Diuretics
Hydrochlorothiazide
Spironolactone
Beta-Adrenergic Blocker
Metoprolol
Inhibitors of the Renin-Angiotensin-Aldosterone System
Captopril (angiotensin-converting enzyme inhibitor)
Losartan (angiotensin II receptor blocker)
Aliskiren (direct renin inhibitor)
Eplerenone (aldosterone antagonist)
Calcium Channel Blockers
Verapamil
Nifedipine
Review of Blood Pressure Control
Before discussing the antihypertensive drugs, we need to review the major mechanisms by which BP is controlled. This information will help you understand the mechanisms by which drugs lower BP.
Principal Determinants of Blood Pressure
The principal determinants of BP are shown in Fig. 39.1. As indicated, arterial pressure is the product of cardiac output and peripheral resistance. An increase in either will increase BP.
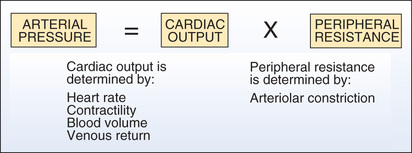
Cardiac Output.
Cardiac output is influenced by four factors: (1) heart rate, (2) myocardial contractility (force of contraction), (3) blood volume, and (4) venous return of blood to the heart. An increase in any of these will increase cardiac output, thereby causing BP to rise. Conversely, a decrease in these factors will make BP fall. Hence, to reduce BP, we might give a beta blocker to reduce cardiac output, or a diuretic to reduce blood volume, or a venodilator to reduce venous return.
Peripheral Vascular Resistance.
Vascular resistance is increased by arteriolar constriction. Accordingly, we can reduce BP with drugs that promote arteriolar dilation.
Systems that Help Regulate Blood Pressure
Having established that BP is determined by heart rate, myocardial contractility, blood volume, venous return, and arteriolar constriction, we can now examine how these factors are regulated. Three regulatory systems are of particular significance: (1) the sympathetic nervous system, (2) the renin-angiotensin-aldosterone system (RAAS), and (3) the kidney.
Sympathetic Baroreceptor Reflex.
The sympathetic nervous system employs a reflex circuit—the baroreceptor reflex—to keep BP at a preset level. This circuit operates as follows:
1. Baroreceptors in the aortic arch and carotid sinus sense BP and relay this information to the brainstem.
2. When BP is perceived as too low, the brainstem sends impulses along sympathetic nerves to stimulate the heart and blood vessels.
3. BP is then elevated by (a) activation of beta1 receptors in the heart, resulting in increased cardiac output; and (b) activation of vascular alpha1 receptors, resulting in vasoconstriction.
4. When BP has been restored to an acceptable level, sympathetic stimulation of the heart and vascular smooth muscle subsides.
The baroreceptor reflex frequently opposes our attempts to reduce BP with drugs. Opposition occurs because the “set point” of the baroreceptors is high in people with hypertension. That is, the baroreceptors are set to perceive excessively high BP as “normal” (i.e., appropriate). As a result, the system operates to maintain BP at pathologic levels. Consequently, when we attempt to lower BP using drugs, the reduced (healthier) pressure is interpreted by the baroreceptors as below what it should be, and, in response, signals are sent along sympathetic nerves to “correct” the reduction. These signals produce reflex tachycardia and vasoconstriction—responses that can counteract the hypotensive effects of drugs. Clearly, if treatment is to succeed, the regimen must compensate for the resistance offered by this reflex. Taking a beta blocker, which will block reflex tachycardia, can be an effective method of compensation. Fortunately, when BP has been suppressed with drugs for an extended time, the baroreceptors become reset at a lower level. Consequently, as therapy proceeds, sympathetic reflexes offer progressively less resistance to the hypotensive effects of medication.
Renin-Angiotensin-Aldosterone System.
The RAAS can elevate BP, negating the hypotensive effects of drugs. The RAAS is discussed in Chapter 36 and reviewed briefly here.
The RAAS elevates BP beginning with the release of renin from juxtaglomerular cells of the kidney. These cells release renin in response to reduced renal blood flow, reduced blood volume, reduced BP, and activation of beta1-adrenergic receptors on the cell surface. After its release, renin catalyzes the conversion of angiotensinogen into angiotensin I, a weak vasoconstrictor. After this, angiotensin-converting enzyme (ACE) acts on angiotensin I to form angiotensin II, a compound that constricts systemic and renal blood vessels. Constriction of systemic blood vessels elevates BP by increasing peripheral resistance. Constriction of renal blood vessels elevates BP by reducing glomerular filtration, which causes retention of salt and water, which in turn increases blood volume and BP. In addition to causing vasoconstriction, angiotensin II causes release of aldosterone from the adrenal cortex. Aldosterone acts on the kidney to further increase retention of sodium and water.
Because drug-induced reductions in BP can activate the RAAS, this system can counteract the effect we are trying to achieve. We have five ways to cope with this problem. First, we can suppress renin release with beta blockers. Second, we can prevent conversion of angiotensinogen to angiotensin I with a direct renin inhibitor (DRI). Third, we can prevent the conversion of angiotensin I into angiotensin II with an ACE inhibitor. Fourth, we can block receptors for angiotensin II with an angiotensin II receptor blocker (ARB). And fifth, we can block receptors for aldosterone with an aldosterone antagonist.
Renal Regulation of Blood Pressure.
As discussed in Chapter 34, the kidney plays a central role in long-term regulation of BP. When BP falls, glomerular filtration rate (GFR) falls too, thereby promoting retention of sodium, chloride, and water. The resultant increase in blood volume increases venous return to the heart, causing an increase in cardiac output, which in turn increases arterial pressure. We can neutralize renal effects on BP with diuretics.
Antihypertensive Mechanisms: Sites of Drug Action and Effects Produced
As discussed previously, drugs can lower BP by reducing heart rate, myocardial contractility, blood volume, venous return, and the tone of arteriolar smooth muscle. In this section we survey the principal mechanisms by which drugs produce these effects.
The major mechanisms for lowering BP are shown in Fig. 39.2 and Table 39.2. The figure depicts the principal sites at which antihypertensive drugs act. The table shows the effects elicited when drugs act at these sites. The numbering system used in the following text corresponds with the system used in Fig. 39.2 and Table 39.2.
