Figure 18-2 The steps of tumor invasion Tumor invasion involves the loss of cell-cell adhesions (cadherins represented by green bars), alterations in cell-matrix adhesion (integrins represented by ovals), proteolysis of the extracellular matrix (blue matrix, degradation demonstrated by clearing of matrix mediated by proteinases represented by scissors), and motility involving alterations in the actin cytoskeleton (intracellular black and gray lines).
Loss of function of cell-cell adhesion molecules other than E-cadherin is associated with the ability of tumor cells to invade and metastasize. Neural cell adhesion molecule (NCAM), a member of the immunoglobulin-like cell adhesion molecule Ig-CAM family, is downregulated in several tumor types, and NCAM loss results in an increased ability of tumor cells to disseminate. 8 Other Ig-CAMs, such as DCC (deleted in colorectal carcinoma), CEACAM1 (carcinoembryonic antigen CAM1), and Mel-CAM (melanoma-CAM), also demonstrate reduced expression in specific cancer types. However, not all cell-cell adhesion molecules can be viewed as potential invasion suppressors. N-cadherin promotes motility in some cell types, and Ig-CAMs such as L1, CEA (carcinoembryonic antigen), and ALCAM (activated leukocyte CAM) are often overexpressed in advanced cancers and have functions associated with cancer progression. This complexity may be explained by signaling functions for these molecules, either direct or indirect, that are distinct from their role in cell-cell adhesion. The interrelatedness of tumor growth and tumor invasion, and limitations of experimental model systems, often does not allow a distinction between growth effects that influence the appearance of an invasive phenotype and an effect on cellular invasion per se.
The extracellular matrix (ECM) provides a scaffold for the organization of cells and spatial cues that dictate cell behavior. 9 The ECM is composed of proteins, primarily triple-helical collagens, glycoproteins such as laminins and fibronectin, and proteoglycans. The basement membrane is an organized ECM that separates polarized epithelial, endothelial, and muscle cells from the underlying tissue. Interstitial matrix provides the structure characteristic of connective tissues. The molecular composition of the ECM varies between tissues and organs and provides contextual information to cellular constituents. In addition, the ECM serves as a repository for secreted regulatory proteins and growth factors. Finally, ECM proteins themselves can be active signaling molecules, activities that frequently are only revealed after proteolysis reveals cryptic sites. Thus, the interaction of cells with ECM molecules determines their capacity for survival, growth, differentiation, and migration.
Cells adhere to ECM via integrins, a family of transmembrane glycoproteins assembled as specific combinations of 18 α and 8 β subunits. 5 Integrins bind to distinct but overlapping subsets of ECM components. During tumor progression, cancer cells tend to undergo a switch in their integrin expression pattern, downregulating the integrins that mediate adhesion and maintain a quiescent, differentiated state, and expressing integrins that promotes survival, migration, and proliferation. 10 Although there is a cell-type dependency on integrin function, in general integrins α2β1 and α3β1 are viewed as suppressors of tumor progression, whereas αvβ3, αβ6, and α6β4 promote cellular proliferation and migration. Integrins mediate both “outside-in” and “inside-out” signaling, so that changes in cellular adhesion can alter cellular phenotype, and changes in intracellular signaling pathways can modulate cellular adhesion. A well-described and important mechanism whereby integrin-ECM interactions modulate cell function is by cooperative signaling with different growth factor receptors. Many of the cellular responses induced by activation of tyrosine kinase growth factor receptors are dependent on the cells being able to adhere to an ECM substrate in an integrin-dependent fashion. Signaling in response to ECM ligation usually activates focal adhesion kinase (FAK) and nonreceptor tyrosine kinases of the src family.
Matrix Degradation
Disruption of basement membrane is a hallmark of malignancy. Degradative enzymes produced by the tumor cells, and by resident and infiltrating cells as a response to the tumor, contribute to matrix degradation and facilitate tumor cell invasion. Proteolytic enzymes of many classes have been implicated in tumor cell invasion, including the serine proteinases plasmin, plasminogen activator, seprase, hepsin, several kallikreins, the cysteine proteinase cathepsin-B, the aspartyl proteinase cathepsin-D, and metal-dependent proteinases of the matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM) families. Other matrix-degrading enzymes such as heparanase, which cleaves heparin sulfate proteoglycans, and hyaluronidase cleavage of its substrate hyaluronic acid have also been causally associated with tumor progression and invasion.
