CHAPTER 11 Iron deficiency anemia, anemia of chronic disorders and iron overload
Introduction
Disturbances of iron metabolism are among the commonest disorders affecting human populations. This high frequency reflects the combination of an essential requirement for iron by all living organisms, with a relatively precarious human iron balance. Iron in heme is maintained in the reduced ferrous state (Fe2+) for oxygen transport by hemoglobin and for storage and use of oxygen in muscles by myoglobin. It is also essential for a wide range of cellular heme and iron-sulfur proteins that are responsible for electron transport and energy generation in mitochondrial respiration and the citric acid cycle, and for ribonucleotide reductase, responsible for DNA synthesis.1 To be available for absorption, dietary iron needs to be in a soluble form, but in an oxygen-rich environment it is readily converted to insoluble ferric (Fe3+) hydroxide (‘rust’),2 or bound as insoluble ferric iron complexes, particularly in a predominantly vegetarian diet. This places an upper limit on the capacity of dietary iron to meet increased iron needs, whether physiologic or due to blood loss. As a result, iron deficiency anemia affects, at a conservative estimate, at least 500 million of the world’s population,3 and this takes no account of the additional, greater numbers of people who will have borderline iron status with depleted iron stores.4,5 In this context it is perhaps not surprising that humans conserve iron rigorously, with no mechanism for active iron excretion.6
Spectrum of pathology related to disorders of iron metabolism
Iron overload
Iron accumulating within macrophages is relatively non-toxic, these cells being specialized to deal with the high throughput of iron derived from hemoglobin in senescent red cells. By contrast, iron loading of parenchymal cells is associated with saturation of the plasma transferrin, the appearance of more labile and toxic non-transferrin-bound iron in the plasma,7 accumulation of iron in hepatocytes, cardiac myocytes and endocrine cells, and increased degradation of ferritin to insoluble hemosiderin within cellular lysosomes:8 the end result is the pattern of organ damage that is characteristic of hemochromatosis. There are no clear direct effects on bone marrow function, but iron-induced liver damage and cirrhosis may have indirect effects on blood cells (e.g. red cell macrocytosis, or cytopenias related to hypersplenism). By contrast, disturbed bone marrow function with massive ineffective erythropoiesis (e.g. in the β-thalassemia intermedia syndromes and some patients with sideroblastic or congenital dyserythropoietic anemias – see Chapters 9, 14, and 15), is definitely associated with inappropriately increased iron absorption and eventual iron overload. The hematologist is likely to be involved in the diagnosis and treatment of iron overload, including phlebotomy in hereditary hemochromatosis and iron chelation in iron-loading anemias.
Major pathways of iron exchange
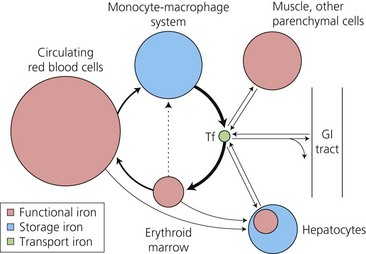
These were delineated many years ago by use of ferrokinetic studies, following the tissue uptake of 59Fe after its intravenous injection bound to plasma transferrin.9 The pathways (Fig. 11.1) are dominated (80–90% of plasma iron turnover) by the supply of plasma iron, bound to the circulating transport protein, transferrin, to bone marrow erythroid precursors for hemoglobin synthesis. At the end of their life span, red cells are phagocytosed by tissue macrophages. Heme oxygenase releases the iron from heme for recycling back to plasma transferrin, or alternatively diversion to intracellular ferritin, a protein cage made up of 24 H- (Heavy) and L- (Light) subunits which can store up to 4500 atoms of iron. A smaller uptake by the liver hepatocytes is the main alternative site of transferrin iron uptake (approximately 10%), and reflects expression of transferrin receptors on hepatocytes as well as erythroblasts. Whereas macrophages gain nearly all their iron in the unidirectional flow from senescent red cells, hepatocytes are able to take up iron in a variety of forms (see below) as well as to release the iron in times of increased need. This makes the liver the major ‘buffer’ within the system, and the prime target for iron loading and damage in iron overload conditions. There is very limited exchange of iron with the exterior. Obligatory losses (skin and gastrointestinal mucosal cell loss) of approximately 1 mg/day in males (rather more in women of child-bearing age with the additional losses of menstruation, pregnancy and lactation)10 are normally balanced by absorption of a similar amount from the diet.
