Chapter 493 Neoplasms of the Kidney
493.1 Wilms Tumor
Peter M. Anderson, Chetan Anil Dhamne, and Vicki Huff
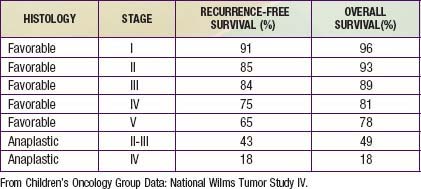
Wilms tumor, also known as nephroblastoma, is the most common primary malignant renal tumor of childhood; other tumors are very rare. Use of multimodality treatment and multi-institutional cooperative group trials has dramatically improved the Wilms tumor cure rate from <30% to ≈90% (Table 493-1).
Etiology: Genetics and Molecular Biology
Wilms tumor is genetically heterogeneous (Table 493-2). Familial Wilms tumors have been linked to FWT1 gene at chromosome 17q and FWT2 gene at chromosome 19q13. However, some families carry neither of these mutations, suggesting that additional Wilms tumor loci exist.
| SYNDROME | CLINICAL CHARACTERISTICS | GENETIC ANOMALIES |
|---|---|---|
| Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation (WAGR syndrome) | Aniridia, genitourinary abnormalities, mental retardation | Del 11p13 (WT1 and PAX6) |
| Denys-Drash syndrome | Early-onset renal failure with renal mesangial sclerosis, male pseudohermaphrodism | WT1 missense mutation |
| Beckwith-Wiedemann syndrome (BWS) | Organomegaly (liver, kidney, adrenal, pancreas) macroglossia, omphalocele, hemihypertrophy | Unilateral paternal disomy, duplication of 11p15.5 loss of imprinting, mutation of p57KIP57 Del 11p15.5 IGF2 and H19 imprinting control region |
Epidemiology
The incidence of Wilms tumor is approximately 8 cases/million children <15 yr of age with about 500 new cases in North America per year. It accounts for nearly 6% of pediatric malignancies and is the second most common malignant abdominal tumor in childhood. Although peak incidence is between 2 and 5 yr of age, Wilms tumor has also been encountered in neonates, adolescents, and adults. It can arise in one or both kidneys. The incidence of bilateral Wilms tumors is 7%, and individuals with horseshoe kidney are at twice the risk for development of Wilms tumor as the general population. Wilms tumor may be associated with hemihypertrophy, aniridia, genitourinary anomalies, and a variety of rare syndromes (see Table 493-2), including BWS and Denys Drash syndrome.





