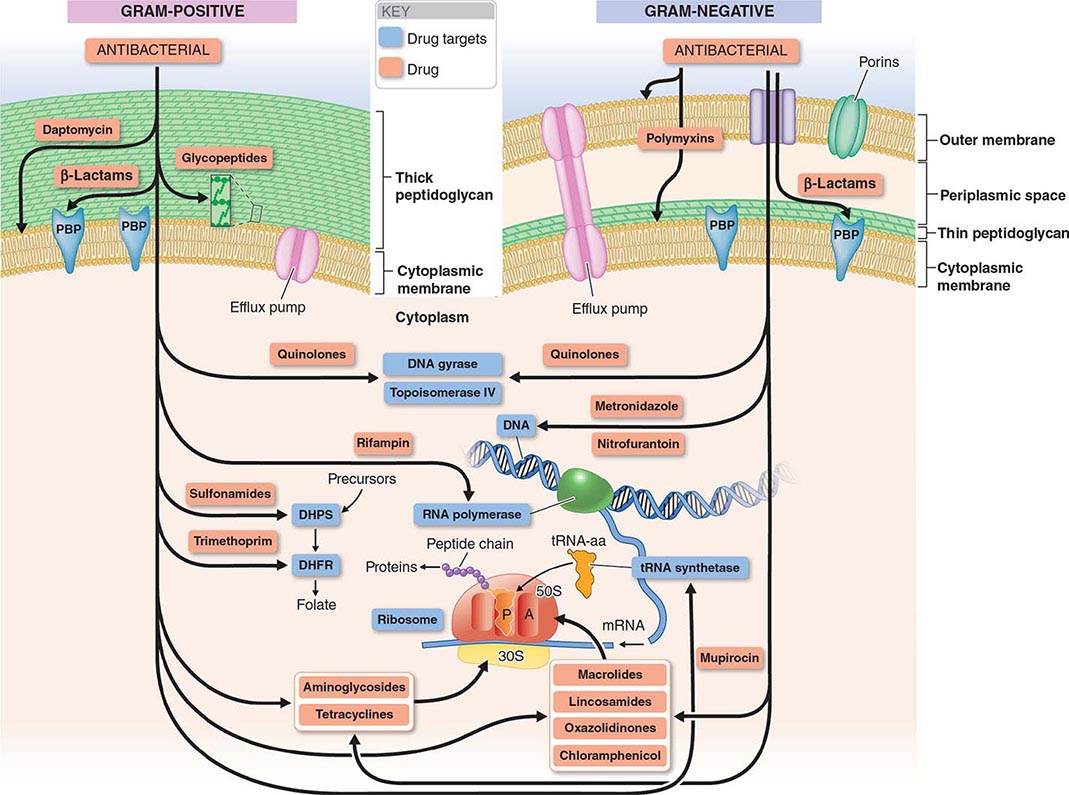
FIGURE 170-1 Antibacterial targets. A, aminoacyl site; DHFR, dihydrofolate reductase; DHPS, dihydropteroate synthetase; P, peptidyl site; PBP, penicillin-binding protein; tRNA-aa, aminoacyl tRNA.
MECHANISMS OF ACTION
Multiple essential components of bacterial cell structures and metabolism have been the targets of antibacterial agents used in clinical medicine, and the interaction of an agent with its target results in either inhibition of bacterial growth and replication (bacteriostatic effect) or bacterial killing (bactericidal effect). In general, targets have been chosen because they either do not exist in mammalian cells and physiology or are sufficiently different from their bacterial counterparts to allow selective bacterial targeting. Treatment with bacteriostatic agents is effective when the patient’s host defenses are sufficient to contribute to eradication of the infecting pathogen. In patients with impaired host defenses (e.g., neutropenia) or infections at body sites with impaired or limited host defenses (e.g., meningitis and endocarditis), bactericidal agents are generally preferred.
Inhibition of Cell Wall Synthesis The bacterial cell wall, which is external to the cytoplasmic membrane and has no counterpart in mammalian cells, protects bacterial cells from lysis under low osmotic conditions. The cell wall is a cross-linked peptidoglycan composed of a polymer of alternating units of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM), four-amino-acid stem peptides linked to each NAM, and a peptide cross-bridge that links adjacent stem peptides to form a netlike structure. Several steps in peptidoglycan synthesis are targets of antibacterial agents. Inhibition of cell wall synthesis generally results in a bactericidal effect that is linked to cell lysis. This effect results not only from the blocking of new cell-wall formation but from the uninhibited action of cell wall–remodeling enzymes called autolysins, which cleave peptidoglycan as part of normal cell-wall growth.
In gram-positive bacteria the peptidoglycan is the most external cell structure, but in gram-negative bacteria an asymmetric lipid outer membrane is external to the peptidoglycan and contains diffusion channels called porins. The space between the cytoplasmic membrane peptidoglycan and the outer membrane is referred to as the periplasmic space. Most antibacterial drugs enter the gram-negative bacterial cell through a porin channel, since the outer membrane is a major diffusion barrier. Although the peptidoglycan layer is thicker in gram-positive (20–80 nm) than in gram-negative (1 nm) bacteria, peptidoglycan itself constitutes only a limited diffusion barrier for antibacterial agents.
β-LACTAMS The β-lactam drugs, including penicillins, cephalosporins, monobactams, and carbapenems, target transpeptidase enzymes (also called penicillin-binding proteins, or PBPs) involved in the stem-peptide cross-linking step.
GLYCOPEPTIDES The glycopeptides, including vancomycin, teicoplanin, telavancin, dalbavancin, and oritavancin, bind the two terminal D-alanine residues of the stem peptide, hindering the glycosyltransferase involved in polymerizing NAG–NAM units. Telavancin also binds to the lipid II intermediate that delivers cell-wall precursor subunits. Likewise, dalbavancin and oritavancin interact with the cell membrane, and oritavancin may also inhibit transpeptidases. Both β-lactams and glycopeptides interact with their targets external to the cytoplasmic membrane.
BACITRACIN (TOPICAL) AND FOSFOMYCIN These agents interrupt enzymatic steps in the production of peptidoglycan precursors in the cytoplasm.
Inhibition of Protein Synthesis Most inhibitors of bacterial protein synthesis target bacterial ribosomes, whose difference from eukaryotic ribosomes allows selective antibacterial action. Some inhibitors bind to the 30S ribosomal subunit and others to the 50S subunit. Most protein synthesis–inhibiting agents are bacteriostatic; aminoglycosides are an exception and are bactericidal.
AMINOGLYCOSIDES Aminoglycosides (amikacin, gentamicin, kanamycin, netilmicin, streptomycin, tobramycin) bind irreversibly to 16S ribosomal RNA (rRNA) of the 30S ribosomal subunit, blocking the translocation of peptidyl transfer RNA (tRNA) from the A (aminoacyl) to the P (peptidyl) site and, at low concentrations, causing misreading of messenger RNA (mRNA) codons and thus causing the introduction of incorrect amino acids into the peptide chain; at higher concentrations, translocation of the peptide chain is blocked. Cellular uptake of aminoglycosides is dependent on the electrochemical gradient across the bacterial membrane. Under anaerobic conditions, this gradient is reduced, with a consequent reduction in the uptake and activity of the aminoglycosides. Spectinomycin is a related aminocyclitol antibiotic that also binds to 16S rRNA of the 30S ribosomal subunit but at a different site. This drug inhibits translocation of the growing peptide chain but does not trigger codon misreading and produces only a bacteriostatic effect.
TETRACYCLINES AND GLYCYLCYCLINES Tetracyclines (doxycycline, minocycline, tetracycline) bind reversibly to the 16S rRNA of the 30S ribosomal subunit and block the binding of aminoacyl tRNA to the ribosomal A site, thereby inhibiting peptide elongation. Active transport of tetracyclines into bacterial but not mammalian cells contributes to the selectivity of these agents. Tigecycline, a derivative of minocycline and the only available glycylcycline, acts similarly to the tetracyclines but is distinctive for its ability to circumvent the most common mechanisms of resistance to the tetracyclines.
MACROLIDES AND KETOLIDES In contrast to the aminoglycosides and tetracyclines, the macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) bind to the 23S rRNA of the 50S ribosomal subunit. These agents block translocation of the growing peptide chain by binding to the tunnel from which the chain exits the ribosome.
LINCOSAMIDES Clindamycin is the only lincosamide in clinical use. It binds to the 23S rRNA of the 50S ribosomal subunit, interacting with both the ribosomal A and P sites and blocking peptide bond formation.
STREPTOGRAMINS The only streptogramin in clinical use is a combination of quinupristin, a group B streptogramin, and dalfopristin, a group A streptogramin. Both components bind to 23S rRNA of the 50S ribosome: dalfopristin binds to both the A and P sites of the peptidyl transferase center, and quinupristin binds to a site that overlaps the macrolide-binding site, blocking the emergence of nascent peptide from the ribosome. The combination is bactericidal, but macrolide-resistant bacteria exhibit cross-resistance to quinupristin, and the remaining activity of dalfopristin alone is bacteriostatic.
CHLORAMPHENICOL Chloramphenicol binds reversibly to the 23S rRNA of the 50S subunit in a manner that interferes with the proper positioning of the aminoacyl component of tRNA in the A site. This site of binding is near those of the macrolides and lincosamides.
OXAZOLIDINONES Linezolid and tedizolid are the only oxazolidinones in clinical use. They bind directly to the A site in the 23S rRNA of the 50S ribosomal subunit and block binding of aminoacyl tRNA, inhibiting the initiation of protein synthesis.
MUPIROCIN Mupirocin (pseudomonic acid) is used topically. It competes with isoleucine for binding to isoleucyl tRNA synthetase, depleting stores of isoleucyl tRNA and thereby inhibiting protein synthesis.
Inhibition of Bacterial Metabolism Available inhibitors (antimetabolites) target the pathway for synthesis of folate, which is a cofactor in a number of one-carbon transfer reactions involved in the synthesis of some nucleic acids, including pyrimidine, thymidine, and all purines (adenine and guanine), as well as some amino acids (methionine and serine) and acetyl coenzyme A (CoA). Two sequential steps in folate synthesis are targeted. The selective antibacterial effect stems from the inability of mammalian cells to synthesize folate; they depend instead on exogenous sources. Antibacterial activity, however, may be reduced in the presence of high exogenous concentrations of the end products of the folate pathway (e.g., thymidine and purines) that may occur in some infections, resulting from local breakdown of leukocytes and host tissues.
SULFONAMIDES Sulfonamides, including sulfadiazine, sulfisoxazole, and sulfamethoxazole, inhibit dihydropteroate synthetase, which adds p-aminobenzoic acid (PABA) to pteridine, producing dihydropteroate. Sulfonamides are structural analogues of PABA and act as competing enzyme substrates.
TRIMETHOPRIM Subsequent steps in folate synthesis are catalyzed by dihydrofolate synthase, which adds glutamate to dihydropteroate, and dihydrofolate reductase, which then generates the final product, tetrahydrofolate. Trimethoprim is a structural analogue of pteridine and inhibits dihydrofolate reductase. Trimethoprim is available alone but is most often used in combination products that also contain sulfamethoxazole and thus block two sequential steps in folate synthesis.
Inhibition of DNA and RNA Synthesis or Activity A variety of antibacterial agents act on these processes.
QUINOLONES The quinolones include nalidixic acid, the first agent in the class, and newer, more widely used fluorinated derivatives (fluoroquinolones), including norfloxacin, ciprofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. The quinolones are synthetic compounds that inhibit bacterial DNA synthesis by interacting with the DNA complexes of two essential enzymes, DNA gyrase and DNA topoisomerase IV, which alter DNA topology. Quinolones trap enzyme–DNA complexes in such a way that they block movement of the DNA replication apparatus and can generate lethal double-strand breaks in DNA, resulting in bactericidal activity. Although mammalian cells also have type II DNA topoisomerases like gyrase and topoisomerase IV, the structures of the mammalian enzymes are sufficiently different from those of the bacterial enzymes that quinolones have substantially selective antibacterial activity.
RIFAMYCINS Rifampin, rifabutin, and rifapentine are semisynthetic derivatives of rifamycin B and bind the β subunit of bacterial RNA polymerase, thereby blocking elongation of mRNA. Their action is highly selective for the bacterial enzyme over mammalian RNA polymerases.
NITROFURANTOIN The reduction of nitrofurantoin, a nitrofuran compound, by bacterial enzymes produces highly reactive derivatives that are thought to cause DNA strand breakage. Nitrofurantoin is used only for the treatment of lower urinary tract infections.
METRONIDAZOLE Metronidazole is a synthetic nitroimidazole with activity limited to anaerobic bacteria and certain anaerobic protozoa. Reduction of its nitro group by the electron-transport system in anaerobic bacteria produces reactive intermediates that damage DNA and result in bactericidal activity. Both nitrofurantoin and metronidazole have selective antibacterial activity because the reducing activity needed to generate active derivatives is generated only by bacterial and not mammalian enzymes.
Disruption of Membrane Integrity The integrity of the bacterial cytoplasmic membrane—and, in gram-negative bacteria, the outer membrane—is important for bacterial viability. Two bactericidal drugs have membrane targets.
POLYMYXINS The polymyxins, including polymyxin B and polymyxin E (colistin), are cationic cyclic polypeptides that disrupt the cytoplasmic membrane and the outer membrane (the latter by binding lipopolysaccharide).
DAPTOMYCIN Daptomycin is a lipopeptide that binds the cytoplasmic membrane of gram-positive bacteria in the presence of calcium, generating a channel that leads to leakage of cytoplasmic potassium ions and membrane depolarization.
MECHANISMS OF RESISTANCE
Bacteria use a wide variety of mechanisms to block or circumvent the activity of antibacterial agents. Although myriad, these mechanisms can generally be grouped into three categories: (1) altered or bypass targets that exhibit reduced binding of the drug, (2) altered access of the drug to its target by reductions in uptake or increases in active efflux, and (3) a modification of the drug that reduces its activity. These mechanisms result from either mutations in bacterial chromosomal genes occurring spontaneously during bacterial DNA replication or the acquisition of new genes by DNA transfer from other bacteria or uptake of exogenous DNA. New genes are most often acquired on self-replicating plasmids or other DNA elements transferred from other bacteria. However, some bacteria, such as Streptococcus pneumoniae and Neisseria gonorrhoeae, can also take up fragments of environmental DNA from related species and recombine that DNA directly into their own chromosomes, a process called transformation. Not uncommonly, resistant bacteria have combinations of resistance mechanisms either within one category or among categories, and many plasmids contain more than one resistance gene. Thus, plasmid acquisition itself can in many cases confer resistance to multiple antibacterial agents.
Many antibacterial drugs are derived from natural products of microbial species. Some genes encoding resistance to these drugs may have evolved and been mobilized onto plasmids from a protection mechanism in the producer organism or in other surviving bacteria in the exposed environment. Exposure to antibacterial agents either in nature or from human or animal use results in the selection of resistant strains within an otherwise susceptible bacterial population. Because the patterns and extent of resistance may differ among settings, initial choices of antibacterial drugs should be based, whenever possible, on local susceptibility data and should be modified as needed as soon as specific microbiology susceptibility data become available.
β-Lactams The most common mechanism of resistance to β-lactams is their degradation by β-lactamases, enzymes that break down the core β-lactam ring and destroy drug activity. Different β-lactamases degrade different β-lactams. Some β-lactamases are encoded on the bacterial chromosome, and their activity contributes to the susceptibility profile of a particular species. Because other β-lactamases are encoded by acquired plasmids, their resistance profiles may be present in some strains of a species but not others. In gram-positive bacteria β-lactamases are secreted into the extracellular environment, whereas in gram-negative bacteria these enzymes are secreted into the periplasmic space between the cytoplasmic and outer membranes. Thus, in gram-negative bacteria, access of β-lactams both to their target PBPs and to β-lactamases requires diffusion across the outer membrane, generally through the porin channels.
Most strains of Staphylococcus aureus produce a plasmid-encoded β-lactamase that degrades penicillin but not semisynthetic penicillins, such as oxacillin and nafcillin. The most common plasmid-encoded β-lactamases of gram-negative bacteria are able to inactivate all penicillins and most earlier-generation cephalosporins. Extended-spectrum β-lactamase (ESBL) variants of these early enzymes that can degrade later-generation cephalosporins (ceftriaxone, cefotaxime, ceftazidime) as well as the monobactam aztreonam have now emerged and are widely disseminated. Some ESBLs also degrade the fourth-generation cephalosporin cefepime. Carbapenems (imipenem, meropenem, ertapenem, doripenem) are not degraded by ESBLs, but additional β-lactamases, called carbapenemases, that degrade carbapenems and most if not all other β-lactams have begun to emerge.
The chromosomal β-lactamase of Klebsiella pneumoniae preferentially degrades penicillins but not cephalosporins. In contrast, the chromosomal β-lactamase of Enterobacter and related genera, AmpC, can degrade almost all cephalosporins but is normally expressed in small amounts. Mutations that cause increased amounts of AmpC to be produced confer full resistance to penicillins and cephalosporins; the exceptions are cefoxitin and cefepime, which are relatively stable to AmpC. Resistance to cefepime can develop, however, through the combined effects of increased AmpC production and decreased porin diffusion channels. Genes encoding AmpC have also been found on plasmids but are less common than plasmid-encoded ESBLs.
Inhibitors of β-lactamases such as clavulanate, sulbactam, and tazobactam have been developed and paired with amoxicillin and ticarcillin, ampicillin, and piperacillin, respectively. These inhibitors have little or no antibacterial activity of their own but inhibit plasmid-mediated β-lactamases, including ESBLs but not AmpC enzymes.
Resistance to β-lactams also occurs through alterations in their target PBPs. In S. pneumoniae, N. gonorrhoeae, and Neisseria meningitidis, resistance to penicillin occurs by recombination of transformed DNA from related species. In staphylococci, resistance to methicillin and other β-lactams occurs by the acquisition of the mec gene, which encodes PBP2a with reduced drug affinity. Ceftaroline is the only β-lactam that has affinity for PBP2a and is thus active against methicillin-resistant staphylococcal strains.
Glycopeptides Resistance to vancomycin in enterococci is due to the acquisition of a set of van genes that result in (1) the production of D-alanine-D-lactate—instead of the normal D-alanine-D-alanine—at the end of the peptidoglycan stem peptide and (2) the reduction of existing D-alanine-D-alanine terminated peptides. Vancomycin binds D-alanine-D-lactate with a thousandfold lower affinity than D-alanine-D-alanine. In a small number of cases, the van gene cassettes have been transferred from enterococci to S. aureus, with the consequent generation of full vancomycin resistance. Particularly in patients receiving prolonged courses of vancomycin, intermediate resistance to this drug has developed in S. aureus by a different mechanism: multiple chromosomal mutations that result in a thickened and poorly cross-linked cell wall in which multiple distant D-alanine-D-alanine stem peptide termini exist and bind vancomycin, impeding its access to the binding sites proximal to the cell membrane where new cell-wall synthesis occurs and where binding would block transpeptidase and transglycosylase enzymes. Susceptibility to telavancin, dalbavancin, and oritavancin is also reduced in strains that exhibit resistance or intermediate susceptibility to vancomycin, although in some cases strains may still be classified as susceptible on the basis of clinical interpretive criteria.
Aminoglycosides The most common mechanism of resistance is due to acquisition of plasmid genes encoding transferase enzymes that modify aminoglycosides by the addition of acetyl, adenyl, or phosphate groups; these added groups decrease the drugs’ binding affinity to their ribosomal target site. Transferases differ in which aminoglycosides they modify, and amikacin resistance occurs less often than resistance to gentamicin or tobramycin. More recently, plasmids encoding methyltransferases that modify the ribosomal site of aminoglycoside binding and confer resistance to all aminoglycosides have been found in enteric gram-negative bacteria. For streptomycin, a ribosomal protein mutation may cause resistance. In Pseudomonas aeruginosa, resistance may also occur through mutations causing increased expression of a chromosomally encoded efflux pump, MexXY.
Tetracyclines and Glycylcyclines For tetracyclines, resistance is most often plasmid mediated and attributable either to active efflux pumps, which are generally specific for tetracyclines, or to proteins that protect the ribosome from tetracycline action. Resistance to the glycylcycline tigecycline, which is not affected by the usual tetracycline resistance mechanisms, can occur through mutations that cause overexpression of certain broad-spectrum efflux pumps in Proteus species.
Macrolides, Ketolides, Lincosamides, and Streptogramins Resistance to macrolides, clindamycin, and quinupristin is most often due to plasmid-acquired methylases that modify the drug binding site on the ribosome. Resistance to quinupristin by this mechanism renders the quinupristin-dalfopristin combination bacteriostatic rather than bactericidal. Telithromycin, a ketolide, has an additional binding site on the ribosome and remains active in the presence of these methylases. Acquired genes encoding active efflux pumps can also contribute to resistance to macrolides in streptococci and resistance to macrolides, clindamycin, and dalfopristin in staphylococci. Plasmid-acquired drug-modifying enzymes in staphylococci can also cause resistance to quinupristin and dalfopristin. Macrolide resistance due to 23S rRNA mutations is uncommon in staphylococci and streptococci because of the multiple copies of the rRNA genes on the chromosomes of these species; such resistance may occur more frequently, however, in mycobacteria and Helicobacter pylori, which have only single chromosomal copies of these rRNA genes.
Chloramphenicol Resistance to chloramphenicol is most often due to a plasmid-encoded drug-modifying acetyltransferase.
Oxazolidinones Linezolid resistance has been seen in enterococci more often than in staphylococci and, in both organisms, is due to mutations in multiple copies of the 23S rRNA genes that reduce drug binding to the ribosome. A plasmid-acquired ribosomal methylase gene that confers resistance to both chloramphenicol and linezolid has also been found in some strains of staphylococci but is not yet widespread. Tedizolid may still be active against some but not all linezolid-resistant strains.
Mupirocin Resistance to mupirocin occurs by either mutation in the target leucyl-tRNA synthetase (low-level resistance) or the acquisition of a plasmid-encoded resistant tRNA synthetase (high-level resistance).
Sulfonamides and Trimethoprim Resistance to both of these antimetabolites is due to plasmid-acquired genes encoding resistant enzymes that bypass the inhibition of the native sensitive enzymes—a resistant dihydropteroate synthetase in the case of sulfonamides and a resistant dihydrofolate reductase in the case of trimethoprim.
Quinolones Resistance to quinolones is most often due either to chromosomal mutations altering the target enzymes DNA gyrase and DNA topoisomerase IV, with consequent reduction in drug binding, or to mutations that increase the expression of native broad-spectrum efflux pumps for which quinolones (among other compounds) are substrates. In addition, three types of genes can confer reduced susceptibility or low-level resistance by protecting target enzymes, modifying some quinolones, or pumping quinolones out of the cell (efflux). These genes are located on multidrug resistance plasmids that have spread worldwide. Their presence can promote the selection of higher levels of quinolone resistance linked to resistance to other antibacterial drugs that is encoded on the same plasmid.
Rifampin and Rifabutin Single mutations in the β subunit of RNA polymerase can cause high-level resistance to rifampin. Thus rifampin and other rifamycins are used for treatment of infections only in combination with other antibacterial drugs in order to prevent resistance.
Metronidazole Acquired resistance to metronidazole in Bacteroides species is rare. Such resistance has been reported in strains that lack endogenous nitroreductase activity or that have acquired nim genes responsible for further reduction of DNA-damaging nitroso intermediates to an inactive derivative. Active efflux and enhanced DNA repair mechanisms also have been associated with resistance.
Nitrofurantoin Resistance to nitrofurantoin in Escherichia coli can emerge through a series of mutations that progressively decrease the nitroreductase activity necessary for generating active nitrofuran metabolites.
Polymyxins Because of emerging multidrug resistance in gram-negative bacteria, colistin and polymyxin B are being used increasingly for infections due to resistant Enterobacteriaceae, P. aeruginosa, and Acinetobacter species. Rates of resistance vary. Resistance can emerge during therapy through mutations that cause reductions in the negative charge of the gram-negative bacterial cell surface, thereby reducing binding of the positively charged colistin.
Daptomycin The mechanisms of resistance to daptomycin are complex and involve mutations in several genes that can alter cell membrane charge and reduce daptomycin binding. Resistance to daptomycin is relatively infrequent but has emerged in some S. aureus strains with intermediate vancomycin susceptibility from patients treated with vancomycin but not with daptomycin. In some methicillin-resistant S. aureus (MRSA) strains, daptomycin resistance has been linked to acquired susceptibility to β-lactams; combinations of daptomycin and nafcillin have been successful for treatment of patients infected with resistant strains when daptomycin alone or in combination with other agents has failed. The mechanism of this effect is not yet clear.
PHARMACOKINETICS AND PHARMACODYNAMICS
The term pharmacokinetics describes the disposition of a drug in the body, whereas pharmacodynamics describes the determinants of drug action on the pathogen in relation to pharmacokinetic factors. An understanding of the principles governing these two areas is required for effective drug selection, dosing, and prevention of toxicities.
PHARMACOKINETICS
The process of drug disposition has four principal phases: absorption, distribution, metabolism, and excretion. These components determine the time course of drug concentrations in serum and subsequently the concentrations in other tissues and body fluids.
Absorption When a drug is given by a particular route, absorption is defined as the percentage of the dose that reaches the systemic circulation. For example, since IV administration provides direct access to the systemic circulation, 100% of a drug dose given IV is usually absorbed. The level of absorption becomes more relevant when non-IV routes are used—e.g., the oral, IM, SC, and topical routes. The percentage of a drug that is absorbed is termed its bioavailability. Examples of antibacterial agents with a high oral bioavailability include metronidazole, levofloxacin, and linezolid. IV administration and oral dosing for highly bioavailable agents usually give equivalent results. Many factors can affect a drug’s oral bioavailability, including the timing of food consumption relative to drug administration, drug-metabolizing enzymes, efflux transporters, concentration-dependent solubility, and acid degradation. Underlying conditions such as diarrhea or ileus can also affect the site of drug absorption and thereby alter bioavailability. Certain orally administered drugs have lower bioavailability because of the first-pass effect—the process by which drugs are absorbed in the small intestine through the portal circulation and then directly transported to the liver for metabolism.
Distribution Distribution describes the process by which a drug transfers reversibly between the general circulation and the tissues. After absorption into the general circulation and the central compartment (the extensively perfused organs), the drug will also distribute into the peripheral compartment (less well-perfused tissues). The volume of distribution (Vd) is a pharmacokinetic parameter that describes the amount of drug in the body at a given time relative to the measured serum concentration. Properties such as the drug’s lipophilicity, partition coefficient within different body tissues, and protein binding; blood flow; and pH can affect the volume of distribution. Drugs with a small volume of distribution are limited to certain areas within the body (typically extracellular fluid), whereas those with a higher volume of distribution penetrate extensively into tissues throughout the body. Antibacterial drugs can bind to serum proteins, and a given drug is usually described as either poorly or highly protein bound. Only the unbound (free) drug is active and available to exert antibacterial effects. For example, because tigecycline is highly protein bound and also has a large volume of distribution, concentrations of free drug in the serum are low.
Metabolism Metabolism is the chemical transformation of a drug by the body. This modification can occur within several areas; the liver is the organ most commonly involved. Drugs are metabolized by enzymes, but enzyme systems have a finite capacity to metabolize a substrate drug. If a drug is given in a dose at which the concentration does not exceed the rate of metabolism, then the metabolic process is generally linear. If the dose exceeds the amount that can be metabolized, drug accumulation and potential toxicity may occur. Drugs are metabolized through phase I or phase II reactions. In phase I reactions, the drug is made more polar through dealkylation, hydroxylation, oxidation, and deamination. Polarity facilitates drug removal from the body. Phase II reactions, which include glucuronidation, sulfation, and acetylation, result in compounds larger and more polar than the parent drug. Both phases usually inactivate the parent drug, but some drugs are rendered more active. The cytochrome P450 (CYP) enzyme system is responsible for phase I reactions and is generally found in the liver. CYP3A4 is a common subfamily within this system that is responsible for the majority of drug metabolism. Antibacterial drugs can be substrates, inhibitors, or inducers of a particular CYP enzyme. Inducers, such as rifampin, can increase the production of CYP enzymes and consequently increase the metabolism of other drugs. Inhibitors, such as quinupristin-dalfopristin, cause a decrease in enzyme activity (or competition for CYP substrate) and therefore an increase in the concentration of the interacting drug.
Excretion Excretion describes the body’s mechanisms of drug elimination. Drugs can be eliminated through more than one mechanism. Renal clearance is the most common route and includes elimination through glomerular filtration, tubular secretion, and/or passive diffusion. Some agents have nonrenal clearance and rely on the biliary tree or the intestine for excretion. Excretion affects the half-life of a drug—i.e., the time it takes for the blood concentration of a drug to decrease by one-half. This value can range from minutes to days. Approximately five to seven half-lives are required for a drug to reach steady state when multiple doses are given in a time frame shorter than the half-life itself. Drug half-life and overall clearance can be extended if the organ responsible for clearance is impaired. Patients with renal or hepatic impairment may require dose adjustments that take delayed clearance into account and prevent toxicities from drug accumulation. For example, imipenem is cleared predominantly through glomerular filtration, and in the presence of renal impairment the dosing interval is typically increased to account for the increased half-life.
PHARMACODYNAMICS
The term pharmacodynamics describes the relationship between the serum concentrations that determine the efficacy of the drug and the serum concentrations that produce the toxic effects of the drug. For an antibacterial agent, the pharmacodynamic focus is the type of drug exposure needed for optimal antibacterial effect in relation to the minimal inhibitory concentration (MIC)—the lowest drug concentration that inhibits the visible growth of a microorganism under standardized laboratory conditions. Antibacterial effect usually correlates with one of the following parameters: (1) ratio of peak serum concentration to the MIC (Cmax/MIC), (2) ratio of the area under the concentration–time curve to the MIC (AUC/MIC), or (3) duration of concentrations above the MIC (T>MIC) (Fig. 170-2).
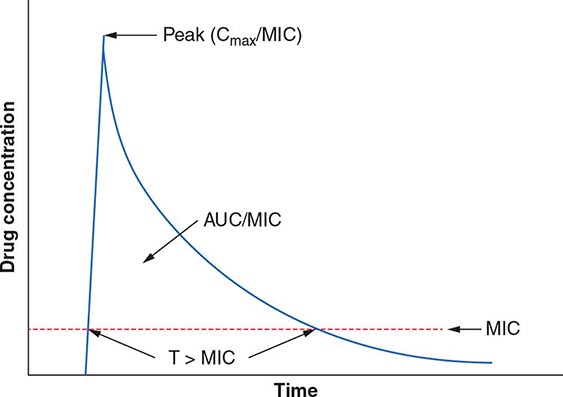
FIGURE 170-2 Pharmacokinetic and pharmacodynamic model predicting efficacy of antibacterial drugs. AUC, area under the time–concentration curve; Cmax, peak serum concentration of drug; MIC, minimal inhibitory concentration; T>MIC, duration of drug concentrations above the MIC.
For concentration-dependent killing agents, as the designation implies, the higher the drug concentration, the higher the rate and extent of bacterial killing. Aminoglycosides fit into the Cmax/MIC model of pharmacodynamics activity, and a particular peak serum concentration is often targeted to achieve optimal killing. Fluoroquinolones exemplify antibacterial agents for which the AUC/MIC is a predictor of efficacy. For example, studies have found that an AUC/MIC ratio of >30 will maximize killing of S. pneumoniae by fluoroquinolones. In contrast, time-dependent killing agents reach a ceiling at which higher concentrations do not result in increased effect. Instead, these agents are active against bacteria only when the drug concentration is above the MIC. The T>MIC predicts clinical efficacy for all β-lactams. The longer the concentration of the β-lactam remains above the MIC for an infecting pathogen during the dosing interval, the greater the killing effect. For some drug classes, such as aminoglycosides, a postantibiotic effect—the delayed regrowth of surviving bacteria after exposure to an antibiotic—supports less frequent dosing.
APPROACH TO THERAPY
The approach to antibiotic therapy is driven by host factors, site of infection, and local resistance profiles of suspected or known pathogens. Further, national and local drug shortages and formulary restrictions can affect available therapies. Regular monitoring of the patient and collection of laboratory data should be undertaken to streamline antibacterial therapy as appropriate and to investigate the possibility of treatment failure if the patient fails to respond appropriately.
EMPIRICAL AND DIRECTED THERAPY
Therapy is considered empirical when the causative agent has yet to be determined and therapeutic decisions are based on the severity of illness and the clinician’s assessment of likely pathogens in light of the clinical syndrome, the patient’s medical conditions and prior therapy, and relevant epidemiologic factors. For patients with severe illness, empirical therapy often takes the form of an antibacterial combination that provides broad coverage of diverse agents and thus ensures adequate treatment of possible pathogens while additional data are being collected. Directed therapy is predicated on identification of the pathogen, determination of its susceptibility profile, and establishment of the extent of the infection. Directed therapy generally allows the use of more targeted and narrower-spectrum antibacterial agents than does empirical therapy.
Information on epidemiology, exposures, and local antibacterial susceptibility patterns can help guide empirical therapy. When empirical treatment is clinically appropriate, care should be taken to obtain clinical specimens for microbiologic analysis before the initiation of therapy and to de-escalate therapy as new information is obtained about the patient’s clinical condition and the causal pathogens. De-escalation to the point of directed therapy can limit unnecessary risks to the patient as well as the risk of emergence of antibacterial resistance.
SITE OF INFECTION
The site of infection is a consideration in antibacterial therapy, largely because of the differing abilities of drugs to penetrate and achieve adequate concentrations at particular body sites. For example, to be effective in the treatment of meningitis, an agent must (1) be able to cross the blood–brain barrier and reach adequate concentrations in the cerebrospinal fluid (CSF) and (2) be active against the relevant pathogen(s). Dexamethasone, administered with or 15–20 min before the first dose of an antibacterial drug, has been shown to improve outcomes in patients with acute bacterial meningitis, but its use may reduce penetration of some antibacterial agents, such as vancomycin, into the CSF. In this case, rifampin is added because its penetration is not reduced by dexamethasone. Infections at other sites where either pathogens are protected from normal host defenses or penetration of an antibacterial drug is suboptimal include osteomyelitis, prostatitis, intraocular infections, and abscesses. In such cases, consideration must be given to the mechanism of drug delivery (e.g., intravitreal injections) as well as to the role of interventions to drain, debride, or otherwise reduce the barriers to effective antibacterial therapy.
HOST FACTORS
Host factors, including immune function, pregnancy, allergies, age, renal and hepatic function, drug–drug interactions, comorbid conditions, and occupational or social exposures, should be considered.
Immune Dysfunction Patients with deficits in immune function that blunt the response to bacterial infection, including neutropenia, deficient humoral immunity, and asplenia (either surgical or functional), are all at increased risk of severe bacterial infection. Such patients should be treated aggressively and often broadly in the early stages of suspected infection pending results of microbiologic tests. For asplenic patients, treatment should include coverage of encapsulated organisms, particularly S. pneumoniae, that may cause rapidly life-threatening infection.
Pregnancy Pregnancy affects decisions regarding antibacterial therapy in two respects. First, pregnancy is associated with an increased risk of particular infections (e.g., those caused by Listeria). Second, the potential risks to the fetus that are posed by specific drugs must be considered. As for other drugs, the safety of the vast majority of antibacterial agents in pregnancy has not been established, and such agents are grouped in categories B and C by the U.S. Food and Drug Administration. Drugs in categories D and × are contraindicated in pregnancy or lactation due to established risks. The risks associated with antibacterial use in pregnancy and during lactation are summarized in Table 170-2.
|
RISKS ASSOCIATED WITH USE OF ANTIBACTERIAL DRUGS IN PREGNANCY AND LACTATION |
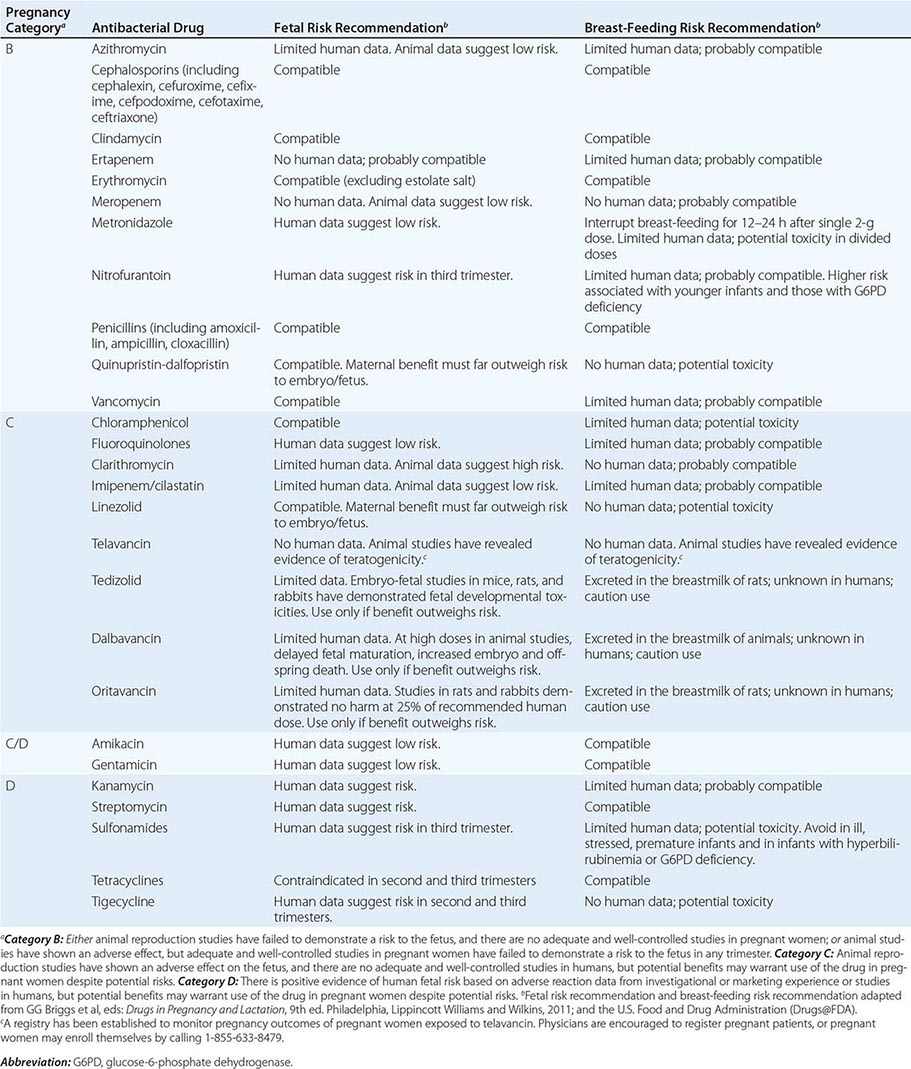
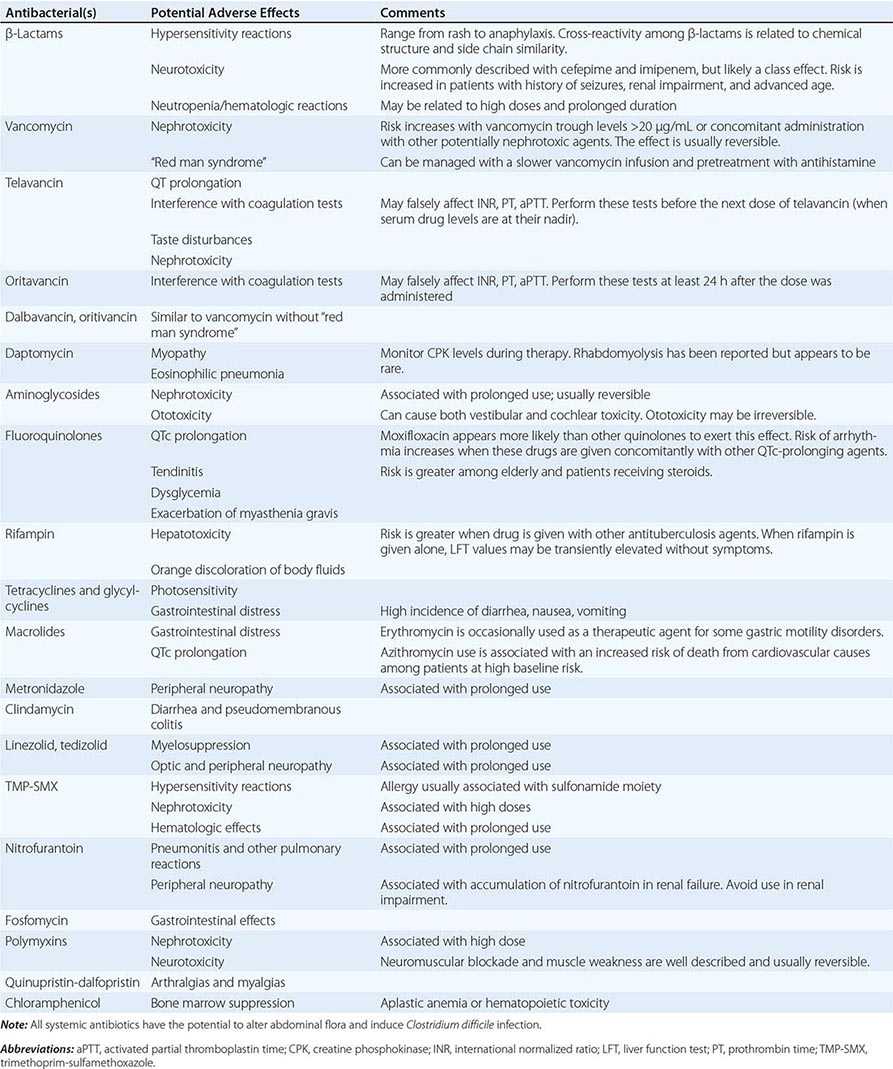
Allergies Allergies to antibiotics are among the most common allergies reported, and an allergy history should be obtained whenever possible before therapy is chosen. A detailed allergy history can shed light on the type of reaction experienced previously and on whether rechallenge with the same or a related medication is advisable (and, if so, under what circumstances). Allergies to the penicillins are most common. Although as many as 10% of patients may report an allergy to penicillin, studies suggest that up to 90% of these patients could tolerate a penicillin or cephalosporin. Adverse effects (Table 170-3) should be distinguished from true allergies to ensure appropriate selection of antibacterial therapy.
|
COMMON ADVERSE REACTIONS TO ANTIBACTERIAL AGENTS |
