99 Hepatorenal Syndrome
An association between advanced liver disease, ascites, and renal failure was described as early as 1861. It is a form of renal failure occurring in the setting of severe liver disease. Helvig and Schutz gave this association its current name of hepatorenal syndrome in 1932.1 Shortly thereafter, hepatorenal syndrome (HRS) was found to be a functional form of renal failure without renal histologic changes.2 Significant advances in understanding the pathogenesis and treatment of the syndrome have been made in the past 2 decades. HRS is characterized by intense renal vasoconstriction, peripheral arterial vasodilation, impaired renal perfusion, and low glomerular filtration rate (GFR).3 The annual incidence of HRS is variably reported at 8% to 40% in patients with cirrhosis.4,5 The variability in incidence is related to the degree of liver dysfunction; the higher the Model for End-stage Liver Disease (MELD) score, the greater the incidence of HRS. HRS has a very high mortality, with nearly half the patients with type 1 HRS dying within 2 weeks of the diagnosis.4,6
 Mechanisms of Renal Dysfunction in Cirrhosis
Mechanisms of Renal Dysfunction in Cirrhosis
Impairment in Renal Sodium Metabolism without Activation of Vasoactive Systems
Chronologically, the first renal functional abnormality in cirrhosis is reduced ability to excrete sodium. When cirrhosis is still compensated (i.e., ascites is absent), subtle abnormalities in renal sodium metabolism already can be detected. Patients may not be capable of escaping from the effect of mineralocorticoids and develop continuous sodium retention. Arterial vasodilatation is already present in compensated cirrhosis with portal hypertension.7
With disease progression, the impairment in sodium handling increases. At a critical point, patients are unable to excrete the amount of sodium normally ingested in the diet. Sodium is retained and accumulates as ascites. Renal perfusion, GFR, the renal ability to excrete a free-water load, plasma renin activity, and the plasma concentrations of aldosterone and norepinephrine are normal.8
Stimulation of the Renin-Angiotensin and Sympathetic Nervous Systems and Antidiuretic Hormone with Preserved Renal Perfusion and Glomerular Filtration Rate
In cases of alcoholic cirrhosis, hepatic, circulatory, and renal function may improve if alcohol consumption is discontinued. In all other forms of cirrhosis and alcoholic cirrhosis with ongoing ethanol abuse, the degree of sodium retention increases progressively with progression of disease. When renal sodium avidity is extremely high, the plasma renin activity and the plasma concentrations of aldosterone and norepinephrine are elevated.7,9 Circulatory dysfunction is greater at this stage of the disease because increased activity of the sympathetic nervous system and the renin-angiotensin system is needed to maintain arterial pressure.
Renal perfusion and GFR are normal or moderately decreased, but renal perfusion is critically dependent on increased renal production of prostaglandins. These lipid mediators are vasodilators that antagonize the vasoconstricting actions of angiotensin II and norepinephrine. A syndrome indistinguishable from HRS can be produced in patients with cirrhosis, ascites, and increased plasma renin activity if prostaglandin synthesis is inhibited with nonsteroidal antiinflammatory drugs (NSAIDs).9,10 In addition, prostacyclin and nitric oxide cooperate to maintain renal perfusion in cirrhosis.11,12
 Pathogenesis
Pathogenesis
Development of HRS represents the terminal phase of the disease. HRS is characterized by low arterial blood pressure; marked increased plasma levels of renin, norepinephrine, and antidiuretic hormone; and very low GFR (<40 mL/min).2 Impairment in GFR in HRS occurs because of decreased renal perfusion secondary to renal vasoconstriction, peripheral vasodilation, and impairment in cardiac function.13 Renal histology is bland. Because renal vascular resistance correlates closely with activity of the renin-angiotensin and sympathetic nervous systems in cirrhosis,14–18 HRS is thought to be related to extreme stimulation of these systems.
Urinary excretion of prostaglandin E2, 6-keto-prostaglandin F1α (a prostacyclin metabolite), and kallikrein is decreased in patients with HRS, indicating that renal production of these substances is reduced.19,20 Renal failure in HRS, therefore, might be the consequence of an imbalance between the activity of vasoconstrictor systems and the renal production of vasodilators. The observation that HRS can be reproduced in nonazotemic, hyperreninemic, cirrhotic patients with ascites with NSAIDs is compatible with this hypothesis.10 Another possibility, however, is that renal vasoconstriction caused by the renin-angiotensin and sympathetic nervous systems is the primary cause of HRS.
