Chapter 18 Pharmacodynamic Interactions of Antiepileptic Drugs
Mechanisms of Actions of AEDs
Despite hundreds of basic studies addressing the mechanisms of action of antiepileptic drugs (AEDs), the relevance of these observations to the clinical effectiveness of these agents remains unclear. This problem is in large part due to the fact that epileptic seizures are manifestations of an abnormal network of neural and glial elements across multiple brain regions, and a mechanistic effect of an AED in one area may or may not be related to blockade of seizure activity.1 Additionally, controlled mechanistic studies can only be performed in vitro, with tissue removed from the milieu in which in vivo effects are observed. Thus, it is not always clear whether a laboratory finding truly translates to the intact epileptic brain. Notwithstanding these limitations, by correlating efficacy studies in humans and in animal models with primarily cellular electrophysiological effects in isolated neurons and in brain slices, a widely accepted conceptual framework has emerged regarding putative mechanisms of AED action.2–5
It should be understood at the outset that no single mechanistic finding is sufficient to explain all the clinical effects of a particular AED. Moreover, it is becoming clearer that every AED possesses multiple potential mechanisms of action, and that such diverse effects are dependent on numerous variables, including brain region, cell type, molecular composition of receptor targets, and drug concentration.2,3,5 And there is an emerging consensus that the multiplicity of molecular action for any given AED is perhaps predictive and correlative with a spectrum of clinical activity across multiple seizure types. For example, topiramate’s actions on voltage-gated sodium and calcium channels, combined with the effects on voltage-gated sodium channels, γ-aminobutyric acidA (GABAA), and glutamate receptors, are consistent with efficacy against both partial and several forms of generalized seizures.5
Dozens of AEDs have been approved for clinical use since the introduction of the barbiturate phenobarbital in 1912. These drugs have been products of extensive testing in a variety of animal seizure models and in human clinical trials.6 However, the paradigms for drug discovery—until very recently—have been sharply biased toward the identification of candidate drugs similar to traditional agents such as phenytoin and phenobarbital. This is in large measure due to testing of compounds in a normal brain against acutely provoked seizures, rather than more appropriate screening in epileptic models that more closely mirror the human condition.6 Moreover, despite their clinical efficacy, current AEDs fail to “cure,” prevent, or modify the disease process. Rather, they eliminate the major symptom (i.e., seizures) by dampening neuronal excitation, synchronization, and spread of seizure activity. No clinical data exist to support the notion that these drugs are truly “antiepileptogenic” or “antiepileptic” (i.e., prevent the development or maintenance of the epileptic state).7
Two traditional models employed in routine screening and identification of new anticonvulsants are the maximal electroshock (MES) and subcutaneous pentylenetetrazol (PTZ or Metrazol) tests, conducted in rodents.6 The former tests the ability of a drug to block tonic extension evoked by an electrical stimulus, whereas the latter tests an agent’s ability to inhibit a generalized clonic seizure induced by subcutaneous administration of PTZ, a GABAA receptor antagonist. MES seizures can be blocked by AEDs such as phenytoin and carbamazepine, which are effective against partial-onset seizures. In contrast, AEDs that are efficacious in the treatment of generalized absence seizures (e.g., ethosuximide) can inhibit PTZ-induced seizures. Valproate, a broad-spectrum agent, is active in both MES and PTZ tests and is clinically effective against most seizure types.8 However, these traditional screening tests may fail to identify drugs that may act through novel mechanisms. An example is that of levetiracetam, a newer AED that is clearly effective in the treatment of partial-onset seizures, but was originally found to be inactive in both MES and PTZ models, and thus initially discarded.6 The later observation that levetiracetam could retard kindling in rodents resurrected this unusual compound as an AED candidate.9
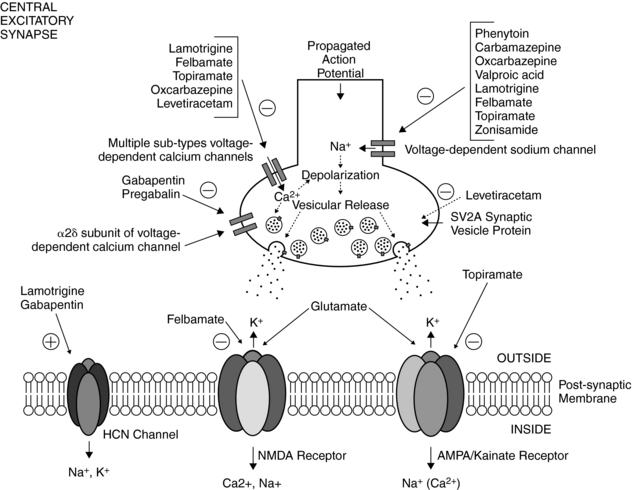
In general, three major classes of molecular targets are believed to be relevant for limiting seizure activity.2 These include: (1) voltage-gated sodium and calcium channels, (2) GABAA receptors, and (3) ionotropic glutamate receptors (i.e., NMDA or N-methyl-D-aspartate, AMPA, or α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid) and kainate receptors. Clinically useful AEDs exert their effects principally on one or more of these targets. Although a host of other targets could affect neuronal excitability (see following discussion), the validity of these voltage-dependent and ligand-gated ion channels toward AED action has stood the test of time.
