CHAPTER 30 PRIMARY AUTONOMIC FAILURE
CLASSIFICATION
A convenient approach to the syndromes of autonomic failure is to distinguish those of the primary variety, in which there is no clear etiological factor or specific disease association, from those with secondary autonomic failure, in which the lesion is defined (anatomically, as in spinal cord injuries, or biochemically, as in dopamine β-hydroxylase deficiency) or is linked to specific disease processes (as in diabetes mellitus). Furthermore, drugs form a major cause of autonomic dysfunction and merit separate categorization. Moreover, another group that probably warrants a separate entity is neurally mediated syncope, in which, between episodic autonomic disturbances, usually no abnormalities can be detected.1
Primary autonomic failure syndromes can be divided into a chronic subgroup and into a rarer subgroup with acute or subacute dysautonomia (Table 30-1). Furthermore, the chronic syndromes can be subdivided into those without (i.e., pure autonomic failure [PAF]) and those with associated neurological deficits. Clinically, the latter belong to at least three categories: Parkinson’s disease associated with autonomic failure, dementia with Lewy bodies (DLB), and multiple-system atrophy (MSA). Patients with parkinsonian features may be responsive to chronic levodopa (L-dopa) therapy, probable as part of treatment for Parkinson’s disease with autonomic failure. Alternatively, there may be no or poorly (transiently) L-dopa–responsive parkinsonism, probable as part of the parkinsonian variant of MSA, i.e. MSA-P. Other patients may have cerebellar features as the predominant motor disorder and may therefore be diagnosed as the cerebellar variant of MSA, i.e., MSA-C. Some patients present initially with autonomic abnormalities, including urogenital and cardiovascular dysfunction, and only later develop the additional neurological manifestations of MSA. Eventually, some patients present with parkinsonian features accompanied by dementia within the first year of disease onset, which leads to a diagnosis of DLB.
TABLE 30-1 Classification of Primary Autonomic Failure Disorders
| Acute/subacute primary autonomic failure disorders | Pure pandysautonomia |
| Pandysautonomia with neurological features | |
| Pure cholinergic dysautonomia | |
| Acute noradrenergic autonomic neuropathy | |
| Chronic primary autonomic failure disorders | Pure autonomic failure |
| Parkinson’s disease with autonomic failure | |
| Dementia with Lewy bodies | |
| Multiple system atrophy |
Actually, it is important to classify patients or assign them to these different disease entities for a number of reasons, including prognosis. Indeed, analyses of two large series in the United Kingdom and the United States indicate that patients with PAF have a substantially better prognosis than those with additional neurological deficits.2 There are differences within this latter group as well, inasmuch as patients with L-dopa–responsive Parkinson’s disease and autonomic failure appear to live longer than do patients with MSA or DLB (personal observations).
NEUROPATHOLOGY
Degeneration of autonomic neurons with disabling dysautonomia is a prominent feature of the Lewy body syndromes and MSA. α-Synuclein is a major component of the Lewy bodies in Parkinson’s disease, DLB, and the glial and neuronal cytoplasmic inclusions of MSA. α-Synuclein also is a major component of Lewy bodies in the brain and peripheral autonomic ganglia in PAF.3 Therefore, these disorders are increasingly being referred to as “synucleinopathies.” Abnormalities in the expression or structure of α-synuclein or associated proteins may cause degeneration of catecholamine-containing neurons.4 However, the function of α-synuclein is not known, but interest in this protein derives from the finding that the gene encoding for α-synuclein is mutated in families with the autosomal-dominant form of Parkinson’s disease.5 To investigate the consequence of α-synuclein overexpression in glia, Stefanova and colleagues6 transfected U373 astrocytoma cells with vectors encoding wild-type human α-synuclein or C-terminally truncated synuclein fused to red fluorescent protein. α-Synuclein immunocytochemistry of transfected astroglial cells revealed diffuse cytoplasmic labeling associated with discrete inclusions within both cell bodies and processes. Susceptibility to oxidative stress was increased in astroglial cells overexpressing α-synuclein, particularly in the presence of cytoplasmic inclusions. However, whether the α-synuclein aggregation is induced by some other factor or factors or whether it is the primary trigger of MSA pathology is unknown. Impairment in the ability of oligodendrocytes to degrade α-synuclein, which they may normally produce at low levels, may promote abnormal subcellular aggregation in MSA.7 Alternatively, ectopic expression of oligodendroglial α-synuclein may result in glial cytoplasmic inclusions. This scenario is supported by experimental studies demonstrating glial cytoplasmic inclusion–like inclusion pathology in transgenic mice overexpressing oligodendroglial α-synuclein.8 More work is necessary to elucidate the cascade of cell death in MSA and to determine exogenous and genetic susceptibility factors, both of which are likely to drive the disease process in this disorder. It is not known what determines whether α-synuclein precipitates in neurons (Parkinson’s disease, PAF) or glial cells (MSA) or on autonomic (PAF) or striatonigral neurons (Parkinson’s disease, MSA). Anyway, there are clear distinctions between the different α-synucleinopathies and little evidence of migration from one clinical form to the other.
