58 Neuroblastoma
Etiology and Pathogenesis
Additionally, a number of genetic aberrations are important in classification and prognosis of neuroblastoma. The DNA content of tumor cells is characterized as near-diploid or hyperdiploid, the latter of which is associated with a more favorable outcome. Examples of other genetic abnormalities associated with prognosis include MYCN amplification, deletion of the short arm of chromosome 1p, and deletion of chromosome 11q. The most important of these, MYCN amplification, is associated with rapid tumor progression and poor prognosis, even in patients with otherwise lower stage disease. Neuroblastoma is most commonly an isolated diagnosis, but it has been associated with other neurocristopathies such as Hirschsprung’s disease, congenital central hypoventilation syndrome (CCHS or Ondine’s curse), and neurofibromatosis type 1. Additionally, a very small number of patients (1%-2%) present as part of a familial neuroblastoma syndrome. In this situation, the most common genetic abnormality is a germline mutation in the anaplastic lymphoma kinase (ALK) gene (Figure 58-1).
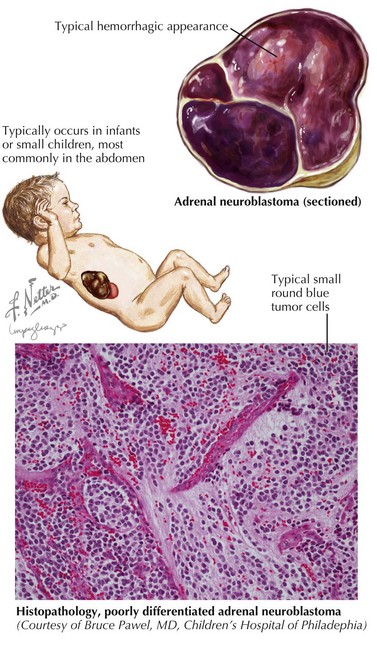
Clinical Presentation
Neuroblastoma can arise anywhere along the sympathetic chain, and symptoms at the time of diagnosis depend on the location and extent of disease. Primary tumors most commonly occur in the abdomen (65%) and usually present as a painless abdominal mass. These patients may also present with vomiting, constipation, or symptoms of intestinal obstruction. Other common sites of primary tumors include the neck, chest, and paraspinal region. Tumors arising from the chest may be found incidentally on chest radiography, and cervical tumors may present as a Horner’s syndrome or more rarely with superior vena cava syndrome (Figure 58-2).
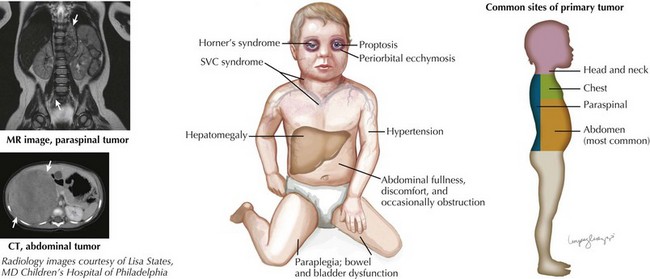
A small percentage of children (5%) present with symptoms of spinal cord compression. This oncologic emergency is associated with paraspinal neuroblastomas, and associated findings include lower extremity weakness, bowel and bladder dysfunction, back pain, and sensory loss. Neurologic function at the time of diagnosis has a strong association with long-term neurologic outcome (see Figure 58-2).
Differential Diagnosis
Because of its varied presentation, the differential diagnosis of a patient with suspected neuroblastoma is broad and should be based on specific signs and symptoms. The clinical presentation can be divided into categories: an abdominal mass or symptoms, a mass in another location, spinal cord compression, and nonspecific neurologic symptoms. Table 58-1 summarizes the differential diagnosis of neuroblastoma using these categories of presenting symptoms.
| Abdominal Mass | ||
|---|---|---|
| Intraabdominal mass | Neuroblastoma | Ovarian torsion |
| Wilms tumor | Ovarian tumor | |
| Omphalocele | Teratoma | |
| Hernia | ||