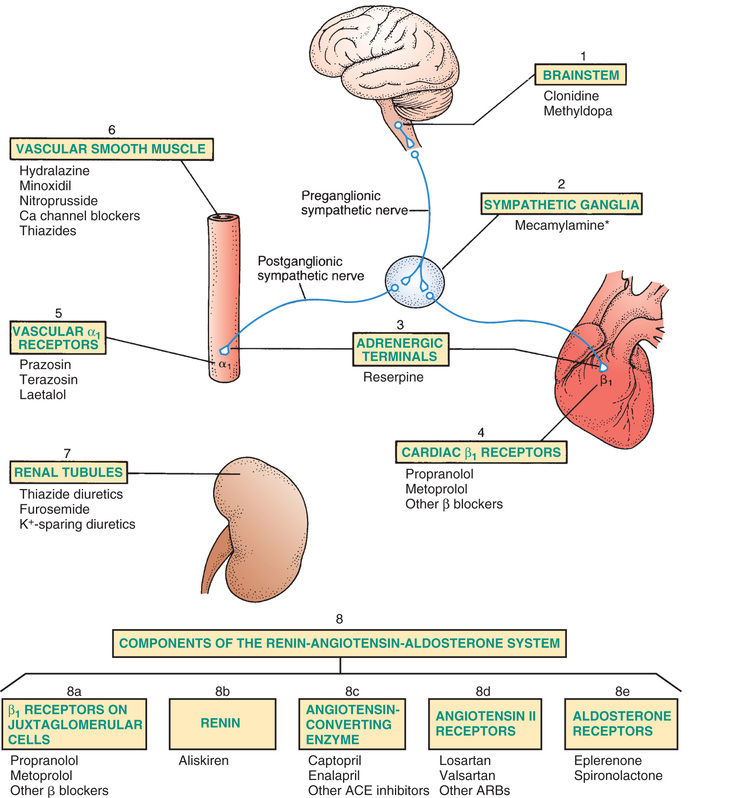
TABLE 39.2
Antihypertensive Effects Elicited by Drug Actions at Specific Sites
| Site of Drug Action* | Representative Drug | Drug Effects |
| Brainstem | Clonidine | Suppression of sympathetic outflow decreases sympathetic stimulation of the heart and blood vessels. |
| Sympathetic ganglia | Mecamylamine† | Ganglionic blockade reduces sympathetic stimulation of the heart and blood vessels. |
| Adrenergic nerve terminals | Reserpine | Reduced norepinephrine release decreases sympathetic stimulation of the heart and blood vessels. |
| Cardiac beta1 receptors | Metoprolol | Beta1 blockade decreases heart rate and myocardial contractility. |
| Vascular alpha1 receptors | Prazosin | Alpha1 blockade causes vasodilation. |
| Vascular smooth muscle | Hydralazine | Relaxation of vascular smooth muscle causes vasodilation. |
| Renal tubules | Hydrochlorothiazide | Promotion of diuresis decreases blood volume. |
| COMPONENTS OF THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM (8A TO 8E) | ||
| 8a.Beta1 receptors on juxtaglomerular cells | Metoprolol | Beta1 blockade suppresses renin release, resulting in (1) vasodilation secondary to reduced production of angiotensin II and (2) prevention of aldosterone-mediated volume expansion. |
| 8b.Renin | Aliskiren | Inhibition of renin suppresses formation of angiotensin I, which in turn decreases formation of angiotensin II and thereby reduces (1) vasoconstriction and (2) aldosterone-mediated volume expansion. |
| 8c.Angiotensin-converting enzyme (ACE) | Captopril | Inhibition of ACE decreases formation of angiotensin II and thereby prevents (1) vasoconstriction and (2) aldosterone-mediated volume expansion. |
| 8d.Angiotensin II receptors | Losartan | Blockade of angiotensin II receptors prevents angiotensin-mediated vasoconstriction and aldosterone-mediated volume expansion. |
| 8e.Aldosterone receptors | Eplerenone | Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water and thereby reduces blood volume. |
1—Brainstem
Antihypertensive drugs acting in the brainstem suppress sympathetic outflow to the heart and blood vessels, resulting in decreased heart rate, decreased myocardial contractility, and vasodilation. Vasodilation contributes the most to reducing BP. Dilation of arterioles reduces BP by decreasing vascular resistance. Dilation of veins reduces BP by decreasing venous return to the heart.
2—Sympathetic Ganglia
Ganglionic blockade reduces sympathetic stimulation of the heart and blood vessels. Antihypertensive effects result primarily from dilation of arterioles and veins. Ganglionic blocking agents produce such a profound reduction in BP that they are used rarely, and then only for hypertensive emergencies. Because use is so limited, the last one available—mecamylamine—was voluntarily withdrawn from the U.S. market in 2009.
3—Terminals of Adrenergic Nerves
Antihypertensive agents that act at adrenergic nerve terminals decrease the release of norepinephrine, resulting in decreased sympathetic stimulation of the heart and blood vessels. These drugs, known as adrenergic neuron blocking agents, are used only rarely. In the United States reserpine is the only drug in this class still on the market.
4—Beta1-Adrenergic Receptors on the Heart
Blockade of cardiac beta1 receptors prevents sympathetic stimulation of the heart. As a result, heart rate and myocardial contractility decline.
5—Alpha1-Adrenergic Receptors on Blood Vessels
Blockade of vascular alpha1 receptors promotes dilation of arterioles and veins. Arteriolar dilation reduces peripheral resistance. Venous dilation reduces venous return to the heart.
6—Vascular Smooth Muscle
Several antihypertensive drugs (see Fig. 39.2) act directly on vascular smooth muscle to cause relaxation. One of these agents—sodium nitroprusside—is used only for hypertensive emergencies. The rest are used for chronic hypertension.
7—Renal Tubules
Diuretics act on renal tubules to promote salt and water excretion. As a result, blood volume declines, causing BP to fall.
Components of the RAAS (8a to 8e)
8a—Beta1 Receptors on Juxtaglomerular Cells.
Blockade of beta1 receptors on juxtaglomerular cells suppresses release of renin. The resultant decrease in angiotensin II levels has three effects: peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8b—Renin.
Inhibition of renin decreases conversion of angiotensinogen into angiotensin I and thereby suppresses the entire RAAS. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8c—Angiotensin-Converting Enzyme.
Inhibitors of ACE suppress formation of angiotensin II. The result is peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8d—Angiotensin II Receptors.
Blockade of angiotensin II receptors prevents the actions of angiotensin II. Hence blockade results in peripheral vasodilation, renal vasodilation, and suppression of aldosterone-mediated volume expansion.