Liotta and colleagues observed that metastatic potential correlates with the degradation of type IV basement membrane collagen and focused attention on the metal-dependent gelatinases. 11 These enzymes are now recognized as MMP2 and MMP9, and many of the 23 members of the MMP family of matrix-degrading metalloproteinases have been associated with tumor progression. Elevated MMP levels correlate with invasion, metastasis, and poor prognosis in many cancer types, and animal models provide evidence for a causal role for MMP activity in cancer progression. 12 The plasminogen activator/plasmin system has also been causally implicated in cancer invasion, 13 and urokinase plasminogen activator (uPA) and plasminogen activator inhibitor-1 (PAI-1) are validated prognostic and predictive markers for breast cancer. 14
The regulation of matrix proteolysis is complex and can involve the concerted action of multiple proteinases and proteinase classes from both tumor cells and adjacent resident and infiltrating cells (Figure 18-3 ). The conversion of pro-MMP2 to active MMP2 requires membrane-type MT1-MMP (MMP14), a transmembrane MMP that is activated intracellularly by the proprotein convertase family member, furin. There is evidence for a cascade of cathepsin-D–cathepsin-B–uPA–plasmin–MMP activation that results in activated enzymes capable of degrading all components of the ECM. Proteolysis is also regulated by the production of specific endogenous protease inhibitors, including the tissue inhibitors of metalloproteinases (TIMPs), serine proteinase inhibitors (serpins), and cysteine protease inhibitors (cystatins). These inhibitory activities are produced and secreted by tumor or stromal cell types, and some proteinase inhibitors are stored in high concentrations in the ECM. Proteinase activity cascades can function via proteolytic degradation of some of these proteinase inhibitors in addition to activation of other proteinases.
The original view that proteolytic enzymes function predominantly to remove physical ECM barriers has been expanded with the realization that proteolysis is a key regulator of multiple steps of tumor progression. For example, MMP substrates in the matrix or on the cell surface that modulate cellular growth, differentiation, apoptosis, angiogenesis, chemotaxis, and migration have been identified. 12 The abundant evidence for a role for MMPs in tumor progression led to the design and testing of synthetic MMP inhibitors for cancer therapy. 15 These inhibitors proved to be ineffective in clinical trials, results that have been explained by problems with inhibitor or clinical trial design and a lack of understanding of the broad range of MMP activities resulting in both cancer-promoting and cancer-inhibitory effects.
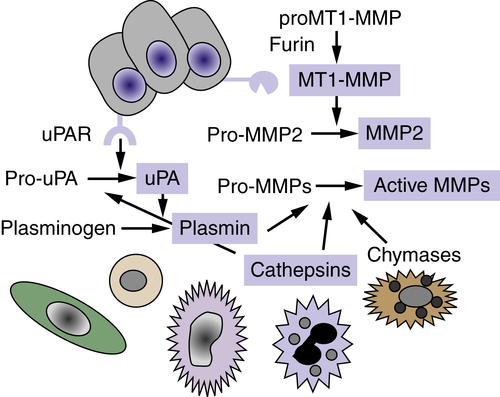
Figure 18-3 Proteolytic cascades Extracellular proteinases are made by tumor cells as well as by stromal fibroblasts and inflammatory cells. Proteolytic cascades result in the conversion of proenzymes to their active form. Enzymes in blue boxes are capable of degrading components of the extracellular matrix (ECM). In many cases, proteolytic cascades are localized to the surface of tumor cells. The urinary plasminogen activator receptor (uPAR) is expressed by many tumor cells and initiates and localizes the conversion of pro-urokinase plasminogen activator (pro-uPA) to its active form, which then converts the serum protein plasminogen into the active serine proteinase, plasmin. The membrane type 1-matrix metalloproteinase (MT1-MMP) is a transmembrane protein that is activated intracellularly by the proprotein convertase furin. MT1-MMP converts pro-MMP2 to its active form, MMP-2. Enzymes of many classes convert pro-MMPs to their active form.
Motility
Cellular locomotion occurs as the result of coordinated polymerization and depolymerization of the actin cytoskeleton to extend a pseudopod at the leading edge of the cell, followed by contraction associated with disassembly of cell-matrix adhesive contacts at the trailing edge. 16 Lamellipodial protrusions at the leading edge are nucleated by a branched actin network involving the Arp2/3 complex and its regulators, the WASp (Wiskott-Aldrich syndrome protein) family, cortactin, and the GTPase Rac. Actin contractility is regulated by myosin light-chain kinase and upstream small GTPases, in particular Rho and its effector Rho-kinase (ROCK). Single cells migrate with a spindle-shaped morphology, referred to as mesenchymal migration, or with the less adhesive ellipsoid shape used by leukocytes and Dictyostelium termed amoeboid migration (Figure 18-4 ). Collective migration can occur when the cells retain cell-cell junctions and clusters of cells move in single file through a tissue.
Tumor cells can secrete factors that stimulate motility in an autocrine fashion. Tumor cell–produced lysophospholipase D (autotaxin) stimulates motility, as does lysophosphatidic acid (LPA), which can be produced by lysophospholipase D activity on lysophosphatidylcholine. Hepatocyte growth factor/scatter factor (HGF/SF) interacts with its receptor, c-met, to induce chemokinetic activity of epithelial cells, resulting in an invasive phenotype. Directional motility is a chemotactic or haptotactic effect in response to a gradient of soluble or localized factors, respectively. Chemotaxis is often the result of growth factors such as IGF, and chemokines of the CCR and CXC family. Haptotaxis is characterized as a response to gradients of ECM components such as laminin-5 and fibronectin and can be modulated positively or negatively by proteolysis.