Peripheral arterial vasodilation has been implicated in HRS, but vasodilation is mainly present in the splanchnic arterial vascular bed. Doppler ultrasonography studies have consistently shown arterial vasoconstriction in renal, brachial, femoral, and cerebral beds.21–22 Several endogenous vasodilators have been implicated as being responsible for splanchnic arteriolar vasodilation, including nitric oxide, carbon monoxide, glucagon, prostacyclin, and endogenous opiates.23–25
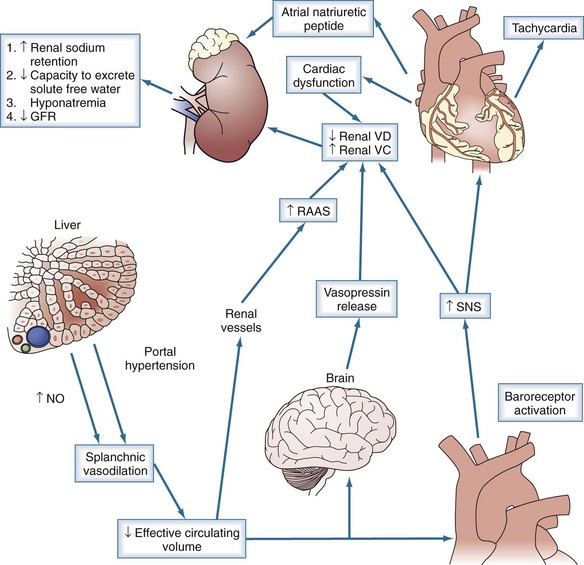
End-stage liver disease is associated with reduced systolic and diastolic response to stress, enlarged cardiac chambers, and repolarization changes, termed cirrhotic cardiomyopathy.26 The development of HRS has been associated with a lower arterial pressure, a marked decrease in cardiac output, and increase in plasma renin activity and plasma norepinephrine.20 The decrease in cardiac output is likely related to decreased effective circulating volume, as evidenced by low filling pressures and improvement with volume expansion; however, further studies are warranted (Figure 99-1).
 Diagnosis
Diagnosis
The first step in the diagnosis of HRS is demonstration of reduced GFR, and this is not easy in advanced cirrhosis.2,27 Muscle mass and, therefore, the release of creatinine is considerably reduced in these patients, and they can have a normal serum creatinine concentration despite having a very low GFR. Similarly, urea is synthesized by the liver, and urea synthesis may be reduced as a consequence of hepatic insufficiency, so failure to appropriately diagnosis HRS is relatively common.28,29
In 1996 and again in 2006, the International Ascites Club proposed different diagnostic criteria of HRS.31,32 Serum creatinine concentration should be greater than 1.5 mg/dL in the absence of other potential causes of renal failure (Table 99-1).30 The diagnosis of HRS requires exclusion of other causes of renal failure in cirrhotic patients. These other causes of renal dysfunction include prerenal failure related to diuretics or lactulose, and acute kidney injury in the setting of shock. Additionally, the use of nephrotoxic medications such as aminoglycosides, NSAIDs, and vasodilators should be excluded as the cause of renal failure. When interpreting results, it is important to note that much of the research cited was performed prior to the revision in definition.
TABLE 99-1 Major Diagnostic Criteria of Hepatorenal Syndrome (International Ascites Club)
Clinical Types
HRS is classified into two types according to the severity and form of presentation of renal failure.32 HRS type 1 is characterized by severe and rapidly progressive renal failure. It has been defined by doubling of the serum creatinine concentration to at least 2.5 mg/dL in less than 2 weeks. Although HRS type 1 may arise spontaneously, it frequently occurs in close relationship with a precipitating factor such as severe bacterial infection, gastrointestinal hemorrhage, major surgical procedure, or acute hepatitis superimposed on cirrhosis. The association of HRS and spontaneous bacterial peritonitis (SBP) has been carefully investigated.33–35 HRS type 1 develops in approximately 30% of patients with SBP despite rapid and successful treatment of the infection with non-nephrotoxic antibiotics. Patients with an intense systemic inflammatory response and high cytokine levels in plasma and ascitic fluid are especially prone to develop HRS type 1 after infection. Patients with HRS type 1 after SBP show signs and symptoms of severe liver failure and circulatory dysfunction that worsen with the impairment in renal function, evolving to multiorgan failure.36 HRS type 1 is the complication of cirrhosis with the poorest prognosis, with a 2-week median survival.
