Risk factors for the development of colorectal cancer are listed in Table 110-1.
|
RISK FACTORS FOR THE DEVELOPMENT OF COLORECTAL CANCER |
Diet The etiology for most cases of large-bowel cancer appears to be related to environmental factors. The disease occurs more often in upper socioeconomic populations who live in urban areas. Mortality from colorectal cancer is directly correlated with per capita consumption of calories, meat protein, and dietary fat and oil as well as elevations in the serum cholesterol concentration and mortality from coronary artery disease. Geographic variations in incidence largely are unrelated to genetic differences, since migrant groups tend to assume the large-bowel cancer incidence rates of their adopted countries. Furthermore, population groups such as Mormons and Seventh Day Adventists, whose lifestyle and dietary habits differ somewhat from those of their neighbors, have significantly lower-than-expected incidence and mortality rates for colorectal cancer. The incidence of colorectal cancer has increased in Japan since that nation has adopted a more “Western” diet. At least three hypotheses have been proposed to explain the relationship to diet, none of which is fully satisfactory.
ANIMAL FATS One hypothesis is that the ingestion of animal fats found in red meats and processed meat leads to an increased proportion of anaerobes in the gut microflora, resulting in the conversion of normal bile acids into carcinogens. This provocative hypothesis is supported by several reports of increased amounts of fecal anaerobes in the stools of patients with colorectal cancer. Diets high in animal (but not vegetable) fats are also associated with high serum cholesterol, which is also associated with enhanced risk for the development of colorectal adenomas and carcinomas.
INSULIN RESISTANCE The large number of calories in Western diets coupled with physical inactivity has been associated with a higher prevalence of obesity. Obese persons develop insulin resistance with increased circulating levels of insulin, leading to higher circulating concentrations of insulin-like growth factor type I (IGF-I). This growth factor appears to stimulate proliferation of the intestinal mucosa.
FIBER Contrary to prior beliefs, the results of randomized trials and case-controlled studies have failed to show any value for dietary fiber or diets high in fruits and vegetables in preventing the recurrence of colorectal adenomas or the development of colorectal cancer.
The weight of epidemiologic evidence, however, implicates diet as being the major etiologic factor for colorectal cancer, particularly diets high in animal fat and in calories.
HEREDITARY FACTORS AND SYNDROMES
Up to 25% of patients with colorectal cancer have a family history of the disease, suggesting a hereditary predisposition. Inherited large-bowel cancers can be divided into two main groups: the well-studied but uncommon polyposis syndromes and the more common nonpolyposis syndromes (Table 110-2).
|
HEREDITABLE (AUTOSOMAL DOMINANT) GASTROINTESTINAL POLYPOSIS SYNDROMES |
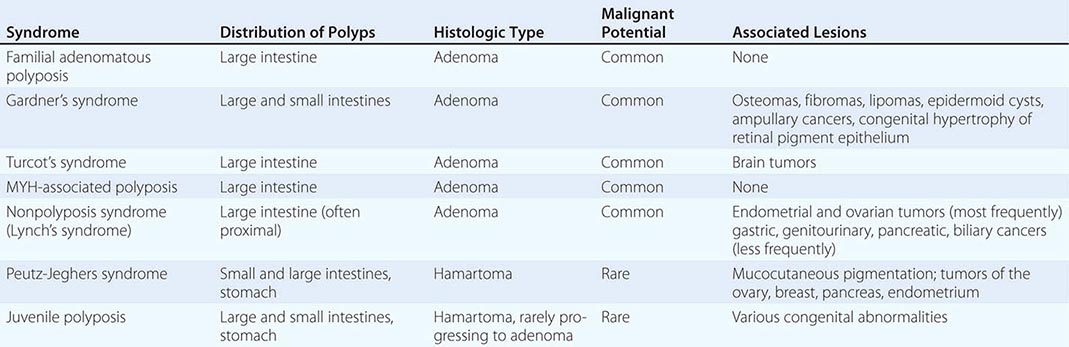
Polyposis Coli Polyposis coli (familial polyposis of the colon) is a rare condition characterized by the appearance of thousands of adenomatous polyps throughout the large bowel. It is transmitted as an autosomal dominant trait; the occasional patient with no family history probably developed the condition due to a spontaneous mutation. Polyposis coli is associated with a deletion in the long arm of chromosome 5 (including the APC gene) in both neoplastic (somatic mutation) and normal (germline mutation) cells. The loss of this genetic material (i.e., allelic loss) results in the absence of tumor-suppressor genes whose protein products would normally inhibit neoplastic growth. The presence of soft tissue and bony tumors, congenital hypertrophy of the retinal pigment epithelium, mesenteric desmoid tumors, and ampullary cancers in addition to the colonic polyps characterizes a subset of polyposis coli known as Gardner’s syndrome. The appearance of malignant tumors of the central nervous system accompanying polyposis coli defines Turcot’s syndrome. The colonic polyps in all these conditions are rarely present before puberty but are generally evident in affected individuals by age 25. If the polyposis is not treated surgically, colorectal cancer will develop in almost all patients before age 40. Polyposis coli results from a defect in the colonic mucosa, leading to an abnormal proliferative pattern and impaired DNA repair mechanisms. Once the multiple polyps are detected, patients should undergo a total colectomy. Medical therapy with nonsteroidal anti-inflammatory drugs (NSAIDs) such as sulindac and selective cyclooxygenase-2 inhibitors such as celecoxib can decrease the number and size of polyps in patients with polyposis coli; however, this effect on polyps is only temporary, and the use of NSAIDs has not been shown to reduce the risk of cancer. Colectomy remains the primary therapy/prevention. The offspring of patients with polyposis coli, who often are prepubertal when the diagnosis is made in the parent, have a 50% risk for developing this premalignant disorder and should be carefully screened by annual flexible sigmoidoscopy until age 35. Proctosigmoidoscopy is a sufficient screening procedure because polyps tend to be evenly distributed from cecum to anus, making more invasive and expensive techniques such as colonoscopy or barium enema unnecessary. Testing for occult blood in the stool is an inadequate screening maneuver. If a causative germline AP C mutation has been identified in an affected family member, an alternative method for identifying carriers is testing DNA from peripheral blood mononuclear cells for the presence of the specific APC mutation. The detection of such a germline mutation can lead to a definitive diagnosis before the development of polyps.
MYH-Associated Polyposis MYH-associated polyposis (MAP) is a rare autosomal recessive syndrome caused by a biallelic mutation in the MUT4H gene. This hereditary condition may have a variable clinical presentation, resembling polyposis coli or colorectal cancer occurring in younger individuals without polyposis. Screening and colectomy guidelines for this syndrome are less clear than for polyposis coli, but annual to biennial colonoscopic surveillance is generally recommended starting at age 25–30.
Hereditary Nonpolyposis Colon Cancer Hereditary nonpolyposis colon cancer (HNPCC), also known as Lynch’s syndrome, is another autosomal dominant trait. It is characterized by the presence of three or more relatives with histologically documented colorectal cancer, one of whom is a first-degree relative of the other two; one or more cases of colorectal cancer diagnosed before age 50 in the family; and colorectal cancer involving at least two generations. In contrast to polyposis coli, HNPCC is associated with an unusually high frequency of cancer arising in the proximal large bowel. The median age for the appearance of an adenocarcinoma is <50 years, 10–15 years younger than the median age for the general population. Despite having a poorly differentiated, mucinous histologic appearance, the proximal colon tumors that characterize HNPCC have a better prognosis than sporadic tumors from patients of similar age. Families with HNPCC often include individuals with multiple primary cancers; the association of colorectal cancer with either ovarian or endometrial carcinomas is especially strong in women, and an increased appearance of gastric, small-bowel, genitourinary, pancreaticobiliary, and sebaceous skin tumors has been reported as well. It has been recommended that members of such families undergo annual or biennial colonoscopy beginning at age 25 years, with intermittent pelvic ultrasonography and endometrial biopsy for afflicted women; such a screening strategy has not yet been validated. HNPCC is associated with germline mutations of several genes, particularly hMSH2 on chromosome 2 and hMLH1 on chromosome 3. These mutations lead to errors in DNA replication and are thought to result in DNA instability because of defective repair of DNA mismatches resulting in abnormal cell growth and tumor development. Testing tumor cells through molecular analysis of DNA or immunohistochemical staining of paraffin-fixed tissue for “microsatellite instability” (sequence changes reflecting defective mismatch repair) in patients with colorectal cancer and a positive family history for colorectal or endometrial cancer may identify probands with HNPCC.
INFLAMMATORY BOWEL DISEASE
(Chap. 351) Large-bowel cancer is increased in incidence in patients with long-standing inflammatory bowel disease (IBD). Cancers develop more commonly in patients with ulcerative colitis than in those with granulomatous (i.e., Crohn’s) colitis, but this impression may result in part from the occasional difficulty of differentiating these two conditions. The risk of colorectal cancer in a patient with IBD is relatively small during the initial 10 years of the disease, but then appears to increase at a rate of ∼0.5–1% per year. Cancer may develop in 8–30% of patients after 25 years. The risk is higher in younger patients with pancolitis.
Cancer surveillance strategies in patients with IBD are unsatisfactory. Symptoms such as bloody diarrhea, abdominal cramping, and obstruction, which may signal the appearance of a tumor, are similar to the complaints caused by a flare-up of the underlying disease. In patients with a history of IBD lasting ≥15 years who continue to experience exacerbations, the surgical removal of the colon can significantly reduce the risk for cancer and also eliminate the target organ for the underlying chronic gastrointestinal disorder. The value of such surveillance techniques as colonoscopy with mucosal biopsies and brushings for less symptomatic individuals with chronic IBD is uncertain. The lack of uniformity regarding the pathologic criteria that characterize dysplasia and the absence of data that such surveillance reduces the development of lethal cancers have made this costly practice an area of controversy.
OTHER HIGH-RISK CONDITIONS
Streptococcus bovis Bacteremia For unknown reasons, individuals who develop endocarditis or septicemia from this fecal bacterium have a high incidence of occult colorectal tumors and, possibly, upper gastrointestinal cancers as well. Endoscopic or radiographic screening appears advisable.
Tobacco Use Cigarette smoking is linked to the development of colorectal adenomas, particularly after >35 years of tobacco use. No biologic explanation for this association has yet been proposed.
PRIMARY PREVENTION
Several orally administered compounds have been assessed as possible inhibitors of colon cancer. The most effective class of chemopreventive agents is aspirin and other NSAIDs, which are thought to suppress cell proliferation by inhibiting prostaglandin synthesis. Regular aspirin use reduces the risk of colon adenomas and carcinomas as well as death from large-bowel cancer; such use also appears to diminish the likelihood for developing additional premalignant adenomas following successful treatment for a prior colon carcinoma. This effect of aspirin on colon carcinogenesis increases with the duration and dosage of drug use. Oral folic acid supplements and oral calcium supplements appear to reduce the risk of adenomatous polyps and colorectal cancers in case-controlled studies. The value of vitamin D as a form of chemoprevention is under study. Antioxidant vitamins such as ascorbic acid, tocopherols, and β-carotene are ineffective at reducing the incidence of subsequent adenomas in patients who have undergone the removal of a colon adenoma. Estrogen replacement therapy has been associated with a reduction in the risk of colorectal cancer in women, conceivably by an effect on bile acid synthesis and composition or by decreasing synthesis of IGF-I.
SCREENING
The rationale for colorectal cancer screening programs is that the removal of adenomatous polyps will prevent colorectal cancer, and that earlier detection of localized, superficial cancers in asymptomatic individuals will increase the surgical cure rate. Such screening programs are particularly important for individuals with a family history of the disease in first-degree relatives. The relative risk for developing colorectal cancer increases to 1.75 in such individuals and may be even higher if the relative was afflicted before age 60. The prior use of proctosigmoidoscopy as a screening tool was based on the observation that 60% of early lesions are located in the rectosigmoid. For unexplained reasons, however, the proportion of large-bowel cancers arising in the rectum has been decreasing during the past several decades, with a corresponding increase in the proportion of cancers in the more proximal descending colon. As such, the potential for proctosigmoidoscopy to detect a sufficient number of occult neoplasms to make the procedure cost-effective has been questioned.
Screening strategies for colorectal cancer that have been examined during the past several decades are listed in Table 110-3.
|
SCREENING STRATEGIES FOR COLORECTAL CANCER |
Many programs directed at the early detection of colorectal cancers have focused on digital rectal examinations and fecal occult blood (i.e., stool guaiac) testing. The digital examination should be part of any routine physical evaluation in adults older than age 40 years, serving as a screening test for prostate cancer in men, a component of the pelvic examination in women, and an inexpensive maneuver for the detection of masses in the rectum. However, because of the proximal migration of colorectal tumors, its value as an overall screening modality for colorectal cancer has become limited. The development of the fecal occult blood test has greatly facilitated the detection of occult fecal blood. Unfortunately, even when performed optimally, the fecal occult blood test has major limitations as a screening technique. About 50% of patients with documented colorectal cancers have a negative fecal occult blood test, consistent with the intermittent bleeding pattern of these tumors. When random cohorts of asymptomatic persons have been tested, 2–4% have fecal occult blood-positive stools. Colorectal cancers have been found in <10% of these “test-positive” cases, with benign polyps being detected in an additional 20–30%. Thus, a colorectal neoplasm will not be found in most asymptomatic individuals with occult blood in their stool. Nonetheless, persons found to have fecal occult blood-positive stool routinely undergo further medical evaluation, including sigmoidoscopy and/or colonoscopy—procedures that are not only uncomfortable and expensive but also associated with a small risk for significant complications. The added cost of these studies would appear justifiable if the small number of patients found to have occult neoplasms because of fecal occult blood screening could be shown to have an improved prognosis and prolonged survival. Prospectively controlled trials have shown a statistically significant reduction in mortality rate from colorectal cancer for individuals undergoing annual stool guaiac screening. However, this benefit only emerged after >13 years of follow-up and was extremely expensive to achieve, because all positive tests (most of which were falsely positive) were followed by colonoscopy. Moreover, these colonoscopic examinations quite likely provided the opportunity for cancer prevention through the removal of potentially premalignant adenomatous polyps because the eventual development of cancer was reduced by 20% in the cohort undergoing annual screening.
With the appreciation that the carcinogenic process leading to the progression of the normal bowel mucosa to an adenomatous polyp and then to a cancer is the result of a series of molecular changes, investigators have examined fecal DNA for evidence of mutations associated with such molecular changes as evidence of the occult presence of precancerous lesions or actual malignancies. Such a strategy has been tested in more than 4000 asymptomatic individuals whose stool was assessed for occult blood and for 21 possible mutations in fecal DNA; these study subjects also underwent colonoscopy. Although the fecal DNA strategy suggested the presence of more advanced adenomas and cancers than did the fecal occult blood testing approach, the overall sensitivity, using colonoscopic findings as the standard, was less than 50%, diminishing enthusiasm for further pursuit of the fecal DNA screening strategy.
The use of imaging studies to screen for colorectal cancers has also been explored. Air contrast barium enemas had been used to identify sources of occult blood in the stool prior to the advent of fiberoptic endoscopy; the cumbersome nature of the procedure and inconvenience to patients limited its widespread adoption. The introduction of computed tomography (CT) scanning led to the development of virtual (i.e., CT) colonography as an alternative to the growing use of endoscopic screening techniques. Virtual colonography was proposed as being equivalent in sensitivity to colonoscopy and being available in a more widespread manner because it did not require the same degree of operator expertise as fiberoptic endoscopy. However, virtual colonography requires the same cathartic preparation that has limited widespread acceptance of endoscopic colonoscopy, is diagnostic but not therapeutic (i.e., patients with suspicious findings must undergo a subsequent endoscopic procedure for polypectomy or biopsy), and, in the setting of general radiology practices, appears to be less sensitive as a screening technique when compared with endoscopic procedures.
With the appreciation of the inadequacy of fecal occult blood testing alone, concerns about the practicality of imaging approaches, and the wider adoption of endoscopic examinations by the primary care community, screening strategies in asymptomatic persons have changed. At present, both the American Cancer Society and the National Comprehensive Cancer Network suggest either fecal occult blood testing annually coupled with flexible sigmoidoscopy every 5 years or colonoscopy every 10 years beginning at age 50 in asymptomatic individuals with no personal or family history of polyps or colorectal cancer. The recommendation for the inclusion of flexible sigmoidoscopy is strongly supported by the recently published results of three randomized trials performed in the United States, the United Kingdom, and Italy, involving more than 350,000 individuals, which consistently showed that periodic (even single) sigmoidoscopic examinations, after more than a decade of median follow-up, lead to an approximate 21% reduction in the development of colorectal cancer and a more than 25% reduction in mortality from the malignant disease. Less than 20% of participants in these studies underwent a subsequent colonoscopy. In contrast to the cathartic preparation required before colonoscopic procedures, which is only performed by highly trained specialists, flexible sigmoidoscopy requires only an enema as preparation and can be accurately performed by nonspecialty physicians or physician-extenders. The randomized screening studies using flexible sigmoidoscopy led to the estimate that approximately 650 individuals needed to be screened to prevent one colorectal cancer death; this contrasts with the data for mammography where the number of women needing to be screened to prevent one breast cancer death is 2500, reinforcing the efficacy of endoscopic surveillance for colorectal cancer screening. Presumably the benefit from the sigmoidoscopic screening is the result of the identification and removal of adenomatous polyps; it is intriguing that this benefit has been achieved using a technique that leaves the proximal half of the large bowel unvisualized.
It remains to be seen whether surveillance colonoscopy, which has gained increasing popularity in the United States for colorectal cancer screening, will prove to be more effective than flexible sigmoidoscopy. Ongoing randomized trials being conducted in Europe are addressing this issue. Although flexible sigmoidoscopy only visualizes the distal half of the large bowel, leading to the assumption that colonoscopy represents a more informative approach, colonoscopy has been reported as being less accurate for screening the proximal rather than the distal colon, perhaps due to technical considerations but also possibly because of a greater frequency of serrated (i.e., “flat”) polyps in the right colon, which are more difficult to identify. At present, colonoscopy performed every 10 years has been offered as an alternative to annual fecal occult blood testing with periodic (every 5 years) flexible sigmoidoscopy. Colonoscopy has been shown to be superior to double-contract barium enema and also to have a higher sensitivity for detecting villous or dysplastic adenomas or cancers than the strategy using occult fecal blood testing and flexible sigmoidoscopy. Whether colonoscopy performed every 10 years beginning at age 50 is medically superior and economically equivalent to flexible sigmoidoscopy remains to be determined.
CLINICAL FEATURES
Presenting Symptoms Symptoms vary with the anatomic location of the tumor. Because stool is relatively liquid as it passes through the ileocecal valve into the right colon, cancers arising in the cecum and ascending colon may become quite large without resulting in any obstructive symptoms or noticeable alterations in bowel habits. Lesions of the right colon commonly ulcerate, leading to chronic, insidious blood loss without a change in the appearance of the stool. Consequently, patients with tumors of the ascending colon often present with symptoms such as fatigue, palpitations, and even angina pectoris and are found to have a hypochromic, microcytic anemia indicative of iron deficiency. Because the cancers may bleed intermittently, a random fecal occult blood test may be negative. As a result, the unexplained presence of iron-deficiency anemia in any adult (with the possible exception of a premenopausal, multiparous woman) mandates a thorough endoscopic and/or radiographic visualization of the entire large bowel (Fig. 110-1).