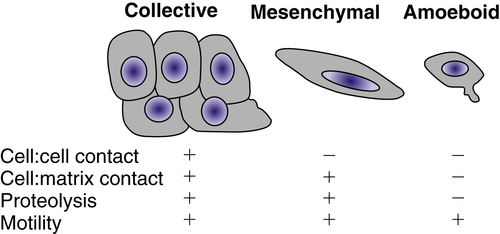
Figure 18-4 Types of cellular invasion Cells can move through matrix barriers as collectives, in which multiple cells remain attached and move together, or as single cells with mesenchymal or amoeboid characteristics. Epithelial-derived tumor cells undergoing collective migration retain cell-cell adhesions, whereas those undergoing mesenchymal or amoeboid movement have reduced or absent cadherin-mediated adhesions. Mesenchymal motility requires proteolysis and integrin-mediated cell-matrix adhesion. In the absence of proteolysis and extracellular matrix (ECM) adhesions, tumor cells can move through ECM using amoeboid movement, similar to that displayed by infiltrating leukocytes. Amoeboid movement is characterized by elevated actin cytoskeleton activity mediated by the small GTPase Rho and its regulator Rho-kinase.
Coordination of Cancer Invasion
The coordination of cell-cell and cell-matrix adhesion, matrix degradation, and cytoskeletal activity is required for cellular invasion. The type of cell migration (i.e., collective, mesenchymal, or amoeboid) is influenced by the relative levels of adhesion mediated by cadherins and integrins, proteolytic activity, and actin contractility. Modulation of any of these factors can convert one type of motility into another. 16
Invadopodia is the name that has been given to structures identified in invading cells that represent the physical convergence of the adhesive, proteolytic, and motility components of invasion (Figure 18-5 ). 4 Invadopodia are actin-rich organelles that protrude from the plasma membrane and contact and locally degrade the ECM. Invadopodia contain adhesion molecules, including several β1 integrins and CD44, the serine proteinases seprase and dipeptidyl dipeptidase IV, and several MMP and ADAM metalloproteinases. Inside the plasma membrane, invadopodia contain actin and actin assembly molecules and multiple signaling molecules including focal adhesion kinase (FAK), src-associated proteins such as p130Cas and Tks5/FISH (tyrosine kinase substrate 5/five SH3 domains), and the small GTPases cdc42, Arf1, and Arf6. Thus, invadopodia are implicated as key cellular structures that are used to coordinate and regulate the various components of the process of cancer invasion.
The Metastatic Cascade
Although invasion is required for metastasis, the ability to invade is not sufficient for metastasis (see Figure 18-1). Some tumors are highly aggressive, forming secondary lesions with high frequency (e.g., small-cell carcinoma of the lung, melanoma, pancreatic carcinoma), whereas others are rarely metastatic despite being locally invasive (e.g., basal cell carcinomas of the skin, glioblastoma multiforme). Fidler and colleagues have proposed an analogy regarding metastasis that is highly illustrative. Metastatic cells are likened to athletes participating in the decathlon. Each cell must be capable of completing every step of the metastatic cascade. If a cell cannot complete any step, it cannot go on to subsequent steps and cannot form a metastasis.
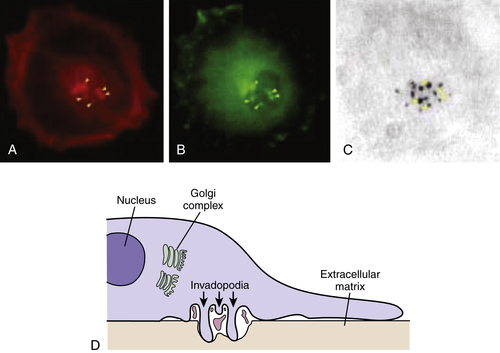
Figure 18-5 Invadopodia Confocal laser image showing triple immunofluorescence labeling of A375MM melanoma cells plated on tetramethylrhodamine isothiocyanate (TRITCJ)-conjugated gelatin. (A) Invadopodial structures marked by actin-binding phalloidin-Alexa 546. (B) Invadopodial structures marked by Alexa 633-conjugated anti-phosphotyrosine antibodies. (C) Degradation areas on the underlying Alexa 488-conjugated gelatin. Image shows colocalization between actin, phosphotyrosine, and patches of degraded extracellular matrix, fulfilling the criteria for the definition of invadopodia. (D) Schematic diagram of the invadopodial complex based on correlative light-electron microscopy reconstructions. Spatial relationships with the nucleus and the Golgi complex are shown. Invadopodial protrusions originate from profound invaginations of the ventral surface of the plasma membrane; within the area delimited by the large invagination, large fragments of gelatin can often be seen. From Ayala I, Baldassarre M, Caldieri G, et al. Invadopodia: a guided tour. Eur J Cell Biol. 2006;85:159-164, with permission.
