Ovarian Sex Cord–Stromal and Steroid Cell Tumors
General Features
Sex cord–stromal tumors account for approximately 8% of all ovarian tumors and are the most common functioning tumors associated with endocrine manifestations.1 These tumors contain granulosa cells, theca cells (and their luteinized derivatives), Sertoli cells, Leydig cells, and fibroblasts of gonadal stromal origin, either separately or in combination and exhibiting varying degrees of differentiation.1
Sex cord–stromal tumors arise from ovarian cells specialized in steroid hormone production. The designation sex cord–stromal tumors (which is favored by the World Health Organization (WHO) over gonadal stromal tumors) simply reflects the uncertainty about the origin of the gonadal sex cords;2 it is still unclear whether they derive from the celomic epithelium (sex cords) or the gonadal mesenchyme (stroma). Sex cords are identified in the developing testis by the fifth week of embryonic life but are not found in the developing ovary where nests of pregranulosa cells (of apparent stromal origin) enveloping the germ cells are evident later in embryonic life.1 Some authors do not accept the term ‘sex cords’ to describe progenitors of granulosa cells. However, the hyphenated term (sex cord–stroma) acknowledges that this category of ovarian tumors may be composed of sex cord derivatives (granulosa and Sertoli cells) that appear as epithelial elements, stromal derivatives (theca, lutein, and Leydig cell) that have the appearance of mesenchymal (gonadal stromal) cells, or derivatives of both precursors.1,2
Tumors composed of ovarian cell types are usually estrogenic, whereas those composed of cells of testicular type are androgenic, but tumors in either group may be nonfunctioning; rarely, tumors made up of female elements are androgenic, and tumors composed of Sertoli and Leydig cells are associated with estrogenic manifestations. Therefore, there is not always a strict correlation between the morphology of the tumor cells and the type of hormone they secrete.2
Because of the great diversity of histologic patterns, evaluation of sex cord–stromal tumors requires awareness of the patient’s age and clinical history, thorough sampling of the tumor, and immunohistochemical validation. The most helpful markers currently available are α-inhibin, calretinin, and epithelial membrane antigen (EMA). The first two are typically positive in sex cord–stromal tumors and the third is typically negative.3–5 Recently, a likely ‘driver’ point mutation of the FOXL2 gene has been identified in the vast majority of adult granulosa cell tumors (97%) but only rarely in juvenile granulosa cell tumors (10%).6 FOXL2, together with α-inhibin and calretinin, forms an immunomarker panel that will result in positive immunoreaction in essentially all cases of sex cord–stromal tumors.7
The distribution of the various sex cord–stromal tumors at the Massachusetts General Hospital over a 25 year period was as follows: tumors in the thecoma–fibroma group (87%), GCTs (12%), Sertoli–Leydig cell tumors (SLCTs) (0.05%), and unclassified (0.05%).1 Most clinically malignant sex cord–stromal tumors are GCTs.
Granulosa Cell Tumors
Granulosa cell tumors (GCTs) comprise two different clinicopathologic subtypes: adult and juvenile.1,2 The adult form occurs more often in middle-aged and postmenopausal women and characteristically contains microfollicles and uniform cells with scanty cytoplasm and pale, grooved nuclei; in contrast, the juvenile subtype occurs mainly in children and younger women, and typically shows large rudimentary follicles and cells with moderate to abundant cytoplasm and darker nuclei usually without grooves.1,2
Adult Granulosa Cell Tumor
Clinical Features
Adult granulosa cell tumors (AGCTs) account for 1–2% of all ovarian tumors and 95% of all GCTs. They occur mainly in menopausal and postmenopausal women of about 50–55 years of age and are the most common ovarian tumors associated with estrogenic symptoms (75% of cases).1, 8–12
Functioning AGCTs (which are typically estrogenic tumors) may be associated with various clinical syndromes depending upon the age of the patient: women in the reproductive age group usually present with irregular and excessive uterine bleeding (metropathia hemorrhagica), but amenorrhea, lasting from months to years, may precede the abnormal bleeding, or may be the only endocrine symptom.1 In postmenopausal women, the most common symptom is postmenopausal bleeding secondary to breakdown of endometrial hyperplasia or carcinoma. Occasionally, swelling, tenderness, and discharge from the breasts are prominent. Vaginal cytology typically shows increased maturation of squamous epithelial cells. Most of the clinical symptoms are related to the continuous estrogen stimulation of the endometrium: luteinizing hormone (LH) secretion from the anterior pituitary stops; ovulation does not occur, and progesterone is not secreted; the estrogenic stimulation of the endometrium results in endometrial hyperplasia (usually simple but occasionally complex atypical hyperplasia). Endometrial carcinoma occurs in approximately 5% of cases1 and is almost always a grade 1 endometrioid adenocarcinoma, which never metastasizes and almost never causes the death of the patient. Rare cases of AGCT appear before puberty and are associated with isosexual pseudoprecocity, due to estrogen production by the ovarian tumor. Although AGCTs are mostly estrogenic, very rare cases, usually large thin-walled cystic tumors, are associated with androgenic manifestations.13
In addition to endocrine symptoms, patients with AGCTs have signs and symptoms related to a pelvic mass such as abdominal pain and swelling; the tumor is palpable on pelvic examination in almost 90% of cases. Approximately 10% of patients present with acute abdominal symptoms due to rupture of the tumor with hemoperitoneum.1 AGCTs are confined to the ovary (International Federation of Gynecology and Obstetrics stage I) in 60–90% of cases.9–11,14 Extraovarian spread occurs to the peritoneum and omentum and occasionally to the liver or lungs.15 Lymph node metastases are uncommon.