FIGURE 110-1 Double-contrast air-barium enema revealing a sessile tumor of the cecum in a patient with iron-deficiency anemia and guaiac-positive stool. The lesion at surgery was a stage II adenocarcinoma.
Because stool becomes more formed as it passes into the transverse and descending colon, tumors arising there tend to impede the passage of stool, resulting in the development of abdominal cramping, occasional obstruction, and even perforation. Radiographs of the abdomen often reveal characteristic annular, constricting lesions (“apple-core” or “napkin-ring”) (Fig. 110-2).
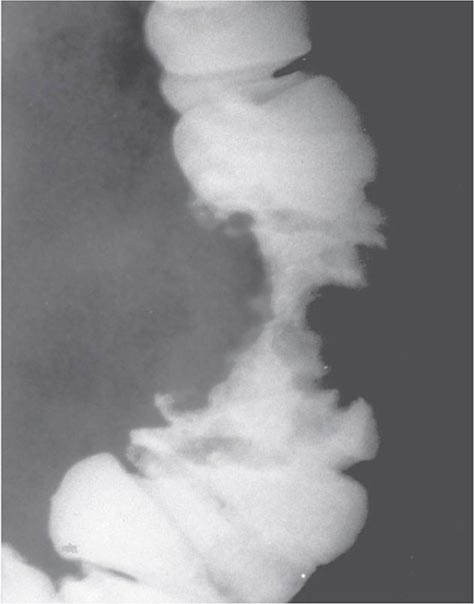
FIGURE 110-2 Annular, constricting adenocarcinoma of the descending colon. This radiographic appearance is referred to as an “apple-core” lesion and is always highly suggestive of malignancy.
Cancers arising in the rectosigmoid are often associated with hematochezia, tenesmus, and narrowing of the caliber of stool; anemia is an infrequent finding. While these symptoms may lead patients and their physicians to suspect the presence of hemorrhoids, the development of rectal bleeding and/or altered bowel habits demands a prompt digital rectal examination and proctosigmoidoscopy.
Staging, Prognostic Factors, and Patterns of Spread The prognosis for individuals having colorectal cancer is related to the depth of tumor penetration into the bowel wall and the presence of both regional lymph node involvement and distant metastases. These variables are incorporated into the staging system introduced by Dukes and subsequently applied to a TNM classification method, in which T represents the depth of tumor penetration, N the presence of lymph node involvement, and M the presence or absence of distant metastases (Fig. 110-3). Superficial lesions that do not involve regional lymph nodes and do not penetrate through the submucosa (T1) or the muscularis (T2) are designated as stage I (T1–2N0M0) disease; tumors that penetrate through the muscularis but have not spread to lymph nodes are stage II disease (T3-4N0M0); regional lymph node involvement defines stage III (TXN1-2M0) disease; and metastatic spread to sites such as liver, lung, or bone indicates stage IV (TXNXM1) disease. Unless gross evidence of metastatic disease is present, disease stage cannot be determined accurately before surgical resection and pathologic analysis of the operative specimens. It is not clear whether the detection of nodal metastases by special immunohistochemical molecular techniques has the same prognostic implications as disease detected by routine light microscopy.
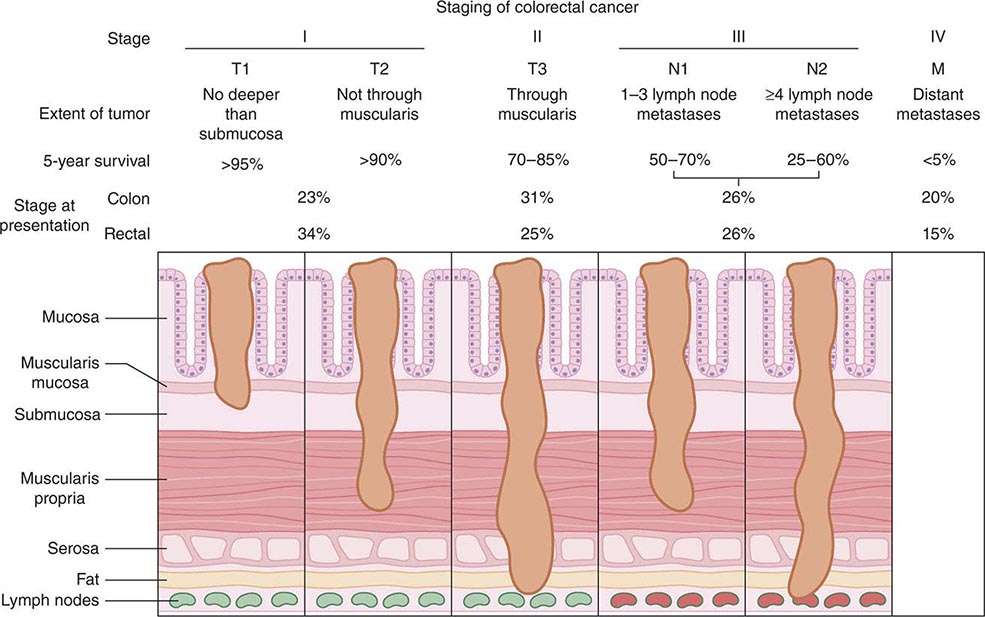
FIGURE 110-3 Staging and prognosis for patients with colorectal cancer.
Most recurrences after a surgical resection of a large-bowel cancer occur within the first 4 years, making 5-year survival a fairly reliable indicator of cure. The likelihood for 5-year survival in patients with colorectal cancer is stage-related (Fig. 110-3). That likelihood has improved during the past several decades when similar surgical stages have been compared. The most plausible explanation for this improvement is more thorough intraoperative and pathologic staging. In particular, more exacting attention to pathologic detail has revealed that the prognosis following the resection of a colorectal cancer is not related merely to the presence or absence of regional lymph node involvement; rather, prognosis may be more precisely gauged by the number of involved lymph nodes (one to three lymph nodes [“N1”] vs four or more lymph nodes [“N2”]) and the number of nodes examined. A minimum of 12 sampled lymph nodes is thought necessary to accurately define tumor stage, and the more nodes examined, the better. Other predictors of a poor prognosis after a total surgical resection include tumor penetration through the bowel wall into pericolic fat, poorly differentiated histology, perforation and/or tumor adherence to adjacent organs (increasing the risk for an anatomically adjacent recurrence), and venous invasion by tumor (Table 110-4). Regardless of the clinicopathologic stage, a preoperative elevation of the plasma carcinoembryonic antigen (CEA) level predicts eventual tumor recurrence. The presence of aneuploidy and specific chromosomal deletions, such as a mutation in the b-raf gene in tumor cells, appears to predict for a higher risk for metastatic spread. Conversely, the detection of microsatellite instability in tumor tissue indicates a more favorable outcome. In contrast to most other cancers, the prognosis in colorectal cancer is not influenced by the size of the primary lesion when adjusted for nodal involvement and histologic differentiation.
|
PREDICTORS OF POOR OUTCOME FOLLOWING TOTAL SURGICAL RESECTION OF COLORECTAL CANCER |
Abbreviation: CEA, carcinoembryonic antigen.
Cancers of the large bowel generally spread to regional lymph nodes or to the liver via the portal venous circulation. The liver represents the most frequent visceral site of metastasis; it is the initial site of distant spread in one-third of recurring colorectal cancers and is involved in more than two-thirds of such patients at the time of death. In general, colorectal cancer rarely spreads to the lungs, supraclavicular lymph nodes, bone, or brain without prior spread to the liver. A major exception to this rule occurs in patients having primary tumors in the distal rectum, from which tumor cells may spread through the paravertebral venous plexus, escaping the portal venous system and thereby reaching the lungs or supraclavicular lymph nodes without hepatic involvement. The median survival after the detection of distant metastases has ranged in the past from 6–9 months (hepatomegaly, abnormal liver chemistries) to 24–30 months (small liver nodule initially identified by elevated CEA level and subsequent CT scan), but effective systemic therapy is significantly improving this prognosis.
Efforts to use gene expression profiles to identify patients at risk of recurrence or those particularly likely to benefit from adjuvant therapy have not yet yielded practice-changing results. Despite a burgeoning literature examining a host of prognostic factors, pathologic stage at diagnosis remains the best predictor of long-term prognosis. Patients with lymphovascular invasion and high preoperative CEA levels are likely to have a more aggressive clinical course.
CANCERS OF THE ANUS
Cancers of the anus account for 1–2% of the malignant tumors of the large bowel. Most such lesions arise in the anal canal, the anatomic area extending from the anorectal ring to a zone approximately halfway between the pectinate (or dentate) line and the anal verge. Carcinomas arising proximal to the pectinate line (i.e., in the transitional zone between the glandular mucosa of the rectum and the squamous epithelium of the distal anus) are known as basaloid, cuboidal, or cloacogenic tumors; about one-third of anal cancers have this histologic pattern. Malignancies arising distal to the pectinate line have squamous histology, ulcerate more frequently, and constitute ∼55% of anal cancers. The prognosis for patients with basaloid and squamous cell cancers of the anus is identical when corrected for tumor size and the presence or absence of nodal spread.
The development of anal cancer is associated with infection by human papillomavirus, the same organism etiologically linked to cervical cancer. The virus is sexually transmitted. The infection may lead to anal warts (condyloma acuminata), which may progress to anal intraepithelial neoplasia and on to squamous cell carcinoma. The risk for anal cancer is increased among homosexual males, presumably related to anal intercourse. Anal cancer risk is increased in both men and women with AIDS, possibly because their immunosuppressed state permits more severe papillomavirus infection. Vaccination against human papilloma viruses may reduce the eventual risk for anal cancer. Anal cancers occur most commonly in middle-aged persons and are more frequent in women than men. At diagnosis, patients may experience bleeding, pain, sensation of a perianal mass, and pruritus.
Radical surgery (abdominal-perineal resection with lymph node sampling and a permanent colostomy) was once the treatment of choice for this tumor type. The 5-year survival rate after such a procedure was 55–70% in the absence of spread to regional lymph nodes and <20% if nodal involvement was present. An alternative therapeutic approach combining external beam radiation therapy with concomitant chemotherapy (5-FU and mitomycin C) has resulted in biopsy-proven disappearance of all tumor in >80% of patients whose initial lesion was <3 cm in size. Tumor recurrences develop in <10% of these patients, meaning that ∼70% of patients with anal cancers can be cured with nonoperative treatment and without the need for a colostomy. Surgery should be reserved for the minority of individuals who are found to have residual tumor after being managed initially with radiation therapy combined with chemotherapy.
111 |
Tumors of the Liver and Biliary Tree |
HEPATOCELLULAR CARCINOMA
INCIDENCE
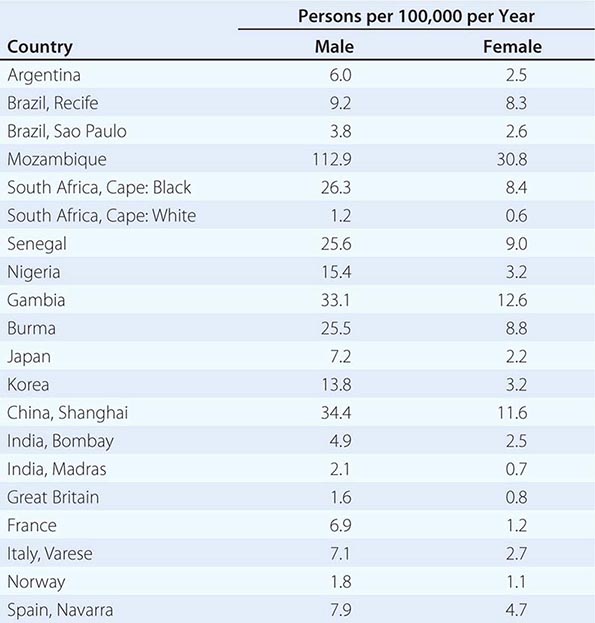
![]() Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. The annual global incidence is approximately 1 million cases, with a male-to-female ratio of approximately 4:1 (1:1 without cirrhosis to 9:1 in many high-incidence countries). The incidence rate equals the death rate. In the United States, approximately 22,000 new cases are diagnosed annually, with 18,000 deaths. The death rates in males in low-incidence countries such as the United States are 1.9 per 100,000 per year; in intermediate areas such as Austria and South Africa, they range from 5.1–20; and in high-incidence areas such as in the Orient (China and Korea), they are as high as 23.1–150 per 100,000 per year (Table 111-1). The incidence of HCC in the United States is approximately 3 per 100,000 persons, with significant gender, ethnic, and geographic variations. These numbers are rapidly increasing and may be an underestimate. Approximately 4 million chronic hepatitis C virus (HCV) carriers are in the United States alone. Approximately 10% of them, or 400,000, are likely to develop cirrhosis. Approximately 5%, or 20,000, of these patients may develop HCC annually. Add to this the two other common predisposing factors—hepatitis B virus (HBV) and chronic alcohol consumption—and 60,000 new HCC cases annually seem possible. Future advances in HCC survival will likely depend in part on immunization strategies for HBV (and HCV) and earlier diagnosis by screening of patients at risk of HCC development.
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. The annual global incidence is approximately 1 million cases, with a male-to-female ratio of approximately 4:1 (1:1 without cirrhosis to 9:1 in many high-incidence countries). The incidence rate equals the death rate. In the United States, approximately 22,000 new cases are diagnosed annually, with 18,000 deaths. The death rates in males in low-incidence countries such as the United States are 1.9 per 100,000 per year; in intermediate areas such as Austria and South Africa, they range from 5.1–20; and in high-incidence areas such as in the Orient (China and Korea), they are as high as 23.1–150 per 100,000 per year (Table 111-1). The incidence of HCC in the United States is approximately 3 per 100,000 persons, with significant gender, ethnic, and geographic variations. These numbers are rapidly increasing and may be an underestimate. Approximately 4 million chronic hepatitis C virus (HCV) carriers are in the United States alone. Approximately 10% of them, or 400,000, are likely to develop cirrhosis. Approximately 5%, or 20,000, of these patients may develop HCC annually. Add to this the two other common predisposing factors—hepatitis B virus (HBV) and chronic alcohol consumption—and 60,000 new HCC cases annually seem possible. Future advances in HCC survival will likely depend in part on immunization strategies for HBV (and HCV) and earlier diagnosis by screening of patients at risk of HCC development.
|
AGE-ADJUSTED INCIDENCE RATES FOR HEPATOCELLULAR CARCINOMA |
Current Directions With the U.S. HCV epidemic, HCC is increasing in most states, and obesity-associated liver disease (nonalcoholic steatohepatitis [NASH]) is increasingly recognized as a cause.
EPIDEMIOLOGY
![]() There are two general types of epidemiologic studies of HCC—those of country-based incidence rates (Table 111-1) and those of migrants. Endemic hot spots occur in areas of China and sub-Saharan Africa, which are associated both with high endemic hepatitis B carrier rates as well as mycotoxin contamination of foodstuffs (aflatoxin B1), stored grains, drinking water, and soil. Environmental factors are important, for example, Japanese in Japan have a higher incidence than Japanese living in Hawaii, who in turn have a higher incidence than those living in California.
There are two general types of epidemiologic studies of HCC—those of country-based incidence rates (Table 111-1) and those of migrants. Endemic hot spots occur in areas of China and sub-Saharan Africa, which are associated both with high endemic hepatitis B carrier rates as well as mycotoxin contamination of foodstuffs (aflatoxin B1), stored grains, drinking water, and soil. Environmental factors are important, for example, Japanese in Japan have a higher incidence than Japanese living in Hawaii, who in turn have a higher incidence than those living in California.
ETIOLOGIC FACTORS
![]() Chemical Carcinogens Causative agents for HCC have been studied along two general lines. First are agents identified as carcinogenic in experimental animals (particularly rodents) that are thought to be present in the human environment (Table 111-2). Second is the association of HCC with various other clinical conditions. Probably the best-studied and most potent ubiquitous natural chemical carcinogen is a product of the Aspergillus fungus, called aflatoxin B1. This mold and aflatoxin product can be found in a variety of stored grains in hot, humid places, where peanuts and rice are stored in unrefrigerated conditions. Aflatoxin contamination of foodstuffs correlates well with incidence rates in Africa and to some extent in China. In endemic areas of China, even farm animals such as ducks have HCC. The most potent carcinogens appear to be natural products of plants, fungi, and bacteria, such as bush trees containing pyrrolizidine alkaloids as well as tannic acid and safrole. Pollutants such as pesticides and insecticides are known rodent carcinogens.
Chemical Carcinogens Causative agents for HCC have been studied along two general lines. First are agents identified as carcinogenic in experimental animals (particularly rodents) that are thought to be present in the human environment (Table 111-2). Second is the association of HCC with various other clinical conditions. Probably the best-studied and most potent ubiquitous natural chemical carcinogen is a product of the Aspergillus fungus, called aflatoxin B1. This mold and aflatoxin product can be found in a variety of stored grains in hot, humid places, where peanuts and rice are stored in unrefrigerated conditions. Aflatoxin contamination of foodstuffs correlates well with incidence rates in Africa and to some extent in China. In endemic areas of China, even farm animals such as ducks have HCC. The most potent carcinogens appear to be natural products of plants, fungi, and bacteria, such as bush trees containing pyrrolizidine alkaloids as well as tannic acid and safrole. Pollutants such as pesticides and insecticides are known rodent carcinogens.
|
FACTORS ASSOCIATED WITH AN INCREASED RISK OF DEVELOPING HEPATOCELLULAR CARCINOMA |
![]() Hepatitis Both case-control and cohort studies have shown a strong association between chronic hepatitis B carrier rates and increased incidence of HCC. In Taiwanese male postal carriers who were hepatitis B surface antigen (HBsAg)-positive, a 98-fold greater risk for HCC was found compared to HBsAg-negative individuals. The incidence of HCC in Alaskan natives is markedly increased related to a high prevalence of HBV infection. HBV-based HCC may involve rounds of hepatic destruction with subsequent proliferation and not necessarily frank cirrhosis. The increase in Japanese HCC incidence rates in the last three decades is thought to be from hepatitis C. A large-scale World Health Organization (WHO)-sponsored intervention study is currently under way in Asia involving HBV vaccination of the newborn. HCC in African blacks is not associated with severe cirrhosis but is poorly differentiated and very aggressive. Despite uniform HBV carrier rates among the South African Bantu, there is a ninefold difference in HCC incidence between Mozambicans living along the coast and inland. These differences are attributed to the additional exposure to dietary aflatoxin B1 and other carcinogenic mycotoxins. A typical interval between HCV-associated transfusion and subsequent HCC is approximately 30 years. HCV-associated HCC patients tend to have more frequent and advanced cirrhosis, but in HBV-associated HCC, only half the patients have cirrhosis, with the remainder having chronic active hepatitis (Chap. 362).
Hepatitis Both case-control and cohort studies have shown a strong association between chronic hepatitis B carrier rates and increased incidence of HCC. In Taiwanese male postal carriers who were hepatitis B surface antigen (HBsAg)-positive, a 98-fold greater risk for HCC was found compared to HBsAg-negative individuals. The incidence of HCC in Alaskan natives is markedly increased related to a high prevalence of HBV infection. HBV-based HCC may involve rounds of hepatic destruction with subsequent proliferation and not necessarily frank cirrhosis. The increase in Japanese HCC incidence rates in the last three decades is thought to be from hepatitis C. A large-scale World Health Organization (WHO)-sponsored intervention study is currently under way in Asia involving HBV vaccination of the newborn. HCC in African blacks is not associated with severe cirrhosis but is poorly differentiated and very aggressive. Despite uniform HBV carrier rates among the South African Bantu, there is a ninefold difference in HCC incidence between Mozambicans living along the coast and inland. These differences are attributed to the additional exposure to dietary aflatoxin B1 and other carcinogenic mycotoxins. A typical interval between HCV-associated transfusion and subsequent HCC is approximately 30 years. HCV-associated HCC patients tend to have more frequent and advanced cirrhosis, but in HBV-associated HCC, only half the patients have cirrhosis, with the remainder having chronic active hepatitis (Chap. 362).
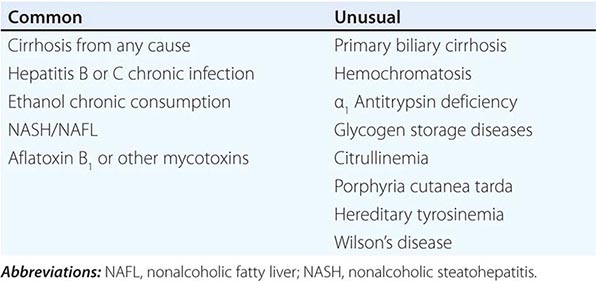
![]() Other Etiologic Conditions The 75–85% association of HCC with underlying cirrhosis has long been recognized, more typically with macronodular cirrhosis in Southeast Asia, but also with micronodular cirrhosis (alcohol) in Europe and the United States (Chap. 365). It is still not clear whether cirrhosis itself is a predisposing factor to the development of HCC or whether the underlying causes of the cirrhosis are actually the carcinogenic factors. However, ∼20% of U.S. patients with HCC do not have underlying cirrhosis. Several underlying conditions are associated with an increased risk for cirrhosis-associated HCC (Table 111-2), including hepatitis, alcohol, autoimmune chronic active hepatitis, cryptogenic cirrhosis, and NASH. A less common association is with primary biliary cirrhosis and several metabolic diseases including hemochromatosis, Wilson’s disease, α1 antitrypsin deficiency, tyrosinemia, porphyria cutanea tarda, glycogenesis types 1 and 3, citrullinemia, and orotic aciduria. The etiology of HCC in those 20% of patients who have no cirrhosis is currently unclear, and their HCC natural history is not well-defined.
Other Etiologic Conditions The 75–85% association of HCC with underlying cirrhosis has long been recognized, more typically with macronodular cirrhosis in Southeast Asia, but also with micronodular cirrhosis (alcohol) in Europe and the United States (Chap. 365). It is still not clear whether cirrhosis itself is a predisposing factor to the development of HCC or whether the underlying causes of the cirrhosis are actually the carcinogenic factors. However, ∼20% of U.S. patients with HCC do not have underlying cirrhosis. Several underlying conditions are associated with an increased risk for cirrhosis-associated HCC (Table 111-2), including hepatitis, alcohol, autoimmune chronic active hepatitis, cryptogenic cirrhosis, and NASH. A less common association is with primary biliary cirrhosis and several metabolic diseases including hemochromatosis, Wilson’s disease, α1 antitrypsin deficiency, tyrosinemia, porphyria cutanea tarda, glycogenesis types 1 and 3, citrullinemia, and orotic aciduria. The etiology of HCC in those 20% of patients who have no cirrhosis is currently unclear, and their HCC natural history is not well-defined.
Current Directions Many patients have multiple etiologies, and the interactions of HBV, HCV, alcohol, smoking, and aflatoxins are just beginning to be explored.
CLINICAL FEATURES
![]() Symptoms These include abdominal pain, weight loss, weakness, abdominal fullness and swelling, jaundice, and nausea (Table 111-3). Presenting signs and symptoms differ somewhat between high- and low-incidence areas. In high-risk areas, especially in South African blacks, the most common symptom is abdominal pain; by contrast, only 40–50% of Chinese and Japanese patients present with abdominal pain. Abdominal swelling may occur as a consequence of ascites due to the underlying chronic liver disease or may be due to a rapidly expanding tumor. Occasionally, central necrosis or acute hemorrhage into the peritoneal cavity leads to death. In countries with an active surveillance program, HCC tends to be identified at an earlier stage, when symptoms may be due only to the underlying disease. Jaundice is usually due to obstruction of the intrahepatic ducts from underlying liver disease. Hematemesis may occur due to esophageal varices from the underlying portal hypertension. Bone pain is seen in 3–12% of patients, but necropsies show pathologic bone metastases in ∼20% of patients. However, 25% of patients may be asymptomatic.
Symptoms These include abdominal pain, weight loss, weakness, abdominal fullness and swelling, jaundice, and nausea (Table 111-3). Presenting signs and symptoms differ somewhat between high- and low-incidence areas. In high-risk areas, especially in South African blacks, the most common symptom is abdominal pain; by contrast, only 40–50% of Chinese and Japanese patients present with abdominal pain. Abdominal swelling may occur as a consequence of ascites due to the underlying chronic liver disease or may be due to a rapidly expanding tumor. Occasionally, central necrosis or acute hemorrhage into the peritoneal cavity leads to death. In countries with an active surveillance program, HCC tends to be identified at an earlier stage, when symptoms may be due only to the underlying disease. Jaundice is usually due to obstruction of the intrahepatic ducts from underlying liver disease. Hematemesis may occur due to esophageal varices from the underlying portal hypertension. Bone pain is seen in 3–12% of patients, but necropsies show pathologic bone metastases in ∼20% of patients. However, 25% of patients may be asymptomatic.
|
HEPATOCELLULAR CARCINOMA CLINICAL PRESENTATION (N = 547) |
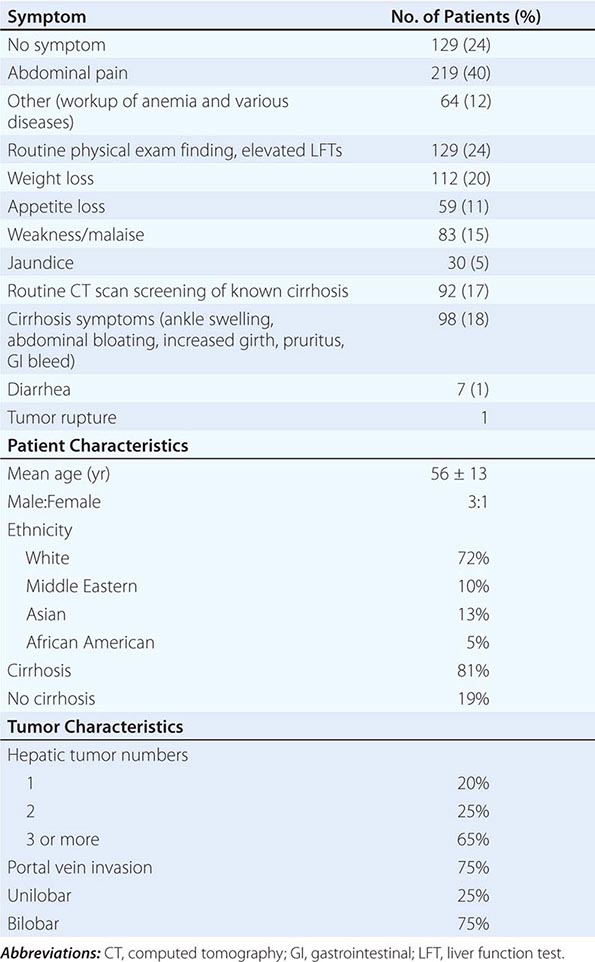
Physical Signs Hepatomegaly is the most common physical sign, occurring in 50–90% of the patients. Abdominal bruits are noted in 6–25%, and ascites occurs in 30–60% of patients. Ascites should be examined by cytology. Splenomegaly is mainly due to portal hypertension. Weight loss and muscle wasting are common, particularly with rapidly growing or large tumors. Fever is found in 10–50% of patients, from unclear cause. The signs of chronic liver disease may often be present, including jaundice, dilated abdominal veins, palmar erythema, gynecomastia, testicular atrophy, and peripheral edema. Budd-Chiari syndrome can occur due to HCC invasion of the hepatic veins, with tense ascites and a large tender liver (Chap. 365).
Paraneoplastic Syndromes Most paraneoplastic syndromes in HCC are biochemical abnormalities without associated clinical consequences. They include hypoglycemia (also caused by end-stage liver failure), erythrocytosis, hypercalcemia, hypercholesterolemia, dysfibrinogenemia, carcinoid syndrome, increased thyroxin-binding globulin, changes in secondary sex characteristics (gynecomastia, testicular atrophy, and precocious puberty), and porphyria cutanea tarda. Mild hypoglycemia occurs in rapidly growing HCC as part of terminal illness, and profound hypoglycemia may occur, although the cause is unclear. Erythrocytosis occurs in 3–12% of patients and hypercholesterolemia in 10–40%. A high percentage of patients have thrombocytopenia associated with their fibrosis or leukopenia, resulting from portal hypertension, and not from cancer infiltration of bone marrow, as in other tumor types. Furthermore, large HCCs have normal or high platelet levels (thrombocytosis), as in ovarian and other gastrointestinal cancers, probably related to elevated interleukin 6 (IL-6) levels.
STAGING
Multiple clinical staging systems for HCC have been described. A widely used one has been the American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) classification. However, the Cancer of the Liver Italian Program (CLIP) system is now popular because it takes cirrhosis into account, based on the original Okuda system (Table 111-4). Patients with Okuda stage III disease have a dire prognosis because they usually cannot be curatively resected, and the condition of their liver typically precludes chemotherapy. Other staging systems have been proposed, and a consensus is needed. They are all based on combining the prognostic features of liver damage with those of tumor aggressiveness and include the Barcelona Clinic Liver Cancer (BCLC) system from Spain (Fig. 111-1), which is externally validated and incorporates baseline survival estimates; the Chinese University Prognostic Index (CUPI); the important and simple Japan Integrated Staging Score (JIS); and SLiDe, which stands for s tage, li ver damage, and de s-γ-carboxy prothrombin. CLIP and BCLC appear most popular in the West, whereas JIS is favored in Japan. Each system has its champions. The best prognosis is for stage I, solitary tumors less than 2 cm in diameter without vascular invasion. Adverse prognostic features include ascites, jaundice, vascular invasion, and elevated α fetoprotein (AFP). Vascular invasion in particular has profound effects on prognosis and may be microscopic or macroscopic (visible on computed tomography [CT] scans). Most large tumors have microscopic vascular invasion, so full staging can usually be made only after surgical resection. Stage III disease contains a mixture of lymph node– positive and–negative tumors. Stage III patients with positive lymph node disease have a poor prognosis, and few patients survive 1 year. The prognosis of stage IV is poor after either resection or transplantation, and 1-year survival is rare.
|
CLIP AND OKUDA STAGING SYSTEMS FOR HEPATOCELLULAR CARCINOMA |
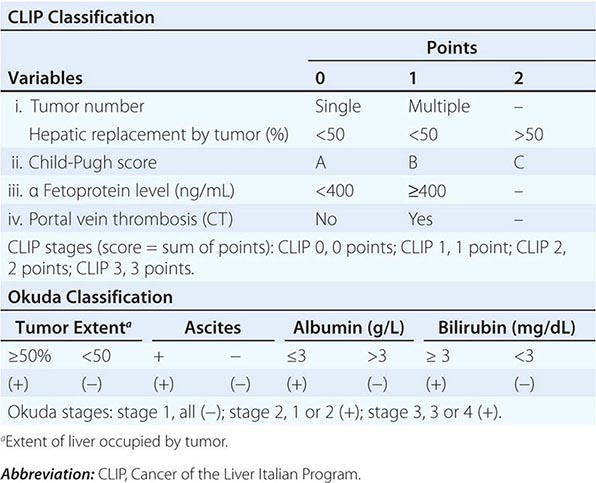
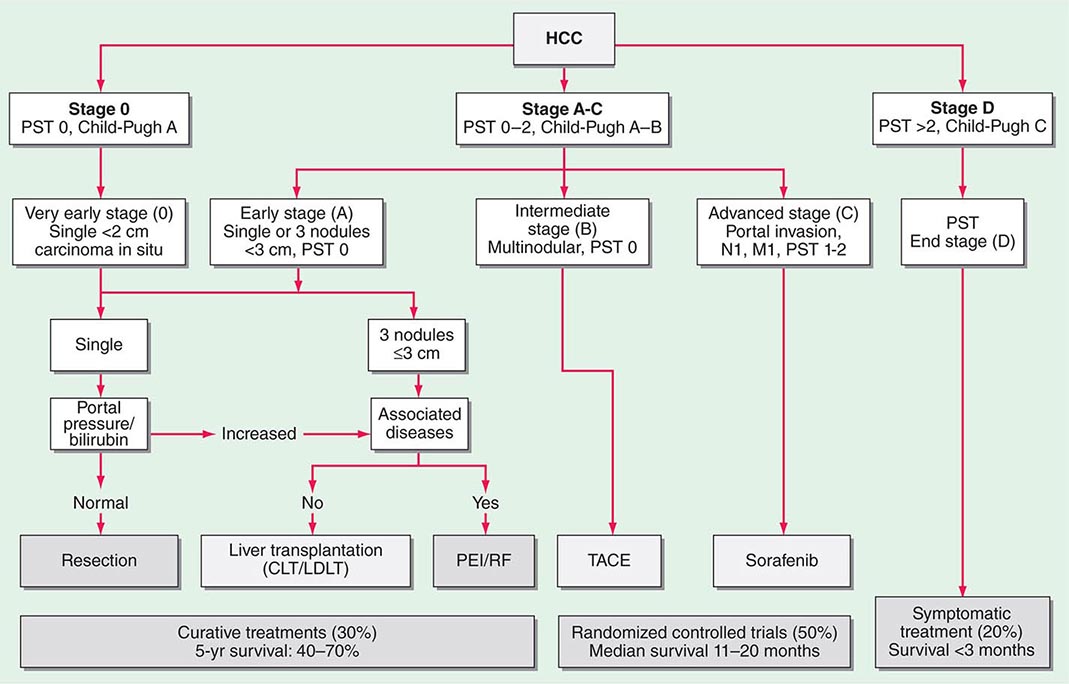
FIGURE 111-1 Barcelona Clinic Liver Cancer (BCLC) staging classification and treatment schedule. Patients with very early hepatocellular carcinoma (HCC) (stage 0) are optimal candidates for resection. Patients with early HCC (stage A) are candidates for radical therapy (resection, liver transplantation [LT], or local ablation via percutaneous ethanol injection [PEI] or radiofrequency [RF] ablation). Patients with intermediate HCC (stage B) benefit from transcatheter arterial chemoembolization (TACE). Patients with advanced HCC, defined as presence of macroscopic vascular invasion, extrahepatic spread, or cancer-related symptoms (Eastern Cooperative Oncology Group performance status 1 or 2) (stage C), benefit from sorafenib. Patients with end-stage disease (stage D) will receive symptomatic treatment. Treatment strategy will transition from one stage to another on treatment failure or contraindications for the procedures. CLT, cadaveric liver transplantation; LDLT, living donor liver transplantation; PST, Performance Status Test. (Modified from JM Llovet et al: JNCI 100:698, 2008.)
New Directions Consensus is needed on staging. These systems will soon be refined or upended by proteomics.
SCREENING HIGH-RISK POPULATIONS
There are two goals of screening, both in patients at increased risk for developing HCC, such as those with cirrhosis. The first goal is to detect smaller tumors that are potentially curable by ablation. The second goal is to enhance survival, compared with patients who were not diagnosed by surveillance. Evidence from Taiwan has shown a survival advantage to population screening in HBV-positive patients, and other evidence has shown its efficacy in diagnosis for HCV. Prospective studies in high-risk populations showed that ultrasound was more sensitive than AFP elevations alone, although most practitioners request both tests at 6-month intervals for HBV and HCV carriers, especially in the presence of cirrhosis or worsening of liver function tests. However, an Italian study in patients with cirrhosis identified a yearly HCC incidence of 3% but showed no increase in the rate of detection of potentially curable tumors with aggressive screening. Prevention strategies including universal vaccination against hepatitis are more likely to be effective than screening efforts. Despite absence of formal guidelines, most practitioners obtain 6-month AFP and ultrasound (cheap and ubiquitous, even in poor countries) or CT (more sensitive, especially in overweight patients, but more costly) studies when following high-risk patients (HBV carriers, HCV cirrhosis, family history of HCC).
Current Directions Cost-benefit analysis is not yet convincing, even though screening is intuitively sound. However, studies from areas with high HBV carrier rates have shown a survival benefit for screening as a result of earlier stage at diagnosis. A definitive clinical trial on screening is unlikely, due to difficulties in obtaining informed consent for patients who are not to be screened. γ-Glutamyl transpeptidase appears useful for detecting small tumors.
PREVENTION
Prevention strategies can only be planned when the causes of a cancer are known or strongly suspected. This is true of few human cancers, with significant exceptions being smoking and lung cancer, papilloma virus and cancer of the cervix uteri, and cirrhosis of any cause or dietary contamination by aflatoxin B1 for HCC. Aflatoxin B1is one of the most potent known chemical carcinogens and is a product of the Aspergillus mold that grows on peanuts and rice when stored in hot and humid climates. The obvious strategy is to refrigerate these foodstuffs when stored and to conduct surveillance programs for elevated aflatoxin B1 levels, as happens in the United States, but not usually in Asia. HBV is commonly transmitted from mother to fetus in Asia (except Japan), and neonatal HBV vaccination programs have resulted in a big decrease in adolescent HBV and, thus, in predicted HCC rates. There are millions of HBV and HCV carriers (4 million with HCV in the United States) who are already infected. Nucleoside analogue–based chemoprevention (entecavir) of HBV-mediated HCC in Japan resulted in a fivefold decrease in HCC incidence over 5 years in cirrhotic but not in noncirrhotic HBV patients. More powerful and effective HCV therapies promise the possibility of prevention of HCV-based HCC in the future.
TREATMENT HEPATOCELLULAR CARCINOMA
Most HCC patients have two liver diseases, cirrhosis and HCC, each of which is an independent cause of death. The presence of cirrhosis usually places constraints on resection surgery, ablative therapies, and chemotherapy. Thus patient assessment and treatment planning have to take the severity of the nonmalignant liver disease into account. The clinical management choices for HCC can be complex (Fig. 111-2, Tables 111-5 and 111-6). The natural history of HCC is highly variable. Patients presenting with advanced tumors (vascular invasion, symptoms, extrahepatic spread) have a median survival of ∼4 months, with or without treatment. Treatment results from the literature are difficult to interpret. Survival is not always a measure of the efficacy of therapy because of the adverse effects on survival of the underlying liver disease. A multidisciplinary team, including a hepatologist, interventional radiologist, surgical oncologist, resection surgeon, transplant surgeon, and medical oncologist, is important for the comprehensive management of HCC patients.
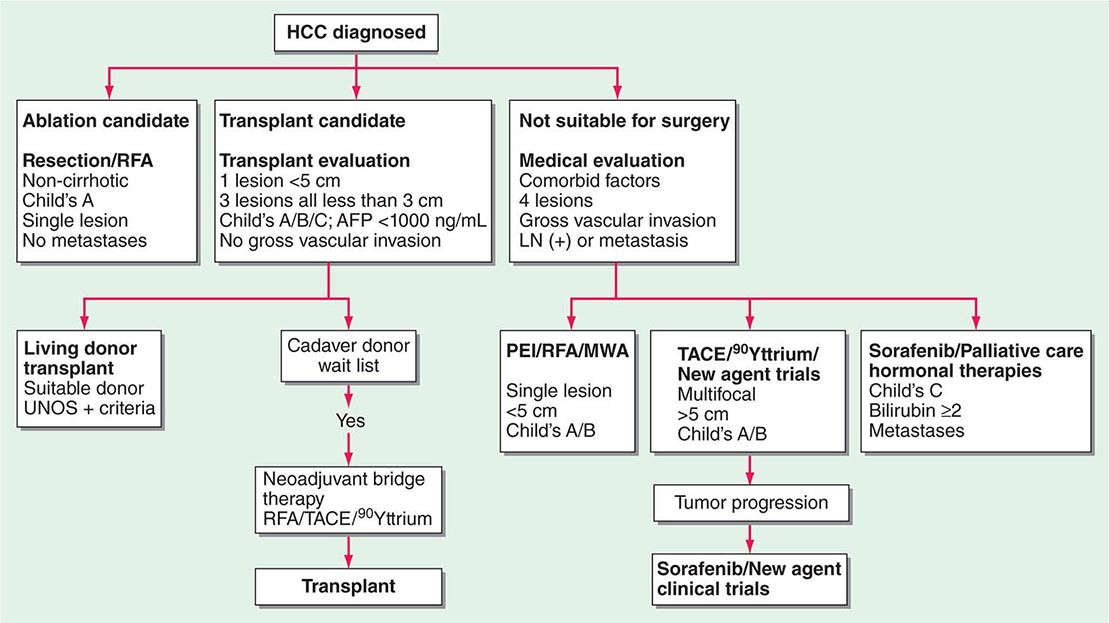
FIGURE 111-2 Hepatocellular carcinoma (HCC) treatment algorithm. The initial clinical evaluation is aimed at assessing the extent of the tumor and the underlying functional compromise of the liver by cirrhosis. Patients are classified as having resectable disease or unresectable disease or as being candidates for transplantation. AFP, α fetoprotein; LN, lymph node; MWA, microwave ablation; OLTX, orthotopic liver transplantation; PEI, percutaneous ethanol injection; RFA, radiofrequency ablation; TACE, transcatheter arterial chemoembolization; UNOS, United Network for Organ Sharing. Child’s A/B/C refers to the Child-Pugh classification of liver failure.
|
TREATMENT OPTIONS FOR HEPATOCELLULAR CARCINOMA |
|
SOME RANDOMIZED CLINICAL TRIALS INVOLVING TRANSHEPATIC ARTERY CHEMOEMBOLIZATION (TACE) FOR HEPATOCELLULAR CARCINOMA |
TNM STAGES I AND II HCC
Early-stage tumors are successfully treated using various techniques, including surgical resection, local ablation (thermal, radiofrequency [RFA], or microwave ablation (MWA]), and local injection therapies (Table 111-6). Because the majority of patients with HCC suffer from a field defect in the cirrhotic liver, they are at risk for subsequent multiple primary liver tumors. Many will also have significant underlying liver disease and may not tolerate major surgical loss of hepatic parenchyma, and they may be eligible for orthotopic liver transplant (OLTX). Living related donor transplants have increased in popularity, resulting in absence of waiting for a transplant. An important principle in treating early-stage HCC in the nontransplant setting is to use liver-sparing treatments and to focus on treatment of both the tumor and the cirrhosis.
Surgical Excision The risk of major hepatectomy is high (5–10% mortality rate) due to the underlying liver disease and the potential for liver failure, but acceptable in selected cases and highly dependent on surgical experience. The risk is lower in high-volume centers. Preoperative portal vein occlusion can sometimes be performed to cause atrophy of the HCC-involved lobe and compensatory hypertrophy of the noninvolved liver, permitting safer resection. Intraoperative ultrasound is useful for planning the surgical approach. The ultrasound can image the proximity of major vascular structures that may be encountered during the dissection. In cirrhotic patients, any major liver surgery can result in liver failure. The Child-Pugh classification of liver failure is still a reliable prognosticator for tolerance of hepatic surgery, and only Child A patients should be considered for surgical resection. Child B and C patients with stages I and II HCC should be referred for OLTX if appropriate, as well as patients with ascites or a recent history of variceal bleeding. Although open surgical excision is the most reliable, the patient may be better served with a laparoscopic approach to resection, using RFA, MWA, or percutaneous ethanol injection (PEI). No adequate comparisons of these different techniques have been undertaken, and the choice of treatment is usually based on physician skill. However, RFA has been shown to be superior to PEI in necrosis induction for tumors <3 cm in diameter and is thought to be equivalent to open resection and, thus, is the treatment of first choice for these small tumors. As tumors get larger than 3 cm, especially ≥5 cm, the effectiveness of RFA-induced necrosis diminishes. The combination of transcatheter arterial chemoembolization (TACE) with RFA has shown superior results to TACE alone in a prospective, randomized trial. Although vascular invasion is a preeminent negative prognostic factor, microvascular invasion in small tumors appears not to be a negative factor.
Local Ablation Strategies RFA uses heat to ablate tumors. The maximum size of the probe arrays allows for a 7-cm zone of necrosis, which would be adequate for a 3- to 4-cm tumor. The heat reliably kills cells within the zone of necrosis. Treatment of tumors close to the main portal pedicles can lead to bile duct injury and obstruction. This limits the location of tumors that are anatomically suited for this technique. RFA can be performed percutaneously with CT or ultrasound guidance, or at the time of laparoscopy with ultrasound guidance.
Local Injection Therapy Numerous agents have been used for local injection into tumors, most commonly ethanol (PEI). The relatively soft HCC within the hard background cirrhotic liver allows for injection of large volumes of ethanol into the tumor without diffusion into the hepatic parenchyma or leakage out of the liver. PEI causes direct destruction of cancer cells, but it is not selective for cancer and will destroy normal cells in the vicinity. However, it usually requires multiple injections (average three), in contrast to one for RFA. The maximum size of tumor reliably treated is 3 cm, even with multiple injections.
CURRENT DIRECTIONS Resection and RFA each obtain similar results. However, a distinction has been made between the causes and prevention strategies needed to prevent early versus late tumor recurrences after resection. Early recurrence has been linked to tumor invasion factors, especially microvascular tumor invasion with elevated transaminases, whereas late recurrence has been associated with cirrhosis and virus hepatitis factors and, thus, the development of new tumors. See the section on virus-directed adjuvant therapy below.
Liver Transplantation (OLTX) A viable option for stages I and II tumors in the setting of cirrhosis is OLTX, with survival approaching that for noncancer cases. OLTX for patients with a single lesion ≤5 cm or three or fewer nodules, each ≤3 cm (Milan criteria), resulted in excellent tumor-free survival (≥70% at 5 years). For advanced HCC, OLTX has been abandoned due to high tumor recurrence rates. Priority scoring for OLTX previously led to HCC patients waiting too long for their OLTX, resulting in some tumors becoming too advanced during the patient’s wait for a donated liver. A variety of therapies were used as a “bridge” to OLTX, including RFA, TACE, and hepatic arterial 90Y-radioembolization. These pre-transplant treatments allow patients to remain on the waiting list longer, giving them greater opportunities to be transplanted, because they can stabilize the tumor and prevent it from growing in the months until a donor liver becomes available. What remains unclear, however, is whether this translates into prolonged survival after transplant. Further, it is not known whether patients who have had their tumor(s) treated preoperatively follow the recurrence pattern predicted by their tumor status at the time of transplant (i.e., post–local ablative therapy), or if they follow the course set by their tumor parameters present before such treatment. The United Network for Organ Sharing (UNOS) point system for priority scoring of OLTX recipients now includes additional points for patients with HCC. The success of living related donor liver transplantation programs has also led to patients receiving transplantation earlier for HCC and often with greater than minimal tumors.
CURRENT DIRECTIONS Expanded criteria for larger HCCs beyond the Milan criteria (one lesion <5 cm or three lesions, each <3 cm), such as the University of California, San Francisco (UCSF) criteria (single lesion ≤6.5 cm or two lesions ≤4.5 cm with a total diameter ≤8 cm; 1- and 5-year survival rates of 90 and 75%, respectively), are being increasingly accepted by various UNOS areas for OLTX with satisfactory longer-term survival comparable to Milan criteria results. Furthermore, downstaging of HCCs that are too large for the Milan criteria by medical therapy (TACE) is increasingly recognized as acceptable treatment before OLTX with equivalent outcomes to patients who originally were within Milan criteria. Within-criteria patients with AFP levels >1000 ng/mL have exceptionally high post-OLTX recurrence rates. Also, the use of “salvage” OLTX after recurrent HCC after resection has produced conflicting outcomes. Shortages of organs combined with advances in resection safety have led to increasing use of resection for patients with good liver function.
Adjuvant Therapy The role of adjuvant chemotherapy for patients after resection or OLTX remains unclear. Both adjuvant and neoadjuvant approaches have been studied, but no clear advantage in disease-free or overall survival has been found. However, a meta-analysis of several trials revealed a significant improvement in disease-free and overall survival. Although analysis of postoperative adjuvant systemic chemotherapy trials demonstrated no disease-free or overall survival advantage, single studies of TACE and neoadjuvant 131I-Ethiodol showed enhanced survival after resection.
Antiviral therapy, instead of anticancer therapy, has been successful in decreasing postresection tumor recurrences in the postresection adjuvant setting. Nucleoside analogues in HBV-based HCC and peg-interferon plus ribavirin for HCV-based HCC have both been effective in reducing recurrence rates.
CURRENT DIRECTIONS A large adjuvant trial examining resection and transplantation, with or without sorafenib (see below) is in progress. The success of viral therapies in decreasing HCC recurrence after resection is part of a broader focus on the tumor microenvironment (stroma, blood vessels, inflammatory cells, and cytokines) as mediators of HCC progression and as targets for new therapies.
TNM STAGES III AND IV HCC
Fewer surgical options exist for stage III tumors involving major vascular structures. In patients without cirrhosis, a major hepatectomy is feasible, although prognosis is poor. Patients with Child A cirrhosis may be resected, but a lobectomy is associated with significant morbidity and mortality rates, and long-term prognosis is poor. Nevertheless, a small percentage of patients will achieve long-term survival, justifying an attempt at resection when feasible. Because of the advanced nature of these tumors, even successful resection can be followed by rapid recurrence. These patients are not considered candidates for transplantation because of the high tumor recurrence rates, unless their tumors can first be downstaged with neoadjuvant therapy. Decreasing the size of the primary tumor allows for less surgery, and the delay in surgery allows for extrahepatic disease to manifest on imaging studies and avoid unhelpful OLTX. The prognosis is poor for stage IV tumors, and no surgical treatment is recommended.
Systemic Chemotherapy A large number of controlled and uncontrolled clinical studies have been performed with most of the major classes of cancer chemotherapy. No single agent or combination of agents given systemically reproducibly leads to even a 25% response rate or has any effect on survival.
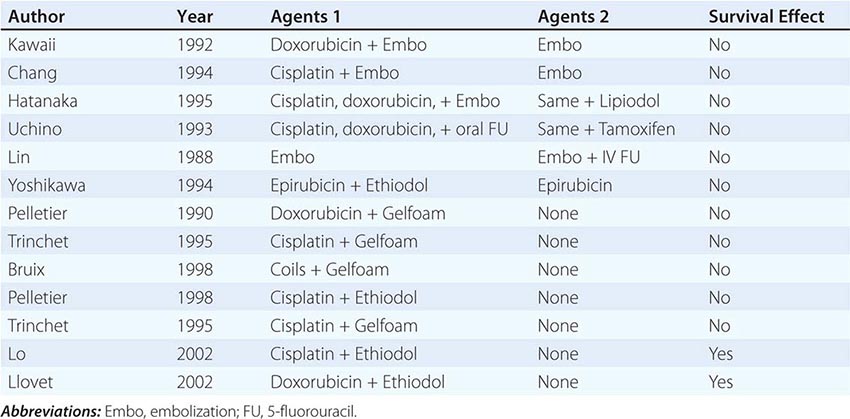
Regional Chemotherapy In contrast to the dismal results of systemic chemotherapy, a variety of agents given via the hepatic artery have activity for HCC confined to the liver (Table 111-6). Two randomized controlled trials have shown a survival advantage for TACE in a selected subset of patients. One used doxorubicin, and the other used cisplatin. Despite the fact that increased hepatic extraction of chemotherapy has been shown for very few drugs, some drugs such as cisplatin, doxorubicin, mitomycin C, and possibly neocarzinostatin, produce substantial objective responses when administered regionally. Few data are available on continuous hepatic arterial infusion for HCC, although pilot studies with cisplatin have shown encouraging responses. Because the reports have not usually stratified responses or survival based on TNM staging, it is difficult to know long-term prognosis in relation to tumor extent. Most of the studies on regional hepatic arterial chemotherapy also use an embolizing agent such as Ethiodol, gelatin sponge particles (Gelfoam), starch (Spherex), or microspheres. Two products are composed of microspheres of defined size ranges—Embospheres (Biospheres) and Contour SE—using particles of 40–120, 100–300, 300–500, and 500–1000 μm in size. The optimal diameter of the particles for TACE has yet to be defined. Consistently higher objective response rates are reported for arterial administration of drugs together with some form of hepatic artery occlusion compared with any form of systemic chemotherapy to date. The widespread use of some form of embolization in addition to chemotherapy has added to its toxicities. These include a frequent but transient fever, abdominal pain, and anorexia (all in >60% of patients). In addition, >20% of patients have increased ascites or transient elevation of transaminases. Cystic artery spasm and cholecystitis are also not uncommon. However, higher responses have also been obtained. The hepatic toxicities associated with embolization may be ameliorated by the use of degradable starch microspheres, with 50–60% response rates. Two randomized studies of TACE versus placebo showed a survival advantage for treatment (Table 111-6). In addition, it is not clear that formal oncologic CT response criteria are adequate for HCC. A loss of vascularity on CT without size change may be an index of loss of viability and thus of response to TACE. A major problem that TACE trials have had in showing a survival advantage is that many HCC patients die of their underlying cirrhosis, not the tumor. Nevertheless, two randomized controlled trials, one using doxorubicin and the other using cisplatin, showed a survival advantage for TACE versus placebo (Table 111-6). However, improving quality of life is a legitimate goal of regional therapy. Drug-eluting beads using doxorubicin (DEB-TACE) have been claimed to produce equivalent survival with less toxicity, but this strategy has not been tested in a randomized trial.
Kinase Inhibitors A survival advantage has been observed for the oral multikinase inhibitor, sorafenib (Nexavar), versus placebo in two randomized trials. It targets both the Raf mitogenic pathway and the vascular endothelial growth factor receptor (VEGFR) endothelial vasculogenesis pathway. However, tumor responses were negligible, and the survival in the treatment arm in Asians was less than the placebo arm in the Western trial (Table 111-7). Sorafenib has considerable toxicity, with 30–40% of patients requiring “drug holidays,” dose reductions, or cessation of therapy. The most common toxicities include fatigue, hypertension, diarrhea, mucositis, and skin changes, such as the painful hand-foot syndrome, hair loss, and itching, each in 20–40% of patients. Several “look-alike” new agents that also target angiogenesis have either proved to be inferior or more toxic. These include sunitinib, brivanib, linifanib, everolimus, and bevacizumab (Table 111-8). The idea of angiogenesis alone as a major HCC therapeutic target may need revision.
|
TARGETED THERAPIES IN HEPATOCELLULAR CARCINOMA: TRIALS |