Metastasis is primarily thought of as developing via dissemination in the bloodstream, although other routes of spread occur. Carcinoma cells tend to escape and spread initially to draining lymph nodes, becoming trapped and proliferating. The thoracic duct links the lymphatic system to the bloodstream, connecting lymphatic to hematogenous spread. Metastases can also develop by spreading across body cavities. For example, ovarian carcinoma cells most frequently establish secondary tumors by dissemination in the peritoneum while rarely forming metastases via hematogenous spread. Other routes of spread also exist but are far less common (e.g., dissemination of melanoma cells along the space between endothelium and basement membrane or perineural spread in pancreatic and prostatic carcinomas). Thus, the route of dissemination is not inherent to a definition of metastasis.
Intravasation
How tumor cells enter the bloodstream is not clearly understood. The growth of a tumor exerts a hydrostatic pressure, and studies imply that tumor-cell invasive cords follow lines of least resistance. Angiogenesis is likely to be a prerequisite for metastasis, but this has not been formally proven (see Chapter 17). Tumor cell entry into intact blood vessels is an active process that requires serine and metalloproteinase activity in an experimental model of intravasation. 17 Tumor blood vessels, however, are highly abnormal, with fewer pericytes and increased permeability compared with normal vessels, and presumably provide an easier route for direct entry into the bloodstream. 18 Lymphatic vessels are also abnormal, but their role in intravasation is unknown. Regardless of the route, tumor cells enter the circulation in great numbers: Estimates are 3 to 4 million cells/day/g of tumor. 19 The number of tumor cells in the peripheral blood, however, does not necessarily predict whether the patient will develop metastases. 20 In contrast, the detection of disseminated tumor cells in lymph nodes and bone marrow does correlate with metastatic relapse, suggesting that, at least in breast cancer, the properties that allow the cells to find their way to these tissues and survive are the same properties that permit distant metastases.
Transport
Once tumor cells enter a circulatory compartment, they can move actively by motility mechanisms or passively, carried or pushed along with fluid flow. Injection of radiolabeled cells directly into circulation reveals that a substantial proportion is lost during the transport phase of the metastatic cascade. Many tumor cells are eliminated by natural killer (NK) cells or monocytes before arrival in a secondary site. Tumor cells that escape immune recognition are frequently killed by exposure to hemostatic shear forces. 21 Bioassays in the lungs, liver, heart, and muscle have been performed following intravenous injection of tumor cells. It is noted that by the time it takes to remove the tissues for assay (2 to 3 minutes), most cells are dead due to mechanical trauma. 21 The average tumor cell diameter ranges from 20 to 30 μm but must navigate through vessels significantly smaller (e.g., 6- to 7-μm capillaries). Even if tumor cells have the ability to deform and squeeze through the passages, they are subjected to significant hydrostatic pressures. Depending on the tumor type and biophysical parameters such as membrane fluidity, cellular elasticity, and cytoskeletal organization, the cells will remain intact or be broken by shear. Deformability is also affected by the pressures found within various tissues. In contrast to the shear forces usually encountered in the vasculature, blood flow in bone sinusoids is sluggish (about 30-fold less than in capillaries and postcapillary venules), and diameter is not a concern. 22
During transport, the behavior of tumor cells is often determined by their presence as single cells or as emboli. Embolization can be homotypic (tumor cell–tumor cell) or heterotypic (tumor cell–leukocyte, tumor cell–platelet, tumor cell–fibrin). The association of tumor cells with blood cells can be the result of altered cell surface glycosylation and expression of sialyl Lewis X/A on the tumor cell that permits interaction with a class of vascular adhesion molecules found on normal leukocytes and endothelium, the selectins. Alterations in the adherence of tumor cells to endothelium via E-selectin, platelets via P-selectin, and leukocytes via L-selectin alter the metastatic potential in animal models. 23 Embolus size can also contribute to protection of the tumor cells from biophysical forces or immune attack. In essence, encapsulation of tumor cells helps to protect them. As a result of the consequence of embolus formation, heparin, an inhibitor of selectin/glycan interactions, has been considered as an antimetastatic agent.
Visualization of tumor cells in the circulation during transport indicates that the cells roll rather than float in a manner analogous to leukocytes. Nonetheless, during this time, tumor cells are weakly adherent and subject to anoikis, a specialized type of apoptosis in which cells that are anchorage dependent are induced to die. 24 In general, metastatic cells are more resistant to anoikis than nonmetastatic cells and are frequently referred to as being anchorage independent. This is somewhat misleading, because some tumor cells will induce apoptosis even if firmly attached to a substrate if that substrate is not the preferred one for the type of cell. It is possible, then, that circulating tumor cells receive sufficient signals from the ECM, other cells, and/or serum proteins to limit their susceptibility to anoikis.
Arrest
It is important to discriminate between the physical trapping and arrest of circulating cells in the microvasculature and selective adhesion to the walls of the microvasculature. Both processes have been observed, and the relative importance of these mechanisms in specific organs is debated.