Lewy Body Disorders
The Lewy body syndromes are characterized by intracytoplasmic eosinophilic neuronal inclusions, so-called Lewy bodies or Lewy neurites, found in the brain, including brainstem, basal ganglia, and cortical neurons, and in the peripheral autonomic nerves of affected patients. Lewy bodies contain abnormally phosphorylated intermediate neurofilament proteins, α-synuclein, ubiquitin, and associated enzymes. There are three different but overlapping phenotypes. In PAF there is early and widespread neuronal degeneration restricted mostly to peripheral autonomic neurons; autonomic failure is the sole clinical finding.9 In fact, in patients with PAF, intracytoplasmic eosinophilic inclusion bodies with the histological appearance of Lewy bodies, similar to those found in Parkinson’s disease, are identified in neurons of the substantia nigra, locus ceruleus, thoracolumbar and sacral spinal cord, and sympathetic ganglia and in peripheral sympathetic and parasympathetic nerves.9,10 Neuropathological reports of patients with PAF showed α-synuclein–positive intraneuronal cytoplasmic inclusions (Lewy bodies) in brainstem nuclei and peripheral autonomic ganglia.3,9 In Parkinson’s disease, there is prominent degeneration of the substantia nigra (Fig. 30-1) and other brainstem nuclei, in addition to peripheral autonomic neurons; clinically, there are motor abnormalities with varying degrees of autonomic failure.11 In DLB there is extensive cortical involvement in addition to degeneration of brainstem nuclei and peripheral autonomic neurons; clinical findings are dominated by severe cognitive impairment in association with parkinsonism and autonomic dysfunction.12 It is likely that the clinical phenotype of Lewy body syndromes depends on the temporal formation and distribution of Lewy bodies and associated neurodegeneration. Individual differences in neuronal susceptibility may determine the manifesting phenotype. Patients with PAF, however, can progress to Parkinson’s disease or DLB, which suggests that phenotypes overlap and that neurodegeneration in the Lewy body syndromes may start in postganglionic autonomic neurons and later affect neurons in the central nervous system. As initially suggested by Oppenheimer,13 PAF may be a “forme fruste” of Parkinson’s disease, with early severe widespread degeneration of peripheral autonomic neurons.9,10
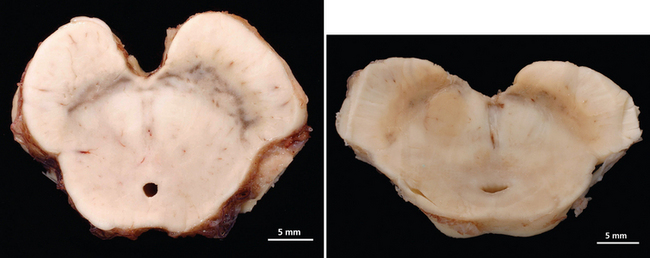
Multiple-System Atrophy
The second type of neurodegeneration with prominent autonomic failure is MSA. The term multiple-system atrophy was introduced in 196914; however, cases of MSA were previously reported under the rubrics of striatonigral degeneration (SND),15–17 olivopontocerebellar atrophy (OPCA),18,19 Shy-Drager syndrome,20 and idiopathic orthostatic hypotension. MSA is a sporadic neurodegenerative disorder characterized clinically by various combinations of parkinsonian, autonomic, cerebellar, or pyramidal symptoms and signs and pathologically by cell loss, gliosis, and glial cytoplasmic inclusions in several brain and spinal cord structures. Indeed, this disorder affects neurons in the basal ganglia, cortex, and spinal cord, but spares peripheral autonomic neurons. Pathologically, cytoplasmic inclusions are located in glial cells and do not form Lewy bodies.21 Clinically, two major motor presentations can be distinguished. Parkinsonian features predominate in 66% of patients (MSA-P), and cerebellar ataxia is the major motor feature in 34% of patients (MSA-C), according to a European survey.22 Severe autonomic failure is prominent in both phenotypes.23
In MSA-P, the striatonigral system is the main site of pathology, but less severe degeneration can be widespread and usually includes the olivopontocerebellar system.24,25 The putamen is shrunken with gray-green discoloration. When putaminal pathology is severe, there may be a cribriform appearance. In early stages, the putaminal lesion shows a distinct topographical distribution with a predilection for the caudal and dorsolateral regions.24 Degeneration of pigmented nerve cells occurs in the substantia nigra pars compacta, whereas cells of the pars reticulata are reported as normal. The topographical patterns of neurodegeneration involving the motor neostriatum, efferent pathways, and nigral neurons reflect their anatomical relationship and suggest a common denominator or “linked” degeneration.24
The location of the α-synuclein precipitates determines not only the presence or absence of movement disorders but also the characteristics of the autonomic cardiovascular abnormality. Autonomic failure in MSA is caused by dysfunction of (1) central and preganglionic efferent autonomic activity, (2) neuronal networks in the brainstem that control cardiovascular and respiratory function, and (3) the neuroendocrine component of the autonomic regulation via the hypothalamopituitary axis. In MSA, cell loss is reported in dorsal motor nucleus of the vagus.26 Catecholaminergic neurons in the rostral (C1 group) and caudal (A1 group) ventrolateral medulla, which are involved in the control of sympathetic outflow to the cardiovascular system and reflex control of vasopressin release, are also affected, as are neurons of the arcuate nucleus that are involved in cardiorespiratory interactions.27–30 Cell loss has also been described for the Edinger-Westphal nucleus and posterior hypothalamus,20 including the tuberomammillary nucleus.31 Papp and Lantos21 demonstrated marked involvement of brainstem pontomedullary reticular formation with glial cytoplasmic inclusions, which represented a supraspinal histological counterpart for impaired visceral function. Autonomic neuronal degeneration affects the locus ceruleus, too.32 It is noteworthy that there is not always a strong correlation between nerve cell depletion or gliosis and the clinical degree of autonomic failure. It is estimated that more than 50% of cells within the intermediolateral column must decay before symptoms become evident.13 Degeneration of sympathetic preganglionic neurons in the intermediolateral column of the thoracolumbar spinal cord is considered contributory to orthostatic hypotension. On the basis of only the reports in which formal