Electrolyte Disorders
Hyperkalemia
Hyperkalemia, defined as serum potassium level greater than 5.0 mEq/L, is the most dangerous acute electrolyte abnormality, potentially leading to life-threatening arrhythmias and death. Although hyperkalemia may have vague and varied symptoms, it is usually totally asymptomatic, with cardiac arrest as its first “symptom.”1 Thus the diagnosis of hyperkalemia depends on paying specific attention to risk factors for impaired potassium excretion, such as dehydration and renal failure, along with an awareness of medications that cause potassium retention. Evaluation of the electrocardiogram (ECG) of patients at risk for this electrolyte disturbance is critical. Hyperkalemia can be rapidly progressive, and lifesaving interventions must be instituted at the earliest suspicion of toxicity.
Upwards of 98% of potassium in the body is contained intracellularly, whereas less than 2% remains circulating in the blood. Serum potassium concentration is normally between 3.5 and 5.0 mEq/L and is tightly regulated by the kidney. In the healthy state, at least 90% of potassium excretion occurs through the kidney; in the renally impaired state, the gastrointestinal tract may account for roughly 25% of excretion. Hyperkalemia usually develops from impaired renal excretion or increased release from cells; however, in advanced chronic kidney disease or end-stage renal disease, dietary intake of potassium may be a significant factor in its development.2
The most common cause of hyperkalemia is spurious elevation due to hemolysis during or after the blood draw. Thus an ECG should be used to assess for true hyperkalemia while another sample is analyzed. Most causes of true hyperkalemia are due to release from cells or renal insufficiency. Renal failure is the most common cause of confirmed hyperkalemia and is often compounded by medications that further impair renal potassium handling. Box 125-1 organizes the most common causes of hyperkalemia. The presence of one of these conditions may be the lone historical clue in hyperkalemia.3
Clinical Features
Hyperkalemia remains a difficult clinical diagnosis to make on clinical grounds alone. It is not uncommon for a patient with mild to moderate hyperkalemia to be identified during routine blood sampling for an unrelated condition. Patients with moderate to severe hyperkalemia may have gastrointestinal effects such as nausea, vomiting, and diarrhea often in association with their underlying disease. Neuromuscular findings, including muscle cramps, generalized weakness, paresthesias, tetany, and focal or global paralysis, may be seen in patients with severe hyperkalemia. The signs and symptoms of progressive muscle weakness, paresthesias, dyspnea, and depressed deep tendon reflexes are neither sensitive nor specific, nor do they appear reliably with a particular serum potassium level.3 Patients with severe hypokalemia may present with hemodynamic instability and cardiac arrhythmias requiring immediate intervention.
Diagnostic Strategies
The ECG is helpful in making the diagnosis of hyperkalemia and can be used in unstable patients to initiate treatment (Figs. 125-1 to 125-3). Classic electrocardiographic changes—the peaked T wave, flattened p wave with prolonged PR interval or a totally absent P wave, wide QRS, and sine wave pattern, portending imminent cardiac arrest—have been well described as appearing sequentially with rising serum potassium levels.4,5 Peaked T waves usually appear as serum potassium levels exceed 5.5 to 6.5 mEq/L; P wave disappearance and PR prolongation are common with levels above 6.5 to 7.5 mEq/L; and QRS prolongation is seen with potassium levels above 7.0 to 8.0 mEq/L. Although these changes may occur in only half the patients, recognition of these patterns when they are present is vital to rapid diagnosis and initiation of lifesaving treatment.6 A serum potassium level above 5.0 mEq/L is diagnostic of hyperkalemia, but the value itself does not always predict electrocardiographic changes or the degree of cardiotoxicity. Subtle electrocardiographic changes consistent with hyperkalemia should not be the sole reason to treat a stable patient not likely to have an elevated potassium concentration until serum levels have returned.3
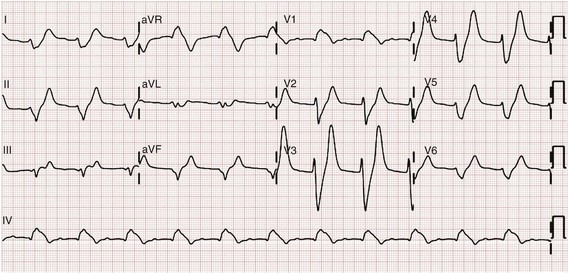
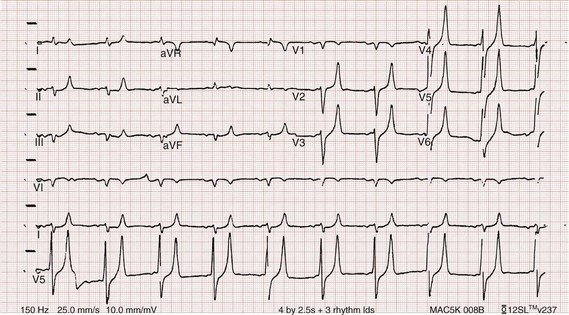
Figure 125-2 Hyperkalemia in the same patient as in Figure 125-1 after potassium-lowering therapy has begun. Tall peaked T waves, decreased P wave.