There are three types of endothelial structures found in higher vertebrates: continuous, discontinuous, and fenestrated. Most endothelial cells form tight junctions with their neighbors and have a continuous, unbroken basement membrane beneath them. However, in certain organs, such as liver and spleen, the endothelial cells and the basement membrane have gaps, or discontinuities, in their structure. In the kidney, a fenestrated endothelium, there are gaps between endothelial cells, but a membrane-like structure connects them and the entire structure overlaps in a continuous basement membrane. The structure of these endothelial/basement membrane barriers contributes to the normal function of the tissues and forms different barriers through which tumor cells must pass.
Adhesion of circulating tumor cells to organ microvessel endothelial cells represents one of the more important steps in metastasis, especially organ-specific metastasis. In general, higher rates of tumor cell–endothelial adhesion correlate well with metastatic potential. In vivo and in vitro kinetic studies indicate that initial attachment of cancer cells occurs preferentially at endothelial cell junctions. 25 Frequently, tumor cells adhere at sites where inflammation is taking place and is most likely related to alterations in cell surface components of endothelial cells at these sites. Tumor cells use many of the same mechanisms to attach to and traverse endothelium as inflammatory cells, including glycan/selectin interactions.
Once tumor cells bind to the endothelium, they induce the endothelial cells to retract and eventually overlap the tumor cell. During this time, there is no loss of electrical resistance, suggesting that tight junction integrity is maintained. Tumor cells then adhere to subendothelial basement membrane components, and a higher rate of tumor cell adhesion to subendothelial basement membrane correlates with metastatic potential. In the case of HT1080 fibrosarcoma cells, the attachment of circulating tumor cells to the lung vasculature is mediated by tumor α3β1 integrin ligation to laminin-5 in the basement membrane. 26 Patches of exposed basement membrane were found to be preexisting using intravital microscopy techniques in isolated, perfused lungs.
Arrested tumor cells can undergo rapid apoptosis. It is envisioned that in some cases this is the result of the lack of suitable survival signals and the initiation of anoikis. In addition, the attachment of tumor cells to endothelium can release nitric oxide (NO) produced by endothelial nitric oxide synthase. 27 NO can induce apoptosis of tumor cells, indicating an active process that contributes to tumor cell loss and metastatic inefficiency.
Extravasation
Extravasation is the process of tumor cells invading from the interior of a vessel into the organ parenchyma. Extravasation was viewed as a rate-limiting step for metastasis formation, but intravital microscopy studies have indicated that extravasation can be a remarkably efficient process, at least in some situations. For example, 87% of B16F1 murine melanoma cells that were injected through the mesenteric vein into the liver were arrested in the liver 90 minutes after injection, and 83% of the injected cells were found in the liver parenchyma by 3 days, indicating that more than 95% of the arrested cells extravasated. 28 The molecular mechanisms underlying extravasation are viewed as being identical to those involved in invasion, and in vitro assays for extravasation reveal a contribution of cellular adhesion molecules, proteinases, and motility factors.
There is controversy as to whether extravasation is required for the formation of metastases. In the case of some pulmonary metastases, there is evidence that tumor cells can attach to the lung endothelium, survive, and grow intravascularly. 29 Extravasation occurs in this model only when the intravascular foci outgrow the vessel.
Colonization
Colonization, the formation of clusters of tumor cells at ectopic sites, represents a highly inefficient step in the metastatic cascade. In the model of B16F1 cells injected into the liver vasculature, only 2% of the injected cells formed micrometastases, and only 0.02% formed lesions that persisted, grew progressively, and threatened the life of the animal. 28 The formation of micrometastatic lesions requires that the tumor cell must first survive and then grow in the foreign environment. In some tumor types (i.e., breast and melanoma), metastases can arise decades after the treatment of the primary tumor, indicating that tumor cells can survive in a state of dormancy for long periods. Tumor cells can persist as solitary cells, or they can grow to a size of several hundred cells in which the rate of growth is balanced by the rate of apoptosis. Conversion to a clinically detectable metastatic lesion requires the subsequent initiation of angiogenesis (see Chapter 17). The growth of the cells is dependent on factors, primarily soluble growth factors, present at the site of colonization. Although it is natural to focus on factors that promote the growth of tumor cells in selective sites, there is ample experimental evidence showing that some tissues are hostile to tumor cells.
A tumor cell’s ability to establish a metastatic lesion is very much dependent on the microenvironment (see Chapter 16). A prime example of this effect is the role of the “vicious cycle” in the propensity for breast carcinoma to metastasize to bone (Figure 18-6 ). 30 The mammary carcinoma cells produce parathyroid hormone–related peptide (PTHrP), which during pregnancy would function to release calcium from bone stores. Using the same molecular pathways, tumor cell-produced PTHrP acts on its receptors on osteoblasts to release the tumor necrosis factor-α (TNF-α) family member, receptor activator of nuclear factor-κB ligand (RANKL). RANKL interacts with its receptor RANK on osteoclasts and activates them to degrade mineralized bone. The bone matrix contains an abundance of growth factors, including PDGF, FGFs, IGF-1, and TGF-β/bone morphogenetic protein family members, which are released during the osteolytic process. It is the release of these growth factors that stimulates the breast cancer cells to grow and to continue to secrete PTHrP and fuel the “vicious cycle.” The colonization of breast cancer cells in the bone is thus facilitated by specific characteristics of the bone microenvironment that promote the growth of breast cancer cells.
