Herpesviruses
Jeffrey I. Cohen, MD
Overview
Eight herpesviruses have been isolated from humans: two of these, Epstein–Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV), are associated with human tumors. EBV has been detected in lesions from patients with post-transplant lymphoproliferative disease, nasopharyngeal and some types of gastric carcinoma, Burkitt lymphoma, Hodgkin lymphoma, and certain other lymphoid tumors. KSHV is associated with Kaposi sarcoma, primary effusion lymphoma, and Castleman disease. Each of these viruses encodes proteins important for establishment of latency, transforming cells, and evading the immune system.
Properties of herpesviruses
Herpesviruses are enveloped DNA viruses that have the capacity to establish latent infection as well as to undergo lytic infection. The ability to establish latent infection in vivo and to reactivate from latency ensures a source of virus to infect previously uninfected individuals. Most adults latently harbor herpes simplex 1, varicella-zoster virus, human herpesviruses 6 and 7, and Epstein–Barr virus (EBV). Several features of herpesvirus replication are important for the maintenance of latency and for oncogenicity; EBV will serve as an example to illustrate the principles of herpesvirus infection relevant to oncogenicity.
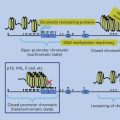
First, viral DNA must be maintained in the cell. The EBV genome is usually maintained in latently infected B cells as a multicopy circular episome in the host cell. Second, a cell transformed by a virus must avoid immune clearance. Replication of EBV requires up to 100 viral proteins; however, latent infection of B cells with EBV results in expression of 12 or fewer genes.1–3 This limited repertoire of gene products prevents frequent virus replication, avoiding death of the infected cell, and restricts the ability of the immune system to recognize and destroy cells latently infected with the virus. Third, specific viral proteins interact with other cell proteins or directly transactivate other cell genes to provide additional functions necessary for cell proliferation and immortalization. Several EBV proteins interact with cellular proteins to activate transcription of viral and cellular genes or to engage signal transduction pathways in the cell.
EBV: an oncogenic human herpesvirus
EBV gene expression in transformed lymphocytes
Infection of primary B cells with EBV in vitro results in transformation of the cells, which can then proliferate indefinitely. Eight EBV proteins and several nontranslated RNAs are expressed in latently infected B lymphocytes that have been growth transformed by EBV in vitro (Table 1). The EBV nuclear proteins EBNA-1, EBNA-2, EBNA-LP, EBNA-3A, EBNA-3B, and EBNA-3C comprise the EBV nuclear antigen complex. EBNA-1 binds to the oriP sequence (origin of viral DNA replication) on EBV DNA and allows the virus genome to be maintained as an episome in transformed B cells.4 EBNA-1 also transactivates its own expression. EBNA-1 transcripts are initiated from one of four different promoters. The Cp and Wp promoters are used to express EBNA-1 in lymphoblastoid cell lines in vitro; the Qp promoter is used in tissues from Burkitt lymphoma, nasopharyngeal carcinoma, and Hodgkin lymphoma; and the Fp promoter is used during lytic replication.5 EBNA-1 inhibits its own protein degradation by proteasomes6 and limits its own translation,7 both of which may reduce its presentation to CD8+ cytotoxic T cells; however, EBNA-1 remains a target for CD4+ cells.8–11 EBNA-1 inhibits apoptosis induced by expression of p53.12
Table 1 Selected EBV genes and their cellular homologs and activities
| Gene | Expression class | Cellular homolog | Activity |
| EBNA-1 | Latent, lytic | None | Episome maintenance, transactivates viral genes, inhibits apoptosis |
| EBNA-2 | Latent | Notch | Transactivates viral and cellular genes, inhibits apoptosis |
| EBNA-3A, B, C | Latent | None | Regulates EBNA-2 activity, transactivates cellular genes |
| EBNA-LP | Latent | None | Increases EBNA-2 activity |
| LMP1 | Latent, lytic | CD40 | Transactivates cellular genes, inhibits apoptosis |
| LMP2 | Latent | None | Prevents EBV reactivation, transactivates Akt |
| EBERs | Latent | None | Upregulates cellular genes |
| BARF-1 | Lytic | CSF-1R | Inhibits IFN-α |
| BCFR1 | Lytic | IL-10 | Inhibits IFN-γ and IL-12 |
| BNLF2a | Lytic | None | Blocks antigen-specific CD8 T-cell recognition |
| BHRF1 | Lytic | Bcl-2 | Inhibits apoptosis |
| BALF1 | Lytic | Bcl-2 | Regulates BHRF1 activity |
| BGFL5 | Lytic | None | Blocks synthesis of MHC class I and II |
| BILF1 | Lytic | GPCR | Removes MHC class I from cell surface |
| BLLF3 | Lytic | None | Upregulates IL-10, TNF-α, and IL-1β |
| BPLFI | Lytic | None | Inhibits toll-like receptor signaling |
| BZLF1 | Lytic | None | Inhibits IFN-γ effects, inhibits function of p53, inhibits TNF-α, initiates lytic infection |
EBNA-2 transactivates expression of EBV LMP113 and LMP2,14 and the cellular genes CD21, CD23, c-myc, and c-fgr.15, 16 EBNA-2 is targeted to the LMP1, LMP2, Cp EBNA, and CD23 promoters by the GTGGGAA-binding protein Jκ, and thereby activates these promoters.17 EBNA-2 is a functional homolog of the Notch receptor, which uses Jκ to regulate gene expression during development.18 EBNA-2 also interacts with the DNA-binding protein PU-1 to transactivate the LMP1 promoter19 and with AUF to transactivate the EBNA Cp promoter.20 The transactivation domain of EBNA-2 is essential for B-lymphocyte transformation.21 This domain interacts with transcription factors TFIIB and the TATA-binding protein-associated factor TAF40.22 EBNA-2 inhibits apoptosis mediated by Nur77.23
EBNA-LP interacts with and enhances the ability of EBNA-2 to transactivate LMP1 and LMP2.24 Although EBNA-LP binds to Rb and p53 in vitro,25 the significance of these interactions is uncertain. Deletion of the carboxy terminus of EBNA-LP markedly reduces the ability of the virus to transform B lymphocytes.26
EBNA-3A, EBNA-3B, and EBNA-3C are distantly related to each other. The EBNA-3 proteins bind to Jκ preventing it from binding DNA, thereby inhibiting transactivation by EBNA-2.27 EBNA-3C upregulates expression of LMP1 and CD21. EBNA-3C binds to Nm23-HI, a human metastasis suppressor protein, and inhibits the protein’s ability to suppress migration of Burkitt lymphoma cells.28 EBNA-3C degrades Rb and enhances kinase activity by disrupting p27.29, 30 EBNA-3A and EBNA-3C are essential for B-lymphocyte transformation in vitro, while EBNA-3B is dispensable.31, 32
LMP1 functions as a transforming oncogene in nude mice.33 Expression of LMP1 in EBV-negative Burkitt lymphoma cells results in B-cell clumping and increased villous projections. Upregulation of bcl-2, bfl-1, and A20, and inhibition of Bax, by LMP1 in B cells protects the cells from apoptosis.34, 35 Expression of LMP1 in epithelial cells inhibits differentiation of the cells.36
LMP1 is a functional homolog of CD40, a member of the tumor necrosis factor receptor (TNFR) family.37 The carboxy terminus of LMP1 interacts with the TNFR-associated factors (TRAFs) 1, 2, 3, and 5, TRADD, RIP, and JAK3 in vitro.38 LMP1 functions as a constitutively active form of CD40 resulting in activation of NF-κB, stress-activated protein kinases, STATs, adhesion molecules, the B7 co-stimulatory molecule, JNK, and B-cell proliferation.39 LMP1 upregulates expression of intracellular adhesion molecules, Fas, CD40, and MMP-940 in B cells and EGF in epithelial cells.41 LMP1 inhibits phosphorylation of Tyk2 resulting in inhibition of interferon (IFN)-α signaling.42 LMP1 is essential for transformation of B lymphocytes by EBV.43 Analysis of EBV-containing human lymphomas shows that LMP1 localizes with TRAF-1, TRAF-3, and that activated NF-κB is present.44