Over recent years, understanding of the processes of cellular iron uptake through transferrin receptors, and regulation of intracellular iron homeostasis, has greatly increased. The discovery of the genetic basis of HFE-related hemochromatosis in the mid 1990s, combined with the use of molecular genetic studies of animal models with altered iron metabolism or erythropoiesis, led to rapid advances, with identification of additional proteins involved in regulating iron absorption and internal exchange.11 An understanding of these molecular processes underpins discussion of the pathophysiology of the iron disorders and their diagnosis.
Molecular mechanisms in iron metabolism
Cellular uptake of iron from transferrin
Cell surface expression of the classical transferrin receptor (TfR1) is greatest on rapidly dividing cells such as erythroid precursors, though it is also required for normal development of lymphoid cells and neuroepithelial differentiation in the developing nervous system. The receptor has a greater affinity for fully saturated, diferric, transferrin than for monoferric transferrin,12 and does not bind apotransferrin at the neutral pH of plasma. These affinities in part account for the importance of measuring the transferrin saturation as a vital part of a screen for the risk of an iron loading condition,13 and the fact that transferrin saturation is a better guide to iron supply to the tissues in iron deficiency than the serum iron value.14 At a high transferrin saturation most plasma iron is present as diferric transferrin, and iron uptake via transferrin receptors is enhanced. By contrast, the increased concentrations of serum transferrin which are found in iron deficiency mean that the small amount of iron present is in the form of monoferric transferrin, with its reduced rate of uptake by transferrin receptors.
After receptor-mediated endocytosis of the transferrin/receptor complex (Fig. 11.2), acidification of the endosome releases the iron from the transferrin, which at low pH even after releasing its iron still has a high affinity for the receptor.15 The apotransferrin recycles with the receptor back to the cell membrane, where it dissociates and is released into the plasma to continue its role in iron delivery to the tissues. The iron is reduced to Fe2+ by a ferrireductase, STEAP3,16 before being transported into the cytosol from the endosome by divalent metal transporter, DMT1 (previously known as NRAMP2, or DCT1).17
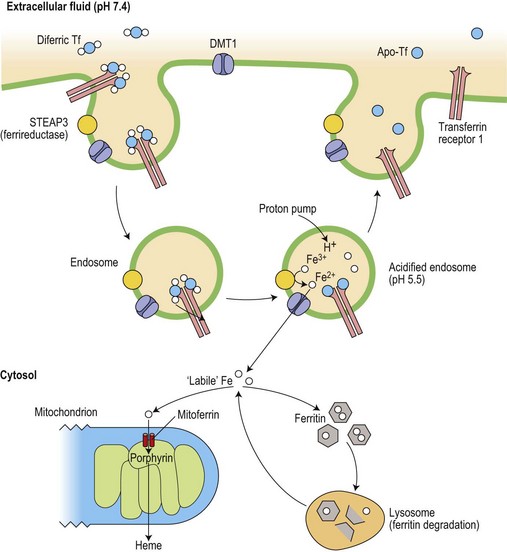
Fig. 11.2 Cellular uptake of transferrin iron and its intracellular utilization. The iron released into the cytoplasm plays a role in the translational regulation of transferrin receptor, ferritin and erythroid ALA-synthase (see Fig. 11.3). Heme synthesis dominates in erythroid cells, whereas ferritin metabolism is crucial in iron storage cells (macrophages and hepatocytes) Ferritin is constantly catabolized and the iron is either released back to the cytosol or remains in lysosomes as insoluble hemosiderin.