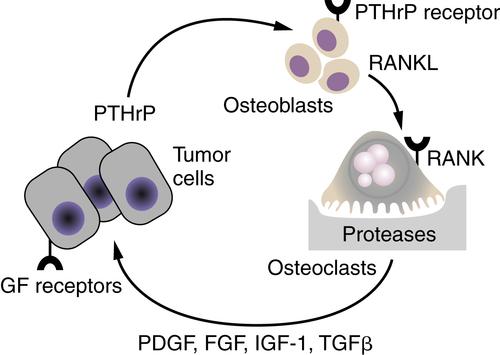
Figure 18-6 The vicious cycle of host-tumor interactions in breast cancer metastasis to bone Breast cancer cells produce parathyroid hormone–related protein (PTHrP), which stimulates bone osteoblasts to express the tumor necrosis factor-α (TNF-α) family member receptor activator of nuclear factor-κB ligand (RANKL). RANKL interacts with its receptor RANK on osteoclast precursors to differentiate into active osteoclasts, resulting in the release of proteases and bone degradation. Growth factors such as platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), insulin-like growth factor (IGF-1), and transforming growth factor-β (TGF-β), which are stored in the bone matrix, are released and stimulate the growth of receptor-containing tumor cells. An increase in tumor cells results in an increase in PTHrP release, leading to a vicious cycle of tumor cell growth and bone degradation.
Organ Selectivity of Metastasis
There is a clear tendency for primary tumors to form metastatic lesions in specific organ sites (Table 18-1 ). Common regional metastatic involvements can often be attributed to anatomic or mechanical considerations (e.g., efferent venous circulation or lymphatic drainage) and explained by arrest of tumor cells in the first capillary bed or lymph node encountered. 31 Because most tumor cells enter the vasculature in small veins or capillaries, the most common site of metastasis is lung and liver. However, distant metastasis patterns are typically more site specific. In 1889, Paget analyzed postmortem data of women who died of breast cancer and noticed a higher frequency of metastasis to skeleton than would be expected based solely on cardiac output to each organ. 32 He concluded that the pattern of organ distribution of metastases was not simply a matter of chance and suggested that metastases develop only when the “seed” (tumor cells with metastatic ability) and the “soil” (organs or tissues providing growth advantages to seeds) are compatible. Importantly, the mechanical theory and the “seed and soil” hypothesis are not mutually exclusive, and both contribute to metastatic dissemination.
Table 18-1
Common Sites of Metastasis
| Primary Tumor Site | Most Common Sites of Metastases |
| Breast | Axillary RLN, contralateral breast via lymphatics, lung, pleura, liver, bone, brain, adrenal, spleen, ovary |
| Colon | RLN, liver, lung, direct extension into urinary bladder or stomach |
| Kidney | Lung, liver, bone |
| Lung | RLN, pleura, diaphragm by direct extension, liver, bone, brain, kidney, adrenal, thyroid, spleen |
| Ovary | Peritoneum, RLN, lung, liver |
| Pancreas | Liver, stomach by direct extension, colon, peritoneum |
| Prostate | Bones of spine and pelvis, RLN |
| Stomach | RLN, liver, lung, bone |
| Testis | RLN, lung, liver |
| Urinary bladder | Direct extension into rectum, colon, prostate, ureter, vagina, bone, RLN, lung, peritoneum, pleura, liver, brain |
| Uterine endometrium | RLN, lung, liver, ovary |
RLN, Regional lymph nodes.
Experimental data supporting the seed and soil hypothesis include preferential invasion and growth of B16 melanoma metastases in specific organs. 33 In addition, palliative treatment of women with advanced ovarian carcinoma has provided an opportunity to test this theory in humans. These patients often have a large ascites burden, but seldom present with disease outside the peritoneal cavity. Tarin and colleagues treated patients with potentially lethal malignant ascites by introducing a tube that drains the peritoneal ascites into the vena cava. 34 In doing so, tumor cells in the ascites were given direct entry into the circulation. Despite continuous entry of billions of viable tumor cells into the circulation, metastases to the lung (i.e., the first capillary bed encountered) were rare. This single clinical observation highlights the inefficiency of the metastatic process and, more important, demonstrates that merely seeding cells in different tissues is not adequate to develop metastases.
The mechanisms responsible for organ selectivity in tissues can be attributed to the arrest and the colonization steps of the metastatic cascade in particular. Tumor cells adhere more selectively to organ-derived microvascular endothelial cells than to large-vessel endothelial cells, and variants of the B16 melanoma previously selected for metastases to brain, lung, ovary, or liver adhere at a more rapid rate to brain, lung, ovary, or liver endothelial cells, respectively. 35 Using phage-display technology, endothelial cells in different tissues have been demonstrated to express unique markers, and tumor cells recognize the molecular “addresses” to adhere to in a selective manner. 35 Tumor cells are also able to recognize subendothelial basement membrane differences. In vitro studies demonstrate the selective growth of tumor cells in organ-derived soluble growth factors or cells. 36 In vivo, breast tumor cells that express the chemokine receptor CXCR4 preferentially metastasized to tissues that expressed the ligand SDF1/CXCL12. 37 There is a concept that tumor cells colonize in a premetastatic niche initiated in target organs by tumor cell–generated soluble factors that induce the expression of fibronectin by resident fibroblast-like cells. 38 Bone marrow–derived cells that express the vascular endothelial cell growth factor receptor 1 and the integrin α4β1 selectively adhere to these regions, produce the proteinase MMP9 and the chemokine SDF1/CXCL12, and provide a permissive niche for colonization by tumor cells.