|
PROMISING TARGETED THERAPIES THAT FAILED THEIR CLINICAL TRIAL GOALS |
New Therapies Although prolonged survival has been reported in phase II trials using newer agents, such as bevacizumab plus erlotinib, the data from a phase III trial were disappointing. Several forms of radiation therapy have been used in the treatment of HCC, including external-beam radiation and conformal radiation therapy. Radiation hepatitis remains a dose-limiting problem. The pure beta emitter 90Yttrium attached to either glass (TheraSphere) or resin (SIR-Spheres) microspheres injected into a major branch hepatic artery has been assessed in phase II trials of HCC and has encouraging tumor control and survival effects with minimal toxicities. Randomized phase III trials comparing it to TACE have yet to be completed. The main attractiveness of 90Yttrium therapy is its safety in the presence of major branch portal vein thrombosis, where TACE is dangerous or contraindicated. Furthermore, external-beam radiation has been reported to be safe and useful in the control of major branch portal or hepatic vein invasion (thrombosis) by tumors. The studies have all been small. Vitamin K has been assessed in clinical trials at high dosage for its HCC-inhibitory actions. This idea is based on the characteristic biochemical defect in HCC of elevated plasma levels of immature prothrombin (DCP or PIVKA-2), due to a defect in the activity of prothrombin carboxylase, a vitamin K–dependent enzyme. Two vitamin K randomized controlled trials from Japan show decreased tumor occurrence, but a major phase III trial aimed at limiting postresection recurrence was not successful.
CURRENT DIRECTIONS A number of new kinase inhibitors are being evaluated for HCC (Tables 111-9 and 111-10). These include the biologicals, such as Raf kinase and vascular endothelial growth factor (VEGF) inhibitors, and agents that target various steps of the cell growth pathway. Current hopes focus particularly on the Met pathway inhibitors such as tivantinib and several IGF receptor antagonists. 90Yttrium looks promising and without chemotherapy toxicities. It is particularly attractive because, unlike TACE, it seems safe in the presence of portal vein thrombosis, a pathognomonic feature of HCC aggressiveness. The bottleneck of liver donors for OLTX is at last widening with increasing use of living donors, and criteria for OLTX for larger HCCs are slowly expanding. Patient participation in clinical trials assessing new therapies is encouraged (www.clinicaltrials.gov).
|
NEW TARGETED AGENTS AND THEIR TARGETS IN CURRENT CLINICAL TRIALS |

