CHAPTER 29 Extracellular Matrix Molecules
Although the extracellular matrix is composed of only five classes of macromolecules—collagens, elastin, proteoglycans, hyaluronan, and adhesive glycoproteins—it can take on a rich variety of different forms with vastly different mechanical properties. This is possible for two reasons. First, each of these classes of macromolecule comes in a number of variants (encoded by different genes or produced by alternative splicing), each with distinctive properties. Second, the cells that constitute the extracellular matrix are versatile with respect to secreting different proportions of these isoforms in different geometrical arrangements. As a result, the extracellular matrix in different tissues is adapted to particular functional requirements, which vary as widely as tendons, blood vessel walls, cartilage, bone, the vitreous body of the eye, and subcutaneous fat. Beyond providing mechanical support, the extracellular matrix also strongly influences embryonic development, provides pathways for cellular migration, provides essential survival signals, and sequesters important growth factors. This chapter introduces the macromolecules of the extracellular matrix.
Collagen
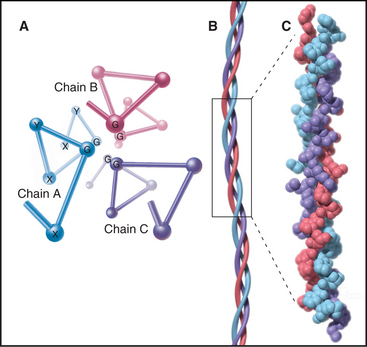
The defining feature of collagens is a rod-shaped domain composed of a triple helix of polypeptides (Fig. 29-1). Each polypeptide folds into a left-handed helix that repeats every third residue with the side chains on the outside. Three of these helices associate to form a triple helix that may be up to 420 nm long. The triple helical domains have a repeating amino acid sequence: glycine-X-Y, where X is most often proline and Y is most often hydroxyproline. The small glycine residues allow tight contact between the polypeptides in the core of the triple helix. Larger residues, even alanine, interfere with packing. Poly-l-proline has a strong tendency to form a left-handed helix like individual collagen chains but does not form a triple helix, owing to steric interference. The triple helix is most stable if all X residues are proline and all Y residues are hydroxyproline, but other residues at some of these positions are essential for collagen to assemble higher-order structures. (Despite their name, α-chains, the collagen polypeptides do not form α-helices.)
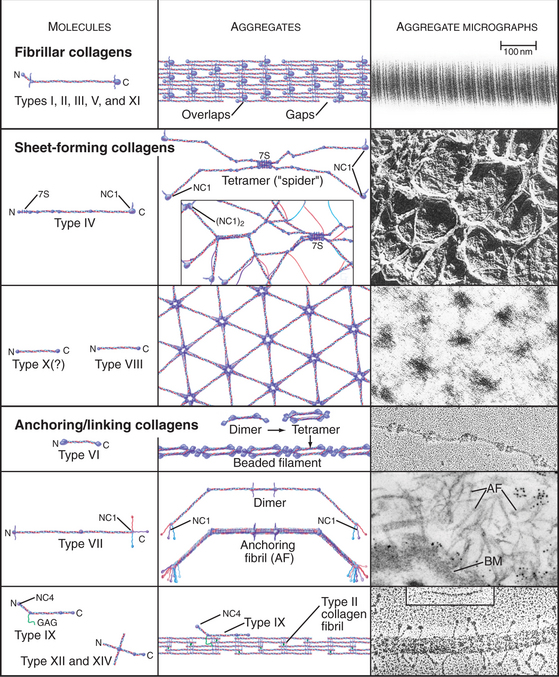
The collagen family is remarkably diverse. Humans have about 100 genes with collagen triple repeats, and more than 20 specialized collagen proteins have been characterized (Fig. 29-2 and Appendix 29-1).
Other proteins, including the extracellular enzyme acetylcholine esterase (see Fig. 11-8) and some cell surface receptors, have similar triple helical domains but are not classified as collagens. To be a collagen, a protein must also form fibrils or other assemblies in the extracellular matrix. Nematodes, which lack connective tissue, seem to have lost the genes for fibrillar collagens but have elaborated a family of 160 genes for collagens that form their cuticle.
The size and shape of collagens vary according to function. Collagens are named numerically (type I, type II, etc.) in the order of their discovery, a nomenclature that bears no relationship to their function. Appendix 29-1 groups collagens according to function. Polypeptides are called α-chains, and Roman numerals in their names correspond to their type number. Some collagens are homotrimers of three identical α-chains. Others are heterotrimers of two or three different α-chains. Some chains (e.g., [a1(II)]) are used in more than one type of collagen.