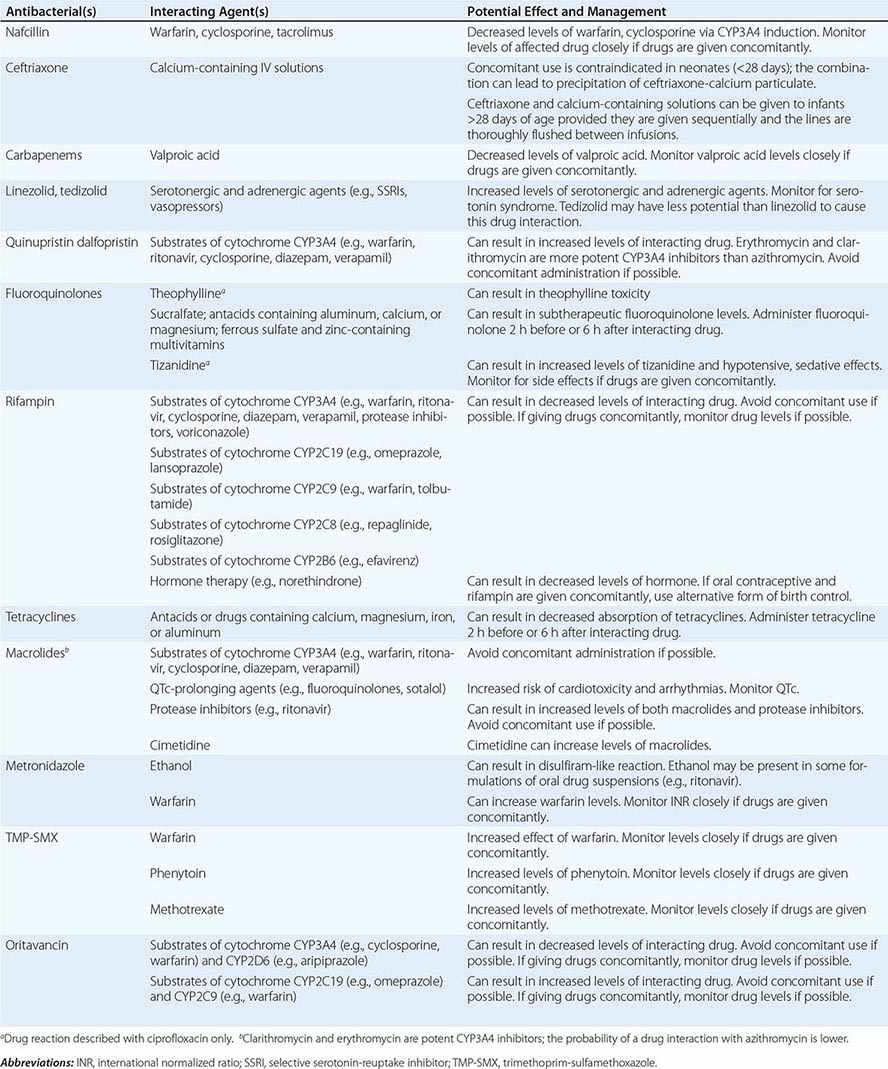
Drug–Drug Interactions Patients commonly receive other drugs that may interact with antibacterial agents. A summary of the most common drug–drug interactions, by antibacterial class, is provided in Table 170-4.
|
SIGNIFICANT ANTIBACTERIAL DRUG INTERACTIONS |
Exposures Exposures, both occupational and social, may provide clues to likely pathogens. When relevant, inquiries about exposure to ill contacts, animals, insects, and water should be included in the history, along with sites of residence and travel.
Other Host Factors Age, renal and hepatic function, and comorbid conditions are all considerations in the choice of and schedule for therapy. Dose adjustments should be made accordingly. In patients with decreased or unreliable oral absorption, IV therapy may be preferred to ensure adequate blood levels of drug and delivery of the antibacterial agent to the site of infection.
DURATION OF THERAPY
Whether empirical or directed, the duration of therapy should be planned in most clinical situations. Guidelines that synthesize available literature and expert opinion provide recommendations on therapy duration that are based on infecting organism, organ system, and patient factors. For example, the American Heart Association has published guidelines endorsed by the Infectious Diseases Society of America (IDSA) on diagnosis, antibacterial therapy, and management of complications of infective endocarditis. Similar guidelines from the IDSA exist for bacterial meningitis, catheter-associated urinary tract infections, intraabdominal infections, community- and hospital-acquired pneumonia, and other infections.
FAILURE OF THERAPY
If a patient does not respond to therapy, investigations often should include the collection of additional specimens for microbiologic testing and imaging as indicated. Failure to respond can be the result of an antibacterial regimen that does not address the underlying causative organism, the development of resistance during therapy, or the existence of a focus of infection at a site poorly penetrated by systemic therapy. Some infections may also require surgical interventions for cure (e.g., large abscesses, myonecrosis). Fever due to allergic drug reactions can sometimes complicate assessment of the patient’s response to antibacterial treatment.
EXPERT GUIDANCE
Selected websites with the most up-to-date information and guidance for the clinician include the following:
• Johns Hopkins ABX Guide (www.hopkins-abxguide.org)
• IDSA Practice Guidelines (www.idsociety.org/IDSA_Practice_Guidelines/)
• Center for Disease Dynamics, Economics and Policy Resistance Map (www.cddep.org/map)
• CDC Antibiotic/Antimicrobial Resistance (www.cdc.gov/drugresistance/)
CLINICAL USE OF ANTIBACTERIAL AGENTS
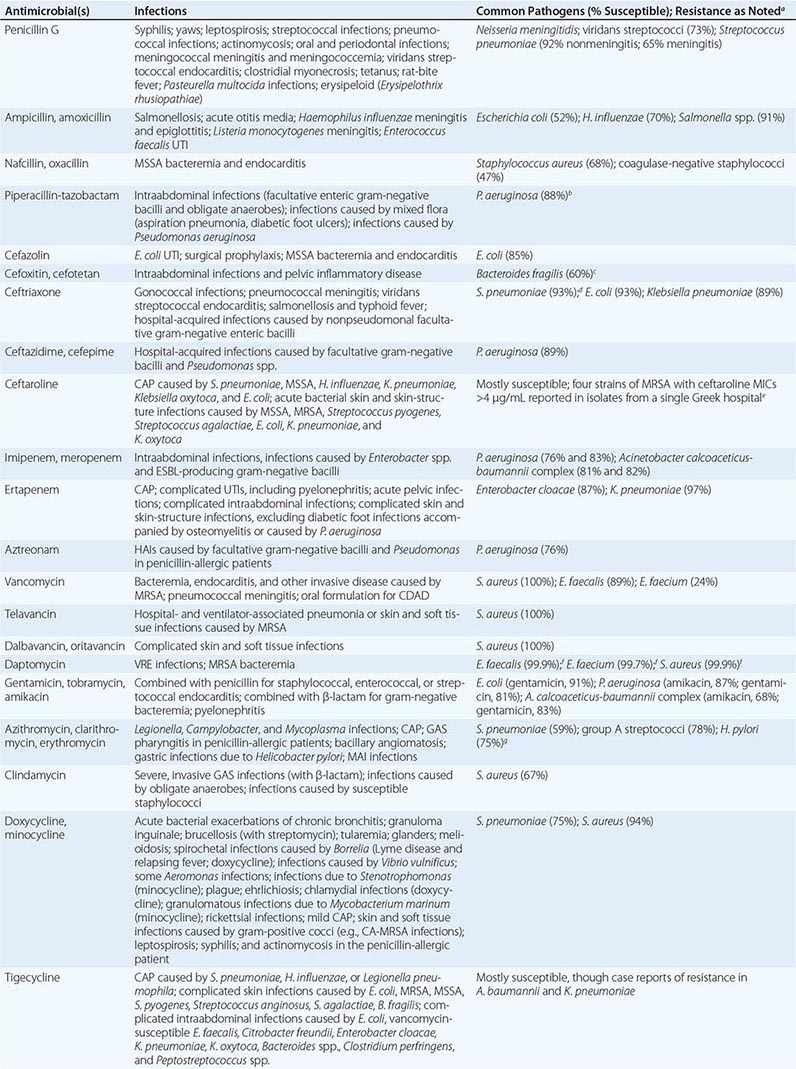
The clinical application of antibacterial therapy is guided by the spectrum of the agent and the suspected or known target pathogen. Infections for which specific antibacterial agents are among the drugs of choice are listed, along with associated pathogens and susceptibility data, in Table 170-5. Resistance rates of specific organisms are dynamic and should be taken into account in the approach to antibacterial therapy. While national resistance rates can serve as a reference, the most useful reference for the clinician is the most recent local laboratory antibiogram, which provides details on local resistance patterns, often on an annual or semiannual basis.
|
DRUG INDICATIONS FOR SPECIFIC INFECTIONS, ASSOCIATED PATHOGENS, AND SAMPLE SUSCEPTIBILITY RATES |
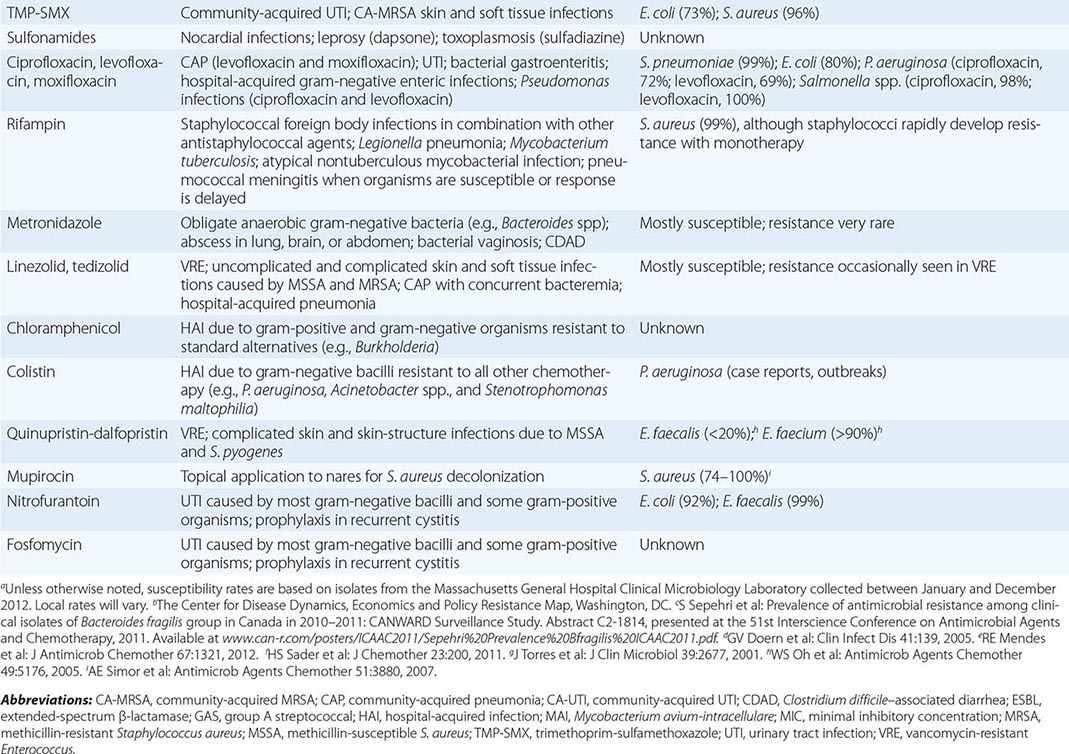
β-LACTAMS
The β-lactam class of antibiotics consists of penicillins, cephalosporins, carbapenems, and monobactams. The term β-lactam reflects the drugs’ four-membered lactam ring, which is their core structure. The differing side chains among the agents of this family determine the spectrum of activity. All β-lactams exert a bactericidal effect by inhibiting bacterial cell-wall synthesis. The β-lactams are classified as time-dependent killing agents; therefore, their clinical efficacy is best correlated with the proportion of the dosing interval during which the drug levels remain above the MIC for the pathogenic organism.
Penicillins and β-Lactamase Inhibitors Penicillin, the first β-lactam, was discovered in 1928 by Alexander Fleming. Natural penicillins, such as penicillin G, are active against non-β-lactamase-producing gram-positive and gram-negative bacteria, anaerobes, and some gram-negative cocci. Penicillin G is used for penicillin-susceptible streptococcal infections, pneumococcal and meningococcal meningitis, enterococcal endocarditis, and syphilis. The antistaphyloccocal penicillins, which have potent activity against methicillin-susceptible S. aureus (MSSA), include nafcillin, oxacillin, dicloxacillin, and flucloxacillin. Aminopenicillins, such as ampicillin and amoxicillin, provide added coverage beyond penicillin against gram-negative cocci, such as Haemophilus influenzae, and some Enterobacteriaceae, including E. coli, Proteus mirabilis, Salmonella, and Shigella. The aminopenicillins are hydrolyzed by many common β-lactamases. These drugs are commonly used for otitis media, respiratory tract infections, intraabdominal infections, endocarditis, meningitis, and urinary tract infections. The antipseudomonal penicillins include ticarcillin and piperacillin. These penicillin groups generally offer adequate anaerobic coverage; the exceptions are Bacteroides species (such as Bacteroides fragilis), which produce β-lactamases and are generally resistant. The rising prevalence of β-lactamase-producing bacteria has led to the increased use of β-lactam/β-lactamase inhibitor combinations, such as ampicillin-sulbactam, amoxicillin-clavulanate, ticarcillin-clavulanate, and piperacillin-tazobactam. The β-lactamase inhibitors themselves do not have antibacterial activity (with the exception of sulbactam, which has activity against Acinetobacter baumannii) but typically inhibit the S. aureus class A β-lactamase, β-lactamases of H. influenzae and Bacteroides species, and a number of plasmid-encoded β-lactamases. These combination agents are typically used when broader-spectrum coverage is needed—e.g., in pneumonia and intraabdominal infections. Piperacillin-tazobactam is a useful agent for broad coverage in febrile neutropenic patients. The combination agents, however, are not effective against organisms that produce AmpC β-lactamases or carbapenemases.
Cephalosporins The cephalosporin drug class encompasses five generations determined by spectrum of antibacterial activity. The first generation (cefazolin, cefadroxil, cephalexin) largely has activity against gram-positive bacteria, with some additional activity against E. coli, P. mirabilis, and K. pneumoniae. First-generation cephalosporins are commonly used for infections caused by MSSA and streptococci (e.g., skin and soft tissue infections). Cefazolin is a popular choice for surgical prophylaxis against skin organisms. The second generation (cefamandole, cefuroxime, cefaclor, cefprozil, cefuroxime axetil, cefoxitin, cefotetan) has additional activity against H. influenzae and Moraxella catarrhalis. Cefoxitin and cefotetan have potent activity against anaerobes as well. Second-generation cephalosporins are used to treat community-acquired pneumonia because of their activity against S. pneumoniae, H. influenzae, and M. catarrhalis. They are also used for other mild or moderate infections, such as acute otitis media and sinusitis. The third-generation cephalosporins are characterized by greater potency against gram-negative bacilli and reduced potency against gram-positive cocci. These cephalosporins, which include cefoperazone, cefotaxime, ceftazidime, ceftriaxone, cefdinir, cefixime, and cefpodoxime, are used for infections caused by Enterobacteriaceae, although resistance is an increasing concern. It is noteworthy that ceftazidime is the only third-generation cephalosporin with activity against P. aeruginosa but lacks activity against gram-positive bacteria. This drug is frequently used for pulmonary infections in cystic fibrosis and febrile neutropenia. Ceftriaxone penetrates the CSF and can be used to treat meningitis caused by H. influenzae, N. meningitidis, and susceptible strains of S. pneumoniae. It is also used for the treatment of later-stage Lyme disease. The fourth generation includes cefepime and cefpirome, broad-coverage agents that provide potent activity against both gram-negative bacilli, including P. aeruginosa, and gram-positive cocci. The fourth generation has clinical applications similar to those of the third generation and can be used in bacteremia, pneumonia, skin and soft tissue infections, and urinary tract infections caused by susceptible bacteria. Cefepime is also commonly used in febrile neutropenia. Ceftaroline, a fifth-generation cephalosporin, differs from the other cephalosporins in its added activity against MRSA, which is resistant to all other β-lactams. Ceftaroline’s gram-negative activity is similar to that of the third-generation cephalosporins but does not include P. aeruginosa. Ceftaroline is efficacious in community-acquired pneumonia and skin infections, but few data are available on its use for more serious infections, such as bacteremia.
Carbapenems With a few exceptions for cefepime, all penicillins and cephalosporins are ineffective in the presence of ESBLs. Carbapenems, including doripenem, imipenem, meropenem, and ertapenem, offer the most reliable coverage for strains containing ESBLs. All carbapenems have broad activity against gram-positive cocci, gram-negative bacilli, and anaerobes. None is active against MRSA, but all are active against MSSA, Streptococcus species, and Enterobacteriaceae. Ertapenem is the only carbapenem that has poor activity against P. aeruginosa and Acinetobacter. Imipenem is active against penicillin-susceptible Enterococcus faecalis but not Enterococcus faecium. Carbapenems are not active against Enterobacteriaceae containing carbapenemases. Stenotrophomonas maltophilia and some Bacillus species are intrinsically resistant to carbapenems because of a zinc-dependent carbapenemase.
Monobactams Aztreonam is the sole monobactam. Its activity is limited to gram-negative bacteria and includes P. aeruginosa and most other Enterobacteriaceae. This drug is inactivated by ESBLs and carbapenemases. The principal use for aztreonam is as an alternative to penicillins, cephalosporins, or carbapenems in patients with serious β-lactam allergy. Aztreonam is structurally related to ceftazidime and should be used cautiously in individuals with a serious ceftazidime allergy. It is commonly used in febrile neutropenia and intraabdominal infections. Aztreonam does not penetrate the CSF and should not be used for treatment of meningitis.
Adverse Reactions to β-Lactam Drugs Agents within the β-lactam class are known for several adverse effects. Gastrointestinal side effects, mainly diarrhea, are common, but hypersensitivity reactions constitute the most common adverse effect of β-lactams. The reactions’ severity can range from rash to anaphylaxis, but the rate of true anaphylactic reactions is only 0.05%. An individual with an accelerated IgE-mediated reaction to one β-lactam agent may still receive another agent within the class, but caution should be taken to choose a β-lactam that has a dissimilar side chain and a low level of cross-reactivity. For example, the second-, third-, and fourth-generation cephalosporins and the carbapenems display very low cross-reactivity in patients with penicillin allergy. Aztreonam is the only β-lactam that has no cross-reactivity with the penicillin group. In cases of severe allergy, desensitization (a graded challenge) to the indicated β-lactam, with close monitoring, may be warranted if other antibacterial options are not suitable.
β-Lactams can rarely cause serum sickness, Stevens-Johnson syndrome, nephropathy, hematologic reactions, and neurotoxicity. Neutropenia appears to be related to high doses or prolonged use. Neutropenia and interstitial nephritis caused by β-lactams generally resolve upon discontinuation of the agent. Imipenem and cefepime are associated with an increased risk of seizure, but this risk is likely a class effect and related to high doses or doses that are not adjusted in renal impairment.
GLYCOPEPTIDES
The glycopeptide antibiotics include vancomycin and telavancin. Vancomycin has activity against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci (including S. pneumoniae), and enterococci. It is not active against gram-negative organisms. Vancomycin also displays activity against Bacillus species, Corynebacterium jeikeium, Listeria monocytogenes, and gram-positive anaerobes such as Peptostreptococcus, Actinomyces, Clostridium, and Propionibacterium species. Vancomycin has several important clinical uses. It is used for serious infections caused by MRSA, including health care–associated pneumonia, bacteremia, osteomyelitis, and endocarditis. It is also commonly used for skin and soft tissue infections. Oral vancomycin is not absorbed systemically and is reserved for the treatment of Clostridium difficile infection. Vancomycin is also an alternative for the treatment of infections caused by MSSA in patients who cannot tolerate β-lactams. Resistance to vancomycin is a rising concern. Strains of vancomycin-intermediate S. aureus (VISA) and vancomycin-resistant enterococci (VRE) are not uncommon. Vancomycin appears to be a concentration-dependent killer, with AUC/MIC ratio being the best predictor of efficacy (Fig. 170-2). Guidelines recommend targeting a vancomycin trough level of 15–20 μg/mL in MRSA infections in order to maintain an AUC/MIC ratio >400. When using vancomycin, clinicians should monitor for nephrotoxicity. The risk increases when trough levels are >20 μg/mL. Concomitant therapy with other nephrotoxic agents, such as aminoglycosides, also increases the risk of nephrotoxicity. Ototoxicity was reported with early formulations of vancomycin but is currently uncommon because purer formulations are available. Both of these adverse effects are reversible upon discontinuation of vancomycin. Clinicians should be aware of the “red man syndrome,” a common reaction that presents as a rapid onset of erythematous rash or pruritus on the head, face, neck, and upper trunk. This reaction is caused by histamine release from basophils and mast cells and can be treated with diphenhydramine and slowing of the vancomycin infusion.
Telavancin, dalbavancin, and oritavancin are structurally similar to vancomycin and are referred to as lipoglycopeptides. They have antibacterial activity against S. aureus (including MRSA and some strains of VISA and vancomycin-resistant S. aureus [VRSA]), streptococci, and enterococci. They also have good activity against anaerobic gram-positive organisms except for Lactobacillus and some Clostridium species. The clinical efficacy of telavancin has been demonstrated in both skin and soft tissue infections and nosocomial pneumonia, and the efficacy of dalbavancin and oritivancin has been shown in skin and soft tissue infections. The vancomycin resistance phenotype may reduce the potency of all three lipoglycopeptides, but the rate of resistance to these drugs among S. aureus and enterococci has been low. Adverse effects of telavancin include insomnia, a metallic taste, nephrotoxicity, and gastrointestinal side effects. Clinicians should be aware of the potential for electrocardiographic QTc prolongation that can increase the risk of cardiac arrhythmias when telavancin is used concomitantly with other QTc-prolonging agents. Telavancin may interfere with certain coagulation tests (e.g., causing false elevations in prothrombin time). Dalbavancin and oritavancin have safety profiles similar to that of vancomycin.
LIPOPEPTIDES
Daptomycin is a lipopeptide antibiotic with activity against a broad range of gram-positive organisms. This drug is active against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci, and enterococci. Daptomycin remains active against enterococci that are resistant to vancomycin. In addition, it exhibits activity against Bacillus, Corynebacterium, Peptostreptococcus, and Clostridium species. Daptomycin’s pharmacodynamic parameter for efficacy is concentration-dependent killing. Resistance to daptomycin is rare, but MICs may be higher for VISA strains. Daptomycin is efficacious in skin and soft tissue infections, bacteremia, endocarditis, and osteomyelitis. It is an important alternative for MRSA and other gram-positive infections when bactericidal therapy is needed and vancomycin cannot be used. Daptomycin is generally well tolerated, and its main toxicity consists of elevation of creatinine phosphokinase (CPK) levels and myopathy. CPK should be monitored during daptomycin treatment, and the drug should be discontinued if muscular toxicities occur. There have also been case reports of reversible eosinophilic pneumonia associated with daptomycin use.
AMINOGLYCOSIDES
The aminoglycosides are a class of antibacterial agents with concentration-dependent activity against most gram-negative organisms. The most commonly used aminoglycosides are gentamicin, tobramycin, and amikacin, although others, such as streptomycin, kanamycin, neomycin, and paromomycin, may be used in special circumstances. Aminoglycosides have a significant dose-dependent postantibiotic effect, meaning that they have an antibacterial effect even after serum drug levels are undetectable. The postantibiotic effect and concentration-dependent killing form the rationale behind extended-interval aminoglycoside dosing, in which a larger dose is given once daily rather than smaller doses multiple times daily. Aminoglycosides are active against gram-negative bacilli, such as Enterobacteriaceae, P. aeruginosa, and Acinetobacter. They also enhance the activity of cell wall–active agents such as β-lactams or vancomycin in some gram-positive bacteria, including staphylococci and enterococci. This combination therapy is termed synergistic because the effect of both agents provides a killing effect greater than would be predicted from the effects of either agent alone. Amikacin and streptomycin have activity against Mycobacterium tuberculosis, and amikacin has activity against Mycobacterium avium-intracellulare. The aminoglycosides do not have activity against anaerobes, S. maltophilia, or Burkholderia cepacia. Aminoglycosides are used in clinical practice in a variety of infections caused by gram-negative organisms, including bacteremia and urinary tract infections. They are frequently used alone or in combination for the treatment of P. aeruginosa infection. When used in combination with a cell wall–active agent, gentamicin and streptomycin are also important for the treatment of gram-positive bacterial endocarditis. All aminoglycosides can cause nephrotoxicity and ototoxicity. The risk of nephrotoxicity is related to the dose and duration of therapy as well as the concomitant use of other nephrotoxic agents. Nephrotoxicity is usually reversible, but ototoxicity can be irreversible.
MACROLIDES AND KETOLIDES
The macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) are classes of antibiotics that inhibit protein synthesis. Compared with erythromycin (the older antibiotic), azithromycin and clarithromycin have better oral absorption and tolerability. Azithromycin, clarithromycin, and telithromycin all have broader spectra of activity than erythromycin, which is less frequently used. These agents are commonly used in the treatment of upper and lower respiratory tract infections caused by S. pneumoniae, H. influenzae, M. catarrhalis, and atypical organisms (e.g., Chlamydia pneumoniae, Legionella pneumophila, and Mycoplasma pneumoniae); group A streptococcal pharyngitis in penicillin-allergic patients; and nontuberculous mycobacterial infections (e.g., caused by M. marinum and M. chelonae) as well as in the prophylaxis and treatment of M. avium-intracellulare infection in patients with HIV/AIDS and in combination therapy for H. pylori infection and bartonellosis. Enterobacteriaceae, Pseudomonas species, and Acinetobacter species are intrinsically resistant to macrolides as a result of decreased membrane permeability, although azithromycin is active against gram-negative diarrheal pathogens. The major adverse effects of this drug class include nausea, vomiting, diarrhea and abdominal pain, prolongation of QTc interval, exacerbation of myasthenia gravis, and tinnitus. Azithromycin specifically has been associated with an increased risk of death, especially among patients with underlying heart disease, because of the risk of QTc interval prolongation and torsades de pointes. Erythromycin, clarithromycin, and telithromycin inhibit the CYP3A4 hepatic drug-metabolizing enzyme and can result in increased levels of coadministered drugs, including benzodiazepines, statins, warfarin, cyclosporine, and tacrolimus. Azithromycin does not inhibit CYP3A4 and lacks these drug–drug interactions.
CLINDAMYCIN
Clindamycin is a lincosamide antibiotic and is bacteriostatic against some organisms and bactericidal against others. It is used most often to treat bacterial infections caused by anaerobes (e.g., B. fragilis, Clostridium perfringens, Fusobacterium species, Prevotella melaninogenicus, and Peptostreptococcus species) and susceptible staphylococci and streptococci. Clindamycin is used for treatment of dental infections, anaerobic lung abscess, and skin and soft tissue infections. It is used together with bactericidal agents (penicillins or vancomycin) to inhibit new toxin synthesis in the treatment of streptococcal or staphylococcal toxic shock syndrome. Other uses include treatment of infections caused by Capnocytophaga canimorsus, a component of combination therapy for malaria and babesiosis, and therapy for toxoplasmosis. Clindamycin has excellent oral bioavailability. Adverse effects include nausea, vomiting, diarrhea, C. difficile–associated diarrhea and pseudomembranous colitis, maculopapular rash, and (rarely) Stevens-Johnson syndrome.
TETRACYCLINES AND GLYCYLCYCLINES
The tetracyclines (doxycycline, minocycline, and tetracycline) and the glycylcyclines (tigecycline) inhibit protein synthesis and are bacteriostatic. These drugs have wide clinical uses. They are used in the treatment of skin and soft tissue infections caused by gram-positive cocci (including MRSA), spirochetal infections (e.g., Lyme disease, syphilis, leptospirosis, and relapsing fever), rickettsial infections (e.g., Rocky Mountain spotted fever), atypical pneumonia, sexually transmitted infections (e.g., Chlamydia trachomatis infection, lymphogranuloma venereum, and granuloma inguinale), infections with Nocardia and Actinomyces, brucellosis, tularemia, Whipple’s disease, and malaria. Tigecycline, the only approved agent in the glycylcycline class, is a derivative of minocycline and is indicated in the treatment of infections due to MRSA, vancomycin-sensitive enterococci, many Enterobacteriaceae, and Bacteroides species. Tigecycline has no activity against P. aeruginosa. It has been used in combination with colistin for the treatment of serious infections with multidrug-resistant gram-negative organisms. A pooled analysis of 13 clinical trials found an increased risk of death and treatment failure among patients treated with tigecycline alone. Tetracyclines have reduced absorption when coadministered with calcium- and iron-containing compounds, including milk, and doses should be spaced at least 2 h apart. The major adverse reactions to both of these classes are nausea, vomiting, diarrhea, and photosensitivity. Tetracyclines have been associated with fetal bone-growth abnormalities and should be avoided during pregnancy and in the treatment of children <8 years old.
TRIMETHOPRIM-SULFAMETHOXAZOLE
Trimethoprim-sulfamethoxazole (TMP-SMX) is an antibiotic whose two components both inhibit folate synthesis and produce antibacterial activity. TMP-SMX is active against gram-positive bacteria such as staphylococci and streptococci; however, its use against MRSA is usually limited to community-acquired infections, and its activity against Streptococcus pyogenes may not be reliable. TMP-SMX is also active against many gram-negative bacteria, including H. influenzae, E. coli, P. mirabilis, N. gonorrhoeae, and S. maltophilia. TMP-SMX does not have activity against anaerobes or P. aeruginosa. It has many uses because of its wide spectrum of activity and high oral bioavailability. Urinary tract infections, skin and soft tissue infections, and respiratory tract infections are among the common uses. Another important indication is for both prophylaxis and treatment of Pneumocysitis jirovecii infections in immunocompromised patients. Resistance to TMP-SMX has limited its use against many Enterobacteriaceae. Resistance rates among urinary isolates of E. coli are almost 25% in the United States. The most common adverse reactions associated with TMP-SMX are gastrointestinal effects such as nausea, vomiting, and diarrhea. In addition, rash is a common allergic reaction and may preclude the subsequent use of other sulfonamides. With prolonged use, leukopenia, thrombocytopenia, and granulocytopenia can develop. TMP-SMX can also cause nephrotoxicity, hyperkalemia, and hyponatremia, which are more common at high doses. TMP-SMX has several important interactions with other drugs (Table 170-4), including warfarin, phenytoin, and methotrexate.
FLUOROQUINOLONES
The fluoroquinolones include norfloxacin, ciprofloxacin, ofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. Ciprofloxacin and levofloxacin have the broadest spectrum of activity against gram-negative bacteria, including P. aeruginosa (similar to that of third-generation cephalosporins). Because of the risk of selection of resistance during fluoroquinolone treatment of serious pseudomonal infections, these agents are usually used in combination with an antipseudomonal β-lactam. Levofloxacin, moxifloxacin, and gemifloxacin have additional gram-positive activity, including that against S. pneumoniae and some strains of MSSA, and are used for treatment of community-acquired pneumonia. Strains of MRSA are commonly resistant to all fluoroquinolones. Moxifloxacin is used as one component of second-line regimens for multidrug-resistant tuberculosis. Fluoroquinolones exhibit concentration-dependent killing, are well absorbed orally, and have elimination half-lives that usually support once- or twice-daily dosing. Oral coadministration with compounds containing high concentrations of aluminum, magnesium, or calcium can reduce fluoroquinolone absorption. Their penetration into prostate tissue supports their use for bacterial prostatitis. Fluoroquinolones are generally well tolerated but can cause CNS stimulatory effects, including seizures; glucose dysregulation; and tendinopathy associated with Achilles tendon rupture, particularly in older patients, organ transplant recipients, and patients taking glucocorticoids. Worsening of myasthenia gravis also has been associated with quinolone use. Moxifloxacin causes modest prolongation of the QTc interval and should be used with caution in patients receiving other QTc-prolonging drugs.
RIFAMYCINS
The rifamycins include rifampin, rifabutin, and rifapentine. Rifampin is the most commonly used rifamycin. For almost all therapeutic indications, it is used in combination with other agents to reduce the likelihood of selection of high-level rifampin resistance. Rifampin is used foremost in the treatment of mycobacterial infections—specifically, as a mainstay of combination therapy for M. tuberculosis infection or as a single agent in the treatment of latent M. tuberculosis infection. In addition, it is often used in the treatment of nontuberculous mycobacterial infection. Rifampin is used in combination regimens for the treatment of staphylococcal infections, particularly prosthetic valve endocarditis and bone infections with retained hardware. It is a component of combination therapy for brucellosis (with doxycycline) and leprosy (with dapsone for tuberculoid leprosy and with dapsone and clofazimine for lepromatous disease). Rifampin can be used alone for prophylaxis in close contacts of patients with H. influenzae or N. meningitidis meningitis. The drug has high oral bioavailability, which is further enhanced when it is taken on an empty stomach. Rifampin has several adverse effects, including elevated aminotransferase levels (14%), rash (1–5%), and gastrointestinal events such as nausea, vomiting, and diarrhea (1–2%). Its many clinically relevant interactions with other drugs mandate the clinician’s careful review of the patient’s medications before rifampin initiation to assess safety and the need for additional monitoring.
METRONIDAZOLE
Metronidazole is used in the treatment of anaerobic bacterial infections as well as infections caused by protozoa (e.g., amebiasis, giardiasis, trichomoniasis). It is the agent of choice as a component of combination therapy for polymicrobial abscesses in the lung, brain, or abdomen, the etiology of which often includes anaerobic bacteria, and for bacterial vaginosis, pelvic inflammatory disease, mild to moderate C. difficile–associated diarrhea, and anaerobic infections, such as those due to Bacteroides, Fusobacterium, and Prevotella species. Metronidazole is bactericidal against anaerobic bacteria and exhibits concentration-dependent killing. It has high oral bioavailability and tissue penetration, including penetration of the blood–brain barrier. The majority of Actinomyces, Propionibacterium, and Lactobacillus species are intrinsically resistant to metronidazole. The major adverse effects include nausea, diarrhea, and a metallic taste. Concomitant ingestion of alcohol may result in a disulfiram-like reaction, and patients are usually instructed to avoid alcohol during treatment. Long-term treatment carries the risk of leukopenia, neutropenia, peripheral neuropathy, and central nervous system toxicity manifesting as confusion, dysarthria, ataxia, nystagmus, and ophthalmoparesis. Through metronidazole’s effect on the CYP2C9 drug-metabolizing enzyme, its coadministration with warfarin can result in decreased metabolism and enhanced anticoagulant effects that require close monitoring. Concomitant administration of metronidazole with lithium can result in increased serum levels of lithium and associated toxicity; coadministration with phenytoin can result in phenytoin toxicity and possibly decreased levels of metronidazole.
OXAZOLIDINONES
Linezolid is a bacteriostatic agent and is indicated for serious infections due to resistant gram-positive bacteria, such as MRSA and VRE. The intrinsic resistance of gram-negative bacteria is mediated primarily by endogenous efflux pumps. Linezolid has excellent oral bioavailability. Adverse effects include myelosuppression and ocular and peripheral neuropathy with prolonged therapy. Peripheral neuropathy may be irreversible. Linezolid is a weak, reversible monoamine oxidase inhibitor, and coadministration with sympathomimetics and foods rich in tyramine should be avoided. Linezolid has been associated with serotonin syndrome when coadministered with selective serotonin-reuptake inhibitors. Tedizolid has properties similar to those of linezolid, but with lower dosing it may be less likely to cause adverse hematologic and neuropathic effects.
NITROFURANTOIN
Nitrofurantoin’s antibacterial activity results from the drug’s conversion to highly reactive intermediates that can damage DNA and other macromolecules. Nitrofurantoin is bactericidal, and its action is concentration dependent. It displays activity against a range of gram-positive bacteria, including S. aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, E. faecalis, Streptococcus agalactiae, group D streptococci, viridans streptococci, and corynebacteria, as well as gram-negative organisms, including E. coli and Enterobacter, Neisseria, Salmonella, and Shigella species. Nitrofurantoin is used primarily in the treatment of urinary tract infections and is preferred in the treatment of such infections in pregnancy. It may be used for the prevention of recurrent cystitis. Recently, there has been interest in the use of nitrofurantoin for treatment of urinary tract infections caused by ESBL-producing Enterobacteriaceae such as E. coli, although resistance has been growing in Latin America and parts of Europe. Coadministration with magnesium should be avoided because of decreased absorption, and patients should be encouraged to take the drug with food to increase its bioavailability and decrease the risk of adverse effects, which include nausea, vomiting, and diarrhea. Nitrofurantoin may also cause pulmonary fibrosis and drug-induced hepatitis. Because the risk of adverse reactions increases with age, the use of nitrofurantoin in elderly patients is not recommended. Patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency are at elevated risk for nitrofurantoin-associated hemolytic anemia.
POLYMYXINS
Colistin and polymyxin B act by disrupting cell membrane integrity and are active against the nonenteric pathogens P. aeruginosa and A. baumannii but not against Burkholderia. These drugs also exhibit activity against many Enterobacteriaceae, with the exceptions of Proteus, Providencia, and Serratia species. They lack activity against gram-positive bacteria. Polymyxins are bactericidal and are available in IV formulations. Colistimethate is converted to the active form (colistin) in plasma. Polymyxins are most often used for infections due to pathogens resistant to multiple other antibacterial agents, including urinary tract infections, hospital-acquired pneumonia, and bloodstream infections. Nebulized formulations have been used for adjunctive treatment of refractory ventilator-associated pneumonia. The most important adverse effect is dose-dependent reversible nephrotoxicity. Neurotoxicity, including paresthesias, muscle weakness, and confusion, is reversible and less common than nephrotoxicity.
QUINUPRISTIN-DALFOPRISTIN
Quinupristin-dalfopristin is a member of the streptogramin class of antibiotics and kills bacteria by inhibiting protein synthesis. The antibacterial spectrum of quinupristin-dalfopristin includes staphylococci (including MRSA), streptococci, and E. faecium (but not E. faecalis). This drug is also active against Corynebacterium species and L. monocytogenes. Quinupristin-dalfopristin is not reliably active against gram-negative organisms. It exhibits concentration-dependent killing, with an AUC/MIC ratio predicting efficacy. The clinical use of quinupristin-dalfopristin is largely for infections due to vancomycin-resistant E. faecium and other gram-positive bacterial infections. The drug has demonstrated efficacy in a variety of infections, including urinary tract infections, bone and joint infections, and bacteremia. Adverse effects associated with quinupristin-dalfopristin include infusion-related reactions, arthralgias, and myalgias. The arthralgias and myalgias may be severe enough to warrant drug discontinuation. Quinupristin-dalfopristin inhibits the CYP3A4 drug-metabolizing enzyme, with consequent drug interactions (Table 170-4).
FOSFOMYCIN
Fosfomycin is a phosphonic acid antibiotic that has greater activity in acidic environments and is excreted in its active form in the urine. Thus, its use is primarily for prophylaxis and treatment of uncomplicated cystitis. The drug is administered as a single 3-g dose that results in high urine concentrations for up to 48 h. Fosfomycin is active against S. aureus, vancomycin-susceptible and vancomycin-resistant enterococci, and a wide range of gram-negative organisms, including E. coli, Enterobacter species, S. marcescens, P. aeruginosa, and K. pneumoniae. Notably, the vast majority of ESBL-producing Enterobacteriaceae are susceptible to fosfomycin. A. baumannii and Burkholderia species are resistant. The emergence of resistance to fosfomycin has not been observed during treatment of cystitis but has been documented during treatment of respiratory tract infections and osteomyelitis. The few adverse effects that have been reported include nausea and diarrhea.
CHLORAMPHENICOL
The use of chloramphenicol is limited by its potentially serious toxicities. When other agents are contraindicated or ineffective, chloramphenicol represents an alternative treatment for infections, including meningitis caused by susceptible bacteria such as N. meningitidis, H. influenzae, and S. pneumoniae. It has also been used for the treatment of anthrax, brucellosis, Burkholderia infections, chlamydial infections, clostridial infections, erlichiosis, rickettsial infections, and typhoid fever. Adverse reactions include aplastic anemia, myelosuppression, and gray baby syndrome. Chloramphenicol inhibits the CYP2C19 and CYP3A4 drug-metabolizing enzymes and consequently increases levels of many classes of drugs.
APPROACH TO PROPHYLAXIS OF INFECTION
Antibacterial prophylaxis is indicated only in selected circumstances (Table 170-6) and should be supported by well-designed studies or expert panel recommendations. In all cases the risk or severity of the infection to be prevented should be greater than the adverse consequences of antibacterial therapy, including the potential for selection of resistance. In addition, the timing and duration of antibacterial treatment should be targeted for maximal effect and minimal required exposure. Prophylaxis of surgical site infections targets bacteria that may contaminate the wound during the surgical procedure, including the skin flora of the patient or operating team and the air in the operating room. Delivery of the antibacterial drug within 1 h before the surgical incision is most effective. For prolonged procedures, redosing may be necessary to maintain effective blood and tissue levels until the wound is closed. In patients with nasal carriage of S. aureus, preoperative decolonization with nasal mupirocin reduces the rate of S. aureus surgical site infections and is generally recommended for high-risk procedures such as cardiac surgery and orthopedic implantation of prosthetic devices. For dental procedures, preprocedure antibacterial drugs are given to prevent transient bacteremia and the seeding of certain high-risk cardiac lesions. Prophylaxis is also used in nonprocedural settings in certain patients who have recurrent infections or who are at risk of serious infection from a specific exposure (e.g., close contact with a patient with meningococcal meningitis). Extension of prophylaxis beyond the period of infection risk (24 h in the case of surgical procedures) does not add further benefit and may increase the risk of resistance selection or C. difficile disease.
|
PROPHYLAXIS OF BACTERIAL INFECTIONS IN ADULTS |

