MicroRNA expression in cancer
Serge Patrick Nana-Sinkam, MD  Mario Acunzo, PhD
Mario Acunzo, PhD  Carlo M. Croce, MD
Carlo M. Croce, MD
Overview
In the last two decades, researchers have identified a novel group of noncoding RNAs (ncRNAs) classified according to their function and size. The best studied of these ncRNAs termed microRNAs (miRNAs) are short noncoding RNAs that are approximately 22 nucleotides in length and play a key role in the regulation of a large number of biological processes and diseases, including cancer. Since the initial description of an association between miRNA and cancer in 2002, miRNAs have emerged as central regulators of processes fundamental to the initiation and progression of cancer. More recently, miRNAs have been detected in bodily fluids including blood, sputum, and urine, thus making them potential noninvasive diagnostic and prognostic biomarkers of disease. The application of miRNAs in human cancer therapy has become a fascinating field of study, representing one of the newest frontiers for cancer treatment. However, our knowledge for these small molecules and their application to human cancers is still growing. In this chapter, we provide an overview of the connection between miRNAs and cancer with a focus on translation to human application.
Background
As investigators increasingly recognize the inherent complexities of cancer, they continue to search for novel molecular pathways in cancer that may be leveraged for the development of novel biomarkers and therapeutics, with the ultimate goal of saving lives. For decades, investigators have held the belief that a large percentage of the human genome was composed of “junk-DNA” or “dark matter” due to its inability to code for proteins. In the last two decades, researchers have now identified functions for regions of the human genome previously considered to be nonfunctional. Some genes located within these regions indeed encode for noncoding RNAs (ncRNAs), with microRNAs (miRNAs) representing the most researched member of this group. Since their initial discovery two decades ago in Caenorhabditis elegans, miRNAs have emerged as key regulators of biological processes fundamental to the initiation and progression of cancers.1, 2 Approximately, 22 nucleotides (nt) in length, miRNAs tend to be highly conserved across species and often demonstrate global deregulation in solid and hematological malignancies.3, 4 By directly binding to either the 3′ or 5′ untranslated region (UTR) of target mRNAs, miRNAs can either degrade target mRNA or inhibit translation. In addition, based on their relatively short size, miRNAs have the capacity for the simultaneous regulation of tens to hundreds of genes, thus interdicting in numerous biological pathways. In fact, the estimate that miRNAs may regulate up to 60% of the human genome is probably wrong and miRNA may regulate over the 90% of the protein-coding genes4, 5). miRNAs are often located in fragile regions of the chromosome and thus susceptible to regulation through chromosomal amplifications, deletions, or rearrangements.6 The mechanisms for the regulation of miRNAs in the setting of cancer are complex and remain only partially understood. However, increasing lines of investigation indicate that miRNA regulation may occur by several mechanisms including alterations in key components of processing, epigenetic silencing, and polymorphisms in either miRNAs or target mRNAs interfering with binding and regulation.7 The mechanisms for miRNA regulation and function are further complicated by their tumor and cell specificity. We are now approaching the identification of nearly 3000 miRNAs. miRNAs may function either as tumor suppressors or oncogenes depending on tumor and cell type and regulate processes fundamental to tumorigenesis (hallmarks of cancer) including differentiation, proliferation, and angiogenesis.8 The mechanisms that regulate miRNAs in cancer, as well as their roles in cancer initiation and progression, are only beginning to be uncovered. While the use of high-throughput profiling strategies for the identification of clinically relevant miRNA-based biomarkers has been useful, this approach to miRNA investigation remains limited by issues of reproducibility and the need for improved algorithms for miRNA-target prediction and validation. Thus, there is still considerable work required to translate miRNAs into markers for clinical decision-making. One must also consider the inherent difficulties in achieving tumor-specific miRNA delivery. As miRNA biology transitions to the clinic, there is encouraging evidence suggesting that human applications for miRNAs, particularly as therapeutics, are in the not-too-distant future. For instance, nanotechnology-based carriers for miRNA delivery represent a new promising tool for the effective shuttling of miRNAs in the human body. Santaris/miRNA Therapeutics Inc. has tested human delivery of an antagomir against miR-122 for the treatment of hepatitis C. More recently, investigators have initiated trials testing human delivery of miR-34 in hepatocellular carcinoma as well as a phase I trial testing miR-16 replacement in recurrent malignant mesothelioma. Such studies represent the first of hopefully a series of future applications for the treatment of human cancer.