For decades, the primary AEDs of choice for the treatment of partial-onset epilepsy have been phenytoin and carbamazepine. Both phenytoin and carbamazepine cause voltage-, frequency-, and use-dependent block of sodium channels in a wide variety of neuronal preparations.10 Sustained, high-frequency, repetitive firing of neurons, is believed to play a significant role in neuronal excitability and is potently inhibited by these AEDs at free plasma concentrations found in patients treated for epilepsy.3 Oxcarbazepine, a structural analog of carbamazepine that is reduced to a monohydroxy-derivative, also blocks sustained repetitive firing.11 And based on this mechanistic profile, oxcarbazepine has been found to be effective only against partial seizures, similar to that of phenytoin and carbamazepine.12
Though structurally unrelated to phenytoin and carbamazepine, lamotrigine can also block sodium channels in a voltage-, frequency-, and use-dependent manner.13 However, lamotrigine possesses a broader spectrum of clinical activity than either phenytoin or carbamazepine, demonstrating efficacy against several forms of generalized seizures (especially absence seizures), in addition to partial seizures.14 The mechanistic basis for this difference remained unclear until recently. Lamotrigine was found to enhance activation of the hyperpolarization-activated cation channel (HCN channel), responsible for the so-called Ih or h-current.15 HCN channels are highly expressed in neuronal dendrites in both thalamus and hippocampus, are activated by hyperpolarization, and tend to stabilize the neuronal membrane potential against both hyperpolarizing and depolarizing inputs.16,17
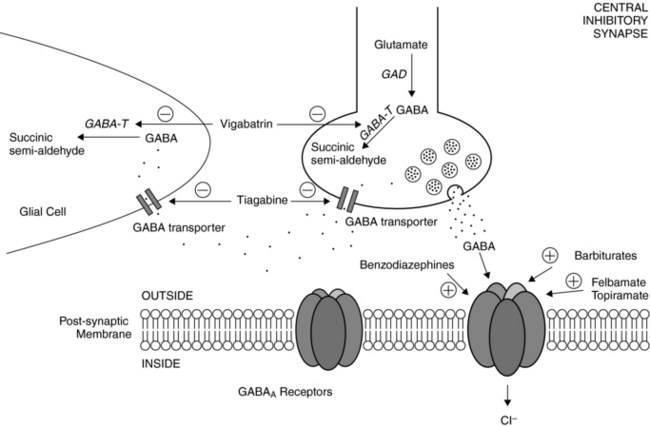
Postsynaptic GABAA receptors are widely regarded as relevant toward the clinical effects of AEDs such as barbiturates and benzodiazepines. Binding of either benzodiazepines or barbiturates to their respective recognition sites on the GABAA receptor results in enhanced chloride influx and, hence, membrane hyperpolarization.18 Benzodiazepines increase the frequency of GABAA receptor channel openings, whereas barbiturates prolong the mean duration of these openings.19
Other agents have been shown to affect the GABAergic system as well. Vigabatrin is an irreversible inhibitor of the major degradative enzyme for GABA (i.e., GABA-transaminase),20 and tiagabine is a potent and selective blocker of the GABA transporter, that which functions to reuptake GABA from the synaptic cleft.21 As predicted from their primary actions, both vigabatrin and tiagabine increase synaptic levels of GABA. Topiramate also increases the GABAA receptor open duration and burst frequency in an allosteric manner,5 and felbamate enhances GABAA receptor-mediated currents in hippocampal and neocortical through a barbiturate-like action.22 Valproate can evoke a wide variety of biochemical and neurophysiological changes in multiple neurotransmitter systems, but despite numerous studies, its precise mechanisms of action remain a mystery.8 Nevertheless, much of the evidence points to valproate’s effects on enhancing GABAergic transmission by enhancing biosynthesis and blocking degradation of GABA, resulting in elevated brain GABA levels.23 At therapeutic serum levels, valproate also inhibits sustained repetitive firing of cultured mouse spinal cord and neocortical neurons, implicating actions on voltage-gated sodium channels as well.24
Gabapentin and pregabalin are structural analogues of GABA, and although it had been predicted that these AEDs might act on the GABAergic system, they do not interact with either GABAA or GABAB receptors, and they do not affect GABA reuptake, synthesis, or metabolism.25 Gabapentin and pregabalin bind uniquely to the α2δ-1 and α2δ-2 auxiliary subunits of the high-voltage activated L-type calcium channel.25 It is believed that this interaction is important in decreasing presynaptic neurotransmitter release at both inhibitory and excitatory glutamatergic synapses. Interestingly, gabapentin was also found to enhance h-currents in hippocampal neurons26 similar to the activity of lamotrigine (see earlier discussion).
Felbamate is the first pharmacological agent to both potentiate GABAA receptor-mediated responses and inhibit NMDA receptor-mediated responses within the same drug concentration range.22 These dual actions are believed to contribute in a synergistic way to protect against seizure activity. Felbamate, like many other AEDs, can also block sustained repetitive firing of neurons, attributable to pathologic firing through voltage-gated sodium channels.27 Similarly, topiramate blocks voltage-gated sodium conductances, but more important, it inhibits AMPA and kainate (specifically, GluR5) receptors.28
Zonisamide is a broad-spectrum agent that has a unique mechanistic profile.29 In cultured spinal cord neurons, zonisamide decreased sustained repetitive firing of action potentials, consistent with actions on voltage-gated sodium channels, and in cultured neurons from rat cerebral cortex, zonisamide blocked low-threshold T-type calcium currents, which predicts efficacy against generalized spike-wave epilepsies—specifically, absence seizures.29
Levetiracetam, one of the newer AEDs, has broadened our conceptual understanding of relevant mechanisms of AED action. As noted earlier, unlike traditional AEDs, levetiracetam failed both MES and PTZ seizure threshold tests, yet had a profound effect in retarding amygdala kindling in rats.6,9 Levetiracetam’s principal molecular target, originally identified as a specific high-affinity neuronal binding site, was recently demonstrated to be a specific synaptic vesicle protein, SV2A, which is involved in neurotransmitter release.30 The functional consequences of such an interaction, although novel and intriguing, remain unclear.