ANTIMICROBIAL STEWARDSHIP
In an era of increasing prevalence of multidrug-resistant bacteria and with a substantial amount of inappropriate antimicrobial use, the need for rational antimicrobial prescribing has never been greater. Antimicrobial stewardship describes the practice of promoting the selection of the appropriate drug, dosage, route, and duration of antimicrobial therapy. Antimicrobial stewardship programs implement a variety of strategies to (1) improve patient care through appropriate antimicrobial use; (2) decrease the development of resistance within patients and populations; (3) reduce the incidence of adverse effects; and (4) control costs.
Infections caused by resistant pathogens result in significant morbidity and mortality as well as increased health care costs. Antimicrobial stewardship programs are typically multidisciplinary and often include infectious disease physicians, clinical pharmacists (usually with special training in infectious disease), clinical microbiologists, information systems specialists, infection prevention and control practitioners, and epidemiologists. These teams employ a variety of approaches to achieving the program’s goals.
Established strategies of antimicrobial stewardship programs include (1) prospective audit of antimicrobial use, with intervention and feedback; (2) formulary restriction; and (3) preauthorization. Prospective audit and feedback are usually undertaken by an infectious disease physician or a pharmacist. In this process, orders for broad-spectrum antimicrobials (e.g., carbapenems) or high-impact agents (e.g., linezolid, daptomycin) are reviewed on a regular basis for appropriateness. In circumstances in which an antimicrobial is used in the absence of an appropriate indication, the stewardship program team intervenes and recommends an alternative to the primary team caring for the patient. This process has been successful in several quasi-experimental studies, resulting in declines in use of broad-spectrum drugs and decreases in adverse events, such as C. difficile infection. Formulary restriction is the inclusion of a limited set of antimicrobial agents in a hospital formulary for the purpose of limiting indiscriminant use of antimicrobials in the absence of demonstrated benefit. Such restriction coincidentally serves to reduce costs. Preauthorization is the practice of requiring clinicians to obtain approval before using selected antimicrobials. Approval may be provided electronically with sophisticated Computerized Provider Order Entry (CPOE) software, after specific criteria for use are met, or after communication with an infectious disease specialist as designated by the stewardship program. These strategies have led to a decrease in C. difficile infections and to improvements in drug susceptibility patterns.
Additional strategies used in specific health-care settings are guidelines and pathways, dose optimization, parenteral-to-oral conversion, and de-escalation of therapy. Antimicrobial stewardship is an evolving area and an increasingly active area of research aimed at identifying the best practices. The IDSA, in collaboration with several other professional organizations, has published guidelines for developing institutional antimicrobial stewardship programs (www.idsociety.org/Antimicrobial_Agents/).
SECTION 5 |
DISEASES CAUSED BY GRAM-POSITIVE BACTERIA |
171 |
Pneumococcal Infections |
In the late nineteenth century, pairs of micrococci were first recognized in the blood of rabbits injected with human saliva by both Louis Pasteur, working in France, and George Sternberg, an American army physician. The important role of these micrococci in human disease was not appreciated at that time. By 1886, when the organism was designated “pneumokokkus” and Diplococcus pneumoniae, the pneumococcus had been isolated by many independent investigators, and its role in the etiology of pneumonia was well known. In the 1930s, pneumonia was the third leading cause of death in the United States (after heart disease and cancer) and was responsible for ~7% of all deaths both in the United States and in Europe. While pneumonia was caused by a host of pathogens, lobar pneumonia—a pattern more likely to be caused by the pneumococcus—accounted for approximately one-half of all pneumonia deaths in the United States in 1929. In 1974, the organism was reclassified as Streptococcus pneumoniae.
MICROBIOLOGY
Etiologic Agent Pneumococci are spherical gram-positive bacteria of the genus Streptococcus. Within this genus, cell division occurs along a single axis, and bacteria grow in chains or pairs—hence the name Streptococcus, from the Greek streptos, meaning “twisted,” and kokkos, meaning “berry.” At least 22 streptococcal species are recognized and are divided further into groups based on their hemolytic properties. S. pneumoniae belongs to the α-hemolytic group that characteristically produces a greenish color on blood agar because of the reduction of iron in hemoglobin (Fig. 171-1). The bacteria are fastidious and grow best in 5% CO2 but require a source of catalase (e.g., blood) for growth on agar plates, where they develop mucoid (smooth/shiny) colonies. Pneumococci without a capsule produce colonies with a rough surface. Unlike that of other α-hemolytic streptococci, their growth is inhibited in the presence of optochin (ethylhydrocupreine hydrochloride), and they are bile soluble.

