CHAPTER 85 MUSCULAR DYSTROPHIES
Muscular dystrophies constitute a clinically and genetically heterogeneous group of skeletal muscle-wasting diseases.1 The molecular causes of the muscular dystrophies remained elusive for many decades, and the muscular dystrophies were classified into relatively few clinical entities. In the late 1980s, major advances in molecular genetics led to the discovery of the dystrophin gene and its protein product, dystrophin.2,3 Mutations in the dystrophin gene result in dystrophin deficiency, which is the pathogenic determinant of the dystrophinopathies.4,5 Several muscular dystrophies are caused by mutations in other genes that cause defects of proteins localized at the sarcolemma, the cytoplasm or the nuclear envelope. Since the mid-1990s, an increasing number of genes have been associated with different forms of muscular dystrophy (Table 85-1, Fig. 85-1). These findings have led to a profound change in the classification of muscular dystrophies, with a new emphasis on molecular genetics rather than on clinical symptoms (which differentiated diseases on the basis of age at onset, severity, mode of inheritance, and muscle groups primarily affected).
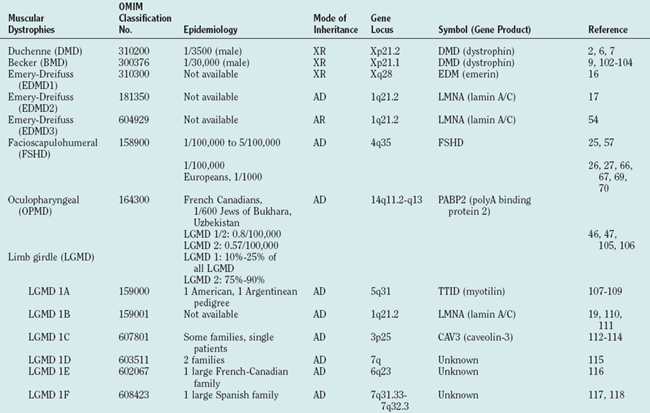

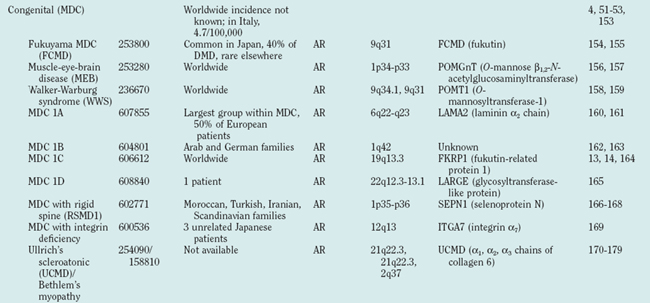
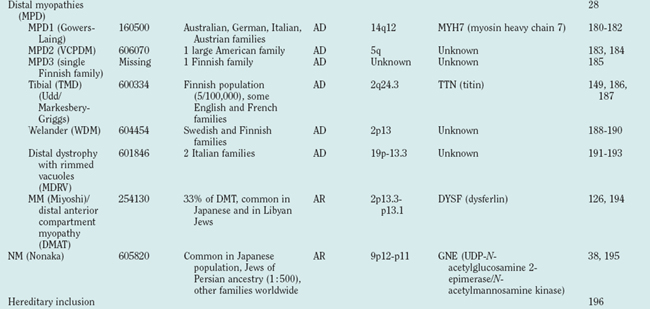

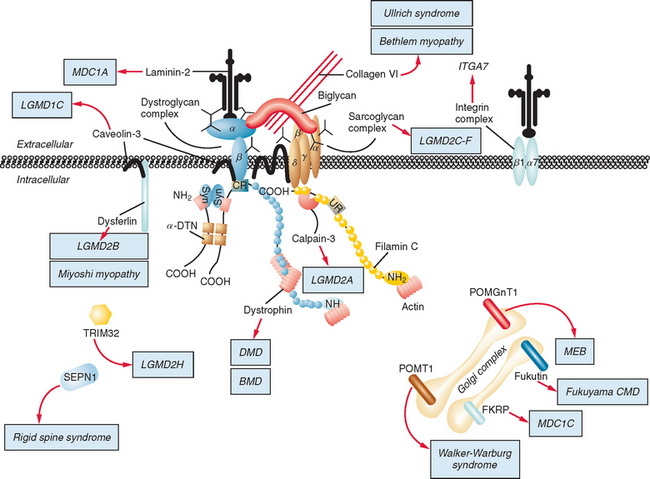
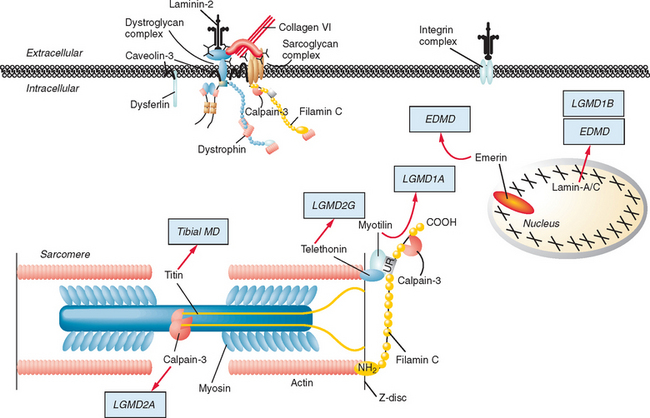
Figure 85-1 Muscular dystrophies and the membrane and enzymatic proteins they are associated with. This schematic shows the locations of various membrane and enzymatic proteins associated with muscular dystrophies. The diseases these molecules cause when mutated are shown in boxes. Dystrophin, through its interaction with the dystroglycan complex, connects the actin cytoskeleton to the extracellular matrix. Intracellularly, it interacts with dystrobrevin (α-DTN) and syntrophins (Syn) (shown in blue). Extracellularly, the sarcoglycan complex (orange) interacts with biglycan, which connects this complex to the dystroglycan complex and the extracellular matrix collagen. Intracellularly, δ- and γ-sarcoglycans interact with filamin C. The majority of filamin C is at the Z-disc of the sarcolemma shown in Figure 85-2. The four proteins shown in the Golgi complex have been demonstrated to affect the glycosylation state of the α-dystroglycan and mediate its binding to the extracellular matrix. Fukutin and fukutin-related protein have been shown to localize to the medial Golgi complex. The localization of POMT1 and POMGnT1 is unknown so far, but the authors hypothesize that they are in the Golgi complex, because they are involved in the glycosylation process. BMD, Becker muscular dystrophy; CMD and MDC, congenital muscular dystrophy; DMD, Duchenne muscular dystrophy; LGMD, limb girdle muscular dystrophy; MEB, muscle-eye-brain disease.
(Reprinted from Dalkilic I, Kunkel LM: Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 2003; 13:231-238.)
The most common form is the X-linked recessive Duchenne muscular dystrophy (DMD), named after Guillaume Benjamin Amand Duchenne, who described it in 1861.6,7 Positional cloning of the gene altered in DMD8 led to the discovery of dystrophin2 and to improved molecular diagnosis of DMD and of its milder allelic variant, Becker muscular dystrophy (BMD).9
About 50 years after Duchenne, Batten10 published the first cases of congenital muscular dystrophy (MDC). Unlike patients with the DMD/BMD phenotype, patients with MDC present with weakness and dystrophic changes in the muscle biopsy at birth, but symptoms are less rapidly progressive than in DMD.
The term limb girdle muscular dystrophy (LGMD) was introduced in the middle of the 20th century, when it became obvious that there was an additional major group of noncongenital muscular dystrophies that differed from both the X-linked DMD and BMD and from the autosomal-dominant facioscapulohumeral forms.11 Nowadays, the term limb girdle muscular dystrophy has changed from a wastebasket designation to an ever-expanding list of subtypes (18 thus far), for which accurate molecular diagnoses are available. Age at onset usually ranges from early childhood to late adulthood12; however, the same gene defect can cause allelic forms of MDCs and LGMDs, as shown for the fukutin-related protein deficiencies.13–15
The finding that mutations in nuclear envelope proteins also cause muscular dystrophies—referred to as nuclear envelopathies or Emery-Dreifuss muscular dystrophies—came as a surprise when emerin, the protein shown to be mutated in X-linked Emery Dreifuss muscular dystrophy (EDMD1, X-EDMD)16 was also found to be an inner nuclear membrane protein (Fig. 85-2). The importance of the nuclear envelope in neuromuscular disease was bolstered by the discoveries that mutations in the LMNA gene can cause autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD2),17 dilated cardiomyopathy with conduction defect (CMD1A),18 limb-girdle muscular dystrophy type 1B (LGMD1B),19 Charcot-Marie-Tooth disorder type 2B1 (CMT2B1),20 and a variety of other, nonneuromuscular diseases, such as Dunnigan’s familial partial lipodystrophy and mandibuloacral dysplasia, or premature aging syndromes, such as Hutchinson-Gilford progeria and atypical Werner’s syndrome.21,22 A new phenotype combining myopathy and progeria has been described.23
Facioscapulohumeral muscular dystrophy (FSHD), first described in 1885,24 is a frequent form of muscular dystrophy that has a distinctive clinical manifestation with autosomal dominant inheritance.11 The gene underlying FSHD was mapped to chromosome 4q35 in 199225 and was shown to be closely linked to locus D4F104S1. Although D4F104S1-associated deletions are closely associated with FSHD, the identity and location of the FSHD gene (or genes) still remain elusive, as does the mechanistic basis of the disease.