|
SOME NOVEL MEDICAL TREATMENTS FOR HEPATOCELLULAR CARCINOMA |
Abbreviations: EGF, epidermal growth factor; mTOR, mammalian target of rapamycin; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
The main effort now is the evaluation of combinations of the compounds listed in Tables 111-7 to 111-9 that target different pathways, as well as the combination of any of these targeted therapies, but especially sorafenib, with TACE or 90Yttrium radioembolization. Combining TACE with sorafenib appears to be safe in phase II studies with promising survival data, but randomized studies are still in progress. The same is true for intra-arterial 90Yttrium plus sorafenib as therapy for HCC and as bridge to transplant therapy.
SIGNIFICANCE AND EVALUATION OF RESPONSES TO NONSURGICAL THERAPIES
Tumor growth or spread is considered a poor prognostic sign and evidence of treatment failure. By contrast, patients receiving chemotherapy are judged to have a response if there is shrinkage of tumor size. Lack of response/size decrease has been thought of as treatment failure. Three considerations in HCC management have completely changed the views concerning nonshrinkage after therapy. First, the correlation between response to chemotherapy and survival is poor in various tumors; in some tumors, such as ovarian cancer and small-cell lung cancer, substantial tumor shrinkage on chemotherapy is followed by rapid tumor regrowth. Second, the Sorafenib HCC Assessment Randomized Protocol (SHARP) phase III trial of sorafenib versus placebo for unresectable HCC showed that survival could be significantly enhanced in the treatment arm with only 2% of the patients having tumor response but 70% of patients having disease stabilization. This observation has led to a reconsideration of the usefulness of response and the significance of disease stability. Third, HCC is a typically highly vascular tumor, and the vascularity is considered to be a measure of tumor viability. As a result, the Response Evaluation Criteria in Solid Tumors (RECIST) have been modified to mRECIST, which requires measurement of vascular/viable tumor on the CT or MRI scan. A partial response is defined as a 30% decrease in the sum of diameters of viable (arterially enhancing) target tumors. The need for semiquantitation of tumor vascularity on scans has led to the introduction of diffusion-weighted MRI imaging. Tissue-specific imaging agents such as gadoxetic acid (Primovist or Eovist) and the move to functional and genetic imaging mark a shift in approaches. Furthermore, plasma AFP response may be a biologic marker of radiologic response.
TREATMENT SUMMARY
Long-term survival is associated with resection or ablation or transplantation, all of which can yield >70% 5-year survival. Liver transplant is the only therapy that can treat the tumor and the underlying liver disease simultaneously and may be the most important advance in HCC therapy in 50 years. Unfortunately, it benefits only patients with limited size tumors without macrovascular portal vein invasion. Untreated patients with multinodular asymptomatic tumors without vascular invasion or extrahepatic spread have a median survival of approximately 16 months. Chemoembolization (TACE) improves their median survival to 19–20 months and is considered standard therapy for these patients, who represent the majority of HCC patients, although 90Yttrium therapy may provide similar results with less toxicity. Patients with advanced-stage disease, vascular invasion, or metastases have a median survival of around 6 months. Among this group, outcomes may vary according to their underlying liver disease. It is this group at which kinase inhibitors are directed.
SUMMARY (Table 111-5)
The Most Common Modes of Patient Presentation
1. A patient with known history of hepatitis, jaundice, or cirrhosis, with an abnormality on ultrasound or CT scan, or rising AFP or DCP (PIVKA-2)
2. A patient with an abnormal liver function test as part of a routine examination
3. Radiologic workup for liver transplant for cirrhosis
4. Symptoms of HCC including cachexia, abdominal pain, or fever
History and Physical Examination
1. Clinical jaundice, asthenia, itching (scratches), tremors, or disorientation
2. Hepatomegaly, splenomegaly, ascites, peripheral edema, skin signs of liver failure
Clinical Evaluation
1. Blood tests: full blood count (splenomegaly), liver function tests, ammonia levels, electrolytes, AFP and DCP (PIVKA-2), Ca2+ and Mg2+; hepatitis B, C, and D serology (and quantitative HBV DNA or HCV RNA, if either is positive); neurotensin (specific for fibrolamellar HCC)
2. Triphasic dynamic helical (spiral) CT scan of liver (if inadequate, then follow with an MRI); chest CT scan; upper and lower gastrointestinal endoscopy (for varices, bleeding, ulcers); and brain scan (only if symptoms suggest)
3. Core biopsy: of the tumor and separate biopsy of the underlying liver
Therapy (Tables 111-5 and 111-6)
1. HCC <2 cm: RFA, PEI, or resection
2. HCC >2 cm, no vascular invasion: liver resection, RFA, or OLTX
3. Multiple unilobar tumors or tumor with vascular invasion: TACE or sorafenib
4. Bilobar tumors, no vascular invasion: TACE with OLTX for patients with tumor response
5. Extrahepatic HCC or elevated bilirubin: sorafenib or bevacizumab plus erlotinib (combination agent trials are in progress)
OTHER PRIMARY LIVER TUMORS
FIBROLAMELLAR HCC (FL-HCC)
This rarer variant of HCC has a quite different biology than adult-type HCC. None of the known HCC causative factors seem important here. It is typically a disease of younger adults, often teenagers and predominantly females. It is AFP-negative, but patients typically have elevated blood neurotensin levels, normal liver function tests, and no cirrhosis. Radiology is similar for HCC, except that characteristic adult-type portal vein invasion is less common. Although it is often multifocal in the liver, and therefore not resectable, metastases are common, especially to lungs and locoregional lymph nodes, but survival is often much better than with adult-type HCC. Resectable tumors are associated with 5-year survival ≥50%. Patients often present with a huge liver or unexplained weight loss, fever, or elevated liver function tests on routine evaluations. These huge masses suggest quite slow growth for many tumors. Surgical resection is the best management option, even for metastases, as these tumors respond much less well to chemotherapy than adult-type HCC. Although several series of OLTX for FL-HCC have been reported, the patients seem to die from tumor recurrences, with a 2- to 5-year lag compared with OLTX for adult-type HCC. Anecdotal responses to gemcitabine plus cisplatin-TACE are reported.
Epithelioid Hemangioendothelioma (EHE) This rare vascular tumor of adults is also usually multifocal and can also be associated with prolonged survival, even in the presence of metastases, which are commonly in the lung. There is usually no underlying cirrhosis. Histologically, these tumors are usually of borderline malignancy and express factor VIII, confirming their endothelial origin. OLTX may produce prolonged survival.
Cholangiocarcinoma (CCC) CCC typically refers to mucin-producing adenocarcinomas (different from HCC) that arise from the biliary tract and have features of cholangiocyte differentiation. They are grouped by their anatomic site of origin, as intrahepatic (IHC), perihilar (central, ∼65% of CCCs), and peripheral (or distal, ∼30% of CCCs). IHC is the second most common primary liver tumor. Depending on the site of origin, they have different features and require different treatments. They arise on the basis of cirrhosis less frequently than HCC, but may complicate primary biliary cirrhosis. However, cirrhosis and both primary biliary cirrhosis and HCV predispose to IHC. Nodular tumors arising at the bifurcation of the common bile duct are called Klatskin tumors and are often associated with a collapsed gallbladder, a finding that mandates visualization of the entire biliary tree. The approach to management of central and peripheral CCC is quite different. Incidence is increasing. Although most CCCs have no obvious cause (etiology unknown), a number of predisposing factors have been identified. Predisposing diseases include primary sclerosing cholangitis (10–20% of primary sclerosing cholangitis [PSC] patients), an autoimmune disease, and liver fluke in Asians, especially Opisthorchis viverrini and Clonorchis sinensis. CCC seems also to be associated with any cause of chronic biliary inflammation and injury, with alcoholic liver disease, choledocholithiasis, choledochal cysts (10%), and Caroli’s disease (a rare inherited form of bile duct ectasia). CCC most typically presents as painless jaundice, often with pruritus or weight loss. Diagnosis is made by biopsy, percutaneously for peripheral liver lesions, or more commonly via endoscopic retrograde cholangiopancreatography (ERCP) under direct vision for central lesions. The tumors often stain positively for cytokeratins 7, 8, and 19 and negatively for cytokeratin 20. However, histology alone cannot usually distinguish CCC from metastases from colon or pancreas primary tumors. Serologic tumor markers appear to be nonspecific, but CEA, CA 19-9, and CA-125 are often elevated in CCC patients and are useful for following response to therapy. Radiologic evaluation typically starts with ultrasound, which is very useful in visualizing dilated bile ducts, and then proceeds with either MRI or magnetic resonance cholangiopancreatography (MRCP) or helical CT scans. Invasive cholangiopancreatography (ERCP) is then needed to define the biliary tree and obtain a biopsy or is needed therapeutically to decompress an obstructed biliary tree with internal stent placement. If that fails, then percutaneous biliary drainage will be needed, with the biliary drainage flowing into an external bag. Central tumors often invade the porta hepatis, and locoregional lymph node involvement by tumor is frequent. Incidence has been increasing in recent decades; few patients survive 5 years. The usual treatment is surgical, but combination systemic chemotherapy may be effective. After complete surgical resection for IHC, 5-year survival is 25–30%. Combination radiation therapy with liver transplant has produced a 5-year recurrence-free survival rate of 65%.
GALLBLADDER CANCER
Gallbladder (GB) cancer has an even worse prognosis than CCC, with a typical survival of ∼6 months or less. Women are affected much more commonly than men (4:1), unlike HCC or CCC, and GB cancer occurs more frequently than CCC. Most patients have a history of antecedent gallstones, but very few patients with gallstones develop GB cancer (∼0.2%). GB cancer presents similarly to CCC and is often diagnosed unexpectedly during gallstone or cholecystitis surgery. Presentation is typically that of chronic cholecystitis, chronic right upper quadrant pain, and weight loss. Useful but nonspecific serum markers include CEA and CA 19-9. CT scans or MRCP typically reveal a GB mass. The mainstay of treatment is surgical, either simple or radical cholecystectomy for stage I or II disease, respectively. Survival rates are near 100% at 5 years for stage I, and range from 60–90% at 5 years for stage II. More advanced GB cancer has worse survival, and many patients are unresectable. Adjuvant radiotherapy, used in the presence of local lymph node disease, has not been shown to enhance survival. Chemotherapy is not useful in advanced or metastatic GB cancer.
CARCINOMA OF THE AMPULLA OF VATER
This tumor arises within 2 cm of the distal end of the common bile duct and is mainly (90%) an adenocarcinoma. Locoregional lymph nodes are commonly involved (50%), and the liver is the most frequent site for metastases. The most common clinical presentation is jaundice, and many patients also have pruritus, weight loss, and epigastric pain. Initial evaluation is performed with an abdominal ultrasound to assess vascular involvement, biliary dilation, and liver lesions. This is followed by a CT scan or MRI and especially MRCP. The most effective therapy is resection by pylorus-sparing pancreaticoduodenectomy, an aggressive procedure resulting in better survival rates than with local resection. Survival rates are ∼25% at 5 years in operable patients with involved lymph nodes and ∼50% in patients without involved nodes. Unlike CCC, approximately 80% of patients are thought to be resectable at diagnosis. Adjuvant chemotherapy or radiotherapy has not been shown to enhance survival. For metastatic tumors, chemotherapy is currently experimental.
TUMORS METASTATIC TO THE LIVER
These are predominantly from colon, pancreas, and breast primary tumors but can originate from any organ primary. Ocular melanomas are prone to liver metastasis. Tumor spread to the liver normally carries a poor prognosis for that tumor type. Colorectal and breast hepatic metastases were previously treated with continuous hepatic arterial infusion chemotherapy. However, more effective systemic drugs for each of these two cancers, especially the addition of oxaliplatin to colorectal cancer regimens, have reduced the use of hepatic artery infusion therapy. In a large randomized study of systemic versus infusional plus systemic chemotherapy for resected colorectal metastases to the liver, the patients receiving infusional therapy had no survival advantage, mainly due to extrahepatic tumor spread. 90Yttrium resin beads are approved in the United States for treatment of colorectal hepatic metastases. The role of this modality, either alone or in combination with chemotherapy, is being evaluated in many centers. Palliation may be obtained from chemoembolization, PEI, or RFA.
BENIGN LIVER TUMORS
Three common benign tumors occur and all are found predominantly in women. They are hemangiomas, adenomas, and focal nodular hyperplasia (FNH). FNH is typically benign, and usually no treatment is needed. Hemangiomas are the most common and are entirely benign. Treatment is unnecessary unless their expansion causes symptoms. Adenomas are associated with contraceptive hormone use. They can cause pain and can bleed or rupture, causing acute problems. Their main interest for the physician is a low potential for malignant change and a 30% risk of bleeding. For this reason, considerable effort has gone into differentiating these three entities radiologically. On discovery of a liver mass, patients are usually advised to stop taking sex steroids, because adenoma regression may then occasionally occur. Adenomas can often be large masses ranging from 8–15 cm. Due to their size and definite, but low, malignant potential and potential for bleeding, adenomas are typically resected. The most useful diagnostic differentiating tool is a triphasic CT scan performed with HCC fast bolus protocol for arterial-phase imaging, together with subsequent delayed venous-phase imaging. Adenomas usually do not appear on the basis of cirrhosis, although both adenomas and HCCs are intensely vascular on the CT arterial phase and both can exhibit hemorrhage (40% of adenomas). However, adenomas have smooth, well-defined edges, and enhance homogeneously, especially in the portal venous phase on delayed images, when HCCs no longer enhance. FNHs exhibit a characteristic central scar that is hypovascular on the arterial-phase and hypervascular on the delayed-phase CT images. MRI is even more sensitive in depicting the characteristic central scar of FNH.
112 |
Pancreatic Cancer |
Pancreatic cancer is the fourth leading cause of cancer death in the United States and is associated with a poor prognosis. Endocrine tumors affecting the pancreas are discussed in Chap. 113. Infiltrating ductal adenocarcinomas, the subject of this Chapter, account for the vast majority of cases and arise most frequently in the head of pancreas. At the time of diagnosis, 85–90% of patients have inoperable or metastatic disease, which is reflected in the 5-year survival rate of only 6% for all stages combined. An improved 5-year survival of up to 24% may be achieved when the tumor is detected at an early stage and when complete surgical resection is accomplished.
EPIDEMIOLOGY
Pancreatic cancer represents 3% of all newly diagnosed malignancies in the United States. The most common age group at diagnosis is 65–84 years for both sexes. Pancreatic cancer was estimated to have been diagnosed in approximately 45,220 patients and accounted for approximately 38,460 deaths in 2013. Although survival rates have almost doubled over the past 35 years for this disease, overall survival remains low.
GLOBAL CONSIDERATIONS
![]() An estimated 278,684 cases of pancreatic cancer occur annually worldwide (the thirteenth most common cancer globally), with up to 60% of these cases diagnosed in more developed countries. It remains the eighth most common cause of cancer death in men and the ninth most common in women. The incidence is highest in the United States and western Europe and lowest in parts of Africa and South Central Asia. However, increasing rates of obesity, diabetes, and tobacco use in addition to access to diagnostic radiology in the developing world are likely to increase incidence rates in these countries. In this situation, consideration of the cost implications of adoption of current treatment paradigms in resource-constrained environments will be necessary. Primary prevention such as limiting tobacco use and avoiding obesity may be more cost effective than improvements in treatment of preexisting disease.
An estimated 278,684 cases of pancreatic cancer occur annually worldwide (the thirteenth most common cancer globally), with up to 60% of these cases diagnosed in more developed countries. It remains the eighth most common cause of cancer death in men and the ninth most common in women. The incidence is highest in the United States and western Europe and lowest in parts of Africa and South Central Asia. However, increasing rates of obesity, diabetes, and tobacco use in addition to access to diagnostic radiology in the developing world are likely to increase incidence rates in these countries. In this situation, consideration of the cost implications of adoption of current treatment paradigms in resource-constrained environments will be necessary. Primary prevention such as limiting tobacco use and avoiding obesity may be more cost effective than improvements in treatment of preexisting disease.
RISK FACTORS
Cigarette smoking may be the cause of up to 20–25% of all pancreatic cancers and is the most common environmental risk factor for this disease. A longstanding history of type 1 or type 2 diabetes also appears to be a risk factor; however, diabetes may also occur in association with pancreatic cancer, possibly confounding this interpretation. Other risk factors may include obesity, chronic pancreatitis, and ABO blood group status. Alcohol does not appear to be a risk factor unless excess consumption gives rise to chronic pancreatitis.
GENETIC AND MOLECULAR CONSIDERATIONS
![]() Pancreatic cancer is associated with a number of well-defined molecular hallmarks. The four genes most commonly mutated or inactivated in pancreatic cancer are KRAS (predominantly codon 12, in 60–75% of pancreatic cancers), the tumor-suppressor genes p16 (deleted in 95% of tumors), p53 (inactivated or mutated in 50–70% of tumors), and SMAD4 (deleted in 55% of tumors). The pancreatic cancer precursor lesion pancreatic intraepithelial neoplasia (PanIN) acquires these genetic abnormalities in a progressive manner associated with increasing dysplasia; initial KRAS mutations are followed by p16 loss and finally p53 and SMAD4 alterations. SMAD4 gene inactivation is associated with a pattern of widespread metastatic disease in advanced-stage patients and poorer survival in patients with surgically resected pancreatic adenocarcinoma.
Pancreatic cancer is associated with a number of well-defined molecular hallmarks. The four genes most commonly mutated or inactivated in pancreatic cancer are KRAS (predominantly codon 12, in 60–75% of pancreatic cancers), the tumor-suppressor genes p16 (deleted in 95% of tumors), p53 (inactivated or mutated in 50–70% of tumors), and SMAD4 (deleted in 55% of tumors). The pancreatic cancer precursor lesion pancreatic intraepithelial neoplasia (PanIN) acquires these genetic abnormalities in a progressive manner associated with increasing dysplasia; initial KRAS mutations are followed by p16 loss and finally p53 and SMAD4 alterations. SMAD4 gene inactivation is associated with a pattern of widespread metastatic disease in advanced-stage patients and poorer survival in patients with surgically resected pancreatic adenocarcinoma.
Up to 16% of pancreatic cancers may be inherited. Germline mutations in the following genes are associated with a significantly increased risk of pancreatic cancer and other cancers: (1) STK11 gene (Peutz-Jeghers syndrome), which carries a 132-fold increased lifetime risk of pancreatic cancer above the general population; (2) BRCA2 (increased risk of breast, ovarian, and pancreatic cancer); (3) p16/CDKN2A (familial atypical multiple mole melanoma), which carries an increased risk of melanoma and pancreatic cancer; (4) PALB2, which confers an increased risk of breast and pancreatic cancer; (5) hMLH1 and MSH2 (Lynch syndrome), which carries an increased risk of colon and pancreatic cancer; and (6) ATM (ataxia-telangiectasia), which carries an increased risk of breast cancer, lymphoma, and pancreatic cancer. Familial pancreatitis and an increased risk of pancreatic cancer are associated with mutations of the PRSS1 (serine protease 1) gene. However, for most familial pancreatic syndromes, the underlying genetic cause remains unexplained. The absolute number of affected first-degree relatives is also correlated with increased cancer risk, and patients with at least two first-degree relatives with pancreatic cancer should be considered to have familial pancreatic cancer until proven otherwise.
The desmoplastic stroma surrounding pancreatic adenocarcinoma functions as a mechanical barrier to chemotherapy and secretes compounds essential for tumor progression and metastasis. Key mediators of these functions include the activated pancreatic stellate cell and the glycoprotein SPARC (secreted protein acidic and rich in cysteine), which is expressed in 80% of pancreatic ductal adenocarcinomas. Targeting this extracellular environment has become increasingly important in the treatment of advanced disease.
SCREENING AND PRECURSOR LESIONS
Screening is not routinely recommended because the incidence of pancreatic cancer in the general population is low (lifetime risk 1.3%), putative tumor markers such as carbohydrate antigen 19-9 (CA19-9) and carcinoembryonic antigen (CEA) have insufficient sensitivity, and computed tomography (CT) has inadequate resolution to detect pancreatic dysplasia. Endoscopic ultrasound (EUS) is a more promising screening tool, and preclinical efforts are focused on identifying biomarkers that may detect pancreatic cancer at an early stage. Consensus practice recommendations based largely on expert opinion have chosen a threshold of greater than fivefold increased risk for developing pancreatic cancer to select individuals who may benefit from screening. This includes people with two or more first-degree relatives with pancreatic cancer, patients with Peutz-Jeghers syndrome, and BRCA 2, p16, and hereditary nonpolyposis colorectal cancer (HNPCC) mutation carriers with one or more affected first-degree relatives.
PanIN represents a spectrum of small (<5 mm) neoplastic but noninvasive precursor lesions of the pancreatic ductal epithelium demonstrating mild, moderate, or severe dysplasia (PanIN 1–3, respectively); however, not all PanIN lesions will progress to frank invasive malignancy. Cystic pancreatic tumors such as intraductal mucinous papillary neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs) are increasingly detected radiologically and are frequently asymptomatic. Main duct IPMNs are more likely to occur in older persons and have higher malignant potential than branched duct IPMNs (invasive cancer in 45% vs 18% of resected lesions, respectively). In contrast, MCNs are solitary lesions of the distal pancreas that do not communicate with the duct system. MCNs have an almost exclusive female distribution (95%). The rate of invasive cancer in resected MCNs is lower (<18%) with increased rates associated with larger tumors or the presence of nodules.
CLINICAL FEATURES
Clinical Presentation Obstructive jaundice occurs frequently when the cancer is located in the head of the pancreas. This may be accompanied by symptoms of abdominal discomfort, pruritus, lethargy, and weight loss. Less common presenting features include epigastric pain, backache, new-onset diabetes mellitus, and acute pancreatitis caused by pressure effects on the pancreatic duct. Nausea and vomiting, resulting from gastroduodenal obstruction, may also be a symptom of this disease.
Physical Signs Patients can present with jaundice and cachexia, and scratch marks may be present. Of patients with operable tumors, 25% have a palpable gallbladder (Courvoisier’s sign). Physical signs related to the development of distant metastases include hepatomegaly, ascites, left supraclavicular lymphadenopathy (Virchow’s node), and periumbilical nodules (Sister Mary Joseph’s nodes).
DIAGNOSIS
Diagnostic Imaging Patients who present with clinical features suggestive of pancreatic cancer undergo imaging to confirm the presence of a tumor and to establish whether the mass is likely to be inflammatory or malignant in nature. Other imaging objectives include the local and distant staging of the tumor, which will determine resectability and provide prognostic information. Dual-phase, contrast-enhanced spiral CT is the imaging modality of choice (Fig. 112-1). It provides accurate visualization of surrounding viscera, vessels, and lymph nodes, thus determining tumor resectability. Intestinal infiltration and liver and lung metastases are also reliably depicted on CT. There is no advantage of magnetic resonance imaging (MRI) over CT in predicting tumor resectability, but selected cases may benefit from MRI to characterize the nature of small indeterminate liver lesions and to evaluate the cause of biliary dilatation when no obvious mass is seen on CT. Endoscopic retrograde cholangiopancreatography (ERCP) is useful for revealing small pancreatic lesions, identifying stricture or obstruction in pancreatic or common bile ducts, and facilitating stent placement; however, it is associated with a risk of pancreatitis (Fig. 112-2). Magnetic resonance cholangiopancreatography (MRCP) is a noninvasive method for accurately depicting the level and degree of bile and pancreatic duct dilatation. EUS is highly sensitive in detecting lesions less than 3 cm in size (more sensitive than CT for lesions <2 cm) and is useful as a local staging tool for assessing vascular invasion and lymph node involvement. Fluorodeoxyglucose positron emission tomography (FDG-PET) should be considered before surgery or radical chemoradiotherapy (CRT), because it is superior to conventional imaging in detecting distant metastases.

FIGURE 112-1 Coronal computed tomography showing pancreatic cancer and dilated intrahepatic and pancreatic ducts (arrows).

FIGURE 112-2 Endoscopic retrograde cholangiopancreatography showing contrast in dilated pancreatic duct (arrows).
Tissue Diagnosis and Cytology Preoperative confirmation of malignancy is not always necessary in patients with radiologic appearances consistent with operable pancreatic cancer. However, EUS-guided fine-needle aspiration is the technique of choice when there is any doubt, and also for use in patients who require neoadjuvant treatment. It has an accuracy of approximately 90% and has a smaller risk of intraperitoneal dissemination compared with the percutaneous route. Percutaneous biopsy of the pancreatic primary or liver metastases is only acceptable in patients with inoperable or metastatic disease. ERCP is a useful method for obtaining ductal brushings, but the sensitivity of ERCP for diagnosis ranges from 35 to 70%.
Serum Markers Tumor-associated CA19-9 is elevated in approximately 70–80% of patients with pancreatic carcinoma but is not recommended as a routine diagnostic or screening test because its sensitivity and specificity are inadequate for accurate diagnosis. Preoperative CA19-9 levels correlate with tumor stage, and postresection CA19-9 level has prognostic value. It is an indicator of asymptomatic recurrence in patients with completely resected tumors and is used as a biomarker of response in patients with advanced disease undergoing chemotherapy. A number of studies have established a high pretreatment CA19-9 level as an independent prognostic factor.
STAGING
The American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) staging of pancreatic cancer takes into account the location and size of the tumor, the involvement of lymph nodes, and distant metastasis. This information is then combined to assign a stage (Fig. 112-3). From a practical standpoint, patients are grouped according to whether the cancer is resectable, locally advanced (unresectable, but without distant spread), or metastatic.