Potassium channels represent an extremely diverse family of ion channels and generally decrease neuronal excitation by causing membrane hyperpolarization. As such, potassium channels represent a natural target for AED development. None of the currently available AEDs is believed to act primarily to enhance potassium channel activity, but recent studies implicate these channels—at least in part—in the anticonvulsant action of a number of AEDs, including phenytoin, carbamazepine, topiramate, levetiracetam, and possibly lamotrigine and zonisamide.3–6 In contrast to these AEDs, retigabine, an investigational compound with broad efficacy in animal seizure models, acts primarily through enhanced activation of KCNQ2 and KCNQ3 potassium channels.31–33 This molecular action is especially intriguing because a rare form of inherited epilepsy—benign familial neonatal convulsions—has been linked to mutations in genes encoding these potassium channel subunits.34,35
Anticonvulsants known to be clinically effective against absence seizures (e.g., ethosuximide and valproate) can block a subtype of voltage-gated calcium channel known as the “low-threshold” or T-type calcium channel.3,5 Although the role of T-type calcium currents in the genesis of absence seizures has been somewhat controversial,36,37 the bulk of evidence supports the involvement of these channels.38 Lamotrigine’s actions on h-currents may also contribute an antiabsence effect, as HCN channels are densely expressed in the thalamus and are critical regulators of pacemaker activity.16,17 However, although it is appealing to think of absence seizures as simply a by-product of T-type calcium channel and/or h-channel dysfunction, the actual pathophysiological mechanisms are much more complex.39
Finally, several AEDs act either principally or in part by inhibiting certain carbonic anhydrase isoforms.40 These include acetazolamide, topiramate, and zonisamide. Carbonic anhydrase inhibitors have been used in epileptic patients for almost 50 years, but it is unclear how clinical effects are achieved with these drugs.41
Pharmacodynamic Interactions Influencing Efficacy
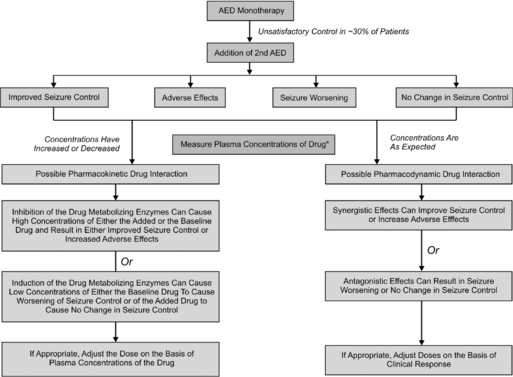
Often in clinical practice, two or more AEDs are combined in an attempt to achieve either seizure reduction or freedom. A number of prospective studies in patients with newly diagnosed epilepsy have shown that the majority will fully respond to trials of two to three AEDs, administered either as monotherapy or in combination.42,43 Thus, when two or more AEDs are combined, an improvement in clinical response may be interpreted as either a positive pharmacodynamic interaction and/or infra-additive toxicity. More often than not, combination AED therapy has resulted in pharmacokinetic and pharmacodynamic toxicity and not necessarily significantly improved seizure control.44 As shown in Figure 18-1, plasma concentrations need to be measured at the time of event to determine whether an interaction is pharmacokinetic or pharmacodynamic.45
Overall, it has been extremely challenging to determine whether a particular AED combination produces a beneficial effect due to additive or synergistic effects stemming from enhanced activity at their molecular sites of action. This is because a number of factors determine—dependently and independently—clinical effects of AEDs, including: (1) the variable and often unpredictable pharmacokinetic interactions that ultimately influence delivery of the drugs to their brain targets; (2) the narrow therapeutic indices of most AEDs, which can predispose the patient to loss of seizure control, toxicity, or perhaps, rarely, an improved response; (3) the chronicity of AED treatment, that can result in induction of various forms of drug tolerance and other adaptive changes; (4) the high incidence of AED cotherapy with nonepilepsy drugs; and (5) evolution of the epileptic state, especially in infants and children.
Despite these limitations, investigators have turned to animal models to study the impact of AED combination therapy. Even in animals, however, despite over a hundred published studies addressing greater than 500 AED interactions, we do not yet have a clear understanding of the pharmacodynamic properties of specific AED combinations.46 Concomitant pharmacokinetic changes of AED cotherapy have not been properly addressed in the majority of preclinical studies to date, and most have been conducted in rodents using acute seizure models, and thus may not be wholly relevant to the chronic epileptic condition. Nevertheless, a couple of intriguing approaches have been taken in preclinical studies.
One novel approach toward analyzing the pharmacodynamic effects of AED combinations has been the use of isobolographic analytical techniques, which is commonly employed to analyze drug combinations. With isobolography, based on careful assessments of efficacy, toxicity, and serum and brain levels of AEDs, one can calculate the equi-effective drug doses for classifying observed interactions as synergistic, additive, neutral, or infra-additive (antagonistic). Although imperfect, and subject to some of the caveats described earlier, isobolography provides a more rigorous approach toward the analysis of drug combinations than can be achieved in clinical studies. Several isobolographic studies of mostly newer-generation AEDs in acute seizure models have suggested that a number of drug combinations may be at least additive from the standpoint of seizure control.47–50 Again, however, because certain fixed doses and AED ratios were used for such studies in MES and PTZ tests only, the clinical applicability of such data remains uncertain. On a final note regarding such preclinical studies, ictal-component analysis (of specific behavioral features) may provide another theoretical basis for the identification of AED combinations that would be useful in treating epileptic patients, particularly those who are classified as medically intractable.51
Far fewer clinical studies have examined pharmacodynamic interactions of AEDs, given the challenges noted earlier. Thus far, no clear clinical data indicate that a particular AED combination results in decreased efficacy due to a pharmacodynamic effect. Although seizure aggravation by certain AEDs has been well described,52 scant data exist for seizure exacerbation due to a paradoxical pharmacodynamic effect. A recent analysis of previously published reports failed to identify consistent evidence of pure pharmacodynamic aggravation of seizures in the absence of modifying factors, such as overdose, encephalopathy, hepatotoxicity, and metabolic derangements.53 However, valproate appeared to a very low potential for pharmacodynamic seizure exacerbation.53
With respect to improved efficacy, several clinical reports suggest that the following combinations yield positive pharmacodynamic effects independent of pharmacokinetic changes induced by additive therapy: (1) valproate and ethosuximide,54 (2) valproate and carbamazepine,54 (3) valproate and lamotrigine,55,56 and (4) topiramate and lamotrigine.57 However, the data suggesting pharmacodynamic synergy are retrospective in nature and hampered by intrinsic difficulties controlling for important variables noted earlier, despite reasonable attempts to account for pharmacokinetic changes by measuring serum blood levels of AEDs.