LMP2 is dispensable for transformation of B cells,45 but induces a transforming phenotype in epithelial cells and promotes their motility.46, 47 LMP2 prevents lytic reactivation of EBV-infected primary B cells in response to activation of the B-cell receptor complex by cross-linking of surface immunoglobulin. LMP2 associates with the src family and syk protein-tyrosine kinases that are coupled to the B-cell receptor complex.48 Binding of LMP2 to these proteins results in their constitutive phosphorylation, which inhibits their ability to mediate signaling for virus reactivation.48, 49 B cells from transgenic mice expressing LMP2 survive even without normal B-cell receptor signaling activity.50 LMP2 activates β-catenin and Ras/PI3K/Akt signaling pathways in epithelial cells resulting in transformation of the cells.51, 52 LMP2 also activates mTOR and increases c-myc expression.53
The two EBV-encoded RNAs, EBER-1 and EBER-2, are the most abundant EBV RNAs in latently infected B cells; however, they are not required for latent or lytic EBV infection, but may contribute to B-cell transformation.54, 55 The EBERs upregulate expression of bcl-2 and IL-1056 and interact with the double-stranded RNA-activated protein kinase, and IFN-inducible oligoadenylate synthetase.57, 58
EBV encodes at least 44 miRNAs from the BART and BHRF1 regions of the genome; both BART and BHRF1 microRNAs downregulate cellular genes and inhibit apoptosis and enhance B-cell proliferation.59 EBV also encodes cellular RNAs that enhance transformation.60 Both EBV microRNAs and LMP1 are secreted from infected cells in exosomes, which may enhance tumorigenicty.61, 62
EBV genes expressed during productive infection
Infection of epithelial cells with EBV results in productive infection, with replication of virus and lysis of infected cells. Immediate-early genes encode regulators of virus gene expression, including the BZLF1 and BRLF1 proteins, which act as switches to initiate lytic infection. The BZLF1 protein inhibits TNF-α signaling63 and helps the virus evade T-cell responses.64 BZLF1 protein also inhibits the expression of the IFN-γ receptor65 and inhibits the function of p53.66 Early genes encode proteins involved in viral DNA synthesis, while late genes encode structural proteins.
Three viral genes expressed during productive infection are functional homologs of cellular genes and are important for the survival of EBV-infected B cells.67, 68 The EBV BCRF-1 protein is homologous to IL-10 and has IL-10 activity.69 BCRF-1 is a B-cell growth factor and inhibits IFN-γ release from activated peripheral blood mononuclear cells and secretion of IL-12 from macrophages.
The EBV BARF-1 protein acts as a soluble receptor for CSF-1.70 BARF-1 inhibits IFN-α secretion by human monocytes. The EBV BNLF2a protein interacts with the TAP complex to block antigen-specific CD8 T-cell recognition.71 The EBV BHRF1 protein is homologous to bcl-2 and protects cells from apoptosis.72 EBV BALF1 is also homologous to bcl-2 and antagonizes the antiapoptotic effect of BHRF1.73
Clinical aspects
EBV infection is usually spread by saliva. The virus infects B cells directly, or oropharyngeal epithelial cells and then spreads to subepithelial B cells.74 During primary infection, up to a few percent of peripheral blood B lymphocytes are infected with EBV and have the capacity to proliferate indefinitely in vitro. Natural killer (NK) cells, CD4 T cells, and HLA- and EBNA- or LMP-restricted cytotoxic T cells control the latently infected B lymphocytes.8 T- and B-cell interactions release lymphokines and cytokines, giving rise to many of the clinical manifestations of acute infectious mononucleosis. After recovery, the fraction of B cells latently infected with EBV in the peripheral blood remains at 1 in 105 to 1 in 106. These lymphocytes are the primary site of EBV persistence and a source of virus for persistent infection of epithelial surfaces.
B-cell tumors that occur early after EBV infection are usually lymphoproliferative processes, in which latent virus infection in B cells is the principal cause of proliferation. In contrast, nasopharyngeal carcinoma occurs long after primary EBV infection and viral gene expression is less important to the growth of the malignant cells.
Lymphoproliferative disease
EBV is associated with B-cell lymphoproliferative disease in patients with congenital or acquired immunodeficiency. X-linked lymphoproliferative syndrome is an inherited immunodeficiency of males; most patients die of a fatal lymphoproliferative disorder or fulminant hepatitis, but some survive with hypogammaglobulinemia or develop EBV-positive lymphomas. The gene mutated in X-linked lymphoproliferative syndrome has been identified as SAP,75 which encodes an SH2-containing protein that interacts with SLAM on B and T cells, and with 2B4 on NK and T cells. Anti-CD20 antibody (rituximab) has been effective in treating some patients with X-linked lymphoproliferative disease and acute EBV infection.76 Mutations in additional cellular proteins including XIAP, ITK, CD27, MagT1, CTP synthase 1, and PI3K 110δ predispose to severe EBV infections.
EBV lymphoproliferative disease occurs in patients who are immunosuppressed as a result of transplantation or AIDS.77–79 Risk factors for development of lymphoproliferative disease include EBV-seronegativity prior to transplant and receipt of T-cell-depleted bone marrow or antilymphocyte antibody. Lymphoproliferative lesions are most commonly seen in the lymph nodes, liver, lungs, kidney, bone marrow, or small intestine. Tumors in transplant patients are usually classified as lymphomas; some patients have hyperplastic lesions. The proliferating lymphocytes in these tumors generally do not have chromosomal translocations.
AIDS-related lymphomas may be systemic (nodal or extranodal) lymphomas or primary central nervous system lymphomas. Although most B-cell tumors in transplant recipients and central nervous system lymphomas in AIDS patients contain EBV, about 50% of other lymphomas in AIDS patients contain EBV. Tumors in patients with AIDS are usually either immunoblastic lymphomas or Burkitt lymphomas; most of the latter have c-myc translocations.
Tissues from transplant recipients or AIDS patients with EBV lymphoproliferative disease show expression of EBERs, EBNA-1, EBNA-2, and LMP1 (Table 2). The EBV viral load in the peripheral blood has been used to predict development of disease and to follow patients after therapy. The expression of EBV genes, which are targets for cytotoxic T cells, has important implications for therapy. Infusion of EBV-specific cytotoxic T cells, nonirradiated donor leukocytes, or HLA-matched allogeneic cytotoxic T cells have been effective in many cases for treatment of EBV lymphoproliferative disease.80–86 Anti-CD20 antibody (rituximab) has induced remissions in some patients and has been used in some studies as pre-emptive therapy when EBV viral DNA in the blood is rising in transplant recipients at risk of lymphoproliferative disease,87 although other studies suggest that pre-emptive therapy may be unnecessary.88
Table 2 Diseases associated with EBV latent gene expression
| Disease | BARTs | EBERs | EBNA-1 | EBNA-2 | LMP1 | LMP2 |
| Burkitt lymphoma | + | + | + | − | − | − |
| Nasopharyngeal carcinoma | + | + | + | − | + | + |
| Hodgkin lymphoma | + | + | + | − | + | + |
| Peripheral T-cell lymphoma | + | + | + | − | + | + |
| Lymphoproliferative disease | + | + | + | + | + | + |
Burkitt lymphoma
Seroepidemiologic studies show a strong association between Burkitt lymphoma and EBV in Africa.89, 90 More than 90% of African Burkitt lymphomas are associated with EBV, whereas only approximately 20% of Burkitt lymphomas in the United States are associated with the virus. African patients with Burkitt lymphoma often have high levels of antibody to EBV antigens, and the virus can be recovered from the tissue.