Macroscopic Features
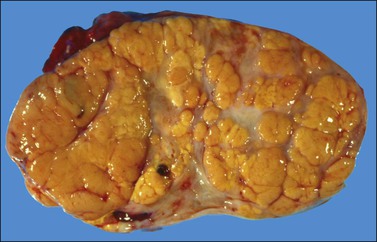
The tumors are unilateral in over 95% of cases. About 10–15% are ruptured. The average diameter is 12 cm but their size ranges from microscopic to very large masses. They are cystic and solid and the cysts characteristically contain blood clots. The solid component varies from yellow (Figure 28.1) to white depending upon the amount of lipid-containing cells, and from soft to firm depending on the quantity of granulosa cells and stromal cells.
Microscopic Features
Histologically, there is a proliferation of granulosa cells often with a stromal component of theca cells, fibroblasts, or both. The granulosa cells grow in a wide variety of patterns, which are commonly admixed and lack prognostic significance. The most common pattern is the diffuse pattern in which the tumor cells grow in sheets with no organized structure (Figure 28.2). More characteristic but less common (30–50%) is the microfollicular pattern, which shows numerous rosette-like structures that simulate the Call–Exner bodies of the Graafian follicle (Figures 28.3 and 28.4). These small spaces contain eosinophilic debris or hyalinized basement membrane material and are surrounded by a circular row of typical granulosa cells. The cells contain scanty cytoplasm and pale, angular, or oval, often grooved, nuclei arranged haphazardly in relation to one another and to the follicles (Figure 28.4). The macrofollicular pattern is uncommon and shows cysts lined by well-differentiated granulosa cells and underlying theca cells (Figures 28.5). In totally cystic tumors, the cells lining the cysts may so closely mimic those of non-neoplastic ovarian follicles that distinction on a purely histologic basis is difficult. Tumor cells grow in anastomosing bands in the trabecular pattern, in undulating ribbons and cords in the gyriform, or in a watered-silk pattern (Figure 28.6), and in circumscribed nests and islands in the insular pattern (Figure 28.7). Some tumors may exhibit a diffuse sarcomatoid pattern characterized by somewhat spindle-shaped cells. Whatever the histologic pattern may be, the best morphologic markers of the AGCT are (1) the presence of Call–Exner bodies (microfollicular) (Figure 28.4) and (2) the cell nuclei, which appear uniform and haphazardly oriented to one another and are oval, round, and angular and pale, often grooved, and lack significant pleomorphism (Figure 28.8).
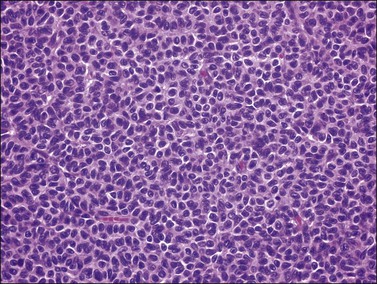
Figure 28.2 Adult granulosa cell tumor. Diffuse pattern. The regular-sized nuclei appear in a disorderly arrangement.
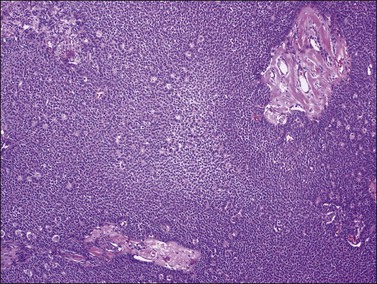
Figure 28.3 Adult granulosa cell tumor. Microfollicular pattern. Numerous Call–Exner bodies are seen.
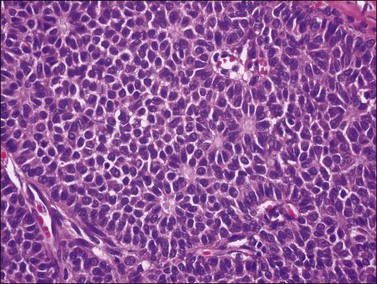
Figure 28.4 Adult granulosa cell tumor. Microfollicular pattern. Numerous Call-Exner bodies containing degenerated material are surrounded by granulosa cells with grooved nuclei.