FIGURE 112-3 Staging of pancreatic cancer, and survival according to stage. AJCC, American Joint Committee on Cancer. (Illustration by Stephen Millward.)
TREATMENT PANCREATIC CANCER
RESECTABLE DISEASE
Approximately 10% of patients present with localized nonmetastatic disease that is potentially suitable for surgical resection. Approximately 30% of patients have R1 resection (microscopic residual disease) following surgery. Those who undergo R0 resection (no microscopic or macroscopic residual tumor) and who receive adjuvant treatment have the best chance of cure, with an estimated median survival of 20–23 months and a 5-year survival of approximately 20%. Outcomes are more favorable in patients with small (<3 cm), well-differentiated tumors and lymph node–negative disease.
Patients should have surgery in dedicated pancreatic centers that have lower postoperative morbidity and mortality rates. The standard surgical procedure for patients with tumors of the pancreatic head or uncinate process is a pylorus-preserving pancreaticoduodenectomy (modified Whipple’s procedure). The procedure of choice for tumors of the pancreatic body and tail is a distal pancreatectomy, which routinely includes splenectomy.
Postoperative treatment improves long-term outcomes in this group of patients. Adjuvant chemotherapy comprising six cycles of gemcitabine is common practice worldwide based on data from three randomized controlled trials (Table 112-1). The Charité Onkologie trial (CONKO 001) found that the use of gemcitabine after complete resection significantly delayed the development of recurrent disease compared with surgery alone. The European Study Group for Pancreatic Cancer 3 (ESPAC-3) trial, which investigated the benefit of adjuvant 5-fluorouracil/folinic acid (5-FU/FA) versus gemcitabine, revealed no survival difference between the two drugs. However, the toxicity profile of adjuvant gemcitabine was superior to 5-FU/FA by virtue of its lower incidence of stomatitis and diarrhea. Adjuvant radiotherapy is not commonly used in Europe based on the negative results of the ESPAC-1 study. Adjuvant 5-FU-based CRT with gemcitabine before and after radiotherapy as used in the Radiation Therapy Oncology Group (RTOG) 97-04 trial is preferred in the United States. This approach may be most beneficial in patients with bulky tumors involving the pancreatic head.
|
PHASE III STUDIES OF ADJUVANT CHEMOTHERAPY IN RESECTED PANCREATIC CANCER |

INOPERABLE LOCALLY ADVANCED DISEASE
Approximately 30% of patients present with locally advanced, unresectable, but nonmetastatic pancreatic carcinoma. The median survival with gemcitabine is 9 months. Patients who respond to chemotherapy or who achieve stable disease after 3–6 months of gemcitabine have frequently been offered consolidation radiotherapy. However, a large, phase III, randomized controlled trial, LAP-07, did not demonstrate any improvement in survival for patients treated with CRT after 4 months of disease control on either gemcitabine or a gemcitabine/erlotinib combination.
METASTATIC DISEASE
Approximately 60% of patients with pancreatic cancer present with metastatic disease. Patients with poor performance status do not usually benefit from chemotherapy. Gemcitabine was the standard treatment with a median survival of 6 months and a 1-year survival rate of only 20%. The addition of nab-paclitaxel (an albumin bound nanoparticle formulation of paclitaxel) to gemcitabine results in significantly improved 1-year survival compared to gemcitabine alone (35% vs 22%, p <.001). Capecitabine, an oral fluoropyrimidine, has also been combined with gemcitabine (GEM-CAP) in a phase III trial that showed an improvement in response rate and progression-free survival over single-agent gemcitabine, but no overall survival benefit. However, pooling of two other randomized controlled trials with this trial in a meta-analysis resulted in a survival advantage with GEM-CAP. Addition of erlotinib, a small-molecule epidermal growth factor receptor inhibitor, produced a statistically significant but clinically marginal benefit when added to gemcitabine in the advanced disease setting. A phase III trial limited to good performance status patients with metastatic pancreatic cancer showed improved survival with the combination of 5-FU/FA, irinotecan, and oxaliplatin (FOLFIRINOX) compared with gemcitabine, but with increased toxicity (Table 112-2).
|
SELECTED PHASE III STUDIES EVALUATING CHEMOTHERAPY TREATMENT IN ADVANCED PANCREATIC CANCER |

FUTURE DIRECTIONS
The early detection and future treatment of pancreatic cancer relies on an improved understanding of molecular pathways involved in the development of this disease. This will ultimately lead to the discovery of novel agents and the identification of patient groups who are likely to benefit most from targeted therapy.
ACKNOWLEDGMENT
Dr. Irene Chong is acknowledged for her work on this chapter in the 18th edition.
113 |
Endocrine Tumors of the Gastrointestinal Tract and Pancreas |
GENERAL FEATURES OF GASTROINTESTINAL NEUROENDOCRINE TUMORS
Gastrointestinal (GI) neuroendocrine tumors (NETs) are tumors derived from the diffuse neuroendocrine system of the GI tract; that system is composed of amine- and acid-producing cells with different hormonal profiles, depending on the site of origin. The tumors historically are divided into GI-NETs (in the GI tract) (also frequently called carcinoid tumors) and pancreatic neuroendocrine tumors (pNETs), although newer pathologic classifications have proposed that they all be classified as GI-NETs. The term GI-NET has been proposed to replace the term carcinoid; however, the term carcinoid is widely used, and many are not familiar with this change. Accordingly, this chapter will use the term GI-NETs (carcinoids). These tumors originally were classified as APUDomas (for amine precursor uptake and decarboxylation), as were pheochromocytomas, melanomas, and medullary thyroid carcinomas, because they share certain cytochemical features as well as various pathologic, biologic, and molecular features (Table 113-1). It was originally proposed that APUDomas had a similar embryonic origin from neural crest cells, but it is now known the peptide-secreting cells are not of neuroectodermal origin. Nevertheless, the concept of APUDomas is useful because these tumors have important similarities as well as some differences (Table 113-1). In this section, the areas of similarity between pNETs and GI-NETs (carcinoids) will be discussed together, and areas in which there are important differences will be discussed separately.
|
GENERAL CHARACTERISTICS OF GASTROINTESTINAL NEUROENDOCRINE TUMORS (GI-NETs [CARCINOIDS], PANCREATIC NEUROENDOCRINE TUMORS [pNETs]) |
Abbreviations: ATRX, alpha-thalassemia X-lined mental retardation protein; DAXX, death domain associated protein; MEN 1, multiple endocrine neoplasia type 1; TNM, tumor, node, metastasis.
CLASSIFICATION/PATHOLOGY/TUMOR BIOLOGY OF NETs
NETs generally are composed of monotonous sheets of small round cells with uniform nuclei, and mitoses are uncommon. They can be identified tentatively on routine histology; however, these tumors are now recognized principally by their histologic staining patterns due to shared cellular proteins. Historically, silver staining was used, and tumors were classified as showing an argentaffin reaction if they took up and reduced silver or as being argyrophilic if they did not reduce it. Currently, immunocytochemical localization of chromogranins (A, B, C), neuron-specific enolase, and synaptophysin, which are all neuroendocrine cell markers, is used (Table 113-1). Chromogranin A is the most widely used.
Ultrastructurally, these tumors possess electron-dense neurosecretory granules and frequently contain small clear vesicles that correspond to synaptic vesicles of neurons. NETs synthesize numerous peptides, growth factors, and bioactive amines that may be ectopically secreted, giving rise to a specific clinical syndrome (Table 113-2). The diagnosis of the specific syndrome requires the clinical features of the disease (Table 113-2) and cannot be made from the immunocytochemistry results alone. The presence or absence of a specific clinical syndrome also cannot be predicted from the immunocytochemistry alone (Table 113-1). Furthermore, pathologists cannot distinguish between benign and malignant NETs unless metastasis or invasion is present.
|
GASTROINTESTINAL NEUROENDOCRINE TUMOR SYNDROMES |

GI-NETs (carcinoids) frequently are classified according to their anatomic area of origin (i.e., foregut, midgut, hindgut) because tumors with similar areas of origin share functional manifestations, histochemistry, and secretory products (Table 113-3). Foregut tumors generally have a low serotonin (5-HT) content; are argentaffin-negative but argyrophilic; occasionally secrete adrenocorticotropic hormone (ACTH) or 5-hydroxytryptophan (5-HTP), causing an atypical carcinoid syndrome (Fig. 113-1); are often multihormonal; and may metastasize to bone. They uncommonly produce a clinical syndrome due to the secreted products. Midgut carcinoids are argentaffin-positive, have a high serotonin content, most frequently cause the typical carcinoid syndrome when they metastasize (Table 113-3, Fig. 113-1), release serotonin and tachykinins (substance P, neuropeptide K, substance K), rarely secrete 5-HTP or ACTH, and less commonly metastasize to bone. Hindgut carcinoids (rectum, transverse and descending colon) are argentaffin-negative, are often argyrophilic, rarely contain serotonin or cause the carcinoid syndrome (Fig. 113-1, Table 113-3), rarely secrete 5-HTP or ACTH, contain numerous peptides, and may metastasize to bone.
|
GI-NET (CARCINOID) LOCATION, FREQUENCY OF METASTASES, AND ASSOCIATION WITH THE CARCINOID SYNDROME |


FIGURE 113-1 Synthesis, secretion, and metabolism of serotonin (5-HT) in patients with typical and atypical carcinoid syndromes. 5-HIAA, 5-hydroxyindolacetic acid.
pNETs can be classified into nine well-established specific functional syndromes (Table 113-2), six additional very rare specific functional syndromes (less than five cases described), five possible specific functional syndromes (pNETs secreting calcitonin, neurotensin, pancreatic polypeptide, ghrelin) (Table 113-2), and nonfunctional pNETs. Other functional hormonal syndromes due to nonpancreatic tumors (usually intraabdominal in location) have been described only rarely and are not included in (Table 113-2). These include secretion by intestinal and ovarian tumors of peptide tyrosine tyrosine (PYY), which results in altered motility and constipation, and ovarian tumors secreting renin or aldosterone causing alterations in blood pressure or somatostatin causing diabetes or reactive hypoglycemia. Each of the functional syndromes listed in Table 113-2 is associated with symptoms due to the specific hormone released. In contrast, nonfunctional pNETs release no products that cause a specific clinical syndrome. “Nonfunctional” is a misnomer in the strict sense because those tumors frequently ectopically secrete a number of peptides (pancreatic polypeptide [PP], chromogranin A, ghrelin, neurotensin, α subunits of human chorionic gonadotropin, and neuron-specific enolase); however, they cause no specific clinical syndrome. The symptoms caused by nonfunctional pNETs are entirely due to the tumor per se. pNETs frequently ectopically secrete PP (60–85%), neurotensin (30–67%), calcitonin (30–42%), and to a lesser degree, ghrelin (5–65%). Whereas a few studies have proposed their secretion can cause a specific functional syndrome, most studies support the conclusion that their ectopic secretion is not associated with a specific clinical syndrome, and thus they are listed in Table 113-2 as possible clinical syndromes. Because a large proportion of nonfunctional pNETs (60–90%) secrete PP, these tumors are often referred to as PPomas (Table 113-2).
GI-NETs (carcinoids) can occur in almost any GI tissue (Table 113-3); however, at present, most (70%) have their origin in one of three sites: bronchus, jejunoileum, or colon/rectum. In the past, GI-NET (carcinoids) most frequently were reported in the appendix (i.e., 40%); however, the bronchus/lung, rectum, and small intestine are now the most common sites. Overall, the GI tract is the most common site for these tumors, accounting for 64%, with the respiratory tract a distant second at 28%. Both race and sex can affect the frequency as well as the distribution of GI-NETs (carcinoids). African Americans have a higher incidence of carcinoids. Race is particularly important for rectal carcinoids, which are found in 41% of Asians/Pacific Islanders with NETs compared to 32% of American Indians/Alaskan natives, 26% of African Americans, and 12% of white Americans. Females have a lower incidence of small intestinal and pancreatic carcinoids.
The term pancreatic neuroendocrine or endocrine tumor, although widely used and therefore retained here, is also a misnomer, strictly speaking, because these tumors can occur either almost entirely in the pancreas (insulinomas, glucagonomas, nonfunctional pNETs, pNETs causing hypercalcemia) or at both pancreatic and extrapancreatic sites (gastrinomas, VIPomas [vasoactive intestinal peptide], somatostatinomas, GRFomas [growth hormone–releasing factor]). pNETs are also called islet cell tumors; however, the use of this term is discouraged because it is not established that they originate from the islets, and many can occur at extrapancreatic sites.
Whereas the classification of GI neuroendocrine tumors into foregut, midgut, or hindgut is widely used and generally useful because the NETs within these areas have many similarities, they also have marked differences, particularly in biologic behavior, and it has not proved useful for prognostic purposes. More general classifications have been developed that allow NETs with similar features in different locations to be compared, have proven prognostic value, and are widely used. New classification systems have been developed for both GI-NETs (carcinoids) and pNETs by the World Health Organization (WHO), European Neuroendocrine Tumor Society (ENETS), and the American Joint Committee on Cancer/International Union Against Cancer (AJCC/UICC). Although there are some differences between these different classification systems, each uses similar information, and it is now recommended that the basic data underlying the classification be included in all standard pathology reports. These classification systems divide NETs from all sites into those that are well differentiated (low grade [G1] or intermediate grade [G2]) and those that are poorly differentiated (high grade [G3] divided into either small-cell carcinoma or large-cell neuroendocrine carcinoma). In these classification systems, both pNETs and GI-NETs (carcinoids) are classified as neuroendocrine tumors, and the old term of carcinoid is equivalent to well-differentiated neuroendocrine tumors of the GI tract. These classification systems are based on not only the differentiation of the NET, but also a grading system assessing proliferative indices (Ki-67 and the mitotic count). NETs are considered low grade (ENETS G1) if the Ki-67 is <3% and the mitotic count is <2 mitoses/high-power field (HPF), intermediate grade (ENETS G2) if the Ki-67 is 3–20% and the mitotic count is 2–20 mitoses/HPF, and high grade (ENETS G3) if the Ki-67 is >20% and the mitotic count is >20 mitoses/HPF. In addition to the grading system, a TNM classification has been proposed that is based on the level of tumor invasion, tumor size, and tumor extent (see Table 113-4 for an example with pNETs and appendiceal GI-NETs [carcinoids]). Because of the proven prognostic value of these classification and grading systems, as well as the fact that NETs with different classifications/grades respond differently to treatments, the systems are now essential for the management of all NETs.
|
COMPARISON OF THE CRITERIA FOR THE TUMOR CATEGORY IN THE ENETS AND SEVENTH EDITION AJCC TNM CLASSIFICATIONS OF PANCREATIC AND APPENDICEAL NETs |

In addition to these classification/grading systems, a number of other factors have been identified that provide important prognostic information that can guide treatment (Table 113-5).
|
PROGNOSTIC FACTORS IN NEUROENDOCRINE TUMORS |
Abbreviations: 5-HIAA, 5-hydroxyindoleacetic acid; AJCC, American Joint Committee on Cancer; chr, chromosome; EGF, epidermal growth factor; FGFR, fibroblast growth factor receptor; GI-NET, gastrointestinal neuroendocrine tumor; IHC, immunohistochemistry; Ki-67, proliferation-associated nuclear antigen recognized by Ki-67 monoclonal antibody; LOH, loss of heterozygosity; MEN, multiple endocrine neoplasia; NET, neuroendocrine tumors; PCNA, proliferating cell nuclear antigen; pNET, pancreatic neuroendocrine tumor; PTEN, phosphatase and tensin homologue deleted from chromosome 10; TGF-α, transforming growth factor α; TNM, tumor, node, metastasis; UICC, International Union Against Cancer; VEGF, vascular endothelial growth factor; WHO, World Health Organization.
The exact incidence of GI-NETs (carcinoids) or pNETs varies according to whether only symptomatic tumors or all tumors are considered. The incidence of clinically significant carcinoids is 7–13 cases/million population per year, whereas any malignant carcinoids at autopsy are reported in 21–84 cases/million population per year. The incidence of GI-NETs (carcinoids) is approximately 25–50 cases per million in the United States, which makes them less common than adenocarcinomas of the GI tract. However, their incidence has increased sixfold in the last 30 years. In an analysis of 35,825 GI-NETs (carcinoids) (2004) from the U.S. Surveillance, Epidemiology, and End Results (SEER) database, their incidence was 5.25/100,000 per year, and the 29-year prevalence was 35/100,000. Clinically significant pNETs have a prevalence of 10 cases/million population, with insulinomas, gastrinomas, and nonfunctional pNETs having an incidence of 0.5–2 cases/million population per year (Table 113-2). pNETs account for 1–10% of all tumors arising in the pancreas and 1.3% of tumors in the SEER database, which consists primarily of malignant tumors. VIPomas are 2–8 times less common, glucagonomas are 17–30 times less common, and somatostatinomas are the least common. In autopsy studies, 0.5–1.5% of all cases have a pNET; however, in less than 1 in 1000 cases was a functional tumor thought to occur.
Both GI-NETs (carcinoids) and pNETs commonly show malignant behavior (Tables 113-2 and 113-3). With pNETs, except for insulinomas in which <10% are malignant, 50–100% in different series are malignant. With GI-NETs (carcinoids), the percentage showing malignant behavior varies in different locations (Table 113-3). For the three most common sites of occurrence, the incidence of metastases varies greatly from the jejunoileum (58%), lung/bronchus (6%), and rectum (4%) (Table 113-3). With both GI-NETs (carcinoids) and pNETs, a number of factors (Table 113-5) are important prognostic factors in determining survival and the aggressiveness of the tumor. Patients with pNETs (excluding insulinomas) generally have a poorer prognosis than do patients with GI-NETs (carcinoids). The presence of liver metastases is the single most important prognostic factor in single and multivariate analyses for both GI-NETs (carcinoids) and pNETs. Particularly important in the development of liver metastases is the size of the primary tumor. For example, with small intestinal carcinoids, which are the most common cause of the carcinoid syndrome due to metastatic disease in the liver (Table 113-2), metastases occur in 15–25% if the tumor is <1 cm in diameter, 58–80% if it is 1–2 cm in diameter, and >75% if it is >2 cm in diameter. Similar data exist for gastrinomas and other pNETs; the size of the primary tumor is an independent predictor of the development of liver metastases. The presence of lymph node metastases or extrahepatic metastases; the depth of invasion; the rapid rate of growth; various histologic features (differentiation, mitotic rates, growth indices, vessel density, vascular endothelial growth factor [VEGF], and CD10 metalloproteinase expression); necrosis; presence of cytokeratin; elevated serum alkaline phosphatase levels; older age; presence of circulating tumor cells; and flow cytometric results, such as the presence of aneuploidy, are all important prognostic factors for the development of metastatic disease (Table 113-5). For patients with GI-NETs (carcinoids), additional associations with a worse prognosis include the development of the carcinoid syndrome (especially the development of carcinoid heart disease), male sex, the presence of a symptomatic tumor or greater increases in a number of tumor markers (5-hydroxyindolacetic acid [5-HIAA], neuropeptide K, chromogranin A), and the presence of various molecular features. With pNETs or gastrinomas, a worse prognosis is associated with female sex, overexpression of the Ha-ras oncogene or p53, the absence of multiple endocrine neoplasia type 1 (MEN 1), higher levels of various tumor markers (i.e., chromogranin A, gastrin), and presence of various histologic features (immunohistochemistry for c-KIT, low cyclin B1, loss of PTEN/TSC-2, expression of fibroblast growth factor-13) and various molecular features (Table 113-5). The TNM classification systems and the grading systems (G1–G3) have important prognostic value.
A number of diseases due to various genetic disorders are associated with an increased incidence of NETs (Table 113-6). Each one is caused by a loss of a possible tumor-suppressor gene. The most important is MEN 1, which is an autosomal dominant disorder due to a defect in a 10-exon gene on 11q13, which encodes for a 610-amino-acid nuclear protein, menin (Chap. 408). Patients with MEN 1 develop hyperparathyroidism due to parathyroid hyperplasia in 95–100% of cases, pNETs in 80–100%, pituitary adenomas in 54–80%, adrenal adenomas in 27–36%, bronchial carcinoids in 8%, thymic carcinoids in 8%, gastric carcinoids in 13–30% of patients with Zollinger-Ellison syndrome, skin tumors (angiofibromas [88%], collagenomas [72%]), central nervous system (CNS) tumors (meningiomas [<8%]), and smooth-muscle tumors (leiomyomas, leiomyosarcomas [1–7%]). Among patients with MEN 1, 80–100% develop nonfunctional pNETs (most are microscopic with 0–13% large/symptomatic), and functional pNETs occur in 20–80% in different series, with a mean of 54% developing Zollinger-Ellison syndrome, 18% insulinomas, 3% glucagonomas, 3% VIPomas, and <1% GRFomas or somatostatinomas. MEN 1 is present in 20–25% of all patients with Zollinger-Ellison syndrome, 4% of patients with insulinomas, and a low percentage (<5%) of patients with other pNETs.
|
GENETIC SYNDROMES ASSOCIATED WITH AN INCREASED INCIDENCE OF NEUROENDOCRINE TUMORS (NETS) (GI-NETS [CARCINOIDS] OR PNETS) |

Three phacomatoses associated with NETs are von Hippel–Lindau disease (VHL), von Recklinghausen’s disease (neurofibromatosis type 1 [NF-1]), and tuberous sclerosis (Bourneville’s disease) (Table 113-6). VHL is an autosomal dominant disorder due to defects on chromosome 3p25, which encodes for a 213-amino-acid protein that interacts with the elongin family of proteins as a transcriptional regulator (Chaps. 118, 339, 407, and 408). In addition to cerebellar hemangioblastomas, renal cancer, and pheochromocytomas, 10–17% develop a pNET. Most are nonfunctional, although insulinomas and VIPomas have been reported. Patients with NF-1 (von Recklinghausen’s disease) have defects in a gene on chromosome 17q11.2 that encodes for a 2845-amino-acid protein, neurofibromin, which functions in normal cells as a suppressor of the ras signaling cascade (Chap. 118). Up to 10% of these patients develop an upper GI-NET (carcinoid), characteristically in the periampullary region (54%). Many are classified as somatostatinomas because they contain somatostatin immunocytochemically; however, they uncommonly secrete somatostatin and rarely produce a clinical somatostatinoma syndrome. NF-1 has rarely been associated with insulinomas and Zollinger-Ellison syndrome. NF-1 accounts for 48% of all duodenal somatostastinomas and 23% of all ampullary GI-NETs (carcinoids). Tuberous sclerosis is caused by mutations that alter either the 1164-amino-acid protein hamartin (TSC1) or the 1807-amino-acid protein tuberin (TSC2) (Chap. 118). Both hamartin and tuberin interact in a pathway related to phosphatidylinositol 3-kinases and mammalian target of rapamycin (mTOR) signaling cascades. A few cases including nonfunctional and functional pNETs (insulinomas and gastrinomas) have been reported in these patients (Table 113-6). Mahvash disease is associated with the development of α-cell hyperplasia, hyperglucagonemia, and the development of NF pNETs and is due to a homozygous P86S mutation of the human glucagon receptor.
Mutations in common oncogenes (ras, myc, fos, src, jun) or common tumor-suppressor genes (p53, retinoblastoma susceptibility gene) are not commonly found in either pNETs or GI-NETs (carcinoids) (Table 113-1). However, frequent (70%) gene amplifications in MDM2, MDM4, and WIPI inactivating the p53 pathway are noted in well-differentiated pNETs, and the retinoblastoma pathway is altered in the majority of pNETs. In addition to these genes, additional alterations that may be important in their pathogenesis include changes in the MEN1 gene, p16/MTS1 tumor-suppressor gene, and DPC4/Smad4 gene; amplification of the HER-2/neu protooncogene; alterations in transcription factors (Hoxc6 [GI carcinoids]), growth factors, and their receptors; methylation of a number of genes that probably results in their inactivation; and deletions of unknown tumor-suppressor genes as well as gains in other unknown genes (Table 113-1). The clinical antitumor activity of everolimus, an mTOR inhibitor, and sunitinib, a tyrosine kinase inhibitor (PDGFR, VEGFR1, VEGFR2, c-KIT, FLT-3), support the importance of the mTOR-AKT pathway and tyrosine kinase receptors in mediating growth of malignant NETs (especially pNETs). The importance of the mTOR pathway in pNET growth is further supported by the finding that a single-nucleotide polymorphism (FGFR4-G388R, in fibroblast growth factor receptor 4) affects selectivity to the mTOR inhibitor and can result in significantly higher risk of advanced pNET stage and liver metastases (Table 113-5). Comparative genomic hybridization, genome-wide allelotyping studies, and genome-wide single-nucleotide polymorphism analyses have shown that chromosomal losses and gains are common in pNETs and GI-NETs (carcinoids), but they differ between these two NETs, and some have prognostic significance (Table 113-5). Mutations in the MEN1 gene are probably particularly important. Loss of heterozygosity at the MEN 1 locus on chromosome 11q13 is noted in 93% of sporadic pNETs (i.e., in patients without MEN 1) and in 26–75% of sporadic GI-NETs (carcinoids). Mutations in the MEN1 gene are reported in 31–34% of sporadic gastrinomas. Exomic sequencing of sporadic pNETs found that the most frequently altered gene was MEN1, occurring in 44% of patients, followed by mutations in 43% of patients in genes encoding for two subunits of a transcription/chromatin remodeling complex consisting of DAXX (death-domain-associated protein) and ATRX (α-thalassemia/mental retardation syndrome X-linked) and in 15% of patients in the mTOR pathway. The presence of a number of these molecular alterations in pNETs or GI-NETs (carcinoids) correlates with tumor growth, tumor size, and disease extent or invasiveness and may have prognostic significance (Table 113-5).
GI-NETs (CARCINOIDS) AND CARCINOID SYNDROME
CHARACTERISTICS OF THE MOST COMMON GI-NETs (CARCINOIDS)
Appendiceal NETs (Carcinoids) Appendiceal NETs (carcinoids) occur in 1 in every 200–300 appendectomies, usually in the appendiceal tip, have an incidence of 0.15/100,000 per year, comprise 2–5% of all GI-NETs (carcinoids), and comprise 32–80% of all appendiceal tumors. Most (i.e., >90%) are <1 cm in diameter without metastases in older studies, but more recently, 2–35% have had metastases (Table 113-3). In the SEER data of 1570 appendiceal carcinoids, 62% were localized, 27% had regional metastases, and 8% had distant metastases. The risk of metastases increases with size, with those <1 cm having a 0 to <10% risk of metastases and those >2 cm having a 25–44% risk. Besides tumor size, other important prognostic factors for metastases include basal location, invasion of mesoappendix, poor differentiation, advanced stage or WHO/ENETS classification, older age, and positive resection margins. The 5-year survival is 88–100% for patients with localized disease, 78–100% for patients with regional involvement, and 12–28% for patients with distal metastases. In patients with tumors <1 cm in diameter, the 5-year survival is 95–100%, whereas it is 29% if tumors are >2 cm in diameter. Most tumors are well-differentiated G1 tumors (87%) (Table 113-4), with the remainder primarily well-differentiated G2 tumors (13%); poorly differentiated G3 tumors are uncommon (<1%). Their percentage of the total number of carcinoids decreased from 43.9% (1950–1969) to 2.4% (1992–1999). Appendiceal goblet cell (GC) NETs (carcinoids)/carcinomas are a rare subtype (<5%) that are mixed adeno-neuroendocrine carcinomas. They are malignant and are thought to comprise a distinct entity; they frequently present with advanced disease and are recommended to be treated as adenocarcinomas, not carcinoid tumors.
SMALL INTESTINAL NETs (CARCINOIDS)
Small intestinal (SI) NETs (carcinoids) have a reported incidence of 0.67/100,000 in the United States, 0.32/100,000 in England, and 1.12/100,000 in Sweden and comprise >50% of all SI tumors. There is a male predominance (1.5:1), and race affects frequency, with a lower frequency in Asians and greater frequency in African Americans. The mean age of presentation is 52–63 years, with a wide range (1–93 years). Familial SI carcinoid families exist but are very uncommon. These are frequently multiple; 9–18% occur in the jeunum, 70–80% are present in the ileum, and 70% occur within 6 cm (2.4 in.) of the ileocecal valve. Forty percent are <1 cm in diameter, 32% are 1–2 cm, and 29% are >2 cm. They are characteristically well differentiated; however, they are generally invasive, with 1.2% being intramucosal in location, 27% penetrating the submucosa, and 20% invading the muscularis propria. Metastases occur in a mean of 47–58% (range 20–100%). Liver metastases occur in 38%, to lymph nodes in 37% and more distant in 20–25%. They characteristically cause a marked fibrotic reaction, which can lead to intestinal obstruction. Tumor size is an important variable in the frequency of metastases. However, even small NETs (carcinoids) of the small intestine (<1 cm) have metastases in 15–25% of cases, whereas the proportion increases to 58–100% for tumors 1–2 cm in diameter. Carcinoids also occur in the duodenum, with 31% having metastases. Duodenal tumors <1 cm virtually never metastasize, whereas 33% of those >2 cm had metastases. SI NETs (carcinoids) are the most common cause (60–87%) of the carcinoid syndrome and are discussed in a later section (Table 113-7). Important prognostic factors are listed in (Table 113-5), and particularly important are the tumor extent, proliferative index by grading, and stage (Table 113-4). The overall survival at 5 years is 55–75%; however, it varies markedly with disease extent, being 65–90% with localized disease, 66–72% with regional involvement, and 36–43% with distant disease.
|
CLINICAL CHARACTERISTICS IN PATIENTS WITH CARCINOID SYNDROME |