8e—Aldosterone Receptors.
Blockade of aldosterone receptors in the kidney promotes excretion of sodium and water and thereby reduces blood volume.
Classes of Antihypertensive Drugs
In this section we consider the principal drugs employed to treat chronic hypertension. Drugs for hypertensive emergencies and hypertensive disorders of pregnancy are considered separately.
Diuretics
Diuretics are a mainstay of antihypertensive therapy. These drugs reduce BP when used alone, and they can enhance the effects of other hypotensive drugs. The basic pharmacology of the diuretics is discussed in Chapter 35.
Thiazide Diuretics.
Thiazide diuretics (e.g., hydrochlorothiazide, chlorthalidone) are first-line drugs for hypertension. They reduce BP by two mechanisms: reduction of blood volume and reduction of arterial resistance. Reduced blood volume is responsible for initial antihypertensive effects. Reduced vascular resistance develops over time and is responsible for long-term antihypertensive effects. The mechanism by which thiazides reduce vascular resistance has not been determined.
Of the thiazides available, hydrochlorothiazide is used most widely. In fact, hydrochlorothiazide is used more widely than any other antihypertensive drug. Nonetheless, other thiazides, especially chlorthalidone, may be more effective.
The principal adverse effect of thiazides is hypokalemia. This can be minimized by consuming potassium-rich foods and using potassium supplements or a potassium-sparing diuretic. Other side effects include dehydration, hyperglycemia, and hyperuricemia.
Thiazides are superior to calcium channel blockers (CCBs) and ACE inhibitors as monotherapy and therefore are preferred.
Loop Diuretics.
Loop diuretics (e.g., furosemide) produce much greater diuresis than the thiazides. For most individuals with chronic hypertension, the amount of fluid loss that loop diuretics can produce is greater than needed or desirable. Consequently, loop diuretics are not used routinely for hypertension. Rather, they are reserved for (1) patients who need greater diuresis than can be achieved with thiazides and (2) patients with a low GFR (because thiazides won’t work when GFR is low). Like the thiazides, the loop diuretics lower BP by reducing blood volume and promoting vasodilation.
Most adverse effects are like those of the thiazides: hypokalemia, dehydration, hyperglycemia, and hyperuricemia. In addition, loop diuretics can cause hearing loss.
Potassium-Sparing Diuretics.
The degree of diuresis induced by the potassium-sparing agents (e.g., spironolactone) is small. Consequently, these drugs have only modest hypotensive effects. However, because of their ability to conserve potassium, these drugs can play an important role in an antihypertensive regimen. Specifically, they can balance potassium loss caused by thiazides or loop diuretics. The most significant adverse effect of the potassium-sparing agents is hyperkalemia. Because of the risk for hyperkalemia, potassium-sparing diuretics must not be used in combination with one another or with potassium supplements. Also, they should not be used routinely with ACE inhibitors, ARBs, or aldosterone antagonists, all of which promote significant hyperkalemia.
Sympatholytics (Antiadrenergic Drugs)
Sympatholytic drugs suppress the influence of the sympathetic nervous system on the heart, blood vessels, and other structures. These drugs are used widely for hypertension.
There are five subcategories of sympatholytic drugs: (1) beta blockers, (2) alpha1 blockers, (3) alpha/beta blockers, (4) centrally acting alpha2 agonists, and (5) adrenergic neuron blockers.
Beta-Adrenergic Blockers.
Like the thiazides, beta blockers (e.g., metoprolol) are widely used antihypertensive drugs. However, despite their efficacy and frequent use, the exact mechanism by which they reduce BP is somewhat uncertain. Beta blockers are less effective in black people than in whites.
The beta blockers have at least four useful actions in hypertension. First, blockade of cardiac beta1 receptors decreases heart rate and contractility, thereby causing cardiac output to decline. Second, beta blockers can suppress reflex tachycardia caused by vasodilators. Third, blockade of beta1 receptors on juxtaglomerular cells of the kidney reduces release of renin and thereby reduces angiotensin II−mediated vasoconstriction and aldosterone-mediated volume expansion. Fourth, long-term use of beta blockers reduces peripheral vascular resistance—by a mechanism that is unknown. This action could readily account for most of their antihypertensive effects.
Three beta blockers have intrinsic sympathomimetic activity: pindolol, penbutolol, and acebutolol. That is, they can produce mild activation of beta receptors while blocking receptor activation by strong agonists (e.g., norepinephrine). As a result, heart rate at rest is slowed less than with other beta blockers. Accordingly, if a patient develops symptomatic bradycardia with another beta blocker, switching to one of these may help.
Beta blockers can produce several adverse effects. Blockade of cardiac beta1 receptors can produce bradycardia, decreased atrioventricular (AV) conduction, and reduced contractility. Consequently, beta blockers should not be used by patients with sick sinus syndrome or second- or third-degree AV block—and must be used with care in patients with heart failure. Blockade of beta2 receptors in the lung can promote bronchoconstriction. Accordingly, beta blockers should be used with caution in patients with asthma. If an asthmatic individual must use a beta blocker, a beta1-selective agent (e.g., metoprolol) should be employed. Beta blockers can mask signs of hypoglycemia and therefore must be used with caution in patients with diabetes. Potential side effects of beta blockers include depression, insomnia, bizarre dreams, and sexual dysfunction; however, a review of older clinical trials has shown that the risk is small or nonexistent.
The basic pharmacology of the beta blockers is discussed in Chapter 14.
Alpha1 Blockers.
The alpha1 blockers (e.g., doxazosin, terazosin) prevent stimulation of alpha1 receptors on arterioles and veins, thereby preventing sympathetically mediated vasoconstriction. The resultant vasodilation reduces both peripheral resistance and venous return to the heart.
The most disturbing side effect of alpha blockers is orthostatic hypotension. Hypotension can be especially severe with the initial dose. Significant hypotension continues with subsequent doses but is less profound.
The American College of Cardiology (ACC) recommends that alpha blockers not be used as first-line therapy for hypertension. In a huge clinical trial known as the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), in which doxazosin was compared with chlorthalidone (a thiazide diuretic), patients taking doxazosin experienced 25% more cardiovascular events and were twice as likely to be hospitalized for heart failure. It is not clear whether doxazosin increased cardiovascular risk or chlorthalidone decreased risk. Either way, the diuretic is clearly preferred to the alpha blocker.
The basic pharmacology of the alpha blockers is discussed in Chapter 14.
Alpha/Beta Blockers: Carvedilol and Labetalol.
