Chapter 53 Generalized Seizures
As reviewed in Chapter 50, seizures are classified into two basic groups: partial and generalized [Commission on Classification and Terminology of the International League Against Epilepsy, 1981]. The International League Against Epilepsy (ILAE) Commission on Classification and Terminology has proposed a modification of the classification system of seizures and epilepsy [Berg et al., 2010]. Generalized seizures are those defined as occurring in bilaterally distributed networks whereas focal (partial) seizures involve networks limited to one hemisphere with either discretely localized or more widely distributed disturbances. While the proposed classification system has some advantages over the 1981 classification, it has not yet been widely used or endorsed by clinicians and in this chapter the older classification system will be used. Partial seizures involve only a portion of the brain at the onset but can spread, resulting in generalized tonic-clonic seizures. Primary generalized seizures are those in which the first clinical changes indicate initial synchronous involvement of both hemispheres without clinical, electroencephalographic (EEG), or other evidence of focal or partial onset. Impairment of consciousness is usual during generalized seizures, although some seizures, such as myoclonic, may be so brief that the level of consciousness cannot be assessed. The generalized seizures span a wide range of clinical presentations, ranging from the rather benign-appearing absence seizure to the dramatic and frightening generalized tonic-clonic seizure.
In most epidemiologic studies, primary generalized seizures are reported to be less common than partial seizures [Hauser and Kurland, 1975; Juul-Jensen and Foldspang, 1983]. For example, Hauser and Kurland [1975] and Juul-Jensen and Foldspang [1983] found that primary generalized seizures accounted respectively for 40.5 percent and 45.8 percent of the seizure types. Studies confined to children have been more variable. Todt [1984], in a study of children with seizures ranging in age from 3 to 16 years, found that 61.5 percent had primary generalized seizures, compared with only 26.6 percent of children between 2 months and 14 years of age in a series reported by Sofijanov [1982]. Of the primary generalized seizures, generalized tonic-clonic are the most common, followed by absence and myoclonic seizures [Hauser and Kurland, 1975].
These epidemiologic studies may be somewhat misleading. With improvement of diagnostic techniques in recent decades, especially with long-term video monitoring using EEG, it has become clear that the prevalence of primary generalized tonic-clonic epilepsy has been overestimated. In fact, most generalized tonic-clonic seizures begin as partial seizures and then generalize [Schmidt et al., 1983; Jobst et al., 2001].
This chapter discusses generalized tonic-clonic seizures and absence, clonic, tonic, and atonic seizures, along with the syndromes in which these are the predominant seizure type. Myoclonic seizures, which are generalized seizures, are reviewed in Chapter 56 and partial seizures in Chapter 54.
Generalized Tonic-Clonic Seizures
Clinical Features
As indicated by the name, generalized tonic-clonic seizures have two distinct phases: tonic and clonic [Gastaut and Broughton, 1972]. Loss of consciousness usually occurs simultaneously with the onset of a generalized stiffening of flexor or extensor muscles – the tonic phase. The loss of consciousness usually is complete, and it is rare for patients to have any partial awareness of what happened during the seizure. During the tonic phase, prolonged extension of the back, neck, and all limbs often occurs. The eyes remain open, and a cry or yell is common. The tonic phase typically lasts 10–30 seconds and is followed by the clonic phase, which usually starts with a rapid tremor and then slows to massive jerks of the extremities and trunk. A decrescendo pattern to the frequency (although not necessarily the strength) of the jerks is seen as the seizure ceases. The clonic phase typically lasts 30–60 seconds.
In a number of syndromes, generalized seizures are a predominant feature. Epilepsy with so-called grand mal seizures on awakening is a syndrome in which the generalized tonic-clonic seizures occur only on awakening [Betting et al., 2006; Unterberger et al., 2001; Beghi et al., 2006]. In generalized epilepsy with febrile seizures plus, patients initially have febrile seizures; later, as the condition evolves, they experience afebrile generalized tonic-clonic or other types of generalized seizures [Abou-Khalil et al., 2001; Baulac et al., 2004; Scheffer and Berkovic, 1997; Wallace et al., 2001, 2002]. Generalized tonic-clonic or clonic-tonic-clonic seizures often can be a prominent component of juvenile myoclonic epilepsy, a condition discussed in Chapter 56. Children with Lennox–Gastaut syndrome may have generalized tonic-clonic seizures, although tonic seizures are the core seizure type in this syndrome.