Rectal NETs (Carcinoids) Rectal NETs (carcinoids) comprise 27% of all GI-NETs (carcinoids) and 16% of all NETs and are increasing in frequency. In the U.S. SEER data, they currently have an incidence of 0.86/100,000 per year (up from 0.2/100,000 per year in 1973) and represent 1–2% of all rectal tumors. They are found in approximately 1 in every 1500/2500 proctoscopies/colonoscopies or 0.05–0.07% of individuals undergoing these procedures. Nearly all occur between 4 and 13 cm above the dentate line. Most are small, with 66–80% being <1 cm in diameter, and rarely metastasize (5%). Tumors between 1 and 2 cm can metastasize in 5–30%, and those >2 cm, which are uncommon, in >70%. Most invade only to the submucosa (75%), with 2.1% confined to the mucosa, 10% to the muscular layer, and 5% to adjacent structures. Histologically, most are well differentiated (98%) with 72% ENETS/WHO grade G1 and 28% grade G2 (Table 113-4). Overall survival is 88%; however, it is very much dependent of the stage, with 5-year survival of 91% for localized disease, 36–49% for regional disease, and 20–32% for distant disease. Risk factors are listed in Table 113-5 and particularly include tumor size, depth of invasion, presence of metastases, differentiation, and recent TNM classification and grade.
Bronchial NETs (Carcinoids) Bronchial NETs (carcinoids) comprise 25–33% of all well-differentiated NETs and 90% of all the poorly differentiated NETs found, likely due to a strong association with smoking. Their incidence ranges from 0.2 to 2/100,000 per year in the United States and European countries and is increasing at a rate of 6% per year. They are slightly more frequent in females and in whites compared with those of Hispanic/Asian/African descent, and are most commonly seen in the sixth decade of life, with a younger age of presentation for typical carcinoids (45 years) compared to atypical carcinoids (55 years).
A number of different classifications of bronchial GI-NETs (carcinoids) have been proposed. In some studies, they are classified into four categories: typical carcinoid (also called bronchial carcinoid tumor, Kulchitsky cell carcinoma I [KCC-I]), atypical carcinoid (also called well-differentiated neuroendocrine carcinoma [KC-II]), intermediate small-cell neuroendocrine carcinoma, and small-cell neuroendocarcinoma (KC-III). Another proposed classification includes three categories of lung NETs: benign or low-grade malignant (typical carcinoid), low-grade malignant (atypical carcinoid), and high-grade malignant (poorly differentiated carcinoma of the large-cell or small-cell type). The WHO classification includes four general categories: typical carcinoid, atypical carcinoid, large-cell neuroendocrine carcinoma, and small-cell carcinoma. The ratio of typical to atypical carcinoids is 8–10:1, with the typical carcinoids comprising 1–2% of lung tumors, atypical 0.1–0.2%, large-cell neuroendocrine tumors 0.3%, and small-cell lung cancer 9.8% of all lung tumors. These different categories of lung NETs have different prognoses, varying from excellent for typical carcinoid to poor for small-cell neuroendocrine carcinomas. The occurrence of large-cell and small-cell lung carcinoids, but not typical or atypical lung carcinoids, is related to tobacco use. The 5-year survival is very much influenced by the classification of the tumor, with survival of 92–100% for patients with a typical carcinoid, 61–88% with an atypical carcinoid, 13–57% with a large-cell neuroendocrine tumor, and 5% with a small-cell lung cancer.
Gastric NET (Carcinoids) Gastric NETs (carcinoids) account for 3 of every 1000 gastric neoplasms and 1.3–2% of all carcinoids, and their relative frequency has increased three- to fourfold over the last five decades (2.2% in 1950 to 9.6% in 2000–2007, SEER data). At present, it is unclear whether this increase is due to better detection with the increased use of upper GI endoscopy or to a true increase in incidence. Gastric NETs (carcinoids) are classified into three different categories, and this has important implications for pathogenesis, prognosis, and treatment. Each originates from gastric enterochromaffin-like (ECL) cells, one of the six types of gastric neuroendocrine cells, in the gastric mucosa. Two subtypes are associated with hypergastrinemic states, either chronic atrophic gastritis (type I) (80% of all gastric NETs [carcinoids]) or Zollinger-Ellison syndrome, which is almost always a part of the MEN 1 syndrome (type II) (6% of all cases). These tumors generally pursue a benign course, with type I uncommonly (<10%) associated with metastases, whereas type II tumors are slightly more aggressive, with 10–30% associated with metastases. They are usually multiple, small, and infiltrate only to the submucosa. The third subtype of gastric NETs (carcinoids) (type III) (sporadic) occurs without hypergastrinemia (14–25% of all gastric carcinoids) and has an aggressive course, with 54–66% developing metastases. Sporadic carcinoids are usually single, large tumors; 50% have atypical histology, and they can be a cause of the carcinoid syndrome. Five-year survival is 99–100% in patients with type I, 60–90% in patients with type II, and 50% in patients with type III gastric NETs (carcinoids).
CLINICAL PRESENTATION OF NETs (CARCINOIDS)
GI/Lung NET (Carcinoid) Without the Carcinoid Syndrome The age of patients at diagnosis ranges from 10 to 93 years, with a mean age of 63 years for the small intestine and 66 years for the rectum. The presentation is diverse and is related to the site of origin and the extent of malignant spread. In the appendix, NETs (carcinoids) usually are found incidentally during surgery for suspected appendicitis. SI NETs (carcinoids) in the jejunoileum present with periodic abdominal pain (51%), intestinal obstruction with ileus/invagination (31%), an abdominal tumor (17%), or GI bleeding (11%). Because of the vagueness of the symptoms, the diagnosis usually is delayed approximately 2 years from onset of the symptoms, with a range up to 20 years. Duodenal, gastric, and rectal NETs (carcinoids) are most frequently found by chance at endoscopy. The most common symptoms of rectal carcinoids are melena/bleeding (39%), constipation (17%), and diarrhea (12%). Bronchial NETs (carcinoids) frequently are discovered as a lesion on a chest radiograph, and 31% of the patients are asymptomatic. Thymic NETs (carcinoids) present as anterior mediastinal masses, usually on chest radiograph or computed tomography (CT) scan. Ovarian and testicular NETs (carcinoids) usually present as masses discovered on physical examination or ultrasound. Metastatic NETs (carcinoids) in the liver frequently presents as hepatomegaly in a patient who may have minimal symptoms and nearly normal liver function test results.
GI-NETs (CARCINOIDS) WITH SYSTEMIC SYMPTOMS DUE TO SECRETED PRODUCTS
GI/lung NETs (carcinoids) immunocytochemically can contain numerous GI peptides: gastrin, insulin, somatostatin, motilin, neurotensin, tachykinins (substance K, substance P, neuropeptide K), glucagon, gastrin-releasing peptide, vasoactive intestinal peptide (VIP), PP, ghrelin, other biologically active peptides (ACTH, calcitonin, growth hormone), prostaglandins, and bioactive amines (serotonin). These substances may or may not be released in sufficient amounts to cause symptoms. In various studies of patients with GI-NETs (carcinoids), elevated serum levels of PP were found in 43%, motilin in 14%, gastrin in 15%, and VIP in 6%. Foregut NETs (carcinoids) are more likely to produce various GI peptides than are midgut NETs (carcinoids). Ectopic ACTH production causing Cushing’s syndrome is seen increasingly with foregut carcinoids (respiratory tract primarily) and, in some series, has been the most common cause of the ectopic ACTH syndrome, accounting for 64% of all cases. Acromegaly due to growth hormone–releasing factor release occurs with foregut NETs (carcinoids), as does the somatostatinoma syndrome, but rarely occurs with duodenal NETs (carcinoids). The most common systemic syndrome with GI-NETs (carcinoids) is the carcinoid syndrome, which is discussed in detail in the next section.
CARCINOID SYNDROME
Clinical Features The cardinal features from a number of series at presentation as well as during the disease course are shown in Table 113-7. Flushing and diarrhea are the two most common symptoms, occurring in a mean of 69–70% of patients initially and in up to 78% of patients during the course of the disease. The characteristic flush is of sudden onset; it is a deep red or violaceous erythema of the upper body, especially the neck and face, often associated with a feeling of warmth and occasionally associated with pruritus, lacrimation, diarrhea, or facial edema. Flushes may be precipitated by stress; alcohol; exercise; certain foods, such as cheese; or certain agents, such as catecholamines, pentagastrin, and serotonin reuptake inhibitors. Flushing episodes may be brief, lasting 2–5 min, especially initially, or may last hours, especially later in the disease course. Flushing usually is associated with metastatic midgut NETs (carcinoids) but can also occur with foregut NETs (carcinoids). With bronchial NETs (carcinoids), the flushes frequently are prolonged for hours to days, reddish in color, and associated with salivation, lacrimation, diaphoresis, diarrhea, and hypotension. The flush associated with gastric NETs (carcinoids) can also be reddish in color, but with a patchy distribution over the face and neck, although the classic flush seen with midgut NETs (carcinoids) can also be seen with gastric NETs (carcinoids). It may be provoked by food and have accompanying pruritus.
Diarrhea usually occurs with flushing (85% of cases). The diarrhea usually is described as watery, with 60% of patients having <1 L/d of diarrhea. Steatorrhea is present in 67%, and in 46%, it is >15 g/d (normal <7 g). Abdominal pain may be present with the diarrhea or independently in 10–34% of cases.
Cardiac manifestations occur initially in 11–40% (mean 26%) of patients with carcinoid syndrome and in 14–41% (mean 30%) at some time in the disease course. The cardiac disease is due to the formation of fibrotic plaques (composed of smooth-muscle cells, myofibroblasts, and elastic tissue) involving the endocardium, primarily on the right side, although lesions on the left side also occur occasionally, especially if a patent foramen ovale exists. The dense fibrous deposits are most commonly on the ventricular aspect of the tricuspid valve and less commonly on the pulmonary valve cusps. They can result in constriction of the valves, and pulmonic stenosis is usually predominant, whereas the tricuspid valve is often fixed open, resulting in regurgitation predominating. Overall, in patients with carcinoid heart disease, 90–100% have tricuspid insufficiency, 43–59% have tricuspid stenosis, 50–81% have pulmonary insufficiency, 25–59% have pulmonary stenosis, and 11% (0–25%) left-side lesions. Up to 80% of patients with cardiac lesions develop heart failure. Lesions on the left side are much less extensive, occur in 30% at autopsy, and most frequently affect the mitral valve. Up to 80% of patients with cardiac lesions have evidence of heart failure. At diagnosis in various series, 27–43% of patients are in New York Heart Association class I, 30–40% are in class II, 13–31% are in class III, and 3–12% are in class IV. At present, carcinoid heart disease is reported to be decreasing in frequency and severity, with mean occurrence in 20% of patients and occurrence in as few as 3–4% in some reports. Whether this decrease is due to the widespread use of somatostatin analogues, which control the release of bioactive agents thought involved in mediating the heart disease, is unclear.
Other clinical manifestations include wheezing or asthma-like symptoms (8–18%), pellagra-like skin lesions (2–25%), and impaired cognitive function. A variety of noncardiac problems due to increased fibrous tissue have been reported, including retroperitoneal fibrosis causing urethral obstruction, Peyronie’s disease of the penis, intraabdominal fibrosis, and occlusion of the mesenteric arteries or veins.
Pathobiology Carcinoid syndrome occurred in 8% of 8876 patients with GI-NETs (carcinoids), with a rate of 1.7–18.4% in different studies. It occurs only when sufficient concentrations of products secreted by the tumor reach the systemic circulation. In 91–100% of cases, this occurs after distant metastases to the liver. Rarely, primary GI-NETs (carcinoids) with nodal metastases with extensive retroperitoneal invasion, pNETs (carcinoids) with retroperitoneal lymph nodes, or NETs (carcinoids) of the lung or ovary with direct access to the systemic circulation can cause the carcinoid syndrome without hepatic metastases. All GI-NETs (carcinoids) do not have the same propensity to metastasize and cause the carcinoid syndrome (Table 113-3). Midgut NETs (carcinoids) account for 57–67% of cases of carcinoid syndrome, foregut NETs (carcinoids) for 0–33%, hindgut for 0–8%, and an unknown primary location for 2–26% (Tables 113-3 and 113-7).
One of the main secretory products of GI-NETs (carcinoids) involved in the carcinoid syndrome is serotonin (5-HT) (Fig. 113-1), which is synthesized from tryptophan. Up to 50% of dietary tryptophan can be used in this synthetic pathway by tumor cells, and this can result in inadequate supplies for conversion to niacin; hence, some patients (2.5%) develop pellagra-like lesions. Serotonin has numerous biologic effects, including stimulating intestinal secretion with inhibition of absorption, stimulating increases in intestinal motility, and stimulating fibrogenesis. In various studies, 56–88% of all GI-NETs (carcinoids) were associated with serotonin overproduction; however, 12–26% of the patients did not have the carcinoid syndrome. In one study, platelet serotonin was elevated in 96% of patients with midgut NETs (carcinoids), 43% with foregut tumors, and 0% with hindgut tumors. In 90–100% of patients with the carcinoid syndrome, there is evidence of serotonin overproduction. Serotonin is thought to be predominantly responsible for the diarrhea. Patients with the carcinoid syndrome have increased colonic motility with a shortened transit time and possibly a secretory/absorptive alteration that is compatible with the known actions of serotonin in the gut mediated primarily through 5-HT3 and, to a lesser degree, 5-HT4 receptors. Serotonin receptor antagonists (especially 5-HT3 antagonists) relieve the diarrhea in many, but not all, patients. A tryptophan 5-hydroxylase inhibitor, LX-1031, which inhibits serotonin synthesis in peripheral tissues, is reported to cause a 44% decrease in bowel movement frequency and a 20% improvement in stool form in patients with the carcinoid syndrome. Additional studies suggest that tachykinins may be important mediators of diarrhea in some patients. In one study, plasma tachykinin levels correlated with symptoms of diarrhea. Serotonin does not appear to be involved in the flushing because serotonin receptor antagonists do not relieve flushing. In patients with gastric carcinoids, the characteristic red, patchy pruritic flush is thought due to histamine release because H1 and H2 receptor antagonists can prevent it. Numerous studies have shown that tachykinins (substance P, neuropeptide K) are stored in GI-NETs (carcinoids) and released during flushing. However, some studies have demonstrated that octreotide can relieve the flushing induced by pentagastrin in these patients without altering the stimulated increase in plasma substance P, suggesting that other mediators must be involved in the flushing. A correlation between plasma tachykinin levels (but not substance P levels) and flushing has been reported. Prostaglandin release could be involved in mediating either the diarrhea or flush, but conflicting data exist. Both histamine and serotonin may be responsible for the wheezing as well as the fibrotic reactions involving the heart, causing Peyronie’s disease and intraabdominal fibrosis.
The exact mechanism of the heart disease remains unclear, although increasing evidence supports a central role for serotonin. Patients with heart disease have higher plasma levels of neurokinin A, substance P, plasma atrial natriuretic peptide (ANP), pro-brain natriuretic peptide, chromogranin A, and activin A as well as higher urinary 5-HIAA excretion.
The valvular heart disease caused by the appetite-suppressant drug dexfenfluramine is histologically indistinguishable from that observed in carcinoid disease. Furthermore, ergot-containing dopamine receptor agonists used for Parkinson’s disease (pergolide, cabergoline) cause valvular heart disease that closely resembles that seen in the carcinoid syndrome. Furthermore, in animal studies, the formation of valvular plaques/fibrosis occurs after prolonged treatment with serotonin as well as in animals with a deficiency of the 5-HIAA transporter gene, which results in an inability to inactivate serotonin. Metabolites of fenfluramine, as well as the dopamine receptor agonists, have high affinity for serotonin receptor subtype 5-HT2B receptors, whose activation is known to cause fibroblast mitogenesis. Serotonin receptor subtypes 5-HT1B,1D,2A,2B normally are expressed in human heart valve interstitial cells. High levels of 5-HT2B receptors are known to occur in heart valves and occur in cardiac fibroblasts and cardiomyocytes. Studies of cultured interstitial cells from human cardiac valves have demonstrated that these valvulopathic drugs induce mitogenesis by activating 5-HT2B receptors and stimulating upregulation of transforming growth factor β and collagen biosynthesis. These observations support the conclusion that serotonin overproduction by GI-NETs (carcinoids) is important in mediating the valvular changes, possibly by activating 5-HT2B receptors in the endocardium. Both the magnitude of serotonin overproduction and prior chemotherapy are important predictors of progression of the heart disease, whereas patients with high plasma levels of ANP have a worse prognosis. Plasma connective tissue growth factor levels are elevated in many fibrotic conditions; elevated levels occur in patients with carcinoid heart disease and correlate with the presence of right ventricular dysfunction and the extent of valvular regurgitation in patients with GI-NETs (carcinoids).
Patients may develop either a typical or, rarely, an atypical carcinoid syndrome (Fig. 113-1). In patients with the typical form, which characteristically is caused by midgut NETs (carcinoids), the conversion of tryptophan to 5-HTP is the rate-limiting step (Fig. 113-1). Once 5-HTP is formed, it is rapidly converted to 5-HT and stored in secretory granules of the tumor or in platelets. A small amount remains in plasma and is converted to 5-HIAA, which appears in large amounts in the urine. These patients have an expanded serotonin pool size, increased blood and platelet serotonin, and increased urinary 5-HIAA. Some GI-NETs (carcinoids) cause an atypical carcinoid syndrome that is thought to be due to a deficiency in the enzyme dopa decarboxylase; thus, 5-HTP cannot be converted to 5-HT (serotonin), and 5-HTP is secreted into the bloodstream (Fig. 113-1). In these patients, plasma serotonin levels are normal but urinary levels may be increased because some 5-HTP is converted to 5-HT in the kidney. Characteristically, urinary 5-HTP and 5-HT are increased, but urinary 5-HIAA levels are only slightly elevated. Foregut carcinoids are the most likely to cause an atypical carcinoid syndrome; however, they also can cause a typical carcinoid syndrome.
One of the most immediate life-threatening complications of the carcinoid syndrome is the development of a carcinoid crisis. This is more common in patients who have intense symptoms or have greatly increased urinary 5-HIAA levels (i.e., >200 mg/d). The crisis may occur spontaneously; however, it is usually provoked by procedures such as anesthesia, chemotherapy, surgery, biopsy, endoscopy, or radiologic examinations such as during biopsies, hepatic artery embolization, and vessel catheterization. It can be provoked by stress or procedures as mild as repeated palpation of the tumor during physical examination. Patients develop intense flushing, diarrhea, abdominal pain, cardiac abnormalities including tachycardia, hypertension, or hypotension, and confusion or stupor. If not adequately treated, this can be a terminal event.
DIAGNOSIS OF THE CARCINOID SYNDROME AND GI-NETs (CARCINOIDS)
The diagnosis of carcinoid syndrome relies on measurement of urinary or plasma serotonin or its metabolites in the urine. The measurement of 5-HIAA is used most frequently. False-positive elevations may occur if the patient is eating serotonin-rich foods such as bananas, pineapples, walnuts, pecans, avocados, or hickory nuts or is taking certain medications (cough syrup containing guaifenesin, acetaminophen, salicylates, serotonin reuptake inhibitors, or L-dopa). The normal range for daily urinary 5-HIAA excretion is 2–8 mg/d. Serotonin overproduction was noted in 92% of patients with carcinoid syndrome in one study, and in another study, 5-HIAA had 73% sensitivity and 100% specificity for carcinoid syndrome. Serotonin overproduction is not synonymous with the presence of clinical carcinoid syndrome because 12–26% of patients with serotonin overproduction do not have clinical evidence of the carcinoid syndrome.
Most physicians use only the urinary 5-HIAA excretion rate; however, plasma and platelet serotonin levels, if available, may provide additional information. Platelet serotonin levels are more sensitive than urinary 5-HIAA but are not generally available. A single plasma 5-HIAA determination was found to correlate with the 24-h urinary values, raising the possibility that this could replace the standard urinary collection because of its greater convenience and avoidance of incomplete or improper collections. Because patients with foregut NETs (carcinoids) may produce an atypical carcinoid syndrome, if this syndrome is suspected and the urinary 5-HIAA is minimally elevated or normal, other urinary metabolites of tryptophan, such as 5-HTP and 5-HT, should be measured (Fig. 113-1).
Flushing occurs in a number of other diseases, including systemic mastocytosis, chronic myeloid leukemia with increased histamine release, menopause, reactions to alcohol or glutamate, and side effects of chlorpropamide, calcium channel blockers, and nicotinic acid. None of these conditions cause increased urinary 5-HIAA.
The diagnosis of carcinoid tumor can be suggested by the carcinoid syndrome, recurrent abdominal symptoms in a healthy-appearing individual, or the discovery of hepatomegaly or hepatic metastases associated with minimal symptoms. Ileal NETs (carcinoids), which make up 25% of all clinically detected carcinoids, should be suspected in patients with bowel obstruction, abdominal pain, flushing, or diarrhea.
Serum chromogranin A levels are elevated in 56–100% of patients with GI-NETs (carcinoids), and the level correlates with tumor bulk. Serum chromogranin A levels are not specific for GI-NETs (carcinoids) because they are also elevated in patients with pNETs and other NETs. Furthermore, a major problem is caused by potent acid antisecretory drugs such as proton pump inhibitors (omeprazole and related drugs) because they almost invariably cause elevation of plasma chromogranin A levels; the elevation occurs rapidly (3–5 days) with continued use, and the elevated levels overlap with the levels seen in many patients with NETs. Plasma neuron-specific enolase levels are also used as a marker of GI-NETs (carcinoids) but are less sensitive than chromogranin A, being increased in only 17–47% of patients. Newer markers have been proposed including pancreastatin (a chromogranin A breakdown product) and activin A. The former is not affected by proton pump inhibitors; however, its sensitivity and specificity are not established. Plasma activin elevations are reported to correlate with the presence of cardiac disease with a sensitivity of 87% and specificity of 57%.
|
TREATMENT |
CARCINOID SYNDROME AND NONMETASTATIC GASTROINTESTINAL NEUROENDOCRINE TUMORS (CARCINOIDS) |
CARCINOID SYNDROME
Treatment includes avoiding conditions that precipitate flushing, dietary supplementation with nicotinamide, treatment of heart failure with diuretics, treatment of wheezing with oral bronchodilators, and control of the diarrhea with antidiarrheal agents such as loperamide and diphenoxylate. If patients still have symptoms, serotonin receptor antagonists or somatostatin analogues (Fig. 113-2) are the drugs of choice.