Pharmacodynamic Interactions Influencing Toxicity
ANTIEPILEPTIC DRUG POLYTHERAPY
It is difficult to separate the role of pharmacokinetic versus pharmacodynamic interactions when evaluating the effect of AED polytherapy on adverse drug reactions. Pharmacokinetic drug interactions are common with the first-generation AEDs.58 To document a pharmacodynamic interaction, a pharmacokinetic interaction must be ruled out as higher drug concentrations or increased formation of reactive metabolites can be associated with an increased incidence of effect. The studies are also confounded as patients receiving polytherapy typically have refractory or treatment-resistant epilepsy, and separating the effects of the seizure disorder from the effects of the AED is often difficult.59
Cognition
The majority of patients receiving first-generation AEDs report adverse events. In a large quality-of-life study in >5000 patients, 40 to 50% reported tiredness, memory problems, difficulty concentrating, sleepiness, and difficulty concentrating.60 Over half (53%) of the patients surveyed were receiving more than one AED. Even with significant controversy regarding the relative effects of the individual AEDs, it is generally accepted that polytherapy is associated with an increased incidence of sedation and cognitive complaints.61–63 For the new AEDs, few controlled studies have evaluated cognition in healthy subjects or patients with epilepsy receiving monotherapy.64
Teratogenicity
The risk of birth defects in healthy, pregnant women is about 2 to 4% and rises to 4 to 6% in pregnant women with epilepsy taking one first-generation AED.65 Malformation rates increased with increasing number of AEDs.66,67 The odds ratio for malformation rate for an infant exposed to one AED compared to two or more AEDs were 2.8 and 4.2, respectively, when compared to nonexposed infants.68 More recently, Morrow et al. found a 3.7% and 6.0% incidence for AED monotherapy versus polytherapy.69 Sufficient data on the effects of monotherapy or polytherapy on the second-generation AEDs is only available for lamotrigine.70 Polytherapy with enzyme-inducing agents does not increase the incidence of malformation over lamotrigine monotherapy alone (2.8% vs. 2.7%). Polytherapy with valproate has significantly higher incidence (11.8%); however, the incidence is approximately the same as valproate alone (10.7%).71
Many different mechanisms for the teratogenicity of the first-generation AEDs have been postulated, and both pharmacokinetic and pharmacodynamic interactions may be involved in the increased teratogenicity due to polytherapy. From a pharmacokinetic viewpoint, phenytoin, phenobarbital, and carbamazepine are metabolized via cytochrome P450-dependent oxidation. Oxidative intermediates are formed and further metabolized via hydroxylation by epoxide hydrolase, a hepatic cytosolic enzyme. Lower levels of this enzyme in fetuses as compared to adults may cause the accumulation of oxidative intermediates. The formation of oxidative intermediates is believed to be partly responsible for birth defects.72 The teratogenic effect is worsened with the addition of valproate to phenytoin, phenobarbital, or carbamazepine. Consistent with the reactive metabolite hypothesis, the addition of phenobarbital and carbamazepine to phenytoin will increase the metabolism of oxidative intermediates. Valproate inhibits the metabolism of oxidative intermediates by inhibition of epoxide hydrolase.73,74
A pharmacodynamics interaction cannot be ruled out. The interference of folic acid metabolism has been widely accepted as a mechanism of teratogenesis.65,75 Folic acid is involved with the biosynthesis of DNA and RNA and with the metabolism of certain amino acids. Phenytoin and valproate inhibit the metabolism of folic acid.65 In pregnant women, serum phenobarbital concentrations were inversely correlated to serum folate concentrations.76 Investigations conducted by Dansky et al. found that spontaneous abortion and developmental anomalies were significantly associated with folate deficiency caused by AED use.75 Therefore, it can be hypothesized that both a pharmacokinetic and a pharmacodynamic effect could be factors in the increased teratogenicity in polytherapy in the first-generation AEDs.
LAMOTRIGINE
Classic signs of carbamazepine neurotoxicity (diplopia, dizziness, ataxia) were reported when lamotrigine was added to carbamazepine therapy.77,78 Even though there were early reports of a possible pharmacokinetic interaction,77 several follow-up studies demonstrated no effect of lamotrigine on plasma concentrations of carbamazepine or its active metabolite, carbamazepine epoxide.79–82 Similar to the carbamazepine and lamotrigine interaction, there were early reports of dizziness when lamotrigine was added to patients receiving oxcarbazepine, the 10-keto analogue of carbamazepine.83 A pharmacokinetic study found that there was not a significant difference in serum concentrations of lamotrigine or plasma concentrations of the pro-drug, oxcarbamazepine and active metabolite, MHD.84 However, subjects receiving lamotrigine plus oxcarbazepine, reported significantly more frequent adverse events (headache, dizziness, nausea, and somnolence) than those receiving lamotrigine or oxcarbazepine monotherapy.