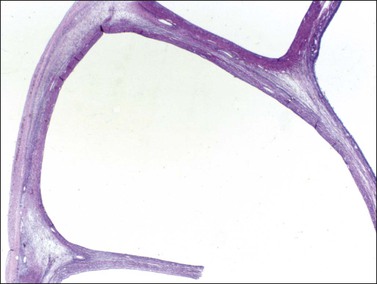
Figure 28.5 Adult granulosa cell tumor. Macrofollicular pattern. The large cystic cavities resemble follicle cysts.
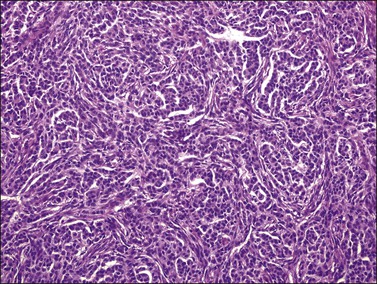
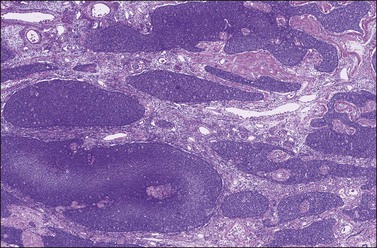
Figure 28.7 Adult granulosa cell tumor. Insular and microfollicular patterns. The tumor cells grow in well-defined islands containing Call–Exner bodies.
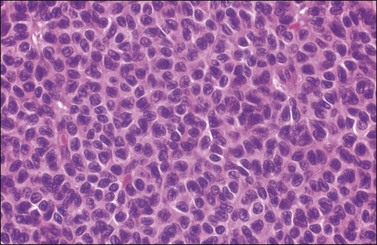
Figure 28.8 Adult granulosa cell tumor. The tumor cells have pale angular nuclei, many of which are grooved.
Approximately 2% of AGCTs contain scattered cells with bizarre, large, hyperchromatic nuclei (Figure 28.9).1 The mitotic activity in AGCTs varies, but is usually low; most tumors have 1–2 mitoses per 10 HPF. The presence of numerous mitoses, particularly abnormal forms, should raise the suspicion of poorly differentiated carcinoma.
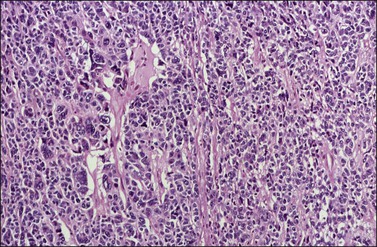
Figure 28.9 Adult granulosa cell tumor with bizarre nuclei. Large, hyperchromatic nuclei, including multinucleated forms, are present.
The stromal component of AGCTs varies from fibromatous to thecomatous (granulosa–theca cell tumor) and it is best enhanced by the reticulin stains (Figure 28.10). In contrast to theca cells and fibroblasts, which are individually invested by fibrils, the granulosa cell component of the tumor contains few fibrils.1 Occasional GCTs with hepatocellular differentiation have been reported.16,17 In contrast to lutein or Leydig cells, the hepatic-type cells are negative for inhibin.17
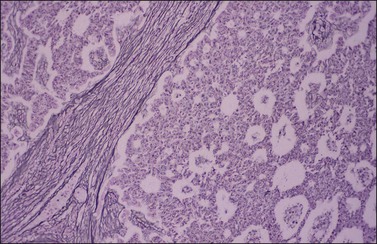
Immunohistochemistry
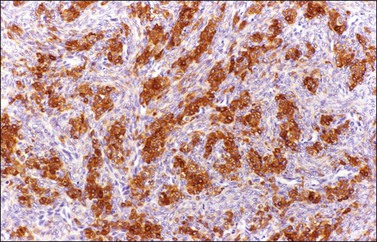
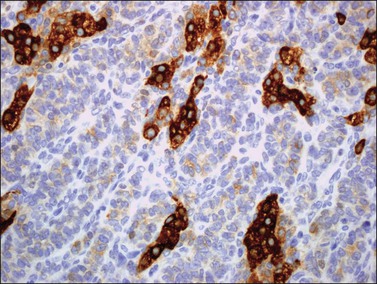
α-Inhibin (Figure 28.11), calretinin, and FOXL2 (Figure 28.12) are the most important positive immunoreactions.7 Calretinin is a more sensitive but less specific marker than α-inhibin.18,19 Steroidogenic factor-1 (SF-1), WT1, and CD56 are also positive.19,20 Almost all GCTs are vimentin positive. Monoclonal antibodies against low molecular weight cytokeratins (CK) 8 and 18 (CAM5.2, AE1/3) stain 30–60% of the tumors.3,21 Most GCTs are immunoreactive for smooth muscle actin but negative for desmin.21 Positive nuclear or cytoplasmic stain for S-100 protein has been reported in 50% of the tumors.3,21 There is membrane staining for CD99 (MIC2 gene product) in 70% of GCTs.22 Positive reaction for müllerian-inhibiting substance (MIS) has also been reported.23 The most significant negative immunoreactions are for EMA and CK7, which are almost always absent in GCTs.3,21
Genetic Profile
Over 80% of the GCTs are diploid. Cytogenetic abnormalities have been detected by comparative genomic hybridization and fluorescence in situ hybridization (FISH) and include trisomy 12 and 14 and monosomy 22.24,25 Over 90% of GCTs contain a missense point mutation 402 C→G (C134W) in the FOXL2 gene at 3q22.3, presumably an early event in tumorigenesis.6
Differential Diagnosis
1. Undifferentiated carcinoma versus diffuse AGCT. The typical nuclear features of AGCT (Figure 28.8) differ from those of undifferentiated carcinoma, namely nuclear hyperchromatism, pleomorphism, and atypical mitotic activity; carcinomas are often associated with higher stage and adverse clinical course. In difficult cases, positive immunostains for α-inhibin, calretinin, and vimentin and lack of staining for EMA favor the diagnosis of AGCT. FOXL2 immunoreaction is helpful (see Chapter 25, and Table 28.1).3,7,26,27