Carvedilol and labetalol are unusual in that they can block alpha1 receptors as well as beta receptors. Blood pressure reduction results from a combination of actions: (1) alpha1 blockade promotes dilation of arterioles and veins, (2) blockade of cardiac beta1 receptors reduces heart rate and contractility, and (3) blockade of beta1 receptors on juxtaglomerular cells suppresses release of renin. Presumably, these drugs also share the ability of other beta blockers to reduce peripheral vascular resistance. Like other nonselective beta blockers, labetalol and carvedilol can exacerbate bradycardia, AV heart block, and asthma. Blockade of venous alpha1 receptors can produce postural hypotension.
Centrally Acting Alpha2 Agonists.
As discussed in Chapter 15 these drugs (e.g., clonidine, methyldopa) act within the brainstem to suppress sympathetic outflow to the heart and blood vessels. The result is vasodilation and reduced cardiac output, both of which help lower BP. All central alpha2 agonists can cause dry mouth and sedation. In addition, clonidine can cause severe rebound hypertension if treatment is abruptly discontinued. Additional adverse effects of methyldopa are hemolytic anemia and liver disorders.
Direct-Acting Vasodilators: Hydralazine and Minoxidil
Hydralazine and minoxidil reduce BP by promoting dilation of arterioles. Neither drug causes significant dilation of veins. Because venous dilation is minimal, the risk for orthostatic hypotension is low. With both drugs, lowering of BP may be followed by reflex tachycardia, renin release, and fluid retention. Reflex tachycardia and release of renin can be prevented with a beta blocker. Fluid retention can be prevented with a diuretic.
The most disturbing adverse effect of hydralazine is a syndrome resembling systemic lupus erythematosus (SLE). Fortunately, this reaction is rare at recommended doses. If an SLE-like reaction occurs, hydralazine should be withdrawn. Hydralazine is considered a third-line drug for chronic hypertension.
A less serious effect is excessive hair growth. Because of its capacity for significant side effects, minoxidil is not used routinely for chronic hypertension. Instead, the drug is reserved for patients with severe hypertension that has not responded to safer drugs.
The basic pharmacology of hydralazine and minoxidil is discussed in Chapter 38.
 Black Box Warning: Minoxidil
Black Box Warning: Minoxidil
Minoxidil can promote pericardial effusion that in some cases progresses to cardiac tamponade.
Calcium Channel Blockers
The CCBs fall into two groups: dihydropyridines (e.g., nifedipine) and nondihydropyridines (verapamil and diltiazem). Drugs in both groups promote dilation of arterioles. In addition, verapamil and diltiazem have direct suppressant effects on the heart.
Like other vasodilators, CCBs can cause reflex tachycardia. This reaction is greatest with the dihydropyridines and minimal with verapamil and diltiazem. Reflex tachycardia is low with verapamil and diltiazem because of cardiosuppression. Because dihydropyridines do not block cardiac calcium channels, reflex tachycardia with these drugs can be substantial.
Because of their ability to compromise cardiac performance, verapamil and diltiazem must be used cautiously in patients with bradycardia, heart failure, or AV heart block. These precautions do not apply to dihydropyridines.
The immediate-release formulation of nifedipine has been associated with increased mortality in patients with MI and unstable angina. As a result, the National Heart, Lung, and Blood Institute has recommended that the use of immediate-release nifedipine be discontinued for treatment of hypertensive emergency.
The basic pharmacology of the CCBs is discussed in Chapter 37.
Drugs that Suppress the RAAS
Because the RAAS plays an important role in controlling BP, drugs that suppress the system—especially the ACE inhibitors—have a significant role in controlling hypertension. The basic pharmacology of these drugs is discussed in Chapter 36.
ACE Inhibitors.
The ACE inhibitors (e.g., captopril, enalapril) lower BP by preventing formation of angiotensin II and thereby prevent angiotensin II–mediated vasoconstriction and aldosterone-mediated volume expansion. In hypertensive diabetic patients with renal damage, these actions slow progression of kidney injury. Like the beta blockers, ACE inhibitors are less effective in blacks than in whites. Principal adverse effects are persistent cough, first-dose hypotension, angioedema, and hyperkalemia (secondary to suppression of aldosterone release). Because of the risk for hyperkalemia, combined use with potassium supplements or potassium-sparing diuretics is generally avoided.
 Black Box Warning: Angiotensin-Converting Enzyme Inhibitors
Black Box Warning: Angiotensin-Converting Enzyme Inhibitors
ACE inhibitors can cause serious fetal harm, especially during the second and third trimesters of pregnancy, and hence must not be given to pregnant women. ACE inhibitors—along with ARBs and DRIs—are the only antihypertensive drugs specifically contraindicated during pregnancy.
Angiotensin II Receptor Blockers.
ARBs lower BP in much the same way as do ACE inhibitors. Like the ACE inhibitors, ARBs prevent angiotensin II–mediated vasoconstriction and release of aldosterone. The only difference is that ARBs do so by blocking the actions of angiotensin II, whereas ACE inhibitors block the formation of angiotensin II. Both groups lower BP to the same extent. In contrast to ACE inhibitors, ARBs have a low incidence of inducing cough or significant hyperkalemia, but they do cause angioedema.
 Black Box Warning: Angiontensin II Receptor Blockers
Black Box Warning: Angiontensin II Receptor Blockers
Like the ACE inhibitors, ARBs can cause fetal harm and must not be used during pregnancy.
Direct Renin Inhibitors.
DRIs act directly on renin to inhibit conversion of angiotensinogen into angiotensin I. As a result, DRIs can suppress the entire RAAS. At this time, only one DRI—aliskiren [Tekturna, Rasilez  ]—is available. Antihypertensive effects equal those of ACE inhibitors, ARBs, and CCBs. Compared with ACE inhibitors, aliskiren causes less hyperkalemia, cough, or angioedema—but poses a similar risk for fetal harm. In addition, aliskiren causes diarrhea in 2.3% of patients. Also, in patients with type 2 diabetes mellitus, use of aliskiren has demonstrated an increased incidence of renal impairment, hypotension, and hyperkalemia. Because of these findings, use of aliskiren is contraindicated in patients with diabetes mellitus who are also taking an ACE inhibitor or ARB. There are conflicting data regarding reduction of stroke, kidney failure, or MI with use of Aliskiren. Accordingly, until experience with the drug is more extensive, other antihypertensives should be considered first.