In one small study of patients with refractory partial seizures with and without secondary generalization, who were receiving carbamazepine and at least one other AED, addition of lamotrigine resulted in a slight improvement in attention during a cognitive task.85
The most common idiosyncratic reaction to lamotrigine is rash affecting 10 to 20% of patients. The rashes typically are maculopapular or morbilliform in appearance and occur generally within 2 to 6 weeks of initiating therapy. Risk factors for the rash are young age (children), concurrent valproate therapy, high starting dose, and rapid escalation.86 Schlienger et al. reviewed the descriptions of the case reports of 26 patients and concluded that the characteristics of the syndrome associated with lamotrigine were consistent with the anticonvulsant hypersensitivity syndrome also induced by carbamazepine, phenytoin, and phenobarbital.87 Studies in rats have demonstrated formation of a reactive arene oxide intermediate of lamotrigine whose formation is blocked by the cytochrome P450 (CYP) inhibitor, ketoconazole.88 In a case report of a patient who developed a febrile maculopapular exanthema and later desquamation of the face 1 month after being placed on lamotrigine therapy (with concurrent sodium valproate), a lymphocyte stimulation test was positive.89 This assay has been used to assess the risk of other anticonvulsant hypersensitivity reactions to carbamazepine, phenytoin, and phenobarbital.90 Based on the formation of the arene epoxide intermediate catalyzed by CYP, inhibition by valproate of the major pathway of elimination of lamotrigine, formation of the N-glucuronides would increase the fraction of the dose metabolized to the reactive metabolite. Valproate is not usually associated with the antiepileptic drug hypersensitive syndrome. However, three case reports suggest that a dual hypersensitivity may occur with valproate and other AEDs.91 Therefore, though a pharmacokinetic interaction can be strongly hypothesized, a pharmacodynamic interaction between lamotrigine and valproate cannot be ruled out.
LEVETIRACETAM
A case report described four patients who experienced intolerable carbamazepine-related adverse effects when levetiracetam was added to their therapy without an alteration in carbamazepine or carbamazepine epoxide concentrations.92 In another case report, increased topiramate adverse events (decreased appetite, weight loss, nervousness) occurred after addition of levetiracetam in four children without a change in topiramate plasma concentrations.93 Using an experimental test of neurological adverse effects (rotarod) in rodents, Luszczki et al. evaluated the acute neurotoxic effects of levetiracetam alone and in combination with carbamazepine, phenytoin, phenobarbital, valproate, lamotrigine, topiramate, oxcarbazepine, and felbamate.94 Levetiracetam significantly reduced the median toxic dose of carbamazepine and topiramate without a significant difference in total brain concentrations of levetiracetam, carbamazepine, or topiramate. There was no pharmacodynamic interaction with the other AEDs tested.
Behavioral disturbances have been reported in 10 to 14% of patients receiving levetiracetam.95,96 Patients with a previous psychiatric history and cotherapy with phenytoin95 were more likely to develop behavioral problems. In contrast, lamotrigine cotherapy had a protective effect.96
TOPIRAMATE
Psychiatric adverse events occur in 10 to 20% of patients receiving topiramate. Risk factors for psychiatric adverse events are family and patient psychiatric history and seizure type, as well as high starting dose and rapid titrations schedule.97 Similar to the protective effect of lamotrigine cotherapy with levetiracetam,96 lamotrigine cotherapy with topiramate was also found to be protective against psychiatric adverse events.97
VALPROATE
Valproate therapy is associated with both transient elevation in liver function test in 15 to 30% of patients and a rare, fatal hepatotoxicity.98 The typical histological findings are microvesicular steatosis accompanied by necrosis of hepatocytes. Most cases of valproate hepatotoxicity occur in children under 2 years of age with preexisting neurological or other physical defects, and many were developmentally delayed. Dreifuss et al. demonstrated that both age and polytherapy were associated with significantly increased prevalence.99 There have been many hypotheses regarding the pathogenesis of the hepatotoxicity including preexisting mitochondrial disease or inborn errors of metabolism,100 valproate inhibition of β-oxidation,101 valproate-induced oxidative stress,102 and toxicity from the unsaturated metabolites of valproate, 4-ene-VPA and 2,4-diene-VPA.103 Infants and children104 and patients treated with polytherapy with enzyme inducers105 have higher concentration ratios of 4-ene-VPA to VPA and the hepatotoxic metabolites, respectively. Patients on VPA enzyme-inducing polytherapy also had higher urinary excretion of thiol conjugates of (E), 2-4-VPA diene evidence of reactivity of the diene metabolite.106 A pharmacokinetic interaction model would propose that polytherapy with enzyme inducers and young age increases the formation of these hepatotoxic metabolites. However, a pharmacodynamic interaction cannot be ruled out. Children treated with valproate had elevated levels of a marker of oxidative stress, 15-F2t-isoprostane, which was not found in children treated with carbamazepine or clobazam or control subjects.107 In an experimental rat model, pretreatment with phenobarbital increased the plasma and hepatic 15-F2t-isoprostane that occurred with valproate treatment.108
Asymptomatic hyperammonemia commonly occurs with valproate treatment with a twofold average increase in ammonia concentration over baseline.109 Both hepatic and renal mechanisms have been hypothesized. A prospective study demonstrated that valproate cotherapy with phenobarbital or phenytoin resulted in a significantly higher increase in ammonia concentrations.110 Rarely, valproate induces a more serious hyperammonemic encephalopathy. Case reports of patients treated with valproate show them developing both hyperammonemia111 or hyperammonemic encephalopathy with the addition of topiramate.112–114 Mechanistically, Hamer et al. hypothesized that topiramate may cause a further increase in ammonia concentrations by inhibition of carbonic anhydrase and cerebral glutamine synthetase.112
Conclusion
Overall, it has been extremely challenging to separate the role of pharmacokinetic versus pharmacodynamic interactions when evaluating the effect of AED polytherapy on efficacy and toxicity. The concept of rational polytherapy is based on knowledge of drug pharmacology and toxicology and the pathophysiology of the disease involved. For the treatment of epilepsy, it has been defined as the selection of combinations of antiepileptic drugs (AEDs) to treat patients in whom two or three drugs have failed as monotherapy.115,116 As recently reviewed by Deckers,117 combinations should consist of drugs with wide therapeutic index, low potential for toxicity, and drug interactions with the selection of the AEDs based on their mechanisms of action.118 Unfortunately, few clinical studies have examined pharmacodynamic interactions of AEDs, and the majority of the reported pharmacodynamic effects are based on case reports or observational studies. Even with the considerable increase in the number of AEDs available since 1990, approximately 30 to 40% patients still have uncontrolled seizures. Unfortunately, the concept of rational polytherapy is still theoretical to some extent. Further experimental models are needed to identify effective rational combinations. More important, the synergetic combinations identified by experimental models need to be evaluated in well-designed clinical studies.