Fibrillar Collagens
Triple helical rod-shaped collagen molecules about 300 nm long self-associate to form banded fibrils (Fig. 29-2). Collagen fibrils provide tensile strength to tendons, ligaments, bones, and dense connective tissue, thus reinforcing most organs. They also form the scaffolding for cartilage and the vitreous body in the eye. Fibrillar collagens are widespread in nature and have been highly conserved during evolution, so the homologs from sponges to vertebrates are similar. Each fibrillar collagen can form homopolymers in vitro; but in vivo, most form heteropolymers with at least one other type of fibrillar collagen (Appendix 29-1). This mix of the fibrillar collagen subunits is one factor that regulates the size of collagen fibers. Proteoglycans also participate (Appendix 29-2).
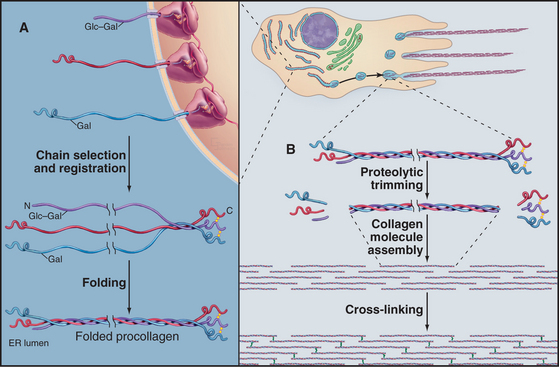
The biosynthesis and assembly of fibrillar collagens involve a remarkable number of posttranslational modifications, including several rounds of precise proteolytic cleavage, glycosylation, catalyzed folding, and chemical cross-linking (Fig. 29-4). The final product is a smooth fibril with staggered molecules that are cross-linked to their neighbors. These strong but flexible collagen fibrils reinforce all the tissues of the body, where they form a variety of higher-order structures. Loose connective tissue (see Fig. 32-1A) has an open network of individual fibrils or small bundles of fibrils that support the cells. In many tissues, the fibrils of type I and associated collagens aggregate to form the so-called collagen fibers that are visible by light microscopy (Fig. 29-3A). In extreme cases, such as in tendons, the extracellular matrix consists almost exclusively of tightly packed, parallel bundles of collagen fibers (see Fig. 32-1B). Layers of orthogonal collagen fibers make the transparent cornea through which one sees (Fig. 29-3C). In bone, type I collagen fibrils form regular layers reinforced by calcium phosphate crystals (see Fig. 32-5). In cartilage and the vitreous body of the eye, type II collagen fibrils trap glycosaminoglycans and proteoglycans, which retain enough water for the matrix to resist compression (see Fig. 32-3) and, in the case of the eye, to provide an optically clear path for light.

Biosynthesis and Assembly of Fibrillar Collagens
All fibrillar collagens are most likely to be produced by similar mechanisms, but type I collagen has been studied the most extensively. Type I collagen is synthesized and secreted by fibroblasts, using the exocytic pathway that is employed for other secretory proteins (see Chapter 21), but the biosynthesis of collagen is noteworthy for the extensive number of processing steps required to prepare the protein for assembly in the extracellular matrix.
The initial transcript, referred to as preprocollagen, translocates into the lumen of the rough endoplasmic reticulum, where intracellular processing begins (Fig. 29-4). First, removal of the N-terminal signal sequence yields procollagen with unfolded α-chains with N- and C-terminal nonhelical propeptides. Second, enzymes hydroxylate some prolines and lysines. Third, enzymes add sugars (gal-glu or gal) to the delta-carbon of some lysines, by a mechanism distinct from the typical glycosylation of asparagine or serine.
Procollagen passes through the Golgi apparatus and moves in vesicles to the cell surface, where it is secreted. Some cells have specialized collagen assembly sites (Fig. 29-4). Like ships laying down communication cables on the ocean floor, fibroblasts help to determine the arrangement of collagen fibrils as they move through tissues (Fig. 29-3C).
Outside the cell, proteolytic enzymes—procollagen proteases—cleave the propeptides from the triple helical domain, forming the mature collagen molecule (formerly called tropocollagen). Relieved of its inhibitory propeptides, collagen self-assembles into fibrils by a classical entropy-driven process (Fig. 29-5). Adjacent collagen molecules are staggered by 67 nm, so a 35-nm gap is required between the ends of the collagen molecules (five staggers at 67 nm = 335 nm = one molecular length of 300 nm + a 35-nm gap).