Although their disorder currently is not classified as a syndrome, a distinct group of patients with photosensitive epilepsy have seizures provoked by flickering lights and have a photoconvulsive response to strobe light on EEG. The photoconvulsive response is characterized by spike-and-wave and multiple spike-and-wave complexes that are bilaterally synchronous, symmetric, and widespread (Figure 53-1). These should be contrasted with spikes that are time-linked with the photic stimulation and confined to the occipital region, which in children are normal. The photoconvulsive response may be self-limited and cease during stimulation or continue beyond the stimulation [Reilly and Peters, 1973]. The most effective frequency of the flash is 10–20 Hz.
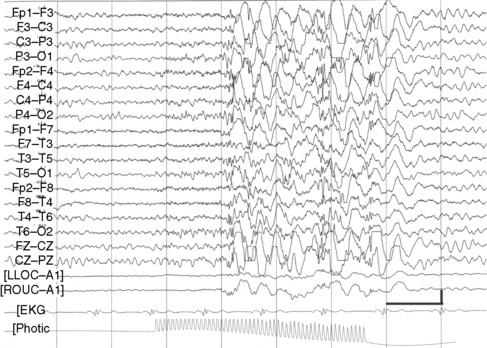
The seizures evoked by photic stimulation usually are primary generalized in type: generalized tonic-clonic, absence, or myoclonic seizures. Although patients with partial seizures may have photoconvulsive responses during an EEG study, it is unusual for a partial seizure to be precipitated by photic stimulation [Jayakar and Chiappa, 1990]. Generalized epileptiform discharges during photic stimulation correlate with generalized seizures, whereas focal discharges are more commonly associated with partial seizures [Gilliam and Chiappa, 1995].
Photosensitive epilepsy can be classified into two major groups: (1) pure photosensitive epilepsy, in which clinical seizures occur only when the patient is exposed to the photic stimulus, and (2) photosensitive epilepsy, in which spontaneous seizures occur in addition to those induced by light stimulation [Binnie and Jeavons, 1992]. Precipitating stimuli that can produce a seizure include sunlight reflected from water, sunlight viewed through leaves of trees in a breeze or while driving along tree-lined streets, discotheque lighting, faulty fluorescent lamp (“nervous ballast”), television, and video games. Photosensitive epilepsy usually appears around puberty, with a mean age at onset of 14 years and a higher incidence in girls than in boys [Jeavons, 1982].
Differential Diagnosis
Difficulty is rarely encountered in correctly diagnosing generalized tonic-clonic seizures (formerly termed grand mal seizures). Generalized tonic-clonic seizures can be divided into those that are primarily generalized from onset and those that begin as partial seizures and then secondarily generalize [Schmidt et al., 1983; Jobst et al., 2001]. Differentiating features are listed in Table 53-1. On very rare occasions, generalized tonic-clonic seizures begin as another generalized seizure type such as absence [Niedermeyer, 1976] before evolving into tonic-clonic activity.
Table 53-1 Comparison of Primary and Secondary Generalized Tonic-Clonic Seizures
| Primary GTC | Secondary GTC | |
|---|---|---|
| Prodrome | Occasionally | Occasionally |
| Aura | Never | Frequently |
| Usual duration | 1–3 min | 1–3 min |
| Family history for seizures | Frequently | Occasionally |
| Acquired lesions | Rarely | Frequently |
| EEG Interictal Photoconvulsive response Hyperventilation |
Normal or generalized spike-wave Frequently May activate spike-wave discharges |
Normal or focal or multifocal spikes Rarely Usually does not activate epileptiform discharges |
Breath-holding attacks, pseudoseizures (nonepileptic events), and syncope occasionally may resemble generalized tonic-clonic seizures. These are discussed in Chapters 64 and 65.
Etiology
The etiology of seizures in children who have secondary generalized tonic-clonic seizures is the same as those described for partial seizures. Patients with primary generalized tonic-clonic seizures usually do not have structural lesions and often have a familial disorder, such as juvenile myoclonic epilepsy. The genetics of generalized tonic-clonic seizures are discussed in Chapter 52.