Table 28.1
Differentiation between AGCT and Undifferentiated Carcinoma
| AGCT | Undifferentiated Carcinoma | |
| Bilaterality | 5% | 15% |
| Stage | I | III |
| Nuclei | Paled, grooved, uniform | Hyperchromatic, pleomorphic |
| Prognosis | Favorable | Poor |
2. Small cell carcinoma of hypercalcemic type versus diffuse AGCT. The small cell carcinoma is often found in advanced stage and has an aggressive clinical course; it is associated with hypercalcemia in two-thirds of patients, lacks estrogenic symptoms, and contains hyperchromatic nuclei and numerous mitoses. Immunohistochemical staining may facilitate the diagnosis (see Chapter 27).
3. Endometrioid stromal sarcoma (primary or metastatic from the uterus) versus diffuse AGCT. The stromal sarcoma lacks endocrine symptoms and is frequently bilateral; microscopically, it shows numerous small arterioles, individual investment of tumor cells by reticulin fibers, and tongue-like pattern of infiltration mainly outside the ovary. Positive immunoreactions for α-inhibin, calretinin, and FOXL2 and negative CD10 staining support the diagnosis of AGCT (see Chapter 27).
4. Thecoma and cellular fibroma versus diffuse AGCT. The distinction may be aided by reticulin stains.
5. Large solitary luteinized follicle cyst of pregnancy and puerperium versus unilocular cystic AGCT. Patients with the former lesion have a history of recent pregnancy. The follicle cyst shows large luteinized cells with large bizarre nuclei. In contrast, the unilocular cystic AGCT is lined by non-luteinized cells with typical granulosa cell nuclei (see Chapter 24).
6. Endometrioid carcinoma (well differentiated and microglandular, resembling a sex cord–stromal tumor; sertoliform carcinoma) versus microfollicular, trabecular, or insular AGCT. Foci of typical endometrioid carcinoma with mucin-secreting glands are usually found in the former; also, carcinomatous (atypical) nuclei with numerous and often abnormal mitoses are seen. Squamous differentiation and adenofibromatous (müllerian) features are common. In carcinomas, α-inhibin and calretinin immunostains are negative (but positive in luteinized stromal cells), whereas cytokeratins and EMA are strongly positive (see Chapter 27).23,26
7. Insular carcinoid tumor versus microfollicular AGCT. The different nuclear features, calcification of acini of carcinoid tumors, and neuroendocrine markers should facilitate the differential diagnosis (see Chapter 29).
8. Sex cord tumor with annular tubules (SCTAT) versus microfollicular AGCT. The former tumor shows larger hyaline deposits than Call–Exner bodies as well as ring-shaped tubules; in cases associated with the Peutz–Jeghers syndrome (PJS) the SCTATs are small, multiple, and bilateral.
9. Gonadoblastoma versus insular or microfollicular AGCT. Background of intersexual condition, presence of germ cells, and larger hyaline deposits than Call–Exner bodies all favor gonadoblastoma (see Chapter 29).
10. Metastatic melanoma versus GCT. Histologic features of melanoma, melanin pigment, positive S-100, and HMB-45 and negative α-inhibin and calretinin all favor the former diagnosis (see Chapter 30).