Biogenesis and production of microRNAs
miRNAs are short ncRNAs with a length of approximately 22 nucleotides encoded by evolutionally conserved genes. miRNAs are more frequently located within the introns or exons of protein-coding genes (about the 70%), or in intergenic regions (30%). The expression of intergenic miRNAs is related to their host gene expression, all intragenic miRNAs having independent transcription units.9 miRNAs are processed and generated through a well-orchestrated series of interrelated steps, each of which is currently being investigated (Figure 1). In the first step, a long primary transcript termed the (pri)-miRNA undergoes transcription by RNA polymerase II. The pri-miRNAs are then bound to the double-stranded RNA-binding domain (dsRBD) protein known as DiGeorge syndrome critical region gene 8 (DGCR8) for vertebrates.10 (An RNase III endonuclease termed The Drosha/DGCR8 complex cleaves the pri-miRNA into a smaller stem-loop ∼70-nucleotide (nt) precursor miRNA (pre-miRNA). Pre-miRNAs are then exported from the nucleus to the cytoplasm using the double-stranded RNA-binding protein, Exportin 5 (XPO5).11 Once in the cytoplasm, the pre-miRNA is cleaved into a mature 18–25 nucleotide miRNA by a complex that includes RNAase Dicer, Argonaute 2 (Ago 2), and transactivation-responsive RNA-binding protein (TRBP). The Ago2 protein belongs to the Argonaute family of proteins that bind fragments to guide RNA (including miRNAs). The resultant miRNA sequence, which consists of two strands, is then loaded into the RNA-induced silencing complex (RISC), with the mature strand being maintained while the complementary strand is degraded. The remaining strand possesses complementarity either to the 3′ or 5′ UTR of a target gene leading to degradation through endonuclease activity or to translational inhibition. Recently, investigators have shown that an miRNA can also bind to the coding sequence of a transcript, leading to translational repression.12 The degree of complementarity between the “seed” sequence of the mature miRNA and the target is the primary determinant of the biological effect. It is important to recognize that investigators have now identified additional mechanisms for miRNA biogenesis, suggesting that we are just beginning to understand the complexities of this process.
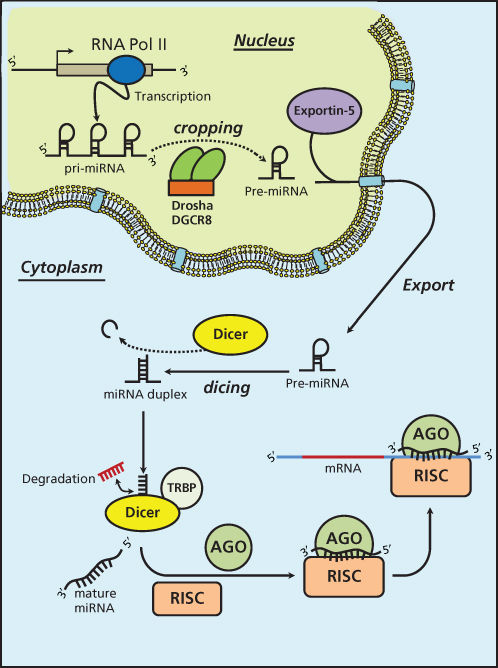
Figure 1 MicroRNA biogenesis.