]—is available. Antihypertensive effects equal those of ACE inhibitors, ARBs, and CCBs. Compared with ACE inhibitors, aliskiren causes less hyperkalemia, cough, or angioedema—but poses a similar risk for fetal harm. In addition, aliskiren causes diarrhea in 2.3% of patients. Also, in patients with type 2 diabetes mellitus, use of aliskiren has demonstrated an increased incidence of renal impairment, hypotension, and hyperkalemia. Because of these findings, use of aliskiren is contraindicated in patients with diabetes mellitus who are also taking an ACE inhibitor or ARB. There are conflicting data regarding reduction of stroke, kidney failure, or MI with use of Aliskiren. Accordingly, until experience with the drug is more extensive, other antihypertensives should be considered first.
 Black Box Warning: Direct Renin Inhibitors
Black Box Warning: Direct Renin Inhibitors
Like the ACE inhibitors, DRIs can cause fetal harm and must not be used during pregnancy.
Aldosterone Antagonists.
Aldosterone antagonists lower BP by promoting renal excretion of sodium and water. Only two agents are available: eplerenone and spironolactone. Both spironolactone and eplerenone promote renal retention of potassium and hence pose a risk for hyperkalemia. Accordingly, they should not be given to patients with existing hyperkalemia and should not be combined with potassium-sparing diuretics or potassium supplements. Combined use with ACE inhibitors, ARBs, and DRIs is permissible but must be done with caution. Spironolactone is discussed in Chapter 35, and eplerenone is discussed in Chapter 36.
Fundamentals of Hypertension Drug Therapy
Treatment Algorithm
The basic approach to treating hypertension was published with the new 2014 guidelines in the Journal of the American Medical Association and can be found on the JAMA Network at jama.jamanetwork.com/article.aspx?articleid=1791497. As shown in the algorithm at this link, lifestyle changes should be instituted first. If these fail to lower BP enough, drug therapy should be started—and the lifestyle changes should continue. Treatment often begins with a single drug. If needed, another drug may be added (if the initial drug was well tolerated but inadequate) or substituted (if the initial drug was poorly tolerated). However, before another drug is considered, possible reasons for failure of the initial drug should be assessed. Among these are insufficient dosage, poor adherence, excessive salt intake, and the presence of secondary hypertension. If treatment with two drugs is unsuccessful, a third and even fourth may be added.
Initial Drug Selection
Initial drug selection is determined by the presence or absence of a compelling indication, defined as a comorbid condition for which a specific class of antihypertensive drugs has been shown to improve outcomes. Initial drugs for patients with and without compelling indications are discussed next.
Patients Without Compelling Indications.
For initial therapy in the absence of a compelling indication, a thiazide diuretic is currently recommended for most patients. This preference is based on long-term controlled trials showing conclusively that thiazides can reduce morbidity and mortality in hypertensive patients and are well tolerated and inexpensive too. Other options for initial therapy—ACE inhibitors, ARBs, and CCBs—equal diuretics in their ability to lower BP. However, they may not be as effective at reducing morbidity and mortality. Accordingly, these drugs should be reserved for special indications and for patients who have not responded to thiazides. Certain other alternatives—centrally acting sympatholytics and direct-acting vasodilators—are associated with a high incidence of adverse effects and hence are not well suited for initial monotherapy. One last alternative—alpha1 blockers—is no longer recommended as first-line therapy. As noted, when the alpha blocker doxazosin was compared with the diuretic chlorthalidone, doxazosin was associated with a much higher incidence of adverse cardiovascular events.
Patients With Compelling Indications.
For patients with hypertension plus certain comorbid conditions (e.g., heart failure, diabetes), there is strong evidence that specific antihypertensive drugs can reduce morbidity and mortality. Drugs shown to improve outcomes for six comorbid conditions are indicated in Table 39.3. Clearly, these drugs should be used for initial therapy. If needed, other antihypertensive agents can be added to the regimen. Management of hypertension in patients with diabetes and renal disease—two specific comorbid conditions—is discussed further under “Individualizing Therapy.”
TABLE 39.3
Classes of Antihypertensive Drugs Recommended for Initial Therapy of Hypertension in Patients With Certain High-Risk Comorbid Conditions
| High-Risk Comorbid Conditions that Constitute Compelling Indications for the Drugs Checked | Drug Classes Recommended for Initial Therapy of Hypertension | |||||
| Diuretic | Beta Blocker | ACE Inhibitor | ARB | CCB | Aldosterone Antagonist | |
| Heart failure | ✔ | ✔ | ✔ | ✔ | ✔ | |
| Post–myocardial infarction | ✔ | ✔ | ✔ | |||
| High coronary disease risk | ✔ | ✔ | ✔ | ✔ | ||
| Diabetes | ✔ | ✔ | ✔ | ✔ | ✔ | |
| Chronic kidney disease | ✔ | ✔ | ||||
| Recurrent stroke prevention | ✔ | ✔ | ||||
Adding Drugs to the Regimen
Rationale for Drug Selection.
When using two or more drugs to treat hypertension, each drug should come from a different class. That is, each drug should have a different mechanism of action. In accord with this guideline, it would be appropriate to combine a beta blocker, a diuretic, and a vasodilator because each lowers BP by a different mechanism. In contrast, it would be inappropriate to combine two thiazide diuretics or two beta blockers or two vasodilators.
Benefits of Multidrug Therapy.
Treatment with multiple drugs offers significant benefits. First, by employing drugs that have different mechanisms, we can increase the chance of success: targeting BP control at several sites is likely to be more effective than targeting at one site. Second, when drugs are used in combination, each can be administered in a lower dosage than would be possible if it were used alone. As a result, both the frequency and the intensity of side effects are reduced. Third, when proper combinations are selected, one agent can offset the adverse effects of another. For example, if a vasodilator is used alone, reflex tachycardia is likely. However, if a vasodilator is combined with a beta blocker, reflex tachycardia will be minimal.
Dosing
For each drug in the regimen, dosage should be low initially and then gradually increased. For most people with chronic hypertension, the disease poses no immediate threat. Hence there is no need to lower BP rapidly using large doses. Also, when BP is reduced slowly, baroreceptors gradually reset to the new, lower pressure. As a result, sympathetic reflexes offer less resistance to the hypotensive effects of therapy. Finally, because there is no need to drop BP rapidly, and because higher doses carry a higher risk for adverse effects, use of high initial doses would needlessly increase the risk for adverse effects.