Comorbidities Associated with Generalized Seizures
While a major goal in the treatment of epilepsy is stopping the seizures, it is not the only treatment goal. Patients with epilepsy are a risk for a number of comorbidities. Comorbidity refers to the co-occurrence of two supposedly separate conditions that occur together more than by chance (also see Chapter 62). Depression occurs more frequently in patients with epilepsy than in the normal population, so that epilepsy and depression are comorbidities. Comorbidities are not necessarily causal. For example, because epilepsy and depression are comorbidities does not mean that epilepsy caused the depression or depression caused the epilepsy. Rather, it is possible that both conditions have a common biological substrate or that another independent variable triggers one of the comorbidities. For example, epilepsy often leads to drug therapy, which could cause depression independently of the epilepsy.
Comorbidities associated with epilepsy include depression [Camfield and Camfield, 2009; Dunn et al., 1999], suicidality [Bridge et al., 2007; Tellez-Zenteno et al., 2007; Donner et al., 2001; McGregor and Wheless, 2006], attention-deficit hyperactivity disorder (ADHD) [Dunn and Kronenberger, 2005], conduct disorders [Dunn et al., 2009], anxiety [Caplan et al., 2005], cognitive impairment and learning disabilities [Fastenau et al., 2004; Aldenkamp et al., 2005], and migraine [Pellock, 2004].
Medical Treatment
One of the issues is that, in many studies, partial seizures with secondary generalization are included along with primary generalized tonic-clonic seizures. In the United States, AEDs that have an indication for monotherapy or adjunctive therapy of generalized seizures include phenytoin and carbamazepine. However, while both drugs are effective in partial seizures, they can exacerbate primary generalized seizures [Perucca et al., 1998; Guerrini et al., 1998]. Studies examining efficacy and safety of AEDs in children with primary generalized epilepsy are very limited. Although a number of the newer AEDs are being used for the treatment of primary generalized tonic-clonic seizures, even in adults definitive evidence for their effectiveness is lacking [Faught, 2003; French et al., 2004]. In addition, data for adults and children often are combined when studies are reported. Antiepileptic drug studies done solely in children with primary generalized tonic-clonic seizures are limited. Although most of the newer antiepileptic drugs have been used in the treatment of generalized tonic-clonic seizures, their usefulness in children has not yet been established [Holmes, 2003; Jarrar and Buchhalter, 2003].
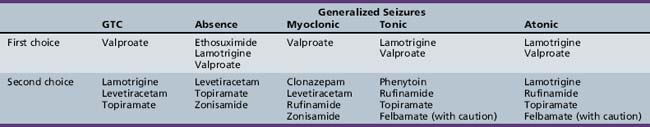
The major drugs typically used to treat primary generalized tonic-clonic seizures include valproate [Murphy and Delanty, 2000; Ramsay and DeToledo, 1997; Marson et al., 2007], lamotrigine [Mikati and Holmes, 1997; Trevathan et al., 2006; Marson et al., 2007], levetiracetam [Rosenfeld et al., 2009;Noachtar et al., 2008;Wheless, 2007], and topiramate [Biton et al., 1999; Marson et al., 2007]. Although phenobarbital is effective in the treatment of primary generalized tonic-clonic seizures, the adverse side effects profile has reduced its use as initial therapy for primary generalized tonic-clonic seizures [Taylor et al., 2001].
The four drugs most commonly used for primary generalized tonic-clonic seizures are reviewed briefly here. Additional information about AEDs is available in Chapter 59.
Valproate
Valproate is approximately 90 percent protein-bound. Protein binding varies with drug plasma concentration, and the free fraction increases with increasing plasma concentration [Davis et al., 1994]. Therefore, with high blood levels of valproate, the free, or active, portion of valproate becomes greater, and toxicity may occur. In addition, drugs or endogenous substances, such as free fatty acids, can alter protein binding. The primary metabolism is by hepatic hydroxylation (mitochondrial beta oxidation) and conjugation with glucuronide. Eighty percent of an administered dose of valproic acid is metabolized through these two pathways and then excreted by the kidney [Levy et al., 2002]. The remainder of a dose is excreted in other oxidized metabolites. Drug interactions are common with valproate [Scheyer, 2002].