Oculopharyngeal muscular dystrophy (OPMD) is unusual among muscular dystrophies because of its manifestation in late adult life, typically in the sixth decade. Symptoms include progressive ptosis and dysphagia, followed by involvement of other cranial and limb muscles. OPMD is usually inherited as an autosomal dominant trait as a result of an expanded guanine-cytosine-guanine (GCG) repeat detectable in the poly A binding protein 2 gene on chromosome 14.26,27
Distal myopathies are frequent in the Nordic countries and are being increasingly recognized elsewhere. To date, six different distal myopathy phenotypes have been identified, and there has been considerable progress in the understanding of the molecular pathophysiology underlying the distal myopathies.28 Mutations in membrane-associated dysferlin cause two different distal phenotypes, allelic to LGMD2B,29 whereas mutations in titin can cause either a distal myopathy (type Udd/Markesbery-Griggs) or LGMD2J,30,31 another example of the diversity between clinical and genetic disease definitions.
Hereditary inclusion body myopathies (HIBM) belong to the heterogeneous group of myopathies with rimmed vacuoles. So far, autosomal recessive (HIBM2)32 and autosomal dominant forms33–37 have been linked to different gene loci. Interestingly, distal myopathy with rimmed vacuoles (Nonaka myopathy) has been attributed to mutations in the GNE gene and is therefore allelic to HIBM2.38
DYSTROPHINOPATHIES
Clinical Features
In DMD, children usually come to medical attention between ages 3 and 5 years because of frequent falls, awkward running, and waddling gait. At this stage, weakness is most pronounced in the hips and proximal leg muscles but over time will affect neck flexors, proximal arm muscles (especially biceps), ankle dorsiflexors, and respiratory muscles, necessitating ventilation at some point during the course of the disease. Calf hypertrophy is a distinctive feature, whereas facial and bulbar muscles are largely spared. Creatine kinase levels are massively elevated, 50 to 100 times normal. The disease progresses relentlessly, and ambulation is lost between 7 and 12 years of age. Prednisone, the primary pharmacological therapy for DMD, may modestly prolong ambulation.
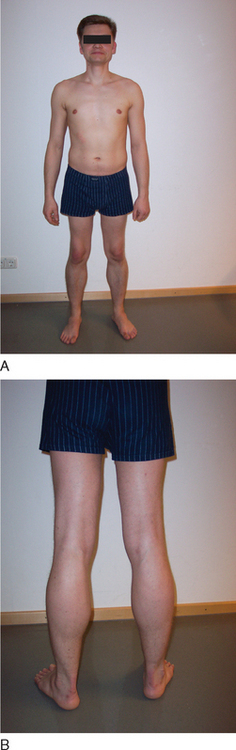
BMD is an allelic variant of DMD and has a more benign and variable manifestation with later onset and slower progression. Onset of symptoms after age 6 or walking after age 13 are typical features. In rare cases, BMD is not diagnosed until adulthood and the patients never lose ambulation (Fig. 85-3).
Female carriers of DMD or BMD are usually asymptomatic but may also have limb weakness, elevated creatine kinase levels, and nonmuscular symptoms such as cardiac involvement. The extent of clinical severity depends in part on the degree of skewed X chromosome inactivation in somatic cells.39
Skeletal muscle weakness in DMD and BMD is frequently accompanied by cardiac muscle dysfunction. Approximately 95% of patients have cardiac involvement by the time of death. This includes electrocardiographic abnormalities, arrhythmias, and myocardial dilatation and thickening. Patients with DMD and BMD may have normal intelligence, but the mean Wechsler full IQ is below average: 25% of BMD and 31% of DMD patients have an IQ of less than 75. Scoliosis is frequently severe. Contractures develop in hips, ankles, and elbows. On occasion, involvement of smooth muscle leads to gastrointestinal complications, including pseudo-obstruction.40
Etiology and Pathophysiology
Dystrophin is the largest human gene, covering 2.5 megabases and including 79 exons. The enormity of the gene, along with the spontaneous mutation rate of each base pair allows a high frequency of novel mutations, which explains how 30% of all DMD and BMD cases result from spontaneous mutations. The gene encodes a 427-kD subsarcolemmal protein. Dystrophin has several well-characterized domains, including an amino-terminal domain with homology to α-actinin, a large “rod” domain with spectrin-like repeats, a cysteine-rich domain, and a carboxy-terminal domain with homology to other dystrophin-related proteins. Dystrophin interacts with and stabilizes a large membrane-associated protein complex, the dystroglycan complex, whose components are involved in other muscular dystrophies (see Fig. 85-1). It links the actin cytoskeleton through its amino-terminal binding domain to the sarcolemma and extracellular matrix through interactions with its carboxy-terminal binding domains. In addition to physically connecting and stabilizing this large protein network, dystrophin may play a role in signal transduction because of its association with nitric oxide synthase and highly phosphorylated postsynaptic proteins.
Approximately two thirds of disease-causing mutations in dystrophin are large deletions; an additional 5% are duplications within hot spots of the dystrophin gene. These common mutations can be detected by commercially available multiplex polymerase chain reaction, which amplifies 18 to 25 of the gene’s 79 exons from genomic DNA obtained from blood samples. Additional deletions and duplications may be detected by amplifying and quantifying all exons. There is no simple relation between size of the deletion and resultant clinical phenotype. For example, deletion of small exons, such as exon 44, often results in classic DMD, whereas large deletions that may involve nearly 50% of the gene have been described in patients with BMD. The central and distal rod domains seem to be functionally almost dispensable, inasmuch as some deletions in this region are associated only with myalgia or muscle cramps, not with weakness. Some patients may even present solely with an isolated increase in creatine kinase level. This has been shown in patients with in-frame deletions in exons 32 to 44, 48 to 51, or 48 to 53, all of whom had normal or near-normal dystrophin levels at the sarcolemma. The effects on the phenotype depend, therefore, not so much on the size of a deletion (or duplication) but on whether it disrupts the reading frame. A further observation is that deletions very different in size and position may produce very similar severe phenotypes. The reason for this effect might be the occurrence of nonsense-mediated RNA decay. This phenomenon may account for the lack of rescue of dystrophin function, as well as for the phenotypic variability related to variations in the efficiency of RNA decay control. Mutations that maintain the reading frame (in-frame mutation) generally result in abnormal but partially functional dystrophin and are associated with BMD. In patients with DMD, deletions and duplications disrupt the reading frame (frame-shift mutations), resulting in unstable RNA and nearly undetectable truncated proteins. The reading frame hypothesis holds true for over 90% of cases and is commonly used both as a diagnostic confirmation of dystrophinopathies and for the differential diagnosis of DMD and BMD.41
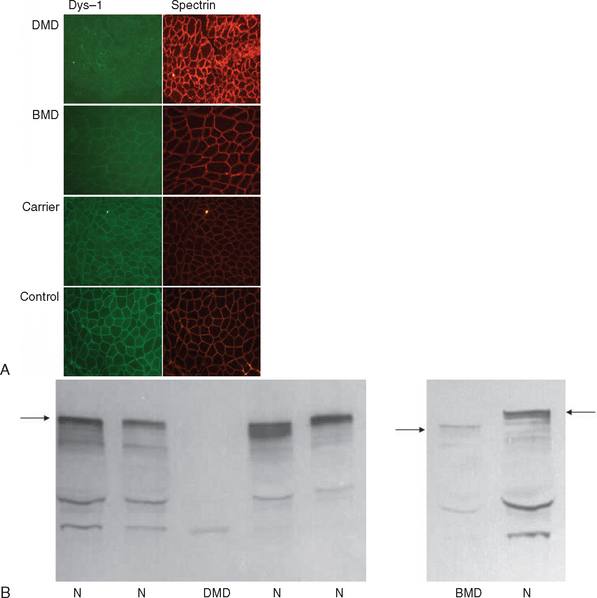
If knowledge of a specific small mutation is desired, complementary DNA may be generated from muscle messenger RNA and sequenced. Single-strand conformation polymorphism analysis42 and single-condition amplification/internal primer sequencing43 of genomic DNA have also been proposed for identification of small mutations. Furthermore, Buzin and associates reported mutation rates in the dystrophin gene with a mutational hot spot at a cytosine-guanine island (CpG) dinucleotide.44 Approximately 30% are point mutations and 10% to 15% are nonsense mutations that produce premature stop codons. If genomic DNA testing does not reveal a deletion or duplication, a muscle biopsy is frequently performed to confirm the diagnosis. Histological features include marked variability of fiber size with aberrant large fibers and hypercontracted muscle fibers along with groups of atrophic, degenerating, and regenerating fibers. Endomysial fibrosis and fatty infiltration are invariably present along with some mononuclear inflammatory cell infiltration. Dystrophin immunostaining shows absence of the protein in DMD and patchy staining in BMD and may reveal a mosaic pattern of expression in female carriers. Dystrophin immunoblotting shows absence of the dystrophin band in DMD, shows weak expression and/or abnormal molecular weight in BMD, and usually yields normal results in female carriers (Fig. 85-4).