Step-Down Therapy
After BP has been controlled for at least 1 year, an attempt should be made to reduce dosages and the number of drugs in the regimen. Of course, lifestyle modifications should continue. When reductions are made slowly and progressively, many patients are able to maintain BP control with less medication—and some can be maintained with no medication at all. If drugs are discontinued, regular follow-up is essential because BP usually returns to hypertensive levels—although it may take years to do so.
Individualizing Therapy
Patients With Comorbid Conditions
Comorbid conditions complicate treatment. Two conditions that are especially problematic—renal disease and diabetes—are discussed here. Preferred drugs for patients with these and other comorbid conditions are shown in Table 39.3. Drugs to avoid in patients with specific comorbid conditions are summarized in Table 39.4.
TABLE 39.4
Comorbid Conditions that Require Cautious Use or Complete Avoidance of Certain Antihypertensive Drugs
| Comorbid Condition | Drugs to Be Avoided or Used With Caution | Reason for Concern |
| CARDIOVASCULAR DISORDERS | ||
| Heart failure | These drugs act on the heart to decrease myocardial contractility and can thereby further reduce cardiac output. | |
| AV heart block | These drugs act on the heart to suppress AV conduction and can thereby intensify AV block. | |
| Coronary artery disease | Hydralazine | Reflex tachycardia induced by hydralazine can precipitate an anginal attack. |
| Post–myocardial infarction | Hydralazine | Reflex tachycardia induced by hydralazine can increase cardiac work and oxygen demand. |
| OTHER DISORDERS | ||
| Dyslipidemia | These drugs may exacerbate dyslipidemia. | |
| Renal insufficiency | Use of these agents can lead to dangerous accumulations of potassium. | |
| Asthma | Beta2 blockade promotes bronchoconstriction. | |
| Depression | Reserpine | Reserpine can cause depression. |
| Diabetes mellitus | Thiazides and furosemide promote hyperglycemia, and beta blockers suppress glycogenolysis and can mask signs of hypoglycemia. | |
| Gout | These diuretics promote hyperuricemia. | |
| Hyperkalemia | These drugs cause potassium accumulation. | |
| Hypokalemia | These drugs cause potassium loss. | |
| Collagen diseases | Hydralazine | Hydralazine can precipitate a lupus erythematosus–like syndrome. |
| Liver disease | Methyldopa | Methyldopa is hepatotoxic. |
| Preeclampsia | These drugs can injure the fetus. | |
Renal Disease.
Nephrosclerosis secondary to hypertension is among the most common causes of progressive renal disease. Pathophysiologic changes include degeneration of renal tubules and fibrotic thickening of the glomeruli, both of which contribute to renal insufficiency. Nephrosclerosis sets the stage for a downward spiral: renal insufficiency causes water retention, which in turn causes BP to rise higher, which in turn promotes even more renal injury, and so forth. Accordingly, early detection and treatment are essential. To slow progression of renal damage, the most important action is to lower BP. As with other populations, the target BP in patients younger than 60 years is 140/90 mm Hg or lower and is 150/90 mm Hg or lower in patients 60 years and older. Although all classes of antihypertensive agents are effective in nephrosclerosis, ACE inhibitors and ARBs work best. Hence, in the absence of contraindications, all patients should get one of these drugs. In most cases, a diuretic is also used. In patients with advanced renal insufficiency, thiazide diuretics are ineffective, hence a loop diuretic should be employed. Potassium-sparing diuretics should be avoided.
Diabetes.
In patients with diabetes, the target BP is the same as with all other populations with special indications (140/90 mm Hg). Preferred antihypertensive drugs are ACE inhibitors, ARBs, CCBs, and diuretics (in low doses). In patients with diabetic nephropathy, ACE inhibitors and ARBs can slow progression of renal damage and reduce albuminuria. In diabetic patients, as in nondiabetic patients, beta blockers and diuretics can decrease morbidity and mortality. Keep in mind, however, that beta blockers can suppress glycogenolysis and mask early signs of hypoglycemia and therefore must be used with caution. Thiazides and loop diuretics promote hyperglycemia and hence should be used with care.
How do ACE inhibitors compare with CCBs in patients with hypertension and diabetes? In one large study, patients taking nisoldipine (a CCB) had a higher incidence of MI than did patients taking enalapril (an ACE inhibitor). Because the study was not placebo controlled, it was impossible to distinguish between two possible interpretations: (1) the CCB increased the risk for MI or (2) the ACE inhibitor protected against MI. Either way, it seems clear that ACE inhibitors are better than CCBs for patients with hypertension and diabetes.
Patients in Special Populations
Blacks.
Hypertension is a major health problem for black adults. Hypertension develops earlier, has a much higher incidence, and is likely to be more severe. As a result, black people face a greater risk for heart disease, end-stage renal disease, and stroke. Compared with the general population, blacks experience a 50% higher rate of death from heart disease, are twice as likely to die of stroke, and are 6 times more likely to experience hypertension-related end-stage renal disease.
With timely treatment, this disparity can be greatly reduced, if not eliminated. We know that blacks and whites respond equally to treatment (although not always to the same drugs). The primary problem is that, among black people, hypertension often goes untreated until after significant organ damage has developed. If hypertension were diagnosed and treated earlier, the prognosis would be greatly improved. Accordingly, it is important that blacks undergo routine monitoring of BP. If hypertension is diagnosed, treatment should begin at once. Because blacks have a high incidence of salt sensitivity and cigarette use, lifestyle modifications are an important component of treatment.
Black people respond better to some antihypertensive drugs than to others. Controlled trials have shown that diuretics can decrease morbidity and mortality in blacks. Accordingly, diuretics are drugs of first choice. CCBs and alpha/beta blockers are also effective. In contrast, monotherapy with beta blockers or ACE inhibitors is less effective in blacks than in whites. Nonetheless, beta blockers and ACE inhibitors should be used if they are strongly indicated for a comorbid condition. For example, ACE inhibitors should be used in black patients who have type 1 diabetes with proteinuria. Also, ACE inhibitors should be used in patients with hypertensive nephrosclerosis, a condition for which ACE inhibitors are superior to CCBs. When BP cannot be adequately controlled with a single drug, several two-drug combinations are recommended: an ACE inhibitor plus a thiazide diuretic, an ACE inhibitor plus a CCB, and a beta blocker plus a thiazide.
In 2014, the International Society on Hypertension in Blacks (ISHIB) issued updated guidelines on managing hypertension in black people. Current goals remain consistent with the JNC 8 Guidelines of maintaining BP less than 140/90 mm Hg in this population.