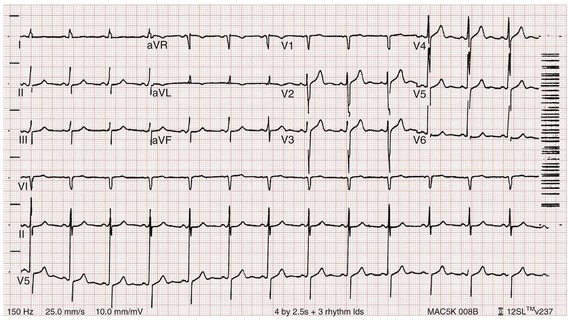
Figure 125-3 The same patient as in Figures 125-1 and 125-2 after dialysis. The electrocardiogram is now normal.
Management
Patients with suspected or known hyperkalemia require intravenous access and continuous cardiac monitoring. The treatment of hyperkalemia is based on the clinical scenario combined with the 12-lead ECG and the laboratory potassium value. The treatment strategy consists of three main steps: stabilization of the cardiac membrane, shifting of potassium into the cells, and then removal of potassium from the body. In patients who do not require urgent treatment, lowering of total body potassium may be the only step necessary. A variety of treatment options are considered for the acute management of hyperkalemia, including calcium, insulin, beta2-adrenergic agonists, sodium bicarbonate, resins, and dialysis (Table 125-1).
Table 125-1
| TREATMENT | MEDICATION | FEATURES |
| Stabilize cardiac membrane | Calcium chloride (10 mL, maximum 20 mL) or calcium gluconate (10-30 mL), IV push | For wide QRS, restores the electrical gradient; does not decrease serum potassium |
| Shift potassium into cells | Insulin, 10 units, IV push, combined with 100 mL of 50% dextrose, IV push High-dose nebulized albuterol by face mask (15-25 mg by continuous inhalation) Bicarbonate 50-100 mL Normal saline 100-250 mL |
If severely acidotic In conjunction with nephrologist if dialysis dependent |
| Remove potassium from the body | Hemodialysis Normal saline and furosemide Ion exchange resin |
Emergently in cardiac arrest, urgently in renal failure; may delay if renal function is normal In patients with rhabdomyolysis or tumor lysis syndrome with intact urine output Not effective acutely |
Intravenous calcium is used to stabilize the cardiac membrane by restoring the electrical gradient. Calcium increases the depolarization threshold and the calcium gradient across the cardiac membrane, quieting myocyte excitability and increasing cardiac conduction speed, thus narrowing the QRS. Calcium does not decrease serum potassium levels, and its effect is rapid but transient. The dose is one ampule, or 10 mL of 10% calcium chloride solution. Some authors prefer calcium gluconate rather than calcium chloride on the basis of the reduced risk of tissue necrosis should it extravasate at the injection site.4,5 More than 10 mL of calcium gluconate will often be required as it contains only one third the calcium contained in calcium chloride. Calcium gluconate is also preferred in pediatric cases as well as in more chronic, less emergent hyperkalemic patients when a slow infusion is desired.