MicroRNA deregulation in cancer
Calin et al.13 made the first observations linking miRNA deregulation to cancer in a study. While investigating the mechanisms for chronic lymphocytic leukemia (CLL), this team of investigators made the startling observation that a pair of miRNAs, miR-15a/16-1 which, along with the deleted in leukemia (DLEU) gene, are located at the chromosomal region 13q14.3, was either deleted or downregulated in 68% of patients with CLL.13 In addition, both of these miRNAs were highly expressed in normal CD5+ B cells. Both findings suggested that these miRNAs were in fact important to the pathogenesis of the disease. This initial observation was corroborated by independent investigators who identified a functional link between miR-15a/16-1 and the prosurvival molecule Bcl2. They validated in vitro that miR-15a/16-1 targeted Bcl2 to induce apoptosis. Since this initial discovery of miRNAs in CLL, investigators have observed patterns of global dysregulation of miRNAs across both solid and hematological malignancies. In the last few years, researchers have determined that the causes for such dysregulation are multifactorial. For instance, the altered expression and/or function of the proteins involved in the biogenesis of miRNAs, such as Drosha and Dicer, can lead to an aberrant expression of miRNAs and thus to cancer.14 A decreased expression level of Drosha and Dicer has been found in a high percentage of ovarian cancer patients.15 Moreover, epigenetic changes within miRNA promoters such as changes in their methylation can also induce changes in miRNA expression levels.16 Like other deregulated genes that cover an important role as oncogenes or tumor suppressors, the epigenetic deregulated expression of a single miRNA can be the triggering event for carcinogenesis. One such example involves the intensely studied miR-155 whose dysregulation can induce leukemia in miR-155 transgenic mice.17 The tumor-suppressor miR-127 in primary prostate cancer and bladder tumors causes the upregulation of the proto-oncogene BCL6, which is a direct target of miR-127.18 On the other hand, miRNAs can also act as protagonists in the control of the global cellular methylation status by acting on the enzymes responsible for epigenetic control. For example, the miR-29 family is able to modulate methylation levels by affecting the ex-novo expression of DNA methyltransferases DNMT3a and DNMT3B in lung cancer.16 Another important and complex regulative mechanism of miRNAs is related to the transcriptional control of gene expression.19 The activation of the miR-17/92 cluster induced by the MYC oncogene modulates the anti-apoptotic action of E2F1, thus mediating the MYC proliferative effect.20 Recently, the effect of the membrane tyrosine-kinase receptors on miRNA expression has been studied. For instance, the hepatocyte growth factor receptor c-MET is able, through the transcriptional factor AP1, to induce the expression of the onco-miRNA miR-221/222 cluster, suggesting that the important effect of deregulated c-MET in cancer is at least in part linked to a deregulated miRNA expression pathway.21 Finally, considering that the loss of p53 is one of the most represented genetic abnormalities in cancer, the link between the miR-34a family and p53 is another important example of miRNA transcriptional regulation.22 p53 stimulates the transcription of the miR-34 family, inducing apoptosis and senescence. The loss of p53 function induces the downregulation of the miR-34 family in a very high percentage of ovarian cancer patients with a p53 mutation.23 Hence, the primary theme is that patterns of miRNA expression are globally deregulated in cancer, this event being potentially a cause as well as a consequence of cancer itself. The global deregulation of miRNA in cancer has a dramatic effect on downstream targets of several cellular pathways.
Recently, investigators have also determined that miRNA function may be altered through mutations in target gene seed sequences. Such mutations can render an miRNA incapable of regulating a given mRNA and have been identified as biomarkers of clinical outcome. Mutations in the 3′ UTR have been identified in several solid malignancies including ovarian, lung, breast, and colon cancers. Conversely, SNPs in miRNA gene sequences can change miRNA functions. We know that mRNA functional regulation by miRNAs is highly sensitive to base pair mismatches within nucleotides 2–8 of the miRNA, which have been defined as the seed region.24 Therefore a single point mutation on an miRNA gene or a post-transcriptional modification such as RNA editing, can change the function or modify the targetome of an miRNA.25, 26 For example, it was determined that a single nucleotide polymorphism (SNP) in a let-7 (lethal-7) miRNA complementary site in the KRAS 3′ UTR increases nonsmall-cell lung cancer risk.27 A mutation of let-7 binding site in the Kras 3′ UTR has been detected also in juvenile myelomonocytic leukemia (JMML).28
MicroRNA as biomarkers in cancer