Although the data strongly support the notion that there are soluble factors produced in different tissues to which tumor cells can respond, the process of homing has not been validated. Strictly speaking, homing would require directed movement throughout the transit of tumor cells as they leave the primary tumor. Rather, tumor cells distribute according to circulatory patterns initially but may “home” once they are more proximate. Many of the mechanisms used by lymphocytes to home to peripheral lymph nodes or sites of inflammation are apparently shared by tumor cells.
Some of the strongest evidence supporting organ selectivity of cancer cells comes from data showing selection of variants that colonize different tissues. The first selections were done by repetitive isolation of lung metastases from the B16 melanoma followed by reinjection and recolonization. 33 Similar approaches have been used for other tumors, most recently using a human breast carcinoma cell line with selection of metastases to bone, lung, and adrenal gland. Using these breast carcinoma cell lines coupled with a comparison by cDNA microarray has highlighted the requirement for coordinated expression of multiple genes for metastasis. 39 Transcriptomes were compared between parental and bone-selective variants, and over- and underexpressed genes were identified. Among the overexpressed genes in the bone metastasis signature were a matrix metalloproteinase, MMP1; the ECM component osteopontin; the cytokine interleukin-11; the chemokine receptor, CXCR4; and connective tissue–derived growth factor. Subpopulations within the parental population expressed one or more of the bone signature genes, but only a few expressed all of them. Transfection of individual cDNAs only modestly increased bone metastatic efficiency, whereas cotransfection of gene combinations into the parental cells resulted in populations as efficient at bone colonization as the bone-selective variants. Similar studies with a lung-selective variant revealed a lung metastasis signature that overlapped only minimally with the bone metastasis signature. 40 These data highlight that there are specific genes that control metastasis in an organ-specific fashion, and coordinated expression of multiple genes is required.
Metastatic Progression
The journey of a metastatic cancer cell involves several steps from the time it leaves the primary site to the moment it reaches a distant organ. So how long does this journey take to commence, and what is the path that these cells take? Are these metastatic cancer cells or “seeds” disseminated early or late in the life of an evolving primary tumor? These questions are important to understand, because they are linked to the functional consequences of genetic and epigenetic changes that are accrued by metastatic cancer cells when compared to their originating primary tumor. If metastatic progression occurs early and takes place in parallel to the primary tumor, the genomic landscape of the primary tumor and metastasis can be predicted to be significantly different. On the other hand, if metastatic progression occurs late through clonal evolution within the primary tumor and the cancer cells that left the tumor early became dead ends, then one can expect the primary tumor and metastases to look genetically similar. These questions have direct clinical implications because several drugs that target the primary tumor fail to treat advanced metastatic disease in the clinic. If the metastases were indeed genetically distinct from the primary tumors, it could be envisioned that drugs that target oncogenic changes in the primary tumor would not work for metastases.
Deciphering both the similarities and distinctions between the primary tumor and the metastases may be important in devising strategies to successfully treat metastatic cancer. What is also necessary is gaining an understanding of the regional differences and heterogeneity within the tumor and metastases. A large part of the intratumoral heterogeneity and metastatic diversity is shaped by the distinct tumor microenvironment in which the cancer cells reside. Therefore it can be envisioned that microenvironment-linked selective pressures influence the genetic and epigenetic heterogeneity of tumors and determine the evolutionary trajectory of cancer cells during metastatic progression. Recognition and characterization of such genetic and phenotypic diversity is therefore key for designing rational therapeutic interventions.
The advent of major technological advances over the past decade in the field of sequencing and high-resolution analysis of disseminated cancer cells has made it possible to begin to address such complex biological questions. Deciphering metastatic progression and phylogeny of metastases is an emerging goal as sequencing data from different cancers accumulate, with several hypotheses and models being discussed and debated. Two hypotheses that tower over the rest in explaining the origin of metastatic cells are the linear progression and clonal evolution model on one hand and the parallel progression model on the other. The two models are not mutually exclusive and could be at play to different degrees, depending on the tumor type and its oncogenetic composition.