Burkitt lymphomas contain chromosomal translocations that result in c-myc dysregulation. The most common chromosomal translocation, t(8;14), places a portion of the c-myc oncogene adjacent to an immunoglobulin heavy chain gene. Less common translocations involve the c-myc oncogene and the κ or λ immunoglobulin light chain genes t(2;8) and t(8;22), respectively. These translocations result in high constitutive expression of c-myc. New approaches to treating Burkitt lymphoma based on inhibition of c-myc are in development.91
EBV-associated endemic Burkitt lymphoma is thought to develop in steps. First, EBV infection may expand the pool of differentiating and proliferating B cells. Second, chronic endemic malaria may cause T-cell suppression and B-cell proliferation. Third, enhanced proliferation of differentiating B cells may favor the chance occurrence of a reciprocal c-myc (t[8;14] or t[8;22]) translocation placing c-myc partially under the control of immunoglobulin-related transcriptional enhancers, with development of a monoclonal tumor.
Nasopharyngeal carcinoma
The nonkeratinizing nasopharyngeal carcinomas are uniformly associated with EBV. Patients with nasopharyngeal carcinoma have high levels of antibodies to EBV antigens. A prospective study of Taiwanese men showed that those with IgA antibodies to EBV viral capsid antigen (VCA) and anti-EBV deoxyribonuclease antibodies had an increased risk for developing nasopharyngeal carcinoma when compared to men without these antibodies.92 These antibodies are useful in screening patients for early detection of nasopharyngeal carcinoma and are prognostic for patients after treatment. Another study showed that quantifying the level of EBV DNA in plasma of patients with advanced nasopharyngeal carcinoma was useful for monitoring patients and predicting outcomes.93 Nasopharyngeal carcinoma tissue contains EBV genomes in every cell. These tumors are monoclonal with regard to EBV infection, indicating that EBV infection precedes malignant cell outgrowth at the cellular level. Unlike Burkitt lymphoma, the association of EBV with nasopharyngeal carcinoma is uniform and universal. Infusions of EBV-specific cytotoxic T cells resulted in remissions in some patients with refractory nasopharyngeal carcinoma.94
Hodgkin lymphoma
Persons with a history of infectious mononucleosis are at a greater risk of developing Hodgkin lymphoma.95 Patients with Hodgkin lymphoma generally have higher titers of antibody to EBV VCA than does the general population. Tissues from ∼40% to 60% of patients with Hodgkin lymphoma contain EBV genomes. Cases of Hodgkin lymphoma in patients with HIV or from developing countries are more likely to contain EBV genomes than persons without HIV or from developed countries.96 The EBV genome is monoclonal and present in Reed–Sternberg cells. EBV is more often associated with aggressive subtypes (especially mixed cellularity) of Hodgkin lymphoma. Tumors from patients with EBV+ Hodgkin lymphoma and some from patients with lymphoproliferative disease arise from postgerminal center cells.97 Infusion of cytotoxic T cells generated from 11 patients with relapsed Hodgkin lymphoma and measurable disease resulted in complete remissions in two patients, partial remission in one patient, stable disease in five patients, and no response in three patients.98 Infusion of arginine butyrate and ganciclovir to induce EBV thymidine kinase expression with phosphorylation of ganciclovir and induction of apoptosis resulted in antitumor responses in some patients with EBV B-cell malignancies.99
Other tumors associated with EBV
EBV has also been detected in non-Hodgkin’s lymphomas. EBV-positive diffuse large B-cell lymphoma has a poorer prognosis than EBV-negative lymphoma.100 Treatment of patients with EBV+ non-Hodgkin’s lymphoma in remission with autologous antigen-presenting cells transduced with LMP2 resulted in increased frequencies of LMP2-specific cytotoxic T cells and tumor responses in several patients with relapsed disease.101 EBV DNA and latency proteins have been detected in tissues from patients with peripheral T-cell lymphomas.102 EBV DNA has also been detected in central nervous system lymphomas from patients with no underlying immunodeficiency, T cells in patients with virus-associated hemophagocytic syndrome, nasal T-cell lymphoma, carcinoma of the palatine tonsil, laryngeal carcinoma, and angioimmunoblastic T-cell lymphoma. EBV DNA and nuclear antigens have been detected in thymic carcinomas and in B-cell lesions from patients with lymphomatoid granulomatosis.
EBV DNA has been found in leiomyosarcomas in AIDS patients,103 and viral RNA and EBNA-2 have been detected in smooth muscle tumors in organ transplant recipients.104 About 7% of primary gastric carcinomas are EBV+, especially in undifferentiated lymphoepithelioma-like carcinomas.
KSHV and malignancies
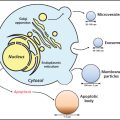
In 1994, Chang et al.105 detected sequences of a new human herpesvirus, Kaposi-sarcoma-associated herpesvirus (KSHV) in Kaposi sarcoma tissues from patients with AIDS. KSHV is present in B cells in asymptomatic persons. B-cell lines derived from primary effusion lymphomas maintain KSHV in a latent state and can be induced to undergo lytic virus replication by the addition of phorbol ester or butyrate. Infection of dermal microvascular endothelial cells in vitro with KSHV results in transformation of the cells with maintenance of long-term infection. The cells become spindle-shaped and show loss of contact inhibition with anchorage-independent growth.106 Although KSHV transforms bone-marrow-derived endothelial cells, virus is present in only a small fraction of the cells.107
Viral proteins
Several KSHV proteins are important for transformation, establishing latency, and modulating the immune response to the virus.108–110 KSHV encodes a large number of cellular homologs (Table 3) that have been grouped into different classes, depending on when they are expressed in primary effusion lymphoma cell lines.111 Expression of the KSHV K1 gene in rodent fibroblasts results in transformation of the cells.112 The K1 protein induces tyrosine phosphorylation in cells113 and activates the Akt signaling pathway.114 The K1 protein inhibits apoptosis115 and results in constitutive calcium-dependent signal activation in B cells.116
Table 3 Selected KSHV genes and their cellular homologs and activities
| Gene | Expression class | Cellular homolog | Activity |
| K1 | II | ITAM motif | Transformation, activates signaling pathways |
| K2 | II | IL-6 | B-cell growth factor, angiogenesis, hematopoiesis |
| K3 | III | None | Reduces surface MHC class I |
| K4 | II | MIP-1α | Chemokine receptor antagonist; angiogenesis; chemotaxis |
| K4.1 | II | MIP-1α | Chemokine receptor agonist; angiogenesis; chemotaxis |
| K5 | II | None | Inhibits NK-cell activity; reduces surface MHC class I; increases monocyte proliferation |
| K6 | II | MIP-1α | Chemokine receptor agonist; angiogenesis; chemotaxis |
| K8 | III | None | Inhibits p53 |
| K9 | II | IRF | Represses IFN activity, transformation, inhibits p53 |
| K10.5 (LANA-2) | II | IRF | Represses IFN activity; inhibits apoptosis; inhibits p53 |
| K11.1 | I | IRF | Represses IFN activity |
| K12 (Kaposin A) | II | None | Transformation |
| K12 (Kaposin B) | II | None | Increases cytokine mRNA stability |
| K14 | II | OX-2 | Induces proinflammatory cytokines |
| K15 (LAMP) | III | None | Binds TRAFs, inhibits B-cell receptor signaling |
| ORF4 | II | CR2 | Complement-binding protein |
| ORF16 | II/III | Bcl-2 | Inhibits apoptosis |
| ORF45 | III | None | Inhibits IRF7 |
| ORF50 | III | None | Increases CD21 and CD23 expression, degrades IRF7 |
| ORF63 | III | NLR proteins | Inhibits NLRP1 |
| ORF71 | I | FLIP | Inhibits apoptosis; activates NF-κB |
| ORF72 | I | Cyclin D | Cell-cycle progression, inhibits Rb |
| ORF73 (LANA-1) | I | None | Episome maintenance, inhibits p53 and Rb |
| ORF74 | II | GPCR | Angiogenesis, transformation, and proliferation |
Expression class I = latent gene, expressed in uninduced primary effusion lymphoma cells, not induced by phorbol ester; class II = expressed in uninduced cells, induced by TPA; class III = lytic gene, expressed only after induction by TPA (includes many structural proteins and DNA replication enzymes). FLIP = FLICE inhibitory protein; GPCR = G-protein-coupled receptor; IRF = interferon regulatory factor; ITAM = immunoreceptor tyrosine-based activation motif; MHC = major histocompatibility complex; MIP = macrophage inflammatory protein; NK = natural killer; Rb = retinoblastoma protein; TRAFs = TNFR-associated factors.