Children and Adolescents.
The incidence of secondary hypertension in children is much higher than in adults. Accordingly, efforts to diagnose and treat an underlying cause should be especially diligent. For children with primary hypertension, treatment is the same as for adults—although doses are lower and should be adjusted with care. Because ACE inhibitors and ARBs can cause fetal harm, they should be avoided in girls who are sexually active or pregnant.
Older Adults.
By age 65 years, most Americans have hypertension. Furthermore, high BP in this group almost always presents as isolated systolic hypertension; DBP is usually normal or low. The good news, as shown in the Hypertension in the Very Elderly Trial (HYVET), is that treatment can reduce the incidence of heart failure, fatal stroke, and all-cause mortality. The bad news is that most older people are not treated.
The new 2014 guidelines advocate for a target BP of less than 150/90 mm Hg in patients 60 years and older. This differs from the 2011 ACC/American Heart Association (AHA) expert consensus document on treatment of hypertension in people 65 years and older, for whom the target systolic pressure is below 140 mm Hg (for patients ages 65–79 years) and below 145 mm Hg (for patients 80 years and older). Data supported improved outcomes with tighter BP control in the older adult population are lacking—hence the likely reason for the change in philosophy. Although the newer guidelines may be more lax, they do not serve as a substitute for clinical judgment.
PATIENT-CENTERED CARE ACROSS THE LIFE SPAN
Hypertension
| Life Stage | Patient Care Concerns |
| Infants | See later entry, “Breastfeeding women.” |
| Children/adolescents | No data are available on the long-term effects of antihypertensive drugs on growth and development of children. Drugs recommended for treatment of hypertension in children 1–18 years old include ACE inhibitors, diuretics, beta blockers, and calcium channel blockers. |
| Pregnant women | Drugs of choice in treating pregnant women with mild preeclampsia include labetalol and methyldopa. Magnesium sulfate is used in the prevention of seizures in severe preeclampsia or for treatment of seizures in eclampsia. |
| Breastfeeding women | Effects of RAAS-blocking drugs have not been studied in breastfeeding. Beta blockers, such as metoprolol, appear safe for the breastfeeding infant. Diuretics appear safe but may suppress lactation. |
| Older adults | Older adults benefit from SBPs <145 mm Hg. Treatment with ACE inhibitors, diuretics, and/or beta blockers is reasonable. Caution must be taken to avoid overdiuresis when using diuretics in the older-adult population. |
Because cardiovascular reflexes are blunted in older adults, treatment carries a significant risk of orthostatic hypotension. Accordingly, initial doses should be low—about one-half those used for younger adults—and dosage escalation should be done slowly. Drugs that are especially likely to cause orthostatic hypotension (e.g., alpha1 blockers, alpha/beta blockers) should be used with caution.
Minimizing Adverse Effects
Antihypertensive drugs can produce many unwanted effects, including hypotension, sedation, and sexual dysfunction. (Although not stressed previously, practically all antihypertensive drugs can interfere with sexual function.)
The fundamental strategy for decreasing side effects is to tailor the regimen to the sensitivities of the patient. Simply put, if one drug causes effects that are objectionable, a more acceptable drug should be substituted. The best way to identify unacceptable responses is to encourage patients to report them.
Adverse effects caused by exacerbation of comorbid diseases are both predictable and avoidable. We know, for example, that beta blockers can intensify AV block and hence should not be taken by people with these disorders. Other conditions that can be aggravated by antihypertensive drugs are listed in Table 39.4. To help avoid drug-disease mismatches, the medical history should identify all comorbid conditions. With this information, the prescriber can choose drugs that are least likely to make the comorbid condition worse.
High initial doses and rapid dosage escalation can increase the incidence and severity of adverse effects. Accordingly, doses should be low at first and then gradually increased. Remember, there is usually no need to reduce BP rapidly. Hence large initial doses that can produce a rapid fall in BP but also produce adverse effects should be avoided.
Promoting Adherence
The major cause of treatment failure in patients with chronic hypertension is lack of adherence to the prescribed regimen. In this section we consider the causes of nonadherence and discuss some solutions.
Why Adherence Is Often Hard to Achieve
Much of the difficulty in promoting adherence stems from the nature of hypertension itself. Hypertension is a chronic, slowly progressing disease that, through much of its course, is devoid of overt symptoms. Because symptoms are absent, it can be difficult to convince patients that they are ill and need treatment. In addition, because there are no symptoms to relieve, drugs cannot produce an obvious therapeutic response. In the absence of such a response, it can be difficult for patients to believe that their medication is doing anything useful.
Because hypertension progresses very slowly, the disease tends to encourage procrastination. For most people, the adverse effects of hypertension will not become manifest for many years. Realizing this, patients may reason (incorrectly) that they can postpone therapy without significantly increasing risk.
The negative aspects of treatment also contribute to nonadherence. Antihypertensive regimens can be complex and expensive. In addition, treatment must continue lifelong. Lastly, antihypertensive drugs can cause a number of adverse effects, ranging from sedation to hypotension to impaired sexual function. It is difficult to convince people who are feeling good to take drugs that may make them feel worse. Some people may decide that exposing themselves to the negative effects of therapy today is paying too high a price to avoid the adverse consequences of hypertension at some indefinite time in the future.
Ways to Promote Adherence
Patient Education.
Adherence requires motivation, and patient education can help provide it. Patients should be taught about the consequences of hypertension and the benefits of treatment. Because hypertension does not cause discomfort, it may not be clear to patients that their condition is indeed serious. Patients must be helped to understand that, left untreated, hypertension can cause heart disease, kidney disease, and stroke. In addition, patients should appreciate that, with proper therapy, the risks for these long-term complications can be minimized, resulting in a longer and healthier life. Lastly, patients must understand that drugs do not cure hypertension—they only control symptoms. Hence, for treatment to be effective, medication must be taken lifelong.
Minimize Side Effects.
If we expect patients to comply with long-term treatment, we must keep adverse effects to a minimum. As discussed earlier, adverse effects can be minimized by (1) encouraging patients to report side effects, (2) discontinuing objectionable drugs and substituting more acceptable ones, (3) avoiding drugs that can exacerbate comorbid conditions, and (4) using doses that are low initially and then gradually increased.
Establish a Collaborative Relationship.