HRS type 2 is characterized by a moderate and steady decrease in renal function (serum creatinine < 2.5 mg/dL). Patients with HRS type 2 show signs of liver failure and arterial hypotension but to a lesser degree than patients with HRS type 1. The dominant clinical feature is severe ascites with poor or no response to diuretics, a condition known as refractory ascites. Patients with HRS type 2 are especially predisposed to develop HRS type 1 after infections or other precipitating events.33–35 The median survival of patients with HRS type 2 is 6 months, and it is worse than for patients with nonazotemic cirrhosis with ascites.
 Treatment
Treatment
Treating the underlying etiology with liver or combined liver-kidney transplant is the goal of therapy. Given the pathophysiology of extreme renal vasoconstriction, splanchnic vasodilation, and decreased cardiac output, many vasoactive drugs have been evaluated as therapeutic agents to reverse HRS. To date, no single therapeutic agent has been found to permanently reverse HRS. As such, the current goals in treatment are as a bridge to hepatic transplantation and possibly improved long-term survival. Dopamine, fenoldopam, endothelin antagonists, natriuretic peptides, and angiotensin-converting enzyme (ACE) inhibitors have been shown to either have no benefit or worsen the outcome of HRS.37
Liver Transplantation
Liver transplantation is the treatment of choice for HRS.38–42 Immediately after transplantation, further impairment in GFR may be observed, and many patients require renal replacement therapy; 35% of patients with HRS compared with 5% of patients without HRS require renal replacement therapy.38 Because cyclosporine or tacrolimus can contribute to impaired renal function, it has been suggested that administration of these drugs should be delayed until renal function begins to recover, usually 48 to 72 hours after transplantation.43 After the initial deterioration in renal function, GFR starts to improve and reaches an average of 30 to 40 mL/min by 1 to 2 months postoperatively. This level of moderate renal failure persists during follow-up and is more marked than is observed after hepatic transplantation in patients without HRS.32 The hemodynamic and neurohormonal abnormalities associated with HRS disappear within the first month after the operation, and patients regain normal sodium and free-water clearance.
Patients with HRS who undergo transplantation have more complications, spend more days in the ICU, and have a higher in-hospital mortality rate than transplantation patients without HRS.38–42 The long-term survival of patients with HRS after liver transplantation, however, is good. The 3-year probability of survival is 60%.38–42 This survival rate is only slightly less than survival rates for liver transplant recipients without HRS (70% and 80%).38,41
Volume Expansion and Vasoconstrictors
Monotherapy with either albumin or vasoconstrictor has not been as effective as combined therapy. Martin-Lalhi and colleagues44 randomized 46 patients to terlipressin plus albumin or albumin alone. Improvement in renal function was better with combination therapy (43.5% versus 8.7% P = .017). Conversely, Ortega and colleagues45 randomized 21 patients to terlipressin with or without albumin, with an improved response with the addition of albumin (77% versus 25% P = .03). One-month survival without transplantation was 87% in patients receiving terlipressin plus albumin and 13% in patients receiving terlipressin alone. Typical dosing of albumin, and as recommended by the consensus panel, is 1 g/kg on day 1 of therapy, using 25% albumin as the preferred formulation of the colloid.