cell counts have been made, it is apparent, with very few exceptions, that all cases of MSA with predominant pathology in either the striatonigral or olivopontocerebellar system show loss of intermediolateral cells.33 Orthostatic hypotension in MSA is caused by blunted autonomic and neuroendocrine reflexes as a result of afferent and central neuronal loss; postganglionic autonomic fibers, however, are spared.34 Disordered bladder, rectal, and sexual function in MSA-P and MSA-C have been associated with cell loss in parasympathetic preganglionic nuclei of the spinal cord. These neurons are localized rostrally in Onuf’s nucleus between sacral segments S2 and S3 and more caudally in the inferior intermediolateral nucleus chiefly in the S3 to S4 segments.35 Loss of corticotrophin-releasing factor neurons in the pontine micturition area may contribute to neurogenic bladder dysfunction.30 In the peripheral component of the autonomic nervous system, Bannister and Oppenheimer36 described atrophy of the glossopharyngeal and vagus nerves. No pathology has been reported in the visceral enteric plexuses or in the innervation of glands, blood vessels, or smooth muscles. Sympathetic ganglia have not often been examined in pathological studies of autonomic failure and have seldom been described quantitatively. In MSA with autonomic failure, there are either no obvious or mild abnormalities in sympathetic ganglia. Any morphological changes reported in sympathetic ganglionic neurons in MSA have tended to be nonspecific,37 exhibiting the normal age-related range of appearances, and published micrographs and counts have indicated at least a moderate density, and sometimes quite a high density, of surviving neurons.38 Enteric and parasympathetic ganglia have been studied only in a few instances.
A variety of other neuronal populations are noted to show cell depletion and gliosis with considerable differences in vulnerability from case to case. Varying degrees of abnormalities in the cerebral hemisphere, including Betz cell loss, were detected in pathologically proved MSA cases.32,33 Furthermore, anterior horn cells may show some depletion, but rarely to the same extent as that occurring in motor neuron disease.39
CLINICAL PRESENTATION
Acute/Subacute Primary Autonomic Failure Disorders
Pure Pandysautonomia and Pandysautonomia with Neurological Features
There is a clinical spectrum of acute autonomic neuropathies. Acute panautonomic neuropathy (pandysautonomia), characterized by severe widespread sympathetic and parasympathetic failure, is at one extreme. Guillain-Barré syndrome is at the other end of the spectrum, in which the brunt of the disorder is borne by the somatic nervous system. Pure acute panautonomic neuropathies are relatively rare. Actually, the majority of acute autonomic neuropathies have some minor somatic features. Dysautonomia may be restricted to the cholinergic system (acute cholinergic neuropathy), the adrenergic system, or other organ systems (e.g., motility disorders).40
In medical history, a definite entity of pure pandysautonomia involving both sympathetic and parasympathetic nervous systems with a subacute onset, monophasic course, and partial recovery without significant features of somatic peripheral neuropathy was first described by Young and colleagues in 1969. Actually, there had been some earlier reports of the condition in the literature, although it was not clearly defined.47 The disorder differs from other neurological causes of autonomic dysfunction in that normal function of the central nervous system is preserved. Furthermore, there are no or only minor features of peripheral somatic nervous system involvement. Since these first descriptions, a number of other cases of acute pandysautonomia have been reported, as well as some cases of pure cholinergic dysautonomia. Some cases of acute dysautonomia with significant sensory disturbances have been described; in some, but not all, there was electrophysiological and pathological evidence of loss of small-diameter myelinated and unmyelinated fibers.41 In 1994, Suarez and colleagues42 clarified the features of acute idiopathic autonomic neuropathy. Both sexes and all ages can be affected. The onset is acute or subacute. In approximately one half of affected patients, there is an antecedent viral infection. Several cases that followed Epstein-Barr virus infection have been described, in one of which Epstein-Barr virus DNA and antibody to the virus were found in the cerebrospinal fluid.43 The most common presenting features are symptomatic orthostatic hypotension (light-headedness, dizziness, syncope) and symptoms of gastrointestinal dysfunction (nausea, vomiting, diarrhea, constipation, and postprandial bloating) or sudomotor dysfunction (failure to sweat, causing heat intolerance and flushing). Other symptoms include numbness, tingling, bladder disturbances, and impotence. Neurological examination findings are normal in about one half the patients; the remainder have depressed reflexes and distal sensory impairment. The clinical course is monophasic. Recovery tends to be gradual and frequently incomplete. The cerebrospinal fluid protein level may be mildly elevated. In rare cases, there may be evidence of sensory neuropathy with sensory symptoms of minimal intensity (mainly thermal and pain hypoesthesia in distal areas). In most cases, nerve conduction studies yield normal results. Sural nerve biopsy in some cases has demonstrated reduction of myelinated fiber density, predominantly of small fibers, and axonal degeneration. Actually, in some cases, there are minimal signs of distal denervation in electromyographic-electroneurographic studies.44 In some acute neuropathies, such as pandysautonomia, small-fiber impairment is relatively pure, but it may also appear in disorders with prominent somatic damage, such as Guillain-Barré syndrome, in which autonomic failure worsens the prognosis.45