Over the last several years, miRNAs have been implicated in virtually every type of cancer. Early studies have focused on applying high-throughput platforms as a means for linking patterns of miRNA deregulation to clinical parameters. One of the first such approaches was made in 2005 by Volinia and colleagues who profiled the miRNA signatures of six human solid tumors, detecting miR-21, miR-17-5p, and miR-191 overexpressed in some tumors.29 Since that initial study, investigators have conducted similar multiple studies with the primary goals of identifying a prognostic miRNA signature. Yanaihara and colleagues conducted high-throughput profiling of cases of stage 1 adenocarcinoma of the lung. They identified over 40 miRNAs that distinguished lung tumors from adjacent uninvolved lung.30 A broader study conducted on 22 different tumor types showed how an miRNA expression profile is able to classify tumors according to tissue of origin with high accuracy.31 Studies have also focused on preselected miRNAs as potential prognostic markers. For example, Nadal et al.32 examined the tumor-suppressive miR-34 as a prognostic biomarker in early-stage adenocarcinoma of the lung. They identified methylation and reduced expression of miR-34b/c in nearly half (46%) of early-stage lung adenocarcinomas and determined that reduced expression and methylation of miR-34b/c correlated with shorter disease-free survival and overall survival. For example, in a separate study, investigators showed that miRNA expression levels may correlate with BCR-ABL kinase activity in chronic myeloid leukemia (CML),33 suggesting, therefore, a potential application in the adjustment of the therapy during treatment to improve the outcome. Another very intriguing application of miRNAs as biomarkers consists of the integration of both protein-coding and noncoding gene expressions in order to develop a prognostic signature in early stages of cancer. Akagi et al.34 examined 148 cases of stage 1 lung adenocarcinoma for 42 preselected genes as predictive biomarkers. Through testing and subsequent validation in independent cohorts, the authors developed a four-gene classifier (DLC1, XPO1, HIF1A, and BRCA1) that correlates to survival in stage 1 lung adenocarcinoma. In addition, they determined that miR-21 expression was independently associated with survival in the same cohorts. Lastly, the combination of the four-gene classifier and miR-21 expression was superior to either biomarker alone. Despite the multitude of very encouraging miRNA profiling studies, investigators have yet to reach consensus on which miRNAs confer the most accurate prognostic information. A primary reason for the lack of reproducibility is that, similarly to other high-throughput analyses, miRNA profiling studies are susceptible to certain biases, including small cohort sizes, varying platforms (array, sequencing, RT-PCR), and variability in data interpretation.
MicroRNAs as noninvasive biomarkers in cancer
The development of noninvasive biomarkers in cancer that may inform clinical decision-making remains elusive and the subject of continued study. Several studies have demonstrated that miRNAs exist in body fluids (serum, plasma, urine, sputum, cerebrospinal fluid, and bronchoalveolar lavage) in a relatively stable form at different conditions of pH and temperature.35–37 miRNAs are also present in blood, where they were detected in plasma, platelets, erythrocytes, and nucleated blood cells. One of the earliest studies revealed that miRNAs were detectable in circulation in prostate cancer.38 Shen et al.39 demonstrated that plasma-based miRNAs could be used as biomarkers to distinguish solitary lung nodules. A subsequent larger study further validated the concept that circulating miRNAs could be used in the setting of lung cancer early detection.40
The compartment specific location of miRNAs in circulation remains the subject of debate. However, circulating miRNAs have been found packaged in extracellular vesicles (EVs) as well as associated with RNA-binding proteins like Argonaute 2 or lipoprotein complexes, which prevent their degradation (Figure 2).41–44 EVs are small membrane-encapsulated fluid particles comprised of a family shedding vesicles and exosomes that are released from a wide variety of cell types by entirely independent cellular mechanisms.45 Investigators to date have focused primarily on exosomes, which are ∼40 to 100 nm vesicles. These particles consist of a lipid bilayer, generated from secretory multivesicular bodies (MVB) that fuse with the plasma membrane for release into the extracellular environment. Exosomes contain various molecular constituents of their cell of origin including lipids, proteins, messenger RNA (mRNA), and miRNA, which may be transferred from donor to target cells to facilitate direct cell-to-cell contact and subsequent reprogramming of the tumor microenvironment.42 In pathological states, such as cancer, exosomes cross-talk, and/or influence major tumor-related pathways such as epithelial-mesenchymal transition (EMT), cancer stemness, angiogenesis, and metastasis involving many cell types within the tumor microenvironment.46–49 Exosomes have also been detected in a number of human body fluids including plasma, urine, breast milk, amniotic fluid, malignant ascites, and bronchoalveolar lavage fluid, suggesting their potential importance as biomarkers of disease.50

Figure 2 Process of extracellular vesicles release from cells. Apoptotic cells release apoptotic bodies.