Vasopressin is an endogenous hormone with three major identified receptors. The V1 receptor, found on vascular smooth muscle, promotes vasoconstriction. The V2 receptor is involved in osmoregulation in the kidney. The V3 receptor affects corticotropin secretion. The V1 receptor has been the target of interest for vasopressin analogs designed to increase splanchnic vasoconstriction.37,46 The original studies were conducted with ornipressin, but the recent focus has been on terlipressin, which has a greater effect on the V1 receptor and fewer side effects.47 Two meta-analyses have shown improved outcome with the use of terlipressin versus placebo.47,48 Fabrizi and colleagues47 analyzed 10 clinical trials and found reversal of HRS in 52% of cases, with a 29% incidence of side effects, most of which responded to reducing the dose of terlipressin. Dobre and colleagues48 identified eight eligible trials which enrolled a total of 320 patients. Four of the studies compared terlipressin to placebo, with an improvement in the terlipressin group with regard to several outcomes, including reversal of HRS (OR of 7.47), improvement in mean arterial pressure, improvement in urine output, and reduction of serum creatinine. Sanyal and colleagues49 conducted a multicenter randomized trial of 112 patients. The terlipressin group received 1 mg of the drug every 6 hours and was more likely to have reversal of HRS compared with placebo (34% versus 12.5%, P = .008). Importantly, a subgroup of patients who received terlipressin for more than 3 days had a greater response to therapy compared to placebo (52.8% versus 18%, P = .002). These data support the contention that length of therapy may contribute to some of the variability in efficacy of therapy. Sanyal et al.50 also showed that earlier therapy increases probability of reversal.
Terlipressin and intravenous (IV) albumin seems to be a promising therapy for type 1 HRS. The dose ranges used vary from 1 to 2 mg every 4 to 6 hours. One algorithm used starts terlipressin at 1 mg every 6 hours until the serum creatinine decreases to less than 1.5 mg/dL on two measurements. If there is no improvement in creatinine concentration after 3 days of therapy, the dose is increased to 2 mg every 6 hours.49 A maximal dose of 12 mg a day has been proposed. The proposed minimum duration of therapy is 3 to 5 days.46 Early initiation of therapy and close monitoring are important. There are varying data on the rates of recurrence of HRS after discontinuation of therapy; estimates range from 5.3% recurrence rate to 50% recurrence rate.36,37,46 However, recurrence with terlipressin is less frequent than with placebo; survival with terlipressin is improved. Further large multicenter studies to evaluate dosing and time for treatment are pending.
α-Adrenergic agonists also have been used in an effort to augment renal perfusion. Duvoux and coworkers51 treated 12 patients with HRS type 1 with IV albumin (to maintain central venous pressure >7 mm Hg) and norepinephrine (0.5 to 3 mg/h) for a minimum of 5 days. A significant improvement in serum creatinine concentration in association with a marked suppression of plasma renin activity was observed in 10 patients. Transient myocardial ischemia was observed in one patient. Three patients underwent transplantation, and three were still alive after 8 months of follow-up. Two randomized control studies have compared norepinephrine to terlipressin.52,53 In the first (n = 22), reversal of HRS occurred in 70% of the norepinephrine group versus 83% of the terlipressin group (P = NS). In the second (n = 20), norepinephrine was effective for increasing mean arterial pressure (MAP), increasing urine output, and decreasing serum creatinine concentration; the efficacy of norepinephrine was not significantly different from the efficacy of terlipressin. There were no differences in outcomes or the incidence of adverse events in either study. Treatment algorithms target an increase in MAP of 10 mm Hg.
Angeli and associates54 used oral midodrine, an α-adrenergic agonist, IV albumin, and subcutaneous octreotide (a somatostatin analog to suppress glucagon) in five patients with HRS type 1. Midodrine dosage was adjusted to increase MAP by more than 15 mm Hg. Patients received treatment for at least 20 days in hospital and subsequently continued treatment at home. In all cases, there was a dramatic improvement in renal perfusion, GFR, blood urea nitrogen concentration, serum creatinine concentration, and serum sodium concentration. Plasma levels of renin, aldosterone, and antidiuretic hormone decreased to normal or near-normal levels. Two patients were transplanted 20 and 64 days after enrollment while on therapy. One patient who was not a candidate for liver transplantation was alive without treatment 472 days after being discharged from the hospital. The remaining two patients died 29 and 75 days after enrollment. The control group received dopamine therapy, with seven of the eight patients dying by day 12. Esrailian and colleagues55 retrospectively evaluated 60 patients, comparing midodrine plus octreotide to untreated controls. Forty percent of treated patients had an improvement in renal function as compared to 10% of controls (P = .03). In addition, treatment with midodrine plus octreotide was associated with an improvement in 30-day mortality (43% versus 71%, P = .03). Their data showed that patients who received the highest dose of 15 mg 3 times a day were more likely to respond to therapy. In a prospective observational study, Skagen56 and colleagues studied midodrine and octreotide in both type 1 and type 2 HRS. The treatment group of 75 patients was compared to historical controls. The 1-month GFR was improved in the treatment group (48 versus 34 mL/min, P = .03). Median survival also was improved for type 1 HRS (40 versus 17 days, P = .007) and type 2 HRS (>12 months versus 22 days, P = .0004). Studies of monotherapy of octreotide have not shown benefit over placebo. The combination of midodrine plus octreotide is a promising regimen for HRS 2 patients and for patients who are treated as outpatients.