11. Metastatic breast carcinoma (particularly of lobular type) versus diffuse AGCT. Bilaterality, history of breast cancer, presence of intracellular mucin, negative α-inhibin, positive EMA, and GCDFP-15 are all features of metastatic breast cancer (see Chapter 30).
Spread and Metastasis
All AGCTs are potentially malignant and capable of extending beyond the ovary, or recurring after apparently complete removal. Spread occurs mainly within the pelvis and lower abdomen; although rare, distant metastases have been reported in many locations.15 Most recurrences appear within 5 years but they can be detected many years postoperatively (occasionally after 20 or 30 years).1
Treatment and Prognosis
In menopausal or postmenopausal women, the optimal treatment is bilateral salpingo-oophorectomy with total hysterectomy. It is reasonable to conserve the opposite ovary and uterus of a young woman with an AGCT confined to one ovary. The recurrence rate is 10–15% for stage IA tumors and 20–30% overall. Recurrence is usually fatal, but reoperation, radiotherapy, and chemotherapy (similar to that used in malignant germ cell tumors) can be helpful.28
The 10 year survival varies from 60% to 90%.8–12 Patients with stage I tumors have much better prognosis than those with higher stage tumors (86% vs 49% 10 year survival).9 The most consistent indicator of aggressive behavior has been the presence of metastases or invasion of structures outside the ovary at the time of diagnosis.9,12 Also unfavorable but less significant factors are large tumor size (>6 cm in diameter)11 and tumor rupture.9 There is no correlation between the microscopic pattern and the clinical outcome. Flow cytometric results regarding DNA ploidy are contradictory.29
Juvenile Granulosa Cell Tumor
Clinical Features
Juvenile granulosa cell tumors (JGCTs) account for only 5% of GCTs and have been so designated because they usually occur in women less than 30 years of age, half of them in the first decade of life.1,30 These tumors differ histologically from the more common AGCTs and contain cells that appear less mature; they are often mistaken for other neoplasms, usually malignant germ cell tumors.
JGCTs occurring before puberty are associated in 80% of cases with isosexual pseudoprecocity, due to estrogen production by the ovarian tumor.30 They account for 10% of cases of sexual precocity. The large majority of cases (90%) of sexual precocity are central (idiopathic or constitutional), and result from premature release of gonadotropins by the hypothalamus (anterior pituitary gland) with no detectable organic lesion. In these cases (true precocity) ovulation occurs and there is a possibility of pregnancy. This does not occur in the cases of pseudoprecocity secondary to estrogen production by a functioning ovarian tumor. The GCTs that occur before puberty are almost always palpable on pelvic or rectal examination.1,30 Noteworthy, other ovarian tumors (of non-endocrine type) secreting human chorionic gonadotropin (hCG) can also be associated with isosexual pseudoprecocity in young females.1 In adult women, however, the most common symptom of the JGCT is uterine bleeding as a result of endometrial hyperplasia.1,30
In the largest series reported,30 which included 125 JGCTs, 44% occurred in the first decade, 34% in the second decade, 18% in the third decade, and only 3% after the age of 30 years. Eighty-two percent of the tumors encountered before the age of normal puberty were associated with isosexual pseudoprecocity. Eight cases of JGCTs that developed in patients with Ollier disease (multiple enchondromatosis),31 and three with Maffuci syndrome (Ollier disease with hemangiomas) have been reported, indicating that the JGCT is a rare component of these disorders.30 At operation, JGCTs are almost always unilateral (98%) and more than 95% of them are confined to the ovary (stage I). Extraovarian spread is found in only 2% of patients, mostly confined to the pelvis.30
Macroscopic Features
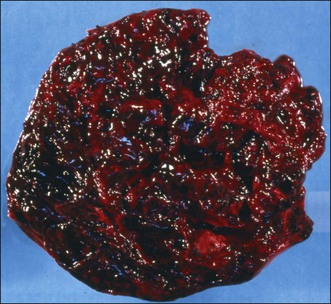
The tumors range from 3 to 32 cm (average 12.5) in diameter and appear similar to the AGCT. They are usually either uniformly solid or partly solid containing cysts that are often filled with blood (Figure 28.13); less commonly the tumors are composed predominantly of one or more thin-walled cysts. The neoplastic tissue is gray or yellow and occasionally large areas of necrosis or hemorrhage suggest a highly malignant tumor.1,30
Microscopic Features
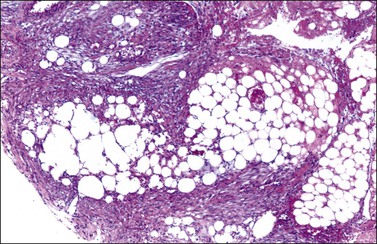
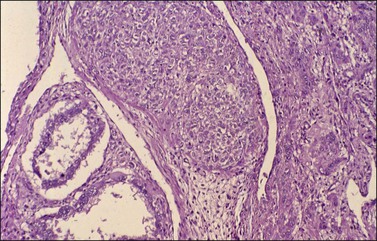
Microscopic examination shows diffuse sheets or nodules of neoplastic granulosa cells, which may be solid but more often are punctured by follicular spaces (Figure 28.14). Generally the follicles do not reach the large size of those encountered in the macrofollicular form of the AGCT; although they vary in size and shape, they are usually round to oval (Figures 28.15 and 28.16). The lumens typically contain eosinophilic or basophilic material that stains with mucicarmine in about two-thirds of cases. Papillae lined by granulosa cells grow into the cystic spaces in some tumors.32 Theca cells may be present in varying amounts and occasionally the granulosa and theca cells are arranged in a disorderly fashion; in such cases reticulin stains may help to differentiate them, with fibrils investing the theca cells individually, but the granulosa cells only in groups. Fat stains (ORO) are positive (Figure 28.17).