FIGURE 113-2 Structure of somatostatin and synthetic analogues used for diagnostic or therapeutic indications.
There are 14 subclasses of serotonin receptors, and antagonists for many are not available. The 5-HT1 and 5-HT2 receptor antagonists methysergide, cyproheptadine, and ketanserin have all been used to control the diarrhea but usually do not decrease flushing. The use of methysergide is limited because it can cause or enhance retroperitoneal fibrosis. Ketanserin diminishes diarrhea in 30–100% of patients. 5-HT3 receptor antagonists (ondansetron, tropisetron, alosetron) can control diarrhea and nausea in up to 100% of patients and occasionally ameliorate the flushing. A combination of histamine H1 and H2 receptor antagonists (i.e., diphenhydramine and cimetidine or ranitidine) may control flushing in patients with foregut carcinoids. The tryptophan 5-hydoxylase inhibitor telotristat etiprate decreased bowel frequency in 44% and improved stool consistency in 20%.
Synthetic analogues of somatostatin (octreotide, lanreotide) are now the most widely used agents to control the symptoms of patients with carcinoid syndrome (Fig. 113-2). These drugs are effective at relieving symptoms and decreasing urinary 5-HIAA levels in patients with this syndrome. Octreotide-LAR and lanreotide-SR/autogel (Somatuline) (sustained-release formulations allowing monthly injections) control symptoms in 74% and 68% of patients, respectively, with carcinoid syndrome and show a biochemical response in 51% and 64%, respectively. Patients with mild to moderate symptoms usually are treated initially with octreotide 100 µg SC every 8 h and then begun on the long-acting monthly depot forms (octreotide-LAR or lanreotide-autogel). Forty percent of patients escape control after a median time of 4 months, and the depot dosage may have to be increased as well as supplemented with the shorter-acting formulation, SC octreotide. Pasireotide (SOM230) is a somatostatin analogue with broader selectivity (high-affinity somatostatin receptors [sst1, sst2, sst3, sst5]) than octreotide/lanreotide (sst2, sst5). In a phase II study of patients with refractory carcinoid syndrome, pasireotide controlled symptoms in 27%.
Carcinoid heart disease is associated with a decreased mean survival (3.8 years), and therefore, it should be sought for and carefully assessed in all patients with carcinoid syndrome. Transthoracic echocardiography remains a key element in establishing the diagnosis of carcinoid heart disease and determining the extent and type of cardiac abnormalities. Treatment with diuretics and somatostatin analogues can reduce the negative hemodynamic effects and secondary heart failure. It remains unclear whether long-term treatment with these drugs will decrease the progression of carcinoid heart disease. Balloon valvuloplasty for stenotic valves or cardiac valve surgery may be required.
In patients with carcinoid crises, somatostatin analogues are effective at both treating the condition and preventing their development during known precipitating events such as surgery, anesthesia, chemotherapy, and stress. It is recommended that octreotide 150–250 µg SC every 6 to 8 h be used 24–48 h before anesthesia and then continued throughout the procedure.
Currently, sustained-release preparations of both octreotide (octreotide-LAR [long-acting release], 10, 20, 30 mg) and lanreotide (lanreotide-PR [prolonged release, lanreotide-autogel], 60, 90, 120 mg) are available and widely used because their use greatly facilitates long-term treatment. Octreotide-LAR (30 mg/month) gives a plasma level ≥1 ng/mL for 25 days, whereas this requires three to six injections a day of the non-sustained-release form. Lanreotide-autogel (Somatuline) is given every 4–6 weeks.
Short-term side effects occur in up to one-half of patients. Pain at the injection site and side effects related to the GI tract (59% discomfort, 15% nausea, diarrhea) are the most common. They are usually short-lived and do not interrupt treatment. Important long-term side effects include gallstone formation, steatorrhea, and deterioration in glucose tolerance. The overall incidence of gallstones/biliary sludge in one study was 52%, with 7% having symptomatic disease that required surgical treatment.
Interferon α is reported to be effective in controlling symptoms of the carcinoid syndrome either alone or combined with hepatic artery embolization. With interferon α alone, the clinical response rate is 30–70%, and with interferon α with hepatic artery embolization, diarrhea was controlled for 1 year in 43% and flushing was controlled in 86%. Side effects develop in almost all patients, with the most frequent being a flu-like syndrome (80–100%), followed by anorexia and fatigue, even though these frequently improve with continued treatment. Other more severe side effects include bone marrow toxicity, hepatotoxicity, autoimmune disorders, and rarely CNS side effects (depression, mental disorders, visual problems).
Hepatic artery embolization alone or with chemotherapy (chemoembolization) has been used to control the symptoms of carcinoid syndrome. Embolization alone is reported to control symptoms in up to 76% of patients, and chemoembolization (5-fluorouracil, doxorubicin, cisplatin, mitomycin) controls symptoms in 60–75% of patients. Hepatic artery embolization can have major side effects, including nausea, vomiting, pain, and fever. In two studies, 5–7% of patients died from complications of hepatic artery occlusion.
Other drugs have been used successfully in small numbers of patients to control the symptoms of carcinoid syndrome. Parachlorophenylanine can inhibit tryptophan hydroxylase and therefore the conversion of tryptophan to 5-HTP. However, its severe side effects, including psychiatric disturbances, make it intolerable for long-term use. α-Methyldopa inhibits the conversion of 5-HTP to 5-HT, but its effects are only partial.
Peptide radioreceptor therapy (using radiotherapy with radiolabeled somatostatin analogues), the use of radiolabeled microspheres, and other methods for treatment of advanced metastatic disease may facilitate control of the carcinoid syndrome and are discussed in a later section dealing with treatment of advanced disease.
GI-NETs (CARCINOIDS) (NONMETASTATIC)
Surgery is the only potentially curative therapy. Because with most GI-NETs (carcinoids), the probability of metastatic disease increases with increasing size, the extent of surgical resection is determined accordingly. With appendiceal NETs (carcinoids) <1 cm, simple appendectomy was curative in 103 patients followed for up to 35 years. With rectal NETs (carcinoids) <1 cm, local resection is curative. With SI NETs (carcinoids) <1 cm, there is not complete agreement. Because 15–69% of SI NETs (carcinoids) this size have metastases in different studies, some recommend a wide resection with en bloc resection of the adjacent lymph-bearing mesentery. If the tumor is >2 cm for rectal, appendiceal, or SI NETs (carcinoids), a full cancer operation should be done. This includes a right hemicolectomy for appendiceal NETs (carcinoids), an abdominoperineal resection or low anterior resection for rectal NETs (carcinoids), and an en bloc resection of adjacent lymph nodes for SI NETs (carcinoids). For appendiceal NETs (carcinoids) 1–2 cm in diameter, a simple appendectomy is proposed by some, whereas others favor a formal right hemicolectomy. For 1–2 cm rectal NETs (carcinoids), it is recommended that a wide, local, full-thickness excision be performed.
With type I or II gastric NETs (carcinoids), which are usually <1 cm, endoscopic removal is recommended. In type I or II gastric carcinoids, if the tumor is >2 cm or if there is local invasion, some recommend total gastrectomy, whereas others recommend antrectomy in type I to reduce the hypergastrinemia, which has led to regression of the carcinoids in a number of studies. For types I and II gastric NETs (carcinoids) of 1–2 cm, there is no agreement, with some recommending endoscopic treatment followed by chronic somatostatin treatment and careful follow-up and others recommending surgical treatment. With type III gastric NETs (carcinoids) >2 cm, excision and regional lymph node clearance are recommended. Most tumors <1 cm are treated endoscopically.
Resection of isolated or limited hepatic metastases may be beneficial and will be discussed in a later section on treatment of advanced disease.
PANCREATIC NEUROENDOCRINE TUMORS
Functional pNETs usually present clinically with symptoms due to the hormone-excess state (Table 113-2). Only late in the course of the disease does the tumor per se cause prominent symptoms such as abdominal pain. In contrast, all the symptoms due to nonfunctional pNETs are due to the tumor per se. The overall result of this is that some functional pNETs may present with severe symptoms with a small or undetectable primary tumor, whereas nonfunctional tumors usually present late in the disease course with large tumors, which are frequently metastatic. The mean delay between onset of continuous symptoms and diagnosis of a functional pNET syndrome is 4–7 years. Therefore, the diagnoses frequently are missed for extended periods.
GASTRINOMA (ZOLLINGER-ELLISON SYNDROME)
A gastrinoma is an NET that secretes gastrin; the resultant hypergastrinemia causes gastric acid hypersecretion (Zollinger-Ellison syndrome [ZES]). The chronic hypergastrinemia results in marked gastric acid hypersecretion and growth of the gastric mucosa with increased numbers of parietal cells and proliferation of gastric ECL cells. The gastric acid hypersecretion characteristically causes peptic ulcer disease (PUD), often refractory and severe, as well as diarrhea. The most common presenting symptoms are abdominal pain (70–100%), diarrhea (37–73%), and gastroesophageal reflux disease (GERD) (30–35%); 10–20% of patients have diarrhea only. Although peptic ulcers may occur in unusual locations, most patients have a typical duodenal ulcer. Important observations that should suggest this diagnosis include PUD with diarrhea; PUD in an unusual location or with multiple ulcers; PUD refractory to treatment or persistent; PUD associated with prominent gastric folds; PUD associated with findings suggestive of MEN 1 (endocrinopathy, family history of ulcer or endocrinopathy, nephrolithiases); and PUD without Helicobacter pylori present. H. pylori is present in >90% of idiopathic peptic ulcers but is present in <50% of patients with gastrinomas. Chronic unexplained diarrhea also should suggest ZES.
Approximately 20–25% of patients with ZES have MEN 1 (MEN1/ZES), and in most cases, hyperparathyroidism is present before the ZES develops. These patients are treated differently from those without MEN 1 (sporadic ZES); therefore, MEN 1 should be sought in all patients with ZES by family history and by measuring plasma ionized calcium and prolactin levels and plasma hormone levels (parathormone, growth hormone).
Most gastrinomas (50–90%) in sporadic ZES are present in the duodenum, followed by the pancreas (10–40%) and other intraabdominal sites (mesentery, lymph nodes, biliary tract, liver, stomach, ovary). Rarely, the tumor may involve extraabdominal sites (heart, lung cancer). In MEN 1/ZES the gastrinomas are also usually in the duodenum (70–90%), followed by the pancreas (10–30%), and are almost always multiple. About 60–90% of gastrinomas are malignant (Table 113-2) with metastatic spread to lymph nodes and liver. Distant metastases to bone occur in 12–30% of patients with liver metastases.
Diagnosis The diagnosis of ZES requires the demonstration of inappropriate fasting hypergastrinemia, usually by demonstrating hypergastrinemia occurring with an increased basal gastric acid output (BAO) (hyperchlorhydria). More than 98% of patients with ZES have fasting hypergastrinemia, although in 40–60% the level may be elevated less than tenfold. Therefore, when the diagnosis is suspected, a fasting gastrin is usually the initial test performed. It is important to remember that potent gastric acid suppressant drugs such as proton pump inhibitors (PPIs) (omeprazole, esomeprazole, pantoprazole, lansoprazole, rabeprazole) can suppress acid secretion sufficiently to cause hypergastrinemia; because of their prolonged duration of action, these drugs have to be tapered or frequently discontinued for a week before the gastrin determination. Withdrawal of PPIs should be performed carefully because PUD complications can rapidly develop in some patients and is best done in consultation with GI units with experience in this area. The widespread use of PPIs can confound the diagnosis of ZES by raising a false-positive diagnosis by causing hypergastrinemia in a patient being treated with idiopathic PUD (without ZES) and lead to a false-negative diagnosis because at routine doses used to treat patients with idiopathic PUD, PPIs control symptoms in most ZES patients and thus mask the diagnosis. If ZES is suspected and the gastrin level is elevated, it is important to show that it is increased when gastric pH is ≤2.0 because physiologically hypergastrinemia secondary to achlorhydria (atrophic gastritis, pernicious anemia) is one of the most common causes of hypergastrinemia. Nearly all ZES patients have a fasting pH ≤2 when off antisecretory drugs. If the fasting gastrin is >1000 pg/mL (increased tenfold) and the pH is ≤2.0, which occurs in 40–60% of patients with ZES, the diagnosis of ZES is established after the possibility of retained antrum syndrome has been ruled out by history. In patients with hypergastrinemia with fasting gastrins <1000 pg/mL (<10-fold increased) and gastric pH ≤2.0, other conditions, such as H. pylori infections, antral G-cell hyperplasia/hyperfunction, gastric outlet obstruction, and, rarely, renal failure, can masquerade as ZES. To establish the diagnosis in this group, a determination of BAO and a secretin provocative test should be done. In patients with ZES without previous gastric acid–reducing surgery, the BAO is usually (>90%) elevated (i.e., >15 mEq/h). The secretin provocative test is usually positive, with the criterion of a >120-pg/mL increase over the basal level having the highest sensitivity (94%) and specificity (100%). Unfortunately the diagnosis of ZES is becoming increasing more difficult. This is due not only to the widespread use of PPIs (leading to false-positive results as well as masking ZES presentation), but also recent studies demonstrate than many of the commercial gastrin kits that are used by most laboratories to measure fasting serum gastrin levels are not reliable. In one study, 7 of the 12 tested commercial gastrin kits inaccurately assessed the true serum concentration of gastrin primarily because the antibodies used had inappropriate specificity for the different circulating forms of gastrin and were not adequately validated. Both underestimation and overestimation of fasting serum gastrin levels occurred using these commercial kits. To circumvent this problem, it is either necessary to use one of the five reliable kits identified or, alternatively, to refer the patient to a center with expertise in making the diagnosis in your area, or if this is not possible, to contact such a center and use the gastrin assay they recommend. An accurate gastrin assay is essential for accurate measurement of fasting serum gastrin level as well as for assessing gastrin levels during the secretin provocative test, and thus, the diagnosis of ZES cannot reliably be made without one.
INSULINOMAS
An insulinoma is an NET of the pancreas that is thought to be derived from beta cells that ectopically secrete insulin, which results in hypoglycemia. The average age of occurrence is 40–50 years old. The most common clinical symptoms are due to the effect of the hypoglycemia on the CNS (neuroglycemic symptoms) and include confusion, headache, disorientation, visual difficulties, irrational behavior, and even coma. Also, most patients have symptoms due to excess catecholamine release secondary to the hypoglycemia, including sweating, tremor, and palpitations. Characteristically, these attacks are associated with fasting.
Insulinomas are generally small (>90% are <2 cm) and usually not multiple (90%); only 5–15% are malignant, and they almost invariably occur only in the pancreas, distributed equally in the pancreatic head, body, and tail.
Insulinomas should be suspected in all patients with hypoglycemia, especially when there is a history suggesting that attacks are provoked by fasting, or with a family history of MEN 1. Insulin is synthesized as proinsulin, which consists of a 21-amino-acid α chain and a 30-amino-acid β chain connected by a 33-amino-acid connecting peptide (C peptide). In insulinomas, in addition to elevated plasma insulin levels, elevated plasma proinsulin levels are found, and C-peptide levels are elevated.
Diagnosis The diagnosis of insulinoma requires the demonstration of an elevated plasma insulin level at the time of hypoglycemia. A number of other conditions may cause fasting hypoglycemia, such as the inadvertent or surreptitious use of insulin or oral hypoglycemic agents, severe liver disease, alcoholism, poor nutrition, and other extrapancreatic tumors. Furthermore, postprandial hypoglycemia can be caused by a number of conditions that confuse the diagnosis of insulinoma. Particularly important here is the increased occurrence of hypoglycemia after gastric bypass surgery for obesity, which is now widely performed. A new entity, insulinomatosis, was described that can cause hypoglycemia and mimic insulinomas. It occurs in 10% of patients with persistent hyperinsulinemic hypoglycemia and is characterized by the occurrence of multiple macro-/microadenomas expressing insulin, and it is not clear how to distinguish this entity from insulinoma preoperatively. The most reliable test to diagnose insulinoma is a fast up to 72 h with serum glucose, C-peptide, proinsulin, and insulin measurements every 4–8 h. If at any point the patient becomes symptomatic or glucose levels are persistently below <2.2 mmol/L (40 mg/dL), the test should be terminated, and repeat samples for the above studies should be obtained before glucose is given. Some 70–80% of patients will develop hypoglycemia during the first 24 h, and 98% by 48 h. In nonobese normal subjects, serum insulin levels should decrease to <43 pmol/L (<6 µU/mL) when blood glucose decreases to <2.2 mmol/L (<40 mg/dL) and the ratio of insulin to glucose is <0.3 (in mg/dL). In addition to having an insulin level >6 µU/mL when blood glucose is <40 mg/dL, some investigators also require an elevated C-peptide and serum proinsulin level, an insulin/glucose ratio >0.3, and a decreased plasma β-hydroxybutyrate level for the diagnosis of insulinomas. Surreptitious use of insulin or hypoglycemic agents may be difficult to distinguish from insulinomas. The combination of proinsulin levels (normal in exogenous insulin/hypoglycemic agent users), C-peptide levels (low in exogenous insulin users), antibodies to insulin (positive in exogenous insulin users), and measurement of sulfonylurea levels in serum or plasma will allow the correct diagnosis to be made. The diagnosis of insulinoma has been complicated by the introduction of specific insulin assays that do not also interact with proinsulin, as do many of the older radioimmunoassays (RIAs), and therefore give lower plasma insulin levels. The increased use of these specific insulin assays has resulted in increased numbers of patients with insulinomas having lower plasma insulin values (<6 µU/mL) than levels proposed to be characteristic of insulinomas by RIA. In these patients, the assessment of proinsulin and C-peptide levels at the time of hypoglycemia is particularly helpful for establishing the correct diagnosis. An elevated proinsulin level when the fasting glucose level is <45 mg/dL is sensitive and specific.
GLUCAGONOMAS
A glucagonoma is NET of the pancreas that secretes excessive amounts of glucagon, which causes a distinct syndrome characterized by dermatitis, glucose intolerance or diabetes, and weight loss. Glucagonomas principally occur between 45 and 70 years of age. The tumor is clinically heralded by a characteristic dermatitis (migratory necrolytic erythema) (67–90%), accompanied by glucose intolerance (40–90%), weight loss (66–96%), anemia (33–85%), diarrhea (15–29%), and thromboembolism (11–24%). The characteristic rash usually starts as an annular erythema at intertriginous and periorificial sites, especially in the groin or buttock. It subsequently becomes raised, and bullae form; when the bullae rupture, eroded areas form. The lesions can wax and wane. The development of a similar rash in patients receiving glucagon therapy suggests that the rash is a direct effect of the hyperglucagonemia. A characteristic laboratory finding is hypoaminoacidemia, which occurs in 26–100% of patients.
Glucagonomas are generally large tumors at diagnosis (5–10 cm). Some 50–80% occur in the pancreatic tail. From 50 to 82% have evidence of metastatic spread at presentation, usually to the liver. Glucagonomas are rarely extrapancreatic and usually occur singly.
Two new entities have been described that can also cause hyperglucagonemia and may mimic glucagonomas. Mahvah disease is due to a homozygous P86S mutation of the human glucagon receptor. It is associated with the development of α-cell hyperplasia, hyperglucagonemia, and the development of nonfunctioning pNETs. A second disease called glucagon cell adenomatosis can mimic glucagonoma syndrome clinically and is characterized by the presence of hyperplastic islets staining positive for glucagon instead of a single glucagonoma.
Diagnosis The diagnosis is confirmed by demonstrating an increased plasma glucagon level. Characteristically, plasma glucagon levels exceed 1000 pg/mL (normal is <150 pg/mL) in 90%; 7% are between 500 and 1000 pg/mL, and 3% are <500 pg/mL. A trend toward lower levels at diagnosis has been noted in the last decade. A plasma glucagon level >1000 pg/mL is considered diagnostic of glucagonoma. Other diseases causing increased plasma glucagon levels include cirrhosis, diabetic ketoacidosis, celiac disease, renal insufficiency, acute pancreatitis, hypercorticism, hepatic insufficiency, severe stress, and prolonged fasting or familial hyperglucagonemia, as well as danazol treatment. With the exception of cirrhosis, these disorders do not increase plasma glucagon >500 pg/mL.
Necrolytic migratory erythema is not pathognomonic for glucagonoma and occurs in myeloproliferative disorders, hepatitis B infection, malnutrition, short-bowel syndrome, inflammatory bowel disease, zinc deficiency, and malabsorption disorders.
SOMATOSTATINOMA SYNDROME
The somatostatinoma syndrome is due to an NET that secretes excessive amounts of somatostatin, which causes a distinct syndrome characterized by diabetes mellitus, gallbladder disease, diarrhea, and steatorrhea. There is no general distinction in the literature between a tumor that contains somatostatin-like immunoreactivity (somatostatinoma) and does (11–45%) or does not (55–90%) produce a clinical syndrome (somatostatinoma syndrome) by secreting somatostatin. In a review of 173 cases of somatostatinomas, only 11% were associated with the somatostatinoma syndrome. The mean age is 51 years. Somatostatinomas occur primarily in the pancreas and small intestine, and the frequency of the symptoms and occurrence of the somatostatinoma syndrome differ in each. Each of the usual symptoms is more common in pancreatic than in intestinal somatostatinomas: diabetes mellitus (95% vs 21%), gallbladder disease (94% vs 43%), diarrhea (92% vs 38%), steatorrhea (83% vs 12%), hypochlorhydria (86% vs 12%), and weight loss (90% vs 69%). The somatostatinoma syndrome occurs in 30–90% of pancreatic and 0–5% of SI somatostatinomas. In various series, 43% of all duodenal NETs contain somatostatin; however, the somatostatinoma syndrome is rarely present (<2%). Somatostatinomas occur in the pancreas in 56–74% of cases, with the primary location being the pancreatic head. The tumors are usually solitary (90%) and large (mean size 4.5 cm). Liver metastases are common, being present in 69–84% of patients. Somatostatinomas are rare in patients with MEN 1, occurring in only 0.65%.
Somatostatin is a tetradecapeptide that is widely distributed in the CNS and GI tract, where it functions as a neurotransmitter or has paracrine and autocrine actions. It is a potent inhibitor of many processes, including release of almost all hormones, acid secretion, intestinal and pancreatic secretion, and intestinal absorption. Most of the clinical manifestations are directly related to these inhibitory actions.
Diagnosis In most cases, somatostatinomas have been found by accident either at the time of cholecystectomy or during endoscopy. The presence of psammoma bodies in a duodenal tumor should particularly raise suspicion. Duodenal somatostatin-containing tumors are increasingly associated with von Recklinghausen’s disease (NF-1) (Table 113-6). Most of these tumors (>98%) do not cause the somatostatinoma syndrome. The diagnosis of the somatostatinoma syndrome requires the demonstration of elevated plasma somatostatin levels.
VIPOMAS
VIPomas are NETs that secrete excessive amounts of vasoactive intestinal peptide (VIP), which causes a distinct syndrome characterized by large-volume diarrhea, hypokalemia, and dehydration. This syndrome also is called Verner-Morrison syndrome, pancreatic cholera, and WDHA syndrome for watery diarrhea, hypokalemia, and achlorhydria, which some patients develop. The mean age of patients with this syndrome is 49 years; however, it can occur in children, and when it does, it is usually caused by a ganglioneuroma or ganglioneuroblastoma.
The principal symptoms are large-volume diarrhea (100%) severe enough to cause hypokalemia (80–100%), dehydration (83%), hypochlorhydria (54–76%), and flushing (20%). The diarrhea is secretory in nature, persisting during fasting, and is almost always >1 L/d and in 70% is >3 L/d. In a number of studies, the diarrhea was intermittent initially in up to half the patients. Most patients do not have accompanying steatorrhea (16%), and the increased stool volume is due to increased excretion of sodium and potassium, which, with the anions, accounts for the osmolality of the stool. Patients frequently have hyperglycemia (25–50%) and hypercalcemia (25–50%).
VIP is a 28-amino-acid peptide that is an important neurotransmitter, ubiquitously present in the CNS and GI tract. Its known actions include stimulation of SI chloride secretion as well as effects on smooth-muscle contractility, inhibition of acid secretion, and vasodilatory effects, which explain most features of the clinical syndrome.
In adults, 80–90% of VIPomas are pancreatic in location, with the rest due to VIP-secreting pheochromocytomas, intestinal carcinoids, and rarely ganglioneuromas. These tumors are usually solitary, 50–75% are in the pancreatic tail, and 37–68% have hepatic metastases at diagnosis. In children <10 years old, the syndrome is usually due to ganglioneuromas or ganglioblastomas and is less often malignant (10%).
Diagnosis The diagnosis requires the demonstration of an elevated plasma VIP level and the presence of large-volume diarrhea. A stool volume <700 mL/d is proposed to exclude the diagnosis of VIPoma. When the patient fasts, a number of diseases can be excluded that can cause marked diarrhea because the high volume of diarrhea is not sustained during the fast. Other diseases that can produce a secretory large-volume diarrhea include gastrinomas, chronic laxative abuse, carcinoid syndrome, systemic mastocytosis, rarely medullary thyroid cancer, diabetic diarrhea, sprue, and AIDS. Among these conditions, only VIPomas caused a marked increase in plasma VIP. Chronic surreptitious use of laxatives/diuretics can be particularly difficult to detect clinically. Hence, in a patient with unexplained chronic diarrhea, screens for laxatives should be performed; they will detect many, but not all, laxative abusers. Elevated plasma levels of VIP should not be the only basis of the diagnosis of VIPomas because they can occur with some diarrheal states including inflammatory bowel disease, post small bowel resection, and radiation enteritis. Furthermore, nesidioblastosis can mimic VIPomas by causing elevated plasma VIP levels, diarrhea, and even false-positive location in the pancreatic region on somatostatin receptor scintigraphy.
NONFUNCTIONAL PANCREATIC NEUROENDOCRINE TUMORS (NF-pNETs)
NF-pNETs are NETs that originate in the pancreas and either secrete no products or their products do not cause a specific clinical syndrome. Their symptoms are due entirely to the tumor per se. NF-pNETs secrete chromogranin A (90–100%), chromogranin B (90–100%), α-HCG (human chorionic gonadotropin) (40%), neuron-specific enolase (31%), and β-HCG (20%), and because 40–90% secrete PP, they are also often called PPomas. Because the symptoms are due to the tumor mass, patients with NF-pNETs usually present late in the disease course with invasive tumors and hepatic metastases (64–92%), and the tumors are usually large (72% >5 cm). NF-pNETs are usually solitary except in patients with MEN 1, in which case they are multiple. They occur primarily in the pancreatic head. Even though these tumors do not cause a functional syndrome, immunocytochemical studies show that they synthesize numerous peptides and cannot be distinguished from functional pNETs by immunocytochemistry. In MEN 1, 80–100% of patients have microscopic NF-pNETs, but they become large or symptomatic in a minority (0–13%) of cases. In VHL, 12–17% develop NF-pNETs, and in 4%, they are ≥3 cm in diameter.
The most common symptoms are abdominal pain (30–80%), jaundice (20–35%), and weight loss, fatigue, or bleeding; 10–35% are found incidentally. The average time from the beginning of symptoms to diagnosis is 5 years.
Diagnosis The diagnosis is established by histologic confirmation in a patient without either the clinical symptoms or the elevated plasma hormone levels of one of the established syndromes. The principal difficulty in diagnosis is to distinguish an NF-pNET from a nonendocrine pancreatic tumor, which is more common, as well as from a functional pNET. Even though chromogranin A levels are elevated in almost every patient, this is not specific for this disease as it can be found in functional pNETs, GI-NETs (carcinoids), and other neuroendocrine disorders. Plasma PP elevations should strongly suggest the diagnosis in a patient with a pancreatic mass because it is usually normal in patients with pancreatic adenocarcinomas. Elevated plasma PP is not diagnostic of this tumor because it is elevated in a number of other conditions, such as chronic renal failure, old age, inflammatory conditions, alcohol abuse, pancreatitis, hypoglycemia, postprandially, and diabetes. A positive somatostatin receptor scan in a patient with a pancreatic mass should suggest the presence of pNET/NF-pNET rather than a nonendocrine tumor.
GRFOMAS
GRFomas are NETs that secrete excessive amounts of growth hormone–releasing factor (GRF) that cause acromegaly. GRF is a 44-amino-acid peptide, and 25–44% of pNETs have GRF immunoreactivity, although it is uncommonly secreted. GRFomas are lung tumors in 47–54% of cases, pNETs in 29–30%, and SI carcinoids in 8–10%; up to 12% occur at other sites. Patients have a mean age of 38 years, and the symptoms usually are due to either acromegaly or the tumor per se. The acromegaly caused by GRFomas is indistinguishable from classic acromegaly. The pancreatic tumors are usually large (>6 cm), and liver metastases are present in 39%. They should be suspected in any patient with acromegaly and an abdominal tumor, a patient with MEN 1 with acromegaly, or a patient without a pituitary adenoma with acromegaly or associated with hyperprolactinemia, which occurs in 70% of GRFomas. GRFomas are an uncommon cause of acromegaly. GRFomas occur in <1% of MEN 1 patients. The diagnosis is established by performing plasma assays for GRF and growth hormone. Most GRFomas have a plasma GRF level >300 pg/mL (normal <5 pg/mL men, <10 pg/mL women). Patients with GRFomas also have increased plasma levels of insulin-like growth factor type I (IGF-I) similar to those in classic acromegaly. Surgery is the treatment of choice if diffuse metastases are not present. Long-acting somatostatin analogues such as octreotide and lanreotide are the agents of choice, with 75–100% of patients responding.
OTHER RARE PANCREATIC NEUROENDOCRINE TUMOR SYNDROMES
Cushing’s syndrome (ACTHoma) due to a pNET occurs in 4–16% of all ectopic Cushing’s syndrome cases. It occurs in 5% of cases of sporadic gastrinomas, almost invariably in patients with hepatic metastases, and is an independent poor prognostic factor. Paraneoplastic hypercalcemia due to pNETs releasing parathyroid hormone–related peptide (PTHrP), a PTH-like material, or unknown factor, is rarely reported. The tumors are usually large, and liver metastases are usually present. Most (88%) appear to be due to release of PTHrP. pNETs occasionally can cause the carcinoid syndrome. A number of very rare pNET syndromes involving a few cases (less than five) have been described; these include a renin-producing pNET in a patient presenting with hypertension; pNETs secreting luteinizing hormone, resulting in masculinization or decreased libido; a pNET secreting erythropoietin, resulting in polycythemia; pNETs secreting IGF-II, causing hypoglycemia; and pNETs secreting enteroglucagon, causing small intestinal hypertrophy, colonic/SI stasis, and malabsorption (Table 113-2). A number of other possible functional pNETs have been proposed, but most authorities classify these as unclear or as a nonfunctional pNET because in each case numerous patients have been described with similar plasma hormone elevations that do not cause any symptoms. These include pNETs secreting calcitonin, neurotensin (neurotensinoma), PP (PPoma), and ghrelin (Table 113-2).
TUMOR LOCALIZATION
Localization of the primary tumor and knowledge of the extent of the disease are essential to the proper management of all GI-NETs (carcinoids) and pNETs. Without proper localization studies, it is not possible to determine whether the patient is a candidate for surgical resection (curative or cytoreductive) or requires antitumor treatment, to determine whether the patient is responding to antitumor therapies, or to appropriately classify/stage the patient’s disease to assess prognosis.
Numerous tumor localization methods are used in both types of NETs, including cross-sectional imaging studies (CT, magnetic resonance imaging [MRI], transabdominal ultrasound), selective angiography, somatostatin receptor scintigraphy (SRS), and positron emission tomography. In pNETs, endoscopic ultrasound (EUS) and functional localization by measuring venous hormonal gradients are also reported to be useful. Bronchial carcinoids are usually detected by standard chest radiography and assessed by CT. Rectal, duodenal, colonic, and gastric carcinoids are usually detected by GI endoscopy. Because of their wide availability, CT and MRI are generally initially used to determine the location of the primary NETs and the extent of disease. NETs are hypervascular tumors, and with both MRI and CT, contrast enhancement is essential for maximal sensitivity, and it is recommended that generally triple-phase scanning be used. The ability of cross-sectional imaging and, to a lesser extent, SRS to detect NETs is a function of NET size. With CT and MRI, <10% of tumors <1 cm in diameter are detected, 30–40% of tumors 1–3 cm are detected, and >50% of tumors >3 cm are detected. Many primary GI-NETs (carcinoids) are small, as are insulinomas and duodenal gastrinomas, and are frequently not detected by cross-sectional imaging, whereas most other pNETs present late in the course of their disease and are large (>4 cm). Selective angiography is more sensitive, localizing 60–90% of all NETs; however, it is now used infrequently. For detecting liver metastases, CT and MRI are more sensitive than ultrasound, and with recent improvements, 5–25% of patients with liver metastases will be missed by CT and/or MRI.
pNETs, as well as GI-NETs (carcinoids), frequently (>80%) overexpress high-affinity somatostatin receptors in both the primary tumors and the metastases. Of the five types of somatostatin receptors (sst1–5), radiolabeled octreotide binds with high affinity to sst2 and sst5, has a lower affinity for sst3, and has a very low affinity for sst1 and sst4. Between 80 and 100% of GI-NETs (carcinoids) and pNETs possess sst2, and many also have the other four sst subtypes. Interaction with these receptors can be used to treat these tumors as well as to localize NETs by using radiolabeled somatostatin analogues (SRS). In the United States, [111In-DTPA-D-Phe1]octreotide (octreoscan) is generally used with gamma camera detection using single-photon emission computed tomography (SPECT) imaging. Numerous studies, primarily in Europe, using gallium-68-labeled somatostatin analogues and positron emission tomography (PET) detection, demonstrate even greater sensitivity than with SRS with 111In-labeled somatostatin analogues. Although not yet approved in the United States, there are a number of centers starting to use this approach. Because of its sensitivity and ability to localize tumor throughout the body, SRS is the initial imaging modality of choice for localizing both the primary tumor and metastatic NETs. SRS localizes tumor in 73–95% of patients with GI-NETs (carcinoids) and in 56–100% of patients with pNETs, except insulinomas. Insulinomas are usually small and have low densities of sst receptors, resulting in SRS being positive in only 12–50% of patients with insulinomas. SRS identifies >90–95% of patients with liver metastases due to NETs. Figure 113-3 shows an example of the increased sensitivity of SRS in a patient with a GI-NET (carcinoid) tumor. The CT scan showed a single liver metastasis, whereas the SRS demonstrated three metastases in the liver in multiple locations. Occasional false-positive responses with SRS can occur (12% in one study) because numerous other normal tissues as well as diseases can have high densities of sst receptors, including granulomas (sarcoid, tuberculosis, etc.), thyroid diseases (goiter, thyroiditis), and activated lymphocytes (lymphomas, wound infections). If liver metastases are identified by SRS, to plan the proper treatment, either a CT or an MRI (with contrast enhancement) is recommended to assess the size and exact location of the metastases because SRS does not provide information on tumor size. For pNETs in the pancreas, EUS is highly sensitive, localizing 77–100% of insulinomas, which occur almost exclusively within the pancreas. Endoscopic ultrasound is less sensitive for extrapancreatic tumors. It is increasingly used in patients with MEN 1, and to a lesser extent VHL, to detect small pNETs not seen with other modalities or for serial pNET assessments to determine size changes or rapid growth in patients in whom surgery is deferred. EUS with cytologic evaluation also is used frequently to distinguish an NF-pNET from a pancreatic adenocarcinoma or another nonendocrine pancreatic tumor. Not infrequently patients present with liver metastases due to an NET and the primary site is unclear. Occult small intestinal NETs (carcinoids) are increasingly detected by double-balloon enteroscopy or capsule endoscopy.

