CHAPTER 45 PROTHROMBOTIC STATES AND RELATED CONDITIONS
THE HEMOSTATIC SYSTEM
The human circulatory system does not act simply as an array of inert, lifeless pipes that convey blood between organs; it is an organ itself with functions beyond those of the various compounds and cells that pass through its conduits. One of these functions is the maintenance of its own structural integrity, essential for transmitting blood pressure, ensuring continuity of flow, and minimizing spread of infection. There has evolved a complex hemostatic system that enables on-line monitoring of a large area of endothelial space (approximately 600 m2, or the equivalent of three tennis courts), with rapid, local restoration of breaches as they occur. This system is composed of multifarious actions and interactions among solid-phase vessel wall, cellular platelets, and humoral coagulation factors; Figure 45-1 is a basic schema of the events that follow vessel injury. It is preferable to talk about this system as a whole rather than as a separate “coagulation system” because of the strong interdependence among these three components. An important attribute of hemostasis is the requirement of both “on knobs” and “off knobs,” delicately adjusted, to prevent a response to vessel wall injury from overshooting and resulting in a sealing of both the defect (which is desired) and the vessel lumen itself (which would cause local hypoperfusion). Endogenous anticoagulant systems are shown in Figure 45-2. As with most metabolic systems, the physiological state is optimized by both prohemostatic and antihemostatic reactions occurring simultaneously (i.e., a dynamic system); the net outcome depends on local vessel integrity.
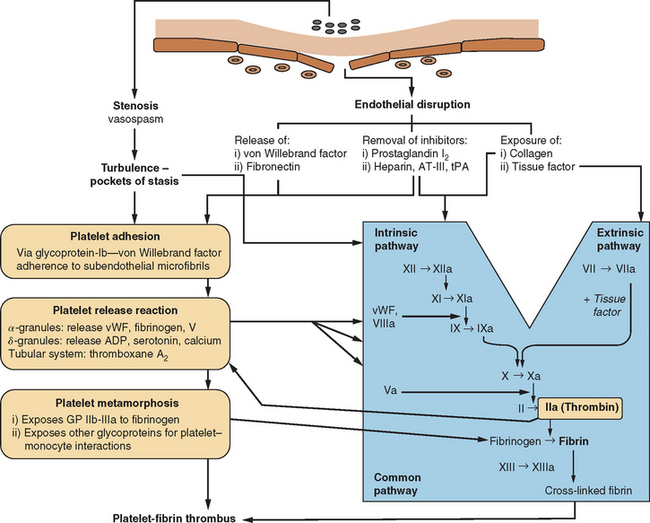
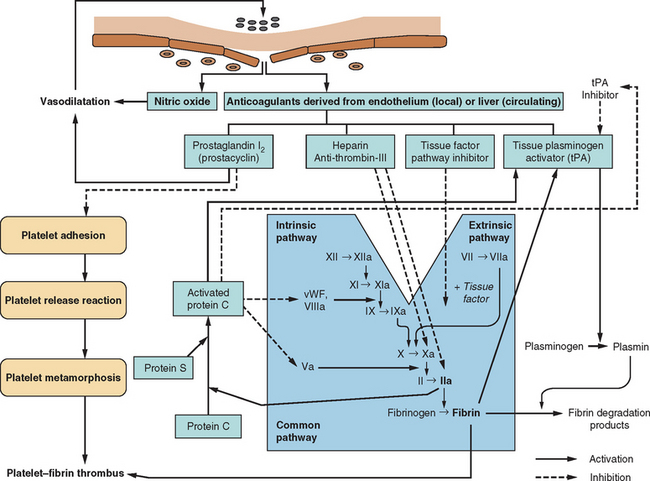
PATHOLOGY OF EXCESSIVE HEMOSTASIS
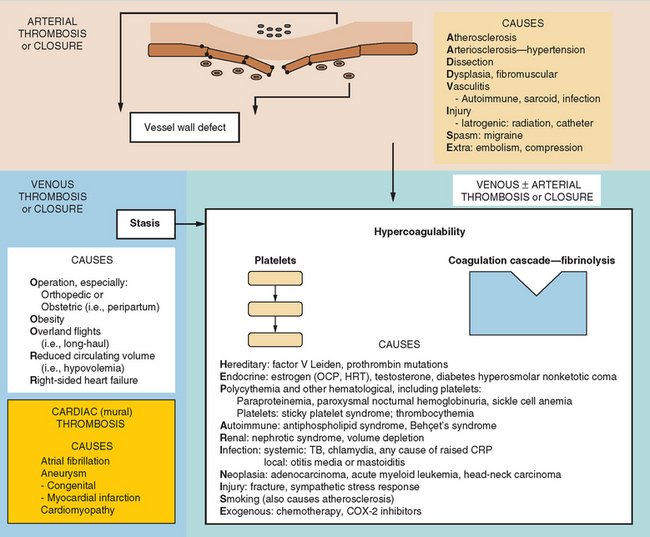
In the same way that the physiology of hemostasis is shared among the trio of vessel wall, platelets, and clotting factors, the pathophysiology of excessive hemostasis can be summarized by its own triad. This so-called Virchow’s triad represents three sets of factors, each of which may give rise to vessel closure, thrombosis being a final common pathway in most cases (Fig. 45-3).