LIMB GIRDLE MUSCULAR DYSTROPHIES
Definition
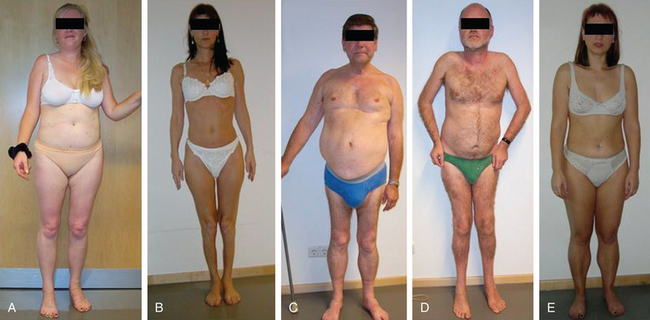
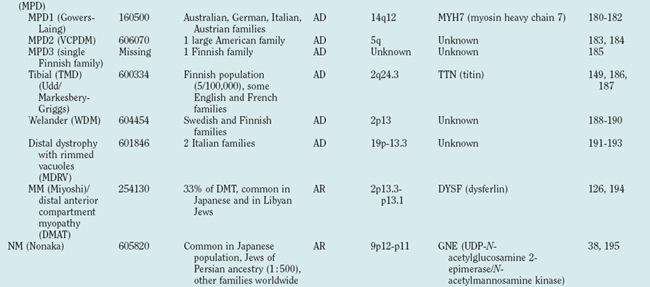
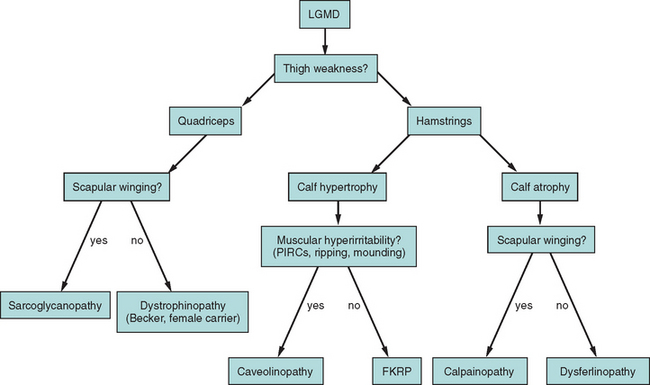
Because of molecular discoveries since the mid-1990s, LGMD has emerged as an entire field within the inherited myopathies; at least six autosomal dominant and ten autosomal recessive gene defects have been identified. Redefined in 1995, the LGMD are face-sparing, predominantly proximal, progressive muscular dystrophies with elevated creatine kinase levels, and dystrophic features are demonstrated on muscle biopsy (Fig. 85-5).12,45–47 In the current classification system, LGMDs are divided into autosomal dominant (LGMD1) and autosomal recessive (LGMD2) disorders with an additional lettering system that denotes the chronological order of chromosomal linkage (thus far, A through G for autosomal dominant LGMD and A through K for autosomal recessive LGMD). Accurate diagnosis is now possible for most LGMDs and is important for genetic counseling. Certain LGMDs are characterized by treatable cardiac and respiratory complications.45,48
The causes of the LGMD include mutations in a wide range of proteins and protein systems. Calpain, mutated in LGMD2A, is a calcium-activated neutral protease, and this is the first known genetic defect in muscular dystrophy stemming from an enzyme defect. Other mutations impair membrane signaling and repair (caveolin, dysferlin), sarcolemmal integrity (sarcoglycans), and contractile dysfunction (myotilin, telethonin, titin). The subcellular localization of these proteins also varies from the nuclear membrane (lamin A/C) to the sarcomere (myotilin, calpain, telethonin, titin), the sarcoplasm (TRIM32, a putative E3-ubiquitin-ligase), the sarcolemma (caveolin, sarcoglycans, dysferlin), and even the extracellular space (fukutin-related protein). Of note, involvement of one nuclear membrane protein, emerin, causes X-linked Emery Dreifuss syndrome (X-EDMD, EDMD1), whereas mutations in the related protein, lamin A/C, cause EDMD2. It remains unclear how mutations in apparently unrelated proteins can cause similar phenotypes, and it is difficult at present to see a common final pathway of action. Likewise, identical genotypes often yield substantially different phenotypes even within the same family. Dysferlinopathies can cause LGMD2B or distal myopathies, and caveolinopathies have been associated with LGMD1C, rippling muscle disease, persistent elevated creatine kinase levels (hyperCKemia), or distal myopathy. Fukutin-related protein gene (FKRP) mutations manifest high clinical variability, with onset at birth or later in adulthood (LGMD2I) (Fig. 85-6). Titinopathies exhibit phenotypic variability depending on the amount of genes involved: A single mutated allele causes a late adult-onset distal myopathy (Finnish tibial muscular dystrophy), whereas two abnormal alleles cause LGMD2J. This remarkable phenotypic variability remains enigmatic, but explanations may be forthcoming through investigations of modifier genes, DNA microarrays, and proteomic studies.49 LGMD2K has been described as an allelic form of Walker-Warburg syndrome in Turkish patients.50 Further phenotypic differentiation of the LGMDs is provided in Table 85-2 and Figure 85-7.

Autosomal Dominant Limb Girdle Muscular Dystrophy
Autosomal dominant LGMDs tend to have an altogether slower course and later onset with less elevation of serum creatine kinase level in comparison with autosomal recessive LGMDs. They are also clinically much more heterogeneous (see Table 85-2 and Fig. 85-7). Except for LGMD1C (caveolinopathy), immunohistochemistry and immunoblotting are of little help in differentiating dominant LGMD subtypes.
Autosomal Recessive Limb Girdle Muscular Dystrophy
The autosomal recessive forms of LGMD for which the genetic bases are known can be further subdivided on the basis of the genes and proteins involved (see Table 85-1). The first autosomal recessive form, LGMD2A, was mapped to chromosome 15q in 1991 in patients from the Reunion Island. Ten additional forms have been mapped since the mid-1990s, most recently LGMD2K. The protein products of these 11 genes have been identified (see Fig. 85-1): calpain-3 for LGMD2A, dysferlin for LGMD2B, γ-sarcoglycan for LGMD2C, α-sarcoglycan for LGMD2D, β-sarcoglycan for LGMD2E, δ-sarcoglycan for LGMD2F, the sarcomeric protein telethonin for LGMD2G, TRIM32 for LGMD2H, fukutin-related protein for LGMD2I, titin for LGMD2J, and POMT1 for LGMD2K. Genotypephenotype correlation studies have been reported for the different forms of LGMD in an attempt to enhance comprehension of the underlying pathological mechanisms, to better characterize each of the subgroups and, of more importance, to identify modifier genes or epigenetic factors that might modulate the clinical course in patients who carry the same pathological mutation. Immunohistochemistry and immunoblotting are pivotal for LGMD2 subtype differentiation and for directing the molecular genetic analysis (see Table 85-2 and Fig. 85-7).
CONGENITAL MUSCULAR DYSTROPHIES
Definition
The MDCs are a group of genetic disorders in which weakness and abnormal muscle histological features are present at birth. Muscle weakness tends to be more stable, but complications can become more prominent over time. A firm diagnosis of MDC requires a muscle biopsy. Pathological findings include variation in fiber size, internal nuclei, and increased endomysial and fatty tissue. Signs of ongoing degeneration and regeneration may be less prominent than in muscular dystrophies of later onset (e.g., DMD, BMD, or sarcoglycanopathies). Creatine kinase concentrations are variable and can be normal. The mode of inheritance for most MDCs is autosomal recessive with significant genetic heterogeneity.51–53 For clinical differentiation, see Table 85-2 and Figs. 85-8 and 85-9.
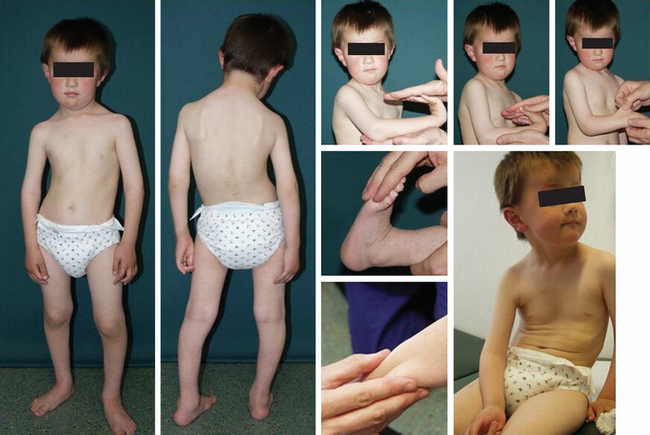
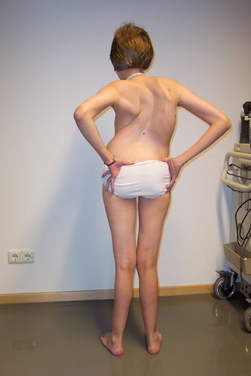
Clinical Features, Etiology, and Pathophysiology
Immunohistochemistry and immunoblotting help to distinguish between merosin-positive and merosin-negative forms of MDC. Reduction or absence of α-dystroglycan immunostaining indicates a defect of one of the glycosylation genes (see Tables 85-1 and 85-2 and Fig. 85-6).
NUCLEAR ENVELOPATHIES
Definition
Emery Dreifuss muscular dystrophies are characterized by early contractures of the elbows, Achilles tendons, and spine; slowly progressive muscle wasting and weakness with a predominantly humeroperoneal distribution; and cardiomyopathy, usually manifesting as heart block. There are two main modes of inheritance: X-linked (X-EDMD, EDMD1) and autosomal dominant (ADEDMD, EDMD2/LGMD1B). However, a rare autosomal recessive mode of inheritance (EDMD3) has also been reported.54 In 1999, the nuclear lamin A/C gene, LMNA, at locus 1q11-q23 was found to be responsible for EDMD2 and EDMD3.17 Whereas mutations in the emerin gene, STA, at locus Xq28, give rise to one phenotype, the X-linked form of Emery Dreifuss muscular dystrophies, mutations in LMNA cause at least nine (and probably more) different neuromuscular and nonneuromuscular phenotypes.55 For clinical differentiation, see Table 85-2.