Nebulized albuterol by face mask is effective in shifting potassium into cells by stimulation of the Na+,K+-ATPase pump.6 Nebulized albuterol begins to take measurable effect after 15 minutes and lowers the serum potassium level by 0.5 to 1 mEq/L, depending on the dose.7 The effective dose is at least four times higher than that typically used for bronchodilation.8 Mild tachycardia is the main side effect but is generally well tolerated. The combination of nebulized albuterol and insulin with glucose appears to be additive, lowering serum potassium by a mean of 1.2 mEq/L.9
Saline infusions also stimulate the Na+,K+-ATPase pump, and only a few hundred milliliters is required for beneficial effects. Saline infusions are given judiciously in anuric patients and usually in consultation with the nephrologist. Sodium bicarbonate is effective only in hyperkalemic patients who are acidotic and has no benefit when it is used for hyperkalemia in nonacidotic patients.10 Sodium bicarbonate buffers hydrogen ions extracellularly while shifting potassium intracellularly but should be reserved for patients with confirmed acidosis. Although intravenous magnesium also can drive potassium intracellularly, it should never be used in hyperkalemia as most patients with elevated potassium levels are also at risk for concomitant hypermagnesemia.
Potassium can best be removed from the body acutely by hemodialysis. Hemodialysis effectively and reliably decreases serum potassium levels by at least 1 mEq/L in the first hour and another 1 mEq/L during the next 2 hours.7,11 It is the only reliable method of potassium removal that has been experimentally studied and should be instituted early in the treatment of life-threatening hyperkalemia in patients with renal failure. In patients with intact renal function, medical management alone is usually sufficient, even in extreme cases, and hemodialysis may not be necessary unless multiple medical modalities fail. There are no randomized trials addressing the use of diuretics such as furosemide in the emergent management of hyperkalemia, but in cases such as rhabdomyolysis or tumor lysis syndrome, it may be appropriate to use a normal saline infusion supplemented by furosemide to enhance diuresis and potassium excretion in the urine.
Cation exchange resins, such as sodium polystyrene sulfonate (Kayexalate), have not been shown to decrease the serum potassium level within the first 4 hours of treatment and should not be used in the acute management of hyperkalemia.12
Hyperkalemia is seen in diabetic ketoacidosis (DKA), although most hyperkalemic patients with DKA are actually total body deficient of potassium. In this insulin-deficient and acidotic state, serum potassium levels rise because of cellular shifts. Simply treating the patient’s underlying DKA will also treat the hyperkalemia. In fact, the mainstay of treatment of DKA—fluids and insulin—closely mirrors the treatment of hyperkalemia itself.13
As hyperkalemia progresses, the end result will be cardiopulmonary arrest due to ventricular fibrillation, pulseless electrical activity, or asystole. In a known or suspected hyperkalemic arrest, an approach beginning with standard advanced cardiac life support combined with the use of multiple potassium-lowering medications is rational.14,15 Epinephrine has been shown to lower potassium by 0.25 mEq/L.16,17 Calcium chloride is given immediately by intravenous push, followed by insulin and glucose. Bicarbonate should be given by bolus dose if the patient is believed to be acidotic. Emergent hemodialysis is recommended if it is readily available. Hemodialysis through central venous access can be used during ongoing cardiopulmonary resuscitation to acutely lower the serum potassium level and may result in return of spontaneous circulation with intact neurologic status despite prolonged resuscitative efforts and failure of conventional medications and defibrillation.18
Hypokalemia
Hypokalemia is the most common electrolyte abnormality encountered in clinical practice. When it is defined as a value of less than 3.5 mEq/L, hypokalemia is found in more than 20% of hospitalized patients and in 10 to 40% of patients treated with thiazide diuretics in the outpatient setting.19 Although hypokalemia is usually asymptomatic, severe cardiac dysrhythmias and rhabdomyolysis can occur secondary to potassium’s effect on the heart and muscle.20 The five most common causes of hypokalemia are renal losses, increased nonrenal losses, decreased potassium intake, intracellular shift, and endocrine etiologies (Box 125-2). Hypokalemia is often seen in association with hypomagnesemia, and patients with low serum potassium levels should be assumed to be hypomagnesemic also.20–22
Increased excretion of potassium, especially coupled with poor intake, is the most common cause of hypokalemia, and patients receiving diuretics represent the single most common patient group encountered in clinical practice. Hypokalemia from thiazide diuretics occurs through increases in distal sodium delivery in the nephron and by activation of the renin-angiotensin-aldosterone system. Thiazide diuretics are more likely than loop or osmotic diuretics to cause hypokalemia, but both the thiazide and loop diuretics block chloride-associated sodium and increase delivery of sodium to the collecting tubules. Hypokalemia is a common adverse effect of treatment with diuretics and may cause fatal arrhythmias and increase the risk of digitalis toxicity.23 In addition to diuretics, other drugs and disorders can cause significant renal potassium losses, including hyperaldosteronism, steroid excess, metabolic acidosis, DKA, renal tubular acidosis, and alcohol consumption. Penicillin and its synthetic derivatives, when they are given in large doses, promote renal potassium excretion by increasing sodium delivery to the distal nephron.