FIGURE 171-1 Pneumococci growing on blood agar, illustrating α hemolysis and optochin sensitivity (zone around optochin disk). Inset: Gram’s stain, illustrating gram-positive diplococci. (Photographs courtesy of Paul Turner, Shoklo Malaria Research Unit, Thailand.)
In common with other gram-positive bacteria, pneumococci have a cell membrane beneath a cell wall, which in turn is covered by a polysaccharide capsule. Pneumococci are divided into serogroups or serotypes based on capsular polysaccharide structure, as distinguished with rabbit polyclonal antisera; capsules swell in the presence of specific antiserum (the Quellung reaction). The most recently discovered serotypes, 6C, 6D, and 11E, have been identified with monoclonal antibodies and by serologic, genetic, and biochemical means, respectively. The currently recognized 93 serotypes fall into 21 serogroups, and each serogroup contains two to five serotypes with closely related capsules. The capsule protects the bacteria from phagocytosis by host cells in the absence of type-specific antibody and is arguably the most important determinant of pneumococcal virulence. Unencapsulated variants tend not to cause invasive disease.
Virulence Factors Within the cytoplasm, cell membrane, and cell wall, many molecules that may play a role in pneumococcal pathogenesis and virulence have been identified (Fig. 171-2). These proteins are often involved in direct interactions with host tissues or in concealment of the bacterial surface from host defense mechanisms. Pneumolysin is a secreted cytotoxin thought to result in cytolysis of cells and tissues, and LytA enhances pathogenesis. A number of cell wall proteins interfere with the complement pathway, thus inhibiting complement deposition and preventing lysis and/or opsonophagocytosis. The pneumococcal H inhibitor (Hic) impedes the formation of C3 convertase, while pneumococcal surface protein C (PspC), also known as choline-binding protein A (CbpA), binds factor H and is thought to accelerate the breakdown of C3. PspA and CbpA inhibit the deposition of or degrade C3b. The numerous pneumococcal proteins thought to be involved in adhesion include the ubiquitous surface-anchored sialidase (neuraminidase) NanA, which cleaves sialic acid on host cells and proteins, and pneumococcal surface adhesin A (PsaA). Pili recently recognized by electron microscopy also may play an important role in binding to cells. Some of the antigens mentioned above are potential vaccine candidates (see “Prevention,” below).

FIGURE 171-2 Schematic diagram of the pneumococcal cell surface, with key antigens and their roles highlighted.
![]() Although the capsule surrounding the cell wall of S. pneumoniae is the basis for categorization by serotype, the behavior and pathogenic potential of a serotype may also be related to the genetic origin of the strain. Molecular typing is therefore of considerable interest. Initially, techniques such as pulsed-field gel electrophoresis were used to determine genetic relatedness; such techniques have been superseded by sequencing of housekeeping genes to define a clone (multilocus sequence typing, MLST). For S. pneumoniae, alleles at each of the loci aroE, gdh, gki, recP, spi, xpt, and ddl are sequenced and compared with all of the known alleles at that locus. Sequences identical to a known allele are assigned the same allele number, whereas those differing from any known allele—even at a single nucleotide site—are assigned new numbers. Software for assignment of alleles at each locus is available on the pneumococcal MLST website (spneumoniae.mlst.net), and the allelic profile of each isolate and its consequent sequence type are generated. With the advent of high-throughput and relatively inexpensive sequencing techniques, whole-genome sequencing will soon supersede MLST.
Although the capsule surrounding the cell wall of S. pneumoniae is the basis for categorization by serotype, the behavior and pathogenic potential of a serotype may also be related to the genetic origin of the strain. Molecular typing is therefore of considerable interest. Initially, techniques such as pulsed-field gel electrophoresis were used to determine genetic relatedness; such techniques have been superseded by sequencing of housekeeping genes to define a clone (multilocus sequence typing, MLST). For S. pneumoniae, alleles at each of the loci aroE, gdh, gki, recP, spi, xpt, and ddl are sequenced and compared with all of the known alleles at that locus. Sequences identical to a known allele are assigned the same allele number, whereas those differing from any known allele—even at a single nucleotide site—are assigned new numbers. Software for assignment of alleles at each locus is available on the pneumococcal MLST website (spneumoniae.mlst.net), and the allelic profile of each isolate and its consequent sequence type are generated. With the advent of high-throughput and relatively inexpensive sequencing techniques, whole-genome sequencing will soon supersede MLST.
EPIDEMIOLOGY
![]() Pneumococcal infections remain a significant global cause of morbidity and death, particularly among children and the elderly. Rapid and dramatic changes in the epidemiology of this disease during the past decade in several developed countries followed the licensure and routine childhood administration of pneumococcal polysaccharide–protein conjugate vaccine (PCV). With PCV introduction in developing and middle-income countries, additional profound changes in pneumococcal ecology and disease epidemiology are likely. The disease burden and serotype distribution in the PCV era may be different than expected because of concomitant secular trends in pneumococcal disease, the impact of antibiotic use on pneumococcal strain ecology, and surveillance system attributes that can themselves affect analysis of epidemiologic features.
Pneumococcal infections remain a significant global cause of morbidity and death, particularly among children and the elderly. Rapid and dramatic changes in the epidemiology of this disease during the past decade in several developed countries followed the licensure and routine childhood administration of pneumococcal polysaccharide–protein conjugate vaccine (PCV). With PCV introduction in developing and middle-income countries, additional profound changes in pneumococcal ecology and disease epidemiology are likely. The disease burden and serotype distribution in the PCV era may be different than expected because of concomitant secular trends in pneumococcal disease, the impact of antibiotic use on pneumococcal strain ecology, and surveillance system attributes that can themselves affect analysis of epidemiologic features.
Serotype Distribution Not all pneumococcal serotypes are equally likely to cause disease; serotype distribution varies by age, disease syndrome, and geography. Geographic differences may be driven by variation in the burden of disease rather than by true serotype distribution differences. Most data on serotype distribution are related to pediatric invasive pneumococcal disease (IPD, defined as infection of a normally sterile site); much less information on global distribution is available for disease in adults. Among children <5 years of age, five to seven serotypes cause >60% of IPD cases in most parts of the world, seven serotypes (1, 5, 6A, 6B, 14, 19F, and 23F) account for ~60% of cases in all areas of the world, but in any given region these seven serotypes may not all rank as the most common disease strains (Fig. 171-3). Some serotypes (e.g., types 1 and 5) not only tend to cause disease in areas with a high disease burden but also cause waves of disease in lower-burden areas (e.g., Europe) or outbreaks (e.g., in military barracks; meningitis in sub-Saharan Africa). The broader range of serotypes causing disease among adults than among children is apparent from a comparison of the coverage of existing multiserotype vaccines in different age groups. For example, data from the United States for 2006–2007 on the serotypes causing IPD indicated that a polysaccharide vaccine containing 23 serotypes (PPSV23) would cover 84% of cases among children <5 years of age and 76% of those among persons 18–64 years of age but only 65% of those among persons ≥65 years of age.

FIGURE 171-3 Meta-analysis of available global pneumococcal serotype data, adjusted for regional disease incidence. The red line shows cumulative incidence, as indicated on the right-hand Y axis. (Source: Global Serotype Project Report for the Pneumococcal Advance Market Commitment Target Product Profile; available at http://www.gavi.org/library/gavi-documents/amc/tpp-codebook/.)
Nasopharyngeal Carriage Pneumococci are intermittent inhabitants of the healthy human nasopharynx and are transmitted by respiratory droplets. In children, pneumococcal nasopharyngeal ecology varies by geographic region, socioeconomic status, climate, degree of crowding, and particularly intensity of exposure to other children, with children in day-care settings having higher rates of colonization. In developed-world settings, children serve as the major vectors of pneumococcal transmission. By 1 year of age, ~50% of children have had at least one episode of pneumococcal colonization. Cross-sectional prevalence data show rates of pneumococcal carriage ranging from 20% to 50% among children <5 years of age and from 5% to 15% among young and middle-aged adults; Fig. 171-4 shows relevant data from the United Kingdom. Data on colonization rates among healthy elderly individuals are limited. In developing-world settings, pneumococcal acquisition occurs much earlier, sometimes within the first few days after birth, and nearly all infants have had at least one episode of colonization by 2 months of age. Cross-sectional studies show that up to the age of 5 years, 70–90% of children carry S. pneumoniae in the nasopharynx, and a significant proportion of adults (sometimes >40%) also are colonized. Their high rates of colonization make adults an important source of transmission and may affect community transmission dynamics.

FIGURE 171-4 Prevalence of pneumococcal carriage in adults and children resident in the United Kingdom who had nasopharyngeal swabs collected monthly for 10 months (no seasonal trend;t test trend, >.05). (Data adapted from D Goldblatt et al: J Infect Dis 192:387, 2005.)
Invasive Disease and Pneumonia IPD develops when S. pneumoniae invades the bloodstream and seeds other organs or directly reaches the cerebrospinal fluid (CSF) by local extension. Pneumonia may follow aspiration of pneumococci, although only 10–30% of such cases are associated with a positive blood culture (and thus contribute to the measured burden of IPD). The dramatic variation of IPD rates with age is illustrated by data from the United States for 1998–1999, a period prior to PCV introduction. Rates of IPD were highest among children <2 years of age and among adults ≥65 years of age (188 and 60 cases/100,000, respectively; Fig. 171-5). Since the introduction of PCV, IPD rates among infants and children in the United States have fallen by >75%, a decrease driven by the near elimination of vaccine-serotype IPD. A similar impact of PCV on vaccine-serotype IPD rates has been consistently observed in countries where PCV has been introduced into the routine pediatric vaccination schedule. However, changes in the non-vaccine-serotype IPD rate in various countries have been heterogeneous; the interpretation of this heterogeneity is a complex issue. In the United States, Canada, and Australia, rates of non-vaccine-serotype IPD have increased but the magnitude of the increase is generally small relative to the substantial reductions in vaccine-serotype IPD. In contrast, in other settings (e.g., Alaska Native communities and the United Kingdom), the reduction in vaccine-serotype IPD has been offset by notable increases in rates of disease caused by non-vaccine serotypes. Explanations for the heterogeneity of findings include replacement disease resulting from vaccine pressure, changes in clinical case investigation, secular trends unrelated to PCV use, antibiotic pressure selecting for resistant organisms, changes in surveillance or reporting systems, rapidity of introduction, and inclusion of a catch-up campaign. A recent systematic review concludes that serotype replacement in IPD follows the use of PCV7 but that the magnitude of this phenomenon is small relative to the reduction in disease from vaccine serotypes. The net effect of PCV is to reduce the rate of pneumococcal disease both in the age group targeted for vaccination and in unvaccinated age groups.