Gastrointestinal toxicity includes anorexia, nausea, and indigestion. These symptoms may be reduced with the divalproex sodium preparations. Dose-related toxicities are action tremor (seen in 40 percent of adults, less frequently in children), elevated plasma transaminase (usually transient but a possible harbinger of serious hepatic disease), and hyperammonemia. Idiosyncratic toxicity includes hepatic necrosis (treatable with l-carnitine), thrombocytopenia, pancreatitis (in 0.5 percent, sometimes fatal), teratogenicity, and stupor and coma. The risk of hepatic fatality is greatest in children younger than 2 years and in patients taking valproic acid in combination with other AEDs [Dreifuss et al., 1987, 1989]. Toxic effects with long-term use are weight gain, hair loss, and platelet dysfunction. Routine monitoring of liver function tests, complete blood count, and valproate blood level determination is recommended.
Lamotrigine
Lamotrigine is a broad-spectrum AED that has a number of attractive features for childhood use. However, there is a risk of rash and the drug must be used with care. Lamotrigine has broad-spectrum efficacy, demonstrating efficacy in partial seizures, absences, tonic, atonic, and generalized tonic-clonic seizures [Besag et al., 1995; Eriksson et al., 1998; Duchowny et al., 2002]. It is indicated as adjunctive therapy for adults and in children with primary generalized seizures and generalized seizures in the Lennox–Gastaut syndrome in children over the age of 2 years. There are indications that the drug is useful in absence seizures.
Lamotrigine is generally well tolerated in children [Duchowny et al., 2002]. The drug is not associated with behavioral or cognitive side effects. With high doses, particularly when used as adjunctive therapy, dizziness, diplopia, headache, ataxia, tremor, and nausea may occur.
Data from clinical trials with lamotrigine indicate that the risk of serious rash in pediatric patients is higher than in adults. The incidence of rash associated with hospitalization among adults treated with lamotrigine is 0.3 percent and the incidence among children is 1.0 percent [Messenheimer, 1998; Hirsch et al., 2006]. The incidence of cases reported as possible Stevens–Johnson syndrome is 0.1 percent for adult patients and 0.5 percent for pediatric patients. The risk of rash is higher in children who are taking valproate, presumably because it decreases the metabolism of lamotrigine. Most rashes occur during the first 6 weeks of therapy. The risk of rash is reduced by slow upward titration of dose.
Levetiracetam
Levetiracetam is a broad-spectrum drug that is widely used in both children and adults for partial and generalized seizures [Carreno, 2007; Wheless, 2007]. It is rapidly and almost completely absorbed after oral administration. The absorption and elimination of levetiracetam are linear. Levetiracetam is not extensively metabolized in humans; the majority of the drug is excreted unchanged in the urine. The major metabolic pathway of levetiracetam is enzymatic hydrolysis of the acetamide group. Levetiracetam produces no inhibition of cytochrome P450 isoforms, epoxide hydrolase, or UDP-glucuronide enzymes. There are no reports of drug interactions between levetiracetam and AEDs or other drugs. The metabolites have no known pharmacological activity and are excreted in the urine. The elimination half-life is 4–8 hours in children. The pharmacodynamic effects of levetiracetam are longer than would be predicted by half-life and the drug can be given twice daily.
There are no reports of serious toxicity with levetiracetam. The drug is well tolerated in children [Wheless and Ng, 2002]. In children, headache, anorexia, somnolence, and mild infection (otitis media, pharyngitis, and gastroenteritis) have been reported [Glauser et al., 2002]. These usually occur early in treatment and can be reduced by slow drug initiation and/or dose reduction. Levetiracetam has caused irritability and aggressiveness [Khurana et al., 2007]. Reversible treatment-emergent psychosis has been associated with levetiracetam therapy [Kossoff et al., 2001]. No hematological or hepatic disturbances have been reported and routine laboratory studies are not recommended.
Topiramate
Topiramate has a broad spectrum of efficacy in children [Ormrod and McClellan, 2001], and has been found to be beneficial in the treatment of medically refractory primary generalized tonic-clonic seizures [Biton et al., 1999; French et al., 2004; Wheless, 2000].
Topiramate is rapidly absorbed and has very low protein binding [Bourgeois, 2000]. In the absence of enzyme-inducing drugs, 80 percent of a dose is excreted unchanged in the urine, with an elimination half-life of 20–30 hours. In the presence of enzyme-inducing drugs, 50–80 percent of a dose is excreted unchanged in the urine, with an elimination half-life of 12–15 hours in teenagers. The metabolic products of topiramate are formed in the liver and do not appear to be biologically active. Prepubescent children have a higher clearance and shorter elimination half-life than in adults [Bourgeois, 2000].