Regulation of cellular iron homeostasis
The intracellular iron content is finely regulated at the level of translation of the mRNA of several key iron-related proteins.18 Two iron-regulatory proteins (IRP1, coded on chromosome 9, and IRP2, coded on chromosome 15), are able to bind to sequences which form stem loop structures called iron responsive elements (IREs) in the untranslated regions of mRNAs for transferrin receptor and both H- and L-subunits of ferritin (Fig. 11.3). When IRP1 contains an iron-sulfur (4Fe-4S) cluster it has a low affinity for the IRE (and functions as cytoplasmic aconitase), while IRP2 is unstable in the presence of iron. However, when intracellular ‘labile’ (metabolically active) iron is at a low level, IRP binding to IREs that are present in the 3′ untranslated region of the mRNA for transferrin receptor protects the message from cytoplasmic degradation and allows its translation: at the same time, binding of IRP to the single stem loop in the 5′ untranslated region of the ferritin mRNA inhibits ferritin protein synthesis. The relative rates of synthesis of the two proteins are reversed in conditions of iron excess, and this reciprocal relationship serves to stabilize the intracellular labile iron content. Other iron related proteins also have IRE structures in their mRNAs.19 Those with 5′ IREs include the cellular iron exporter protein, ferroportin, (see below) and erythroid delta-aminolevulinic acid synthase (ALAS). IRP binding to the latter enables heme synthesis to be matched to the iron supply: the initial part of the heme synthetic pathway is switched off when the cell is deficient in iron. At least one isoform of DMT1 has an IRE in its 3′ untranslated mRNA, and IRP binding may stabilize its mRNA in an iron depleted cell, leading to increased expression and enhanced iron uptake.
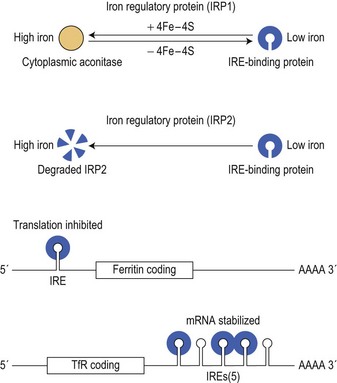
Fig. 11.3 Coordinate regulation of synthesis of ferritin and transferrin receptor-1 (TfR1) by the interaction of iron binding proteins (IRPs) with mRNA iron responsive elements (IRE). When cytoplasmic iron (see Fig. 11.2) is low, IRPs bind to IRE stem-loop structures to inhibit ferritin translation but to increase translation of TfR1 by preventing degradation of its mRNA. When iron levels are high, IRP1 functions as a cytoplasmic aconitase and no longer binds to IREs and IRP2 is degraded: this allows increased ferritin synthesis but reduces TfR1 synthesis.
Iron entering the cell cytosol becomes available for a variety of functional iron compounds or can be taken up by the iron storage protein, ferritin (Fig. 11.2). However, control of these intracellular movements is only just beginning to be explored. The H-subunits of ferritin have an intrinsic ferroxidase activity which is important for their storage function. Mitochondrial iron importers (mitoferrin-1 and -2) have been shown to be essential for heme and iron-sulfur cluster synthesis in the mitochondria of a variety of cells. Mitoferrin-1, itself stabilized by a mitochondrial ATP binding cassette transporter protein,20 is essential for iron utilization by the mitochondria of erythroid cells.21 The amount of mitoferrin may thus regulate the delivery of iron for incorporation into protoporphyrin by the enzyme ferrochelatase in the final step of heme synthesis.