FIGURE 171-5 Rates of invasive pneumococcal disease before the introduction of pneumococcal conjugate vaccine, by age group: United States, 1998. (Source: CDC, Active Bacterial Core Surveillance/Emerging Infectious Program Network, 2000. Data adapted from MMWR 49[RR-9], 2000.)
Pneumonia is the most common of the serious pneumococcal disease syndromes and poses special challenges from a clinical and public health perspective. Most cases of pneumococcal pneumonia are not associated with bacteremia, and in these cases a definitive etiologic diagnosis is difficult. As a result, estimates of disease burden focus primarily on IPD rates and fail to include the major portion of the burden of serious pneumococcal disease. Among children, PCV trials designed to collect efficacy data on syndrome-based outcomes (e.g., radiographically confirmed pneumonia, clinically diagnosed pneumonia) have revealed the burden of culture-negative pneumococcal pneumonia.
The case-fatality ratios (CFRs) for pneumococcal pneumonia and IPD vary by age, underlying medical condition, and access to care. In addition, the CFR for pneumococcal pneumonia varies with the severity of disease at presentation (rather than according to whether the pneumonia episode is associated with bacteremia) and with the patient’s age (from <5% among hospitalized patients 18–44 years old to >12% among those >65 years old, even when appropriate and timely management is available). Notably, the likelihood of death in the first 24 h of hospitalization did not change substantially with the introduction of antibiotics; this surprising observation highlights the fact that the pathophysiology of severe pneumococcal pneumonia among adults reflects a rapidly progressive cascade of events that often unfolds irrespective of antibiotic administration. Management in an intensive care unit can provide critical support for the patient through the acute period, with lower CFRs.
Rates of pneumococcal disease vary by season, with higher rates in colder than in warmer months in temperate climates; by sex, with males more often affected than females; and by risk group, with risk factors including underlying medical conditions, behavioral issues, and ethnic group. In the United States, some Native American populations (including Alaska natives) and African Americans have higher rates of disease than the general population; the increased risk is probably attributable to socioeconomic conditions and the prevalence of underlying risk factors for pneumococcal disease. Medical conditions that increase the risk of pneumococcal infection are listed in Table 171-1. Outbreaks of disease are well recognized in crowded settings with susceptible individuals, such as infant day-care facilities, military barracks, and nursing homes. Furthermore, there is a clear association between preceding viral respiratory disease (especially but not exclusively influenza) and risk of secondary pneumococcal infections. The significant role of pneumococcal pneumonia in the morbidity and mortality associated with seasonal and pandemic influenza is increasingly recognized.
|
CLINICAL RISK GROUPS FOR PNEUMOCOCCAL INFECTION |

![]() Antibiotic Resistance Reduced pneumococcal susceptibility to penicillin was first noted in 1967, but not until the 1990s did reduced antibiotic susceptibility emerge as a significant clinical and public health issue, with an increasing prevalence of pneumococcal isolates resistant to single or multiple classes of antibiotics and a rising absolute magnitude of minimal inhibitory concentrations (MICs). Strains with reduced susceptibility to penicillin G, cefotaxime, ceftriaxone, macrolides, and other antibiotics are now found worldwide and account for a significant proportion of disease-causing strains in many locations, especially among children. Vancomycin resistance has not yet been observed in clinical pneumococcal strains. Lack of antimicrobial susceptibility is clearly related to a subset of serotypes, many of which disproportionately cause disease among children. The vicious cycle of antibiotic exposure, selection of resistant organisms in the nasopharynx, and transmission of these organisms within the community, leading to difficult-to-treat infections and increased antibiotic exposure, has been interrupted to some extent by the introduction and routine use of PCV. The clinical implications of pneumococcal antimicrobial nonsusceptibility are addressed below in the section on treatment.
Antibiotic Resistance Reduced pneumococcal susceptibility to penicillin was first noted in 1967, but not until the 1990s did reduced antibiotic susceptibility emerge as a significant clinical and public health issue, with an increasing prevalence of pneumococcal isolates resistant to single or multiple classes of antibiotics and a rising absolute magnitude of minimal inhibitory concentrations (MICs). Strains with reduced susceptibility to penicillin G, cefotaxime, ceftriaxone, macrolides, and other antibiotics are now found worldwide and account for a significant proportion of disease-causing strains in many locations, especially among children. Vancomycin resistance has not yet been observed in clinical pneumococcal strains. Lack of antimicrobial susceptibility is clearly related to a subset of serotypes, many of which disproportionately cause disease among children. The vicious cycle of antibiotic exposure, selection of resistant organisms in the nasopharynx, and transmission of these organisms within the community, leading to difficult-to-treat infections and increased antibiotic exposure, has been interrupted to some extent by the introduction and routine use of PCV. The clinical implications of pneumococcal antimicrobial nonsusceptibility are addressed below in the section on treatment.
PATHOGENESIS
Pneumococci colonize the human nasopharynx from an early age; colonization acquisition events are generally described as asymptomatic, but evidence exists to associate acquisition with mild respiratory symptoms, especially in the very young. From the nasopharynx, the bacteria spread either via the bloodstream to distant sites (e.g., brain, joint, bones, peritoneal cavity) or locally to mucosal surfaces where they can cause otitis media or pneumonia. Direct spread from the nasopharynx to the central nervous system (CNS) can occur in rare cases of skull base fracture, although most cases of pneumococcal meningitis are secondary to hematogenous spread. Pneumococci can cause disease in almost any organ or part of the body; however, otitis media, pneumonia, bacteremia, and meningitis are most common. Colonization is a relatively frequent event, yet disease is rare. In the nasopharynx, pneumococci survive in mucus secreted by epithelial cells, where they can avoid local immune factors such as leukocytes and complement. The mucus itself is a component of local defense mechanisms, and the flow of mucus (driven in part by cilia in what is known as the mucociliary escalator) effects mechanical clearance of pneumococci. While many colonization episodes are of short duration, longitudinal studies in adults and children have revealed persistent colonization with a specific serotype over many months. Colonization eventually results in the development of capsule-specific serum IgG, which is thought to play a role in mediating clearance of bacteria from the nasopharynx. IgG antibodies to surface-exposed cell wall or secreted proteins also appear in the circulation in an age-dependent fashion or after colonization; the biologic role of these antibodies is less clear. Recent acquisition of a new colonizing serotype is more likely to be associated with subsequent invasion, presumably as a result of the absence of type-specific immunity. Intercurrent viral infections make the host more susceptible to pneumococcal colonization, and pneumococcal disease in a colonized individual often follows perturbation of the nasopharyngeal mucosa by such infections. Local cytokine production after a viral infection is thought to upregulate adhesion factors in the respiratory epithelium, allowing pneumococci to adhere via a variety of surface adhesin molecules, including PsaA, PspA, CbpA, PspC, Hyl, pneumolysin, and the neuraminidases (Fig. 171-2). Adhesion coupled with inflammation induced by pneumococcal factors such as peptidoglycans and teichoic acids results in invasion. It is the inflammation induced by various bacterium-derived factors that is responsible for the pathology associated with pneumococcal infection. Cell wall–derived teichoic acids and peptidoglycans induce a variety of cytokines, including the proinflammatory cytokines interleukin (IL) 1, IL-6, and tumor necrosis factor, and activate complement via the alternative pathway. Polymorphonuclear leukocytes are thus attracted, and an intense inflammatory response is initiated. Pneumolysin also is important in local pathology, inducing proinflammatory cytokine production by local monocytes.
The pneumococcal capsule, consisting of polysaccharides with antiphagocytic properties due to resistance to the deposition of complement, plays an important role in pathogenesis. While most capsular types can cause human disease, certain capsular types are more commonly isolated from sites of infection. The reason for the dominance of some serotypes over others in IPD, as depicted in Fig. 171-3, is unclear.
HOST DEFENSE MECHANISMS
Innate Immunity As described above, intact respiratory epithelium and a host of nonspecific or innate immune factors (e.g., mucus, splenic function, complement, neutrophils, and macrophages) constitute the first line of defense against pneumococci. Physical factors such as the cough reflex and the mucociliary escalator are important in clearing bacteria from the lungs. Immunologic factors are critical as well: C-reactive protein (CRP) binds phosphorylcholine in the pneumococcal cell wall, inducing complement activation and leading to bacterial clearance; Toll-like receptor 2 (TLR2) recognizes both pneumococcal lipoteichoic acid and cell wall peptidoglycan; and in animal models, the absence of host TLR2 leads to more severe infection and impaired clearance of nasopharyngeal colonization. TLR4 appears to be necessary for the proinflammatory effect of pneumolysin on macrophages. The importance of TLR recognition is underlined by descriptions of an inherited deficiency of human IL-1 receptor–associated kinase 4 (IRAK-4) that manifests as an unusual susceptibility to infection with bacteria, including S. pneumoniae. IRAK-4 is essential for the normal functioning of several TLRs. Other factors that interfere with these nonspecific mechanisms (e.g., viral infections, cystic fibrosis, bronchiectasis, complement deficiency, and chronic obstructive pulmonary disease) all predispose to the development of pneumococcal pneumonia. Patients who lack a spleen or have abnormal splenic function (e.g., persons with sickle cell disease) are at high risk of developing overwhelming pneumococcal disease.
Acquired Immunity Acquired immunity induced via contact following colonization or through cross-reactive antigens rests largely on the development of serum IgG antibody specific for the pneumococcal capsular polysaccharide. Nearly all polysaccharides are T cell–independent antigens; B cells can make antibodies to such antigens without T cell help. However, in children <1–2 years old, such B cell responses are poorly developed. This delayed ontogeny of capsule-specific IgG in young children is associated with susceptibility to pneumococcal infection (Fig. 171-5). The extremely high risk of pneumococcal infection in the absence of serum immunoglobulin (i.e., in conditions such as agammaglobulinemia) highlights the important role of capsular antibody in protection against disease. Each serotype’s capsule is chemically distinct; thus immunity tends to be serotype specific, although some cross-immunity exists. For example, conjugate vaccine–induced antibodies to serotype 6B prevent infection due to serotype 6A. However, cross-protection against serotypes within serogroups is not universal; for instance, antibodies to serotype 19F do not appear to confer protection against disease caused by serotype 19A. Antibodies to surface-exposed or secreted pneumococcal proteins (such as pneumolysin, PsaA, and PspA) also appear in the circulation with increasing age of the host, but their functional significance remains unclear. Data from murine models suggest that CD4+ T cells may play a role in preventing pneumococcal colonization and disease, and recent experimental data derived from humans suggest that IL-17-secreting CD4+ T cells may be relevant.
CLINICAL MANIFESTATIONS
The clinical manifestations of pneumococcal disease depend on the site of infection and the duration of illness. Clinical syndromes are classified as noninvasive (e.g., otitis media and nonbacteremic pneumonia) or invasive (e.g., bacteremic pneumonia). The pathogenesis of noninvasive illness involves contiguous spread from the nasopharynx or skin; invasive disease involves infection of a normally sterile body fluid or follows bacteremia.
Pneumonia Pneumonia is the most common serious pneumococcal syndrome and is considered invasive when associated with a positive blood culture. Pneumococcal pneumonia can present as a mild community-acquired infection at one extreme and as a life-threatening disease requiring intubation and intensive support at the other.
PRESENTING MANIFESTATIONS The presentation of pneumococcal pneumonia does not reliably distinguish it from pneumonia of other etiologies. In a subset of cases, pneumococcal pneumonia is recognized at the outset as associated with a viral upper respiratory infection and is characterized by the abrupt onset of cough and dyspnea accompanied by fever, shaking chills, and myalgias. The cough evolves from nonpurulent to productive of sputum that is purulent and sometimes tinged with blood. Patients may describe stabbing pleuritic chest pain and significant dyspnea indicating involvement of the parietal pleura. Among the elderly, the presenting clinical symptoms may be less specific, with confusion or malaise but without fever or cough. In such cases, a high index of suspicion is required because failure to treat pneumococcal pneumonia promptly in an elderly patient is likely to result in rapid evolution of the infection, with increased severity, morbidity, and risk of death.
FINDINGS ON PHYSICAL EXAMINATION The clinical signs associated with pneumococcal pneumonia among adults include tachypnea (>30 breaths/min) and tachycardia, hypotension in severe cases, and fever in most cases (although not in all elderly patients). Respiratory signs are varied, including dullness to percussion in areas of the chest with significant consolidation, crackles on auscultation, reduced expansion of the chest in some cases as a result of splinting to reduce pain, bronchial breathing in a minority of cases, pleural rub in occasional cases, and cyanosis in cases with significant hypoxemia. Among infants with severe pneumonia, chest wall indrawing and nasal flaring are common. Nonrespiratory findings can include upper abdominal pain if the diaphragmatic pleura is involved as well as mental status changes, particularly confusion in elderly patients.
DIFFERENTIAL DIAGNOSIS The differential diagnosis of pneumococcal pneumonia includes cardiac conditions such as myocardial infarction and heart failure with atypical pulmonary edema; pulmonary conditions such as atelectasis; and pneumonia caused by viral pathogens, mycoplasmas, Haemophilus influenzae, Klebsiella pneumoniae, Staphylococcus aureus, Legionella, or (in HIV-infected and otherwise immunocompromised hosts) Pneumocystis. In cases with abdominal symptoms, the differential diagnosis includes cholecystitis, appendicitis, perforated peptic ulcer disease, and subphrenic abscesses. The challenge in cases with abdominal symptoms is to remember to include pneumococcal pneumonia—a nonabdominal process—in the differential diagnosis.
DIAGNOSIS Some authorities advocate treating uncomplicated, nonsevere, community-acquired pneumonia without determining the microbiologic etiology, given that this information is unlikely to alter clinical management. However, efforts to identify the cause of pneumonia are important when the disease is more severe and when the diagnosis of pneumonia is not clearly established. The gold standard for etiologic diagnosis of pneumococcal pneumonia is pathologic examination of lung tissue. In lieu of that procedure, evidence of an infiltrate on chest radiography warrants a diagnosis of pneumonia. However, cases of pneumonia without radiographic evidence do occur. An infiltrate can be absent either early in the course of the illness or with dehydration; upon rehydration, an infiltrate usually appears. The radiographic appearance of pneumococcal pneumonia is varied; it classically consists of lobar or segmental consolidation (Fig. 171-6) but in some cases is patchy. More than one lobe is involved in ~30% of cases. Consolidation may be associated with a small pleural effusion or empyema in complicated cases. In children, “round pneumonia,” a distinctly spherical consolidation on chest radiography, is associated with a pneumococcal etiology. Round pneumonia is uncommon in adults. S. pneumoniae is not the only cause of such lesions; other causes, especially cancer, should be considered.

FIGURE 171-6 Chest radiograph depicting classic lobar pneumococcal pneumonia in the right lower lobe of an elderly patient’s lung.
Blood drawn from patients with suspected pneumococcal pneumonia can be used for supportive or definitive diagnostic tests. Blood cultures are positive for pneumococci in a minority (<30%) of cases of pneumococcal pneumonia. Nonspecific findings include an elevated polymorphonuclear leukocyte count (>15,000/μL in most cases and upward of 40,000/μL in some), leukopenia in <10% of cases (a poor prognostic sign associated with a fatal outcome), and elevated values in liver function tests (e.g., both conjugated and unconjugated hyperbilirubinemia). Anemia, low serum albumin levels, hyponatremia, and elevated serum creatinine levels are all found in ~20–30% of patients.
Urinary pneumococcal antigen assays have facilitated etiologic diagnosis. In adults, among whom the prevalence of pneumococcal nasopharyngeal colonization is relatively low, a positive pneumococcal urinary antigen test has a high predictive value. The same is not true for children, in whom a positive urinary antigen test can reflect the mere presence of S. pneumoniae in the nasopharynx.
Most cases of pneumococcal pneumonia are diagnosed by Gram’s staining and culture of sputum. The utility of a sputum specimen is directly related to its quality and the patient’s antibiotic treatment status.
COMPLICATIONS Empyema is the most common focal complication of pneumococcal pneumonia, occurring in <5% of cases. When fluid in the pleural space is accompanied by fever and leukocytosis (even low-grade) after 4–5 days of appropriate antibiotic treatment for pneumococcal pneumonia, empyema should be considered. Parapneumonic effusions are more common than empyema, representing a self-limited inflammatory response to pneumonia. Pleural fluid with frank pus, bacteria (detected by microscopic examination), or a pH of ≤7.1 indicates empyema and demands aggressive and complete drainage, usually through chest tube insertion.
Meningitis Pneumococcal meningitis typically presents as a pyogenic condition that is clinically indistinguishable from meningitis of other bacterial etiologies. Meningitis can be the primary presenting pneumococcal syndrome or a complication of other conditions such as skull fracture, otitis media, bacteremia, or mastoiditis. Now that H. influenzae type b vaccine is routinely used, S. pneumoniae and Neisseria meningitidis are the most common bacterial causes of meningitis in both adults and children. Pyogenic meningitis, including that due to S. pneumoniae, is associated clinically with findings that include severe, generalized, gradual-onset headache, fever, and nausea as well as specific CNS manifestations such as stiff neck, photophobia, seizures, and confusion. Clinical signs include a toxic appearance, altered consciousness, bradycardia, and hypertension indicative of increased intracranial pressure. A small proportion of adult patients have Kernig’s or Brudzinski’s sign or cranial nerve palsies (particularly of the third and sixth cranial nerves).
A definitive diagnosis of pneumococcal meningitis rests on the examination of CSF for (1) evidence of turbidity (visual inspection); (2) elevated protein level, elevated white blood cell count, and reduced glucose concentration (quantitative measurement); and (3) specific identification of the etiologic agent (culture, Gram’s staining, antigen testing, or polymerase chain reaction [PCR]). A blood culture positive for S. pneumoniae in conjunction with clinical manifestations of meningitis also is considered confirmatory. Among adults, detection of pneumococcal antigen in urine is considered highly specific because of the low prevalence of nasopharyngeal colonization in this age group.
The mortality rate for pneumococcal meningitis is ~20%. In addition, up to 50% of survivors experience acute or chronic complications, including deafness, hydrocephalus, and mental retardation in children and diffuse brain swelling, subarachnoid bleeding, hydrocephalus, cerebrovascular complications, and hearing loss in adults.
Other Invasive Syndromes S. pneumoniae can cause other invasive syndromes involving virtually any body site. These syndromes include primary bacteremia without other sites of infection (bacteremia without a source; occult bacteremia), osteomyelitis, septic arthritis, endocarditis, pericarditis, and peritonitis. The essential diagnostic approach is collection of fluid from the site of infection by sterile technique and examination by Gram’s staining, culture, and—when relevant—capsular antigen assay or PCR. Hemolytic-uremic syndrome can complicate invasive pneumococcal disease.
Noninvasive Syndromes The two major noninvasive syndromes caused by S. pneumoniae are sinusitis and otitis media; the latter is the most common pneumococcal syndrome and most often affects young children. The manifestations of otitis media include the acute onset of severe pain, fever, deafness, and tinnitus, most frequently in the setting of a recent upper respiratory tract infection. Clinical signs include a red, swollen, often bulging tympanic membrane with reduced movement on insufflation or tympanography. Redness of the tympanic membrane is not sufficient for the diagnosis of otitis media.
Pneumococcal sinusitis is also a complication of upper respiratory tract infections and presents with facial pain, congestion, fever, and—in many cases—persistent nighttime cough. A definitive diagnosis is made by aspiration and culture of sinus material; however, presumptive treatment is most commonly initiated after application of a strict set of clinical diagnostic criteria.
|
TREATMENT |
PNEUMOCOCCAL INFECTIONS |
Historically, the activity of penicillin against pneumococci made parenteral penicillin G the drug of choice for disease caused by susceptible organisms, including community-acquired pneumonia. For susceptible strains, penicillin G remains the most commonly used agent, with daily doses ranging from 50,000 U/kg for minor infections to 300,000 U/kg for meningitis. Other parenteral β-lactam drugs, such as ampicillin, cefotaxime, ceftriaxone, and cefuroxime, can be used against penicillin-susceptible strains but offer little advantage over penicillin. Macrolides and cephalosporins are alternatives for penicillin-allergic patients. While agents such as clindamycin, tetracycline, and trimethoprim-sulfamethoxazole exhibit some activity against pneumococci, resistance to these agents is frequently encountered in different parts of the world.
Penicillin-resistant pneumococci were first described in the mid-1960s, at which point tetracycline- and macrolide-resistant strains had already been reported. Multidrug-resistant strains were first described in the 1970s, but it was during the 1990s that pneumococcal drug resistance reached pandemic proportions. The use of antibiotics selects for resistant pneumococci, and strains resistant to β-lactam agents and to multiple drugs are now found all over the world. The emergence of high rates of macrolide and fluoroquinolone resistance also has been described.
The molecular basis of penicillin resistance in S. pneumoniae is the alteration of penicillin-binding protein (PBP) genes by transformation and horizontal transfer of DNA from related streptococcal species. Such alteration of PBPs results in lower affinity for penicillins. Depending on the specific PBP(s) and the number of PBPs altered, the level of resistance ranges from intermediate to high. For many years, penicillin susceptibility breakpoints have been defined by MICs as follows: susceptible, ≤0.06 μg/mL; intermediate, 0.12–1.0 μg/mL; and resistant, ≥2.0 μg/mL. However, in vitro results often were not predictive of the response of a patient to treatment for pneumococcal diseases other than meningitis. New recommendations have been based on the revised penicillin G breakpoints established in 2008 by the Clinical and Laboratory Standards Institute. For IV treatment of meningitis with at least 24 million units per day in 8 divided doses, the susceptibility breakpoint remains ≤0.06 μg/mL, and MICs of ≥0.12 μg/mL indicate resistance. For IV treatment of nonmeningeal infections with 12 million units per day in 6 divided doses, the breakpoints are ≤2 μg/mL for susceptible organisms, 4 μg/mL for intermediate organisms, and ≥8 μg/mL for resistant organisms; a dosage of 18–24 million units per day is recommended for strains with MICs in the intermediate category. The original breakpoints remain the same for oral treatment of nonmeningeal infections with penicillin V.
Although guidelines for antibiotic therapy should be driven in part by local patterns of resistance, guidelines from national organizations in many countries (e.g., the Infectious Diseases Society of America/American Thoracic Society, the British Thoracic Society, and the European Respiratory Society) lay out evidence-based approaches. The following guidelines for the treatment of individual sepsis syndromes are based on those advocated by the American Academy of Pediatrics and published in the 2012 Red Book.
MENINGITIS LIKELY OR PROVEN TO BE DUE TO S. PNEUMONIAE
As a result of the increased prevalence of resistant pneumococci, first-line therapy for persons ≥1 month of age is a combination of vancomycin (adults, 30–60 mg/kg per day; infants and children, 60 mg/kg per day) and cefotaxime (adults, 8–12 g/d in 4–6 divided doses; children, 225–300 mg/kg per day in 1 dose or 2 divided doses) or ceftriaxone (adults, 4 g/d in 1 dose or 2 divided doses; children, 100 mg/kg per day in 1 dose or 2 divided doses). If children are hypersensitive to β-lactam agents (penicillins and cephalosporins), rifampin (adults, 600 mg/d; children, 20 mg/d in 1 dose or 2 divided doses) can be substituted for cefotaxime or ceftriaxone. A repeat lumbar puncture should be considered after 48 h if the organism is not susceptible to penicillin and information on cephalosporin sensitivity is not yet available, if the patient’s clinical condition does not improve or deteriorates, or if dexamethasone has been administered and may be compromising clinical evaluation. When antibiotic sensitivity data become available, treatment should be modified accordingly. If the isolate is sensitive to penicillin, vancomycin can be discontinued and penicillin can replace the cephalosporin, or cefotaxime or ceftriaxone can be continued alone. If the isolate displays any resistance to penicillin but is susceptible to the cephalosporins, vancomycin can be discontinued and cefotaxime or ceftriaxone continued. If the isolate exhibits any resistance to penicillin and is not susceptible to cefotaxime and ceftriaxone, vancomycin and high-dose cefotaxime or ceftriaxone can be continued; rifampin may be added as well if the isolate is susceptible and the patient’s clinical condition is worsening, if the CSF remains positive for bacteria, or if the MIC of the cephalosporin in question against the infecting strain is high. Some physicians advocate the use of glucocorticoids in children >6 months old, but this recommendation remains controversial and is not universally considered the standard of care. Glucocorticoids significantly reduce rates of mortality, severe hearing loss, and neurologic sequelae in adults and should be administered to those with community-acquired bacterial meningitis. If dexamethasone is given to either adults or children, it should be administered before or in conjunction with the first antibiotic dose.
INVASIVE INFECTIONS (EXCLUDING MENINGITIS)
In previously well children with noncritical illness, therapy with a recommended antibiotic should be instigated at the following dosages: penicillin G, 250,000–400,000 units/kg per day (in divided doses 4–6 h apart); cefotaxime, 75–100 mg/d (doses 8 h apart); or ceftriaxone, 50–75 mg/d (doses 12–24 h apart). For critically ill children, including those who have myocarditis or multilobular pneumonia with hypoxia or hypotension, vancomycin may be added if the isolate may possibly be resistant to β-lactam drugs, with its use reviewed once susceptibility data become available. If the organism is resistant to β-lactam agents, therapy should be modified on the basis of clinical response and susceptibility to other antibiotics. Clindamycin or vancomycin can be used as a first-line agent for children with severe β-lactam hypersensitivity, but vancomycin should not be continued if the organism is shown to be sensitive to other non-β-lactam antibiotics.
For outpatient management, amoxicillin (1 g every 8 h) provides effective treatment for virtually all cases of pneumococcal pneumonia. Neither cephalosporins nor quinolones, which are far more expensive, offer any advantage over amoxicillin. Levofloxacin (500–750 mg/d as a single dose) and moxifloxacin (400 mg/d as a single dose) also are highly likely to be effective in the United States except in patients who come from closed populations where these drugs are used widely or who have themselves been treated recently with a quinolone. Clindamycin (600–1200 mg/d every 6 h) is effective in 90% of cases and azithromycin (500 mg on day 1 followed by 250–500 mg/d) or clarithromycin (500–750 mg/d as a single dose) in 80% of cases. Treatment failure resulting in bacteremic disease due to macrolide-resistant isolates has been amply documented in patients given azithromycin empirically. As noted above, rates of resistance to all these antibiotics are relatively low in some countries and much higher in others; high-dose amoxicillin remains the best option worldwide.
The optimal duration of treatment for pneumococcal pneumonia is uncertain, but its continuation for at least 5 days once the patient becomes afebrile appears to be a prudent approach. Cases with a second focus of infection (e.g., empyema or septic arthritis) require longer therapy.
ACUTE OTITIS MEDIA
Amoxicillin (80–90 mg/kg per day) is recommended for children with acute otitis media except in situations where observation and symptom-based treatment without antibiotics are advocated. These situations include nonsevere illness and an uncertain diagnosis in children 6 months to 2 years of age and nonsevere illness (even if the diagnosis seems certain) in children >2 years of age. Although the optimal duration of therapy has not been conclusively established, a 10-day course is recommended for younger children and for children with severe disease at any age. For children >6 years old who have mild or moderate disease, a course of 5–7 days is considered adequate. Patients whose illness fails to respond should be reassessed at 48–72 h. If acute otitis media is confirmed and antibiotic treatment has not been started, administration of amoxicillin should be commenced. If antibiotic therapy fails, a change is indicated. Failure to respond to second-line antibiotics as well indicates that myringotomy or tympanocentesis may need to be undertaken in order to obtain samples for culture.
The above recommendations can also be followed for the treatment of sinusitis. Detailed information on the further management of these conditions in children has been published by the American Academy of Pediatrics and the American Academy of Family Physicians.
PREVENTION
Measures to prevent pneumococcal disease include vaccination against S. pneumoniae and influenza viruses, reduction of comorbidities that increase the risk of pneumococcal disease, and prevention of antibiotic overuse, which fuels pneumococcal resistance.
Capsular Polysaccharide Vaccines The 23-valent pneumococcal polysaccharide vaccine (PPSV23), containing 25 μg of each capsular polysaccharide, has been licensed for use since 1983. Recommendations for its use vary by country. The U.S. Advisory Committee on Immunization Practices recommends PPSV23 for all persons ≥65 years of age and for those 2–64 years of age who have underlying medical conditions that put them at increased risk for pneumococcal disease or, if infected, disease of increased severity (Table 171-1; see also www.cdc.gov/vaccines/schedules/). The committee recently updated their recommendations to include the combined use of PPSV23 and a conjugate vaccine in at-risk individuals (see “Polysaccharide–Protein Conjugate Vaccines,” below). Revaccination 5 years after the first dose is recommended for persons >2 years of age who have underlying medical conditions but not routinely for those whose only indication is an age of ≥65 years. PPSV23 does not induce an anamnestic response, and antibody concentrations wane over time; thus revaccination is particularly important for individuals with conditions resulting in loss of antibody. Concerns about repeated revaccination have focused on safety (i.e., local reactions) and the induction of immune hyporesponsiveness. Neither the clinical relevance nor the biologic basis of hyporesponsiveness is clear, but, given the possibility of its occurrence, more than one revaccination has not been recommended.
The effectiveness of PPSV23 against IPD, pneumococcal pneumonia, all-cause pneumonia, and death is controversial, with wide variation in observations. The many published meta-analyses of PPSV efficacy have often reached opposing conclusions with regard to a given clinical entity. Generally, observational studies cite greater effectiveness than do controlled clinical trials. The consensus is that PPSV is effective against IPD but is less effective or ineffective against nonbacteremic pneumococcal pneumonia. However, published trials, observational studies, and meta-analyses contradict this view. Efficacy is often lower in the elderly and in immunodeficient patients whose condition is associated with reduced antibody responses to vaccines than in younger, healthier populations. When PPSV is effective, the duration of protection following a single dose of vaccine is estimated to be ~5 years.
What is not disputed is that improved pneumococcal vaccines are needed for adults. Even in the setting of routine pneumococcal conjugate vaccination of infants (which indirectly protects adults from vaccine-serotype strains), disease caused by serotypes not represented in the conjugate vaccine continues to be a significant burden among adults.
Polysaccharide–Protein Conjugate Vaccines Infants and young children respond poorly to PPSV, which contains T cell–independent antigens. Consequently, another class of pneumococcal vaccines, the PCVs, were developed specifically for infants and young children. The first product, a 7-valent PCV, was licensed in 2000 in the United States. Three PCV products—containing 7, 10, and 13 serotypes, respectively—are currently (2014) commercially available. The serotypes included in these PCV formulations are important causes of IPD and antibiotic resistance among young children. Randomized controlled trials have demonstrated a high degree of efficacy of PCVs against vaccine-serotype IPD as well as efficacy against pneumonia, otitis media, nasopharyngeal colonization, and all-cause mortality. PCVs are recommended by the World Health Organization for inclusion in routine childhood immunization schedules worldwide, especially in countries with high infant mortality rates.
The United States was the first country to introduce PCV and therefore has the longest experience with its community-wide effects. The introduction of PCV in the United States has resulted in a >90% reduction in vaccine-serotype IPD among the whole population (Fig. 171-7). This decline has been noted not only in those age groups immunized but also in adults and is attributable to the near elimination of vaccine-serotype nasopharyngeal colonization in immunized infants, which reduces spread to adults. This protection of unimmunized community members through vaccination of a subset of the community is termed the indirect effect. Increases in colonization with—and concomitantly in disease due to—non-vaccine-serotype strains (i.e., replacement colonization and disease) have been seen; however, the absolute rate increases in IPD caused by non-vaccine serotypes are generally small, especially relative to decreases in vaccine-serotype IPD (see “Epidemiology,” above). Since vaccine-serotype strains are more commonly resistant to antibiotics than are non-vaccine serotypes, use of PCV has also resulted in dramatic declines in the proportion and absolute rates of drug-resistant pneumococcal disease. The recommendations of the Advisory Committee on Immunization Practices for the use of conjugate vaccines can be found at www.cdc.gov/MMWR/pdf/wk/mm5909.pdf. Recently, PCV has been shown to prevent pneumococcal infection in HIV-infected adults. In the United States, PCV13 followed by a dose of PPSV23 is now recommended for all immunocompromised children and adults.

FIGURE 171-7 Changes in invasive pneumococcal disease (IPD) incidence, by serotype group, among children <5 years old (top) and adults >65 years old (bottom), 1998–2009. 7-Valent pneumococcal conjugate vaccine (PCV7) was introduced in the United States for routine administration to infants and young children during the second half of 2000, while PCV13 was introduced in 2010, the year following this surveillance period. PCV7 serotypes include serotypes 4, 6B, 9V, 14, 18C, 19F, and 23F as well as cross-reactive serotype 6A. PCV13 serotypes include the PCV7 serotypes as well as serotypes 1, 3, 5, 6A, 7F, and 19A. (Reprinted with permission from Dr. M. Moore, Centers for Disease Control and Prevention.)
Other Prevention Strategies Pneumococcal disease can also be averted through the prevention of illnesses that predispose individuals to pneumococcal infections. Relevant measures include influenza vaccination and improved management and control of diabetes, HIV infection, heart disease, and lung disease. Finally, the reduction of antibiotic misuse is a strategy for the prevention of pneumococcal disease in that antimicrobial resistance directly and indirectly perpetuates organism transmission and disease in the community.
WEBSITES
American Academy of Pediatrics RED BOOK. The report of the Committee on Infectious Diseases: aapredbook.aappublications.org; Pneumococcal Regional Serotype Distribution for Pneumococcal AMC TPP: www.gavialliance.org/library/documents/amc/tpp-codebook/
172 |
Staphylococcal Infections |
Staphylococcus aureus, the most virulent of the many staphylococcal species, has demonstrated its versatility by remaining a major cause of morbidity and mortality worldwide despite the availability of numerous effective antistaphylococcal antibiotics. S. aureus is a pluripotent pathogen, causing disease through both toxin- and non-toxin-mediated mechanisms. This organism is responsible for numerous nosocomial and community-based infections that range from relatively minor skin and soft tissue infections to life-threatening systemic infections.
The “other” staphylococci, collectively designated coagulase-negative staphylococci (CoNS), are considerably less virulent than S. aureus but remain important pathogens in infections that are primarily associated with prosthetic devices.
MICROBIOLOGY AND TAXONOMY
Staphylococci, gram-positive cocci in the family Micrococcaceae, form grapelike clusters on Gram’s stain (Fig. 172-1). These organisms (~1 μm in diameter) are catalase-positive (unlike streptococcal species), nonmotile, aerobic, and facultatively anaerobic. They are capable of prolonged survival on environmental surfaces under varying conditions. Some species have a relatively broad host range, including mammals and birds, whereas for others the host range is quite narrow—i.e., limited to one or two closely related animals.