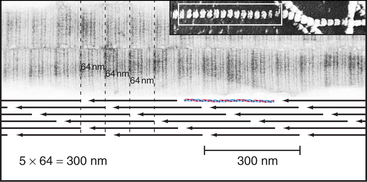
Figure 29-5 structure of collagen fibrils. Electron micrographs and drawing of molecular packing.
(Micrographs courtesy of Alan Hodges, Marine Biological Laboratory, Woods Hole, Massa-chusetts.)
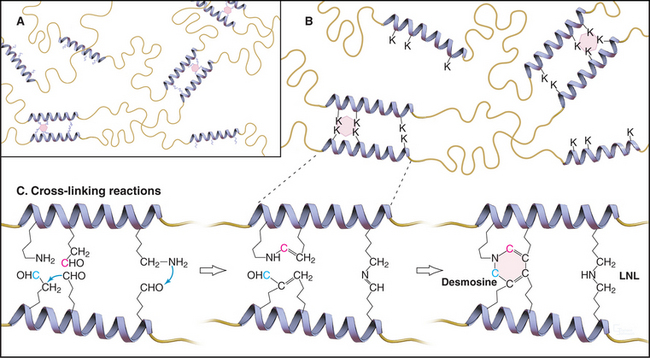
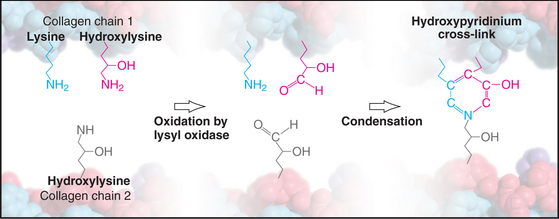
Weak, noncovalent bonds between collagen molecules specify the self-assembly of fibrils but provide little tensile strength, so covalent cross-linking is required for reinforcement. For most fibrillar collagens, the enzyme lysyl oxidase catalyzes the formation of covalent bonds between the ends of collagen molecules (Figs. 29-4 and 29-6). The enzyme oxidizes the e amino groups of selected lysines and hydroxylysines to aldehydes. These aldehydes react spontaneously with nearby lysine and hydroxylysine side chains to form a variety of covalent cross-links between two or three polypeptides. Disulfide bonds, rather than modified lysine side chains, cross-link type III collagen fibrils. Covalent bonds between the inextensible triple helices give mature collagen fibrils their great tensile strength.
Point mutations or deletions in collagen genes or lack of function of one of the enzymes that processes collagen (lysyl hydroxylase, lysyl oxidase, or procollagen proteases) can each cause defective collagen fibrils (Appendix 29-1). These defects cause a remarkable variety of deforming and even lethal human diseases: brittle bones (osteogenesis imperfecta), fragile cartilage (several forms of dwarfism), and weak connective tissue (Ehlers-Danlos syndrome). Chapter 34 covers these diseases in more detail.
Sheet-Forming Collagens
A second group of collagens polymerizes into sheets rather than fibrils (Fig. 29-2). These sheets surround organs, epithelia, or even whole animals. Six different human genes for type IV collagen encode proteins that form net-like polymers that assemble into the basal lamina beneath epithelia (Fig. 29-7) and around muscle and nerve cells. The concluding section of this chapter provides details about basal lamina structure, function, and diseases. Hexagonal nets of type VIII collagen form a special basement membrane (Descemet’s membrane) under the endothelium of the cornea. Related collagens form the cuticle of earthworms and the organic skeleton of sponges.
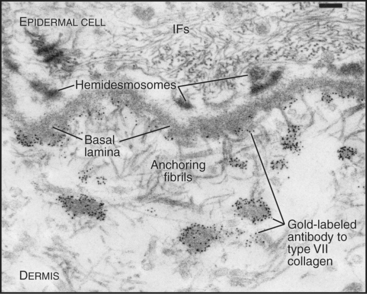
Figure 29-7 anchoring fibrils of type vii collagen. Electron micrograph of a thin section of human skin reacted with a gold-labeled antibody to the C-terminal domain of type VII collagen. Top to bottom, Basal epithelial cell with keratin intermediate filaments (IFs) attached to hemidesmosomes, which link to the basal lamina. Short fibrils of type VII collagen link the basal lamina to plaques in the dermis. Both ends of these bipolar fibrils (Fig. 29-2) are labeled with gold. Bar is 0.1 mm.