Administration of insulin may cause a reduction in serum potassium because of insulin’s ability to stimulate the Na+,K+-ATPase pump and move potassium intracellularly; hypokalemia can be a dangerous complication with intentional overdoses of insulin and during treatment of DKA. Although most patients with DKA present with high-normal or mildly elevated serum potassium levels, patients are usually 2 to 3 mEq/kg body weight deficient in total body potassium.24 Failure to appreciate this total body deficit—and to not begin potassium infusion once significant hyperkalemia has been ruled out and intact renal function confirmed—may lead to otherwise unexplained arrhythmias or cardiac arrest in patients hours after their initial therapy has begun.
Dietary potassium deficiency should be considered in the severely malnourished patient and the chronic alcoholic.25 When poor potassium intake is combined with increased nonrenal losses, severe hypokalemia can result. Hypokalemia can also result from an acute shift of potassium from the extracellular compartment into cells. This is most commonly seen in patients with metabolic alkalosis, in patients with hyperventilation, and in those patients taking medications such as beta-agonists or decongestants. Beta receptor stimulation can lead to hypokalemia, especially in patients using repetitive and high doses of beta-agonists for chronic obstructive pulmonary disease or asthma. Albuterol-induced hypokalemia can occur even at normal therapeutic doses.26 A standard dose of nebulized albuterol reduces serum potassium by 0.2 to 0.4 mEq/L, and a second dose taken within 1 hour has the potential to reduce it by almost 1 mEq/L. Patients with starvation or near-starvation may suffer from hypokalemia when they are fed because insulin secretion and increased cellular uptake can cause an acute intracellular migration of potassium.
Clinical Features
Hypokalemia is usually asymptomatic but can be manifested with nonspecific complaints, including palpitations, skeletal muscle weakness, easy fatigability, depression, and muscle pain. Although short periods of mild potassium depletion are typically well tolerated in healthy individuals, severe potassium depletion can result in serious cardiovascular instability, neurologic dysfunction, glucose intolerance, gastrointestinal symptoms, and renal failure as well as affect the acid-base balance in the body.27 The likelihood of symptoms appears to correlate with the rapidity of the decrease in serum potassium. In patients without underlying heart disease, abnormalities in cardiac conduction are extremely unusual, even when the serum potassium concentration is below 3.0 mEq/L. Paresthesias, depressed deep tendon reflexes, fasciculations, muscle weakness, and confusion can occur when the serum potassium level is less than 2.5 mEq/L. However, in patients with cardiac ischemia or heart failure, even mild to moderate hypokalemia increases the likelihood of cardiac arrhythmias secondary to potassium’s effect on the action potential. The data linking hypokalemia with arrhythmias and cardiac arrest in acute myocardial infarction are fairly strong, but the direct myocardial stimulatory effects of increased circulating epinephrine is a possible confounder.20 Hypokalemia is an independent risk factor contributing to reduced survival of cardiac patients and increased incidence of arrhythmic death. On the basis of available evidence, it appears best to attempt to maintain a serum potassium concentration above 4.5 mEq/L in patients having an acute myocardial infarction. Hypokalemic patients can demonstrate first- and second-degree heart block, atrial fibrillation, ventricular fibrillation, and asystole. Hypokalemia can also promote metabolic acidosis.