The KSHV K2 gene encodes an IL-6 homolog (vIL-6). IL-6 is a B-cell growth factor and acts as an autocrine growth factor for lymphoid tumors resulting in proliferation.117 vIL-6 prevents death of IL-6-dependent B9 cells in vitro,118 promotes hematopoiesis, and induces VEGF to promote angiogenesis.119 The K3 [modulator of immune recognition 1 (MIR1)] and K5 (MIR2) proteins induce rapid endocytosis of MHC (major histocompatibility complex) class I molecules and IFN-γ receptor 1 from the surface of cells by ubiquitination of these proteins.120–122 The K5 protein also downregulates ICAM-1 and B7.2, which results in inhibition of NK-cell-mediated cytotoxicity,123 and removes CD31 from the surface of endothelial cells.124
The KSHV K4, K4.1, and K6 genes encode three chemokines: the viral macrophage inflammatory proteins (MIP)-II, -III, and -I, respectively. vMIP-I inhibits replication of HIV strains dependent on CCR5.118 vMIP-I and vMIP-III are chemokine receptor agonists for CCR8125 and CCR4,126 respectively, while vMIP-II is a broad-spectrum chemokine receptor antagonist. vMIP-II is a chemoattractant for eosinophils,127 binds to both CC and CXC chemokines, and blocks calcium mobilization induced by chemokines.128
The KSHV K9, K11.1, and K10.5 proteins are referred to as viral IFN regulatory factor (vIRF)-1, -2, and -3, respectively. Each of these proteins inhibits virus-mediated activation of the IFN-α promoter.129 vIRF1 inhibits MHC-1 transcription and surface expression.130 vIRF1 and vIRF3 inhibit p53-mediated apoptosis131, 132 and transform NIH 3T3 cells.133 vIRF3 (also called LANA2) protects cells from p53-induced apoptosis.134, 135 KSHV ORF45 blocks the activity of IRF7 and inhibits the activation of IFN-α and -β.136
The KSHV K12 locus encodes several proteins termed kaposins. Kaposin A induces transformation of cells.137 Kaposin B increases expression of cytokines by activating MAP-kinase-associated protein kinase 2 and inhibiting the degradation of cytokine mRNA.138, 139 The KSHV K14 protein, a homolog of the cellular OX2 protein, stimulates monocytes to produce proinflammatory cytokines such as TNF-α, IL-1β, and IL-6.140 KSHV ORF4 protein inhibits the complement system.141 KSHV ORF16 encodes a homolog of bcl-2 and inhibits bax-induced apoptosis.142 The ORF71 gene encodes a homolog of cellular FLIP that blocks apoptosis. ORF71 binds to Atg3 and protects cells from autophagy. KSHV ORF71 activates NF-κB, promotes tumor growth, and is required for survival of KSHV-infected lymphoma cells.143–147 KSHV ORF72 encodes a cyclin D homolog that binds to and activates cdk6 and phosphorylates p27, and stimulates cell-cycle progression in normally quiescent fibroblasts.148, 149 The viral cyclin protein phosphorylates and thereby inactivates Rb.150
KSHV ORF73 encodes LANA1 that localizes with viral DNA episomes and tethers them to chromosomes during cell division.151 KSHV LANA1 is required for persistence of the episome in dividing cells and transactivates its own promoter. In addition, LANA1 inhibits the activity of both p53 and Rb,152, 153 upregulates expression and stabilizes β-catenin,154 upregulates and activates survivin,155 induces nuclear accumulation of HIF,156 and activates c-myc.157 Expression of LANA in transgenic mice results in development of lymphomas.158 LANA inhibits TGF-β signaling.159 KSHV ORF74 encodes a G-protein-coupled receptor that is homologous to the cellular IL-8 receptor; unlike the latter protein, however, the KSHV receptor is constitutively active and induces cellular proliferation.160 ORF74 protein induces angiogenesis,161 activates the Akt signaling pathway,162 and induces proliferation and vascular permeability of endothelial cells.163–165 ORF74 activates NF-κB and JNK, and upregulates IL-1, IL-8, TNF-α, and FGF, and inhibits viral lytic gene expression.166
KSHV K15 encodes the latency-associated membrane protein (LAMP) and interacts with TRAFs 1, 2, and 3.167 K15 suppresses tyrosine phosphorylation and intracellular calcium mobilization, inhibiting B-cell receptor signaling.168 KSHV encodes several microRNAs located between K12 and ORF71 or within K12 that are expressed during latency and detected in patient plasma.169 These microRNAs are important for NF-κB activation and blocking cell-cycle arrest of latently infected cells.109, 170 KSHV microRNAs also target cellular genes.171
Clinical aspects
Seroprevalence rates for KSHV vary from <5% in normal blood donors in the United States or United Kingdom to 30–35% in HIV-positive homosexual men.172 Antibody to KSHV is more common in African and Mediterranean populations. At least 85% of patients with Kaposi sarcoma have antibodies to KSHV.173 The prevalence of Kaposi sarcoma is lower in women than in men, and HIV-seropositive women have a much lower incidence of antibody to KSHV than do seropositive men. KSHV seropositivity in HIV-positive homosexual men is predictive of subsequent development of Kaposi sarcoma.174 Levels of KSHV DNA are higher in patients with active Kaposi sarcoma or multicentric Castleman disease, than in those in remission, and are also elevated in patients with primary effusion lymphoma.175 The virus is not thought to be pathogenic in most healthy individuals and can persist in a latent phase for life; however, in immunocompromised persons, it is strongly associated with Kaposi sarcoma. Thus, although infection with KSHV appears to be required for development of Kaposi sarcoma, it is probably not sufficient by itself and other cofactors, such as HIV and impaired cellular immunity, are important. KSHV is thought to be sexually transmitted in homosexual men172 and has been associated with sexual transmission and intravenous drug use in women.176 In endemic populations (e.g., Africa), KSHV may be transmitted vertically from mother to child and between siblings. KSHV has been transmitted by renal allografts.177, 178
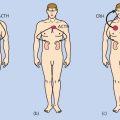
Kaposi sarcoma
KSHV has been found in nearly all biopsies of classic Kaposi sarcoma, African endemic Kaposi sarcoma, Kaposi sarcoma in HIV-seronegative transplant recipients and homosexual men, and Kaposi sarcoma in patients with AIDS.179, 180 KSHV is present in the endothelial and spindle cells of the tumor, but not in normal endothelium.181 Most of the tumor cells are latently infected with the virus, but 1–5% of the spindle cells in HIV-positive Kaposi sarcoma show lytic KSHV infection. Kaposi sarcoma can be polyclonal, oligoclonal, or monoclonal. KSHV is also present in the peripheral blood mononuclear cells of approximately 50% of patients with Kaposi sarcoma, and its presence is predictive of development of the malignancy.172 KSHV has also been detected in the saliva of patients with Kaposi sarcoma, and, infrequently, in semen. Several KSHV proteins are expressed in Kaposi’s tissues (Table 4). Foscarnet and ganciclovir reportedly reduce the frequency of new Kaposi sarcoma lesions in some, but not all, studies.182, 183 Cidofovir had no effect on treatment of established lesions.184 In contrast, HIV protease inhibitors have been reported to induce regression of Kaposi sarcoma lesions.185 IL-12, in combination with liposomal doxorubicin, resulted in tumor responses in AIDS patients with Kaposi sarcoma receiving HAART; responses were maintained with IL-12 therapy.186 Sirolimus,187 imatinib,188 bevacizumab,189 and paclitaxel190 have all been reported to have activity against Kaposi sarcoma.