1. Faingold CL. Emergent properties of CNS neuronal networks as targets for pharmacology: application to anticonvulsant drug action. Prog Neurobiol. 72:55-85.
2. Rho JM, Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40:1471-1483.
3. Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553-564.
4. Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurother. 2007;4:18-61.
5. White HS, Smith MD, Wilcox KS. Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol. 2007;81:85-110.
6. Smith M, Wilcox KS, White HS. Discovery of antiepileptic drugs. Neurother. 2007;4:12-17.
7. Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515-524.
8. Loscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669-694.
9. Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284:474-479.
10. Catterall WA. Molecular properties of brain sodium channels: an important target for anticonvulsant drugs. Adv Neurol. 1999;79:441-456.
11. Schmutz M, Brugger F, Gentsch C, et al. Oxcarbazepine: preclinical anticonvulsant profile and putative mechanisms of action. Epilepsia. 1994;35(Suppl 5):S47-S50.
12. Schachter SC. Oxcarbazepine: current status and clinical applications. Expert Opin Investig Drugs. 1999;8:1103-1112.
13. Kuo CC, Lu L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol. 1997;121:1231-1238.
14. Choi H, Morrell MJ. Review of lamotrigine and its clinical applications in epilepsy. Expert Opin Pharmacother. 2003;4:243-251.
15. Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767-774.
16. Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453-480.
17. Poolos NP. H-channel dysfunction in generalized epilepsy: it takes two. Epilepsy Curr. 2006;6:88-90.
18. MacDonald RL, Twyman RE. Kinetic properties and regulation of GABAA receptor channels. Ion Channels. 1992;3:315-343.
19. Twyman RE, Rogers CJ, Macdonald RL. Differential regulation of gamma-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann Neurol. 1989;25:213-220.
20. Sabers A, Gram L. Pharmacology of vigabatrin. Pharmacol Toxicol. 1992;70:237-243.
21. Suzdak PD, Jansen JA. A review of the preclinical pharmacology of tiagabine: a potent and selective anticonvulsant GABA uptake inhibitor. Epilepsia. 1995;36:612-626.
22. Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and γ-aminobutyric acidA receptors. Ann Neurol. 1994;35:229-234.
23. Loscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog Neurobiol. 1999;58:31-59.
24. Zona C, Avoli M. Effects induced by the antiepileptic drug valproic acid upon the ionic currents recorded in rat neocortical neurons in cell culture. Exp Brain Res. 1990;81:313-317.
25. Taylor CP, Angelotti T, Fauman E. Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007;73:137-150.
26. Surges R, Freiman TM, Feuerstein TJ. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia. 2006;44:150-156.
27. White HS, Wolf HH, Swinyard EA, et al. A neuropharmacological evaluation of felbamate as a novel anticonvulsant. Epilepsia. 1992;33:564-572.
28. Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069-7074.
29. Biton V. Clinical pharmacology and mechanism of action of zonisamide. Clin Neuropharmacol. 2007;30:230-240.
30. Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861-9866.
31. Wickenden AD, Yu W, Zou A, et al. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591-600.
32. Tatulian L, Brown DA. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol. 2003;549:57-63.
33. Schenzer A, Friedrich T, Pusch M, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051-5060.
34. Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25-29.
35. Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403-406.
36. Leresche N, Parri HR, Erdemli G, et al. On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. J Neurosci. 1998;18:4842-4853.
37. Song I, Kim D, Choi S, et al. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004;24:5249-5257.
38. Shin HS. T-type Ca2+ channels and absence epilepsy. Cell Calcium. 2006;40:191-196.
39. McCormick DA, Contreras D. On the cellular and network bases of epileptic seizures. Annu Rev Physiol. 2001;63:815-846.
40. Rho JM, Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia. 1999;40:1471-1483.
41. Thiry A, Dogne JM, Supuran CT, et al. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem. 2007;7:855-864.
42. Mattson RH, Cramer JA, Collins JF, et al. Comparison of carbamazepine, phenobarbital, phenytoin and primidone in partial and secondarily generalized tonic-clonic seizures. N Engl J Med. 1985;313:145-151.
43. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314-319.
44. Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs. 2006;66:1817-1829.
45. Patsalos PN, Froscher W, Pisani F, et al. The importance of drug interactions in epilepsy therapy. Epilepsia. 2002;43:365-385.
46. Jonker DM, Voskuyl RA, Danhof M. Synergistic combinations of anticonvulsant agents: what is the evidence from animal experiments? Epilepsia. 2007;48:412-434.
47. Luszczki JJ, Czuczwar M, Kis J, et al. Interactions of lamotrigine with topiramate and first-generation antiepileptic drugs in the maximal electroshock test in mice: an isobolographic analysis. Epilepsia. 2003;44:1003-1013.
48. Luszczki JJ, Andres MM, Czuczwar P, et al. Pharmacodynamic and pharmacokinetic characterization of interactions between levetiracetam and numerous antiepileptic drugs in the mouse maximal electroshock seizure model: an isobolographic analysis. Epilepsia. 2006;47:10-20.
49. Borowicz KK, Swiader M, Luszczki J, et al. Effect of gabapentin on the anticonvulsant activity of antiepileptic drugs against electroconvulsions in mice: an isobolographic analysis. Epilepsia. 2002;43:956-963.
50. Borowicz KK, Luszczki JJ, Czuczwar SJ. Isobolographic and subthreshold analysis of interactions among felbamate and four conventional antiepileptic drugs in pentylenetetrazole-induced seizures in mice. Epilepsia. 2004;45:1176-1183.
51. Jonker DM, Voskuyl RA, Danhof M. Pharmacodynamic analysis of the anticonvulsant effects of tiagabine and lamotrigine in combination in the rat. Epilepsia. 2004;45:424-435.
52. Genton P. When antiepileptic drugs aggravate epilepsy. Brain Dev. 2000;22:75-80.
53. Hirsch E, Genton P. Antiepileptic drug-induced pharmacodynamic aggravation of seizures: does valproate have a lower potential? CNS Drugs. 2003;17:633-640.