FIGURE 113-3 Ability of computed tomography (CT) scanning (top) or somatostatin receptor scintigraphy (SRS) (bottom) to localize metastatic carcinoid in the liver.
Insulinomas frequently overexpress receptors for glucagon-like peptide-1 (GLP-1), and radiolabeled GLP-1 analogues have been developed that can detect occult insulinomas not localized by other imaging modalities. Functional localization by measuring hormonal gradients is now uncommonly used with gastrinomas (after intra-arterial secretin injections) but is still frequently used in insulinoma patients in whom other imaging studies are negative (assessing hepatic vein insulin concentrations post-intra-arterial calcium injections). Functional localization measuring hormone gradients in insulinomas or gastrin gradients in gastrinoma is a sensitive method, being positive in 80–100% of patients. The intra-arterial calcium test may also allow differentiation of the cause of the hypoglycemia and indicate whether it is due to an insulinoma or a nesidioblastosis. The latter entity is becoming increasingly important because hypoglycemia after gastric bypass surgery for obesity is increasing in frequency, and it is primarily due to nesidioblastosis, although it can occasionally be due to an insulinoma.
PET and use of hybrid scanners such as CT and SRS may have increased sensitivity. PET scanning with 18F-fluoro-DOPA in patients with carcinoids or with 11C-5-HTP in patients with pNETs or GI-NETs (carcinoids) has greater sensitivity than cross-sectional imaging studies and may be used increasingly in the future. PET scanning for GI-NETs is not currently approved in the United States.
|
TREATMENT |
ADVANCED DISEASE (DIFFUSE METASTATIC DISEASE) |
The single most important prognostic factor for survival is the presence of liver metastases (Fig. 113-4). For patients with foregut carcinoids without hepatic metastases, the 5-year survival in one study was 95%, and with distant metastases, it was 20% (Fig. 113-4). With gastrinomas, the 5-year survival without liver metastases is 98%; with limited metastases in one hepatic lobe, it is 78%; and with diffuse metastases, 16% (Fig. 113-4). In a large study of 156 patients (67 pNETs, rest carcinoids), the overall 5-year survival rate was 77%; it was 96% without liver metastases, 73% with liver metastases, and 50% with distant disease. Another very important prognostic factor is whether the NET is well-differentiated (G1/G2) or poorly differentiated (<1% of all NETs) (G3). Well-differentiated NETs have a 5-year survival of 50–80%, whereas poorly differentiated NETs have a 5-year survival of only 0–15%.

FIGURE 113-4 Survival (Kaplan-Meier plots) of patients with pancreatic neuroendocrine tumors (pNETs; n = 1072) (A–C) or gastrointestinal neuroendocrine tumors (GI-NETs; carcinoids) (appendix, n = 138; midgut, n = 238) (D–F) stratified according to recent proposed classification and grading systems. (Panels A–C are drawn from data in G Rindi et al: J Natl Cancer Inst 104:764, 2012; panels D and E are drawn from data in M Volante et al: Am J Surg Pathol 37:606, 2013; and panel F is drawn from data in MS Khan: Br J Cancer 108:1838, 2013.)
Therefore, treatment for advanced metastatic disease is an important challenge. A number of different modalities are reported to be effective, including cytoreductive surgery (surgically or by radiofrequency ablation [RFA]), treatment with chemotherapy, somatostatin analogues, interferon α, hepatic embolization alone or with chemotherapy (chemoembolization), molecular targeted therapy, radiotherapy with radiolabeled beads/microspheres, peptide radioreceptor therapy (PRRT), and liver transplantation.
SPECIFIC ANTITUMOR TREATMENTS
Cytoreductive surgery is considered if either all of the visible metastatic disease or at last 90% is thought resectable; however, unfortunately, this is possible in only the 9–22% of patients who present with limited hepatic metastases. Although no randomized studies have proven that it extends life, results from a number of studies suggest that it may increase survival; therefore, it is recommended, if possible. RFA can be applied to NET liver metastases if they are limited in number (usually less than five) and size (usually <3.5 cm in diameter). It can be used at the time of surgery (either general or laparoscopic) or using radiologic guidance.
Response rates are >80%, the responses can last up to 3 years, the morbidity rate is low, and this procedure may be particularly helpful in patients with functional pNETs that are difficult to control medically. Although RFA has not been established in a controlled trial, both the European and North American Neuroendocrine Tumor Society guidelines (ENETS, NANETS) state it can be an effective antitumor treatment for both refractory functional syndromes and for palliative treatment.
Chemotherapy plays a different role in the treatment of patients with pNETs and GI-NETs (carcinoids). Chemotherapy continues to be widely used in the treatment of patients with advanced pNETs with moderate success (response rates 20–70%); however, in general, its results in patients with metastatic GI-NETs (carcinoids) has been disappointing, with response rates of 0–30% with various two- and three-drug combinations, and thus, it is infrequently used in these patients. An important distinction in patients with pNETs is whether the tumor is well differentiated (G1/G2) or poorly differentiated (G3). The chemotherapeutic approach is different for these two groups. The current regimen of choice for patients with well-differentiated pNETs is the combination of streptozotocin and doxorubicin with or without 5-fluorouracil. Streptozotocin is a glucosamine nitrourea compound originally found to have cytotoxic effects on pancreatic islets, and later in studies with doxorubicin with or without 5-fluorouracil, it produced response rates of 20–45% in advanced pNETs. Streptozotocin causes considerable morbidity, with 70–100% of patients developing side effects (most prominent being nausea/vomiting in 60–100% or leukopenia/thrombocytopenia) and 15–40% of patients developing some degree of renal dysfunction (proteinuria in 40–50%, decreased creatine clearance). The combination of temozolomide (TMZ) with capecitabine produces partial response rates as high as 70% in patients with advance pNETs and a 2-year survival of 92%. The use of TMZ or another alkylating agent in advanced pNETs is supported by studies that show low levels of the DNA repair enzyme O6-methylguanine DNA methyltransferase in pNETs, but not in GI-NETs (carcinoids), which increases the sensitivity of pNETs to TMZ. In poorly differentiated NETs (G3), chemotherapy with a cisplatin-based regimen with etoposide or other agents (vincristine, paclitaxel) is the recommended treatment, with response rates of 40–70%; however, responses are generally short-lived (<12 months). This chemotherapy regimen can be associated with significant toxicity including GI toxicities (nausea, vomiting), myelosuppression, and renal toxicity.
In addition to the effectiveness in controlling the functional hormonal state, long-acting somatostatin analogues such as octreotide and lanreotide are increasingly used for their antiproliferative effects. Whereas somatostatin analogues rarely decrease tumor size (i.e., 0–17%), these drugs have tumoristatic effects, stopping additional growth in 26–95% of patients with NETs. In a randomized, double-blind study in patients with metastatic midgut carcinoids (PROMID study) octreotide-LAR demonstrated a marked lengthening of time to progression (14.3 vs 6 months, p = .000072). This improvement was seen in patients with limited liver involvement. This study did not assess whether such treatment will extend survival. A double-blind, randomized, placebo-controlled, phase III study in patients with well-differentiated, metastatic, inoperable pNETs (45%) or GI-NETs (carcinoids) (55%) (CLARINET study) showed that monthly treatment with lanreotide-autogel reduced tumor progression or death by 53%. Somatostatin analogues can induce apoptosis in GI-NETs (carcinoids), which probably contributes to their tumoristatic effects. Treatment with somatostatin analogues is generally well-tolerated, with most side effects being mild and uncommonly leading to stopping the drug. Potential long-term side effects include diabetes/glucose intolerance, steatorrhea, and the development of gallbladder sludge/gallstones (10–80%), although only 1% of patients develop symptomatic gallbladder disease. Because of these phase III studies, somatostatin analogues are generally recommended as first-line treatment for patients with well-differentiated metastatic NETs.
Interferon α, similar to somatostatin analogues, is effective at controlling the hormonal excess symptoms of NETs and has antiproliferative effects in NETs, which primarily result in disease stabilization (30–80%), with a decrease in tumor size in <15% of patients. Interferon can inhibit DNA synthesis, block cell cycle progression in the G1 phase, inhibit protein synthesis, inhibit angiogenesis, and induce apoptosis. Interferon α treatment results in side effects in the majority of patients, with the most frequent being a flu-like syndrome (80–100%), anorexia with weight loss, and fatigue. These side effects frequently decrease in severity with continued treatment. In addition, patients become accommodated to the symptoms. More serious side effects include hepatotoxicity (31%), hyperlipidemia (31%), bone marrow toxicity, thyroid disease (19%), and rarely CNS side effects (depression, mental/visual disorders). ENETS 2012 guidelines conclude that in patients with well-differentiated NETs that are slowly progressive, interferon α treatment should be considered if the tumor is somatostatin receptor negative or if somatostatin treatment fails.
Selective internal radiation therapy (SIRT) using yttrium-90 (90Y) glass or resin microspheres is a relatively newer approach being evaluated in patients with unresectable NET liver metastases, with approximately 500 NET patients treated. The treatment requires careful evaluation for vascular shunting before treatment and a pretreatment angiogram to evaluate placement of the catheter and is generally is reserved for patients without extrahepatic metastatic disease and with adequate hepatic reserve. One of two types of 90Y microspheres are used: either microspheres with a 20- to 60-µm diameter and 50 Bq/sphere (SIR-Spheres) or glass microspheres (TheraSpheres) with a 20- to 30-µm diameter and 2500 Bq/sphere. The 90Y-microspheres are delivered to the liver by intra-arterial injection from percutaneously placed catheters. In four studies involving metastatic NETs, the response rate varied from 50–61% (partial or complete), tumor stabilization occurred in 22–41%, 60–100% had symptomatic improvement, and overall survival varied from 25–70 months. Side effects include postembolization syndrome (pain, fever, nausea/vomiting [frequent]), which is usually mild, although grade 2 (43%) or grade 3 (1%) symptoms can occur; radiation-induced liver disease (<1%); and radiation pneumonitis (<1%). Contraindications to use include excess shunting to the GI tract or lung, inability to isolate the liver arterial supply, and inadequate liver reserve. Because of the limited data available in the ENETS 2012 guidelines, treatment with SIRTs is considered experimental.
Molecular targeted medical treatment with either an mTOR inhibitor (everolimus) or a tyrosine kinase inhibitor (sunitinib) is now approved treatment in the United States and Europe for patients with metastatic unresectable pNET, each supported by a phase III, double-blind, prospective, placebo-controlled trial. mTOR is a serine-threonine kinase that plays an important role in proliferation, cell growth, and apoptosis in both normal and neoplastic cells. Activation of the mTOR cascade is important in mediating NET cell growth, especially in pNETs. A number of mTOR inhibitors have shown promising antitumor activity in NETs including everolimus and temsirolimus, with the former undergoing a phase III trial (RADIANT-3) involving 410 patients with advance progressive pNETs. Everolimus caused significant improvement in progression-free survival (11 vs 4.6 months, p <.001) and increased by a factor of 3.7 the proportion of patients progression-free at 18 months (37% vs 9%). Everolimus treatment was associated with frequent side effects, causing a twofold increase in adverse events, with the most frequent being grade 1 or 2. Grade 3 or 4 side effects included hematologic, GI (diarrhea), stomatitis, or hypoglycemia occurring in 3–7% of patients. Most grade 3 or 4 side effects were controlled by dose reduction or drug interruption. The ENETS 2012 guidelines conclude that everolimus, similar to sunitinib (below), should be considered as a first-line treatment in selected cases of well-differentiated pNETs that are unresectable. NETs, like other normal and neoplastic cells, frequently possess multiple types of the 20 different tyrosine kinase (TK) receptors that are known and mediate the action of different growth factors. Numerous studies demonstrate that TK receptors in normal and neoplastic tissues as well as NETs are especially important in mediating cell growth, angiogenesis, differentiation, and apoptosis. Whereas a number of TK inhibitors show antiproliferative activity in NETs only sunitinib has undergone a phase III controlled trial. Sunitinib is an orally active small-molecule inhibitor of TK receptors (PDGFRs, VEGFR-1, VEGFR-2, c-KIT, FLT-3). In a phase III study in which 171 patients with progressive, metastatic, nonresectable pNETs were treated with sunitinib (37.5 mg/d) or placebo, sunitinib treatment caused a doubling of progression-free survival (11.4 vs 4.5 months, p <.001), an increase in objective tumor response rate (9% vs 0%, p = .007), and an increase in overall survival. Sunitinib treatment was associated with an overall threefold increase in side effects, although most were grade 1 or 2. The most frequent grade 3 or 4 side effects were neutropenia (12%) and hypertension (9.6%), which were controlled by dose reduction or temporary interruption. There is no consensus regarding the order of sunitinib or everolimus use in patients with advanced, well-differentiated, progressive pNETs.
PRRT for NETs involves treatment with radiolabeled somatostatin analogues. The success of this approach is based on the finding that somatostatin receptors (sst) are overexpressed or ectopically expressed by 60–100% of all NETs, which allows the targeting of cytotoxic, radiolabeled somatostatin receptor ligands.
Three different radionuclides are being used. High doses of [111In-DTPA-D-Phe1]octreotide, which emits γ-rays, internal conversion, and Auger electrons; 90yttrium, which emits high-energy β-particles coupled by a DOTA chelating group to octreotide or octreotate; and 177lutetium-coupled analogues, which emit both, are all in clinical studies. At present, the 177lutetium-coupled analogues are the most widely used. 111Indium-, 90yttrium-, and 177lutetium-labeled compounds caused tumor stabilization in 41–81%, 44–88%, and 23–40%, respectively, and a decrease in tumor size in 8–30%, 6–37%, and 38%, respectively, of patients with advanced metastatic NETs. In one large study involving 504 patients with malignant NETs, 177lutetium-labeled analogues produced a reduction of tumor size of >50% in 30% of patients (2% complete) and tumor stabilization in 51% of patients. An effect on survival has not been established. At present, PRRT is not approved for use in either the United States or Europe, but because of the above promising results, a large phase III study is now being conducted in both the United States and Europe. The ENETS 2012, NANETS 2010, Nordic 2010, and European Society for Medical Oncology (ESMO) guidelines list PRRT as an experimental or investigational treatment at present.
The use of liver transplantation has been abandoned for treatment of most metastatic tumors to the liver. However, for metastatic NETs, it is still a consideration. Among 213 European patients with NETs (50% functional NETs) who had liver transplantation from 1982 to 2009, the overall 5-year survival was 52% and disease free-survival was 30%. In various studies, the postoperative mortality rate is 10–14%. These results are similar to the United Network for Organ Sharing data in the United States in which 150 NET patients had liver transplants and the 5-year survival was 49%. In various studies, important prognostic factors for a poor outcome include a major resection performed in addition at the time of the liver transplant; poor tumor differentiation; hepatomegaly; age >45 years; a primary NET in the duodenum or pancreas; the presence of extrahepatic metastatic disease or extensive liver involvement (>50%); Ki-67 proliferative index >10%; and abnormal E-cadherin staining. The ENETS 2012 guidelines conclude that liver transplantation should be viewed as providing palliative care, with cure an exception, and recommend it be reserved for patients with life-threatening hormonal disturbances refractory to other treatments or for selected patients with a nonfunctional tumor with diffuse liver metastatic disease refractory to all other treatments.