Table 4 Diseases associated with KSHV gene expression
| Gene | Kaposi sarcoma | Primary effusion lymphoma | Castleman disease |
| LANA (ORF73) | + | + | + |
| K12 (Kaposin) | + | + | − |
| ORF72 (v-cyclin) | + | + | − |
| ORF71 (v-FLIP) | + | + | + |
| ORF74 (GPCR) | + | − | − |
| K10.5 (vIRF3) | − | + | + |
| K9 (vIRF1) | − | − | + |
| K2 (vIL-6) | − | ± | + |
Primary effusion lymphoma
KSHV has also been found in primary effusion lymphomas in patients with AIDS.180, 181, 191 These body-cavity-based lymphomas of B-cell lineage are located in the pleural, peritoneal, or pericardial space and usually contain EBV as well as KHSV genomes. Some KSHV-positive lymphomas have been found in patients without AIDS.
Multicentric Castleman disease
KSHV has also been detected in biopsies from some patients with multicentric Castleman disease, especially in the variant known as the plasma cell type.180, 181, 192–194 This disease is usually polyclonal and presents as generalized lymphadenopathy, fever, and hypergammaglobulinemia. Symptoms are thought to be due to increased levels of IL-6 and vIL-6.195 KSHV is detected more frequently in biopsies from HIV-positive patients than in biopsies from those patients without HIV. KSHV is present in the immunoblastic B cells of the mantle zone of the lesions. Zidovudine plus valganciclovir showed activity in some patients with KSHV-associated Castleman disease.196
Summary
- Two herpesviruses, Epstein–Barr virus (EBV) and Kaposi sarcoma-associated herpesvirus (KSHV), are associated with human tumors.
- EBV is associated with Burkitt lymphoma, Hodgkin and non-Hodgkin lymphoma, post-transplant lymphoproliferative disease, T-cell lymphoma, nasopharyngeal carcinoma, and certain types of gastric carcinoma.
- EBV transformed B cells and EBV post-transplant lymphoproliferative lesions express EBNA-1, -2, -3s, and -LP and LMP1 and LMP2 and RNAs including EBERs and BARTs in culture. Burkitt lymphomas express only EBNA-1, EBERs, and BARTs. Hodgkin lymphomas and nasopharyngeal carcinomas express EBNA-1, LMP1, LMP2, EBERs, and BARTs.
- EBV EBNA-1 is important for maintaining the viral episome during replication of the cells. EBNA-2 transactivates several virus and cellular promoters and is a functional homolog of the Notch receptor. EBNA-3’s regulate the activity of EBNA-2 and also transactivate viral genes. LMP1 is a functional homolog of CD40 and binds to TNFR-associated factors to upregulate NF-κB, STATs, JNK, and stress-activated protein kinases.
- KSHV is associated with primary effusion lymphoma, Kaposi sarcoma, and Castleman disease.
- Each of the three KSHV-associated tumors express LANA (ORF73) and v-FLIP (ORF71). Other KSHV proteins, including Kaposin (K12), v-cyclin (ORF72), a G-protein-coupled receptor (GPCR, ORF74), v-IRF3 (K10.5), v-IRF1 (K9), and v-IL-6 (K2) are expressed in some, both not all virus-associated malignancies.
- LANA maintains the viral episome during replication and inhibits the activity of Rb and p53. KSHV v-FLIP is a homolog of cellular FLIP and inhibits apoptosis and activates NF-κB. Kaposin increases the activity of cytokines, v-cyclin inhibits Rb and helps virus-infected cells to progress through the cell cycle, and the viral GPCR contributes to proliferation of KSHV-infected cells. v-IRF1 and v-IRF3 inhibit the activity of interferon and p53, while v-IL-6 functions as a B-cell growth factor.
References
- 1 Longnecker R, Kieff E, Cohen JI. Epstein-Barr virus. In: Knipe DM, Howley PM, Cohen JI, Griffith DE, Lamb RA, Martin MA, Racaniello V, Roizman B, eds. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2013:1898–1959.
- 2 Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–1337.
- 3 Price AM, Luftig MA. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv Virus Res. 2014;88:279–313.
- 4 Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A. 1984;81:3806–3810.
- 5 Deacon EM, Pallesen G, Niedobitek G, et al. Epstein-Barr virus and Hodgkin’s disease; transcriptional analysis of virus latency in the malignant cells. J Exp Med. 1993;177:339–349.
- 6 Levitskaya J, Shapiro A, Leonchiks A, et al. Inhibition of ubiquitin/proteosome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc Natl Acad Sci U S A. 1997;94:12616–12621.
- 7 Yin Y, Manoury B, Fahraeus R. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science. 2003;301:1371–1374.
- 8 Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007;25:587–617.
- 9 Voo KS, Fu T, Wang HY, et al. Evidence for the presentation of major histocompatibility complex class I-restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J Exp Med. 2004;199:459–470.
- 10 Lee SP, Brooks JM, Al-Jarrah H, et al. CD8 T cell recognition of endogenously expressed Epstein-Barr virus nuclear antigen 1. J Exp Med. 2004;199:1409–1420.
- 11 Heller KN, Upshaw J, Seyoum B, et al. Distinct memory CD4+ T-cell subsets mediate immune recognition of Epstein Barr virus nuclear antigen 1 in healthy virus carriers. Blood. 2007;109:1138–1146.
- 12 Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc Natl Acad Sci U S A. 2003;100:14269–14274.
- 13 Wang F, Tsang SF, Kurilla MG, et al. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein. J Virol. 1990;64:3407–3416.
- 14 Zimber-Strobl U, Kremmer E, Grasser F, et al. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. 1993;12:167–175.
- 15 Wang F, Gregory C, Sample C, et al. Epstein-Barr virus latent membrane protein (LMP-1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP-1 cooperatively induce CD23. J Virol. 1990;64:2309–2318.
- 16 Knutson JC. The level of c-fgr RNA is increased by EBNA-2, an Epstein-Barr virus gene required for B-cell immortalization. J Virol. 1990;64:2530–2536.
- 17 Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA-2 transactivation by recombination signal-binding protein Jkappa. Science. 1994;265:92–95.
- 18 Gordadze AV, Peng R, Tan J, et al. Notch IIC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J Virol. 2001;75:5899–5912.
- 19 Johannsen E, Koh E, Mosialos G, et al. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by Jkappa and PU.1. J Virol. 1995;69:253–262.
- 20 Fuentes-Panana EM, Peng R, Brewer G, et al. Regulation of the Epstein-Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J Virol. 2000;74:8166–8175.
- 21 Cohen JI. A region of herpes simplex virus VP16 can substitute for a transforming domain of Epstein-Barr virus nuclear protein 2. Proc Natl Acad Sci U S A. 1992;89:8030–8034.
- 22 Tong X, Wang F, Thut CJ, Kieff E. The Epstein-Barr virus nuclear protein 2 acidic domain can interact with TFIIB, TAF40, and RPA70 but not with TATA-binding protein. J Virol. 1995;69:585–588.
- 23 Lee JM, Lee KH, Weidner M, et al. Epstein-Barr virus EBNA2 blocks Nur77-mediated apoptosis. Proc Natl Acad Sci U S A. 2002;99:11878–11883.
- 24 Peng CW, Xue Y, Zhao B, et al. Direct interactions between Epstein-Barr virus leader protein LP and the EBNA2 acidic domain underlie coordinate transcriptional regulation. Proc Natl Acad Sci U S A. 2004;101:1033–1038.
- 25 Szekely L, Selivanova G, Magnusson KP, et al. EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma and p53 proteins. Proc Natl Acad Sci U S A. 1993;90:5455–5459.