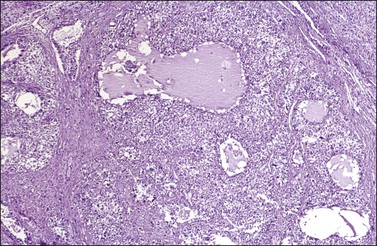
Figure 28.14 Juvenile granulosa cell tumor. Numerous oval to round follicles containing eosinophilic fluid.
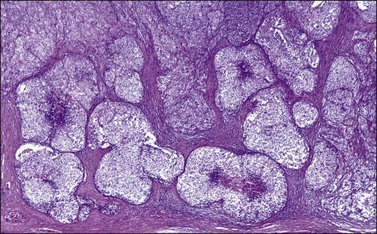
Figure 28.15 Juvenile granulosa cell tumor. The tumor cells form solid nodules, some of which exhibit central necrosis.
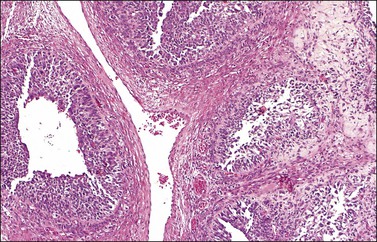
Figure 28.16 Juvenile granulosa cell tumor. Well-defined follicles of varying sizes separated by fibrovascular tissue.
The cytologic features of the JGCT also differ from those of the AGCT. The nuclei of both the granulosa and theca cells appear less mature in the former than the latter, showing hyperchromatism (Figure 28.18) and often considerable mitoses, which may be atypical; nuclear atypia varies from minimal to severe and nuclear grooves are absent. Rare cases of anaplastic JGCT exhibit a sheet-like growth with striking nuclear atypia resembling undifferentiated carcinoma. Another cytologic feature of these tumors is luteinization of both granulosa and theca cells (Figure 28.19) with fat stains typically showing abundant intracytoplasmic lipid in the two cell types.
Immunohistochemistry and Genetic Profile
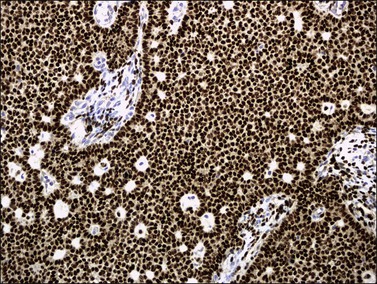
Like the adult variant, JGCTs show immunoreaction for inhibin (Figure 28.20), calretinin, MIS, FOXL2, and SF-1.7,20 There is strong membrane reaction for CD99 (Figure 28.21) and CD56.22 The tumor cells are vimentin positive and react for low molecular weight cytokeratin in 25–50% of cases.21 EMA is usually negative, as is typical of sex cord–stromal tumors, but sometimes may be focally positive.33 Trisomy 12 has been demonstrated in many cases by FISH.34
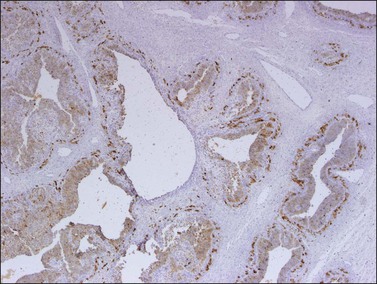
Figure 28.20 Juvenile granulosa cell tumor. The tumor cells are immunoreactive for α-inhibin, particularly the theca cells that surround rudimentary follicles.
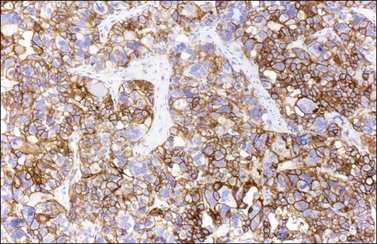
Figure 28.21 Juvenile granulosa cell tumor. The immunostaining for CD99 shows a characteristic membranous pattern.