Selected miRNAs implicated in cancer
Following a multitude of miRNA in cancer studies, several miRNAs, including miR-155, let-7, miR-21, and miR-34, for example, have emerged as fundamental contributors to tumor development. These miRNAs are now being integrated into clinical trials as biomarkers for clinical diagnosis and therapy.
LET-7
The let-7 gene was first discovered in C. elegans as a key regulator of development.51 The mature form of let-7 family members is highly conserved across species. let-7 miRs family plays an essential role in regulating cell proliferation and differentiation during development in different species. In addition, let-7 is a marker of fully differentiated cells while undetectable in stem cells.52 The human let-7 family contains 12 members located on nine different chromosomes, they map to fragile sites associated with different types of solid cancers.53 Indeed, the deregulation of these miRs has been shown as a feature of many types of cancer.54–56 One of the most studied let-7 targets and regulators is the c-Myc oncogene, given, moreover, the well-known double-negative feedback between them.
MiR-21
MiR-21 is one of most studied oncomiRs, as most of its validated targets are tumor suppressors (i.e., PTEN, Bcl2, and Sprouty1 and 2). This miR is probably one of the most dynamic miRNAs, being responsive to various stimuli given its involvement in positive and negative feedback loops. miR-21 is one of the most altered miRNAs in solid tumors including breast, ovaries, cervix, colon, lung, liver, brain, esophagus, prostate, pancreas, and thyroid.29, 57–59 miR-21 is also upregulated in leukemic cancers,60 indicating an important key role for this miRNA in cancer development and progression. Moreover, a recent study established circulating miR-21 as a biomarker of various carcinomas, unveiling its potential as a tool for cancer diagnosis.61
MiR-34
The miR-34 family consists of three highly related miRNAs: miR-34a, miR-34b, and miR-34c. miR-34a is expressed at higher levels than miR-34b/c in all tissues, particularly brain, with the exception being the lung.62 These miRNAs are directly induced by p53 in response to DNA damage or oncogenic stress63 and contribute to the p53 downstream effect by targeting c-Myc, Bcl2, C-Met, and Src.64 MiR-34a plays a fundamental role not only in tumor suppression but also in modulation of drug response in a number of cellular models, including HCC, breast, bladder, HNSCC, and NSCLC (e.g., an overexpression of miR-34a, downregulating PDGFR, restores TRAIL-induced apoptosis in NSCLC cell lines).65
MiR-200 family
The miR-200 family consists of five members: miR-200a, miR-200b, miR-200c, miR-141, and miR-429. These miRs are highly expressed in epithelial tissues and are involved in tumor suppression by inhibiting EMT, migration, invasion, tumor cell adhesion, and metastasis.66 Among miR-200 family targets, ZEB1 and ZEB2, two central mediators of EMT, are two of the most studied. Moreover, there is a double-negative ZEB/miR-200 feedback loop, given the ability of ZEB1 to suppress the expression of miR-200 family members.67 Furthermore, there is evidence that breast cancer metastases may be under the control of the Akt-miR200-E-cadhenin axis. Specifically, the balance of the three Akt isoforms can control the expression of miR-200 and that of the E-cadherin mRNA in primary and metastatic human breast cancers.68
Therapeutic targeting and miRNA
Given that deregulation of a single or group of miRNAs can drive malignancy, it is hypothesized that through directed targeting of their deregulation, at least in vitro, one can attenuate carcinogenesis. Alternatively, patterns of miRNA deregulation may drive chemoresistance to traditional agents. For example, strategies aimed at using global miRNA expression profiling of selectively created drug-resistant cell lines have been utilized to identify which specific miRNAs are responsible for the acquired resistance. These findings have translated to miRNAs serving both as directed targets and predictors of response to chemotherapeutic agents. Importantly, the contribution of specific miRNAs to chemoresistance can be highly cell tumor specific. As mentioned, miRNAs have the capacity to simultaneously target and regulate multiple biological pathways. In the case of CLL, selected patterns of miRNA expression could clinically predict which patients would respond to the selected agent fludarabine.69 In solid tumors such as lung cancer, miRNAs including miR-21, miR-30, miR-221, and miR-222 have been associated with response to chemotherapeutic agents. Recently, Vecchione et al.70 profiled an miRNA signature that is able to define chemoresistance in ovarian cancer. It has been reported that an miRNA profiling is useful to identify a subtype of temozolomide-resistant glioblastoma.71