These studies show the following:
Transjugular Intrahepatic Portosystemic Shunt
Because portal hypertension is the initial abnormality with regard to circulatory dysfunction in cirrhosis, decreasing portal pressure by portosystemic anastomosis is a rational approach for the treatment of HRS. There are several case reports showing reversal of HRS after surgical portosystemic shunt.57,58 However, major surgical procedures in patients with HRS are not likely to be tolerated well. The development of transjugular intrahepatic portosystemic shunt (TIPS) has reinvigorated the idea of treating HRS by reducing portal pressure.
Several studies assessing TIPS in the management of HRS type 1 have been reported.59–61 The first study included 14 patients with type 1 HRS who were not candidates for transplantation. At 3, 6, and 12 months after TIPS, survival rates were 54%, 50%, and 20%, respectively.59 In one study, which specifically investigated the effect of TIPS on neurohormonal systems, improvement in GFR and serum creatinine concentration was related to marked suppression of the plasma levels of renin and antidiuretic hormone.60 The most recent study included 14 patients with type 1 HRS who were initially treated with midodrine and octreotide.62 Ten of the 14 patients responded to vasoconstrictor therapy, and five subsequently underwent TIPS. These patients had an improvement in renal function, sodium excretion, and portosystemic gradient. These studies strongly suggest that TIPS is useful in the management of HRS type 1 and may be beneficial in combination with vasoconstrictor therapy.
Other Therapeutic Methods
Hemodialysis and arteriovenous or venovenous hemofiltration are frequently used in patients with HRS. Extracorporeal albumin dialysis uses an albumin-containing dialysate that is recirculated and perfused through charcoal and anion-exchanger columns. This modality has been shown to improve systemic hemodynamics and reduce plasma levels of renin in patients with HRS type 1.63,64 In a small series of patients, improved survival was reported.62 Newer modalities such as the Prometheus system and single-pass albumin dialysis have been used in a few patients with some success.65,66 Further studies are needed to confirm these findings.
 Prevention
Prevention
Three randomized controlled studies enrolling large series of patients have shown that HRS can be prevented in specific clinical settings. In the first study,67 albumin (1.5 g/kg IV at infection diagnosis and 1 g/kg IV 48 hours later) together with cefotaxime was compared to cefotaxime alone in patients with cirrhosis and SBP. Treatment with albumin markedly reduced the incidence of impaired circulatory function and the occurrence of HRS type 1. Moreover, the hospital mortality rate (10% versus 29%) and the 3-month mortality rate (22% versus 41%) were lower in patients receiving albumin plus antibiotics versus antibiotics alone. The second study showed that oral prophylaxis using norfloxacin decreased the 1-year probability of developing SBP and type 1 HRS and improved survival.68 In a third study,68 administration of the tumor necrosis factor synthesis inhibitor, pentoxifylline (400 mg TID), to patients with severe acute alcoholic hepatitis reduced the occurrence of HRS (8% in the pentoxifylline group versus 35% in the placebo group) and hospital mortality (24% versus 46%, respectively). Because bacterial infections and acute alcoholic hepatitis are two important precipitating factors of HRS type 1, these prophylactic measures may decrease the incidence of this complication.
 Conclusion
ConclusionKey Points
Salerno F, Gerbes A, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310-1318.
Ginés A, Escorsell A, Ginés P, et al. Incidence, predictive factors, and prognosis of hepatorenal syndrome in cirrhosis. Gastroenterology. 1993;105:229-236.
Sanyal A, Boyer T, et al. A randomized, prospective, double-blind, placebo controlled trial of terlipressin for type 1 hepatorenal syndrome. Dig Dis Sci. 2008;53:830-835.
Sort P, Navasa M, Arroyo V, et al. Effect of plasma volume expansion on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341:403-409.