The cause of the condition remains uncertain. Pathological features include the presence of a small inflammatory mononuclear cell infiltrate in the epineurium. It is probably a form of acute idiopathic polyneuritis restricted to autonomic nerves with an immune-mediated pathogenesis similar to that of the Guillain-Barré syndrome. Together, the acute onset, frequent antecedent viral infection, selectivity of involvement by fiber type and autonomic level, and presence of perivascular mononuclear cell infiltration suggest that the underlying mechanism is likely to be immune mediated. The following differential diagnoses have to be kept in mind: botulism, acute autonomic neuropathy associated with Guillain-Barré syndrome, porphyria, diabetes, toxic causes, systemic lupus erythematosus, and other connective tissue diseases.41
Pure Cholinergic Dysautonomia
In pure cholinergic dysautonomia, clinical and laboratory features indicate only a cholinergic failure. A number of cases of pure cholinergic dysautonomia have been described in children. Clinical features include blurred vision, impaired lacrimation, dry mouth, constipation, urinary retention and incontinence, and absence of sweating. There is no postural hypotension. Excessive salivation and sweat secretion have been described in early disease stages. Cerebrospinal fluid findings are normal.41
Chronic Primary Autonomic Failure Disorders
Primary Autonomic Failure
Bradbury and Eggleston46 were the first to describe PAF in 1925. They used the term idiopathic orthostatic hypotension. Actually, the name pure autonomic failure was introduced by Oppenheimer as one of the primary autonomic failure syndromes. It is a sporadic, adult-onset, slowly progressive, neurodegenerative disorder of the autonomic nervous system.92 Clinically, it is characterized by an isolated impairment of the autonomic nervous system with no other neurological deficits.47 PAF affects men slightly more often than women, usually in their sixth decade. Its onset is slow, and symptoms begin developing insidiously for years as minimal impairment (nonspecific weakness and orthostatic intolerance). The patient may recall that symptoms first manifested several years before he or she sought medical treatment. Common symptoms causing the patient to seek medical advice include unsteadiness, lightheadedness, or faintness on standing. Questioning often elicits descriptions of aching in the neck or occiput only when standing; lying down relieves all symptoms. In general, orthostatic symptoms are more prominent after prolonged recumbency, as in the morning hours. Moreover, postural hypotension is exacerbated after mealtimes and physical exertion. Other contributory factors are heat, alcohol ingestion, coughing, and defecation.48,49 In fact, straining during evacuation or micturition elevates intrathoracic pressure and may result in symptomatic hypotension. Mathias and colleagues50 investigated the frequency of symptoms associated with orthostatic hypotension in PAF and MSA and found that more patients with PAF had syncope (91% vs. 45%), visual disturbances (75% vs. 53%), and suboccipital/paracervical “coat hanger” neck pain (81 vs. 53%) than did the patients with MSA. The reasons for this are unclear. Patients with PAF may also develop supine hypertension. Moreover, a decreased ability to sweat may be apparent, particularly in hot climates. Men found to have PAF may have sought advice about urinary tract symptoms (hesitancy, urgency, dribbling, and occasional incontinence). Other signs of dysautonomia, including erectile and ejaculatory dysfunction, an inability to appreciate orgasm, and retrograde ejaculation may be present, too. Women may experience urinary retention or incontinence as early symptoms. In patients with neurally mediated syncope, nausea and pallor, which are prominent signs of autonomic activation, occur before loss of consciousness. In contrast, in patients with PAF, these signs are noticeably absent, and consciousness is lost with little or no warning.51
Autonomic tests are abnormal: orthostatic hypotension, cardiovagal dysfunction, and hypo- or anhidrosis of the postganglionic type (see “Laboratory Assessment” section). A definitive diagnosis of orthostatic hypotension as the cause of symptoms is made when symptoms are reproduced while declines in systolic blood pressure of at least 20 mm Hg and diastolic pressure of at least 10 mm Hg are documented, within 3 minutes of standing. The diagnosis cannot be excluded with a single measurement of upright blood pressure that does not fulfill these criteria. Several measurements of orthostatic blood pressure, preferably early in the morning or after a meal, may be necessary. Patients with PAF also have decreased sinus arrhythmia and absent blood pressure overshoot during phase IV of the Valsalva maneuver, which indicates parasympathetic and sympathetic efferent dysfunction. PAF affects mainly efferent postganglionic neurons; afferent pathways and somatic neurons are not affected. Nevertheless, there is evidence of a preganglionic disorder in 22% of patients with PAF, which suggests that such patients actually may have some central component.44
In terms of differential diagnosis, PAF should be distinguished from other forms of neurogenic orthostatic hypotension, including peripheral somatic neuropathies with autonomic involvement (e.g., diabetes and amyloid), MSA, Parkinson’s disease, and DLB. There are no symptoms or signs of sensory, cerebellar, pyramidal, or extrapyramidal dysfunction in patients with PAF. In general, this allows a clinical distinction from other forms of neurogenic orthostatic hypotension. However, it cannot be determined whether a single patient with PAF will eventually develop more widespread, nonautonomic neuronal damage that leads to a diagnosis of MSA or, in rare cases, DLB. A number of warning signs, or “red flags,” for a clinical diagnosis of MSA have been operationally defined, and their frequency has been determined in a large cohort of European patients with MSA in a natural history study conducted by the European MSA-Study Group. Some of these features that are, if present, suggestive of MSA can be attributed, at least in part, to autonomic nervous system abnormalities. Abnormal respiration occurred in 42% to 60% of patients; its manifestations included inspiratory stridor (19% to 33%), involuntary deep sighs and/or gasps (34% to 37%), sleep apnea (13% to 18%), and excessive snoring (22% to 33%). Rapid eye movement (REM) sleep behavior disorder was present in 35% to 39%. Cold hands and/or feet were noted in 26% to 34%, whereas Raynaud’s phenomenon was recorded in only 6% to 7%.52,53 Although the specificity and positive predictive value of the red flags for a diagnosis of MSA have not been determined yet, they may serve as useful “soft signs” pointing toward a diagnosis of MSA. Because of the slow disease progression in PAF, most patients probably die before central nervous system involvement can become clinically evident. Apart from dysautonomia, these patients are otherwise normal and have a relatively good prognosis. Complications are usually related to falls and associated disorders.54