Etiology and Pathophysiology
One of the most intriguing questions regarding the role of emerin and lamins A and C in disease is how mutations in nearly ubiquitously expressed proteins can cause different tissue-specific illnesses. Several hypothetical pathophysiological mechanisms have been proposed. Protein complexes of the nuclear envelope, which include lamins and emerin, may function in the regulation of gene expression, inasmuch as there are several examples of nuclear envelope proteins that interact with chromatin proteins. The functional effects of these interactions are not yet understood, but they suggest a potential role for the nuclear envelope in chromatin organization. Therefore, alterations in the nuclear envelope that result from mutations in emerin and lamins A and C may change the expression of genes involved in striated muscle function. Another hypothesis is that mutations in nuclear envelope proteins cause neuromuscular disease by altering the structural integrity of the nucleus. This so-called “mechanical stress” hypothesis could partly explain the muscle-specific phenotypes of some of these diseases. Nuclei with mutant structural proteins may be more fragile. During muscle contraction, nuclei in muscle cells may be under more stress than are nuclei of other cells and therefore may be sensitive to defects in the nucleoskeleton.56
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Definition
FSHD is a highly variable disorder in which weakness appears from infancy to late life but typically in the second decade. Usually, the disease initially involves the face and scapulae, followed by foot dorsiflexors and hip girdles. Typical features include the often striking asymmetry of muscle involvement and the sparing of bulbar extraocular and respiratory muscles.57
Clinical Features
Clinical diagnosis rests on the distinctive pattern of muscle involvement (Fig. 85-10). Although several inherited myopathies can include facial and shoulder girdle weakness, the following features are specific to FSHD: (1) Facial weakness is prominent and typically more severe in the lower facial musculature. (2) Extraocular, eyelid, and bulbar muscles are spared. (3) The shoulders anteriorly have a typical contour with straight clavicles, forward sloping, and rounding. (4) Pectoral, biceps, and triceps muscles are typically involved, whereas deltoid and forearm muscles are spared. (5) Lower abdominal muscles are selectively involved, resulting in lumbar lordosis. (6) Leg involvement frequently starts with foot dorsiflexor weakness, but quadriceps weakness can also be manifest early in the disease.57 (7) Contractures are rarely present, but during the course of the disease, patients may develop additional weakness of wrist extensors. (8) Muscle weakness is often asymmetrical.
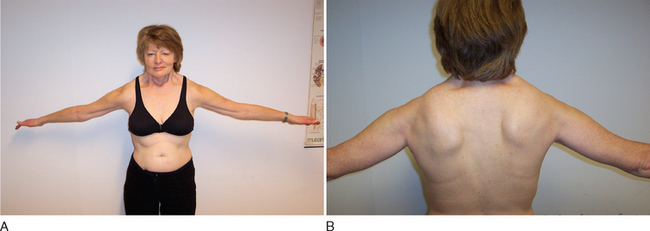
Extramuscular manifestations of FSHD include high-frequency hearing loss, retinal telangiectasias, and a tendency to develop atrial arrhythmias.58 These manifestations, if present, are mostly mild. Symptomatic cardiac involvement, present in approximately 5% of patients with molecularly confirmed FSHD, consists of conduction abnormalities with frequent supraventricular tachyarrhythmias.
Inheritance is autosomal dominant with a high degree of penetrance, but sporadic cases caused by de novo mutations account for up to 30% of cases.59
Etiology and Pathophysiology
Understanding the pathophysiology of FSHD is hampered by the unique molecular genetic lesion. Despite the identification of a causal deletion on chromosome 4q35 in 1990, the molecular pathophysiology of FSHD remains unclear. When FSHD was linked to chromosome 4q35, subtelomeric rearrangements consisting of deletions of an integral number of copies of a 3.3kb DNA repeat named D4Z4 were identified.25 Whereas normal individuals have 15 or more repeats on each copy of 4q35, individuals with FSHD have 12 or fewer repeats on each copy. The deletions do not appear do disrupt an expressed gene. Rare FSHD kindreds fail to show linkage to 4q35, but alternative genetic loci have not been identified thus far.
Whereas it is clear that a critical deletion within the 4q35 D4Z4 repeats results in FSHD, no expressed sequences are present within the deleted segments. Investigators have found 4q35 genes located upstream of D4Z4 to be inappropriately overexpressed in FSHD muscle. An element within D4Z4 has been shown to behave as a silencer that provides a binding site for a transcriptional repressing complex. These results are suggestive of a model in which deletion of D4Z4 leads to inappropriate transcriptional repression of 4q35 genes, resulting in disease.60 FRG2, an FSHD candidate gene, has been found to be transcriptionally upregulated in differentiating primary myoblast cultures from patients with FSHD and is an attractive candidate culprit.61 In most cases, diagnosis can be achieved by blood genetic testing without a prior muscle biopsy.
OCULOPHARYNGEAL MUSCULAR DYSTROPHY
Definition
OPMD was first clearly described by Taylor in 1915 in four members of a French Canadian family.62 They had late-onset ptosis in association with progressive swallowing difficulties (dysphagia), which led to starvation and death. This paper and a few other case reports63–65 were largely overlooked until the classic publication by Victor and associates in 1962.66 They were the first to refer to this myopathy as oculopharyngeal muscular dystrophy.
Clinical Features
OPMD usually manifests in the fifth or sixth decade with two cardinal symptoms: ptosis of the eyelid and dysphagia, both of which have a slowly progressive course (Fig. 85-11). Later, all extraocular and other voluntary muscles may become affected. In most cases, ptosis is the first symptom. As the disease evolves, there may be impairment of eye movements and, occasionally, diplopia; nevertheless, complete external ophthalmoplegia is infrequent.26,67,68 With time, the dysphagia results in undernutrition and may result in death from aspiration pneumonia. Other symptoms and signs are caused by involvement of other muscles. In most patients, the voice becomes nasal as a result of palatal weakness. Weakness and atrophy of the tongue are common.69 Mild facial, temporal, and masseter muscle involvement may become apparent after the ptosis has become obvious. Weakness often affects pelvic girdle muscles and, to a lesser extent, the shoulder girdle. The disease has a slowly progressive course. Life expectancy is not diminished by the condition.70 Until recently, death usually occurred at an advanced age as a result of starvation or aspiration pneumonia. With progress in the treatment of pharyngeal dysfunction and better nutrition, the life spans of patients have become longer, and the prognosis and quality of life have improved.

The pathological changes of extraocular and other voluntary muscles vary according to the stage of the disease and the muscle examined. Probably all skeletal muscles are affected, but extraocular, lingual, pharyngeal, and diaphragmatic muscles are found to be more severely involved in autopsy studies.67 Muscles studied by classic histological methods show changes that are common to many muscular dystrophies. Histochemical studies reveal two particular changes: (1) small angulated fibers that often react strongly for oxidative enzymes and are more frequently type 1 than type 2 and (2) rimmed vacuoles within the muscle fibers. Rimmed vacuoles have been observed in several other disorders, particularly inclusion body myositis. However, their presence supports the clinical diagnosis of OPMD. The most significant ultrastructural change is the presence of intranuclear tubular filaments with an 8.5-nm outer diameter and a 3-nm inner diameter.
Etiology and Pathophysiology
Autosomal dominant and recessive forms of OPMD have been described. The OPMD locus was first mapped to chromosome 14q11.1 by linkage analysis in large French Canadian families.27 A positional cloning strategy led to the identification of short (GCG)8 to (GCG)13 expansions in the PABPN1 gene in all autosomal dominant OPMD cases.26 They consist of mitotically and meiotically stable expansions of a (GCG)n repeat in the first exon of the gene. Dominant mutations consist of the addition of two to seven (GCG) repeats to the usual (GCG)6 stretch. In populations with founder effects, such as French Canadians or Jews from Bukhara, Uzbekistan, most families share the same historical mutation, which only rarely change in size.26 OPMD is one of the few triplet repeat diseases in which it is not known whether there is an inverse correlation between the severity of the phenotype and the size of the mutation. It is clear, however, that the severity of the phenotype varies even among carriers of the same size (GCG)n PABPN1 mutation.26,69 Only carriers of the smallest (GCG)8 PABPN1 mutation appear to have a milder phenotype, with an age at onset in the seventh decade and only mild dysphagia. Persons who are compound heterozygotes for dominant and recessive mutations have more severe phenotypes.26 Diagnosis is usually achieved by direct blood genetic testing.
DISTAL MYOPATHIES
Definition
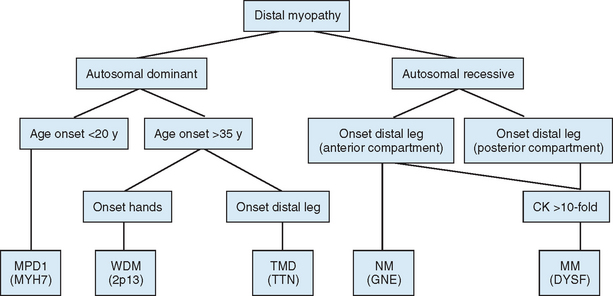
Distal myopathies are a heterogeneous group of genetic disorders characterized clinically by progressive muscular weakness and atrophy beginning in the hands or in the feet and pathologically by myopathic changes in skeletal muscles. Eight distinct distal myopathies have been identified, and four have been defined at the molecular level. They are classified according to age at onset, mode of inheritance, and muscle groups initially involved. Since 2000, much has been learned about the molecular etiologies of these myopathies. All have been linked to specific chromosome regions. One of the most interesting conclusions of the molecular studies is that the same gene can be responsible for diverse clinical phenotypes.71
The diagnostic process for the common forms of distal myopathy is simplified in Figure 85-12. Except for Miyoshi myopathy, in which the serum creatine kinase level is markedly elevated and the pathological findings are typical of muscular dystrophy, most distal myopathies are characterized by normal or mildly elevated creatine kinase levels and share in common the pathological feature of rimmed vacuoles.