FIGURE 172-1 Gram’s stain of S. aureus in a sputum sample. (From ASM MicrobeLibrary.org.© Pfizer, Inc.)
More than 30 staphylococcal species are pathogenic. Identification of the more clinically important species has generally relied on a series of biochemical tests. Automated diagnostic systems, kits for biochemical characterization, and DNA-based assays are available for species identification. With few exceptions, S. aureus is distinguished from other staphylococcal species by its production of coagulase, a surface enzyme that converts fibrinogen to fibrin. Latex kits that detect both protein A and clumping factor also distinguish S. aureus from most other staphylococcal species. S. aureus ferments mannitol, is positive for protein A, and produces DNAse. On blood agar plates, S. aureus tends to form golden β-hemolytic colonies; in contrast, CoNS produce small white nonhemolytic colonies. Increasingly, sequence-based methods (e.g., 16S rRNA) are being used to identify different staphylococcal species.
Determining whether multiple staphylococcal isolates from different patients are the same or different is often relevant when there is concern that a nosocomial outbreak is due to a common point source (e.g., a contaminated medical instrument). Molecular typing methods, such as pulsed-field gel electrophoresis and sequence-based techniques (e.g., staphylococcal protein A [SpA] typing), have increasingly been used for this purpose. More recently, whole-genome sequencing has enhanced the ability to discriminate among clinical isolates.
S. AUREUS INFECTIONS
EPIDEMIOLOGY
S. aureus is both a commensal and an opportunistic pathogen. Approximately 30% of healthy persons are colonized with S. aureus, with a smaller percentage (~10%) persistently colonized. The rate of colonization is elevated among insulin-dependent diabetics, HIV-infected patients, patients undergoing hemodialysis, injection drug users, and individuals with skin damage. The anterior nares and oropharynx are frequent sites of human colonization, although the skin (especially when damaged), vagina, axilla, and perineum may also be colonized. These colonization sites serve as a reservoir for future infections.
Transmission of S. aureus most frequently results from direct personal contact. Colonization of different body sites allows transfer from one person to another during contact. Spread of staphylococci in aerosols of respiratory or nasal secretions from heavily colonized individuals has also been reported. Most individuals who develop S. aureus infections become infected with a strain that is already a part of their own commensal flora. Breaches of the skin or mucosal membrane allow S. aureus to initiate infection.
Some diseases increase the risk of S. aureus infection; diabetes, for example, combines an increased rate of S. aureus colonization and the use of injectable insulin with the possibility of impaired leukocyte function. Individuals with congenital or acquired qualitative or quantitative defects of polymorphonuclear leukocytes (PMNs) are at increased risk of S. aureus infections; this group includes neutropenic patients (e.g., those receiving chemotherapeutic agents), those with chronic granulomatous disease, and those with Job’s or Chédiak-Higashi syndrome. Other groups at risk include individuals with end-stage renal disease, HIV infection, skin abnormalities, or prosthetic devices.
S. aureus is a leading cause of health care–associated infections (Chap. 168). It is the most common cause of surgical wound infections and is second only to CoNS as a cause of primary bacteremia. These isolates are generally resistant to multiple antibiotics; thus available therapeutic options are limited. In the community, S. aureus remains an important cause of skin and soft tissue infections, respiratory infections, and (among injection drug users) infective endocarditis. The increasing use of home infusion therapy is another cause of community-acquired staphylococcal infections.
![]() In the past two decades, there has been a dramatic change in the epidemiology of infections due to methicillin-resistant S. aureus (MRSA). In addition to its major role as a nosocomial pathogen, MRSA has become an established community-based pathogen. Numerous outbreaks of community-associated MRSA (CA-MRSA) infections have been reported in both rural and urban settings in widely separated regions throughout the world. The outbreaks have occurred among such diverse groups as children, prisoners, athletes, Native Americans, and drug users. Risk factors common to these outbreaks include poor hygienic conditions, close contact, contaminated material, and damaged skin. These infections have been caused by a limited number of MRSA strains. In the United States, strain USA300 (defined by pulsed-field gel electrophoresis) has been the predominant clone. In other geographic regions of the world, different strains of CA-MRSA have been responsible for these community-based outbreaks. Although the majority of infections caused by these strains have involved the skin and soft tissue, 5–10% have been invasive and potentially life-threatening. CA-MRSA strains have also been responsible for an increasing number of nosocomial infections. Of concern has been the apparent capacity of CA-MRSA to cause disease in immunocompetent individuals.
In the past two decades, there has been a dramatic change in the epidemiology of infections due to methicillin-resistant S. aureus (MRSA). In addition to its major role as a nosocomial pathogen, MRSA has become an established community-based pathogen. Numerous outbreaks of community-associated MRSA (CA-MRSA) infections have been reported in both rural and urban settings in widely separated regions throughout the world. The outbreaks have occurred among such diverse groups as children, prisoners, athletes, Native Americans, and drug users. Risk factors common to these outbreaks include poor hygienic conditions, close contact, contaminated material, and damaged skin. These infections have been caused by a limited number of MRSA strains. In the United States, strain USA300 (defined by pulsed-field gel electrophoresis) has been the predominant clone. In other geographic regions of the world, different strains of CA-MRSA have been responsible for these community-based outbreaks. Although the majority of infections caused by these strains have involved the skin and soft tissue, 5–10% have been invasive and potentially life-threatening. CA-MRSA strains have also been responsible for an increasing number of nosocomial infections. Of concern has been the apparent capacity of CA-MRSA to cause disease in immunocompetent individuals.
PATHOGENESIS
General Concepts S. aureus is a pyogenic pathogen known for its capacity to induce abscess formation at sites of both local and metastatic infections. This classic pathologic response to S. aureus defines the framework within which the infection will progress. The bacteria elicit an inflammatory response characterized by an initial intense infiltration of PMNs and a subsequent infiltration of macrophages and fibroblasts. Either the host cellular response (including the deposition of fibrin and collagen) contains the infection, or infection spreads to the adjoining tissue or the bloodstream.
In toxin-mediated staphylococcal disease, infection is not invariably present. For example, once toxin has been elaborated into food, staphylococcal food poisoning can develop in the absence of viable bacteria. In staphylococcal toxic shock syndrome (TSS), conditions allowing toxin elaboration at colonization sites (e.g., the presence of a superabsorbent tampon) suffice for initiation of clinical illness.
![]() The S. aureus Genome The complete genomes of numerous strains of S. aureus have now been fully sequenced. Among the interesting revelations are (1) the high degree of nucleotide sequence similarity of the core genomes of different strains; (2) acquisition of a relatively large amount of genetic information by horizontal transfer from other bacterial species; and (3) the presence of unique “pathogenicity” or “genomic” islands—mobile genetic elements that contain clusters of enterotoxin and exotoxin genes and/or antimicrobial resistance determinants. Among the genes in these islands are those carrying mecA, the gene responsible for methicillin resistance. Methicillin resistance–containing islands have been designated staphylococcal cassette chromosome mec (SCCmec) types and range in size from ~20 to 60 kb. To date, 11 SCCmec types have been identified. Among the more common types, types 1–3 are traditionally associated with nosocomial MRSA isolates, whereas types 4–6 have been associated with the epidemic CA-MRSA strains.
The S. aureus Genome The complete genomes of numerous strains of S. aureus have now been fully sequenced. Among the interesting revelations are (1) the high degree of nucleotide sequence similarity of the core genomes of different strains; (2) acquisition of a relatively large amount of genetic information by horizontal transfer from other bacterial species; and (3) the presence of unique “pathogenicity” or “genomic” islands—mobile genetic elements that contain clusters of enterotoxin and exotoxin genes and/or antimicrobial resistance determinants. Among the genes in these islands are those carrying mecA, the gene responsible for methicillin resistance. Methicillin resistance–containing islands have been designated staphylococcal cassette chromosome mec (SCCmec) types and range in size from ~20 to 60 kb. To date, 11 SCCmec types have been identified. Among the more common types, types 1–3 are traditionally associated with nosocomial MRSA isolates, whereas types 4–6 have been associated with the epidemic CA-MRSA strains.
![]() A limited number of MRSA clones have been responsible for most community- and hospital-associated infections worldwide. A comparison of these strains with those from earlier outbreaks (e.g., the phage 80/81 strains from the 1950s) has revealed preservation of the nucleotide sequence over time. This observation suggests that these strains possess determinants that facilitate survival and spread.
A limited number of MRSA clones have been responsible for most community- and hospital-associated infections worldwide. A comparison of these strains with those from earlier outbreaks (e.g., the phage 80/81 strains from the 1950s) has revealed preservation of the nucleotide sequence over time. This observation suggests that these strains possess determinants that facilitate survival and spread.
![]() Regulation of Virulence Gene Expression In both toxin-mediated and non-toxin-mediated diseases due to S. aureus, the expression of virulence determinants associated with infection depends on a series of regulatory genes (e.g., accessory gene regulator [agr] and staphylococcal accessory regulator [sar]) that coordinately control the expression of many virulence genes. The regulatory gene agr is part of a quorum-sensing signal transduction pathway that senses and responds to bacterial density. Staphylococcal surface proteins are synthesized during the bacterial exponential growth phase in vitro. In contrast, many secreted proteins, such as α toxin, the enterotoxins, and assorted enzymes, are released during the postexponential growth phase in response to transcription of the effector molecule of agr, RNAIII.
Regulation of Virulence Gene Expression In both toxin-mediated and non-toxin-mediated diseases due to S. aureus, the expression of virulence determinants associated with infection depends on a series of regulatory genes (e.g., accessory gene regulator [agr] and staphylococcal accessory regulator [sar]) that coordinately control the expression of many virulence genes. The regulatory gene agr is part of a quorum-sensing signal transduction pathway that senses and responds to bacterial density. Staphylococcal surface proteins are synthesized during the bacterial exponential growth phase in vitro. In contrast, many secreted proteins, such as α toxin, the enterotoxins, and assorted enzymes, are released during the postexponential growth phase in response to transcription of the effector molecule of agr, RNAIII.
It has been hypothesized that these regulatory genes serve a similar function in vivo. Successful invasion requires the sequential expression of these different bacterial elements. Bacterial adhesins are needed to initiate colonization of host tissue surfaces. The subsequent release of various enzymes enables the colony to obtain nutritional support and permits bacteria to spread to adjacent tissues. Studies with strains in which these regulatory genes are inactivated show reduced virulence in several animal models of S. aureus infection.
Pathogenesis of Invasive S. aureus Infection Staphylococci are opportunists. For these organisms to invade the host and cause infection, some or all of the following steps are necessary: contamination and colonization of host tissue surfaces, breach of cutaneous or mucosal barriers, establishment of a localized infection, invasion, evasion of the host response, and metastatic spread. Colonizing strains or strains transferred from other individuals are introduced into damaged skin, a wound, or the bloodstream. Recurrences of S. aureus infections are common, apparently because of the capacity of these pathogens to survive, to persist in a quiescent state in various tissues, and then to cause recrudescent infections when suitable conditions arise.
S. AUREUS COLONIZATION OF BODY SURFACES The anterior nares is one of the primary sites of staphylococcal colonization in humans. Colonization appears to involve the attachment of S. aureus to keratinized epithelial cells of the anterior nares. Other factors that may contribute to colonization include the influence of other resident nasal flora and their bacterial density, host factors, and nasal mucosal damage (e.g., that resulting from inhalational drug use). Other colonized body sites, such as damaged skin, the groin, and the oropharynx, may be particularly important reservoirs for CA-MRSA strains.
INOCULATION AND COLONIZATION OF TISSUE SURFACES Staphylococci may be introduced into tissue as a result of minor abrasions, administration of medications such as insulin, or establishment of IV access with catheters. After their introduction into a tissue site, bacteria replicate and colonize the host tissue surface. A family of structurally related S. aureus surface proteins referred to as MSCRAMMs (microbial surface components recognizing adhesive matrix molecules) plays an important role in mediating adherence to these sites. By adhering to exposed matrix molecules (e.g., fibrinogen, fibronectin), MSCRAMMs such as clumping factor and collagen-binding protein enable the bacteria to colonize different tissue surfaces; these proteins contribute to the pathogenesis of invasive infections such as endocarditis and septic arthritis by facilitating the adherence of S. aureus to surfaces with exposed fibrinogen or collagen.
Although CoNS are classically known for their ability to elaborate biofilms and to colonize prosthetic devices, S. aureus also possesses the genes responsible for biofilm formation, such as the intercellular adhesion (ica) locus. Binding to these devices occurs in a stepwise fashion, involving staphylococcal adherence to serum constituents that have coated the device surface and subsequent biofilm elaboration. S. aureus is thus a frequent cause of biomedical-device infections.
INVASION After colonization, staphylococci replicate at the initial site of infection, elaborating enzymes that include serine proteases, hyaluronidases, thermonucleases, and lipases. These enzymes facilitate bacterial survival and local spread across tissue surfaces, although their precise role in infections is not well defined. The lipases may facilitate survival in lipid-rich areas such as the hair follicles, where S. aureus infections are often initiated. The S. aureus toxin Panton-Valentine leukocidin is cytolytic to PMNs, macrophages, and monocytes. Strains elaborating this toxin have been epidemiologically linked with cutaneous and more serious infections caused by strains of CA-MRSA. MSCRAMMs also appear to play an important role in the ability of S. aureus to spread and cause disease at other tissue sites.
Constitutional findings may result from either localized or systemic infections. The staphylococcal cell wall—consisting of alternating N-acetyl muramic acid and N-acetyl glucosamine units in combination with an additional cell wall component, lipoteichoic acid—can initiate an inflammatory response that includes the sepsis syndrome. Staphylococcal α toxin, which causes pore formation in various eukaryotic cells, can also initiate an inflammatory response with findings suggestive of sepsis.
EVASION OF HOST DEFENSE MECHANISMS Staphylococci have a multitude of immune evasion strategies that are critical to their success as invasive pathogens. They possess an antiphagocytic polysaccharide microcapsule. Most human S. aureus infections are due to capsular types 5 and 8. The zwitterionic (both negatively and positively charged) S. aureus capsule plays a critical role in the induction of abscess formation. Protein A, an MSCRAMM unique to S. aureus, acts as an Fc receptor, binding the Fc portion of IgG subclasses 1, 2, and 4 and preventing opsonophagocytosis by PMNs. Both chemotaxis inhibitory protein of staphylococci (CHIPS, a secreted protein) and extracellular adherence protein (EAP, a surface protein) interfere with PMN migration to sites of infection.
An additional potential mechanism of S. aureus evasion is its capacity for intracellular survival. Both professional and nonprofessional phagocytes internalize staphylococci. Internalization by these cells may provide a sanctuary that protects bacteria against the host’s defenses. The intracellular environment favors the phenotypic expression of S. aureus small-colony variants. Small-colony variants are found in patients receiving antimicrobial therapy (e.g., with aminoglycosides) and in those with cystic fibrosis or osteomyelitis. These variants, whether intra- or extracellular, may facilitate prolonged staphylococcal survival in different tissue sites and enhance the likelihood of recurrences. Finally, S. aureus can survive within PMNs and may use these cells to spread and to seed other tissue sites.
PATHOGENESIS OF COMMUNITY-ACQUIRED MRSA INFECTIONS A number of virulence determinants have been identified as contributing to the pathogenesis of CA-MRSA infections. There is a strong epidemiologic association linking the presence of the gene for the Panton-Valentine leukocidin with skin and soft tissue infections as well as with necrotizing postinfluenza pneumonia. Other determinants that play a role in the pathogenesis of these infections include the arginine catabolic mobile element (ACME), a cluster of unique genes that may facilitate evasion of host defense mechanisms; phenol-soluble modulins, a family of cytolytic peptides; and α toxin.
Host Response to S. aureus Infection The primary host response to S. aureus infection is the recruitment of PMNs. These cells are attracted to infection sites by bacterial components such as formylated peptides or peptidoglycan as well as by the cytokines tumor necrosis factor (TNF) and interleukins (ILs) 1 and 6, which are released by activated macrophages and endothelial cells.
Although most individuals have antibodies to staphylococci, it is not clear that antibody levels are qualitatively or quantitatively sufficient to protect against infection. Although anticapsular and anti-MSCRAMM antibodies facilitate opsonization in vitro and have been protective against infection in several animal models, they have not yet successfully prevented staphylococcal infections in clinical trials.
Pathogenesis of Toxin-Mediated Disease S. aureus produces three types of toxin: cytotoxins, pyrogenic toxin superantigens, and exfoliative toxins. Both epidemiologic data and studies in animals suggest that antitoxin antibodies are protective against illness in TSS, staphylococcal food poisoning, and staphylococcal scalded-skin syndrome (SSSS). Illness develops after toxin synthesis and absorption and the subsequent toxin-initiated host response.
ENTEROTOXIN AND TOXIC SHOCK SYNDROME TOXIN 1 (TSST-1) The pyrogenic toxin superantigens are a family of small-molecular-size, structurally similar proteins that are responsible for two diseases: TSS and food poisoning. TSS results from the ability of enterotoxins and TSST-1 to function as T cell mitogens. In the normal process of antigen presentation, the antigen is first processed within the cell, and peptides are then presented in the major histocompatibility complex (MHC) class II groove, initiating a measured T cell response. In contrast, enterotoxins bind directly to the invariant region of MHC—outside the MHC class II groove. The enterotoxins can then bind T cell receptors via the vβ chain; this binding results in a dramatic overexpansion of T cell clones (up to 20% of the total T cell population). The consequence of this T cell expansion is a “cytokine storm,” with the release of inflammatory mediators that include interferon γ, IL-1, IL-6, TNF-α, and TNF-β. The resulting multisystem disease produces a constellation of findings that mimic those in endotoxin shock; however, the pathogenic mechanisms differ. The release of endotoxin from the gastrointestinal tract may synergistically enhance the toxin’s effects.
A different region of the enterotoxin molecule is responsible for the symptoms of food poisoning. The enterotoxins are heat stable and can survive conditions that kill the bacteria. Illness results from the ingestion of preformed toxin. As a result, the incubation period is short (1–6 h). The toxin stimulates the vagus nerve and the vomiting center of the brain. It also appears to stimulate intestinal peristaltic activity.
EXFOLIATIVE TOXINS AND SSSS The exfoliative toxins are responsible for SSSS. The toxins that produce disease in humans are of two serotypes: ETA and ETB. These toxins are serine proteases, which cleave desmosomal cadherins in the superficial layer of the skin, triggering exfoliation. The result is a split in the epidermis at the granular level, which is responsible for the superficial desquamation of the skin that typifies this illness.
DIAGNOSIS
Staphylococcal infections are readily diagnosed by Gram’s stain (Fig. 172-1) and microscopic examination of abscess contents or of infected tissue. Routine culture of infected material usually yields positive results, and blood cultures are sometimes positive even when infections are localized to extravascular sites. S. aureus is rarely a blood culture contaminant. Polymerase chain reaction (PCR)–based assays have been applied to the rapid diagnosis of S. aureus infection and are increasingly used in clinical microbiology laboratories. A number of point-of-care tests are now available to screen patients for colonization with MRSA. Determining whether patients with documented S. aureus bacteremia also have infective endocarditis or a metastatic focus of infection remains a diagnostic challenge. Uniformly positive blood cultures suggest an endovascular infection such as endocarditis (see “Bacteremia, Sepsis, and Infective Endocarditis,” below).
CLINICAL SYNDROMES
|
COMMON ILLNESSES CAUSED BY STAPHYLOCOCCUS AUREUS |
Skin and Soft Tissue Infections S. aureus causes a variety of cutaneous infections, many of which can also be caused by group A streptococci or (less commonly) other streptococcal species. Common factors predisposing to S. aureus cutaneous infection include chronic skin conditions (e.g., eczema), skin damage (e.g., insect bites, minor trauma), injections (e.g., in diabetes, injection drug use), and poor personal hygiene. These infections are characterized by the formation of pus-containing blisters, which often begin in hair follicles and spread to adjoining tissues. Folliculitis is a superficial infection that involves the hair follicle, with a central area of purulence (pus) surrounded by induration and erythema. Furuncles (boils) are more extensive, painful lesions that tend to occur in hairy, moist regions of the body and extend from the hair follicle to become a true abscess with an area of central purulence. Carbuncles are most often located in the lower neck and are even more severe and painful, resulting from the coalescence of other lesions that extend to a deeper layer of the subcutaneous tissue. In general, furuncles and carbuncles are readily apparent, with pus often expressible or discharging from the abscess. Other cutaneous S. aureus infections include impetigo and cellulitis. S. aureus is one of the most common causes of surgical wound infection.
Mastitis develops in 1–3% of nursing mothers. This infection of the breast, which generally presents within 2–3 weeks after delivery, is characterized by findings that range from cellulitis to abscess formation. Systemic signs, such as fever and chills, are often present in more severe cases.
Musculoskeletal Infections S. aureus is among the most common causes of bone infections—both those resulting from hematogenous dissemination and those arising from contiguous spread from a soft tissue site. Hematogenous osteomyelitis in children most often involves the long bones. Infections present as fever and bone pain or with a child’s reluctance to bear weight. The white blood cell count and erythrocyte sedimentation rate are often elevated. Blood cultures are positive in ~50% of cases. When necessary, bone biopsies for culture and histopathologic examination are usually diagnostic. Routine x-rays may be normal for up to 14 days after the onset of symptoms. 99mTc-phosphonate scanning often detects early evidence of infection. MRI is more sensitive than other techniques in establishing a radiologic diagnosis.
In adults, hematogenous osteomyelitis involving the long bones is less common. However, vertebral osteomyelitis is among the more common clinical presentations. Vertebral bone infections are most often seen in patients with endocarditis, those undergoing hemodialysis, diabetics, and injection drug users. These infections may present as intense back pain and fever but may also be clinically occult, presenting as chronic back pain and low-grade fever. S. aureus is the most common cause of epidural abscess, a complication that can result in neurologic compromise. Patients complain of difficulty voiding or walking and of radicular pain in addition to the symptoms associated with their osteomyelitis. Surgical intervention in this setting often constitutes a medical emergency. MRI most reliably establishes the diagnosis (Fig. 172-2).

FIGURE 172-2 S. aureus vertebral osteomyelitis and epidural abscess involving the thoracic disk between T9 and T10. Sagittal postcontrast MRI of the spine illustrates destruction of the T9–T10 intervertebral space with enhancement (arrow). There is impingement on the thoracic cord and an epidural collection extending from T9 through T11 (short arrows).
Bone infections that result from contiguous spread tend to develop from soft tissue infections, such as those associated with diabetic or vascular ulcers, surgery, or trauma. Exposure of bone, a draining fistulous tract, failure to heal, or continued drainage suggests involvement of underlying bone. Bone involvement is established by bone culture and histopathologic examination (revealing evidence of PMN infiltration). Contamination of culture material from adjacent tissue can make the diagnosis of osteomyelitis difficult in the absence of pathologic confirmation. In addition, it is sometimes hard to distinguish radiologically between osteomyelitis and overlying soft tissue infection with underlying osteitis.
In both children and adults, S. aureus is the most common cause of septic arthritis in native joints. This infection is rapidly progressive and may be associated with extensive joint destruction if left untreated. It presents as intense pain on motion of the affected joint, swelling, and fever. Aspiration of the joint reveals turbid fluid, with >50,000 PMNs/μL and gram-positive cocci in clusters on Gram’s stain (Fig. 172-1). In adults, septic arthritis may result from trauma, surgery, or hematogenous dissemination. The most commonly involved joints include the knees, shoulders, hips, and phalanges. Infection frequently develops in joints previously damaged by osteoarthritis or rheumatoid arthritis. Iatrogenic infections resulting from aspiration or injection of agents into the joint also occur. In these settings, the patient experiences increased pain and swelling in the involved joint in association with fever.
Pyomyositis is an unusual infection of skeletal muscles that is seen primarily in tropical climates but also occurs in immunocompromised and HIV-infected patients. It is believed to arise from occult bacteremia. Pyomyositis presents as fever, swelling, and pain overlying the involved muscle. Aspiration of fluid from the involved tissue yields pus. Although a history of trauma may be associated with the infection, its pathogenesis is poorly understood.
Respiratory Tract Infections Respiratory tract infections caused by S. aureus occur in selected clinical settings. S. aureus is a cause of serious respiratory tract infections in newborns and infants; these infections present with shortness of breath, fever, and respiratory failure. Chest x-ray may reveal pneumatoceles (shaggy, thin-walled cavities). Pneumothorax and empyema are recognized complications.
In adults, nosocomial S. aureus pulmonary infections are common among intubated patients in intensive care units. Nasally colonized patients are at increased risk of these infections. The clinical presentation is no different from that encountered in pulmonary infections of other bacterial etiologies. Patients produce increased volumes of purulent sputum and develop respiratory distress, fever, and new pulmonary infiltrates. Distinguishing bacterial pneumonia from respiratory failure or other causes of new pulmonary infiltrates in critically ill patients is often difficult and relies on a constellation of clinical, radiologic, and laboratory findings.
Community-acquired respiratory tract infections due to S. aureus usually follow viral infections—most commonly influenza. Patients may present with fever, bloody sputum production, and midlung-field pneumatoceles or multiple, patchy pulmonary infiltrates. Diagnosis is made by sputum Gram’s stain and culture. Blood cultures, although useful, are usually negative.
Bacteremia, Sepsis, and Infective Endocarditis S. aureus bacteremia may be complicated by sepsis, endocarditis, vasculitis, or metastatic seeding (establishment of suppurative collections at other tissue sites). The frequency of metastatic seeding during bacteremia has been estimated to be as high as 31%. Among the more commonly seeded tissue sites are bones, joints, kidneys, and lungs.
Recognition of these complications by clinical and laboratory diagnostic methods alone is often difficult. Comorbid conditions that are frequently seen in association with S. aureus bacteremia and that increase the risk of complications include diabetes, HIV infection, and renal insufficiency. Other host factors associated with an increased risk of complications include presentation with community-acquired S. aureus bacteremia (except in injection drug users), lack of an identifiable primary focus of infection, and the presence of prosthetic devices or material.
Clinically, S. aureus sepsis presents in a manner similar to that documented for sepsis due to other bacteria. The well-described progression of hemodynamic changes—beginning with respiratory alkalosis and clinical findings of hypotension and fever—is commonly seen. The microbiologic diagnosis is established by positive blood cultures.
The overall incidence of S. aureus endocarditis has increased over the past 20 years. S. aureus is now the leading cause of endocarditis worldwide, accounting for 25–35% of cases. This increase is due, at least in part, to the increased use of intravascular devices. Studies of patients with S. aureus bacteremia and intravascular catheters that used transesophageal echocardiography found an infective endocarditis incidence of ~25%. Other factors associated with an increased risk of endocarditis are injection drug use, hemodialysis, the presence of intravascular prosthetic devices at the time of bacteremia, and immunosuppression. Patients with implantable cardiac devices (e.g., permanent pacemakers) are at increased risk of endocarditis or device-related infections. Despite the availability of effective antibiotics, mortality rates from these infections continue to range from 20% to 40%, depending on both the host and the nature of the infection. Complications of S. aureus endocarditis include cardiac valvular insufficiency, peripheral emboli, metastatic seeding, and central nervous system (CNS) involvement (e.g., mycotic aneurysms, embolic strokes).
S. aureus endocarditis is encountered in four clinical settings: (1) right-sided endocarditis in association with injection drug use, (2) left-sided native-valve endocarditis, (3) prosthetic-valve endocarditis, and (4) nosocomial endocarditis. In each of these settings, the diagnosis is suspected by recognition of clinical stigmata suggestive of endocarditis. These findings include cardiac manifestations, such as new or changing cardiac valvular murmurs; cutaneous evidence, such as vasculitic lesions, Osler’s nodes, or Janeway lesions; evidence of right- or left-sided embolic disease; and a history suggesting a risk for S. aureus bacteremia. In the absence of antecedent antibiotic therapy, blood cultures are almost uniformly positive. Transthoracic echocardiography, while less sensitive than transesophageal echocardiography, is less invasive and may establish the presence of valvular vegetations. The Duke criteria (see Table 155-3) are now commonly used to help establish the likelihood of this diagnosis.
Acute right-sided tricuspid valvular S. aureus endocarditis is most often seen in injection drug users. The classic presentation includes a high fever, a toxic clinical appearance, pleuritic chest pain, and the production of purulent (sometimes bloody) sputum. Chest x-rays or CT scans reveal evidence of septic pulmonary emboli (small, peripheral, circular lesions that may cavitate with time) (Fig. 172-3). A high percentage of affected patients have no history of antecedent valvular damage. At the outset of their illness, patients may present with fever alone, without cardiac or other localizing findings. As a result, a high index of clinical suspicion is essential for diagnosis.

FIGURE 172-3 CT scan illustrating septic pulmonary emboli in a patient with methicillin-resistant Staphylococcus aureus bacteremia.
Individuals with antecedent cardiac valvular damage more commonly present with left-sided native-valve endocarditis involving the damaged valve. These patients tend to be older than those with right-sided endocarditis, their prognosis is worse, and their incidence of complications (including peripheral emboli, cardiac decompensation, and metastatic seeding) is higher.
S. aureus is one of the more common causes of prosthetic-valve endocarditis. This infection is especially fulminant in the early postoperative period and is associated with a high mortality rate. In most instances, medical therapy alone is not sufficient and urgent valve replacement is necessary. Patients are prone to develop valvular insufficiency or myocardial abscesses originating from the region of valve implantation.
The increased frequency of nosocomial endocarditis (15–30% of cases, depending on the series) reflects in part the increased use of intravascular devices. This form of endocarditis is most commonly caused by S. aureus. Because patients often are critically ill, are receiving antibiotics for various other indications, and have comorbid conditions, the diagnosis is often missed.
Urinary Tract Infections Urinary tract infections (UTIs) are infrequently caused by S. aureus. The presence of S. aureus in the urine generally suggests hematogenous dissemination. Ascending S. aureus infections occasionally result from instrumentation of the genitourinary tract.
Prosthetic Device–Related Infections S. aureus accounts for a large proportion of prosthetic device–related infections. These infections often involve intravascular catheters, prosthetic valves, orthopedic devices, peritoneal catheters, pacemakers, left-ventricular-assist devices, and vascular grafts. In contrast with the more indolent presentation of CoNS infections, S. aureus device-related infections are often more acute, with both localized and systemic manifestations. The latter infections also tend to progress more rapidly. It is relatively common for a pyogenic collection to be present at the device site. Aspiration of these collections and performance of blood cultures are important components in establishing a diagnosis. S. aureus infections tend to occur more commonly soon after implantation unless the device is used for access (e.g., intravascular or hemodialysis catheters). In the latter instance, infections can occur at any time. As in most prosthetic-device infections, successful therapy usually involves removal of the device. Left in place, the device is a potential nidus for either persistent or recurrent infections.
Infections Associated with Community-Acquired MRSA Although the skin and soft tissues are the most common sites of infection associated with CA-MRSA, 5–10% of these infections are invasive and can even be life-threatening. The latter unique infections, including necrotizing fasciitis, necrotizing pneumonia, and sepsis with Waterhouse-Friderichsen syndrome or purpura fulminans, were rarely associated with S. aureus prior to the emergence of CA-MRSA. These life-threatening infections reflect the increased virulence of CA-MRSA strains.
Toxin-Mediated Diseases • FOOD POISONING S. aureus is among the most common causes of foodborne outbreaks of infection in the United States. Staphylococcal food poisoning results from the inoculation of toxin-producing S. aureus into food by colonized food handlers. Toxin is then elaborated in such growth-promoting food as custards, potato salad, or processed meats. Even if the bacteria are killed by warming, the heat-stable toxin is not destroyed. The onset of illness is rapid, occurring within 1–6 h of ingestion. The illness is characterized by nausea and vomiting, although diarrhea, hypotension, and dehydration may also occur. The differential diagnosis includes diarrhea of other etiologies, especially that caused by similar toxins (e.g., the toxins elaborated by Bacillus cereus). The rapidity of onset, the absence of fever, and the epidemic nature of the presentation (without second-degree spread) arouse suspicion of staphylococcal food poisoning. Symptoms generally resolve within 8–10 h. The diagnosis can be established by the demonstration of bacteria or the documentation of enterotoxin in the implicated food. Treatment is entirely supportive.
TOXIC SHOCK SYNDROME TSS gained attention in the early 1980s, when a nationwide outbreak occurred in the United States among young, otherwise healthy, menstruating women. Epidemiologic investigation demonstrated that these cases were associated with the use of a highly absorbent tampon that had recently been introduced to the market. Subsequent studies established the role of TSST-1 in these illnesses. Withdrawal of the tampon from the market resulted in a rapid decline in the incidence of this disease. However, menstrual and nonmenstrual cases continue to be reported. Nonmenstrual cases are frequently seen in patients with surgical or postpartum wound infections.
The clinical presentation is similar in menstrual and nonmenstrual TSS. Evidence of clinical S. aureus infection is not a prerequisite. TSS results from the elaboration of an enterotoxin or the structurally related enterotoxin-like TSST-1. More than 90% of menstrual cases are caused by TSST-1, whereas a high percentage of nonmenstrual cases are caused by enterotoxins. TSS begins with relatively nonspecific flulike symptoms. In menstrual cases, the onset usually comes 2 or 3 days after the start of menstruation. Patients present with fever, hypotension, and erythroderma of variable intensity. Mucosal involvement is common (e.g., conjunctival hyperemia). The illness can rapidly progress to symptoms that include vomiting, diarrhea, confusion, myalgias, and abdominal pain. These symptoms reflect the multisystemic nature of the disease, with involvement of the liver, kidneys, gastrointestinal tract, and/or CNS. Desquamation of the skin occurs during convalescence, usually 1–2 weeks after the onset of illness. Laboratory findings may include azotemia, leukocytosis, hypoalbuminemia, thrombocytopenia, and liver function abnormalities.
Diagnosis of TSS still depends on a constellation of findings rather than one specific finding and on a lack of evidence of other possible infections (Table 172-2). Other diagnoses to be considered are drug toxicities, viral exanthems, Rocky Mountain spotted fever, sepsis, and Kawasaki disease. Illness occurs only in persons who lack antibody to TSST-1. Recurrences are possible if antibody fails to develop after the illness.
|
CASE DEFINITION OF S. AUREUS TOXIC SHOCK SYNDROME |
Source: M Wharton et al: Case definitions for public health surveillance. MMWR 39:1, 1990; with permission.
STAPHYLOCOCCAL SCALDED-SKIN SYNDROME SSSS primarily affects newborns and children. The illness may vary from a localized blister to exfoliation of much of the skin surface. The skin is usually fragile and often tender, with thin-walled, fluid-filled bullae. Gentle pressure results in rupture of the lesions, leaving denuded underlying skin (Nikolsky’s sign; Fig. 172-4). The mucous membranes are usually spared. In more generalized infection, there are often constitutional symptoms, including fever, lethargy, and irritability with poor feeding. Significant amounts of fluid can be lost in more extensive cases. Illness usually follows localized infection at one of a number of possible sites. SSSS is much less common among adults but can follow infections caused by exfoliative toxin–producing strains.