The FOXL2 (C402G) mutation that is present in AGCTs is absent in JGCTs.6,35
Differential Diagnosis
1. Adult granulosa cell tumor. There are regular follicles, Call–Exner bodies, nuclear features (nuclear grooves), and lack of luteinization (except in pregnancy).
2. Malignant germ cell tumor (yolk sac tumor, embryonal carcinoma). Although the nuclei of JGCTs may occasionally appear severely atypical, they do not look as uniformly malignant (embryonal) as those of a primitive germ cell tumor. The latter lacks the typical follicular pattern and shows positive immunoreactions for hCG, α-fetoprotein, and cytokeratins, as well as nuclear transcription factors SALL4, LIN28, and OCT-4 or glypican-3 (see Chapter 29).
3. Thecoma. The patients are older and there is absence of follicles, predominance of theca cells, mild nuclear atypia, and characteristic reticulin stain.
4. Clear cell or undifferentiated carcinoma. Symptoms include older age of the patient, carcinomatous cells throughout the specimen, and negative α-inhibin in almost all cases.
5. Small cell carcinoma of hypercalcemic type. There is a lack of estrogenic symptoms, hypercalcemia, predominance of small cells (although it may contain large, rhabdoid-appearing cells focally), absence of the luteinized theca cell component, and higher mitotic rate. There are also negative α-inhibin and calretinin, and positive cytokeratin stains (see Chapter 27 and Table 28.2).
Table 28.2
Differentiation between JGCT and Small Cell Carcinoma
| JGCT | Small Cell Carcinoma |
| Rarely malignant | Highly malignant |
| No ↑ Ca2+ | ↑ Ca2+ frequent |
| Estrogenic | Never estrogenic |
| Follicles + + + | Follicles + |
| Thecomatous stroma | Nonspecific stroma |
| Abundant cytoplasm | Scanty cytoplasm |
6. Metastatic malignant melanoma. It is rare in young patients; there is a clinical history of melanoma, bilaterality, positive S-100 protein, and HMB-45 stains (Melan-A immunostaining can be positive in sex cord–stromal tumors; see Chapter 30).
Prognosis
Although the JGCT usually appears histologically more malignant than the AGCT, the follow-up data in four series of cases indicate a high cure rate.30,36–38 The stage of the tumor correlates best with prognosis. In the largest series reported, only 2 of 80 stage I tumors with follow-up information were clinically malignant. Both patients had stage IC tumors. All three stage II tumors were fatal.30 Contrary to the AGCT, almost all recurrences of the JGCT appear within 3 years postoperatively. The clinically malignant tumors were on average larger than those with a favorable outcome (20 vs 12 cm).30 Even if the FOXL2 (C402G) mutation, which is consistently present in AGCTs, is absent in JGCTs, patients with the latter tumors and higher FOXL2 protein expression had worse overall survival and disease-free survival than those with negative or weakly immunoreactive tumors.35 Neither DNA ploidy nor the S-phase fraction are predictive of the outcome. Some patients with advanced or recurrent disease respond to platinum-based combination chemotherapy.39
Thecoma
Thecomas are stromal tumors composed of lipid-containing cells resembling theca cells, lutein cells, and fibroblasts. These tumors can be divided into typical and luteinized variants.1
Typical Form
General Features
Typical thecomas are one-third as common as GCTs.1 They occur in older patients than those with GCTs (mean age, 59 years) and are estrogenic in almost all cases.1,40 About 60% of the patients present with uterine bleeding and 21% have an endometrial carcinoma.40
Macroscopic Features
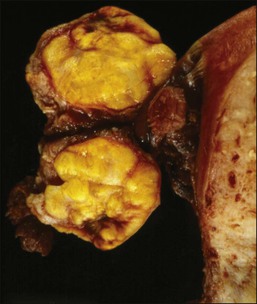
Like GCTs, thecomas are unilateral in 97% of cases.1 Grossly, they are typically solid yellow masses (Figure 28.22) of about 7 cm in diameter. Cystic change and calcification may occur.
Microscopic Features
Typical thecomas are composed of sheets of round to spindle cells with ill-defined pale cytoplasm, often vacuolated and lipid-rich, alternating with collagen-producing fibroblasts (Figure 28.23). The nuclei vary from round to spindle shape and typically lacks atypia and mitoses. A fibromatous component often intersects sheets of theca-like cells. Hyaline plaques or even large zones of hyalinization may be present. Foci of calcification may also be found. Extensively calcified thecomas occasionally occur, especially in young women.41 Typical thecomas may also contain scattered ‘sex cord elements.’1 Contrary to GCTs, reticulin fibrils typically invest individual thecoma cells (Figure 28.24).