HEREDITARY INCLUSION BODY MYOPATHIES
Definition
The morphological hallmarks of inclusion body myopathies are the rimmed vacuoles and characteristic inclusion bodies. Inclusion bodies contain a variety of proteins, such as amyloid and the prion protein. Ultrastructurally, there are typical 15- to 18-nm filaments. Clinically, most cases of inclusion body myopathies are sporadic, manifest at an older age than HIBM, and demonstrate inflammatory muscle infiltrations similar to those of polymyositis. Although the degree of inflammation is variable, sporadic inclusion body myopathy is considered an autoimmune disorder. In contrast to the sporadic cases, muscle biopsy specimens of patients with HIBM usually (but not always) lack inflammatory infiltrates, and HIBM belongs to the heterogeneous group of myopathies with rimmed vacuoles, characterized clinically by progressive muscular weakness and atrophy beginning in the hands or feet. However, there is considerable phenotypic and genotypic overlap with distal myopathies and muscular dystrophies (see Tables 85-1 and 85-2).
Etiology and Pathophysiology
The unusual constellation of proteins present in the inclusions of this condition is characteristic and does not occur in vacuolated or nonvacuolated fibers of other muscle diseases. In the various inclusion body myopathies, different etiologies may lead to a common pathogenic cascade, which is ultimately responsible for the characteristic muscle fiber degeneration.72
TREATMENT OF MUSCULAR DYSTROPHIES
In contrast to most compounds that were tested in clinical trials for muscular dystrophies, corticosteroids, such as prednisone and deflazacort, proved to be of some real benefit in terms of muscle strength.73–77 However, side effects (weight gain, fluid retention, growth retardation, cataract) are prominent and often prohibitive of long-term treatment. There is still no general consensus among specialists on corticosteroid treatment, which varies among and within different countries. In general, European clinicians prescribe corticosteroids more restrictively but pay more attention to supportive treatment. Even in the United States, where most of the positive trials were performed, the use of corticosteroids is not uniform. Some patients respond dramatically well to corticosteroids with minimal side effects. Low-dosage and alternate-day regimens may help reduce side effects.78,79 Many uncertainties remain about the best regimen and the type of corticosteroid to obtain maximal efficacy with minimal side effects. Corticosteroid treatment with deflazacort appears to cause fewer side effects, particularly less weight gain, in comparison with prednisone.80,81 The efficacy of corticosteroids in other muscular dystrophies has not yet been tested systematically, but steroids are also used in patients with BMD82 and sarcoglycanopathies.83,84 An international European Neuromuscular Centre workshop has combined efforts to define the “gold standards” in the use of steroids for DMD.85
Aminoglycosides, including gentamicin, have been known to promote “read-through” of premature stop codons and thus suppression of nonsense mutations. Therefore, considerable interest followed a report showing gentamicin-induced read-through in mdx mice that generated full-length dystrophin and raised hopes that DMD may be treatable by a conventional drug.86 Unfortunately, several human and animals trials were unable to replicate the beneficial results of gentamicin treatment.87,88
Creatine supplementation results in increased high-intensity strength in patients with different types of neuromuscular diseases. A controlled study showed significant improvement of strength and daily life activities in patients with different types of muscular dystrophies (DMD, BMD, LGMD, FSHD) during short-term creatine supplementation.89 Short-term administration of creatine produces few side effects. Minor adverse effects result from increase of muscle mass, muscular cramps, and entrapment syndromes that were seen in athletes but not in patients with muscle diseases so far.
In a Cochrane Review on drug treatment for FSHD,90 researchers did not find evidence from two randomized controlled trials to support any drug treatment for FSHD, but only two high-quality trials that fulfilled the selection criteria have been published. In one, creatine supplementation was compared with placebo, and in the other, high- and low-dose albuterol was compared with placebo. (Another randomized controlled trial of albuterol in FSHD has not been published.) The creatine trial demonstrated a nonsignificant difference in favor of creatine. The albuterol trial demonstrated no significant difference in muscle strength at 1 year, but some secondary measures such as lean body mass and hand grip strength did improve.89,91
Potentially causative treatment strategies are conducted mainly for dystrophinopathy and include cellular approaches, such as myoblast transplantation and stem cell transplantation; gene therapy, such as direct delivery of dystrophin or other therapeutic genes; and new techniques, such as exon skipping by antisense oligonucleotides. Introduction of normal muscle precursor cells (myoblasts) into dystrophic muscle results in their incorporation into myofibers, so that a proportion of the nuclei in each newly formed myofiber carry a functional copy of the dystrophin gene. This approach was successful in demonstrating relocalization of dystrophin to the sarcolemma in mdx muscle and expression of dystrophin transcripts in DMD patients by reverse-transcriptase polymerase chain reaction. However, clinical trials have not shown any objective benefit in patients injected with donor myoblasts. The ability of cell populations such as transplanted stem cells to adapt to a tissue phenotype is of potential therapeutic use, inasmuch as previous studies have shown that such cells can contribute to muscle repair in mice that have undergone bone marrow transplantation. Stem cells derived from a dystrophin-positive donor have also been shown to contribute to dystrophin-positive muscle and cardiac tissue in mdx mice in vivo. In addition, bone marrow transplantation from a dystrophin-positive donor to a patient with DMD has been shown to be a viable means by which to deliver these cells to muscle. However, the poor efficiency of the procedure currently precludes clinical use.92
Gene therapy approaches aim to deliver DNA encoding dystrophin or other therapeutic genes (e.g., utrophin) to muscle. Results of studies with mdx mice indicate that this approach is effective in principle and that the level of dystrophin expression required is not critical as long as threshold levels of therapeutic genes are achieved. To date, most dystrophin-delivery approaches are hampered by the large size of the gene (which is larger than the cargo capacity of most current viral vectors), by the immune responses to protein and viral antigens, and by vector delivery to skeletal and heart muscle. “Mini-dystrophin” genes (in which a large proportion of the rod domain is deleted) retain some function and have the advantage of being within viral cloning capacities, similar to “mini-utrophin” to provoke a less vigorous immune reaction, but systemic delivery remains a formidable difficulty. Use of naked plasmid DNA and plasmid DNA-liposome complexes has been described for the delivery of genes to skeletal muscle. The advantage of this technique is that plasmid DNA appears to be nonantigenic. Postmitotic myofibers can sustain episomal expression of plasmids for long periods of time, but this approach is hampered by transfection inefficiency, with the exception of neonatal muscle, and the difficulty of introducing plasmid DNA into a sufficiently high proportion of myofibers to effect phenotypic recovery in vivo. Nevertheless, there is remarkable progress in this direction. It is encouraging to note that in the relatively short time since the DMD gene was identified, this approach was used for the first gene therapy trial in patients with DMD, demonstrating that exogenous dystrophin expression can be obtained in patients with DMD or BMD after intramuscular transfer of plasmid, without adverse effects, and hence paving the way for future developments in gene therapy.93
One technique appears to reestablish an open reading frame mutant dystrophin messenger RNA through modification of endogenous dystrophin. In this approach, antisense oligonucleotides complementary to intron/exon boundaries or exonic sequences are used to induce exon skipping of mutant regions of the dystrophin protein (point mutations), which leads to in-frame translation. In cultured muscle cells of six patients with DMD who carried different deletions and a nonsense mutation, this approach removed the targeted exon, restoring the reading frame and thereby dystrophin synthesis in 75% of the cells.94 In 2005, Lu and colleagues showed that systemic delivery of antisense oligoribonucleotide restores dystrophin expression in bodywide skeletal muscles of mdx mice.95 Similarly, persistent exon skipping was achieved in mdx mice with a single administration of an adenovirus-associated virus expressing antisense sequences linked to a modified U7 small nuclear RNA.96
Surgery is an important treatment modality in patients with DMD, but patients with other types of muscular dystrophy may also profit from similar techniques. In specific types of muscular dystrophy, surgical fixation of joints or limbs with instability and secondary muscle weakness can greatly improve the patient’s quality of life. In FSHD, scapulothoracic arthrodesis and scapulopexy may improve upper limb function and performance of activities of daily living, and they may also fulfill esthetic purposes. According to a Cochrane Database review, surgical interventions appear to produce significant benefits, although these have to be balanced against postoperative immobilization, need for physiotherapy, and potential complications.97 Patients with OPMD may profit from ptosis correction.
Cardiorespiratory involvement is frequent in most muscular dystrophies and a dominant symptom in some of them. Therefore, close monitoring of pulmonary function, electrocardiography, 24-hour electrocardiography, and echocardiography are necessary. If pulmonary function deteriorates, noninvasive methods of ventilation should be offered to the patient, thereby improving quality of life and survival.98 Early recognition of cardiac involvement is warranted as adequate, and timely cardiac therapy may be pivotal in the survival and disease course of patients with muscular dystrophy.99 Therapeutic options in impulse generation and conduction abnormalities range from drugs (digitalis, amiodarone, β blockers, Ca2+ antagonists, anticoagulants) to cardioversion or insertion of a pacemaker or implanted defibrillator. In heart failure caused by diastolic or systolic dysfunction, angiotensin-converting enzyme inhibitors play an important role. Successful cardiac transplantation has been described in patients with DMD, BMD, Emery Dreifuss muscular dystrophies, LGMD, and myotonic dystrophy.