Alessandria C, Ottobrelli A, et al. Noradrenalin vs. terlipressin in patients with hepatorenal syndrome: a prospective, randomized, unblinded, pilot study. J Hepatol. 2007;47:499-505.
1 Helvig FC, Shutz CB. A liver and kidney syndrome: clinical, pathological, and experimental studies. Surg Gynecol Obstet. 1932;55:570-582.
2 Hecker R, Sherlock S. Electrolyte and circulatory changes in terminal liver failure. Lancet. 1956;2:1121-1129.
3 Turban S, Thuluvath P, Atta M. Hepatorenal syndrome. World J Gastroenterol. 2007;13:4046-4055.
4 Gines A, Escorell A, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993;105:229-236.
5 Fernandez J, Navasa M, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-824.
6 Nguyen GC, Sergev D. Nationwide increase in hospitalizations and hepatitis C among inpatients with cirrhosis and sequelae of portal hypertension. Clin Gastroenterol Hepatol. 2007;5:1092-1099.
7 Bosch J, Arroyo V, Betriu A, et al. Hepatic hemodynamics and the renin-angiotensin aldosterone system in cirrhosis. Gastroenterology. 1980;78:92-99.
8 Saló J, Ginés A, Anibarro L, et al. Effect of upright posture and physical exercise on endogenous neurohormonal systems in cirrhotic patients with sodium retention and normal plasma renin, aldosterone and norepinephrine levels. Hepatology. 1995;22:479-487.
9 Arroyo V, Planas R, Gaya J, et al. Sympathetic nervous activity, renin-angiotensin system and renal excretion of prostaglandin E2 in cirrhosis: Relationship to functional renal failure and sodium and water excretion. Eur J Clin Invest. 1983;13:271-278.
10 Arroyo V, Ginés P, Rimola A, Gaya J. Renal function abnormalities, prostaglandins, and effects of nonsteroidal anti-inflammatory drugs in cirrhosis with ascites: An overview with emphasis on pathogenesis. Am J Med. 1986;81:104-122.
11 Ros J, Claria J, Jiménez W, et al. Role of nitric oxide and prostacyclin in the control of renal perfusion in experimental cirrhosis. Hepatology. 1995;22:915-920.
12 Angeli P, Jiménez W, Arroyo V, et al. Renal effects of endogenous natriuretic peptide receptors blockade in cirrhotic rats with ascites. Hepatology. 1994;20:948-954.
13 Epstein M, Berk DP, Hollenberg NK, et al. Renal failure in the patient with cirrhosis: The role of active vasoconstriction. Am J Med. 1970;49:175-185.
14 Schroeder ET, Eich RH, Smulyan H, et al. Plasma renin levels in hepatic cirrhosis: Relationship to functional renal failure. Am J Med. 1970;49:186-191.
15 Maroto A, Ginés A, Salo J, et al. Diagnosis of functional renal failure of cirrhosis by Doppler sonography: Prognostic value of resistive index. Hepatology. 1994;20:839-844.
16 Fernández-Seara J, Prieto J, Quiroga J, et al. Systemic and regional hemodynamics in patients with liver cirrhosis and ascites with and without functional renal failure. Gastroenterology. 1989;97:1304-1312.
17 DiBona GF. Renal nerve activity in hepatorenal syndrome. Kidney Int. 1984;25:841-853.
18 Henriksen JH, Ring-Larsen H. Hepatorenal disorders: role of the sympathetic nervous system. Semin Liver Dis. 1994;116:446-455.
19 Rimola A, Ginés P, Arroyo V, et al. Urinary excretion of 6-keto-prostaglandin F1-alpha, thromboxane B2 and prostaglandin E2 in cirrhosis with ascites: Relationship to functional renal failure (hepatorenal syndrome). J Hepatol. 1986;3:111-117.
20 Ruiz del Arbol, Monescillo A, et al. Circulatory function and hepato-renal syndrome. Hepatology. 2005;42:439-447.
21 Angeli P, Merkel C. Pathogenesis and management of hepatorenal syndrome in patients with cirrhosis. J Hepatol. 2008;4:S93-103.
22 Sugano S, Yamamoto H, et al. Postprandial middle cerebral arterial vasoconstriction in cirrhotic patients. A placebo controlled evaluation. J Hepatol. 2001;34:373-377.