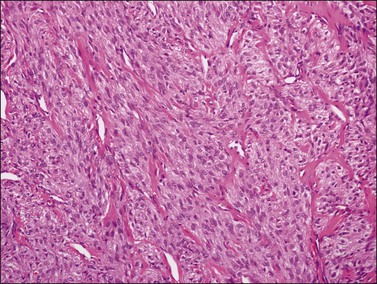
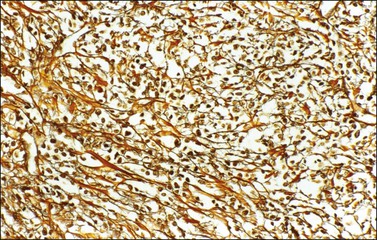
Rarely, mild nuclear atypia or even bizarre nuclei are seen in otherwise typical tumors, but severe atypia, high mitotic activity, and malignant behavior are extremely rare. Most of the tumors reported as ‘malignant thecomas’ probably represent examples of fibrosarcomas or diffuse GCTs.1
Immunohistochemistry and Cytogenetics
Thecomas are immunoreactive for vimentin, inhibin, calretinin, and other sex cord markers. Immunoreactions for cytokeratin are negative.18,26 Trisomy 12 can often be identified in thecomas.34
Differential Diagnosis
Thecomas and fibromas have overlapping features. They are essentially similar neoplasms, and typical thecomas are most frequently functioning tumors and contain lipid-rich cells that immunoreact for α-inhibin. Fat stains, however, are not helpful in distinguishing between thecomas and fibromas because otherwise typical-appearing fibromas can contain abundant lipid and some thecomas contain very little. The term ‘thecoma’ should be reserved for stromal tumors either containing moderate to large amounts of cytoplasmic lipid or associated with clinical or laboratory evidence of steroid hormone secretion. In contrast, fibromas are nonfunctioning tumors composed almost exclusively of spindle cells producing collagen and containing only small amounts of cytoplasmic lipid. Tumors with intermediate features are classified as ‘tumors in the thecoma–fibroma group.’
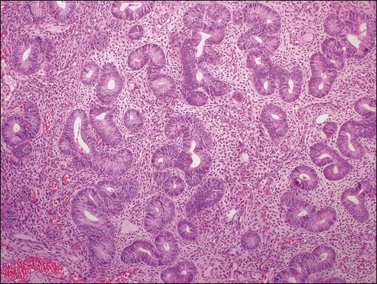
Luteinized Thecoma
General Features
Luteinized thecomas occur in a younger age group than typical thecomas. Although they are most common in postmenopausal women, 30% develop in women under 30 years of age.42 Approximately half of them are estrogenic, 40% nonfunctioning, and 10% androgenic.42 A rare variant of the luteinized thecoma is that associated with sclerosing peritonitis, which may be complicated by intestinal obstruction and can be fatal.43,44 Like luteinized thecomas of the usual type, these tumors occur mainly in young patients who present with abdominal swelling and ascites. The cause of the sclerosing peritonitis is unknown.
Macroscopic Features
Except for the lesions associated with sclerosing peritonitis, the gross features of luteinized thecomas are similar to those of typical thecomas. The former lesions are often bilateral ranging from normal-sized or slightly enlarged ovaries with a striking cerebriform appearance (Figure 28.26) to large tumors up to 31 cm in diameter.43 On section, there is prominent cortical edema and cystic change. The lack of a discrete mass in some cases suggests that they may represent a hyperplastic rather than neoplastic process. The sclerosing peritonitis involves the omentum, peritoneum, and intestinal serosa. There is fibroblastic proliferation, collagen, and inflammation with fibrin deposition.
Microscopic Features
Luteinized thecomas have the appearance of a fibroma or a typical thecoma but contain sharply defined lutein (steroid-type) cells, singly or forming nests (Figures 28.27 and 28.28) or masses. These cells are polyhedral or rounded cells with abundant eosinophilic cytoplasm and central, round nuclei with single prominent nucleoli.1,42,45,46 Occasionally, the steroid-type cells contain crystals of Reinke and these rare tumors, which may cause virilization, have been designated ‘stromal–Leydig cell tumors.’42 The variant associated with sclerosing peritonitis (Figure 28.29) is characterized by a dense proliferation of spindle cells admixed with lutein cells. There is minimal nuclear atypia but mitotic figures may be numerous (up to 40 per 10 HPF). In some cases there is diffuse edema with microcystic change.43
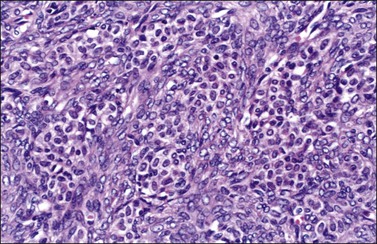
Figure 28.27 Luteinized thecoma in a patient with sclerosing peritonitis. Clusters of luteinized cells are admixed with spindle cells.