CONCLUSIONS AND RECOMMENDATIONS
Curative therapy is not currently available, although the development of promising new treatment modalities is under way.92,100 In the future, molecular therapy may be the best way to reverse the molecular defects that cause muscular dystrophies, but practical and effective treatment in humans may still be years away. For evaluating different treatment modalities, symptomatic strategies, such as pharmacological therapies and supportive treatment, are differentiated from causative treatment schedules, such as gene or cell therapy.
Although no major therapeutic breakthrough has been achieved and curative treatment modalities are not yet applicable, life expectancy and quality of life in patients with muscular dystrophy have steadily improved since the 1960s. This achievement has been reached by improved symptomatic treatment and care, such as assisted ventilation, drug therapy for heart failure, and surgical therapy to prevent complications. However, there are still few pharmacological options to specifically prevent or delay the dystrophic process in muscle fibers.101
The exact mechanism of how steroids work in muscular dystrophies remains unknown. Future studies should attempt to unravel this mechanism; it is hoped that this will lead to an equally potent but less toxic drug. Creatine seems to be of limited efficacy without remarkable side effects; therefore, short-term administration may be helpful in individual cases. Long-term studies are required for evaluation of long-term safety and efficacy. New molecular therapeutic approaches have been invented and are currently investigated in cell and animal models of muscular dystrophies. The first phase I clinical trials for plasmid-based gene therapy of dystrophinopathies have yielded promising results.93 Although molecular therapies promise causal intervention and curative treatment, a large number of technical and methodological problems need to be solved. Furthermore, molecular therapies should be applied to patients with muscular dystrophy only if they are considered reasonably safe. Therefore, the majority of patients with muscular dystrophy today must rely on standard symptomatic therapy, while molecular approaches continue to hold promise for the future.
Bushby K, Muntoni F, Urtizberea A, et al. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:526-534.
Kirschner J, Bonnemann CGL. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch Neurol. 2004;61:189-199.
Lu QL, Rabinowitz A, Chen YC, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in bodywide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102:198-203.
Muntoni F, Valero de Bernabe B, Bittner R, et al. 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD) 17–19 January 2003, Naarden, The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE). Neuromuscul Disord. 2003;13:579-588.
1 Kissel JT, Mendell JR. Muscular dystrophy: historical overview and classification in the genetic era. Semin Neurol. 1999;19:5-7.
2 Hoffman EP, Brown RHJr, Kunkel LM. Dystrophin:the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919-928.
3 Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509-517.
4 Mendell JR, Sahenk Z, Prior TW. The childhood muscular dystrophies: diseases sharing a common pathogenesis of membrane instability. J Child Neurol. 1995;10:150-159.
5 van den Bergh PY, Tome FM, Fardeau M. Etiology and pathogenesis of the muscular dystrophies. Acta Neurol Belg. 1995;95:123-141.
6 Duchenne G. De l’Electrisation Localisée et de Son Application à la Pathologie et à la Therapeutique. Paris: Bailliere et Fils, 1861.
7 Duchenne G. Recherches surla paralysis musculaire pseudo-hypertrophique on paralysis myosclerosique. Ach Gen Med. 1868;11:5-25.
8 Monaco AP, Neve RL, Colletti-Feener C, et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646-650.
9 Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90-95.
10 Batten F. Three cases of myopathy, infantile type. Brain. 1903;26:147-148.
11 Bell J. On pseudohypertrophic and allied types of progressive muscular dystrophy. In: Fischer RA, editor. The Treasury of Human Inheritance. London: Cambridge University Press; 1943:283-342.
12 Bushby KM. Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC Consortium on Limb-Girdle Dystrophies. Neuromuscul Disord. 1995;5:71-74.
13 Brockington M, Blake DJ, Brown SC, et al. The gene for a novel glycosyltransferase is mutated in congenital muscular dystrophy MDC1C and limb girdle muscular dystrophy 2I. Neuromuscul Disord. 2002;12:233-234.
14 Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am J Hum Genet. 2001;69:1198-1209.
15 Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851-2859.
16 Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323-327.
17 Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285-288.
18 Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715-1724.
19 Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453-1459.
20 de Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726-736.
21 Bonne G, Levy N. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003;362:1585-1586. author reply, Lancet 2003; 362:1586.
22 Bonne G, Yaou RB, Beroud C, et al. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13–15 September 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:508-515.
23 Kirschner J, Brune T, Wehnert M, et al. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. 2005;57:148-151.
24 Landouzy L, Dejerine J. De la myopathie atrophique progressive. Rev Med. 1885;81–117:253-366.
25 Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26-30.
26 Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164-167.
27 Brais B, Xie YG, Sanson M, et al. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13. Hum Mol Genet. 1995;4:429-434.
28 Penisson-Besnier I. Distal myopathies. Rev Neurol (Paris). 2004;160:211-216.
29 Ueyama H, Kumamoto T, Horinouchi H, et al. Clinical heterogeneity in dysferlinopathy. Intern Med. 2002;41:532-536.
30 Hackman JP, Vihola AK, Udd AB. The role of titin in muscular disorders. Ann Med. 2003;35:434-441.
31 Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492-500.
32 Argov Z, Tiram E, Eisenberg I, et al. Various types of hereditary inclusion body myopathies map to chromosome 9p1-q1. Ann Neurol. 1997;41:548-551.
33 Clark JR, D’Agostino AN, Wilson J, et al. Autosomal dominant myofibrillar inclusion body myopathy: clinical, histologic, histochemical, and ultrastructural characteristics. Neurology. 1978;28:399.
34 Abe K, Kobayashi K, Chida K, et al. Dominantly inherited cytoplasmic body myopathy in a Japanese kindred. Tohoku J Exp Med. 1993;170:261-272.
35 Kovach MJ, Waggoner B, Leal SM, et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. 2001;74:458-475.
36 Martinsson T, Darin N, Kyllerman M, et al. Dominant hereditary inclusion-body myopathy gene (IBM3) maps to chromosome region 17p13.1. Am J Hum Genet. 1999;64:1420-1426.
37 Martinsson T, Oldfors A, Darin N, et al. Autosomal dominant myopathy: missense mutation (Glu-706→Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S A. 2000;97:14614-14619.
38 Nishino I, Noguchi S, Murayama K, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689-1693.
39 Hoffman EP, Arahata K, Minetti C, et al. Dystrophinopathy in isolated cases of myopathy in females. Neurology. 1992;42:967-975.
40 Wagner KR. Genetic diseases of muscle. Neurol Clin. 2002;20:645-678.
41 Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731-740.
42 Mendell JR, Buzin CH, Feng J, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology. 2001;57:645-650.
43 Flanigan KM, von Niederhausern A, Dunn DM, et al. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72:931-939.
44 Buzin CH, Feng J, Yan J, et al. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum Mutat. 2005;25:177-188.
45 Bushby KM, Beckmann JS. The 105th ENMC sponsored workshop: pathogenesis in the non-sarcoglycan limb-girdle muscular dystrophies, Naarden, April 12–14, 2002. Neuromuscul Disord. 2003;13:80-90.
46 Zatz M, de Paula F, Starling A, et al. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord. 2003;13:532-544.
47 Zatz M, Vainzof M, Passos-Bueno MR. Limb-girdle muscular dystrophy: one gene with different phenotypes, one phenotype with different genes. Curr Opin Neurol. 2000;13:511-517.
48 Beckmann JS, Brown RH, Muntoni F, et al. 66th/67th ENMC sponsored international workshop: the limb-girdle muscular dystrophies, 26–28 March 1999, Naarden, The Netherlands. Neuromuscul Disord. 1999;9:436-445.
49 Wicklund MP, Hilton-Jones D. The limb-girdle muscular dystrophies: genetic and phenotypic definition of a disputed entity. Neurology. 2003;60:1230-1231.
50 Balci B, Uyanik G, Dincer P, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord. 2005;15:271-275.
51 Kirschner J, Bonnemann CG. The congenital and limb-girdle muscular dystrophies: sharpening the focus, blurring the boundaries. Arch Neurol. 2004;61:189-199.
52 Muntoni F, Valero de Bernabe B, Bittner R, et al. 114th ENMC International Workshop on Congenital Muscular Dystrophy (CMD) 17–19 January 2003, Naarden, The Netherlands: (8th Workshop of the International Consortium on CMD; 3rd Workshop of the MYO-CLUSTER project GENRE). Neuromuscul Disord. 2003;13:579-588.
53 Muntoni F, Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635-649.
54 Di Barletta MR, Ricci E, Galluzzi G, et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2000;66:1407-1412.
55 Walter MC, Witt TN, Weigel BS, et al. Deletion of the LMNA initiator codon leading to a neurogenic variant of autosomal dominant Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2005;15:40-44.
56 Ostlund C, Worman HJ. Nuclear envelope proteins and neuromuscular diseases. Muscle Nerve. 2003;27:393-406.
57 Tawil R. Facioscapulohumeral muscular dystrophy. Curr Neurol Neurosci Rep. 2004;4:51-54.
58 Padberg GW, Brouwer OF, de Keizer RJ, et al. On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:S73-S80.
59 Padberg GW, Frants RR, Brouwer OF, et al. Facioscapulohumeral muscular dystrophy in the Dutch population. Muscle Nerve. 1995;2:S81-S84.
60 Tupler R, Gabellini D. Molecular basis of facioscapulohumeral muscular dystrophy. Cell Mol Life Sci. 2004;61:557-566.
61 Rijkers T, Deidda G, van Koningsbruggen S, et al. FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J Med Genet. 2004;41:826-836.