- 26 Mannick JB, Cohen JI, Birkenbach M, et al. The Epstein Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B lymphocyte transformation. J Virol. 1991;65:6826–6837.
- 27 Robertson ES, Lin J, Kieff E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJkappa. J Virol. 1996;70:3068–3074.
- 28 Subramanian C, Cotter MA, Robertson ES. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-HI: a molecular link to cancer metastasis. Nat Med. 2001;7:350–355.
- 29 Knight JS, Sharma N, Robertson ES. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc Natl Acad Sci U S A. 2005;102:18562–18566.
- 30 Knight JS, Sharma N, Robertson ES. SCFSkp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol Cell Biol. 2005;25:1749–1763.
- 31 Tomkinson B, Robertson E, Kieff E. Epstein-Barr virus nuclear proteins (EBNA) 3A and 3C are essential for B lymphocyte growth transformation. J Virol. 1993;67:2014–2025.
- 32 Maruo S, Wu Y, Ishikawa S, Kanda T, Iwakiri D, Takada K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc Natl Acad Sci U S A. 2006;103:19500–19505.
- 33 Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840.
- 34 Fries KL, Miller WE, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J Virol. 1996;70:8653–8659.
- 35 Grimm T, Schneider S, Naschberger E, et al. EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood. 2005;105:3263–3269.
- 36 Dawson CW, Rickinson AB, Young LS. Epstein-Barr virus latent membrane protein inhibits human epithelial cell differentiation. Nature. 1990;344:777–780.
- 37 Uchida J, Yasui T, Takaoka-Shichijo Y, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP-1 in B lymphocyte responses. Science. 1999;286:300–303.
- 38 Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-κB. Proc Natl Acad Sci U S A. 1997;94:12592–12597.
- 39 Gires O, Kohlhuber F, Kilger E, et al. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999;18:3064–3073.
- 40 Yoshizaki T, Sato H, Furukawa M, et al. The expression of matrix metalloproteinase 9 is enhanced by Epstein-Barr virus latent membrane protein 1. Proc Natl Acad Sci U S A. 1998;95:3621–3626.
- 41 Miller WE, Cheshire JL, Raab-Traub N. Interaction of tumor necrosis factor receptor-associated factor signaling proteins with the latent membrane protein 1 PXQXT motif is essential for induction of epidermal growth factor receptor expression. Mol Cell Biol. 1998;18:2835–2844.
- 42 Geiger TR, Martin JM. The Epstein-Barr virus-encoded LMP-1 oncoprotein negatively affects Tyk2 phosphorylation and interferon signaling in human B cells. J Virol. 2006;80:11638–11650.
- 43 Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A. 1993;90:9150–9154.
- 44 Liebowitz D. Epstein-Barr virus and a cellular signaling pathway in lymphomas from immunosuppressed patients. N Engl J Med. 1998;338:1413–1421.
- 45 Longnecker R, Miller CL, Tomkinson B, et al. Deletion of DNA encoding the first five transmembrane domains of Epstein-Barr virus latent membrane proteins 2A and 2B. J Virol. 1993;67:5068–5074.
- 46 Scholle F, Bendt KM, Raab-Traub N. Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol. 2000;74:10681–10689.
- 47 Allen MD, Young LS, Dawson CW. The Epstein-Barr virus-encoded LMP2A and LMP2B proteins promote epithelial cell spreading and motility. J Virol. 2005;79:1789–1802.
- 48 Miller CL, Burkhardt AL, Lee JH, et al. Integral membrane protein 2 of Epstein-Barr virus regulates reactivation from latency through dominant negative effects on protein-tyrosine kinases. Immunity. 1995;2:155–166.
- 49 Miller CL, Lee JH, Kieff E, Longnecker R. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc Natl Acad Sci U S A. 1994;91:772–776.
- 50 Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–411.
- 51 Morrison JA, Klingelhutz AJ, Raab-Traub N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J Virol. 2003;77:12276–12284.
- 52 Fukuda M, Longnecker R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J Virol. 2007;81:9299–9306.
- 53 Moody CA, Scott RS, Amirghahari N, et al. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol. 2005;79:5499–5506.
- 54 Swaminathan S, Tomkinson B, Kieff E. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc Natl Acad Sci U S A. 1991;88:1546–1550.
- 55 Wu Y, Maruo S, Yajima M, et al. Epstein-Barr virus (EBV)-encoded RNA 2 (EBER2) but not EBER1 plays a critical role in EBV-induced B-cell growth transformation. J Virol. 2007;81:11236–11245.
- 56 Kitagawa N, Goto M, Kurozumi K, et al. Epstein-Barr virus-encoded poly(A)(−) RNA supports Burkitt’s lymphoma growth through interleukin-10 induction. EMBO J. 2000;19:6742–6750.
- 57 Clarke PA, Schwemmle M, Schickinger J, et al. Binding of the Epstein-Barr virus small RNA EBER-1 to double-stranded RNA-activated protein kinase. Nucleic Acids Res. 1991;19:243–248.
- 58 Sharp TV, Raine DA, Gewert DR, et al. Activation of the interferon-inducible (2′-5′) oligoadenylate synthetase by the Epstein-Barr virus RNA, EBER-1. Virology. 1999;257:303–313.
- 59 Klinke O, Feederle R, Delecluse HJ. Genetics of Epstein-Barr virus microRNAs. Semin Cancer Biol. 2014;26:52–59.
- 60 Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR. Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. J Virol. 2010;84:11670–11678.
- 61 Meckes DG Jr Shair KH, Marquitz AR, Kung CP, Edwards RH, Raab-Traub N. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20370–20375.
- 62 Meckes DG Jr Gunawardena HP, Dekroon RM, et al. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc Natl Acad Sci U S A. 2013;110:E2925–E2933.
- 63 Morrison TE, Mauser A, Klingelhutz A, et al. Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J Virol. 2004;78:544–549.
- 64 Zuo J, Thomas WA, Haigh TA, et al. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 2011;7:e1002455.
- 65 Morrison TE, Mauser A, Wong A, et al. Inhibition of interferon-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity. 2001;15:787–799.
- 66 Mauser A, Saito S, Appella E, et al. The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J Virol. 2002;76:12503–12512.
- 67 Horst D, Verweij MC, Davison AJ, Ressing ME, Wiertz EJ. Viral evasion of T cell immunity: ancient mechanisms offering new applications. Curr Opin Immunol. 2011;23:96–103.
- 68 Jochum S, Moosmann A, Lang S, Hammerschmidt W, Zeidler R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012;8:e1002704.
- 69 Hsu D-H, de Waal MR, Fiorentino DF, et al. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science. 1990;250:830–832.
- 70 Cohen JI, Lekstrom K. The Epstein-Barr virus BARF1 protein is dispensable for B cell transformation and inhibits interferon alpha secretion from mononuclear cells. J Virol. 1999;73:7627–7632.
- 71 Hislop AD, Ressing ME, van Leeuwen D, et al. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med. 2007 Aug 6;204(8):1863–1873.
- 72 Henderson S, Huen D, Rowe M, et al. Epstein-Barr virus-coded BHRF1 protein, a viral homolog of bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A. 1993;90:8479–8483.
- 73 Bellows DS, Howell M, Pearson C, et al. Epstein-Barr virus BALF1 is a bcl-2-like antagonist of the herpesvirus antiapoptotic bcl-2 proteins. J Virol. 2002;76:2469–2479.
- 74 Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–492.
- 75 Sayos J, Wu C, Morra N, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469.
- 76 Milone MC, Tsai DE, Hodinka RL. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell-directed therapy. Blood. 2005;105:994–996.
- 77 Cohen JI. Epstein-Barr virus lymphoproliferative disease associated with acquired immunodeficiency. Medicine. 1991;70:137–160.
- 78 Paya CV, Fung JI, Nalesnik MA, et al. Epstein-Barr virus induced posttransplant lymphoproliferative disorders. Transplantation. 1999;68:1517–1525.