54. Pisani F, Perucca E, Di Perri R. Clinically relevant anti-epileptic drug interactions. J Int Med Res. 1990;18:1-15.
55. Brodie MJ, Yuen AWC, Group S. Lamotrigine substitution study: evidence for synergism with sodium valproate. Epilepsy Res. 1997;26:423-432.
56. Pisani F, Oteri G, Russo MF, et al. The efficacy of valproate-lamotrigine comedication in refractory complex partial seizures: evidence for a pharmacodynamic interaction. Epilepsia. 1999;40:1141-1146.
57. Stephen LJ, Sills GJ, Brodie MJ. Lamotrigine and topiramate may be a useful combination. Lancet. 1998;351:958-963.
58. Anderson G. A mechanistic approach to antiepileptic drug interactions. Ann Pharmacoth. 1998;32:554-563.
59. Vermeulen J, Aldenkamp A. Cognitive side-effects of chronic antiepileptic drug treatment: a review of 25 years of research. Epilepsy Res. 1995;22:65-95.
60. Baker GA, Jacoby A, Buck D, et al. Quality of life of people with epilepsy: a European study. Epilepsia. 1997;38:353-362.
61. Trimble MR. Anticonvulsant drugs and cognitive function: a review of the literature. Epilepsia. 1987;28(Suppl 3):S37-S45.
62. Brunbech L, Sabers A. Effect of antiepileptic drugs on cognitive function in individuals with epilepsy: a comparative review of newer versus older agents. Drugs. 2002;62:593-604.
63. Meador KJ. Current discoveries on the cognitive effects of antiepileptic drugs. Pharmacotherapy. 2000;20:185S-190S.
64. Aldenkamp AP, De Krom M, Reijs R. Newer antiepileptic drugs and cognitive issues. Epilepsia. 44(Suppl 4):21-29.
65. Kaneko S, Kondo T. Antiepileptic agents and birth defects. Incidence, mechanisms and prevention. CNS Drugs. 1995;3:41-55.
66. Kaneko S, Battino D, Andermann E, et al. Congenital malformations due to antiepileptic drugs. Epilepsy Res. 1999;33:145-158.
67. Nakane Y, Okuma T, Takahashi R, et al. Multi-institutional study on the teratogenicity and fetal toxicity of antiepileptic drugs: a report of a collaborative study group in Japan. Epilepsia. 1980;21:663-680.
68. Holmes LB, Harvey EA, Coull BA, et al. The teratogenicity of anticonvulsant drugs. N Engl J Med. 2001;344:1132-1138.
69. Morrow J, Russell A, Guthrie E, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. J Neurol Neurosurg Psychiatry. 2006;77:193-198.
70. Cunnington M, Tennis P. Lamotrigine and the risk of malformations in pregnancy. Neurology. 2005;64:955-960.
71. Wyszynski DF, Nambisan M, Surve T, et al. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64:961-965.
72. Finnell RH, Buehler BA, Kerr BM, et al. Clinical and experimental studies linking oxidative metabolism to phenytoin-induced teratogenesis. Neurology. 1992;42(Suppl.5):25-31.
73. Raymond GV, Buehler BA, Holmes LB. Placental epoxide hydrolase activity: correlation with features of fetal anticonvulsant syndrome. Teratology Abstracts. 1992;45:461.
74. Kerr BM, Rettie AE, Eddy AC, et al. Inhibition of human liver microsomal epoxide hydrolase by valproate and valpromide: in vitro/in vivo correlation. Clin Pharmacol Ther. 1989;46:82-93.
75. Dansky LV, Rosenblatt DS, Andermann E. Mechanisms of teratogenesis: folic acid and antiepileptic therapy. Neurology. 1992;42(Suppl.5):32-42.
76. Hiilesmaa VK, Teramo K, Granstrom ML, et al. Serum folate concentrations during pregnancy in women with epilepsy: relation to antiepileptic drug concentrations, number of seizures, and fetal outcome. Br Med J (Clin Res Ed). 1983;287:577-579.
77. Warner T, Patsalos PN, Prevett M, et al. Lamotrigine-induced carbamazepine toxicity: an interaction with carbamazepine-10,11-epoxide. Epilepsy Res. 1992;11:147-150.
78. Wolf P. Lamotrigine: preliminary clinical observations on pharmacokinetics and interactions with traditional antiepileptic drugs. J Epilepsy. 1991;5:73-79.
79. Gidal BE, Rutecki P, Shaw R, et al. Effect of lamotrigine on carbamazepine epoxide/carbamazepine serum concentration ratios in adult patients with epilepsy. Epilepsy Res. 1997;28:207-211.
80. Besag FM, Berry DJ, Pool F, et al. Carbamazepine toxicity with lamotrigine: pharmacokinetic or pharmacodynamic interaction? Epilepsia. 1998;39:183-187.
81. Eriksson AS, Boreus LO. No increase in carbamazepine-10,11-epoxide during addition of lamotrigine treatment in children. Ther Drug Monit. 1997;19:499-501.
82. Pisani F, Xiao B, Fazio A, et al. Single dose pharmacokinetics of carbamazepine-10,11-epoxide in patients on lamotrigine monotherapy. Epilepsy Res. 1994;19:245-248.
83. Sabers A, Gram L. Newer anticonvulsants: comparative review of drug interactions and adverse effects. Drugs. 2000;60:23-33.
84. Theis JG, Sidhu J, Palmer J, et al. Lack of pharmacokinetic interaction between oxcarbazepine and lamotrigine. Neuropsychopharmacology. 2005;30:2269-2274.
85. Marciani MG, Stanzione P, Mattia D, et al. Lamotrigine add-on therapy in focal epilepsy: electroencephalographic and neuropsychological evaluation. Clin Neuropharmacol. 1998;21:41-47.
86. Guberman AH, Besag FM, Brodie MJ, et al. Lamotrigine-associated rash: risk/benefit considerations in adults and children. Epilepsia. 1999;40:985-991.