Disturbances of intracellular iron distribution resulting from abnormal mitochondrial iron homeostasis are associated with a number of inherited diseases. For example, mutations in erythroid ALAS are responsible for X-linked sideroblastic anemia, with iron accumulation within mitochondria related to impaired protoporphyrin synthesis.22 Mutations in two other mitochondrial proteins give rise to mitochondrial iron accumulation, and decreased cytosolic iron with defects in iron-sulfur cluster formation and heme synthesis. Mutations in the gene for ATP-binding cassette 7 (ABCB7) underlie the sideroblastic anemia with spinocerebellar ataxia that is linked to Xq13 (see Chapter 14).23 Autosomal recessive inheritance of increased numbers of trinucleotide repeats in the gene coding for the mitochondrial iron chaperone, frataxin, is accompanied by the neurological and cardiac problems of Friedreich’s ataxia and by up-regulation of the mitochondrial iron importer, mitoferrin, with mitochondrial iron loading.24
Regulation of iron uptake in specific tissues
Developmental (transcriptional) regulation of iron uptake in the erythron
Committed erythroid progenitors express erythropoietin receptors maximally at the late BFU-E and CFU-E stages, declining from the proerythroblast stage. Erythropoietin prevents apoptosis of these progenitors and allows their proliferation. It also activates IRP and thus up-regulates synthesis of transferrin receptors25 which reach a peak in the basophilic erythroblasts. Uptake of iron thus precedes the onset of maximum heme synthesis in later polychromatic erythroblasts.26 Any iron not subsequently used appears in cytoplasmic ferritin and siderotic granules (giving rise to normal sideroblasts on Perls’ staining of marrow smears, or to Pappenheimer bodies in mature red cells on Romanowsky staining of peripheral blood films). These are more prominent where transferrin saturation, and thus the proportion of diferric transferrin, which has a high affinity for the receptor (see above), is increased, and are absent when a low saturation gives rise to iron-deficient erythropoiesis.
Hepatocyte iron uptake
The hepatocyte is able to take up iron in many forms, including transferrin- and non-transferrin-bound iron, hemoglobin–haptoglobin and heme–hemopexin complexes (which provide a potential direct shunt from the erythron to the hepatocyte in conditions associated with hemolysis or ineffective erythropoiesis), and any tissue ferritin which has been released into the circulation as the result of cell damage. The hepatocyte has a low level of expression of classical transferrin receptors (TfR1), but expresses a homolog, transferrin receptor 2 (TfR2).27,28 This differs from TfR1 in having no 3′ IRE in its mRNA, and it is thus not down-regulated in the presence of iron overload.29 Homozygous inheritance of a mutation in the TfR2 gene underlies some cases of hemochromatosis, implicating this receptor in the pathway that regulates iron absorption in relation to iron stores (see below).30 Iron uptake by hepatocytes from transferrin may occur by the receptor-mediated endocytosis described above or, following release of the iron at the hepatocyte surface, by the route taken by non-transferrin-bound iron.15 Although the detailed mechanisms of iron uptake (and release) by hepatocytes remain somewhat uncertain, it is clear that the liver may continue to take in iron from various sources, even when there are already increased iron stores, and it is thus highly vulnerable to damage in iron-loading disorders.
Macrophage iron uptake
The phagocytic cells of the reticuloendothelial system normally recycle approximately 20 mg iron a day from hemoglobin in senescent red cells and the normal small proportion of ineffective erythropoiesis.9,26 Although they express transferrin receptors, these are at a low level, and do not account for a significant proportion of the plasma iron turnover derived from transferrin. Increased uptake of iron from lactoferrin produced by neutrophils is no longer thought to be a significant source of the increased macrophage storage iron that is seen with inflammation.31
Uptake of iron by duodenal mucosal cells
Duodenal mucosal cells are specialized to take up soluble iron at their apical brush border and to transfer this across their basolateral membrane to circulating plasma transferrin (Fig. 11.4). Stomach acid has an important role in the solubilization of non-heme iron, and the detailed make-up of the diet influences its availability for absorption. For example, phytates and phosphates in vegetarian diets inhibit non-heme iron absorption by forming insoluble complexes, whereas ascorbate and meat protein digestion products form soluble complexes and enhance absorption. Heme iron from animal products is also relatively well absorbed.