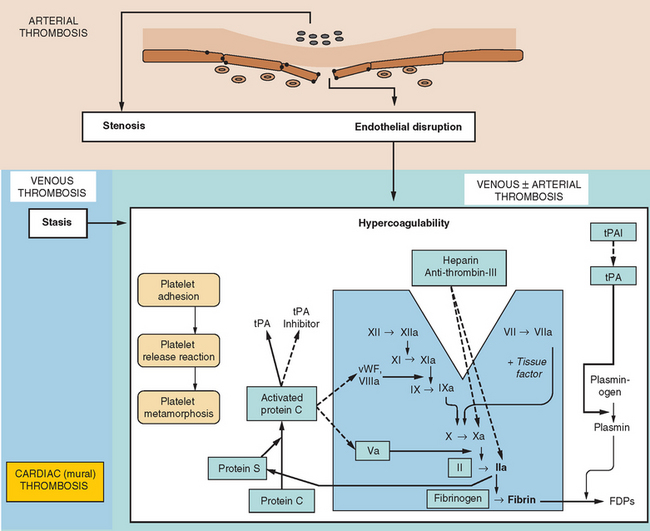
A modern-day listing of Virchow’s triad has changed little since its original formulation in 1860:
Figure 45-4 lists the various causes within each of these categories, including states predisposing to thrombosis or vessel closure by other means (e.g., vasospasm). These causes can be remembered by the mnemonic “ADVISE OR HEPARINISE.”
A thrombosis consists of a solid aggregation of platelets and erythrocytes within a fibrin mesh adherent to the vessel wall. Microscopic observation reveals that thrombi within arteries have a higher platelet composition (in relation to fibrin) than do those in veins. Because fibrin tends to adhere to red blood cells, arterial thrombi appear relatively white, whereas venous thromboses, with a higher fibrin content, appear red.
NEUROLOGICAL CONSEQUENCES OF EXCESSIVE HEMOSTASIS
Thromboembolism represents the commonest cause of ischemic stroke; other pathological processes may also result in vessel closure to produce regional ischemia within the nervous system. Examples of such pathological processes are vasospasm (that in the brain results in migrainous auras), arteriosclerotic lipohyalinosis (commonly caused by hypertension and resulting in isolated “lacunar” strokes or, when diffuse, a subcortical dementia), and vasculitis. Figure 45-5 depicts the varied neurological effects of ischemia secondary to thromboembolism or other processes resulting in vessel closure.
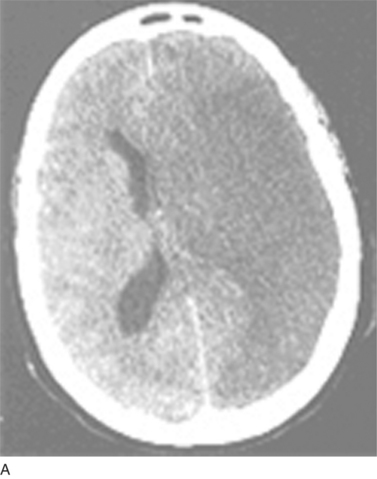




(B, From http://www.aic.cuhk.edu.hk/web8/0294_Cerebral_venous_haemorrhagic_infarction.jpg. Copyright © Chanles Gomersall. C, From http://www.kup.at/kup/images/thumbs/336.jpg. E, From The Robert Bendheim Digital Atlas of Ophthalmology, The New York Eye and Ear Infirmary, http://www.nyee.edu/page_deliv.html?page_no=50. F, From http://medicine.ucsd.edu/clinicalmed/eyes-cn6-palsy3.jpg.)
VESSEL WALL DEFECTS
Atherosclerosis
Postmortem appearances of arteries from people who died from nonischemic causes have shown that atherosclerotic lesions are virtually universal after middle age, especially in residents of industrialized nations. In fact, the strong geographical dependence of atherosclerosis risk corresponds to recognized environmental factors that may initiate atherogenesis (Fig. 45-6). The most important predisposing state is a high circulating triglyceride (lipid) load, especially during the postprandial period—which for many residents of the industrialized world represents most of the waking day. Because triglyceride-rich particles (very-low-density lipoprotein) continuously exchange their triglycerides with cholesterol found within high-density lipoprotein particles, the level of high-density lipoprotein-cholesterol is inversely correlated with, and an accurate predictor of, atherosclerosis risk. The level of low-density lipoprotein (LDL)-cholesterol is less predictive of atherosclerosis, although a subset of LDL-cholesterol particles formed under high triglyceride conditions, called small, dense LDL-cholesterol,







{kind=link}
{kind=link}
{kind=link}