23 Wiest R, Groaszmann RJ, et al. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology. 2002;35:478-491.
24 Bolognesi M, Sacredoti D, et al. Carbon monoxide-mediated activation of large conductance calcium-activated potassium channels contributes to mesenteric vasodilation in cirrhotic rats. J Pharmacol Exp Ther. 2007;321:187-194.
25 Angeli P, Volpin R, et al. The role of nitric oxide in the pathogenesis of systemic and splanchnic vasodilation in cirrhotic rats before and after the onset of ascites. Liver. 2005;29:429-437.
26 Tsai M, Peng Y, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: effect of treatment with hydrocortisone on survival. Hepatology. 2006;44:1288-1295.
27 Orr TG, Helwing FC. Liver trauma and the hepatorenal syndrome. Ann Surg. 1939;110:683-692.
28 Servin-abad L, Regev A, et al. retrospective analysis of 140 patients labeled as hepatorenal syndrome in a referral centr. Hepatology. 2005;42:543.
29 Watt K, Uhanova J, et al. Hepatorenal syndrome: diagnostic accuracy, clinical features and outcome in a tertiary care center. Am J Gastroenterol. 97, 2002. 2056–2050
30 Wadei HM, Marin M, et al. Hepatorenal syndrome: pathophysiology and management. Clin. J Am Soc Nephrol.. 2006;1:1066-1079.
31 Arroyo V, Ginés P, Gerbes A, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. Hepatology. 1996;23:164-176.
32 Salerno F, Gerbes A, et al. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut. 2007;56:1310-1318.
33 Toledo C, Salmerón JM, Rimola A, et al. Spontaneous bacterial peritonitis in cirrhosis: Predictive factors of infection resolution and survival in patients treated with cefotaxime. Hepatology. 1993;17:251-257.
34 Follo A, Llovet JM, Navasa M, et al. Renal impairment following spontaneous bacterial peritonitis in cirrhosis: Incidence, clinical course, predictive factors and prognosis. Hepatology. 1994;20:1495-1501.
35 Navasa M, Follo A, Filella X, et al. Tumor necrosis factor and inter-leukin-6 in spontaneous bacterial peritonitis in cirrhosis: Relationship with the development of renal impairment and mortality. Hepatology. 1998;27:1227-1232.
36 Arroyo V, Terra C, Gens P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol. 2007;46:935-946.
37 Kiser T, MacLaren R, et al. Treatment of hepatorenal syndrome. Pharmacotherapy. 2009;29:1196-1211.
38 Gonwa TA, Morris CA, Goldstein RM, et al. Long-term survival and renal function following liver transplantation in patients with and without hepatorenal syndrome—experience in 300 patients. Transplantation. 1991;91:428-430.
39 Lerut J, Goffette P, Laterre PF, et al. Sequential treatment of hepatorenal syndrome and posthepatic cirrhosis by intrahepatic portosystemic shunt (TIPS) and liver transplantation. Hepatogastroenterology. 1995;42:985-987.
40 Gonwa TA, Klintmalm GB, Jennings LS, et al. Impact of pretransplant renal function on survival after liver transplantation. Transplantation. 1995;59:361-365.
41 Seu P, Wilkinson AH, Shaked A, Busuttil BW. The hepatorenal syndrome in liver transplant recipients. Am Surg. 1991;57:806-809.
42 Rimola A, Gavaler JS, Schade RR, et al. Effects of renal impairment on liver transplantation. Gastroenterology. 1987;93:148-156.
43 Lopez L, Villanueva F, et al. Evolution of hepatorenal syndrome after orthotopic liver transplantation: comparative analysis with patients who developed acute renal failure in the early postoperative period of liver transplantation. Transplant Proc. 2007;39:2318-2319.
44 Martin-Llahi M, Pepin MN, et al. Terlipressin and albumin vs albumin in patients with cirrhosis and hepatorenal syndome: a randomized study. Gastroenterology. 2008;134:1352-1359.
45 Ortega R, Gines P, et al. Terlipressin therapy with and without albumin for patients with hepatorenal syndome: results of a prospective, nonrandomized study. Hepatology. 2002;36:941-948.
46 Munoz S. The hepatorenal syndrome. Med Clin N Am. 2008;92:813-837.
47 Fabrizi F, Dixit V, et al. Meta-analysis: terlipressin therapy for the hepatorenal syndrome. Aliment Pharmacol Ther. 2006;24:935-944.