- 79 Nourse JP, Jones K, Gandhi MK. Epstein-Barr virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transplant. 2011 May;11(5):888–895.
- 80 O’Reilly RJ, Small TN, Papadopoulos E, et al. Biology and adoptive cell therapy of Epstein-Barr virus-associated lymphoproliferative disorders in recipients of marrow allografts. Immunol Rev. 1997;157:195–216.
- 81 Rooney CM, Smith CA, Ng YC, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphomas in allogeneic transplant recipients. Blood. 1998;5:1549–1555.
- 82 Haque T, Wilkie GM, Taylor C, et al. Treatment of Epstein-Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet. 2002;360:436–442.
- 83 Haque T, Wilkie GM, Jones MM, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007;110:1123–1131.
- 84 Savoldo B, Goss JA, Hammer MM, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood. 2006;108:2942–2949.
- 85 Leen AM, Bollard CM, Mendizabal AM, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123.
- 86 Bollard CM, Rooney CM, Heslop HE. T-cell therapy in the treatment of post-transplant lymphoproliferative disease. Nat Rev Clin Oncol. 2012;9:510–519.
- 87 van Esser JW, Niesters HG, van der Holt B, et al. Prevention of Epstein-Barr virus-lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood. 2002;99:4364–4369.
- 88 Wagner HJ, Cheng YC, Huls MH, et al. Prompt versus preemptive intervention for EBV lymphoproliferative disease. Blood. 2004;103:3979–3981.
- 89 Grömminger S, Mautner J, Bornkamm GW. Burkitt lymphoma: the role of Epstein-Barr virus revisited. Br J Haematol. 2012;156:719–729.
- 90 Molyneux EM, Rochford R, Griffin B, et al. Burkitt’s lymphoma. Lancet. 2012;379:1234–1244.
- 91 Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–120.
- 92 Chien Y-C, Chen J-Y, Liu M-Y, et al. Serological markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N Engl J Med. 2001;345:1877–1882.
- 93 Lin JC, Wang WY, Chen KY, et al. Quantification of plasma Epstein-Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N Engl J Med. 2004;350:2461–2470.
- 94 Louis CU, Straathof K, Bollard CM, et al. Adoptive transfer of EBV-specific T cells results in sustained clinical responses in patients with locoregional nasopharyngeal carcinoma. J Immunother. 2010;33:983–990.
- 95 Hjalgrim H, Askling J, Rostgaard K, et al. Characteristics of Hodgkin’s lymphoma after infectious mononucleosis. N Engl J Med. 2003;349:1324–1332.
- 96 Glaser SL, Lin RJ, Stewart SL, et al. Epstein-Barr virus associated Hodgkin’s disease: epidemiologic characteristics in international data. Int J Cancer. 1997;70:375–382.
- 97 Timms JM, Bell A, Flavell JR, et al. Target cells of Epstein-Barr-virus (EBV)-positive post-transplant lymphoproliferative disease: similarities to EBV-positive Hodgkin’s lymphoma. Lancet. 2003;361:217–223.
- 98 Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808.
- 99 Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood. 2007;109:2571–2578.
- 100 Park S, Lee J, Ko YH, et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood. 2007;110:972–978.
- 101 Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808.
- 102 Chen C-L, Sadler RH, Walling DM, et al. Epstein-Barr virus gene expression in EBV-positive peripheral lymphomas. J Virol. 1993;67:6303–6308.
- 103 McClain KL, Leach CT, Jensen HB, et al. Association of Epstein-Barr virus with leiomyosarcomas in young people with AIDS. N Engl J Med. 1995;332:12–18.
- 104 Lee ES, Locker J, Nalesnik M, et al. The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation. N Engl J Med. 1995;332:19–25.
- 105 Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869.
- 106 Moses AV, Fish KN, Ruhl R, et al. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol. 1999;73:6892–6902.
- 107 Flore O, Rafii S, Ely S, et al. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature. 1998;394:588–592.
- 108 Damania B, Cesarman E. Kaposi’s sarcoma-associated herpesvirus. In: Knipe DM, Howley PM, Cohen JI, Griffith DE, Lamb RA, Martin MA, Racaniello V, Roizman B, eds. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2013:2080–2128.
- 109 Giffin L, Damania B. KSHV: pathways to tumorigenesis and persistent infection. Adv Virus Res. 2014;88:111–159.
- 110 Cesarman E. How do viruses trick B cells into becoming lymphomas? Curr Opin Hematol. 2014;21:358–368.
- 111 Sarid R, Flore O, Bohenzky RA, et al. Transcription map-ping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J Virol. 1998;72:1005–1012.
- 112 Lee H, Guo J, Li M, et al. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol Cell Biol. 1998;18:5219–5228.
- 113 Lee H, Veazey R, Williams K, et al. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat Med. 1998;4:435–440.
- 114 Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–1927.
- 115 Wang S, Maeng H, Young DP, et al. K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood. 2007;109:2174–2182.
- 116 Lagunoff M, Majeti R, Weiss A, Ganem D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1999;96:5704–5709.
- 117 Chatterjee M, Osborne J, Bestetti G, et al. Viral IL-6-induced cell proliferation and immune evasion of interferon activity. Science. 2002;298:1432–1435.
- 118 Moore PS, Bashoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response path-way genes by KSHV. Science. 1996;274:1739–1744.
- 119 Aoki Y, Jaffe ES, Chang Y, et al. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93:4034–4043.
- 120 Ishido S, Wang C, Lee B-S. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol. 2000;74:5300–5309.
- 121 Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–130.
- 122 Li Q, Means R, Lang S, et al. Downregulation of gamma interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J Virol. 2007 Mar;81(5):2117–2127.
- 123 Ishido S, Cook J-K, Lee B-S. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity. 2000;13:365–374.
- 124 Mansouri M, Douglas J, Rose PP. Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood. 2006;108:1932–1940.
- 125 Dairaghi DJ, Fan RA, McMaster BE, et al. HHV-8-encoded vMIP-1 selectively engages chemokine receptor CCR8: agonist and antagonist profiles of viral chemokines. J Biol Chem. 1999;274:21569–21574.
- 126 Stine JT, Wood C, Hill M, et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood. 2000;95:1151–1157.
- 127 Bashoff C, Endo Y, Collins PD, et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278:290–294.
- 128 Kledal TN, Rosenkilde MM, Coulin F, et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science. 1997;277:1656–1659.
- 129 Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J Virol. 2006;80:3092–3097.
- 130 Lagos D, Trotter MW, Vart RJ, et al. Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells. Blood. 2007;109:1550–1558.
- 131 Nakamura H, Li M, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J Virol. 2001;75:7572–7582.
- 132 Rivas C, Thlick AE, Parravicini C, et al. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B cell specific latent viral protein that inhibits p53. J Virol. 2001;75:429–438.
- 133 Li M, Lee H, Guo J, et al. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J Virol. 1998;72:5433–5440.
- 134 Burysek L, Pitha PM. Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J Virol. 2001;75:2345–2352.
- 135 Rivas C, Thlick A, Parravicini C, et al. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell–specific latent viral protein that inhibits p53. J Virol. 2001;75:429–438.
- 136 Zhu FX, King SM, Smith EJ, et al. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc Natl Acad Sci U S A. 2002;99:5573–5578.
- 137 Muralidhar S, Pumferry AM, Hassani M, et al. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J Virol. 1998;72:4980–4988.
- 138 McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307:739–741.
- 139 Yoo J, Kang J, Lee HN, et al. Kaposin-B enhances the PROX1 mRNA stability during lymphatic reprogramming of vascular endothelial cells by Kaposi’s sarcoma herpes virus. PLoS Pathog. 2010;6:e1001046.
- 140 Chung YH, Means RE, Choi JK, et al. Kaposi’s sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J Virol. 2002;76:4688–4698.