62 Taylor EW. Progressive vagus-glossopharyngeal paralysis with ptosis: a contribution to the group of family diseases. J Nerv Ment Dis. 1915;42:129-139.
63 Amyot R. Hereditary, familial and acquired ptosis of late onset. CMAJ. 1948;59:434-438.
64 Amyot R. Ptosis héréditaire familiale et tardif des paupières supérieures, in Pharyngoplégie également héréditaire et familiales concomittante. Union Med Can. 1948;77:1287.
65 Saucier J. The clinical significance of ptosis with special reference to ptosis of late onset. J Nerv Ment Dis. 1954;119:148-158.
66 Victor M, Hayes R, Adams RD. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the evelids. N Engl J Med. 1962;267:1267-1272.
67 Brais B. Oculopharyngeal muscular dystrophy: a late-onset polyalanine disease. Cytogenet Genome Res. 2003;100:252-260.
68 Brais B, Rouleau GA, Bouchard JP, et al. Oculopharyngeal muscular dystrophy. Semin Neurol. 1999;19:59-66.
69 Bouchard JP, Brais B, Brunet D, et al. Recent studies on oculopharyngeal muscular dystrophy in Quebec. Neuromuscul Disord. 1997;7(Suppl 1):S22-S29.
70 Becher MW, Morrison L, Davis LE, et al. Oculopharyngeal muscular dystrophy in Hispanic New Mexicans. JAMA. 2001;286:2437-2440.
71 Udd B, Bushby K, Nonaka I, et al. 104th European Neuromuscular Centre (ENMC) International Workshop: distal myopathies, 8–10th March 2002 in Naarden, The Netherlands. Neuromuscul Disord. 2002;12:897-904.
72 Askanas V, Engel WK. Unfolding story of inclusion-body myositis and myopathies: role of misfolded proteins, amyloidbeta, cholesterol, and aging. J Child Neurol. 2003;18:185-190.
73 Brooke MH, Fenichel GM, Griggs RC, et al. Clinical investigation of Duchenne muscular dystrophy. Interesting results in a trial of prednisone. Arch Neurol. 1987;44:812-817.
74 DeSilva S, Drachman DB, Mellits D, et al. Prednisone treatment in Duchenne muscular dystrophy. Long-term benefit. Arch Neurol. 1987;44:818-822.
75 Griggs RC, Moxley RT3rd, Mendell JR, et al. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch Neurol. 1991;48:383-388.
76 Mendell JR, Moxley RT, Griggs RC, et al. Randomized, doubleblind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592-1597.
77 Fenichel GM, Florence JM, Pestronk A, et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology. 1991;41:1874-1877.
78 Fenichel GM, Mendell JR, Moxley RT3rd, et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol. 1991;48:575-579.
79 Sansome A, Royston P, Dubowitz V. Steroids in Duchenne muscular dystrophy; pilot study of a new low-dosage schedule. Neuromuscul Disord. 1993;3:567-569.
80 Bonifati MD, Ruzza G, Bonometto P, et al. A multicenter, doubleblind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23:1344-1347.
81 Campbell C, Jacob P. Deflazacort for the treatment of Duchenne dystrophy: a systematic review. BMC Neurol. 2003;3:7.
82 Johnsen SD. Prednisone therapy in Becker’s muscular dystrophy. J Child Neurol. 2001;16:870-871.
83 Angelini C, Fanin M, Menegazzo E, et al. Homozygous α-sarcoglycan mutation in two siblings: one asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve. 1998;21:769-775.
84 Connolly AM, Pestronk A, Mehta S, et al. Primary α-sarcoglycan deficiency responsive to immunosuppression over three years. Muscle Nerve. 1998;21:1549-1553.
85 Bushby K, Muntoni F, Urtizberea A, et al. Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids. 2–4 April 2004, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:526-534.
86 Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375-381.
87 Dunant P, Walter MC, Karpati G, et al. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve. 2003;27:624-627.
88 Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706-711.
89 Walter MC, Lochmüller H, Reilich P, et al. Creatine monohydrate in muscular dystrophies: A doubleblind, placebocontrolled clinical study. Neurology. 2000;54:1848-1850.
90 Rose MR, Tawil R. Drug treatment for facioscapulohumeral muscular dystrophy. Cochrane Database Syst Rev. (2):2004. CD002276
91 Kissel JT, McDermott MP, Mendell JR, et al. Randomized, doubleblind, placebocontrolled trial of albuterol in facioscapulohumeral dystrophy. Neurology. 2001;57:1434-1440.
92 Bogdanovich S, Perkins KJ, Krag TO, et al. Therapeutics for Duchenne muscular dystrophy: current approaches and future directions. J Mol Med. 2004;82:102-115.
93 Romero NB, Braun S, Benveniste O, et al. Phase I study of dystrophin plasmid-based gene therapy in Duchenne/Becker muscular dystrophy. Hum Gene Ther. 2004;15:1065-1076.
94 Aartsma-Rus A, Janson AA, Kaman WE, et al. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum Mol Genet. 2003;12:907-914.
95 Lu QL, Rabinowitz A, Chen YC, et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in bodywide skeletal muscles. Proc Natl Acad Sci U S A. 2005;102:198-203.
96 Goyenvalle A, Vulin A, Fougerousse F, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796-1799.
97 Mummery CJ, Copeland SA, Rose MR. Scapular fixation in muscular dystrophy. Cochrane Database Syst Rev. (3):2003. CD003278
98 Wallgren-Pettersson C, Bushby K, Mellies U, et al. 117th ENMC workshop: ventilatory support in congenital neuromuscular disorders—congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4–6 April 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:56-69.
99 Bushby K, Muntoni F, Bourke JP. 107th ENMC International Workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:166-172.
100 van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001;10:1547-1554.
101 Walter MC, Lochmüller H. Novel approaches to treat muscular dystrophies. Expert Opin Investig Drugs. 2001;10:695-707.
102 Becker PE. Two new families of benign sex-linked recessive muscular dystrophy. Rev Can Biol. 1962;21:551-566.
103 Becker PE. Eine neue X-chromosomale Muskeldystrophie. Acta Psychiat Neurol Scand. 1955;193:427.
104 Fadda S, Mochi M, Roncuzzi L, et al. Definitive localization of Becker muscular dystrophy in Xp by linkage to a cluster of DNA polymorphisms (DXS43 and DXS9). Hum Genet. 1985;71:33-36.
105 Mathews KD, Moore SA. Limb-girdle muscular dystrophy. Curr Neurol Neurosci Rep. 2003;3:78-85.
106 Urtasun M, Saenz A, Roudaut C, et al. Limb-girdle muscular dystrophy in Guipuzcoa (Basque Country, Spain). Brain. 1998;121(Pt 9):1735-1747.
107 Hauser MA, Conde CB, Kowaljow V, et al. myotilin Mutation found in second pedigree with LGMD1A. Am J Hum Genet. 2002;71:1428-1432.
108 Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004;62:1363-1371.
109 Speer MC, Yamaoka LH, Gilchrist JH, et al. Confirmation of genetic heterogeneity in limb-girdle muscular dystrophy: linkage of an autosomal dominant form to chromosome 5q. Am J Hum Genet. 1992;50:1211-1217.
110 van der Kooi AJ, Ledderhof TM, de Voogt WG, et al. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol. 1996;39:636-642.
111 van der Kooi AJ, van Meegen M, Ledderhof TM, et al. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. Am J Hum Genet. 1997;60:891-895.
112 Betz RC, Schoser BG, Kasper D, et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. 2001;28:218-219.
113 de Paula F, Vainzof M, Bernardino AL, et al. Mutations in the caveolin-3 gene: when are they pathogenic? Am J Med Genet. 2001;99:303-307.
114 Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18:365-368.
115 Speer MC, Vance JM, Grubber JM, et al. Identification of a new autosomal dominant limb-girdle muscular dystrophy locus on chromosome 7. Am J Hum Genet. 1999;64:556-562.
116 Messina DN, Speer MC, Pericak-Vance MA, et al. Linkage of familial dilated cardiomyopathy with conduction defect and muscular dystrophy to chromosome 6q23. Am J Hum Genet. 1997;61:909-917.
117 Gamez J, Navarro C, Andreu AL, et al. Autosomal dominant limb-girdle muscular dystrophy: a large kindred with evidence for anticipation. Neurology. 2001;56:450-454.
118 Palenzuela L, Andreu AL, Gamez J, et al. A novel autosomal dominant limb-girdle muscular dystrophy (LGMD 1F) maps to 7q32.1–32.2. Neurology. 2003;61:404-406.
119 Starling A, Kok F, Passos-Bueno MR, et al. A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21. Eur J Hum Genet. 2004;12:1033-1040.
120 Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain. 1996;119(Pt 1):295-308.
121 Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27-40.
122 Richard I, Roudaut C, Saenz A, et al. Calpainopathy—a survey of mutations and polymorphisms. Am J Hum Genet. 1999;64:1524-1540.
123 Saenz A, Leturcq F, Cobo AM, et al. LGMD2A: genotypephenotype correlations based on a large mutational survey on the calpain 3 gene. Brain. 2005;128(Pt 4):732-742.
124 Bansal D, Miyake K, Vogel SS, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168-172.
125 Bashir R, Britton S, Strachan T, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37-42.
126 Illa I, Serrano-Munuera C, Gallardo E, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130-134.
127 Miyoshi K, Saijo K, Kuryu T, et al. Four cases of distal myopathy in two families. Jpn J Hum Genet. 1967;12:113.
128 Walter MC, Braun C, Vorgerd M, et al. Variable reduction of caveolin-3 in patients with LGMD2B/MM. J Neurol. 2003;250:1431-1438.