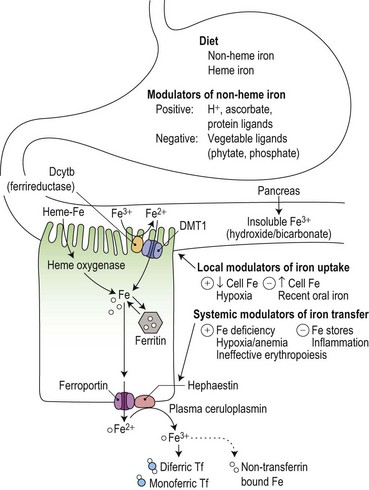
Reduction from Fe3+ to Fe2+ is an essential initial step in the uptake of dietary non-heme iron.32 It is mediated by a heme-containing ferrireductase, duodenal cytochrome b (Dcytb) which is expressed in the apical brush border membrane of duodenal enterocytes and induced by iron deficiency.33 Ascorbate could have a role as an electron donor in this process, contributing to its ability to promote the availability of iron for absorption. An apical mucosal iron transporter, DMT1, then transfers the Fe2+ into the enterocyte.34 Alternative splicing generates two isoforms of intestinal DMT1, only one of which has a 3′ IRE that could potentially be regulated by the IRE/IBP mechanism: the presence of further isoforms containing an additional 5′ exon may be required for up-regulation in iron deficiency.35 Dietary heme is taken up at the apical surface by a separate ill-understood mechanism, and the iron released by intracellular heme oxygenase is thought to join the same metabolic pool as that derived from non-heme iron.36 Once inside the cell, the iron may either be incorporated into ferritin stores (to be shed with the enterocyte into the gut lumen at the end of the cell’s life span) or transferred across the basolateral membrane by another iron transporter protein, ferroportin (also known as Ireg1).37
Duodenal enterocytes also express TfRs at their basolateral membrane, and iron uptake by this route during their maturation from duodenal crypt cells into absorptive enterocytes may contribute to modulation of the expression of transporter proteins through IRE/IBP mechanisms or through transcriptional regulation.38 The observation that a large oral dose of iron may result in a ‘mucosal block’ to absorption of a subsequent oral dose has been attributed to a rapid and selective down-regulation of Dcytb and DMT1 with no effect on the mRNA of basolateral iron transport molecules.39
Iron release from ‘donor’ cells
As well as being found in the basolateral membrane of the duodenal enterocyte, ferroportin is also found in macrophages and hepatocytes.40 Ferroportin transports reduced (Fe2+) iron and is coupled with the copper oxidases, plasma ceruloplasmin and its membrane bound homolog, hephaestin:41 the iron released as Fe3+ is taken up by plasma transferrin. A ceruloplasmin defect impairs macrophage and hepatocyte iron release,42 giving rise to liver iron overload that is resistant to phlebotomy.43 Transferrin is not essential for release of iron at the plasma membrane, since its absence in rare cases of congenital atransferrinemia does not prevent increased iron uptake from the duodenum and subsequent development of liver parenchymal iron overload.44
Regulation of iron absorption and internal iron exchange
Iron absorption in health and disease
Iron absorption is normally extremely sensitive to changes in body iron status, and both heme and non-heme iron absorption show an inverse relationship to iron stores.45 However, pathological disturbances also influence iron absorption (Fig. 11.4). Inflammatory disease reduces absorption contributing to the impaired iron supply which is characteristic of the anemia of chronic disorders.46 It has also long been recognized that grossly expanded erythropoiesis, particularly when the latter is ineffective as, for example, in the β-thalassemia disorders, can be associated with a marked increase in iron absorption, even in the face of pre-existing iron overload and an increased transferrin saturation.47,48 Hypoxia is known to potentiate mucosal iron uptake in animal studies,49 and in normal humans, stimulation of erythropoiesis by injection of recombinant erythropoietin markedly enhanced non-heme iron absorption.50 Iron absorption studies carried out sequentially during treatment with erythropoietin and phlebotomy of iron-loaded patients with chronic renal failure suggested that anemic hypoxia enhanced mucosal iron uptake, while reduction in iron stores and increased erythropoiesis independently increased the transfer of iron to the plasma.51 The concept of ‘iron store’ and ‘erythroid’ regulators of iron absorption gained currency, but their mechanisms remained obscure.52 It is now known that hepcidin, a 25 amino-acid peptide hormone derived from a propeptide produced by the liver,53 plays a key role in mediating these effects.