The most common side effects with topiramate are central nervous system-related: drowsiness, dizziness, decreased attention or impaired concentration, paresthesia, nervousness, confusion, and impaired memory. These side effects usually are mild to moderate, develop during the first weeks of therapy, and may decline over time. The central nervous system side effects appear to be fewer when topiramate is used for monotherapy than when it is used in polytherapy, and are less common in children than in adults. Weight loss [Ormrod and McClellan, 2001] and acidosis [Izzedine et al., 2004; Takeoka et al., 2001] can occur with topiramate therapy. Renal stones [Kuo et al., 2002; Lamb et al., 2004] and acute myopia [Coats, 2003] associated with secondary angle-closure glaucoma [Browne et al., 1974] have been reported. These side effects appear to be very rare in children.
Table 53-2 summarizes the AEDs used in the treatment of generalized tonic-clonic seizures and other generalized seizures in childhood. This listing is based on both efficacy and tolerability, and reflects the author’s personal preferences. AED therapy should always be tailored to the needs of the particular patient.
In addition to treating with AEDs, it is important to counsel the patient and parents about factors that could exacerbate seizures, such as sleep deprivation. Drugs that can lower seizure threshold, such as bupropion, should be avoided [Foley et al., 2006].
Absence Seizures
Clinical Features
Absence seizures, formerly termed petit mal seizures, are characterized by an abrupt cessation of activity, change in facial expression, and impairment of consciousness [Browne et al., 1974; Pearl and Holmes, 2008; Porter et al., 1973]. Absence seizures are not common, accounting for less than 10 percent of all seizure types [Hauser and Hersdorffer, 1990; Hauser and Kurland, 1975; Sofijanov, 1982]. Absence seizures may be the most common seizure type to go undetected. The prevalence of absence seizures is highest during the first 10 years of life and then drops dramatically to a very low level [Hauser and Kurland, 1975; Hertoft, 1963; Sato et al., 1976]. Absence seizures are more common in girls than in boys [Hertoft, 1963; Sato et al., 1976]. Typical absence seizures rarely start before the age of 2 years or after the teenage years [Sato et al., 1983; Sato, 1983]. In a study of 83 patients with absence seizures, Sato and co-workers [Sato et al., 1983] found the average age at onset was 3.8 years.
Absence seizures are classified as typical or atypical in type (Table 53-3). Typical absence seizures are short, rarely lasting over 30 seconds; as with other generalized seizures, they are never associated with an aura or postictal impairment. The sudden onset of impaired consciousness, usually associated with a blank facial appearance without other motor or behavioral phenomena, is characteristic. The degree of impairment of consciousness is variable. Some children remember virtually everything that is said during the seizure, whereas for others, the entire duration of the seizure is “time lost.”
| Clinical Seizure Type | EEG – Interictal | EEG – Ictal |
|---|---|---|
| Typical absence |
Although the absence seizure is commonly thought to consist only of staring, in fact the behavioral changes associated with the seizure type usually are more complex. Most absence seizures are accompanied by motor, behavioral, or autonomic phenomena, and seizures characterized by only staring and altered consciousness are unusual. Penry et al. [1975] reviewed 374 absence seizures recorded on videotape from 48 patients and found simple absences, characterized by staring and cessation of activities, to constitute only 9.4 percent of the seizures. A majority of the patients had other clinical manifestations.
Automatisms, semipurposeful behaviors of which the patient is unaware and subsequently cannot recall, are very common with absence seizures [Holmes et al., 1987; Penry et al., 1975]. They either may be perseverative, reflecting continuation of preictal activities, or may arise de novo. Simple behaviors, such as rubbing the face or hands, licking the lips, chewing, grimacing, scratching, or fumbling with clothes, tend to be de novo automatisms. Complex activities, such as dealing cards, moving a chess piece, or handling a toy, are generally perseverative. Speech, if it occurs during the seizure, usually is perseverative and may be slow and slurred, but also may be totally normal [McKeever et al., 1983]. The longer the absence seizure lasts, the more likely automatisms are to occur [Penry et al., 1975].