In the last few years, the majority of applications of miRNAs as directed therapies in human disease have taken place in in vitro and murine models of disease settings. The primary goal of directed therapeutics is to manipulate miRNAs that are known to be deregulated in tumors and thus alter their downstream targets and biological pathways. This approach may occur through the selected targeting of an miRNA or as a strategy for augmenting the effects of an established therapeutic agent. The manipulation of miRNAs via either selective gain of function (e.g., mimics) for the purposes of repletion or silencing (e.g., antagonists) has been applied with variable results. miRNAs may be delivered by several modalities including viral vectors and nanoparticles (NPs). Viral-based carriers have been effectively used to deliver miRNAs in solid tumors including let-7 in lung cancer.72 However, viral carriers for small molecule delivery are not without limitations including the potential immunogenic and toxic effects of the carrier to the host. More recently, investigators have employed lipid-based NPs as carriers for small molecules including miRNA. NPs represent smaller engineered particles that are particularly suitable for drug delivery based on their modifiable composition allowing for optimal binding and absorption. Several recent studies have demonstrated the efficacy of NPs as vehicles for miRNA delivery in vivo. For example, Wu et al. recently showed that lipid-based NPs could be utilized to effectively deliver miR-29 in lung cancer both in vitro and in vivo.73 In an independent study, Trang et al. showed that delivery of let-7 could reduce tumor growth and many oncogenes.74 Issues of off-target effects, stability of carriers, and toxicity all remain germane to miRNA-based therapeutics. The use of miRNA sponges represents an alternative strategy to antagomirs. RNA sponges are able to simultaneously repress a large number of miRNA molecules. The existence of circular RNA (circRNA) has been already established in nature and can represent an example of miRNA sponge. circRNA is a type of RNA, which forms a covalently closed continuous loop, forming a circRNA whereby the 3′ and 5′ ends are attached together forming a round RNA molecule. This structural characteristic confers stability to circRNAs in the cytosol, while also ensuring the ability to bind simultaneously a variable number of miRNA molecules thus inhibiting their action. circRNAs are derived from protein-coding genes but do not encode for any protein and are thus classified as ncRNA.75 Recently, investigators determined that a circRNA called R1as/CiRS-7 could serve as sponge for miR-7. This circRNA was able to downregulate miR-7 by acting as a specific sponge for it through 63 miR-7 binding sites.76 The design of synthetic sponges able to mimic natural circRNA action represents a novel approach for the modulation of aberrant miRNA expression in cancer. This technology is very useful in satisfying the need to simultaneously downregulate several miRNAs or miRNA families.77
Recently, investigators have developed novel computational tools for the design of synthetic miRNAs capable of effectively and simultaneously targeting multiple specific mRNAs of choice (multitarget, multisite targeting). Lagana et al.78 in 2014 developed and validated a bioinformatic tool termed “miR-Synth,” which represents a single synthetic miRNA able to simultaneously target MET and EGFR. Interestingly, this tool can be applied to create synthetic miRNAs for a wide variety of different target combination choices. The concept of modulating miRNA in cancer through the reintroduction or the repression of deregulated miRNAs combined with the use of synthetic technology for the modulation of miRNA expression (miRNA sponges) and, finally, the employment of synthetic miRNA that can modulate simultaneously the expression of different genes of choice does not represent a strategy void of issues. The off-target problem and difficulties in delivery make miRNA-centered cancer therapy a promising technology not yet applicable.