Parkinson’s Disease and Autonomic Failure
In Parkinson’s disease, extrapyramidal motor problems are the presenting features. Later in the disease process, patients may also suffer severe autonomic failure, which makes the clinical distinction from MSA difficult. As in Parkinson’s disease, some patients with MSA display motor deficits before autonomic failure is apparent, which complicates the distinction further. However, dysautonomia in Parkinson’s disease is rarely as severe as that in MSA. The uncommonly encountered patients with both Parkinson’s disease and autonomic failure are usually older and are often responsive to L-dopa. Although in most cases autonomic failure occurs late, there is a subgroup of patients with Parkinson’s disease who have clinically significant autonomic failure early in the course of the disease. Orthostatic hypotension is often the key clinical feature suggestive of autonomic failure. However, there are many causes of orthostatic hypotension, including side effects of antiparkinsonian therapy (such as L-dopa or selegiline), coincidental disease causing autonomic dysfunction (e.g., diabetes mellitus), or concomitant administration of drugs for an allied condition (e.g., antihypertensives or α-adrenoceptor blockers).55 Studies on patients with Parkinson’s disease indicate that selegiline can cause orthostatic hypotension independently of autonomic failure through mechanisms that are not clearly defined.56 Together, the confounding effect of antiparkinsonian drugs that often worsens orthostatic hypotension and difficulties in the differential diagnosis (particularly between Parkinson’s disease and MSA) make it difficult to estimate accurately the prevalence of autonomic dysfunction in patients with Parkinson’s disease. Studies that mistakenly include patients with MSA-P may overestimate the frequency of autonomic dysfunction in Parkinson’s disease or underestimate it if patients with both Parkinson’s disease and autonomic dysfunction are diagnosed as MSA-P.54
In a retrospective study, almost one third of patients with Parkinson’s disease confirmed with post mortem examination had autonomic dysfunction documented in their clinical records.57 However, it has to be kept in mind that this retrospective method may underestimate the frequency of autonomic failure. Actually, bladder dysfunction (such as urgency, frequency, and incontinence) and decreased gastrointestinal motility represent the most frequent autonomic problems in Parkinson’s disease. Constipation is extremely common. Moreover, intestinal pseudo-obstruction and toxic megacolon may occur. Sexual dysfunction (loss of libido and erectile failure) is common in this disorder.54
In a study of patients whose Parkinson’s disease was diagnosed by means of clinical criteria, almost two thirds of subjects had orthostatic hypotension with symptoms of cerebral hypoperfusion, including syncope, when tested on a tilt table for 40 minutes or until symptoms developed.58 Because patients with normal responses and with orthostatic hypotension were taking similar drug regimens, antiparkinsonian medication was not the main cause of orthostatic hypotension. Senard and colleagues59 found a fall of at least 20 mm Hg of systolic blood pressure in almost 60% of patients with Parkinson’s disease. There was symptomatic orthostatic hypotension in 20% of the patients. It was related to duration and severity of the disorder, as well as with the use of higher daily L-dopa and bromocriptine dosages.59 A higher prevalence of symptomatic orthostatic hypotension (78%) was found in a retrospective study on patients with neuropathologically confirmed Parkinson’s disease.60 In an earlier study, vagal control of the heart and hemodynamic response to standing were impaired and related to duration of the clinical features of Parkinson’s disease.61
Between 20% and 40% of patients with Parkinson’s disease become demented in the course of their illness.62 Operational criteria defining the clinical boundaries between Parkinson’s disease and Parkinson’s disease with dementia (PDD) are lacking, although this distinction may have profound clinical implications for prognosis and treatment strategies.63 The criteria in Diseases and Statistical Manual of Mental Disorders (Fourth Edition, DSM-IV™) are incomplete and descriptive and do not describe several core clinical features associated with dementia in Parkinson’s disease. Peralta and colleagues showed that orthostatic hypotension is more frequent and more severe in patients with PDD than in those with Parkinson’s disease. Attentional scores during tilt testing were also more reduced in patients with PDD in comparison with those with Parkinson’s disease, which suggests that orthostatic hypotension may exacerbate cognitive dysfunction in patients with PDD.64
Dementia with Lewy Bodies
DLB is the most frequent cause of degenerative dementia after Alzheimer’s disease. Whether DLB and PDD are the same or different disorders is uncertain.65 Clinically, the central feature required for a diagnosis of DLB is progressive cognitive decline, severe enough to cause social and occupational functional impairment. Core features of DLB are fluctuating cognition, recurrent and persistent visual hallucinations, and extrapyramidal motor symptoms. Supportive features may increase diagnostic sensitivity. They include repeated falls, syncope, transient loss of consciousness, neuroleptic sensitivity, systematized delusions, and hallucinations in other modalities.