(Courtesy of D. R. Keene, Portland Shriners Hospital, Oregon.)
Linking Collagens
Connecting and anchoring collagens link fibrillar and sheet-forming collagens to other structures (Fig. 29-2). The type VII collagen homotrimer has an exceptionally long triple-helix domain with nonhelical domains at the N-terminus of each chain. Type VII molecules self-associate tail to tail to form antiparallel dimers. In the process, proteases remove the C-terminal globular domain. Several dimers associate laterally to form so-called anchoring fibrils that link type IV collagen of the basal lamina of stratified epithelia to plaques in the underlying connective tissue (Fig. 29-7). Mutations in type VII collagen cause both the dominant and recessive forms of a severe blistering disease, dystrophic epidermolysis bullosa. In heterozygotes, mutated chains interfere with the assembly of anchoring fibrils by normal type VII collagen chains. Without anchoring fibrils, the basal lamina adheres weakly to the connective tissue matrix. Even mild physical trauma to the skin causes the epithelium to pull away from the connective tissue, forming a blister. Related diseases are caused by mutations in intermediate filaments (see Fig. 35-6).
Type IX collagen links glycosaminoglycans to type II collagen fibrils (Fig. 29-2). This collagen heterotrimer has a serine modified with a glycosaminoglycan chain of variable length. Type IX collagens do not polymerize, but they associate laterally with type II collagen fibrils. The N-terminal helical segment and associated glycosaminoglycan project from the surface of the type II collagen fibril. In the vitreous body of the eye, these polysaccharides fill most of the extracellular space.
Elastic Fibers
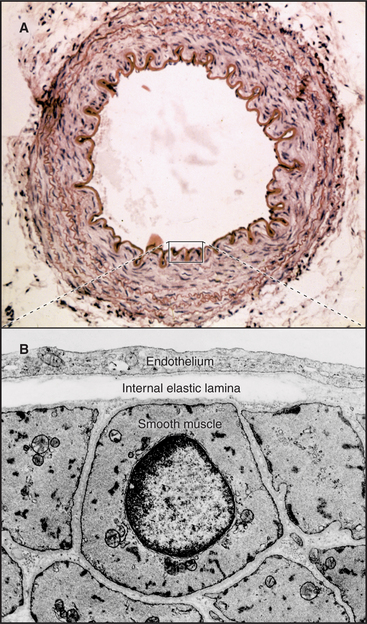
In contrast to inextensible collagen fibrils, elastic fibers are similar to rubber. They are found throughout the body but are prominent in the connective tissue of skin, the walls of arteries (Fig. 29-8), and the lung. They recoil passively after tissues are stretched. Every time the heart beats, pressurized blood flows into and stretches the large arteries. Energy stored in elastic fibers pushes blood through the circulation between heartbeats.
Elastic fibers are a composite material: A network of fibrillin microfibrils is embedded in an amorphous core of cross-linked elastin, which makes up 90% of the organic mass (Fig. 29-9). Fibroblasts produce both components. Loose bundles of microfibrils initiate assembly. A third protein, called fibulin, is required for elastin subunits to assemble between the micro-fibrils.
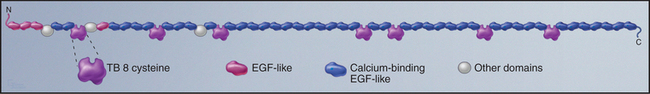
Fibrillin is the primordial component of elastic fibers, having arisen in Cnidarians (see Fig. 2-9). It is a long, floppy protein consisting of a tandem array of domains (Fig. 29-10). Humans have two fibrillin genes, and both fibrillin-1 and fibrillin-2 are components of 10-nm microfibrils, along with several glycoproteins. In microfibrils, fibrillin molecules interact head to tail with a reinforcing disulfide bond, but their arrangement is still being investigated. Microfibrils are about 100 times stiffer than elastin, and they stretch by rearrangement of molecules and domains rather than unfolding.
Elastin subunits are a family of closely related 60-kD proteins called tropoelastins, the products of alternative splicing from a single elastin gene. They have long sequences that are rich in hydrophobic residues interrupted by short sequences with pairs of lysines separated by two or three small amino acids (Fig. 29-11). Lysine-rich sequences are thought to form α-helices with pairs of lysines adjacent on the surface.