Human applications for miRNAs
While the majority of miRNA-focused lines of investigation have been laboratory based, an increasing number of human clinical trials have begun to incorporate miRNAs as with predictive/therapeutic biomarkers or as directed therapeutics. Currently, there are over 100 clinical trials incorporating miRNAs that are either actively recruiting or completed recruiting. The majority of such studies are utilizing miRNAs as potential clinical biomarkers. However, in the last few years, we have witnessed the emergence if clinical studies directed at utilizing miRNAs as therapeutics in humans. The most recognized such study involves the application of an antagomir for miR-122 to treat hepatitis C.79 Both phase I and phase II trials have been completed using the agent SPC3649 (Miravirsen) in both healthy volunteers and those with chronic Hepatitis C.80 Currently, studies are ongoing to examine the utility of Miravirsen in chronic Hepatitis C nonresponders. In the area of cancers, there are two exciting studies on the horizon that have potential for clinical application. The first such study is an ongoing phase I trial, which is a multicenter phase I trial using a liposomal formulation of miR-34 (MRX34) for primary unresectable liver cancer or advanced metastatic solid malignancies with or without metastases. The second study identified as MesomiR-1 is a phase I trial involving the use of an EGFR-targeting delivery vehicle harboring miR-16 in individuals with malignant pleural mesothelioma or advanced nonsmall-cell lung cancer who have failed previous therapies.
References
- 1 Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854.
- 2 Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355.
- 3 Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714.
- 4 Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858.
- 5 Miranda KC, Huynh T, Tay Y, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217.
- 6 Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866.
- 7 Nana-Sinkam SP, Hunter MG, Nuovo GJ, et al. Integrating the MicroRNome into the study of lung disease. Am J Respir Crit Care Med. 2009;179:4–10.
- 8 Nana-Sinkam SP, Croce CM. Non-coding RNAs in cancer initiation and progression and as novel biomarkers. Mol Oncol. 2011;5:483–491.
- 9 Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910.
- 10 Gregory RI, Yan KP, Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240.
- 11 Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–191.
- 12 Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665.
- 13 Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529.
- 14 Karube Y, Tanaka H, Osada H, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–115.
- 15 Merritt WM, Lin YG, Han LY, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650.
- 16 Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810.
- 17 Costinean S, Sandhu SK, Pedersen IM, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–1382.
- 18 Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443.
- 19 Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060.
- 20 O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843.
- 21 Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18:74–82.
- 22 He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822.
- 23 Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and microRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 2007;67:8433–8438.
- 24 Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85.
- 25 Nigita G, Alaimo S, Ferro A, Giugno R, Pulvirenti A. Knowledge in the investigation of A-to-I RNA editing signals. Front Bioeng Biotechnol. 2015;3:18.
- 26 Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140.
- 27 Chin LJ, Ratner E, Leng S, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540.
- 28 Steinemann D, Tauscher M, Praulich I, Niemeyer CM, Flotho C, Schlegelberger B. Mutations in the let-7 binding site – a mechanism of RAS activation in juvenile myelomonocytic leukemia? Haematologica. 2010;95:1616.
- 29 Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261.
- 30 Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198.
- 31 Rosenfeld N, Aharonov R, Meiri E, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26:462–469.
- 32 Nadal E, Chen G, Gallegos M, et al. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2013;19:6842–6852.
- 33 Ferreira AF, Moura LG, Tojal I, et al. ApoptomiRs expression modulated by BCR-ABL is linked to CML progression and imatinib resistance. Blood Cells Mol Dis. 2014;53:47–55.
- 34 Akagi I, Okayama H, Schetter AJ, et al. Combination of protein coding and noncoding gene expression as a robust prognostic classifier in stage I lung adenocarcinoma. Cancer Res. 2013;73:3821–3832.
- 35 Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110:483–495.
- 36 Shen J, Liao J, Guarnera MA, et al. Analysis of microRNAs in sputum to improve computed tomography for lung cancer diagnosis. J Thorac Oncol. 2014;9:33–40.
- 37 Xing L, Todd NW, Yu L, Fang H, Jiang F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod Pathol. 2010;23:1157–1164.