The two main differential diagnoses are Alzheimer’s disease and PDD. In order to improve the differential diagnosis of DLB, consensus criteria that establish possible and probable levels of diagnostic accuracy have been developed.12,66 In general, their sensitivity is variable and low, but their specificity is high. Current consensus is to restrict a diagnosis of DLB only to patients with parkinsonism who develop dementia within 12 months of the onset of motor symptoms. With the use of operationally defined criteria, DLB can be clinically diagnosed with an accuracy similar to that achieved for Alzheimer’s disease or Parkinson’s disease.
Autonomic failure is frequent in DLB. A retrospective analysis of autonomic symptoms in neuropathologically diagnosed DLB showed that 62% of affected patients had significant autonomic failure.67 Patients with DLB may suffer vocal cord palsy, which results in sudden death. However, autonomic function has not been well documented in patients with DLB. Some of the supportive features, including repeated falls, syncope, and transient loss of consciousness, can be attributed in part to autonomic nervous system abnormalities. Orthostasis, either asymptomatic or associated with syncope, may be observed in these patients, although symptomatic orthostatic hypotension has been found in a lower frequency (15%) than in other types of parkinsonism.60
Mean age at onset is 75 years; the age range is 50 to 80 years, with a slight male predominance.68 Although dementia is the most frequent presenting feature, psychiatric symptoms or transient alterations of consciousness are other early features. Indeed, affected patients may present with recurrent visual hallucinations even without exposure to dopaminergic antiparkinsonian agents and may have marked diurnal fluctuations in cognitive performance, which have been the most difficult feature of the disease to define but are often conspicuous in the environment. Although parkinsonism is common in DLB, occurring at some point during the course of the illness in 75% to 80% of cases,69,70 a minority of patients present with parkinsonism alone. In general, autonomic features occur later in the course of the disease, but some cases have been described in which dysautonomia was the initial and prominent feature, leading to an initial misdiagnosis of MSA.71 Fluctuating cognition, probably related to fluctuations in attention, is characteristic of DLB, occurring in 58% of cases at the time of presentation and observed during the disease course in 75%.72 The natural history of the neuropsychological changes in DLB is not well characterized, although differences with Alzheimer’s disease appear particularly pronounced in the early stages and lessen as the disease progresses. A rapidly progressive dementia, accompanied by aphasia, dyspraxia, and spatial disorientation suggestive of temporoparietal dysfunction can be seen as the disease progresses. Disability in DLB progresses at a rate similar to that in Parkinson’s disease (approximately 10% decline per year) or even at a significantly faster rate. The latency to onset of orthostatic hypotension in a postmortem series of the National Institute of Neurologic Disorders and Stroke were short in MSA patients, intermediate in patients with DLB, corticobasal degeneration, and progressive supranuclear palsy (PSP) and long in those with Parkinson’s disease.60 These data underpin the rapidly progressive nature of the disease process in DLB in comparison with that of Parkinson’s disease. As a result, mean length of survival in a series of patients with DLB confirmed with post mortem examination has been less than 10 years. It is similar to that for Alzheimer’s disease, although some patients with DLB show rapid symptom progression and die within 1 to 2 years of onset. Risk factors for increased mortality in DLB that are present at disease onset include older age, dementia, fluctuating cognition, and hallucinations.73 Strikingly, patients with DLB with neuroleptic sensitivity reactions show a twofold to threefold increase in mortality.
Multiple-System Atrophy
This disease affects both men and women, usually starts in the sixth decade, and progresses relentlessly, with a mean survival length of 6 to 9 years.74–77 There is considerable variation in disease progression, with survival lengths of more than 15 years in some instances.