87. Schlienger RG, Knowles SR, Shear NH. Lamotrigine-associated anticonvulsant hypersensitivity syndrome. Neurology. 1998;51:1172-1175.
88. Maggs JL, Nasibitt DJ, Tettey JNA, et al. Metabolism of lamotrigine to a reactive arene oxide intermediate. Chem Resarch Toxicol. 2000;13:1075-1081.
89. Schaub N, Bircher AJ. Severe hypersensitivity syndrome to lamotrigine confirmed by lymphocyte stimulation in vitro. Allergy. 2000;55:191-193.
90. Shear NH, Spielberg SP. Anticonvulsant hypersensitivity syndrome: in vitro assessment of risk. J Clin Invest. 1988;82:1826-1832.
91. Arevalo-Lorido JC, Carretero-Gomez J, Bureo-Dacal JC, et al. Antiepileptic drug hypersensitivity syndrome in a patient treated with valproate. Br J Clin Pharmacol. 2003;55:415-416.
92. Sisodiya SM, Sander JW, Patsalos PN. Carbamazepine toxicity during combination therapy with levetiracetam: a pharmacodynamic interaction. Epilepsy Res. 2002;48:217-219.
93. Glauser TA, Pellock JM, Bebin EM, et al. Efficacy and safety of levetiracetam in children with partial seizures: an open-label trial. Epilepsia. 2002;43:518-524.
94. Luszczki JJ, Andres MM, Czuczwar P, et al. Levetiracetam selectively potentiates the acute neurotoxic effects of topiramate and carbamazepine in the rotarod test in mice. Eur Neuropsychopharmacol. 2005;15:609-616.
95. French J, Edrich P, Cramer JA. A systematic review of the safety profile of levetiracetam: a new antiepileptic drug. Epilepsy Res. 2001;47:77-90.
96. Mula M, Trimble MR, Yuen A, et al. Psychiatric adverse events during levetiracetam therapy. Neurology. 2003;61:704-706.
97. Mula M, Trimble MR, Lhatoo SD, et al. Topiramate and psychiatric adverse events in patients with epilepsy. Epilepsia. 2003;44:659-663.
98. Eadie MJ, Hooper WD, Dickinson RG. Valproate-associated hepatotoxicity and its biochemical mechanisms. Med Toxicol Adverse Drug Exp. 1988;3:85-106.
99. Dreifuss FE, Santilli N, Langer DH, et al. Valproic acid hepatic fatalities: a retrospective review. Neurology. 1987;37:379-385.
100. Appleton RE, Farrell K, Applegarth DA, et al. The high incidence of valproate hepatotoxicity in infants may relate to familial metabolic defects. Can J Neurol Sci. 1990;17:145-148.
101. Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101-154.
102. Chang TK, Abbott FS. Oxidative stress as a mechanism of valproic acid-associated hepatotoxicity. Drug Metab Rev. 2006;38:627-639.
103. Kesterson JW, Granneman GR, Machinist JM. The hepatotoxicity of valproic acid and its metabolites in rats. I. Toxicologic, biochemical and histopathologic studies. Hepatology. 1984;4:1143-1152.
104. Shen DD, Pollack GM, Cohen ME, et al. Effect of age on the serum metabolite pattern of valproic acid in epileptic children (abstract). Epilepsia. 1984;5:674.
105. Levy RH, Rettenmeier AW, Anderson GD, et al. Effects of polytherapy with phenytoin, carbamazepine, and stiripentol on formation of 4-ene-valproate, a hepatotoxic metabolite of valproic acid. Clin Pharmacol Ther. 1990;48:225-235.
106. Gopaul S, Farrell K, Abbott F. Effects of age and polytherapy, risk factors of valproic acid (VPA) hepatotoxicity, on the excretion of thiol conjugates of (E)-2,4-diene VPA in people with epilepsy taking VPA. Epilepsia. 2003;44:322-328.
107. Michoulas A, Tong V, Teng XW, et al. Oxidative stress in children receiving valproic acid. J Pediatr. 2006;149:692-696.
108. Tong V, Chang TK, Chen J, et al. The effect of valproic acid on hepatic and plasma levels of 15-F2t-isoprostane in rats. Free Radic Biol Med. 2003;34:1435-1446.
109. Chicharro AV, de Marinis AJ, Kanner AM. The measurement of ammonia blood levels in patients taking valproic acid: looking for problems where they do not exist? Epilepsy Behav. 2008;12(3):497-498.
110. Zaccara G, Paganini M, Campostrini R, et al. Effect of associated antiepileptic treatment on valproate-induced hyperammonemia. Ther Drug Monit. 1985;7:185-190.
111. Longin E, Teich M, Koelfen W, et al. Topiramate enhances the risk of valproate-associated side effects in three children. Epilepsia. 2002;43:451-454.
112. Hamer HM, Knake S, Schomburg U, et al. Valproate-induced hyperammonemic encephalopathy in the presence of topiramate. Neurology. 2000;54:230-232.
113. Cheung E, Wong V, Fung CW. Topiramate-valproate-induced hyperammonemic encephalopathy syndrome: case report. J Child Neurol. 2005;20:157-160.
114. Latour P, Biraben A, Polard E, et al. Drug induced encephalopathy in six epileptic patients: topiramate? valproate? or both? Hum Psychopharmacol. 2004;19:193-203.
115. Ferrendelli JA. Relating pharmacology to clinical practice: the pharmacologic basis of rational polypharmacy. Neurology. 1995;45:S12-S16.
116. Ferrendelli JA. Rational polypharmacy. Epilepsia. 1995;36(Suppl 2):S115-S118.
117. Deckers CL. Place of polytherapy in the early treatment of epilepsy. CNS Drugs. 2002;16:155-163.
118. Deckers CL, Czuczwar SJ, Hekster YA, et al. Selection of antiepileptic drug polytherapy based on mechanisms of action: the evidence reviewed. Epilepsia. 2000;41:1364-1374.