Role of hepcidin
In 2001 it was reported that expression of hepcidin was increased in iron loaded mice,54 and that mice in which HAMP, coding for hepcidin, had been knocked out developed hepatic but not macrophage iron overload.55 This pattern of disturbed iron metabolism in hepcidin deficient mice mirrors that seen in human hemochromatosis56 and it became clear that the various types of hemochromatosis (see Table 11.3, below) are associated with inappropriately low production of hepcidin except in rare cases with a ferroportin mutation that prevents a response to hepcidin. Conversely, hepcidin over-expression in mice led to iron deficiency,57 including in mice with inactivation of TMPRSS6 (coding for matriptase-2, a liver transmembrane serine protease).58,59 A human inherited iron-refractory iron deficiency anemia (IRIDA) was then shown to be associated with over-expression of hepcidin resulting from recessive inheritance of inactivating mutations of TMPRSS6.60,61
It is now clear that hepcidin is the major physiological regulator of iron absorption and internal iron exchange (Fig. 11.5), and that inappropriate hepcidin production underlies much iron pathophysiology.11 Hepcidin binds to ferroportin at the ‘donor’ cell surface and promotes the transporter’s internalization followed by ubiquitin-mediated lysosomal degradation.62,63,64 Hepcidin-induced degradation of ferroportin prevents iron release to circulating plasma transferrin and leads to reduced iron absorption, a block on the release of iron derived from senescent red cells within macrophages, and reduced serum iron concentration. It is therefore a negative regulator of iron release from cells. Hepcidin is excreted rapidly into the urine consistent with its regulation at the level of production. Decreased hepcidin production is seen with hypoxia65 and in association with ineffective erythropoiesis,66 as well as in iron deficiency. Conversely increased hepcidin production, occurs with inflammation65 as well as with increased iron stores.
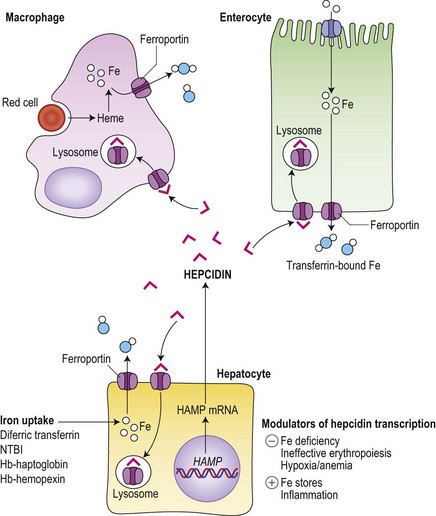
Fig. 11.5 Central role of hepcidin in iron metabolism. Hepcidin produced by hepatocytes down-regulates iron export to circulating transferrin from iron ‘donor’ cells (hepatocytes, macrophages and duodenal enterocytes) by promoting the internalization and lysosomal degradation of ferroportin. Hepatocytes take up iron in a number of forms, whereas enterocytes obtain their iron predominantly from the gut lumen (see Fig. 11.4) and macrophages are specialized to deal with the high throughput of iron from senescent red cells.
Regulation of hepcidin production
Uncovering the genetic basis of the various types of hemochromatosis (see Table 11.3, below) and of IRIDA stimulated exploration of signal pathways that act on the liver to regulate hepcidin production.67 Iron status signals via the bone morphogenetic protein (BMP) pathway (Fig. 11.6).68,69 Hemojuvelin (HJV), mutated in most cases of juvenile hemochromatosis, is a glycosylphosphoinositol (GPI)-linked cell surface protein which acts as a co-receptor for BMP6 (a key endogenous regulator of hepcidin synthesis).69 It is thought that cell surface HFE (commonly mutated in adult hemochromatosis) and TfR2 (mutated in other cases of hemochromatosis) associates with HJV on the cell surface to allow signaling via BMP receptors and a SMAD pathway to the hepcidin promoter. It is possible that the link to iron status may be provided by competitive interactions between HFE and diferric transferrin for binding to TfR1: only the HFE remaining would then be available to associate with TfR2, the complex being stabilized by diferric transferrin.70 A physiological increase in diferric transferrin would signal increased hepcidin transcription, reducing iron absorption and release from macrophages, while pathological disruption of HFE, HJV or TfR2 (in hemochromatosis) would be accompanied by reduced hepcidin synthesis and increased iron absorption. Negative regulation of hepcidin production by matriptase-2 (TMPRSS6)71 is mediated by proteolysis of membrane HJV,72 and its absence accounts for the over-expression of hepcidin in familial iron-refractory iron deficiency anemia.