- 141 Spiller OB, Blackbourn DJ, Mark L, et al. Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J Biol Chem. 2003;278:9283–9289.
- 142 Cheng EH-Y, Nicholas J, Bellows DS, et al. A bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with bax or bak. Proc Natl Acad Sci U S A. 1997;94:690–694.
- 143 Sun Q, Zachariah S, Chaudhary PM. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-κB activation. J Biol Chem. 2003;278:52437–52445.
- 144 Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199:993–1003.
- 145 Djerbi M, Screpati V, Catrina AL, et al. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J Exp Med. 1999;190:1025–1031.
- 146 Chugh P, Matta H, Schamus S, et al. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc Natl Acad Sci U S A. 2005;102:12885–12890.
- 147 Grossmann C, Podgrabinska S, Skobe M, et al. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80:7179–7185.
- 148 Swanton C, Mann DJ, Fleckenstein B, et al. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. 1997;390:184–187.
- 149 Sarek G, Jarviluoma A, Ojala PM, et al. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood. 2006;107:725–732.
- 150 Chang Y, Moore PS, Talbot SJ, et al. Cyclin encoded by KS herpesvirus. Nature. 1996;382:410.
- 151 Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284:641–644.
- 152 Friborg J, Kong W, Hottiger MO, et al. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894.
- 153 Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene h-ras transforms primary rat cells. Nat Med. 2000;6:1121–1127.
- 154 Fujimuro M, Wu FY, ApRhys C, et al. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat Med. 2003;9:300–306.
- 155 Lu J, Jha HC, Verma SC, Sun Z, et al. Kaposi’s sarcoma-associated herpesvirus-encoded LANA contributes to viral latent replication by activating phosphorylation of survivin. J Virol. 2014;88:4204–4217.
- 156 Cai Q, Murakami M, Si H, Robertson ES. A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposi’s sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J Virol. 2007;81:10413–10423.
- 157 Liu J, Martin HJ, Liao G, et al. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol. 2007;81:10451–10459.
- 158 Fakhari FD, Jeong JH, Kanan Y, et al. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest. 2006;116:735–742.
- 159 Di Bartolo DL, Cannon M, Liu YF, et al. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood. 2008;111:4731–4740.
- 160 Arvanitakis L, Geras-Raaka E, Varma A, et al. Human herpesvirus KSHV encodes a constitutively active G-protein–coupled receptor linked to cell proliferation. Nature. 1997;385:347–350.
- 161 Bais C, Santomasso B, Coso O, et al. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391:86–89.
- 162 Sodhi A, Montaner S, Patel V, et al. Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor. Proc Natl Acad Sci U S A. 2004;101:4821–4826.
- 163 Ma T, Jham BC, Hu J, et al. Viral G protein-coupled receptor up-regulates Angiopoietin-like 4 promoting angiogenesis and vascular permeability in Kaposi’s sarcoma. Proc Natl Acad Sci U S A. 2010;107:14363–14368.
- 164 Yang BT, Chen SC, Leach MW. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191:445–454.
- 165 Grisotto MG, Garin A, Martin AP, et al. The human herpesvirus 8 chemokine receptor vGPCR triggers autonomous proliferation of endothelial cells. J Clin Invest. 2006;116:1264–1273.
- 166 Cannon M, Cesarman E, Boshoff C. KSHV G protein-coupled receptor inhibits lytic gene transcription in primary-effusion lymphoma cells via p21-mediated inhibition of Cdk2. Blood. 2006;107:277–284.
- 167 Glenn M, Rainbow L, Aurade F, et al. Identification of a spliced gene from Kaposi’s sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-Barr virus. J Virol. 1999;73:6953–6963.
- 168 Choi J-K, Lee B-S, Shim SN, et al. Identification of the novel K15 gene at the rightmost end of the Kaposi’s sarcoma-associated herpesvirus genome. J Virol. 2000;74:436–446.
- 169 Chugh PE, Sin SH, Ozgur S, et al. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013;9:e1003484.
- 170 Ganem D, Ziegelbauer J. MicroRNAs of Kaposi’s sarcoma-associated herpes virus. Semin Cancer Biol. 2008 Dec;18(6):437–440.
- 171 Samols MA, Skalsky RL, Maldonado AM, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. Plos Pathogens. 2007;35:e65.
- 172 Martin JN, Ganem DE, Osmond DH, et al. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med. 1998;338:948–954.
- 173 Simpson GR, Schulz TF, Whitby D, et al. Prevalence of Kaposi’s sarcoma-associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet. 1996;348:1133–1138.
- 174 Gao SJ, Kingsley L, Hoover DR, et al. Seroconversion of antibodies to Kaposi’s sarcoma-associated herpesvirus related latent nuclear antigens prior to onset of Kaposi’s sarcoma. N Engl J Med. 1996;335:233–241.
- 175 Marcelin AG, Motol J, Guihot A. Relationship between the quantity of Kaposi sarcoma-associated herpesvirus (KSHV) in peripheral blood and effusion fluid samples and KSHV-associated disease. J Infect Dis. 2007;196:1163–1166.
- 176 Cannon MJ, Dollard SC, Smith DK, et al. Blood-borne and sexual transmission of human herpesvirus 8 in women with or at risk for human immunodeficiency virus infection. N Engl J Med. 2001;344:637–743.
- 177 Regamey N, Tamm M, Wernli M, et al. Transmission of human herpesvirus 8 infection from renal-transplant donors to recipients. N Engl J Med. 1998;339:1358–1363.
- 178 Barozzi P, Luppi M, Facchetti F, et al. Post-transplant Kaposi sarcoma originates from the seeding of donor-derived progenitors. Nat Med. 2003;9:554–561.
- 179 Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and those without HIV infection. N Engl J Med. 1995;332:1181–1185.
- 180 Sullivan RJ, Pantanowitz L, Casper C, Stebbing J, Dezube BJ. HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma-associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47:1209–1215.
- 181 Dupin N, Fisher C, Kellam P, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96:4546–4551.
- 182 Glesby MJ, Hoover DR, Weng S, et al. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: data from the Multicenter AIDS Cohort Study. J Infect Dis. 1996;173:1477–1480.
- 183 Martin DF, Kuppermann BD, Wolitz RA, et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with ganciclovir implantRoche Ganciclovir Study Group. N Engl J Med. 1999;340:1063–1070.
- 184 Little RF, Merced-Galindez F, Staskus K, et al. A pilot study of cidofovir in patients with Kaposi sarcoma. J Infect Dis. 2003;187:149–153.
- 185 Cattelan AM, Calabro’ ML, Aversa SML, et al. Regression of AIDS-related Kaposi’s sarcoma following antiretroviral therapy with protease inhibitors: biological correlates of clinical outcome. Eur J Cancer. 1999;35:1809–1815.
- 186 Little RF, Aleman K, Kumar P. Phase 2 study of pegylated liposomal doxorubicin in combination with interleukin-12 for AIDS-related Kaposi sarcoma. Blood. 2007;110:4165–4171.
- 187 Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–1323.
- 188 Koon HB, Krown SE, Lee JY, et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol. 2014;32:402–408.
- 189 Uldrick TS, Wyvill KM, Kumar P, et al. Phase II study of bevacizumab in patients with HIV-associated Kaposi’s sarcoma receiving antiretroviral therapy. J Clin Oncol. 2012;30:1476–1483.
- 190 Cianfrocca M, Lee S, Von Roenn J, et al. Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer. 2010;116:3969–3977.
- 191 Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191.
- 192 Soulier J, Grollety L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–1280.
- 193 Katano H, Sato Y, Kurata T, et al. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology. 2000;269:335–344.
- 194 Uldrick TS, Polizzotto MN, Yarchoan R. Recent advances in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Curr Opin Oncol. 2012 Sep;24(5):495–505.
- 195 Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood. 2013;122:4189–4198.
- 196 Uldrick TS, Polizzotto MN, Aleman K, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117:6977–6986.