Clinically, cardinal features include autonomic failure, parkinsonism, cerebellar ataxia, and pyramidal signs in various combinations. Previous studies suggest that 29% to 33% of patients with isolated late-onset cerebellar ataxia and 8% of patients with parkinsonism eventually develop MSA.78–80 Of importance, both motor presentations of MSA are associated with similar survival times.76 However, patients with MSA-P have a more rapid functional deterioration than do patients with MSA-C.74
MSA-P associated parkinsonism is characterized by progressive akinesia and rigidity. Jerky postural tremor and, less commonly, tremor at rest may be superimposed. Frequently, patients exhibit orofacial or craniocervical dystonia in association with a characteristic quivering, high-pitched dysarthria. Postural stability is compromised early on; however, recurrent falls at disease onset are unusual, in contrast to PSP. Differentiating between MSA-P and Parkinson’s disease may be exceedingly difficult in the early stages because of a number of overlapping features such as rest tremor or asymmetrical akinesia and rigidity. Furthermore, L-dopa–induced improvement of parkinsonism may be seen in 30% of MSA-P patients. However, the benefit is transient in most of these subjects, leaving 90% of the MSA-P patients unresponsive to L-dopa in the long term. L-Dopa–induced dyskinesias affecting orofacial and neck muscles occur in 50% of MSA-P patients, sometimes in the absence of motor benefit.81 In most instances, a fully developed clinical picture of MSA-P evolves within 5 years of disease onset, allowing a clinical diagnosis during follow-up.82
The cerebellar disorder of MSA-C comprises gait ataxia, limb kinetic ataxia, and scanning dysarthria, as well as cerebellar oculomotor disturbances. Patients with MSA-C usually develop additional noncerebellar symptoms and signs but, before doing so, may be indistinguishable from other patients with idiopathic late-onset cerebellar ataxia, many of whom have a disease restricted clinically to cerebellar signs and pathologically to degeneration of the cerebellum and olives.78
Dysautonomia is characteristic of both MSA motor presentations, comprising primarily urogenital and orthostatic dysfunction. During the early stages of MSA, autonomic deficits may be the sole clinical manifestation, thus resembling PAF, but after a variable period of time (sometimes 2 or 3 years, always less than 5), extrapyramidal or cerebellar deficits or both invariably develop. Early impotence (erectile dysfunction) is virtually universal in men with MSA, and urinary incontinence or retention, often early in the course or as presenting symptoms, are frequent.77 Disorders of micturition in MSA are caused by changes in the complex peripheral and central innervation of the bladder83 and generally occur more commonly, earlier, and to a more severe degree than in Parkinson’s disease. In fact, patients with MSA have early dysuria with or without chronic retention, frequently associated with a hypoactive detrusor muscle and low urethral pressure. In contrast, patients with Parkinson’s disease have urgency to void, with or without difficulty, but without chronic retention, in association with detrusor hyperreflexia and normal urethral sphincter function. Constipation occurs in equal percentages of patients in Parkinson’s disease and MSA. Symptomatic orthostatic hypotension is present in 68% of patients with clinical diagnoses of MSA, but recurrent syncope emerges in only 15%.77 L-Dopa or dopamine agonists may provoke or worsen orthostatic hypotension.
LABORATORY ASSESSMENTS
In addition to the clinical presentation, several laboratory investigations have been used to distinguish among Parkinson’s disease, PAF, and MSA (Table 30-2). Basically, most of these tests exploit the anatomopathological distinction between Lewy body syndromes, which affect postganglionic autonomic neurons, and MSA, which affects preganglionic, central autonomic neurons.
TABLE 30-2 Laboratory Investigations in Primary Autonomic Failure
| Examined Body Domain/Function | Parameters/Techniques |
|---|---|
| Cardiovascular | |
| Physiological | Head-up tilt (60 degrees); standing; Valsalva maneuver |
| Pressor stimuli (isometric exercise, cutaneous cold, mental arithmetic) | |
| Heart rate responses-deep breathing, hyperventilation, standing, head-up tilt, 30:15 R-R interval ratio | |
| Liquid meal challenge | |
| Modified exercise testing | |
| Carotid sinus massage | |
| Biochemical | Plasma noradrenaline: supine and head-up tilt or standing; urinary catecholamines; plasma renin activity, and aldosterone |
| Pharmacological | Noradrenaline: α-adrenoceptors, vascular |
| Isoprenaline: β-adrenoceptors, vascular and cardiac | |
| Tyramine: pressor and noradrenaline responses | |
| Edrophonium: noradrenaline response | |
| Atropine: parasympathetic cardiac blockade | |
| Imaging | Cardiac [123I]MIBG SPECT, 6-[18F] fluorodopamine PET |
| Brain | |
| Imaging | MRI (1.5 Tesla), diffusion-weighted imaging, voxel-based morphometry, [(123)I]β-CIT, [123I]iodobenzamide SPECT, 18F-fluorodopa PET, [11C]diprenorphine PET, 18F-fluorodeoxyglucose PET, 99mTc-hexamethylpropyleneamine oxime, [123I]FP-CIT |
| CSF studies | Neurofilament protein levels |
| Endocrine | Clonidine–α2-adrenoceptor agonist: noradrenaline suppression; growth hormone stimulation |
| Sudomotor | Thermoregulatory sweat test |
| Sweat gland response to intradermal acetylcholine, QSART, localized sweat test | |
| Sympathetic skin response | |
| Gastrointestinal | External anal sphincter EMG, video-cinefluoroscopy, barium studies, endoscopy, gastric emptying studies, transit time, lower gut studies |
| Renal function and urinary tract | Day and night urine volumes and sodium/potassium excretion measurements |
| Urodynamic studies, intravenous urography, ultrasonographic examination, sphincter electromyography | |
| Sexual | Penile plethysmography |
| Intracavernosal papaverine | |
| Respiratory | Laryngoscopy |
| Sleep studies to assess apnea and oxygen desaturation | |
| Eye and lacrimal glands | Pupillary function, pharmacological and physiological |
| Schirmer’s test | |
11C, carbon 11; β-CIT, 2β-carboxymethoxy-3β-(4-iodophenyl)tropane; CSF, cerebrospinal fluid; EMG, electromyography; 18F, fluorine 18; 123I, iodine 123; FP-CIT, 2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; MIBG, meta-iodobenzylguanedine; MRI, magnetic resonance imaging; PET, positron emission tomography; QSART, quantitative sudomotor axon reflex test; SPECT, single photon emission computed tomography.