FIGURE 172-4 Evidence of staphylococcal scalded-skin syndrome in a 6-year-old boy. Nikolsky’s sign, with separation of the superficial layer of the outer epidermal layer, is visible. (Reprinted with permission from LA Schenfeld et al: N Engl J Med 342:1178, 2000. © 2000 Massachusetts Medical Society. All rights reserved.)
PREVENTION
Primary prevention of S. aureus infections in the hospital setting involves hand washing and careful attention to appropriate isolation procedures. Through careful screening for MRSA carriage and strict isolation practices, several Scandinavian countries have been remarkably successful at preventing the introduction and dissemination of MRSA in hospitals.
Decolonization strategies, using both universal and targeted approaches with topical agents (e.g., mupirocin) to eliminate nasal colonization and/or chlorhexidine to eliminate cutaneous colonization with S. aureus, have been successful in some clinical settings (e.g., intensive care units) where the risk of infection is high. An analysis of clinical trials suggests that there may also be a reduction in the incidence of postsurgical infections among persons who are nasally colonized with S. aureus.
“Bundling” (the application of selected medical interventions in a sequence of prescribed steps) has reduced rates of nosocomial infections related to such procedures as the insertion of intravenous catheters, in which staphylococci are among the most common pathogens (see Table 168-4). A number of immunization strategies to prevent S. aureus infections—both active (e.g., capsular polysaccharide–protein conjugate vaccine) and passive (e.g., clumping factor antibody)—have been investigated. However, none has been successful for either prophylaxis or therapy in clinical trials.
COAGULASE-NEGATIVE STAPHYLOCOCCAL INFECTIONS
Although considerably less virulent than S. aureus, CoNS are among the most common causes of prosthetic-device infections. Approximately half of the identified CoNS species have been associated with human infections. Of these species, Staphylococcus epidermidis is the most common human pathogen. This component of the normal human flora is found on the skin (where it is the most abundant bacterial species) as well as in the oropharynx and vagina. Staphylococcus saprophyticus, a novobiocin-resistant species, is a common pathogen in UTIs.
PATHOGENESIS
S. epidermidis is the CoNS species most often associated with prosthetic-device infections. Infection is a two-step process, with initial adhesion to the device followed by colonization. S. epidermidis is uniquely adapted to colonize these devices by its capacity to elaborate the extracellular polysaccharide (glycocalyx or slime) that facilitates formation of a protective biofilm on the device surface.
Implanted prosthetic material is rapidly coated with host serum or tissue constituents such as fibrinogen or fibronectin. These molecules serve as potential bridging ligands, facilitating initial bacterial attachment to the device surface. A number of staphylococcal surface-associated proteins, such as autolysin (AtlE), fibrinogen-binding protein, and accumulation-associated protein (AAP), may play a role in attachment to either modified or unmodified prosthetic surfaces. The polysaccharide intercellular adhesin facilitates subsequent staphylococcal colonization and accumulation on the device surface. In S. epidermidis, intercellular adhesin (ica) genes are more commonly found in strains associated with device infections than in strains associated with colonization of mucosal surfaces. Biofilm appears to act as a barrier protecting bacteria from host defense mechanisms as well as from antibiotics, while providing a suitable environment for bacterial survival. Poly-γ-DL-glutamic acid is secreted by S. epidermidis and provides protection against neutrophil phagocytosis.
Two additional staphylococcal species, Staphylococcus lugdunensis and Staphylococcus schleiferi, produce more serious infections (native-valve endocarditis and osteomyelitis) than do other CoNS. The basis for this enhanced virulence is not known, although both species appear to share more virulence determinants with S. aureus (e.g., clumping factor and lipase) than do other CoNS.
The capacity of S. saprophyticus to cause UTIs in young women appears to be related to its enhanced capacity to adhere to uroepithelial cells. A 160-kDa hemagglutinin/adhesin may contribute to this affinity.
DIAGNOSIS
Although the detection of CoNS at sites of infection or in the bloodstream is not difficult by standard microbiologic culture methods, interpretation of these results is frequently problematic. Because these organisms are present in large numbers on the skin, they often contaminate cultures. It has been estimated that only 10–25% of blood cultures positive for CoNS reflect true bacteremia. Similar problems arise with cultures obtained from other sites. Among the clinical findings suggestive of true bacteremia are fever, evidence of local infection (e.g., erythema or purulent drainage at the IV catheter site), leukocytosis, and systemic signs of sepsis. Laboratory findings suggestive of true bacteremia include multiple isolations of the same strain (i.e., the same species with the same antibiogram or a closely related DNA fingerprint) from separate cultures, growth of the strain within 48 h, and bacterial growth in both aerobic and anaerobic bottles.
CLINICAL SYNDROMES
CoNS cause a diverse array of prosthetic device–related infections, including those that involve prosthetic cardiac valves and joints, vascular grafts, intravascular devices, and CNS shunts. In all of these settings, the clinical presentation is similar. The signs of localized infection are often subtle, the rate of disease progression is slow, and the systemic findings are often limited. Signs of infection, such as purulent drainage, pain at the site, or loosening of prosthetic implants, are sometimes evident. Fever is frequently but not always present, and there may be mild leukocytosis. Acute-phase reactant levels, the erythrocyte sedimentation rate, and the C-reactive protein concentration may be elevated.
Infections that are not associated with prosthetic devices are infrequent, although native-valve endocarditis due to CoNS has accounted for ~5% of cases in some reviews. S. lugdunensis appears to be a more aggressive pathogen in this setting, causing greater mortality and rapid valvular destruction with abscess formation.
|
TREATMENT |
STAPHYLOCOCCAL INFECTIONS |
GENERAL PRINCIPLES OF THERAPY
Surgical incision and drainage of all suppurative collections constitute the most important therapeutic intervention for staphylococcal infections. The emergence of MRSA in the community has increased the importance of culturing all collections in order to identify pathogens and to determine antimicrobial susceptibility. Successful therapy for prosthetic-device infections generally requires device removal. In situations in which removal is not possible or the infection is due to CoNS, an initial attempt at medical therapy without device removal may be warranted. Because of the well-recognized risk of complications associated with S. aureus bacteremia (e.g., endocarditis, metastatic foci of infection), therapy is generally prolonged (4–6 weeks) unless the patient is identified as being among those individuals who are at low risk for complications.
DURATION OF ANTIMICROBIAL THERAPY
Debate continues regarding the duration of therapy for bacteremic S. aureus infections. Patients with “complicated” bacteremia are at increased risk of endocarditis and metastatic infections. Among the findings associated with an increased risk of complicated bacteremia are persistently positive blood cultures 96 h after institution of therapy, acquisition of the infection in the community, failure to remove a removable focus of infection (i.e., an intravascular catheter), and infection with cutaneous or embolic manifestations. For immunocompetent patients in whom short-course therapy is planned, transesophageal echocardiography to rule out endocarditis is warranted because neither clinical nor laboratory findings can reliably detect cardiac involvement. In addition, an aggressive radiologic investigation to identify potential metastatic collections is indicated. All symptomatic body sites must be carefully evaluated.
CHOICE OF ANTIMICROBIAL AGENTS
The choice of antimicrobial agents to treat both coagulase-positive and coagulase-negative staphylococcal infections has become increasingly problematic because of the prevalence of multidrug-resistant strains. Staphylococcal resistance to most antibiotic families, including β-lactams, aminoglycosides, fluoroquinolones, and (to a lesser extent) glycopeptides, has increased. This trend is more apparent with CoNS: >80% of nosocomial isolates are resistant to methicillin, and these methicillin-resistant strains are usually resistant to most other antibiotics as well. Because the selection of antimicrobial agents for S. aureus infections is similar to that for CoNS infections, treatment options for these pathogens are discussed together and are summarized in Table 172-3.
|
ANTIMICROBIAL THERAPY FOR STAPHYLOCOCCAL INFECTIONSa |

As a result of the widespread dissemination of plasmids containing the enzyme penicillinase, few strains of staphylococci (≤5%) remain susceptible to penicillin. However, penicillin remains the drug of choice against susceptible strains if the laboratory can reliably test for penicillin susceptibility. Penicillin-resistant isolates are treated with semisynthetic penicillinase-resistant penicillins (SPRPs), such as oxacillin or nafcillin. Methicillin, the first of the SPRPs, is now used infrequently. Cephalosporins are alternative therapeutic agents for these infections. Second- and third-generation cephalosporins do not offer a therapeutic advantage over first-generation cephalosporins for the treatment of staphylococcal infections. The carbapenems have excellent activity against methicillin-sensitive S. aureus but not against MRSA.
The isolation of MRSA was reported within 1 year of the introduction of methicillin. Since then, the prevalence of MRSA has steadily increased. In many hospitals, 40–50% of S. aureus isolates are now resistant to methicillin. Resistance to methicillin indicates resistance to all SPRPs as well as to all cephalosporins (except ceftaroline). Production of a novel penicillin-binding protein (PBP2a) is responsible for methicillin resistance. This protein is synthesized by the mecA gene, which (as stated above) is part of a large mobile genetic element—a pathogenicity or genomic island—called SCCmec. It is hypothesized that this genetic material was acquired via horizontal transfer from a related staphylococcal species, such as Staphylococcus sciuri. Phenotypic expression of methicillin resistance may be constitutive (i.e., expressed in all organisms in a population) or heterogeneous (i.e., displayed by only a proportion of the total organism population). Detection of methicillin resistance in the clinical microbiology laboratory can be difficult if the strain expresses heterogeneous resistance. Therefore, susceptibility studies are routinely performed at reduced temperatures (≤35°C for 24 h), with increased concentrations of salt in the medium to enhance the expression of resistance. In addition to PCR-based techniques, a number of rapid methods for the detection of methicillin resistance have been developed.
In light of decreasing susceptibility of MRSA isolates to vancomycin, both vancomycin and daptomycin are now recommended as the drugs of choice for the treatment of MRSA infections. Vancomycin is less effective than SPRPs for the treatment of infections due to methicillin-susceptible strains. Alternatives to SPRPs should be used only after careful consideration in patients with a history of serious β-lactam allergies.
Three types of staphylococcal resistance to vancomycin have emerged. (1) Minimal inhibitory concentration (MIC) “creep” refers to the incremental increase in vancomycin MICs that has been detected in various geographic areas. Studies suggest that infections due to S. aureus strains with vancomycin MICs of >1 μg/mL may not respond as well to vancomycin therapy as those due to strains with MICs of <1 μg/mL. Some authorities (e.g., The Medical Letter) have recommended choosing an alternative agent in this setting. (2) In 1997, an S. aureus strain with reduced susceptibility to vancomycin (vancomycin-intermediate S. aureus [VISA]) was reported from Japan. Subsequently, additional VISA clinical isolates were reported. These strains were all resistant to methicillin and many other antimicrobial agents. The VISA strains appear to evolve (under vancomycin selective pressure) from strains that are susceptible to vancomycin but are heterogeneous, with a small proportion of the bacterial population expressing the resistance phenotype. The mechanism of VISA resistance is in part due to an abnormally thick cell wall. Vancomycin is trapped by the abnormal peptidoglycan cross-linking and is unable to gain access to its target site. (3) In 2002, the first clinical isolate of fully vancomycin-resistant S. aureus was reported. Resistance in this and several additional clinical isolates was due to the presence of vanA, the gene responsible for expression of vancomycin resistance in enterococci. This observation suggested that resistance was acquired as a result of horizontal conjugal transfer from a vancomycin-resistant strain of Enterococcus faecalis. Several patients had both MRSA and vancomycin-resistant enterococci cultured from infection sites. The vanA gene is responsible for the synthesis of the dipeptide D-Ala-D-Lac in place of D-Ala-D-Ala. Vancomycin cannot bind to the altered peptide.
Daptomycin, a parenteral bactericidal agent with antistaphylococcal activity, is approved for the treatment of bacteremia (including right-sided endocarditis) and complicated skin infections. It is not effective in respiratory infections. This drug has a novel mechanism of action: it disrupts the cytoplasmic membrane. Staphylococcal resistance to daptomycin, sometimes developing during therapy, has been reported.
Linezolid—the first oxazolidinone—is bacteriostatic against staphylococci and offers the advantage of comparable bioavailability after oral or parenteral administration. Cross-resistance with other inhibitors of protein synthesis has not been detected. However, resistance to linezolid has been reported. Serious adverse reactions to linezolid include thrombocytopenia, occasional cases of neutropenia, and rare instances of peripheral and optic neuropathy.
Tedizolid, a second oxazolidinone released in 2014, is available as both oral and parenteral preparations. It has enhanced in vitro activity against antibiotic-resistant gram-positive bacteria, including staphylococci. Tedizolid is administered once a day.
Ceftaroline is a fifth-generation cephalosporin with bactericidal activity against MRSA (including strains with reduced susceptibility to vancomycin and daptomycin). It is approved for use in nosocomial pneumonias and for skin and soft tissue infections.
The parenteral streptogramin antibiotic quinupristin/dalfopristin displays bactericidal activity against all staphylococci, including VISA strains. This drug has been used successfully to treat serious MRSA infections. In cases of resistance to erythromycin or clindamycin, quinupristin/dalfopristin is bacteriostatic against staphylococci. There are limited data on the efficacy of either quinupristin/dalfopristin or linezolid for the treatment of infective endocarditis.
Telavancin is a parenteral lipoglycopeptide derivative of vancomycin that is approved for the treatment of complicated skin and soft tissue infections and for nosocomial pneumonia. The drug has two targets: the cell wall and the cell membrane. It remains active against VISA strains. Because of its nephrotoxicity, it should be avoided in patients with renal disease.
Dalbavancin is a long-acting, parenterally administered lipoglycopeptide that has been used to treat skin and soft tissue infections. Because of its long half-life, it can be administered on a weekly basis. There are limited data on its use in the treatment of invasive staphylococcal infections.
Although the quinolones are active against staphylococci in vitro, the frequency of staphylococcal resistance to these agents has increased progressively, especially among methicillin-resistant isolates. Of particular concern in MRSA is the possible emergence of quinolone resistance during therapy. Therefore, quinolones are not recommended for the treatment of MRSA infections. Resistance to the quinolones is most commonly chromosomal and results from mutations of the topoisomerase IV or DNA gyrase genes, although multidrug efflux pumps may also contribute. Although the newer quinolones exhibit increased in vitro activity against staphylococci, it is uncertain whether this increase translates into enhanced in vivo activity.
Tigecycline, a broad-spectrum minocycline analogue, has bacteriostatic activity against MRSA and is approved for use in skin and soft tissue infections as well as intraabdominal infections caused by S. aureus. Other antibiotics, such as minocycline and trimethoprim-sulfamethoxazole, have been used successfully to treat MRSA infections in cases of vancomycin toxicity or intolerance.
Combinations of antistaphylococcal agents have been used to enhance bactericidal activity in the treatment of serious infections such as endocarditis or osteomyelitis. In selected instances (e.g., right-sided endocarditis), drug combinations are also used to shorten the duration of therapy. Among the antimicrobial agents used in combinations are rifampin, aminoglycosides (e.g., gentamicin), and fusidic acid (not readily available in the United States). To date, clinical studies have not documented a therapeutic benefit; recent reports have raised concern about the potential nephrotoxicity of gentamicin and about adverse drug reactions from the addition of rifampin. As a result, the use of gentamicin in combination with β-lactams or other antimicrobial agents is no longer routinely recommended for the treatment of endocarditis. Rifampin continues to be used for the treatment of prosthetic device–related infections and for osteomyelitis.
The combination of daptomycin with a β-lactam antibiotic has been successfully used to treat patients with persistent MRSA bacteremia, even those infected with isolates that exhibit reduced susceptibility to daptomycin. The combination appears to enhance the bactericidal activity of daptomycin, perhaps by reducing the bacterial cell surface charge and thus allowing more daptomycin binding.
ANTIMICROBIAL THERAPY FOR SELECTED SETTINGS
When necessary, the use of oral antistaphylococcal agents for uncomplicated skin and soft tissue infections is usually successful. For other infections, parenteral therapy is indicated.
S. aureus endocarditis is usually an acute, life-threatening infection. Thus, prompt collection of blood for cultures must be followed immediately by empirical antimicrobial therapy. For life-threatening S. aureus native-valve endocarditis, therapy with a β-lactam is recommended. If a MRSA strain is isolated, vancomycin (15–20 mg/kg every 8–12 h, given in equal doses up to a total of 2 g) or daptomycin (6 mg/kg every 24 h) is recommended. The vancomycin dose should be adjusted on the basis of trough vancomycin levels. Patients are generally treated for 4–6 weeks, with duration depending on whether there are complications. For prosthetic-valve endocarditis, surgery in addition to antibiotic therapy is often necessary. The combination of a β-lactam agent—or, if the isolate is β-lactam-resistant, vancomycin (30 mg/kg every 24 h, given in doses up to a total of 2 g) or daptomycin (6 mg/kg every 24 h)—with an aminoglycoside (gentamicin, 1 mg/kg IV every 8 h) and rifampin (300 mg orally or IV every 8 h) for ≥6 weeks is recommended.
For hematogenous osteomyelitis or septic arthritis in children, a 4-week course of therapy is usually adequate. In adults, treatment is often more prolonged. For chronic forms of osteomyelitis, surgical debridement is necessary in combination with antimicrobial therapy. For joint infections, a critical component of therapy is the repeated aspiration or arthroscopy of the affected joint to prevent damage from leukocytes. The combination of rifampin with ciprofloxacin has been used successfully to treat prosthetic joint infections, especially when the device cannot be removed. The efficacy of this combination may reflect enhanced activity against staphylococci in biofilms as well as the attainment of effective intracellular concentrations.
The choice of empirical therapy for staphylococcal infections depends in part on susceptibility data for the local geographic area. Increasingly, vancomycin and daptomycin are the drugs of choice for both community- and hospital-acquired infections. The increase in CA-MRSA skin and soft tissue infections has drawn attention to the need for initiation of appropriate empirical therapy. Oral agents that have been effective against these isolates include clindamycin, trimethoprim-sulfamethoxazole, doxycycline, linezolid, and tedizolid.
THERAPY FOR TOXIC SHOCK SYNDROME
Supportive therapy with reversal of hypotension is the mainstay of therapy for TSS. Both fluids and pressors may be necessary. Tampons or other packing material should be promptly removed. The role of antibiotics is less clear. Some investigators recommend a combination of clindamycin and a semisynthetic penicillin or vancomycin (if the isolate is resistant to methicillin). Clindamycin is advocated because, as a protein synthesis inhibitor, it reduces toxin synthesis in vitro. Linezolid also appears to be effective. A semisynthetic penicillin or glycopeptide is suggested to eliminate any potential focus of infection as well as to eradicate persistent carriage that might increase the likelihood of recurrent illness. Anecdotal reports document the successful use of IV immunoglobulin to treat TSS. The role of glucocorticoids in the treatment of this disease is uncertain.
THERAPY FOR OTHER TOXIN-MEDIATED DISEASES
Therapy for staphylococcal food poisoning is entirely supportive. For SSSS, antistaphylococcal therapy targets the primary site of infection.
173 |
Streptococcal Infections |
Many varieties of streptococci are found as part of the normal flora colonizing the human respiratory, gastrointestinal, and genitourinary tracts. Several species are important causes of human disease. Group A Streptococcus (GAS, Streptococcus pyogenes) is responsible for streptococcal pharyngitis, one of the most common bacterial infections of school-age children, and for the postinfectious syndromes of acute rheumatic fever (ARF) and poststreptococcal glomerulonephritis (PSGN). Group B Streptococcus (GBS, Streptococcus agalactiae) is the leading cause of bacterial sepsis and meningitis in newborns and a major cause of endometritis and fever in parturient women. Viridans streptococci are the most common cause of bacterial endocarditis. Enterococci, which are morphologically similar to streptococci, are now considered a separate genus on the basis of DNA homology studies. Thus, the species previously designated as Streptococcus faecalis and Streptococcus faecium have been renamed Enterococcus faecalis and Enterococcus faecium, respectively. The enterococci are discussed in Chap. 174.
Streptococci are gram-positive, spherical to ovoid bacteria that characteristically form chains when grown in liquid media. Most streptococci that cause human infections are facultative anaerobes, although some are strict anaerobes. Streptococci are relatively fastidious organisms, requiring enriched media for growth in the laboratory. Clinicians and clinical microbiologists identify streptococci by several classification systems, including hemolytic pattern, Lancefield group, species name, and common or trivial name. Many streptococci associated with human infection produce a zone of complete (β) hemolysis around the bacterial colony when cultured on blood agar. The β-hemolytic streptococci can be classified by the Lancefield system, a serologic grouping based on the reaction of specific antisera with bacterial cell-wall carbohydrate antigens. With rare exceptions, organisms belonging to Lancefield groups A, B, C, and G are all β-hemolytic, and each is associated with characteristic patterns of human infection. Other streptococci produce a zone of partial (α) hemolysis, often imparting a greenish appearance to the agar. These α-hemolytic streptococci are further identified by biochemical testing and include Streptococcus pneumoniae (Chap. 171), an important cause of pneumonia, meningitis, and other infections, and the several species referred to collectively as the viridans streptococci, which are part of the normal oral flora and are important agents of subacute bacterial endocarditis. Finally, some streptococci are nonhemolytic, a pattern sometimes called γ hemolysis. Among the organisms classified serologically as group D streptococci, the enterococci are classified as a distinct genus (Chap. 174). The classification of the major streptococcal groups causing human infections is outlined in Table 173-1.
|
CLASSIFICATION OF STREPTOCOCCI |

GROUP A STREPTOCOCCI
Lancefield’s group A consists of a single species, S. pyogenes. As its species name implies, this organism is associated with a variety of suppurative infections. In addition, GAS can trigger the postinfectious syndromes of ARF (which is uniquely associated with S. pyogenes infection; Chap. 381) and PSGN (Chap. 338).
![]() Worldwide, GAS infections and their postinfectious sequelae (primarily ARF and rheumatic heart disease) account for an estimated 500,000 deaths per year. Although data are incomplete, the incidence of all forms of GAS infection and that of rheumatic heart disease are thought to be tenfold higher in resource-limited countries than in developed countries (Fig. 173-1).
Worldwide, GAS infections and their postinfectious sequelae (primarily ARF and rheumatic heart disease) account for an estimated 500,000 deaths per year. Although data are incomplete, the incidence of all forms of GAS infection and that of rheumatic heart disease are thought to be tenfold higher in resource-limited countries than in developed countries (Fig. 173-1).

FIGURE 173-1 Prevalence of rheumatic heart disease in children 5–14 years old. The circles within Australia and New Zealand represent indigenous populations (and also Pacific Islanders in New Zealand). (From JR Carapetis et al: Lancet Infect Dis 5:685, 2005, with permission.)
PATHOGENESIS
GAS elaborates a number of cell-surface components and extracellular products important in both the pathogenesis of infection and the human immune response. The cell wall contains a carbohydrate antigen that may be released by acid treatment. The reaction of such acid extracts with group A–specific antiserum is the basis for definitive identification of a streptococcal strain as S. pyogenes. The major surface protein of GAS is M protein, which occurs in more than 100 antigenically distinct types and is the basis for the serotyping of strains with specific antisera. The M protein molecules are fibrillar structures anchored in the cell wall of the organism that extend as hairlike projections away from the cell surface. The amino acid sequence of the distal or amino-terminal portion of the M protein molecule is quite variable, accounting for the antigenic variation of the different M types, while more proximal regions of the protein are relatively conserved. A newer technique for assignment of M type to GAS isolates uses the polymerase chain reaction to amplify the variable region of the emm gene, which encodes M protein. DNA sequence analysis of the amplified gene segment can be compared with an extensive database (developed at the Centers for Disease Control and Prevention [CDC]) for assignment of emm type. This method eliminates the need for typing sera, which are available in only a few reference laboratories. The presence of M protein on a GAS isolate correlates with its capacity to resist phagocytic killing in fresh human blood. This phenomenon appears to be due, at least in part, to the binding of plasma fibrinogen to M protein molecules on the streptococcal surface, which interferes with complement activation and deposition of opsonic complement fragments on the bacterial cell. This resistance to phagocytosis may be overcome by M protein–specific antibodies; thus individuals with antibodies to a given M type acquired as a result of prior infection are protected against subsequent infection with organisms of the same M type but not against that with different M types.
GAS also elaborates, to varying degrees, a polysaccharide capsule composed of hyaluronic acid. The production of large amounts of capsule by certain strains imparts a characteristic mucoid appearance to the colonies. The capsular polysaccharide plays an important role in protecting GAS from ingestion and killing by phagocytes. In contrast to M protein, the hyaluronic acid capsule is a weak immunogen, and antibodies to hyaluronate have not been shown to be important in protective immunity. The presumed explanation is the apparent structural identity between streptococcal hyaluronic acid and the hyaluronic acid of mammalian connective tissues. The capsular polysaccharide may also play a role in GAS colonization of the pharynx by binding to CD44, a hyaluronic acid–binding protein expressed on human pharyngeal epithelial cells.
GAS produces a large number of extracellular products that may be important in local and systemic toxicity and in the spread of infection through tissues. These products include streptolysins S and O, toxins that damage cell membranes and account for the hemolysis produced by the organisms; streptokinase; DNAses; SpyCEP, a serine protease that cleaves and inactivates the chemoattractant cytokine interleukin 8, thereby inhibiting neutrophil recruitment to the site of infection; and several pyrogenic exotoxins. Previously known as erythrogenic toxins, the pyrogenic exotoxins cause the rash of scarlet fever. Since the mid-1980s, pyrogenic exotoxin–producing strains of GAS have been linked to unusually severe invasive infections, including necrotizing fasciitis and the streptococcal toxic shock syndrome (TSS). Several extracellular products stimulate specific antibody responses useful for serodiagnosis of recent streptococcal infection. Tests for these antibodies are used primarily for detection of preceding streptococcal infection in cases of suspected ARF or PSGN.
CLINICAL MANIFESTATIONS
Pharyngitis Although seen in patients of all ages, GAS pharyngitis is one of the most common bacterial infections of childhood, accounting for 20–40% of all cases of exudative pharyngitis in children; it is rare among those under the age of 3. Younger children may manifest streptococcal infection with a syndrome of fever, malaise, and lymphadenopathy without exudative pharyngitis. Infection is acquired through contact with another individual carrying the organism. Respiratory droplets are the usual mechanism of spread, although other routes, including food-borne outbreaks, have been well described. The incubation period is 1–4 days. Symptoms include sore throat, fever and chills, malaise, and sometimes abdominal complaints and vomiting, particularly in children. Both symptoms and signs are quite variable, ranging from mild throat discomfort with minimal physical findings to high fever and severe sore throat associated with intense erythema and swelling of the pharyngeal mucosa and the presence of purulent exudate over the posterior pharyngeal wall and tonsillar pillars. Enlarged, tender anterior cervical lymph nodes commonly accompany exudative pharyngitis.
The differential diagnosis of streptococcal pharyngitis includes the many other bacterial and viral etiologies (Table 173-2). Streptococcal infection is an unlikely cause when symptoms and signs suggestive of viral infection are prominent (conjunctivitis, coryza, cough, hoarseness, or discrete ulcerative lesions of the buccal or pharyngeal mucosa). Because of the range of clinical presentations of streptococcal pharyngitis and the large number of other agents that can produce the same clinical picture, diagnosis of streptococcal pharyngitis on clinical grounds alone is not reliable. The throat culture remains the diagnostic gold standard. Culture of a throat specimen that is properly collected (i.e., by vigorous rubbing of a sterile swab over both tonsillar pillars) and properly processed is the most sensitive and specific means of definitive diagnosis. A rapid diagnostic kit for latex agglutination or enzyme immunoassay of swab specimens is a useful adjunct to throat culture. While precise figures on sensitivity and specificity vary, rapid diagnostic kits generally are >95% specific. Thus a positive result can be relied upon for definitive diagnosis and eliminates the need for throat culture. However, because rapid diagnostic tests are less sensitive than throat culture (relative sensitivity in comparative studies, 55–90%), a negative result should be confirmed by throat culture.
|
INFECTIOUS ETIOLOGIES OF ACUTE PHARYNGITIS |

|
TREATMENT |
GAS PHARYNGITIS |
In the usual course of uncomplicated streptococcal pharyngitis, symptoms resolve after 3–5 days. The course is shortened little by treatment, which is given primarily to prevent suppurative complications and ARF. Prevention of ARF depends on eradication of the organism from the pharynx, not simply on resolution of symptoms, and requires 10 days of penicillin treatment (Table 173-3). A first-generation cephalosporin, such as cephalexin or cefadroxil, may be substituted for penicillin in cases of penicillin allergy if the nature of the allergy is not an immediate hypersensitivity reaction (anaphylaxis or urticaria) or another potentially life-threatening manifestation (e.g., severe rash and fever).
|
TREATMENT OF GROUP A STREPTOCOCCAL INFECTIONS |