- 38 Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–10518.
- 39 Shen J, Liu Z, Todd NW, et al. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer. 2011;11:374.
- 40 Boeri M, Verri C, Conte D, et al. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc Natl Acad Sci U S A. 2011;108:3713–3718.
- 41 Arroyo JD, Chevillet JR, Kroh EM, et al. Argonaute 2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108:5003–5008.
- 42 Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659.
- 43 Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433.
- 44 Zernecke A, Bidzhekov K, Noels H, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81.
- 45 Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39:7223–7233.
- 46 Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891.
- 47 Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer – the emerging science of cellular ‘debris’. Semin Immunopathol. 2011;33:455–467.
- 48 Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A. 2009;106:3794–3799.
- 49 Garnier D, Magnus N, Lee TH, et al. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J Biol Chem. 2012;287:43565–43572.
- 50 Cazzoli R, Buttitta F, Di Nicola M, et al. microRNAs derived from circulating exosomes as noninvasive biomarkers for screening and diagnosing lung cancer. J Thorac Oncol. 2013;8:1156–1162.
- 51 Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906.
- 52 Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, Hannon GJ. A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes Dev. 2007;21:3238–3243.
- 53 Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004.
- 54 Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756.
- 55 Dahiya N, Sherman-Baust CA, Wang TL, et al. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One. 2008;3:e2436.
- 56 O’Hara AJ, Wang L, Dezube BJ, Harrington WJ Jr Damania B, Dittmer DP. Tumor suppressor microRNAs are underrepresented in primary effusion lymphoma and Kaposi sarcoma. Blood. 2009;113:5938–5941.
- 57 Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070.
- 58 Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–8707.
- 59 Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–6043.
- 60 Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109:4944–4951.
- 61 Wu K, Li L, Li S. Circulating microRNA-21 as a biomarker for the detection of various carcinomas: an updated meta-analysis based on 36 studies. Tumour Biol. 2015;36:1973–1981.
- 62 Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199.
- 63 Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314.
- 64 Misso G, Di Martino MT, De Rosa G, et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014;3:e194.
- 65 Garofalo M, Jeon YJ, Nuovo GJ, et al. MiR-34a/c-dependent PDGFR-alpha/beta downregulation inhibits tumorigenesis and enhances TRAIL-induced apoptosis in lung cancer. PLoS One. 2013;8:e67581.
- 66 Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther. 2010;10:219–222.
- 67 Brabletz S, Bajdak K, Meidhof S, et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770–782.
- 68 Iliopoulos D, Polytarchou C, Hatziapostolou M, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009;2:ra62.
- 69 Ferracin M, Zagatti B, Rizzotto L, et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol Cancer. 2010;9:123.
- 70 Vecchione A, Belletti B, Lovat F, et al. A microRNA signature defines chemoresistance in ovarian cancer through modulation of angiogenesis. Proc Natl Acad Sci U S A. 2013;110:9845–9850.
- 71 Yan W, Liu Y, Yang P, Wang Z, You Y, Jiang T. MicroRNA profiling of Chinese primary glioblastoma reveals a temozolomide-chemoresistant subtype. Oncotarget. 2015;6:11676–11682.
- 72 Esquela-Kerscher A, Trang P, Wiggins JF, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764.
- 73 Wu Z, Huang X, Huang X, Zou Q, Guo Y. The inhibitory role of Mir-29 in growth of breast cancer cells. J Exp Clin Canc Res. 2013;32:98.
- 74 Trang P, Wiggins JF, Daige CL, et al. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. 2011;19:1116–1122.
- 75 Li J, Yang J, Zhou P, et al. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res. 2015;5:472–480.
- 76 Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338.
- 77 Ebert MS, Sharp PA. MicroRNA sponges: progress and possibilities. RNA. 2010;16:2043–2050.
- 78 Lagana A, Acunzo M, Romano G, et al. miR-Synth: a computational resource for the design of multi-site multi-target synthetic miRNAs. Nucleic Acids Res. 2014;42:5416–5425.
- 79 Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694.
- 80 Janssen HL, Kauppinen S, Hodges MR. HCV infection and miravirsen. N Engl J Med. 2013;369:878.