Hypokalemic periodic paralysis is a rare disorder characterized by potentially fatal episodes of muscle weakness through the involvement of the respiratory muscles. Life-threatening cardiac arrhythmias are managed by restoration of serum potassium levels into the normal range.28
Diagnostic Strategies
Hypokalemia is rarely suspected on the basis of clinical presentation, and the diagnosis is typically made by measurement of the serum potassium concentration during routine laboratory study. If there is any suspicion for hypokalemia or a patient presents with generalized weakness, palpitations, or arrhythmias, an ECG should be obtained. Just as a tall-peaked T wave is characteristic of hyperkalemia, a flattened T wave can be seen in hypokalemia. U waves, which are small deflections after the T wave, may also be seen (Figs. 125-4 and 125-5). The real danger of hypokalemia is that it may also cause a prolonged QT interval. Once the QT interval becomes longer than 500 milliseconds, the risk of malignant ventricular arrhythmias and torsades de pointes increases dramatically.28 Hypokalemia is also notorious for causing nonspecific ST and T wave changes. In addition, prolonged potassium depletion of even modest proportion can provoke or exacerbate kidney injury or hypertension. A severe degree of hypokalemia with paralysis is a potentially life-threatening medical emergency; measurement of relative urinary potassium excretion and an assessment of the acid-base status might help narrow the differential diagnosis in the emergency setting.
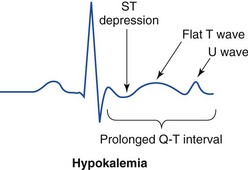
Figure 125-5 Electrocardiographic changes in hypokalemia.
Management
Potassium is an intracellular cation, so a low serum potassium level almost always reflects a significant total potassium deficit. When treating hypokalemia, one should remember that each 0.3 mEq potassium drop below normal correlates with an approximately 100 mEq total body deficit.29 Patients who have mild or moderate hypokalemia are usually asymptomatic or present with minor symptoms. These patients may only need oral potassium replacement therapy if they do not have nausea or vomiting as the cause of their hypokalemia. Oral replacement is available in liquid, powder, and tablet form. Potassium chloride is the most commonly used supplementation, and 40 to 60 mEq orally every 2 to 4 hours is typically well tolerated. If the cause of hypokalemia is not clear or the hypokalemia is severe and associated with profound weakness, obtain a spot urine potassium level before starting therapy to assess whether the patient’s kidneys are inappropriately wasting potassium from a renal or endocrine cause. Patients in whom severe hypokalemia is suspected should be immediately placed on a cardiac monitor and intravenous access secured.
Treatment of hypokalemia is essential in multiple populations of patients. Hypokalemia is arrhythmogenic, especially in the settings of acute myocardial infarction, high catecholamine states, and hypertrophied or dilated ventricles. Hypokalemia is an important independent risk factor for morbidity and mortality in patients with heart failure. Correction of serum potassium levels to between 4.0 and 5.0 mEq/L is important in these patients.30
Hypokalemia is associated with hypomagnesemia, and the severity of the hypokalemia correlates with a similar degree of hypomagnesemia.31 Magnesium replacement should usually accompany potassium repletion. Unless the patient receives at least 0.5 g/hr of magnesium sulfate along with potassium replacement, potassium will not move intracellularly and the patient will lose potassium through excretion.22 Correction of large potassium deficits may require several days, and oral and intravenous replacement can occur simultaneously.
Hypernatremia
Hypernatremia is defined as a serum sodium concentration above 145 mEq/ L and is usually associated with a poor prognosis. It is uncommon in previously normal patients, and in adults it is almost exclusively due to a total body water deficit.32 Most hypernatremic patients have either an impaired sense of thirst or no access to water. Thus elders, infants, patients in coma or with mental impairment, and those who are intubated and paralyzed are at highest risk for this disorder.33,34 Hypernatremia can be divided into three physiologic pairings (Box 125-3). Diabetes insipidus, a condition that results in insufficient production of or lack of response to antidiuretic hormone, can lead to life-threatening hypernatremia35 (Box 125-4).