![]() Alternative agents are erythromycin and azithromycin. Azithromycin is more expensive but offers the advantages of better gastrointestinal tolerability, once-daily dosing, and a 5-day treatment course. Resistance to erythromycin and other macrolides is common among isolates from several countries, including Spain, Italy, Finland, Japan, and Korea. Macrolide resistance may be becoming more prevalent elsewhere with the increasing use of this class of antibiotics. In areas with resistance rates exceeding 5–10%, macrolides should be avoided unless results of susceptibility testing are known.
Alternative agents are erythromycin and azithromycin. Azithromycin is more expensive but offers the advantages of better gastrointestinal tolerability, once-daily dosing, and a 5-day treatment course. Resistance to erythromycin and other macrolides is common among isolates from several countries, including Spain, Italy, Finland, Japan, and Korea. Macrolide resistance may be becoming more prevalent elsewhere with the increasing use of this class of antibiotics. In areas with resistance rates exceeding 5–10%, macrolides should be avoided unless results of susceptibility testing are known.
Follow-up culture after treatment is no longer routinely recommended but may be warranted in selected cases, such as those involving patients or families with frequent streptococcal infections or those occurring in situations in which the risk of ARF is thought to be high (e.g., when cases of ARF have recently been reported in the community).
COMPLICATIONS Suppurative complications of streptococcal pharyngitis have become uncommon with the widespread use of antibiotics for most symptomatic cases. These complications result from the spread of infection from the pharyngeal mucosa to deeper tissues by direct extension or by the hematogenous or lymphatic route and may include cervical lymphadenitis, peritonsillar or retropharyngeal abscess, sinusitis, otitis media, meningitis, bacteremia, endocarditis, and pneumonia. Local complications, such as peritonsillar or parapharyngeal abscess formation, should be considered in a patient with unusually severe or prolonged symptoms or localized pain associated with high fever and a toxic appearance. Nonsuppurative complications include ARF (Chap. 381) and PSGN (Chap. 338), both of which are thought to result from immune responses to streptococcal infection. Penicillin treatment of streptococcal pharyngitis has been shown to reduce the likelihood of ARF but not that of PSGN.
BACTERIOLOGIC TREATMENT FAILURE AND THE ASYMPTOMATIC CARRIER STATE Surveillance cultures have shown that up to 20% of individuals in certain populations may have asymptomatic pharyngeal colonization with GAS. There are no definitive guidelines for management of these asymptomatic carriers or of asymptomatic patients who still have a positive throat culture after a full course of treatment for symptomatic pharyngitis. A reasonable course of action is to give a single 10-day course of penicillin for symptomatic pharyngitis and, if positive cultures persist, not to re-treat unless symptoms recur. Studies of the natural history of streptococcal carriage and infection have shown that the risk both of developing ARF and of transmitting infection to others is substantially lower among asymptomatic carriers than among individuals with symptomatic pharyngitis. Therefore, overly aggressive attempts to eradicate carriage probably are not justified under most circumstances. An exception is the situation in which an asymptomatic carrier is a potential source of infection to others. Outbreaks of food-borne infection and nosocomial puerperal infection have been traced to asymptomatic carriers who may harbor the organisms in the throat, vagina, or anus or on the skin.
|
TREATMENT |
ASYMPTOMATIC PHARYNGEAL COLONIZATION WITH GAS |
When a carrier is transmitting infection to others, attempts to eradicate carriage are warranted. Data are limited on the best regimen to clear GAS after penicillin alone has failed. Regimens reported to have efficacy superior to that of penicillin alone for eradication of carriage include (1) oral clindamycin (7 mg/kg; 300 mg maximum) three times daily for 10 days or (2) penicillin (as recommended for treatment of pharyngitis in Table 173-3) plus oral rifampin (10 mg/kg; 300 mg maximum) twice daily for the first 4 days of treatment. A 10-day course of oral vancomycin (250 mg four times daily) and rifampin (600 mg twice daily) has eradicated rectal colonization.
Scarlet Fever Scarlet fever consists of streptococcal infection, usually pharyngitis, accompanied by a characteristic rash (Fig. 173-2). The rash arises from the effects of one of several toxins, currently designated streptococcal pyrogenic exotoxins and previously known as erythrogenic or scarlet fever toxins. In the past, scarlet fever was thought to reflect infection of an individual lacking toxin-specific immunity with a toxin-producing strain of GAS. Susceptibility to scarlet fever was correlated with results of the Dick test, in which a small amount of erythrogenic toxin injected intradermally produced local erythema in susceptible individuals but elicited no reaction in those with specific immunity. Subsequent studies have suggested that development of the scarlet fever rash may reflect a hypersensitivity reaction requiring prior exposure to the toxin. For reasons that are not clear, scarlet fever has become less common in recent years, although strains of GAS that produce pyrogenic exotoxins continue to be prevalent in the population. The symptoms of scarlet fever are the same as those of pharyngitis alone. The rash typically begins on the first or second day of illness over the upper trunk, spreading to involve the extremities but sparing the palms and soles. The rash is made up of minute papules, giving a characteristic “sandpaper” feel to the skin. Associated findings include circumoral pallor, “strawberry tongue” (enlarged papillae on a coated tongue, which later may become denuded), and accentuation of the rash in skinfolds (Pastia’s lines). Subsidence of the rash in 6–9 days is followed after several days by desquamation of the palms and soles. The differential diagnosis of scarlet fever includes other causes of fever and generalized rash, such as measles and other viral exanthems, Kawasaki disease, TSS, and systemic allergic reactions (e.g., drug eruptions).
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
(See also Chap. 148) In addition to receiving antibiotic prophylaxis, transplant recipients should be vaccinated against likely pathogens (Table 169-6). In the case of HSC transplant recipients, optimal responses cannot be achieved until after immune reconstitution, despite previous immunization of both donor and recipient. Recipients of an allogeneic HSC transplant must be reimmunized if they are to be protected against pathogens. The situation is less clear-cut in the case of autologous transplantation. T and B cells in the peripheral blood may reconstitute the immune response if they are transferred in adequate numbers. However, cancer patients (particularly those with Hodgkin’s disease, in whom vaccination has been extensively studied) who are undergoing chemotherapy do not respond normally to immunization, and titers of antibodies to infectious agents fall more rapidly than in healthy individuals. Therefore, even immunosuppressed patients who have not undergone HSC transplantation may need booster vaccine injections. If memory cells are specifically eliminated as part of a stem cell “cleanup” procedure, it will be necessary to reimmunize the recipient with a new primary series. Optimal times for immunizations of different transplant populations are being evaluated. Yearly immunization of household and other contacts (including health care personnel) against influenza benefits the patient by preventing local spread.
|
VACCINATION OF HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) AND SOLID ORGAN TRANSPLANT (SOT) RECIPIENTS |
In the absence of compelling data as to optimal timing, it is reasonable to administer the pneumococcal and H. influenzae type b conjugate vaccines to both autologous and allogeneic HSC transplant recipients beginning 12 months after transplantation. A series that includes both the 13-valent pneumococcal conjugate vaccine (Prevnar) and the 23-valent pneumococcal polysaccharide vaccine (Pneumovax) is now recommended (according to CDC guidelines). The pneumococcal and H. influenzae type b vaccines are particularly important for patients who have undergone splenectomy. The Neisseria meningitidis polysaccharide conjugate vaccine (Menactra or Menveo) also is recommended. In addition, diphtheria, tetanus, acellular pertussis, and inactivated polio vaccines can all be given at these same intervals (12 months and, as required, 24 months after transplantation). Some authorities recommend a new primary series for tetanus/diphtheria/pertussis and inactivated poliovirus vaccines beginning 12 months after transplantation. Vaccination to prevent hepatitis B and hepatitis A (both killed vaccines) also seems advisable. A formal recommendation regarding immunization with the tetravalent HPV virus-like particle vaccine (Gardasil) after HSC transplantation has not been issued. However, HPV vaccination, which can prevent genital warts as well as specific cancers, is recommended through age 26 for healthy young adults who previously have not been vaccinated or have not received the full series. Live-virus measles/mumps/rubella (MMR) vaccine can be given to autologous HSC transplant recipients 24 months after transplantation and to most allogeneic HSC transplant recipients at the same point if they are not receiving maintenance therapy with immunosuppressive drugs and do not have ongoing GVHD. The risk of spread from a household contact is low for MMR vaccine. In parts of the world where live poliovirus vaccine is used, patients as well as contacts should be advised to receive only the killed vaccine. In the rare setting where both donor and recipient are VZV naïve and the recipient is no longer receiving acyclovir or ganciclovir prophylaxis, the patient should be counseled to receive varicella-zoster immune globulin (VariZIG) up to 10 days after an exposure to a person with chickenpox or uncovered zoster; such patients should avoid close contact with persons recently vaccinated with Varivax. A formal recommendation regarding Varivax immunization of such patients is not currently available. Neither patients nor their household contacts should receive vaccinia vaccine unless they have been exposed to smallpox virus. Among patients who have active GVHD and/or are taking high maintenance doses of glucocorticoids, it may be prudent to avoid all live-virus vaccines.
In the case of SOT recipients, administration of all the usual vaccines and of the indicated booster doses should be completed before immunosuppression, if possible, to maximize responses. For patients taking immunosuppressive agents, the administration of pneumococcal vaccine should be repeated every 5 years. No data are available for the meningococcal vaccine, but it is probably reasonable to administer it along with the pneumococcal vaccine. H. influenzae conjugate vaccine is safe and should be efficacious in this population; therefore, its administration before transplantation is recommended. Booster doses of this vaccine are not recommended for adults. SOT recipients who continue to receive immunosuppressive drugs should not receive live-virus vaccines. A person in this group who is exposed to measles should be given measles immune globulin. Similarly, an immunocompromised patient who is seronegative for varicella and who comes into contact with a person who has chickenpox should be given varicella-zoster immune globulin as soon as possible (optimally within 96 h; up to 10 days after contact); if this is not possible, a 10- to 14-day course of acyclovir therapy should be started immediately. Upon the discontinuation of treatment, clinical disease may still occur in a small number of patients; thus vigilance is indicated. Rapid re-treatment with acyclovir should limit the symptoms of disease. Household contacts of transplant recipients can receive live attenuated VZV vaccine, but vaccinees should avoid direct contact with the patient if a rash develops. Virus-like particle vaccines have been licensed for the prevention of infection with several HPV serotypes most commonly implicated in cervical and anal carcinomas and in anogenital and laryngeal warts. These vaccines are not live; however, no information is yet available about their immunogenicity or efficacy in transplant recipients.
Immunocompromised patients who travel may benefit from some but not all vaccines (Chaps. 148 and 149). In general, these patients should receive any killed or inactivated vaccine preparation appropriate to the area they are visiting; this recommendation includes the vaccines for Japanese encephalitis, hepatitis A and B, poliomyelitis, meningococcal infection, and typhoid. The live typhoid vaccines are not recommended for use in most immunocompromised patients, but an inactivated or purified polysaccharide typhoid vaccine can be used. Live yellow fever vaccine should not be administered. On the other hand, primary immunization or boosting with the purified-protein hepatitis B vaccine is indicated. Inactivated hepatitis A vaccine should also be used in the appropriate setting (Chap. 148). A vaccine is now available that provides dual protection against hepatitis A and hepatitis B. If hepatitis A vaccine is not administered, travelers should consider receiving passive protection with immune globulin (the dose depending on the duration of travel in the high-risk area).
SECTION 4 |
APPROACH TO THERAPY FOR BACTERIAL DISEASES |
170 |
Treatment and Prophylaxis of Bacterial Infections |
Antimicrobial agents have had a major impact on human health. Together with vaccines, they have contributed to reduced mortality, extended lifespan, and enhanced quality of life. Among drugs used in human medicine, however, they are distinctive in that their use promotes the occurrence of drug resistance in the pathogens they are designed to treat as well as in other “bystander” organisms. Indeed, the history of antimicrobial development has been driven in large part by the medical need engendered by the emergence of resistance to each generation of agents. Thus, the careful and appropriate use of antimicrobial drugs is particularly important not only for optimizing efficacy and minimizing adverse effects but also for minimizing the risk of resistance and preserving the value of existing agents. Although this chapter focuses on antibacterial agents, the optimal use of all antimicrobials depends on an understanding of each drug’s mechanism of action, spectrum of activity, mechanisms of resistance, pharmacology, and adverse effect profile. This information is then applied in the context of the patient’s clinical presentation, underlying conditions, and epidemiology to define the site and likely nature of the infection or other condition and thus to choose the best therapy. Gathering of microbiologic information is important for refining therapeutic choices on the basis of documented pathogen and susceptibility data whenever possible; this information also makes it possible to choose more targeted therapy, thereby reducing the risk of selection of resistant bacteria. Durations of therapy are chosen according to the nature of the infection and the patient’s response to treatment and are informed by clinical studies when they are available, with the understanding that shorter courses are less likely than longer ones to promote the emergence of resistance. This chapter provides specific information that is necessary for making informed choices among antibacterial agents.
MECHANISMS OF ACTION AND RESISTANCE
The mechanisms of action of and resistance to antibacterial agents are discussed in detail in the text and are summarized for the most commonly used groups of agents in Table 170-1. A schematic of antibacterial targets is provided in Fig. 170-1.
|
MECHANISMS OF ACTION OF AND RESISTANCE TO ANTIBACTERIAL AGENTS |
FIGURE 170-1 Antibacterial targets. A, aminoacyl site; DHFR, dihydrofolate reductase; DHPS, dihydropteroate synthetase; P, peptidyl site; PBP, penicillin-binding protein; tRNA-aa, aminoacyl tRNA.
MECHANISMS OF ACTION
Multiple essential components of bacterial cell structures and metabolism have been the targets of antibacterial agents used in clinical medicine, and the interaction of an agent with its target results in either inhibition of bacterial growth and replication (bacteriostatic effect) or bacterial killing (bactericidal effect). In general, targets have been chosen because they either do not exist in mammalian cells and physiology or are sufficiently different from their bacterial counterparts to allow selective bacterial targeting. Treatment with bacteriostatic agents is effective when the patient’s host defenses are sufficient to contribute to eradication of the infecting pathogen. In patients with impaired host defenses (e.g., neutropenia) or infections at body sites with impaired or limited host defenses (e.g., meningitis and endocarditis), bactericidal agents are generally preferred.
Inhibition of Cell Wall Synthesis The bacterial cell wall, which is external to the cytoplasmic membrane and has no counterpart in mammalian cells, protects bacterial cells from lysis under low osmotic conditions. The cell wall is a cross-linked peptidoglycan composed of a polymer of alternating units of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM), four-amino-acid stem peptides linked to each NAM, and a peptide cross-bridge that links adjacent stem peptides to form a netlike structure. Several steps in peptidoglycan synthesis are targets of antibacterial agents. Inhibition of cell wall synthesis generally results in a bactericidal effect that is linked to cell lysis. This effect results not only from the blocking of new cell-wall formation but from the uninhibited action of cell wall–remodeling enzymes called autolysins, which cleave peptidoglycan as part of normal cell-wall growth.
In gram-positive bacteria the peptidoglycan is the most external cell structure, but in gram-negative bacteria an asymmetric lipid outer membrane is external to the peptidoglycan and contains diffusion channels called porins. The space between the cytoplasmic membrane peptidoglycan and the outer membrane is referred to as the periplasmic space. Most antibacterial drugs enter the gram-negative bacterial cell through a porin channel, since the outer membrane is a major diffusion barrier. Although the peptidoglycan layer is thicker in gram-positive (20–80 nm) than in gram-negative (1 nm) bacteria, peptidoglycan itself constitutes only a limited diffusion barrier for antibacterial agents.
β-LACTAMS The β-lactam drugs, including penicillins, cephalosporins, monobactams, and carbapenems, target transpeptidase enzymes (also called penicillin-binding proteins, or PBPs) involved in the stem-peptide cross-linking step.
GLYCOPEPTIDES The glycopeptides, including vancomycin, teicoplanin, telavancin, dalbavancin, and oritavancin, bind the two terminal D-alanine residues of the stem peptide, hindering the glycosyltransferase involved in polymerizing NAG–NAM units. Telavancin also binds to the lipid II intermediate that delivers cell-wall precursor subunits. Likewise, dalbavancin and oritavancin interact with the cell membrane, and oritavancin may also inhibit transpeptidases. Both β-lactams and glycopeptides interact with their targets external to the cytoplasmic membrane.
BACITRACIN (TOPICAL) AND FOSFOMYCIN These agents interrupt enzymatic steps in the production of peptidoglycan precursors in the cytoplasm.
Inhibition of Protein Synthesis Most inhibitors of bacterial protein synthesis target bacterial ribosomes, whose difference from eukaryotic ribosomes allows selective antibacterial action. Some inhibitors bind to the 30S ribosomal subunit and others to the 50S subunit. Most protein synthesis–inhibiting agents are bacteriostatic; aminoglycosides are an exception and are bactericidal.
AMINOGLYCOSIDES Aminoglycosides (amikacin, gentamicin, kanamycin, netilmicin, streptomycin, tobramycin) bind irreversibly to 16S ribosomal RNA (rRNA) of the 30S ribosomal subunit, blocking the translocation of peptidyl transfer RNA (tRNA) from the A (aminoacyl) to the P (peptidyl) site and, at low concentrations, causing misreading of messenger RNA (mRNA) codons and thus causing the introduction of incorrect amino acids into the peptide chain; at higher concentrations, translocation of the peptide chain is blocked. Cellular uptake of aminoglycosides is dependent on the electrochemical gradient across the bacterial membrane. Under anaerobic conditions, this gradient is reduced, with a consequent reduction in the uptake and activity of the aminoglycosides. Spectinomycin is a related aminocyclitol antibiotic that also binds to 16S rRNA of the 30S ribosomal subunit but at a different site. This drug inhibits translocation of the growing peptide chain but does not trigger codon misreading and produces only a bacteriostatic effect.
TETRACYCLINES AND GLYCYLCYCLINES Tetracyclines (doxycycline, minocycline, tetracycline) bind reversibly to the 16S rRNA of the 30S ribosomal subunit and block the binding of aminoacyl tRNA to the ribosomal A site, thereby inhibiting peptide elongation. Active transport of tetracyclines into bacterial but not mammalian cells contributes to the selectivity of these agents. Tigecycline, a derivative of minocycline and the only available glycylcycline, acts similarly to the tetracyclines but is distinctive for its ability to circumvent the most common mechanisms of resistance to the tetracyclines.
MACROLIDES AND KETOLIDES In contrast to the aminoglycosides and tetracyclines, the macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) bind to the 23S rRNA of the 50S ribosomal subunit. These agents block translocation of the growing peptide chain by binding to the tunnel from which the chain exits the ribosome.
LINCOSAMIDES Clindamycin is the only lincosamide in clinical use. It binds to the 23S rRNA of the 50S ribosomal subunit, interacting with both the ribosomal A and P sites and blocking peptide bond formation.
STREPTOGRAMINS The only streptogramin in clinical use is a combination of quinupristin, a group B streptogramin, and dalfopristin, a group A streptogramin. Both components bind to 23S rRNA of the 50S ribosome: dalfopristin binds to both the A and P sites of the peptidyl transferase center, and quinupristin binds to a site that overlaps the macrolide-binding site, blocking the emergence of nascent peptide from the ribosome. The combination is bactericidal, but macrolide-resistant bacteria exhibit cross-resistance to quinupristin, and the remaining activity of dalfopristin alone is bacteriostatic.
CHLORAMPHENICOL Chloramphenicol binds reversibly to the 23S rRNA of the 50S subunit in a manner that interferes with the proper positioning of the aminoacyl component of tRNA in the A site. This site of binding is near those of the macrolides and lincosamides.
OXAZOLIDINONES Linezolid and tedizolid are the only oxazolidinones in clinical use. They bind directly to the A site in the 23S rRNA of the 50S ribosomal subunit and block binding of aminoacyl tRNA, inhibiting the initiation of protein synthesis.
MUPIROCIN Mupirocin (pseudomonic acid) is used topically. It competes with isoleucine for binding to isoleucyl tRNA synthetase, depleting stores of isoleucyl tRNA and thereby inhibiting protein synthesis.
Inhibition of Bacterial Metabolism Available inhibitors (antimetabolites) target the pathway for synthesis of folate, which is a cofactor in a number of one-carbon transfer reactions involved in the synthesis of some nucleic acids, including pyrimidine, thymidine, and all purines (adenine and guanine), as well as some amino acids (methionine and serine) and acetyl coenzyme A (CoA). Two sequential steps in folate synthesis are targeted. The selective antibacterial effect stems from the inability of mammalian cells to synthesize folate; they depend instead on exogenous sources. Antibacterial activity, however, may be reduced in the presence of high exogenous concentrations of the end products of the folate pathway (e.g., thymidine and purines) that may occur in some infections, resulting from local breakdown of leukocytes and host tissues.
SULFONAMIDES Sulfonamides, including sulfadiazine, sulfisoxazole, and sulfamethoxazole, inhibit dihydropteroate synthetase, which adds p-aminobenzoic acid (PABA) to pteridine, producing dihydropteroate. Sulfonamides are structural analogues of PABA and act as competing enzyme substrates.
TRIMETHOPRIM Subsequent steps in folate synthesis are catalyzed by dihydrofolate synthase, which adds glutamate to dihydropteroate, and dihydrofolate reductase, which then generates the final product, tetrahydrofolate. Trimethoprim is a structural analogue of pteridine and inhibits dihydrofolate reductase. Trimethoprim is available alone but is most often used in combination products that also contain sulfamethoxazole and thus block two sequential steps in folate synthesis.
Inhibition of DNA and RNA Synthesis or Activity A variety of antibacterial agents act on these processes.
QUINOLONES The quinolones include nalidixic acid, the first agent in the class, and newer, more widely used fluorinated derivatives (fluoroquinolones), including norfloxacin, ciprofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. The quinolones are synthetic compounds that inhibit bacterial DNA synthesis by interacting with the DNA complexes of two essential enzymes, DNA gyrase and DNA topoisomerase IV, which alter DNA topology. Quinolones trap enzyme–DNA complexes in such a way that they block movement of the DNA replication apparatus and can generate lethal double-strand breaks in DNA, resulting in bactericidal activity. Although mammalian cells also have type II DNA topoisomerases like gyrase and topoisomerase IV, the structures of the mammalian enzymes are sufficiently different from those of the bacterial enzymes that quinolones have substantially selective antibacterial activity.
RIFAMYCINS Rifampin, rifabutin, and rifapentine are semisynthetic derivatives of rifamycin B and bind the β subunit of bacterial RNA polymerase, thereby blocking elongation of mRNA. Their action is highly selective for the bacterial enzyme over mammalian RNA polymerases.
NITROFURANTOIN The reduction of nitrofurantoin, a nitrofuran compound, by bacterial enzymes produces highly reactive derivatives that are thought to cause DNA strand breakage. Nitrofurantoin is used only for the treatment of lower urinary tract infections.
METRONIDAZOLE Metronidazole is a synthetic nitroimidazole with activity limited to anaerobic bacteria and certain anaerobic protozoa. Reduction of its nitro group by the electron-transport system in anaerobic bacteria produces reactive intermediates that damage DNA and result in bactericidal activity. Both nitrofurantoin and metronidazole have selective antibacterial activity because the reducing activity needed to generate active derivatives is generated only by bacterial and not mammalian enzymes.
Disruption of Membrane Integrity The integrity of the bacterial cytoplasmic membrane—and, in gram-negative bacteria, the outer membrane—is important for bacterial viability. Two bactericidal drugs have membrane targets.
POLYMYXINS The polymyxins, including polymyxin B and polymyxin E (colistin), are cationic cyclic polypeptides that disrupt the cytoplasmic membrane and the outer membrane (the latter by binding lipopolysaccharide).
DAPTOMYCIN Daptomycin is a lipopeptide that binds the cytoplasmic membrane of gram-positive bacteria in the presence of calcium, generating a channel that leads to leakage of cytoplasmic potassium ions and membrane depolarization.
MECHANISMS OF RESISTANCE
Bacteria use a wide variety of mechanisms to block or circumvent the activity of antibacterial agents. Although myriad, these mechanisms can generally be grouped into three categories: (1) altered or bypass targets that exhibit reduced binding of the drug, (2) altered access of the drug to its target by reductions in uptake or increases in active efflux, and (3) a modification of the drug that reduces its activity. These mechanisms result from either mutations in bacterial chromosomal genes occurring spontaneously during bacterial DNA replication or the acquisition of new genes by DNA transfer from other bacteria or uptake of exogenous DNA. New genes are most often acquired on self-replicating plasmids or other DNA elements transferred from other bacteria. However, some bacteria, such as Streptococcus pneumoniae and Neisseria gonorrhoeae, can also take up fragments of environmental DNA from related species and recombine that DNA directly into their own chromosomes, a process called transformation. Not uncommonly, resistant bacteria have combinations of resistance mechanisms either within one category or among categories, and many plasmids contain more than one resistance gene. Thus, plasmid acquisition itself can in many cases confer resistance to multiple antibacterial agents.
Many antibacterial drugs are derived from natural products of microbial species. Some genes encoding resistance to these drugs may have evolved and been mobilized onto plasmids from a protection mechanism in the producer organism or in other surviving bacteria in the exposed environment. Exposure to antibacterial agents either in nature or from human or animal use results in the selection of resistant strains within an otherwise susceptible bacterial population. Because the patterns and extent of resistance may differ among settings, initial choices of antibacterial drugs should be based, whenever possible, on local susceptibility data and should be modified as needed as soon as specific microbiology susceptibility data become available.
β-Lactams The most common mechanism of resistance to β-lactams is their degradation by β-lactamases, enzymes that break down the core β-lactam ring and destroy drug activity. Different β-lactamases degrade different β-lactams. Some β-lactamases are encoded on the bacterial chromosome, and their activity contributes to the susceptibility profile of a particular species. Because other β-lactamases are encoded by acquired plasmids, their resistance profiles may be present in some strains of a species but not others. In gram-positive bacteria β-lactamases are secreted into the extracellular environment, whereas in gram-negative bacteria these enzymes are secreted into the periplasmic space between the cytoplasmic and outer membranes. Thus, in gram-negative bacteria, access of β-lactams both to their target PBPs and to β-lactamases requires diffusion across the outer membrane, generally through the porin channels.
Most strains of Staphylococcus aureus produce a plasmid-encoded β-lactamase that degrades penicillin but not semisynthetic penicillins, such as oxacillin and nafcillin. The most common plasmid-encoded β-lactamases of gram-negative bacteria are able to inactivate all penicillins and most earlier-generation cephalosporins. Extended-spectrum β-lactamase (ESBL) variants of these early enzymes that can degrade later-generation cephalosporins (ceftriaxone, cefotaxime, ceftazidime) as well as the monobactam aztreonam have now emerged and are widely disseminated. Some ESBLs also degrade the fourth-generation cephalosporin cefepime. Carbapenems (imipenem, meropenem, ertapenem, doripenem) are not degraded by ESBLs, but additional β-lactamases, called carbapenemases, that degrade carbapenems and most if not all other β-lactams have begun to emerge.
The chromosomal β-lactamase of Klebsiella pneumoniae preferentially degrades penicillins but not cephalosporins. In contrast, the chromosomal β-lactamase of Enterobacter and related genera, AmpC, can degrade almost all cephalosporins but is normally expressed in small amounts. Mutations that cause increased amounts of AmpC to be produced confer full resistance to penicillins and cephalosporins; the exceptions are cefoxitin and cefepime, which are relatively stable to AmpC. Resistance to cefepime can develop, however, through the combined effects of increased AmpC production and decreased porin diffusion channels. Genes encoding AmpC have also been found on plasmids but are less common than plasmid-encoded ESBLs.
Inhibitors of β-lactamases such as clavulanate, sulbactam, and tazobactam have been developed and paired with amoxicillin and ticarcillin, ampicillin, and piperacillin, respectively. These inhibitors have little or no antibacterial activity of their own but inhibit plasmid-mediated β-lactamases, including ESBLs but not AmpC enzymes.
Resistance to β-lactams also occurs through alterations in their target PBPs. In S. pneumoniae, N. gonorrhoeae, and Neisseria meningitidis, resistance to penicillin occurs by recombination of transformed DNA from related species. In staphylococci, resistance to methicillin and other β-lactams occurs by the acquisition of the mec gene, which encodes PBP2a with reduced drug affinity. Ceftaroline is the only β-lactam that has affinity for PBP2a and is thus active against methicillin-resistant staphylococcal strains.
Glycopeptides Resistance to vancomycin in enterococci is due to the acquisition of a set of van genes that result in (1) the production of D-alanine-D-lactate—instead of the normal D-alanine-D-alanine—at the end of the peptidoglycan stem peptide and (2) the reduction of existing D-alanine-D-alanine terminated peptides. Vancomycin binds D-alanine-D-lactate with a thousandfold lower affinity than D-alanine-D-alanine. In a small number of cases, the van gene cassettes have been transferred from enterococci to S. aureus, with the consequent generation of full vancomycin resistance. Particularly in patients receiving prolonged courses of vancomycin, intermediate resistance to this drug has developed in S. aureus by a different mechanism: multiple chromosomal mutations that result in a thickened and poorly cross-linked cell wall in which multiple distant D-alanine-D-alanine stem peptide termini exist and bind vancomycin, impeding its access to the binding sites proximal to the cell membrane where new cell-wall synthesis occurs and where binding would block transpeptidase and transglycosylase enzymes. Susceptibility to telavancin, dalbavancin, and oritavancin is also reduced in strains that exhibit resistance or intermediate susceptibility to vancomycin, although in some cases strains may still be classified as susceptible on the basis of clinical interpretive criteria.
Aminoglycosides The most common mechanism of resistance is due to acquisition of plasmid genes encoding transferase enzymes that modify aminoglycosides by the addition of acetyl, adenyl, or phosphate groups; these added groups decrease the drugs’ binding affinity to their ribosomal target site. Transferases differ in which aminoglycosides they modify, and amikacin resistance occurs less often than resistance to gentamicin or tobramycin. More recently, plasmids encoding methyltransferases that modify the ribosomal site of aminoglycoside binding and confer resistance to all aminoglycosides have been found in enteric gram-negative bacteria. For streptomycin, a ribosomal protein mutation may cause resistance. In Pseudomonas aeruginosa, resistance may also occur through mutations causing increased expression of a chromosomally encoded efflux pump, MexXY.
Tetracyclines and Glycylcyclines For tetracyclines, resistance is most often plasmid mediated and attributable either to active efflux pumps, which are generally specific for tetracyclines, or to proteins that protect the ribosome from tetracycline action. Resistance to the glycylcycline tigecycline, which is not affected by the usual tetracycline resistance mechanisms, can occur through mutations that cause overexpression of certain broad-spectrum efflux pumps in Proteus species.
Macrolides, Ketolides, Lincosamides, and Streptogramins Resistance to macrolides, clindamycin, and quinupristin is most often due to plasmid-acquired methylases that modify the drug binding site on the ribosome. Resistance to quinupristin by this mechanism renders the quinupristin-dalfopristin combination bacteriostatic rather than bactericidal. Telithromycin, a ketolide, has an additional binding site on the ribosome and remains active in the presence of these methylases. Acquired genes encoding active efflux pumps can also contribute to resistance to macrolides in streptococci and resistance to macrolides, clindamycin, and dalfopristin in staphylococci. Plasmid-acquired drug-modifying enzymes in staphylococci can also cause resistance to quinupristin and dalfopristin. Macrolide resistance due to 23S rRNA mutations is uncommon in staphylococci and streptococci because of the multiple copies of the rRNA genes on the chromosomes of these species; such resistance may occur more frequently, however, in mycobacteria and Helicobacter pylori, which have only single chromosomal copies of these rRNA genes.
Chloramphenicol Resistance to chloramphenicol is most often due to a plasmid-encoded drug-modifying acetyltransferase.
Oxazolidinones Linezolid resistance has been seen in enterococci more often than in staphylococci and, in both organisms, is due to mutations in multiple copies of the 23S rRNA genes that reduce drug binding to the ribosome. A plasmid-acquired ribosomal methylase gene that confers resistance to both chloramphenicol and linezolid has also been found in some strains of staphylococci but is not yet widespread. Tedizolid may still be active against some but not all linezolid-resistant strains.
Mupirocin Resistance to mupirocin occurs by either mutation in the target leucyl-tRNA synthetase (low-level resistance) or the acquisition of a plasmid-encoded resistant tRNA synthetase (high-level resistance).
Sulfonamides and Trimethoprim Resistance to both of these antimetabolites is due to plasmid-acquired genes encoding resistant enzymes that bypass the inhibition of the native sensitive enzymes—a resistant dihydropteroate synthetase in the case of sulfonamides and a resistant dihydrofolate reductase in the case of trimethoprim.
Quinolones Resistance to quinolones is most often due either to chromosomal mutations altering the target enzymes DNA gyrase and DNA topoisomerase IV, with consequent reduction in drug binding, or to mutations that increase the expression of native broad-spectrum efflux pumps for which quinolones (among other compounds) are substrates. In addition, three types of genes can confer reduced susceptibility or low-level resistance by protecting target enzymes, modifying some quinolones, or pumping quinolones out of the cell (efflux). These genes are located on multidrug resistance plasmids that have spread worldwide. Their presence can promote the selection of higher levels of quinolone resistance linked to resistance to other antibacterial drugs that is encoded on the same plasmid.
Rifampin and Rifabutin Single mutations in the β subunit of RNA polymerase can cause high-level resistance to rifampin. Thus rifampin and other rifamycins are used for treatment of infections only in combination with other antibacterial drugs in order to prevent resistance.
Metronidazole Acquired resistance to metronidazole in Bacteroides species is rare. Such resistance has been reported in strains that lack endogenous nitroreductase activity or that have acquired nim genes responsible for further reduction of DNA-damaging nitroso intermediates to an inactive derivative. Active efflux and enhanced DNA repair mechanisms also have been associated with resistance.
Nitrofurantoin Resistance to nitrofurantoin in Escherichia coli can emerge through a series of mutations that progressively decrease the nitroreductase activity necessary for generating active nitrofuran metabolites.
Polymyxins Because of emerging multidrug resistance in gram-negative bacteria, colistin and polymyxin B are being used increasingly for infections due to resistant Enterobacteriaceae, P. aeruginosa, and Acinetobacter species. Rates of resistance vary. Resistance can emerge during therapy through mutations that cause reductions in the negative charge of the gram-negative bacterial cell surface, thereby reducing binding of the positively charged colistin.
Daptomycin The mechanisms of resistance to daptomycin are complex and involve mutations in several genes that can alter cell membrane charge and reduce daptomycin binding. Resistance to daptomycin is relatively infrequent but has emerged in some S. aureus strains with intermediate vancomycin susceptibility from patients treated with vancomycin but not with daptomycin. In some methicillin-resistant S. aureus (MRSA) strains, daptomycin resistance has been linked to acquired susceptibility to β-lactams; combinations of daptomycin and nafcillin have been successful for treatment of patients infected with resistant strains when daptomycin alone or in combination with other agents has failed. The mechanism of this effect is not yet clear.
PHARMACOKINETICS AND PHARMACODYNAMICS
The term pharmacokinetics describes the disposition of a drug in the body, whereas pharmacodynamics describes the determinants of drug action on the pathogen in relation to pharmacokinetic factors. An understanding of the principles governing these two areas is required for effective drug selection, dosing, and prevention of toxicities.
PHARMACOKINETICS
The process of drug disposition has four principal phases: absorption, distribution, metabolism, and excretion. These components determine the time course of drug concentrations in serum and subsequently the concentrations in other tissues and body fluids.
Absorption When a drug is given by a particular route, absorption is defined as the percentage of the dose that reaches the systemic circulation. For example, since IV administration provides direct access to the systemic circulation, 100% of a drug dose given IV is usually absorbed. The level of absorption becomes more relevant when non-IV routes are used—e.g., the oral, IM, SC, and topical routes. The percentage of a drug that is absorbed is termed its bioavailability. Examples of antibacterial agents with a high oral bioavailability include metronidazole, levofloxacin, and linezolid. IV administration and oral dosing for highly bioavailable agents usually give equivalent results. Many factors can affect a drug’s oral bioavailability, including the timing of food consumption relative to drug administration, drug-metabolizing enzymes, efflux transporters, concentration-dependent solubility, and acid degradation. Underlying conditions such as diarrhea or ileus can also affect the site of drug absorption and thereby alter bioavailability. Certain orally administered drugs have lower bioavailability because of the first-pass effect—the process by which drugs are absorbed in the small intestine through the portal circulation and then directly transported to the liver for metabolism.
Distribution Distribution describes the process by which a drug transfers reversibly between the general circulation and the tissues. After absorption into the general circulation and the central compartment (the extensively perfused organs), the drug will also distribute into the peripheral compartment (less well-perfused tissues). The volume of distribution (Vd) is a pharmacokinetic parameter that describes the amount of drug in the body at a given time relative to the measured serum concentration. Properties such as the drug’s lipophilicity, partition coefficient within different body tissues, and protein binding; blood flow; and pH can affect the volume of distribution. Drugs with a small volume of distribution are limited to certain areas within the body (typically extracellular fluid), whereas those with a higher volume of distribution penetrate extensively into tissues throughout the body. Antibacterial drugs can bind to serum proteins, and a given drug is usually described as either poorly or highly protein bound. Only the unbound (free) drug is active and available to exert antibacterial effects. For example, because tigecycline is highly protein bound and also has a large volume of distribution, concentrations of free drug in the serum are low.
Metabolism Metabolism is the chemical transformation of a drug by the body. This modification can occur within several areas; the liver is the organ most commonly involved. Drugs are metabolized by enzymes, but enzyme systems have a finite capacity to metabolize a substrate drug. If a drug is given in a dose at which the concentration does not exceed the rate of metabolism, then the metabolic process is generally linear. If the dose exceeds the amount that can be metabolized, drug accumulation and potential toxicity may occur. Drugs are metabolized through phase I or phase II reactions. In phase I reactions, the drug is made more polar through dealkylation, hydroxylation, oxidation, and deamination. Polarity facilitates drug removal from the body. Phase II reactions, which include glucuronidation, sulfation, and acetylation, result in compounds larger and more polar than the parent drug. Both phases usually inactivate the parent drug, but some drugs are rendered more active. The cytochrome P450 (CYP) enzyme system is responsible for phase I reactions and is generally found in the liver. CYP3A4 is a common subfamily within this system that is responsible for the majority of drug metabolism. Antibacterial drugs can be substrates, inhibitors, or inducers of a particular CYP enzyme. Inducers, such as rifampin, can increase the production of CYP enzymes and consequently increase the metabolism of other drugs. Inhibitors, such as quinupristin-dalfopristin, cause a decrease in enzyme activity (or competition for CYP substrate) and therefore an increase in the concentration of the interacting drug.
Excretion Excretion describes the body’s mechanisms of drug elimination. Drugs can be eliminated through more than one mechanism. Renal clearance is the most common route and includes elimination through glomerular filtration, tubular secretion, and/or passive diffusion. Some agents have nonrenal clearance and rely on the biliary tree or the intestine for excretion. Excretion affects the half-life of a drug—i.e., the time it takes for the blood concentration of a drug to decrease by one-half. This value can range from minutes to days. Approximately five to seven half-lives are required for a drug to reach steady state when multiple doses are given in a time frame shorter than the half-life itself. Drug half-life and overall clearance can be extended if the organ responsible for clearance is impaired. Patients with renal or hepatic impairment may require dose adjustments that take delayed clearance into account and prevent toxicities from drug accumulation. For example, imipenem is cleared predominantly through glomerular filtration, and in the presence of renal impairment the dosing interval is typically increased to account for the increased half-life.
PHARMACODYNAMICS
The term pharmacodynamics describes the relationship between the serum concentrations that determine the efficacy of the drug and the serum concentrations that produce the toxic effects of the drug. For an antibacterial agent, the pharmacodynamic focus is the type of drug exposure needed for optimal antibacterial effect in relation to the minimal inhibitory concentration (MIC)—the lowest drug concentration that inhibits the visible growth of a microorganism under standardized laboratory conditions. Antibacterial effect usually correlates with one of the following parameters: (1) ratio of peak serum concentration to the MIC (Cmax/MIC), (2) ratio of the area under the concentration–time curve to the MIC (AUC/MIC), or (3) duration of concentrations above the MIC (T>MIC) (Fig. 170-2).
FIGURE 170-2 Pharmacokinetic and pharmacodynamic model predicting efficacy of antibacterial drugs. AUC, area under the time–concentration curve; Cmax, peak serum concentration of drug; MIC, minimal inhibitory concentration; T>MIC, duration of drug concentrations above the MIC.
For concentration-dependent killing agents, as the designation implies, the higher the drug concentration, the higher the rate and extent of bacterial killing. Aminoglycosides fit into the Cmax/MIC model of pharmacodynamics activity, and a particular peak serum concentration is often targeted to achieve optimal killing. Fluoroquinolones exemplify antibacterial agents for which the AUC/MIC is a predictor of efficacy. For example, studies have found that an AUC/MIC ratio of >30 will maximize killing of S. pneumoniae by fluoroquinolones. In contrast, time-dependent killing agents reach a ceiling at which higher concentrations do not result in increased effect. Instead, these agents are active against bacteria only when the drug concentration is above the MIC. The T>MIC predicts clinical efficacy for all β-lactams. The longer the concentration of the β-lactam remains above the MIC for an infecting pathogen during the dosing interval, the greater the killing effect. For some drug classes, such as aminoglycosides, a postantibiotic effect—the delayed regrowth of surviving bacteria after exposure to an antibiotic—supports less frequent dosing.
APPROACH TO THERAPY
The approach to antibiotic therapy is driven by host factors, site of infection, and local resistance profiles of suspected or known pathogens. Further, national and local drug shortages and formulary restrictions can affect available therapies. Regular monitoring of the patient and collection of laboratory data should be undertaken to streamline antibacterial therapy as appropriate and to investigate the possibility of treatment failure if the patient fails to respond appropriately.
EMPIRICAL AND DIRECTED THERAPY
Therapy is considered empirical when the causative agent has yet to be determined and therapeutic decisions are based on the severity of illness and the clinician’s assessment of likely pathogens in light of the clinical syndrome, the patient’s medical conditions and prior therapy, and relevant epidemiologic factors. For patients with severe illness, empirical therapy often takes the form of an antibacterial combination that provides broad coverage of diverse agents and thus ensures adequate treatment of possible pathogens while additional data are being collected. Directed therapy is predicated on identification of the pathogen, determination of its susceptibility profile, and establishment of the extent of the infection. Directed therapy generally allows the use of more targeted and narrower-spectrum antibacterial agents than does empirical therapy.
Information on epidemiology, exposures, and local antibacterial susceptibility patterns can help guide empirical therapy. When empirical treatment is clinically appropriate, care should be taken to obtain clinical specimens for microbiologic analysis before the initiation of therapy and to de-escalate therapy as new information is obtained about the patient’s clinical condition and the causal pathogens. De-escalation to the point of directed therapy can limit unnecessary risks to the patient as well as the risk of emergence of antibacterial resistance.
SITE OF INFECTION
The site of infection is a consideration in antibacterial therapy, largely because of the differing abilities of drugs to penetrate and achieve adequate concentrations at particular body sites. For example, to be effective in the treatment of meningitis, an agent must (1) be able to cross the blood–brain barrier and reach adequate concentrations in the cerebrospinal fluid (CSF) and (2) be active against the relevant pathogen(s). Dexamethasone, administered with or 15–20 min before the first dose of an antibacterial drug, has been shown to improve outcomes in patients with acute bacterial meningitis, but its use may reduce penetration of some antibacterial agents, such as vancomycin, into the CSF. In this case, rifampin is added because its penetration is not reduced by dexamethasone. Infections at other sites where either pathogens are protected from normal host defenses or penetration of an antibacterial drug is suboptimal include osteomyelitis, prostatitis, intraocular infections, and abscesses. In such cases, consideration must be given to the mechanism of drug delivery (e.g., intravitreal injections) as well as to the role of interventions to drain, debride, or otherwise reduce the barriers to effective antibacterial therapy.
HOST FACTORS
Host factors, including immune function, pregnancy, allergies, age, renal and hepatic function, drug–drug interactions, comorbid conditions, and occupational or social exposures, should be considered.
Immune Dysfunction Patients with deficits in immune function that blunt the response to bacterial infection, including neutropenia, deficient humoral immunity, and asplenia (either surgical or functional), are all at increased risk of severe bacterial infection. Such patients should be treated aggressively and often broadly in the early stages of suspected infection pending results of microbiologic tests. For asplenic patients, treatment should include coverage of encapsulated organisms, particularly S. pneumoniae, that may cause rapidly life-threatening infection.
Pregnancy Pregnancy affects decisions regarding antibacterial therapy in two respects. First, pregnancy is associated with an increased risk of particular infections (e.g., those caused by Listeria). Second, the potential risks to the fetus that are posed by specific drugs must be considered. As for other drugs, the safety of the vast majority of antibacterial agents in pregnancy has not been established, and such agents are grouped in categories B and C by the U.S. Food and Drug Administration. Drugs in categories D and × are contraindicated in pregnancy or lactation due to established risks. The risks associated with antibacterial use in pregnancy and during lactation are summarized in Table 170-2.
|
RISKS ASSOCIATED WITH USE OF ANTIBACTERIAL DRUGS IN PREGNANCY AND LACTATION |
Allergies Allergies to antibiotics are among the most common allergies reported, and an allergy history should be obtained whenever possible before therapy is chosen. A detailed allergy history can shed light on the type of reaction experienced previously and on whether rechallenge with the same or a related medication is advisable (and, if so, under what circumstances). Allergies to the penicillins are most common. Although as many as 10% of patients may report an allergy to penicillin, studies suggest that up to 90% of these patients could tolerate a penicillin or cephalosporin. Adverse effects (Table 170-3) should be distinguished from true allergies to ensure appropriate selection of antibacterial therapy.
|
COMMON ADVERSE REACTIONS TO ANTIBACTERIAL AGENTS |
Drug–Drug Interactions Patients commonly receive other drugs that may interact with antibacterial agents. A summary of the most common drug–drug interactions, by antibacterial class, is provided in Table 170-4.
|
SIGNIFICANT ANTIBACTERIAL DRUG INTERACTIONS |
Exposures Exposures, both occupational and social, may provide clues to likely pathogens. When relevant, inquiries about exposure to ill contacts, animals, insects, and water should be included in the history, along with sites of residence and travel.
Other Host Factors Age, renal and hepatic function, and comorbid conditions are all considerations in the choice of and schedule for therapy. Dose adjustments should be made accordingly. In patients with decreased or unreliable oral absorption, IV therapy may be preferred to ensure adequate blood levels of drug and delivery of the antibacterial agent to the site of infection.
DURATION OF THERAPY
Whether empirical or directed, the duration of therapy should be planned in most clinical situations. Guidelines that synthesize available literature and expert opinion provide recommendations on therapy duration that are based on infecting organism, organ system, and patient factors. For example, the American Heart Association has published guidelines endorsed by the Infectious Diseases Society of America (IDSA) on diagnosis, antibacterial therapy, and management of complications of infective endocarditis. Similar guidelines from the IDSA exist for bacterial meningitis, catheter-associated urinary tract infections, intraabdominal infections, community- and hospital-acquired pneumonia, and other infections.
FAILURE OF THERAPY
If a patient does not respond to therapy, investigations often should include the collection of additional specimens for microbiologic testing and imaging as indicated. Failure to respond can be the result of an antibacterial regimen that does not address the underlying causative organism, the development of resistance during therapy, or the existence of a focus of infection at a site poorly penetrated by systemic therapy. Some infections may also require surgical interventions for cure (e.g., large abscesses, myonecrosis). Fever due to allergic drug reactions can sometimes complicate assessment of the patient’s response to antibacterial treatment.
EXPERT GUIDANCE
Selected websites with the most up-to-date information and guidance for the clinician include the following:
• Johns Hopkins ABX Guide (www.hopkins-abxguide.org)
• IDSA Practice Guidelines (www.idsociety.org/IDSA_Practice_Guidelines/)
• Center for Disease Dynamics, Economics and Policy Resistance Map (www.cddep.org/map)
• CDC Antibiotic/Antimicrobial Resistance (www.cdc.gov/drugresistance/)
CLINICAL USE OF ANTIBACTERIAL AGENTS
The clinical application of antibacterial therapy is guided by the spectrum of the agent and the suspected or known target pathogen. Infections for which specific antibacterial agents are among the drugs of choice are listed, along with associated pathogens and susceptibility data, in Table 170-5. Resistance rates of specific organisms are dynamic and should be taken into account in the approach to antibacterial therapy. While national resistance rates can serve as a reference, the most useful reference for the clinician is the most recent local laboratory antibiogram, which provides details on local resistance patterns, often on an annual or semiannual basis.
|
DRUG INDICATIONS FOR SPECIFIC INFECTIONS, ASSOCIATED PATHOGENS, AND SAMPLE SUSCEPTIBILITY RATES |
β-LACTAMS
The β-lactam class of antibiotics consists of penicillins, cephalosporins, carbapenems, and monobactams. The term β-lactam reflects the drugs’ four-membered lactam ring, which is their core structure. The differing side chains among the agents of this family determine the spectrum of activity. All β-lactams exert a bactericidal effect by inhibiting bacterial cell-wall synthesis. The β-lactams are classified as time-dependent killing agents; therefore, their clinical efficacy is best correlated with the proportion of the dosing interval during which the drug levels remain above the MIC for the pathogenic organism.
Penicillins and β-Lactamase Inhibitors Penicillin, the first β-lactam, was discovered in 1928 by Alexander Fleming. Natural penicillins, such as penicillin G, are active against non-β-lactamase-producing gram-positive and gram-negative bacteria, anaerobes, and some gram-negative cocci. Penicillin G is used for penicillin-susceptible streptococcal infections, pneumococcal and meningococcal meningitis, enterococcal endocarditis, and syphilis. The antistaphyloccocal penicillins, which have potent activity against methicillin-susceptible S. aureus (MSSA), include nafcillin, oxacillin, dicloxacillin, and flucloxacillin. Aminopenicillins, such as ampicillin and amoxicillin, provide added coverage beyond penicillin against gram-negative cocci, such as Haemophilus influenzae, and some Enterobacteriaceae, including E. coli, Proteus mirabilis, Salmonella, and Shigella. The aminopenicillins are hydrolyzed by many common β-lactamases. These drugs are commonly used for otitis media, respiratory tract infections, intraabdominal infections, endocarditis, meningitis, and urinary tract infections. The antipseudomonal penicillins include ticarcillin and piperacillin. These penicillin groups generally offer adequate anaerobic coverage; the exceptions are Bacteroides species (such as Bacteroides fragilis), which produce β-lactamases and are generally resistant. The rising prevalence of β-lactamase-producing bacteria has led to the increased use of β-lactam/β-lactamase inhibitor combinations, such as ampicillin-sulbactam, amoxicillin-clavulanate, ticarcillin-clavulanate, and piperacillin-tazobactam. The β-lactamase inhibitors themselves do not have antibacterial activity (with the exception of sulbactam, which has activity against Acinetobacter baumannii) but typically inhibit the S. aureus class A β-lactamase, β-lactamases of H. influenzae and Bacteroides species, and a number of plasmid-encoded β-lactamases. These combination agents are typically used when broader-spectrum coverage is needed—e.g., in pneumonia and intraabdominal infections. Piperacillin-tazobactam is a useful agent for broad coverage in febrile neutropenic patients. The combination agents, however, are not effective against organisms that produce AmpC β-lactamases or carbapenemases.
Cephalosporins The cephalosporin drug class encompasses five generations determined by spectrum of antibacterial activity. The first generation (cefazolin, cefadroxil, cephalexin) largely has activity against gram-positive bacteria, with some additional activity against E. coli, P. mirabilis, and K. pneumoniae. First-generation cephalosporins are commonly used for infections caused by MSSA and streptococci (e.g., skin and soft tissue infections). Cefazolin is a popular choice for surgical prophylaxis against skin organisms. The second generation (cefamandole, cefuroxime, cefaclor, cefprozil, cefuroxime axetil, cefoxitin, cefotetan) has additional activity against H. influenzae and Moraxella catarrhalis. Cefoxitin and cefotetan have potent activity against anaerobes as well. Second-generation cephalosporins are used to treat community-acquired pneumonia because of their activity against S. pneumoniae, H. influenzae, and M. catarrhalis. They are also used for other mild or moderate infections, such as acute otitis media and sinusitis. The third-generation cephalosporins are characterized by greater potency against gram-negative bacilli and reduced potency against gram-positive cocci. These cephalosporins, which include cefoperazone, cefotaxime, ceftazidime, ceftriaxone, cefdinir, cefixime, and cefpodoxime, are used for infections caused by Enterobacteriaceae, although resistance is an increasing concern. It is noteworthy that ceftazidime is the only third-generation cephalosporin with activity against P. aeruginosa but lacks activity against gram-positive bacteria. This drug is frequently used for pulmonary infections in cystic fibrosis and febrile neutropenia. Ceftriaxone penetrates the CSF and can be used to treat meningitis caused by H. influenzae, N. meningitidis, and susceptible strains of S. pneumoniae. It is also used for the treatment of later-stage Lyme disease. The fourth generation includes cefepime and cefpirome, broad-coverage agents that provide potent activity against both gram-negative bacilli, including P. aeruginosa, and gram-positive cocci. The fourth generation has clinical applications similar to those of the third generation and can be used in bacteremia, pneumonia, skin and soft tissue infections, and urinary tract infections caused by susceptible bacteria. Cefepime is also commonly used in febrile neutropenia. Ceftaroline, a fifth-generation cephalosporin, differs from the other cephalosporins in its added activity against MRSA, which is resistant to all other β-lactams. Ceftaroline’s gram-negative activity is similar to that of the third-generation cephalosporins but does not include P. aeruginosa. Ceftaroline is efficacious in community-acquired pneumonia and skin infections, but few data are available on its use for more serious infections, such as bacteremia.
Carbapenems With a few exceptions for cefepime, all penicillins and cephalosporins are ineffective in the presence of ESBLs. Carbapenems, including doripenem, imipenem, meropenem, and ertapenem, offer the most reliable coverage for strains containing ESBLs. All carbapenems have broad activity against gram-positive cocci, gram-negative bacilli, and anaerobes. None is active against MRSA, but all are active against MSSA, Streptococcus species, and Enterobacteriaceae. Ertapenem is the only carbapenem that has poor activity against P. aeruginosa and Acinetobacter. Imipenem is active against penicillin-susceptible Enterococcus faecalis but not Enterococcus faecium. Carbapenems are not active against Enterobacteriaceae containing carbapenemases. Stenotrophomonas maltophilia and some Bacillus species are intrinsically resistant to carbapenems because of a zinc-dependent carbapenemase.
Monobactams Aztreonam is the sole monobactam. Its activity is limited to gram-negative bacteria and includes P. aeruginosa and most other Enterobacteriaceae. This drug is inactivated by ESBLs and carbapenemases. The principal use for aztreonam is as an alternative to penicillins, cephalosporins, or carbapenems in patients with serious β-lactam allergy. Aztreonam is structurally related to ceftazidime and should be used cautiously in individuals with a serious ceftazidime allergy. It is commonly used in febrile neutropenia and intraabdominal infections. Aztreonam does not penetrate the CSF and should not be used for treatment of meningitis.
Adverse Reactions to β-Lactam Drugs Agents within the β-lactam class are known for several adverse effects. Gastrointestinal side effects, mainly diarrhea, are common, but hypersensitivity reactions constitute the most common adverse effect of β-lactams. The reactions’ severity can range from rash to anaphylaxis, but the rate of true anaphylactic reactions is only 0.05%. An individual with an accelerated IgE-mediated reaction to one β-lactam agent may still receive another agent within the class, but caution should be taken to choose a β-lactam that has a dissimilar side chain and a low level of cross-reactivity. For example, the second-, third-, and fourth-generation cephalosporins and the carbapenems display very low cross-reactivity in patients with penicillin allergy. Aztreonam is the only β-lactam that has no cross-reactivity with the penicillin group. In cases of severe allergy, desensitization (a graded challenge) to the indicated β-lactam, with close monitoring, may be warranted if other antibacterial options are not suitable.
β-Lactams can rarely cause serum sickness, Stevens-Johnson syndrome, nephropathy, hematologic reactions, and neurotoxicity. Neutropenia appears to be related to high doses or prolonged use. Neutropenia and interstitial nephritis caused by β-lactams generally resolve upon discontinuation of the agent. Imipenem and cefepime are associated with an increased risk of seizure, but this risk is likely a class effect and related to high doses or doses that are not adjusted in renal impairment.
GLYCOPEPTIDES
The glycopeptide antibiotics include vancomycin and telavancin. Vancomycin has activity against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci (including S. pneumoniae), and enterococci. It is not active against gram-negative organisms. Vancomycin also displays activity against Bacillus species, Corynebacterium jeikeium, Listeria monocytogenes, and gram-positive anaerobes such as Peptostreptococcus, Actinomyces, Clostridium, and Propionibacterium species. Vancomycin has several important clinical uses. It is used for serious infections caused by MRSA, including health care–associated pneumonia, bacteremia, osteomyelitis, and endocarditis. It is also commonly used for skin and soft tissue infections. Oral vancomycin is not absorbed systemically and is reserved for the treatment of Clostridium difficile infection. Vancomycin is also an alternative for the treatment of infections caused by MSSA in patients who cannot tolerate β-lactams. Resistance to vancomycin is a rising concern. Strains of vancomycin-intermediate S. aureus (VISA) and vancomycin-resistant enterococci (VRE) are not uncommon. Vancomycin appears to be a concentration-dependent killer, with AUC/MIC ratio being the best predictor of efficacy (Fig. 170-2). Guidelines recommend targeting a vancomycin trough level of 15–20 μg/mL in MRSA infections in order to maintain an AUC/MIC ratio >400. When using vancomycin, clinicians should monitor for nephrotoxicity. The risk increases when trough levels are >20 μg/mL. Concomitant therapy with other nephrotoxic agents, such as aminoglycosides, also increases the risk of nephrotoxicity. Ototoxicity was reported with early formulations of vancomycin but is currently uncommon because purer formulations are available. Both of these adverse effects are reversible upon discontinuation of vancomycin. Clinicians should be aware of the “red man syndrome,” a common reaction that presents as a rapid onset of erythematous rash or pruritus on the head, face, neck, and upper trunk. This reaction is caused by histamine release from basophils and mast cells and can be treated with diphenhydramine and slowing of the vancomycin infusion.
Telavancin, dalbavancin, and oritavancin are structurally similar to vancomycin and are referred to as lipoglycopeptides. They have antibacterial activity against S. aureus (including MRSA and some strains of VISA and vancomycin-resistant S. aureus [VRSA]), streptococci, and enterococci. They also have good activity against anaerobic gram-positive organisms except for Lactobacillus and some Clostridium species. The clinical efficacy of telavancin has been demonstrated in both skin and soft tissue infections and nosocomial pneumonia, and the efficacy of dalbavancin and oritivancin has been shown in skin and soft tissue infections. The vancomycin resistance phenotype may reduce the potency of all three lipoglycopeptides, but the rate of resistance to these drugs among S. aureus and enterococci has been low. Adverse effects of telavancin include insomnia, a metallic taste, nephrotoxicity, and gastrointestinal side effects. Clinicians should be aware of the potential for electrocardiographic QTc prolongation that can increase the risk of cardiac arrhythmias when telavancin is used concomitantly with other QTc-prolonging agents. Telavancin may interfere with certain coagulation tests (e.g., causing false elevations in prothrombin time). Dalbavancin and oritavancin have safety profiles similar to that of vancomycin.
LIPOPEPTIDES
Daptomycin is a lipopeptide antibiotic with activity against a broad range of gram-positive organisms. This drug is active against staphylococci (including MRSA and coagulase-negative staphylococci), streptococci, and enterococci. Daptomycin remains active against enterococci that are resistant to vancomycin. In addition, it exhibits activity against Bacillus, Corynebacterium, Peptostreptococcus, and Clostridium species. Daptomycin’s pharmacodynamic parameter for efficacy is concentration-dependent killing. Resistance to daptomycin is rare, but MICs may be higher for VISA strains. Daptomycin is efficacious in skin and soft tissue infections, bacteremia, endocarditis, and osteomyelitis. It is an important alternative for MRSA and other gram-positive infections when bactericidal therapy is needed and vancomycin cannot be used. Daptomycin is generally well tolerated, and its main toxicity consists of elevation of creatinine phosphokinase (CPK) levels and myopathy. CPK should be monitored during daptomycin treatment, and the drug should be discontinued if muscular toxicities occur. There have also been case reports of reversible eosinophilic pneumonia associated with daptomycin use.
AMINOGLYCOSIDES
The aminoglycosides are a class of antibacterial agents with concentration-dependent activity against most gram-negative organisms. The most commonly used aminoglycosides are gentamicin, tobramycin, and amikacin, although others, such as streptomycin, kanamycin, neomycin, and paromomycin, may be used in special circumstances. Aminoglycosides have a significant dose-dependent postantibiotic effect, meaning that they have an antibacterial effect even after serum drug levels are undetectable. The postantibiotic effect and concentration-dependent killing form the rationale behind extended-interval aminoglycoside dosing, in which a larger dose is given once daily rather than smaller doses multiple times daily. Aminoglycosides are active against gram-negative bacilli, such as Enterobacteriaceae, P. aeruginosa, and Acinetobacter. They also enhance the activity of cell wall–active agents such as β-lactams or vancomycin in some gram-positive bacteria, including staphylococci and enterococci. This combination therapy is termed synergistic because the effect of both agents provides a killing effect greater than would be predicted from the effects of either agent alone. Amikacin and streptomycin have activity against Mycobacterium tuberculosis, and amikacin has activity against Mycobacterium avium-intracellulare. The aminoglycosides do not have activity against anaerobes, S. maltophilia, or Burkholderia cepacia. Aminoglycosides are used in clinical practice in a variety of infections caused by gram-negative organisms, including bacteremia and urinary tract infections. They are frequently used alone or in combination for the treatment of P. aeruginosa infection. When used in combination with a cell wall–active agent, gentamicin and streptomycin are also important for the treatment of gram-positive bacterial endocarditis. All aminoglycosides can cause nephrotoxicity and ototoxicity. The risk of nephrotoxicity is related to the dose and duration of therapy as well as the concomitant use of other nephrotoxic agents. Nephrotoxicity is usually reversible, but ototoxicity can be irreversible.
MACROLIDES AND KETOLIDES
The macrolides (azithromycin, clarithromycin, erythromycin) and ketolides (telithromycin) are classes of antibiotics that inhibit protein synthesis. Compared with erythromycin (the older antibiotic), azithromycin and clarithromycin have better oral absorption and tolerability. Azithromycin, clarithromycin, and telithromycin all have broader spectra of activity than erythromycin, which is less frequently used. These agents are commonly used in the treatment of upper and lower respiratory tract infections caused by S. pneumoniae, H. influenzae, M. catarrhalis, and atypical organisms (e.g., Chlamydia pneumoniae, Legionella pneumophila, and Mycoplasma pneumoniae); group A streptococcal pharyngitis in penicillin-allergic patients; and nontuberculous mycobacterial infections (e.g., caused by M. marinum and M. chelonae) as well as in the prophylaxis and treatment of M. avium-intracellulare infection in patients with HIV/AIDS and in combination therapy for H. pylori infection and bartonellosis. Enterobacteriaceae, Pseudomonas species, and Acinetobacter species are intrinsically resistant to macrolides as a result of decreased membrane permeability, although azithromycin is active against gram-negative diarrheal pathogens. The major adverse effects of this drug class include nausea, vomiting, diarrhea and abdominal pain, prolongation of QTc interval, exacerbation of myasthenia gravis, and tinnitus. Azithromycin specifically has been associated with an increased risk of death, especially among patients with underlying heart disease, because of the risk of QTc interval prolongation and torsades de pointes. Erythromycin, clarithromycin, and telithromycin inhibit the CYP3A4 hepatic drug-metabolizing enzyme and can result in increased levels of coadministered drugs, including benzodiazepines, statins, warfarin, cyclosporine, and tacrolimus. Azithromycin does not inhibit CYP3A4 and lacks these drug–drug interactions.
CLINDAMYCIN
Clindamycin is a lincosamide antibiotic and is bacteriostatic against some organisms and bactericidal against others. It is used most often to treat bacterial infections caused by anaerobes (e.g., B. fragilis, Clostridium perfringens, Fusobacterium species, Prevotella melaninogenicus, and Peptostreptococcus species) and susceptible staphylococci and streptococci. Clindamycin is used for treatment of dental infections, anaerobic lung abscess, and skin and soft tissue infections. It is used together with bactericidal agents (penicillins or vancomycin) to inhibit new toxin synthesis in the treatment of streptococcal or staphylococcal toxic shock syndrome. Other uses include treatment of infections caused by Capnocytophaga canimorsus, a component of combination therapy for malaria and babesiosis, and therapy for toxoplasmosis. Clindamycin has excellent oral bioavailability. Adverse effects include nausea, vomiting, diarrhea, C. difficile–associated diarrhea and pseudomembranous colitis, maculopapular rash, and (rarely) Stevens-Johnson syndrome.
TETRACYCLINES AND GLYCYLCYCLINES
The tetracyclines (doxycycline, minocycline, and tetracycline) and the glycylcyclines (tigecycline) inhibit protein synthesis and are bacteriostatic. These drugs have wide clinical uses. They are used in the treatment of skin and soft tissue infections caused by gram-positive cocci (including MRSA), spirochetal infections (e.g., Lyme disease, syphilis, leptospirosis, and relapsing fever), rickettsial infections (e.g., Rocky Mountain spotted fever), atypical pneumonia, sexually transmitted infections (e.g., Chlamydia trachomatis infection, lymphogranuloma venereum, and granuloma inguinale), infections with Nocardia and Actinomyces, brucellosis, tularemia, Whipple’s disease, and malaria. Tigecycline, the only approved agent in the glycylcycline class, is a derivative of minocycline and is indicated in the treatment of infections due to MRSA, vancomycin-sensitive enterococci, many Enterobacteriaceae, and Bacteroides species. Tigecycline has no activity against P. aeruginosa. It has been used in combination with colistin for the treatment of serious infections with multidrug-resistant gram-negative organisms. A pooled analysis of 13 clinical trials found an increased risk of death and treatment failure among patients treated with tigecycline alone. Tetracyclines have reduced absorption when coadministered with calcium- and iron-containing compounds, including milk, and doses should be spaced at least 2 h apart. The major adverse reactions to both of these classes are nausea, vomiting, diarrhea, and photosensitivity. Tetracyclines have been associated with fetal bone-growth abnormalities and should be avoided during pregnancy and in the treatment of children <8 years old.
TRIMETHOPRIM-SULFAMETHOXAZOLE
Trimethoprim-sulfamethoxazole (TMP-SMX) is an antibiotic whose two components both inhibit folate synthesis and produce antibacterial activity. TMP-SMX is active against gram-positive bacteria such as staphylococci and streptococci; however, its use against MRSA is usually limited to community-acquired infections, and its activity against Streptococcus pyogenes may not be reliable. TMP-SMX is also active against many gram-negative bacteria, including H. influenzae, E. coli, P. mirabilis, N. gonorrhoeae, and S. maltophilia. TMP-SMX does not have activity against anaerobes or P. aeruginosa. It has many uses because of its wide spectrum of activity and high oral bioavailability. Urinary tract infections, skin and soft tissue infections, and respiratory tract infections are among the common uses. Another important indication is for both prophylaxis and treatment of Pneumocysitis jirovecii infections in immunocompromised patients. Resistance to TMP-SMX has limited its use against many Enterobacteriaceae. Resistance rates among urinary isolates of E. coli are almost 25% in the United States. The most common adverse reactions associated with TMP-SMX are gastrointestinal effects such as nausea, vomiting, and diarrhea. In addition, rash is a common allergic reaction and may preclude the subsequent use of other sulfonamides. With prolonged use, leukopenia, thrombocytopenia, and granulocytopenia can develop. TMP-SMX can also cause nephrotoxicity, hyperkalemia, and hyponatremia, which are more common at high doses. TMP-SMX has several important interactions with other drugs (Table 170-4), including warfarin, phenytoin, and methotrexate.
FLUOROQUINOLONES
The fluoroquinolones include norfloxacin, ciprofloxacin, ofloxacin, levofloxacin, moxifloxacin, and gemifloxacin. Ciprofloxacin and levofloxacin have the broadest spectrum of activity against gram-negative bacteria, including P. aeruginosa (similar to that of third-generation cephalosporins). Because of the risk of selection of resistance during fluoroquinolone treatment of serious pseudomonal infections, these agents are usually used in combination with an antipseudomonal β-lactam. Levofloxacin, moxifloxacin, and gemifloxacin have additional gram-positive activity, including that against S. pneumoniae and some strains of MSSA, and are used for treatment of community-acquired pneumonia. Strains of MRSA are commonly resistant to all fluoroquinolones. Moxifloxacin is used as one component of second-line regimens for multidrug-resistant tuberculosis. Fluoroquinolones exhibit concentration-dependent killing, are well absorbed orally, and have elimination half-lives that usually support once- or twice-daily dosing. Oral coadministration with compounds containing high concentrations of aluminum, magnesium, or calcium can reduce fluoroquinolone absorption. Their penetration into prostate tissue supports their use for bacterial prostatitis. Fluoroquinolones are generally well tolerated but can cause CNS stimulatory effects, including seizures; glucose dysregulation; and tendinopathy associated with Achilles tendon rupture, particularly in older patients, organ transplant recipients, and patients taking glucocorticoids. Worsening of myasthenia gravis also has been associated with quinolone use. Moxifloxacin causes modest prolongation of the QTc interval and should be used with caution in patients receiving other QTc-prolonging drugs.
RIFAMYCINS
The rifamycins include rifampin, rifabutin, and rifapentine. Rifampin is the most commonly used rifamycin. For almost all therapeutic indications, it is used in combination with other agents to reduce the likelihood of selection of high-level rifampin resistance. Rifampin is used foremost in the treatment of mycobacterial infections—specifically, as a mainstay of combination therapy for M. tuberculosis infection or as a single agent in the treatment of latent M. tuberculosis
[/not-level-membership-for-internal-medicine-category]