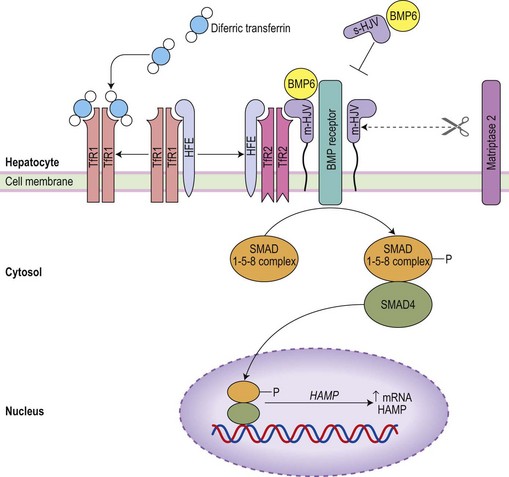
The increase in hepcidin in response to inflammation is predominantly mediated by interleukin-6 (IL-6)73 and activation of signal transduction via STAT3.74 Suppression of hepcidin production in association with ineffective erythropoiesis in thalassemia major occurs independently of iron stores.75 A number of potential mechanisms for the ‘erythroid regulator’ are emerging. Erythropoietin can directly down-regulate hepcidin production through the erythropoietin receptor and C/EBPα transcription factor,76,77 but this cannot account for a dependence of the erythroid regulator on the presence of an expanded erythroid marrow. Growth differentiation factor 15 (a member of the transforming growth factor β family) is up-regulated in serum from patients with thalassemia and congenital dyserythropoietic anemia type 1, and suppresses hepcidin production in vitro.78,79 Its release from apoptotic erythroblasts would explain the greater iron loading in anemias with expanded erythroid marrow due to ineffective erythropoiesis rather than hemolysis. A further candidate molecule derived from erythroblasts is ‘twisted gastrulation’ which has been shown to interfere with BMP signaling via its receptor80 and would thus suppress hepcidin synthesis. Finally, hypoxia may inhibit hepcidin production through involvement of the hypoxia inducible factor (HIF)/von Hippel–Lindau pathway. HIF-1α, induced by hypoxia and stabilized by iron deficiency, up-regulates furin-mediated cleavage of HJV to produce a soluble form of HJV: it is likely that this competes with membrane HJV for the co-receptors for BMP, thus down-regulating hepcidin production.81
Assessment of iron status
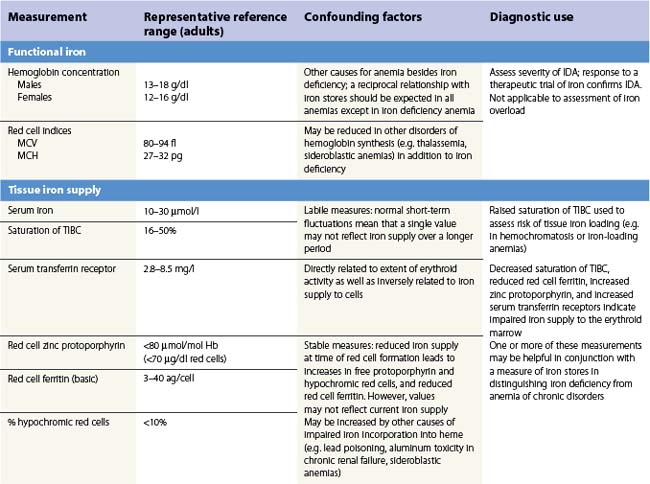
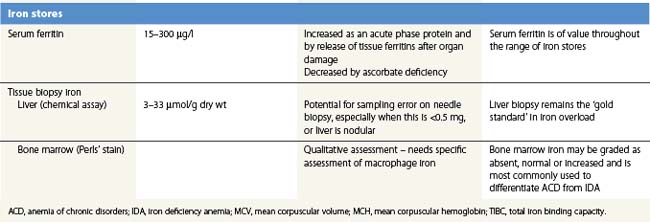
There is no single measure of iron status that is applicable in every situation. Combinations of measures of iron stores (macrophage and hepatocyte), iron supply to the tissues, and functional hemoglobin iron are often needed to arrive at a clear assessment of iron status.82 The measures used are summarized in Table 11.1. All are subject to potential confounding factors.