The patient who feels like a collaborative partner in the treatment program is more likely to comply than is the patient who feels that treatment is being imposed. Collaboration allows the patient to help set treatment goals, create the treatment program, and evaluate progress. In addition, a collaborative relationship facilitates communication about side effects.
Simplify the Regimen.
Antihypertensive regimens may consist of several drugs taken multiple times a day. Such complex regimens deter adherence. Therefore, to promote adherence, the dosing schedule should be as simple as possible. After an effective regimen has been established, dosing just once or twice daily should be tried. If an appropriate combination product is available (e.g., a fixed-dose combination of a thiazide diuretic plus an ACE inhibitor), the combination product may be substituted for its components.
Other Measures.
Adherence can be promoted by giving positive reinforcement when therapeutic goals are achieved. Involvement of family members can be helpful. Also, adherence can be promoted by scheduling office visits at convenient times and by following up when appointments are missed. For many patients, antihypertensive therapy represents a significant economic burden; devising a regimen that is effective but inexpensive will help.
Drugs for Hypertensive Disorders of Pregnancy
Hypertension is the most common complication of pregnancy, with an incidence of about 10%. When hypertension develops, it is essential to distinguish between chronic hypertension and preeclampsia. Chronic hypertension is relatively benign, whereas preeclampsia can lead to life-threatening complications for the patient and the fetus.
Chronic Hypertension
Chronic hypertension, seen in 5% of pregnancies, is defined as hypertension that was present before pregnancy or that developed before the 20th week of gestation. Persistent severe hypertension carries a risk to both the patient and the fetus. Potential adverse outcomes include placental abruption, maternal cardiac decompensation, premature birth, fetal growth delay, central nervous system hemorrhage, and renal failure. The goal of treatment is to minimize the risk for hypertension to the patient and fetus while avoiding drug-induced harm to the fetus. With the exception of ACE inhibitors, ARBs, and DRIs, antihypertensive drugs that were being taken before pregnancy can be continued. ACE inhibitors, ARBs, and DRIs are contraindicated owing to their potential for harm (fetal growth delay, congenital malformations, neonatal renal failure, neonatal death). When drug therapy is initiated during pregnancy, methyldopa and labetalol are the traditional agents of choice. These drugs have limited effects on uteroplacental and fetal hemodynamics and do not adversely affect the fetus or neonate. Regardless of the drug selected, treatment should not be too aggressive because an excessive drop in BP could compromise uteroplacental blood flow.
According to guidelines issued in 2012 by the American College of Obstetricians and Gynecologists (ACOG), “severe” hypertension requires treatment, whereas “mild” hypertension generally does not. (The ACOG defines severe hypertension as SBP >160 mm Hg or DBP >110 mm Hg, and mild hypertension as SBP 140–159 mm Hg or DBP 90–109 mm Hg.) There is good evidence that treating severe hypertension reduces risk. In contrast, there is little evidence that treating mild hypertension offers significant benefit.
Patients who have chronic hypertension during pregnancy are at increased risk for developing preeclampsia (see later). Unfortunately, reducing BP does not lower this risk.
Preeclampsia and Eclampsia
Preeclampsia is a multisystem disorder characterized by the combination of elevated BP (>140/90 mm Hg) and proteinuria (≥300 mg in 24 hours) that develops after the 20th week of gestation. The disorder occurs in about 5% of pregnancies. Rarely, women with preeclampsia develop seizures. If seizures do develop, the condition is then termed eclampsia. Risk factors for preeclampsia include black race, chronic hypertension, diabetes, collagen vascular disorders, and previous preeclampsia. The etiology of preeclampsia is complex and incompletely understood.
Preeclampsia poses serious risks for the fetus and mother. Risks for the fetus include intrauterine growth restriction, premature birth, and even death. The mother is at risk for seizures (eclampsia), renal failure, pulmonary edema, stroke, and death.
Management of preeclampsia is based on the severity of the disease, the status of mother and fetus, and the length of gestation. The objective is to preserve the health of the mother and deliver an infant who will not require intensive and prolonged neonatal care. Success requires close maternal and fetal monitoring. Although drugs can help reduce BP, delivery is the only cure.
Management of mild preeclampsia is controversial and depends on the duration of gestation. If preeclampsia develops near term, and if fetal maturity is certain, induction of labor is advised. However, if mild preeclampsia develops earlier in gestation, experts disagree about what to do. Suggested measures include bed rest, prolonged hospitalization, treatment with antihypertensive drugs, and prophylaxis with an anticonvulsant. Studies to evaluate these strategies have generally failed to demonstrate benefits from any of them, including treatment with antihypertensive drugs.
The definitive intervention for severe preeclampsia is delivery. However, making the choice to induce labor presents a dilemma. Because preeclampsia can deteriorate rapidly, with grave consequences for the patient and fetus, immediate delivery is recommended. However, if the fetus is not sufficiently mature, immediate delivery could threaten its life. Do we deliver the fetus immediately, which would eliminate risk for the patient but present a serious risk for the fetus—or do we postpone delivery, which would reduce risk for the fetus but greatly increase risk for the patient? If the patient elects to postpone delivery, then BP can be lowered with drugs. Because severe preeclampsia can be life threatening, treatment must be done in a tertiary care center to permit close monitoring. The major objective is to prevent maternal cerebral complications (e.g., hemorrhage, encephalopathy). The drug of choice for lowering BP is labetalol (20 mg by intravenous [IV] bolus over 2 minutes); dosing may be repeated at 10-minute intervals up to a total of 300 mg.
Because severe preeclampsia can evolve into eclampsia, an antiseizure drug may be given for prophylaxis. Magnesium sulfate is the drug of choice. In one study, prophylaxis with magnesium sulfate reduced the risk for eclampsia by 58% and the risk for death by 45%. Dosing is the same as for treating eclampsia.
If eclampsia develops, magnesium sulfate is the preferred drug for seizure control. Initial dosing consists of a 4- to 6-g IV loading dose followed by a continuous IV infusion of 1 to 2 g for the next 24 hours. To ensure therapeutic effects and prevent toxicity, blood levels of magnesium, as well as presence of patellar reflex, should be monitored. The target range for serum magnesium is 4 to 7 mEq/L (the normal range for magnesium is 1.5–2 mEq/L).
When started before 16 weeks of gestation, low-dose aspirin reduces the risk for preeclampsia by about 50%. Similarly, L-arginine (combined with antioxidant vitamins) can also help. By contrast, several other preparations—magnesium, zinc, vitamin C, vitamin E, fish oil, and diuretics—appear to offer no protection at all.