129 Weiler T, Bashir R, Anderson LV, et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum Mol Genet. 1999;8:871-877.
130 Noguchi S, McNally EM, Ben Othmane K, et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819-822.
131 Roberds SL, Leturcq F, Allamand V, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625-633.
132 Bonnemann CG, Modi R, Noguchi S, et al. β-Sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11:266-273.
133 Nigro V, de Sa Moreira E, Piluso G, et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nat Genet. 1996;14:195-198.
134 Moreira ES, Vainzof M, Marie SK, et al. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11–12. Am J Hum Genet. 1997;61:151-159.
135 Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24:163-166.
136 Frosk P, Weiler T, Nylen E, et al. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet. 2002;70:663-672.
137 Weiler T, Greenberg CR, Zelinski T, et al. A gene for autosomal recessive limb-girdle muscular dystrophy in Manitoba Hutterites maps to chromosome region 9q31-q33: evidence for another limb-girdle muscular dystrophy locus. Am J Hum Genet. 1998;63:140-147.
138 de Paula F, Vieira N, Starling A, et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur J Hum Genet. 2003;11:923-930.
139 Driss A, Amouri R, Ben Hamida C, et al. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscul Disord. 2000;10:240-246.
140 Driss A, Noguchi S, Amouri R, et al. Fukutin-related protein gene mutated in the original kindred limb-girdle MD 2I. Neurology. 2003;60:1341-1344.
141 Esapa CT, McIlhinney RA, Blake DJ. Fukutin-related protein mutations that cause congenital muscular dystrophy result in ER-retention of the mutant protein in cultured cells. Hum Mol Genet. 2005;14:295-305.
142 Frosk P, Greenberg CR, Tennese AA, et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat. 2005;25:38-44.
143 Karpati G, Holland P. Sweetening the pot in muscle: genetic defects of protein glycosylation causing muscle disease. Neurology. 2002;59:1674-1676.
144 Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537-542.
145 Poppe M, Bourke J, Eagle M, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004;56:738-741.
146 Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. 2003;60:1246-1251.
147 Walter MC, Petersen JA, Stucka R, et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet. 2004;41:e50.
148 Udd B. Limb-girdle type muscular dystrophy in a large family with distal myopathy: homozygous manifestation of a dominant gene? J Med Genet. 1992;29:383-389.
149 Udd B, Vihola A, Sarparanta J, et al. Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD2J. Neurology. 2005;64:636-642.
150 Dincer P, Balci B, Yuva Y, et al. A novel form of recessive limb girdle muscular dystrophy with mental retardation and abnormal expression of α-dystroglycan. Neuromuscul Disord. 2003;13:771-778.
151 Koss-Harnes D, Hoyheim B, Jonkman MF, et al. Life-long course and molecular characterization of the original Dutch family with epidermolysis bullosa simplex with muscular dystrophy due to a homozygous novel plectin point mutation. Acta Derm Venereol. 2004;84:124-131.
152 Shimizu H, Takizawa Y, Pulkkinen L, et al. Epidermolysis bullosa simplex associated with muscular dystrophy: phenotype-genotype correlations and review of the literature. J Am Acad Dermatol. 1999;41:950-956.
153 Mostacciuolo ML, Miorin M, Martinello F, et al. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet. 1996;97:277-279.
154 Fukuyama Y, Osawa M, Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type—clinical, genetic and pathological considerations. Brain Dev. 1981;3:1-29.
155 Silan F, Yoshioka M, Kobayashi K, et al. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol. 2003;53:392-396.
156 Santavuori P, Somer H, Sainio K, et al. Muscle-eye-brain disease (MEB). Brain Dev. 1989;11:147-153.
157 Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717-724.
158 Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033-1043.
159 Dobyns WB, Pagon RA, Armstrong D, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32:195-210.
160 Helbling-Leclerc A, Zhang X, Topaloglu H, et al. Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11:216-218.
161 Jones KJ, Morgan G, Johnston H, et al. The expanding phenotype of laminin α2 chain (merosin) abnormalities: case series and review. J Med Genet. 2001;38:649-657.
162 Brockington M, Sewry CA, Herrmann R, et al. Assignment of a form of congenital muscular dystrophy with secondary merosin deficiency to chromosome 1q42. Am J Hum Genet. 2000;66:428-435.
163 Muntoni F, Taylor J, Sewry CA, et al. An early onset muscular dystrophy with diaphragmatic involvement, early respiratory failure and secondary α2 laminin deficiency unlinked to the LAMA2 locus on 6q22. Eur J Paediatr Neurol. 1998;2:19-26.
164 Topaloglu H, Brockington M, Yuva Y, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology. 2003;60:988-992.
165 Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of α-dystroglycan. Hum Mol Genet. 2003;12:2853-2861.
166 Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desminrelated myopathy with Mallory body–like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. 2004;55:676-686.
167 Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739-749.
168 Moghadaszadeh B, Desguerre I, Topaloglu H, et al. Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigidity of the spine, on chromosome 1p35–36. Am J Hum Genet. 1998;62:1439-1445.
169 Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin α7 gene cause congenital myopathy. Nat Genet. 1998;19:94-97.
170 Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14:279-293.
171 Camacho Vanegas O, Bertini E, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A. 2001;98:7516-7521.
172 Demir E, Ferreiro A, Sabatelli P, et al. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics. 2004;35:103-112.
173 Demir E, Sabatelli P, Allamand V, et al. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet. 2002;70:1446-1458.
174 Higuchi I, Shiraishi T, Hashiguchi T, et al. Frameshift mutation in the collagen VI gene causes Ullrich’s disease. Ann Neurol. 2001;50:261-265.
175 Mercuri E, Yuva Y, Brown SC, et al. Collagen VI involvement in Ullrich syndrome: a clinical, genetic, and immunohisto-chemical study. Neurology. 2002;58:1354-1359.
176 Pan TC, Zhang RZ, Sudano DG, et al. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355-369.
177 Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100th ENMC International Workshop, 23–24 November 2001, Naarden, The Netherlands. Neuromuscul Disord. 2002;12:984-993.
178 Pepe G, de Visser M, Bertini E, et al. Bethlem myopathy (BETHLEM) 86th ENMC International Workshop, 10–11 November 2000, Naarden, The Netherlands. Neuromuscul Disord. 2002;12:296-305.
179 Ullrich O. Kongenitale atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegenerativen Erkrankungen des neuromuskulären Systems. Z Ges Neurol Psychiatr. 1930;126:171-201.
180 Bohlega S, Abu-Amero SN, Wakil SM, et al. Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology. 2004;62:1518-1521.
181 Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am J Hum Genet. 2004;75:703-708.
182 Tajsharghi H, Thornell LE, Lindberg C, et al. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003;54:494-500.
183 Feit H, Silbergleit A, Schneider LB, et al. Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am J Hum Genet. 1998;63:1732-1742.
184 Young ID, Harper PS. Hereditary distal spinal muscular atrophy with vocal cord paralysis. J Neurol Neurosurg Psychiatry. 1980;43:413-418.
185 Mahjneh I, Haravuori H, Paetau A, et al. A distinct phenotype of distal myopathy in a large Finnish family. Neurology. 2003;61:87-92.
186 Markesbery WR, Griggs RC, Leach RP, et al. Late onset hereditary distal myopathy. Neurology. 1974;24:127-134.
187 Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol. 1993;50:604-608.
188 Ahlberg G, von Tell D, Borg K, et al. Genetic linkage of Welander distal myopathy to chromosome 2p13. Ann Neurol. 1999;46:399-404.
189 Welander L. Homozygous appearance of distal myopathy. Acta Genet Stat Med. 1957;7:321-325.
190 Welander L. Myopathia distalis tarda hereditaria. Acta Med Scand. 1951;141(Suppl 265):1-124.
191 Di Blasi C, Moghadaszadeh B, Ciano C, et al. Abnormal lysosomal and ubiquitin-proteasome pathways in 19p13.3 distal myopathy. Ann Neurol. 2004;56:133-138.
192 Sangiuolo F, Bruscia E, Capon F, et al. Fine mapping of a distinctive autosomal dominant vacuolar neuromyopathy using 11 novel microsatellite markers from chromosome band 19p13.3. Eur J Hum Genet. 2000;8:809-812.
193 Servidei S, Capon F, Spinazzola A, et al. A distinctive autosomal dominant vacuolar neuromyopathy linked to 19p13. Neurology. 1999;53:830-837.
194 Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31-36.
195 Nonaka I, Sunohara N, Ishiura S, et al. Familial distal myopathy with rimmed vacuoles and lamellar (myeloid) body formation. J Neurol Sci. 1981;51:141-155.
196 Argov Z, Eisenberg I, Grabov-Nardini G, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. 2003;60:1519-1523.
197 Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83-87.
198 Eisenberg I, Grabov-Nardini G, Hochner H, et al. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat. 2003;21:99.
199 Eisenberg I, Hochner H, Shemesh M, et al. Physical and transcriptional map of the hereditary inclusion body myopathy locus on chromosome 9p12-p13. Eur J Hum Genet. 2001;9:501-509.
200 Eisenberg I, Thiel C, Levi T, et al. Fine-structure mapping of the hereditary inclusion body myopathy locus. Genomics. 1999;55:43-48.
201 Krause S, Hinderlich S, Amsili S, et al. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304:365-379.
202 Chinnery PF, Johnson MA, Walls TJ, et al. A novel autosomal dominant distal myopathy with early respiratory failure: clinico-pathologic characteristics and exclusion of linkage to candidate genetic loci. Ann Neurol. 2001;49:443-452.
203 Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377-381.