Modified from Mathias CJ: Autonomic diseases: clinical features and laboratory evaluation. J Neurol Neurosurg Psychiatry 2003; 74(Suppl 3):iii31-iii41.
Cardiovascular Function Testing
A history of postural faintness or other evidence of orthostatic hypotension, such as neck ache on rising in the morning or posturally related changes of visual perception, should be sought in all patients in whom MSA is suspected. After a comprehensive history is documented, cardiovascular function should be tested according to consensus recommendations.47,84 A drop in systolic blood pressure of 20 mm Hg or more or in diastolic blood pressure of 10 mm Hg or more, in comparison with baseline within a standing time of 3 minutes, is defined as orthostatic hypotension and must lead to more specific assessment. This is based on continuous noninvasive measurements of blood pressure and heart rate during tilt-table testing.85–87 Although abnormal cardiovascular test results may provide evidence of sympathetic and/or parasympathetic failure, they do not differentiate autonomic failure associated with Parkinson’s disease from that associated with MSA.88
The autonomic abnormality of MSA can be distinguished biochemically from that of PAF. In MSA, during supine rest, norepinephrine (noradrenaline) levels (representing postganglionic sympathetic efferent activity) are normal,89,90 and there is no denervation hypersensitivity, which indicates a lack of increased expression of adrenergic receptors on peripheral neurons.90 In contrast to this normal or only slightly decreased plasma norepinephrine level during recumbency in MSA and varying levels in patients with Parkinson’s disease, patients with PAF have very low plasma norepinephrine levels when recumbent.90,91 On standing or tilt-table testing, patients with PAF, those with MSA, and some with Parkinson’s disease with autonomic failure do not have the expected increase in plasma norepinephrine levels, which indicates an inability to normally stimulate the release of catecholamines by baroreflex activation in all these disorders. When norepinephrine is infused into patients with PAF, there is an exaggerated increase in blood pressure. This reflects an excessive sensitivity of postsynaptic α-adrenergic receptors to exogenous catecholamines. In contrast, patients with MSA and Parkinson’s disease show only a mildly increased blood pressure response to infused norepinephrine, without leftward shift in the dose-response curve.92 Similarly, there is a greater degree of β-adrenergic receptor supersensitivity in PAF than in MSA, as shown by Baser and associates93 in a study with intravenous isoproterenol.
Sympathetic cardiac innervation is selectively affected in Parkinson’s disease and PAF but is intact in MSA. Imaging studies that measure catecholamine uptake by cardiac sympathetic neurons have confirmed that peripheral sympathetic nerves are preserved in MSA but greatly reduced in PAF.94 Visualization of sympathetic cardiac neurons through scintigraphy with norepinephrine analogue iodine 123–metaiodobenzylguanidine ([123I]MIBG) has revealed loss of binding in patients with Parkinson’s disease, regardless of disease severity, which reflects postganglionic sympathetic denervation; in comparison, cardiac binding is preserved in MSA95–100 and PSP.101 Pooled data from several studies showed that MIBG scintigraphy occurately discriminated a total of 246 cases of Parkinson’s disease from 45 of MSA with high sensitivity (90%) and specificity (95%).96 Similarly, (18F) fluorodopamine positron emission tomography (PET) is able to demonstrate cardiac sympathetic denervation in PAF and Parkinson’s disease in contrast with intact cardiac sympathetic innervation in MSA.94 6-[18F] Fluorodopamine is a catecholamine taken up by sympathetic post ganglionic neurons and handled similarly to norepinephrine. Together, these types of imaging of sympathetic cardiac neurons may turn out to be useful diagnostic tests to distinguish between Parkinson’s disease and MSA because sympathetic innervation of the heart is impaired in Parkinson’s disease and not in MSA. Moreover, in a patient with apparent PAF, finding normal sympathetic cardiac innervation should indicate a likely development of MSA.4 A caveat of this approach to be kept in mind is that published studies have compared patients with established diagnoses of MSA and PAF and, therefore, probably in later disease stages. It is not known whether these differences are apparent in patients during earlier stages of the disorder, when a diagnostic method would be more useful in the workup of patients in clinical practice.92