Clinical Features
Hypernatremia is a disease seen predominantly in elders, but it can also be seen in patients who depend on others to provide them with water, including infants, intubated patients, and persons with mental debilitation.36,37 In addition, patients will also have multifactorial causes leading to severe hypernatremia.38 Patients may complain of polyuria or polydipsia or have obvious causes of extrarenal fluid losses; others may have no complaints at all.
Diagnostic Strategies
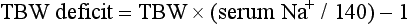
A patient’s total body water is usually calculated by multiplying the patient’s body weight in kilograms times 0.6. However, because of percentage body fat differences based on the age and sex of the patient, it is more accurate to use the correction factors listed in Table 125-2.
Table 125-2
| POPULATION | TOTAL BODY WATER |
| Children and adult men | Body weight (kg) × 0.6 |
| Adult women | Body weight (kg) × 0.5 |
| Elderly men | Body weight (kg) × 0.5 |
| Elderly women | Body weight (kg) × 0.45 |
Management
The treatment of hypernatremia has three interdependent goals: first, to quickly correct underlying shock, hypoperfusion, or significant hypovolemia with normal saline; second, to treat the underlying cause of hypernatremia, such as fever, vomiting, or diabetes insipidus; and third, to carefully lower the serum sodium level, usually by replacement of the body’s total water deficit.39 Until hypoperfusion and hypovolemia are corrected, homeostatic mechanisms for sodium balance will promote sodium resorption to maintain intravascular volume, even at the expense of the serum sodium concentration.
The rate of correction in hypernatremia is extremely important to minimize morbidity and mortality. In adult patients who have had hypernatremia during a short time as a result of sodium loading, “rapid correction” at 1 to 2 mEq/hr lowering of serum sodium appears relatively safe.39,40 However, most adult patients have hypernatremia during days to weeks. In this group of patients, serum sodium concentration should be slowly corrected at no more than 0.5 mEq/hr or 10 to 12 mEq/day.
Normal saline can typically be started for volume replacement until the patient is hemodynamically stable and then changed to half-normal saline at 100 mL/hr once vital signs have normalized. The treatment of central diabetes insipidus with desmopressin (DDAVP) is an effective means of improving polyuria and hypernatremia; initial doses in the acute setting range from 1 to 2 µg.33
Hyponatremia
Hyponatremia, defined as serum sodium concentration of less than 135 mEq/L, is the second most common electrolyte abnormality encountered in clinical practice.37 It is important to recognize hyponatremia because of its potential morbidity and also because it can be a marker of underlying disease. The most common causes of severe hyponatremia in adults are therapy with thiazides, the postoperative state including transurethral prostatectomy, the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), polydipsia in psychiatric patients, and unintentional water intoxication. Gastrointestinal fluid loss, ingestion of overly dilute formula, accidental ingestion of excessive water, and receipt of multiple tap-water enemas are the main causes of severe hyponatremia in infants and children. Most patients presenting to the emergency department (ED) with hyponatremia are asymptomatic and do not require emergent therapy. If symptoms are present, they are typically based on the degree of hyponatremia and how acutely the hyponatremia developed. Symptoms range from headache, nausea, and vomiting to confusion, seizures, and coma. There are two groups of hyponatremic patients that will require treatment with either normal saline or hypertonic saline: (1) severe but asymptomatic hyponatremia with a sodium level of 110 mEq/L or less and (2) acute symptomatic hyponatremia with a sodium level below 120 mEq/L.
Central nervous system (CNS) damage due to hyponatremia may be caused by cerebral edema and increased intracranial pressure, by osmotic fluid shifts during overly aggressive treatment, or by both. When they are subjected to a hyponatremic environment, neurons become depleted of sodium and potassium in an attempt to limit their own osmolarity to prevent intracellular fluid shifts that would lead to cerebral edema. If fluid therapy raises extracellular sodium levels too quickly, fluids shift out of neurons and diffuse demyelination may occur, leading to flaccid paralysis and often death due to a syndrome most commonly referred to as central pontine myelinolysis, although it is more accurately labeled the osmotic demyelinating syndrome.41
Although hyponatremia has many causes, they fall into four general categories: pseudohyponatremia, hyponatremia with dehydration and decreased extracellular volume, hyponatremia with increased extracellular volume, and euvolemic hyponatremia with increased total body water (Box 125-5).