Linear Progression Model
Pioneering work by the group of Isaiah Fidler has shown that only a subset of preexisting cells within a heterogeneous primary tumor is competent to metastasize. 41 The linear progression paradigm has been the prevalent model to explain this observation. According to this model, cancer cells undergo multiple rounds of mutation and selection in the primary tumor, and only a small subset of these malignant clones acquire the genetic and epigenetic alterations necessary for metastasis. 42,43 A clinical correlation between tumor size and frequency of metastasis is in line with this model. 44 The model also suggests that larger tumors growing over time would have a higher likelihood of containing metastasis-competent clones within their heterogeneous population. The reduced probability of metastasis in cases where primary tumors of less than 2 cm in size are surgically resected in the clinic lends support to this model. New insights into genomic evolution during tumor progression came from Aparicio and colleagues, who sequenced a breast cancer metastasis and its corresponding primary tumor that was removed 9 years earlier. 45 Eleven of the 30 mutations detected in the metastatic lesion were present in the primary tumor. This study revealed that preexisting mutations in the primary tumor do get selected in the metastases, but there is also considerable genomic evolution that occurs during the metastatic process. Comparing the frequency of these somatic mutations in the primary tumors and metastases revealed two discernible trends. Five of the 11 shared mutations were prevalent in the primary tumor, whereas 6 were present in low frequencies. In contrast, all these mutations were prevalent in the metastases, indicating less heterogeneity than the primary tumors. These data are compatible with the clonal expansion model and suggest that a clone from the heterogeneous primary tumor is selected, expands, and generates the distant metastasis. More recently, analysis of tumor evolution using single-cell sequencing technology in two breast cancer cases, by Michael Wigler and colleagues, suggested that a single clonal expansion both formed the primary tumor and seeded the metastasis in the examined cases. 46 It remains to be seen how general these findings are as more matched primary tumors and their metastases in different cancers are sequenced.
In the context of metastasis, what is the time line of metastatic progression? The linear progression model would predict metastatic spread to be a late event in tumor progression, given the time required for the accumulation of sufficient genetic and epigenetic alterations within the primary tumor that permits metastasis. Also implicit in the assumptions from this model and discussed earlier, primary tumors and metastasis are expected to be genetically similar, and a majority of the mutations detected in metastasis should preexist in the primary tumor. Indeed, new insights from genomic sequencing of pancreatic cancer patients suggest that metastasis is a late event in the genetic evolution of pancreatic cancer. 47 Clonal populations that give rise to metastasis preexist within the primary tumor but are more genetically evolved than the original parental, nonmetastatic clone. Using mathematical modeling, Yachida and colleagues showed that a decade passes between the occurrence of an initiating mutation and the birth of a parental, nonmetastatic cancer cell clone in a pancreatic tumor. Thereafter, at least 5 years pass before metastatic ability is gained in these clones and an average of 2 years elapses until the patient’s death. In an analogous manner, colorectal cancer progression was calculated to be 17 years between the birth of an adenoma founder cell and advanced carcinoma; however, only a further 1.8 years pass until the evolution of a metastatic founder cell appears. 48 These studies provide several key insights into the time scale of evolution of metastases in the setting of pancreatic and colon cancer. They suggest that clones in certain advanced cancer already harbor most of the mutations needed for metastatic competence and it takes a relatively short time to develop into metastases. We have to keep in mind that the temporal course of acquisition of metastatic traits might vary greatly between different types of cancers. It is thought that in the case of estrogen receptor–positive breast cancer, there is usually a prolonged latency period, often up to a decade, from gaining infiltration potential and seeding until competence to colonize and outgrow in a distant organ is gained. However, in the case of lung cancer, there is a relatively short interval (often measured in months) between gaining infiltration ability and successful colonization. 49 Large-scale sequencing efforts are under way to reveal the genetic landscape of primary tumors and their metastases in other cancer types, and this could provide insights into the generality of these observations.
Parallel Progression Model
Although there is supportive evidence for the linear progression model, single-cell comparative genomic hybridization analysis of isolated disseminated tumor cells (DTCs) in the bone marrow and primary breast tumors provides an alternative explanation. Christoph Klein and colleagues observed that the genetic changes in the DTCs in the bone marrow did not resemble those in their corresponding primary breast tumor, which led to the conception of the “parallel progression model.” According to this model, quasinormal cells disseminate relatively early in the course of tumor progression and evolve independently from the cells in the primary tumor. 50 As a result, genetic and epigenetic evolution occurs through multiple rounds of genetic diversification and clonal selection mostly at the distant organ site(s), which ultimately gives rise to overt metastasis. Parallel evolution thus predicts divergence for many mutations and genetic alterations for the selected cells at the distant sites. 51 Based on the mathematical modeling of tumor growth rates, the parallel progression model questions how large sizes of metastases can be explained by the linear model if they arise only at the advanced stages of tumor progression. Work emerging from several laboratories has shown that tumor cells can indeed disseminate early at the pre-invasive stages in breast cancer progression, both in human breast cancer patients and in experimental mammary tumor models. 52–54 Moreover, Podsypanina and colleagues 54 showed that even untransformed mouse mammary epithelial progenitor cells are able to extravasate and survive at the distant site for prolonged periods and start to grow again on oncogene induction. Collectively, these findings in breast cancer lend support to the parallel progression model and underscore the need for a deeper understanding of metastatic progression in different cancers.
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading. Learn more here