48 Dobre M, Demirjian S, et al. Terlipressin in hepatorenal syndrome: a systematic review and meta-analysis. Int Urol Nephrol. 2010. March Epub
49 Sanyal A, Boyer T, et al. A randomized, prospective, double-blind, placebo controlled trial of terlipressin for type 1 hepatorenal syndrome. Dig Dis Sci. 2008;53:830-835.
50 Sanyal A, Boyer T, et al. Prognostic factors for hepatorenal syndrome reversal in patients with type 1 HRS enrolled in a randomized double blind placebo controlled trial. Hepatology. 2007;46:564.
51 Duvoux C, Zanditenas D, Hezode C, et al. Effects of noradrenalin and albumin in patients with hepatorenal syndrome: A pilot study. Hepatology. 2002;36:374-380.
52 Alessandria C, Ottobrelli A, et al. Noradrenalin vs terlipressin in patients with hepatorenal syndrome: a prospective, randomized, unblinded, pilot study. J Hepatol. 2007;47:499-505.
53 Sharma P, Kumar A, et al. An open label, pilot, randomized controlled trial of noradrenaline versus terlipressin in the treatment of type-1 hepatorenal syndrome and predictors of response. Am J Gastroenterol. 2008;103:1689-1697.
54 Angeli P, Volpin R, Gerunda G, et al. Reversal of type 1 hepatorenal syndrome with the administration of midodrine and octreotide. Hepatology. 1999;29:1690-1697.
55 Esrailian E, Pantangco ER, et al. Octreotide/midodrine therapy significantly improves renal function and 30-day survival in patients with type 1 hepatorenal syndrome. Dig Dis Sci. 2007;52:742-748.
56 Skagen C, Einstein M, et al. Combination treatment with octreotide, midodrine, and albumin improves survival in patients with type 1 and type 2 hepatorenal syndrome. J Clin Gastroenterol. 2009;43:680-685.
57 Schroeder ET, Numann PJ, Chamberlain BE. Functional renal failure in cirrhosis: Recovery after portacaval shunt. Ann Intern Med. 1970;72:293-298.
58 Ariyan S, Sweeney T, Kerstein MD. The hepatorenal syndrome: Recovery after portacaval shunt. Ann Surg. 1975;181:847-849.
59 Brensing KA, Textro J, Perz J, et al. Long-term outcome after trans-jugular intrahepatic portosystemic stent-shunt in non-transplant patients with hepatorenal syndrome: A phase II study. Gut. 2000;47:288-295.
60 Guevara M, Ginés P, Bandi JC, et al. Transjugular intrahepatic portosystemic shunt in hepatorenal syndrome: Effects on renal function and vasoactive systems. Hepatology. 1998;28:416-422.
61 Wong F, Pantea L, et al. Midodrine, octreotide, albumin and TIPS in selected patients with cirrhosis and type 1 hepatorenal syndrome. Hepatology. 2004;40:55-64.
62 Mitzner SR, Stange J, Klammt S, et al. Improvement of hepatorenal syndrome with extracorporeal albumin dialysis MARS: Results of a prospective, randomized controlled clinical trial. Liver Transpl. 2000;6:277-286.
63 Sorkine P, Abraham RB, Szold O, et al. Role of molecular adsorbent recycling system (MARS) in the treatment of acute exacerbation of chronic liver failure. Crit Care Med. 2001;29:1332-1336.
64 Rifai K, Ernst T, et al. The Prometheus device for extracorporeal support of combined liver and renal failure. Blood Purif. 2005;23:298-302.
65 Rahman E, Al Suwaida AK, Askar A. Single-pass albumin dialysis in hepatorenal syndrome. Saudi J Kidney Dis Transpl. 2008;19:479-484.
66 Sort P, Navasa M, Arroyo V, et al. Effect of plasma volume expansion on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341:403-409.
67 Navasa M, Fernandez J, et al. Randomized, double-blind placebo-controlled trial evaluating norfloxacin the primary prophylaxis of spontaneous bacterial peritonitis in cirrhotics with renal impairment, hyponatremia or severe liver failure. J Hepatol. 2006;44:S51.
68 Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: A double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637-1648.