Serum transferrin receptors
Serum transferrin receptors are truncated soluble receptors that are shed into the circulation mainly from the erythroblasts in the marrow.83 The measurement reflects both the iron status of individual eythroblasts and the total mass of the erythron.84 It is likely to be of most value in distinguishing iron deficiency from the anemia of chronic disorders.85
Serum ferritin
Serum ferritin is apoferritin made up from glycosylated ferritin light chains, the release of which from cells reflects current ferritin protein synthesis.86 It is thus related to the intracellular labile iron that determines IRP affinity for the IRE, and only indirectly to iron stores through the release of ferritin iron either within the cytosol or through lysosomal degradation.87 Ferritin protein synthesis also increases in response to inflammatory cytokines, behaving as an acute phase protein independently of iron stores. Damage to ferritin-rich tissues can release iron-containing ferritin into the circulation giving high ferritin values, e.g. in hepatitis, splenic infarction, or bone marrow infarction in sickle cell disease. Its use as a guide to the presence of increased iron stores is thus limited,88 though a low serum ferritin is a clear indication that iron stores are absent. The dependence of ferritin protein synthesis on translational regulation by the IRP/IRE mechanism is illustrated by the rare hereditary hyperferritinemia/cataract syndrome, where autosomal dominant mutations in critical parts of the IRE stem loop are accompanied by uncontrolled synthesis of ferritin light chain: high serum ferritin values are seen with the development of cataracts, but there is no iron overload and transferrin saturations are not increased.89 Other uncommon autosomal dominant causes of a high serum ferritin but normal transferrin saturation include loss of function ‘ferroportin disease’ and a benign hyperferritinemia associated with a point mutation in the coding sequence of L-ferritin.90
The reciprocal relationship between the synthesis of transferrin receptors and ferritin within cells that is mediated by the IRE/IBP mechanism (Fig. 11.3) has its counterpart in values for serum transferrin receptors (increased) and serum ferritin (reduced) during the development of iron deficiency. The sensitivity of these measures for assessing iron status is increased by expressing them as a ratio.91,92
Tissue biopsy
Bone marrow biopsy is used to examine macrophage iron stores. It is primarily used for supporting a diagnosis of the anemia of chronic disorders rather than iron deficiency. Bone marrow biopsy touch preparations may give results comparable to aspirates.93 Positive identification of the absence of iron stores may be inaccurate94 unless careful examination of macrophages is made.
Liver biopsy provides the opportunity for histological examination of increased hepatocyte iron and any fibrotic or cirrhotic changes. The periportal distribution of iron accumulation in hemochromatosis may reflect selective expression of ferroportin in periportal hepatocytes and its degradation when hepcidin production is low.64 In addition, chemical iron determination allows a quantitative assessment of the degree of iron loading. Quantitative phlebotomy in patients with thalassemia major who had undergone curative allogeneic bone marrow transplantation has confirmed that the liver iron concentration is a reliable indicator of total body iron stores in secondary iron overload resulting from red cell transfusions.95 Magnetic resonance quantitative measurements (particularly T2*) of liver (and cardiac) iron96 may reduce the future need for liver biopsy in iron overload disorders.






